
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA metal–organic framework/polymer derived catalyst containing single-atom nickel species for electrocatalysis†
Shuliang
Yang‡
a,
Jie
Zhang‡
 ab,
Li
Peng
c,
Mehrdad
Asgari
a,
Dragos
Stoian
a,
Ilia
Kochetygov
a,
Wen
Luo
*ab,
Emad
Oveisi
d,
Olga
Trukhina
a,
Adam H.
Clark
e,
Daniel T.
Sun
a and
Wendy L.
Queen
*a
ab,
Li
Peng
c,
Mehrdad
Asgari
a,
Dragos
Stoian
a,
Ilia
Kochetygov
a,
Wen
Luo
*ab,
Emad
Oveisi
d,
Olga
Trukhina
a,
Adam H.
Clark
e,
Daniel T.
Sun
a and
Wendy L.
Queen
*a
aInstitute of Chemical Sciences and Engineering, École Polytechnique Fédérale de Lausanne (EPFL), EPFL-ISIC-Valais, Sion, 1950, Switzerland. E-mail: wendy.queen@epfl.ch
bEmpa Materials Science & Technology, CH-8600 Dübendorf, Switzerland. E-mail: wen.luo@epfl.ch
cCollege of Chemistry and Chemical Engineering, Xiamen University, Xiamen, 361005, China
dInterdiciplinary Center for Electron Microscopy, École Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland
ePaul Scherrer Institute, Forschungsstrasse 111, 5232 Villigen, Switzerland
First published on 25th September 2020
Abstract
While metal–organic frameworks (MOF) alone offer a wide range of structural tunability, the formation of composites, through the introduction of other non-native species, like polymers, can further broaden their structure/property spectrum. Here we demonstrate that a polymer, placed inside the MOF pores, can support the collapsible MOF and help inhibit the aggregation of nickel during pyrolysis; this leads to the formation of single atom nickel species in the resulting nitrogen doped carbons, and dramatically improves the activity, CO selectivity and stability in electrochemical CO2 reduction reaction. Considering the vast number of multifarious MOFs and polymers to choose from, we believe this strategy can open up more possibilities in the field of catalyst design, and further contribute to the already expansive set of MOF applications.
Introduction
During the last three decades, metal–organic frameworks (MOFs) have demonstrated applications in many areas coupled to catalysis, gas adsorption/separation, water remediation, sensing, drug delivery and so on. This is a direct result of their vast structural and chemical tunability, unprecedented porosity, diverse set of potential topologies, etc.1–7 Despite this, the use of MOFs directly in the field of electrochemical catalysis of reduction reactions remains elusive,8 a result of the relatively weak coordination bond and reducible metal species found in MOF structures.9–11 To alleviate this, many have resorted to the pyrolysis of MOF precursors, which results in the formation of high surface area, metal doped carbons.12–14 These MOF-derived materials have not only been impactful in electrochemical catalysis, but have also found use in photocatalysis, batteries, and supercapacitors, among others.15–19Unfortunately, the list of MOFs employed as precursors for pyrolysis is to date not extensive due to several structural aspects. First, many MOFs are constructed by carbon-based ligands with oxygen-coordination sites, and most of the oxygen naturally exits the structure during pyrolysis in the form of CO2.20 This, combined with very low densities of other types of potential metal-coordinating species, such as nitrogen, limits accessible lone pairs on the ligand backbone. As such, many MOF structures have limited capacity to inhibit metal aggregation during pyrolysis. This leads to MOF-derived carbons with nanoparticles (NPs) rather than small clusters or single-atom sites, a feature that effectively reduces the number of active sites limiting the activity and longevity of the resulting catalysts.
To date, there are a few MOF precursors that are regularly employed to make MOF-derived carbons, such as ZIF-8 and ZIF-67.21–24 ZIFs, a subset of MOFs, are constructed by imidazole ligands having a relatively high density of N-based coordination sites.25 However, ZIFs are predominately constructed by Zn and Co cations limiting the type of the metallic species that are subsequently incorporated into the carbons.26 To expand the list of potential pyrolysis precursors, one could make a number of structural modifications to MOFs that might improve metal dispersion and hence the resulting performance of MOF-derived catalysts. For instance, ligands having higher densities of non-oxygen coordinating species could be designed. However, after the painstaking task of ligand synthesis, one must also elucidate the conditions for MOF formation. Such a laborious effort is unappealing for one who aims to subsequently destroy the material.27,28 Rather than making new ligands and their corresponding MOFs, another alternative and effective strategy to increase the density of Lewis base functionality is to introduce a second component that can help to immobilize metal species during thermal treatment.29–32 For instance, our group recently introduced nitrogen-containing polymers into MOF channels,33–37 and demonstrated that these can inhibit the aggregation of Pd NPs during thermal catalysis, an effort that was found to extend the catalyst lifetime significantly.38 The results of this work inspired us to test whether such polymers can also improve metal dispersion and stability during MOF pyrolysis. We hypothesize that such an effort can help to extend the scope of MOF materials employed as precursors, thereby significantly broadening the structural features and compositions of the resulting carbons.39,40
As a promising process to help achieve a carbon neutral energy cycle, the electrochemically driven CO2 reduction reaction (CO2RR) has attracted much attention; however, this process is still suffering from the lack of efficient electrocatalysts.41,42 Although some MOFs and MOF-derived catalysts have been developed for CO2RR, their selectivity and stability require further improvement.43,44
Herein, we show that MOF-polymer composites are a platform for the preparation of MOF-derived carbons having highly dispersed metal species. The introduced polymer not only greatly improves metal dispersion, but also the accessible surface area, and thus provides better electrochemical activity for CO2RR than the parent MOF. If one considers the plethora of MOFs and polymers to choose from, this approach to the design of porous carbons, could have a host of potential applications coupled to energy conversion and storage. It should be noted that during the preparation of this work, a MOF-polymer derived carbon was employed for CO2RR.32 In the present manuscript, we note the polymer introduction is easy, does not require MOF activation or excessive amounts of monomer, and upon polymer introduction, the surface area of the composite is markedly improved because the polymer serves as a support for the MOF walls.
Results and discussion
The preparation method is shown in Scheme 1. First, a MOF known as Ni2(NDISA) was prepared using previously reported procedures, using nickel nitrate hexahydrate and naphthalene diimide salicylic acid (NDISA) as the metal salt and organic linker, respectively.45 After collecting the as-synthesized MOF, polydopamine (PDA) was introduced into the MOF using an in situ aerobic oxidation reaction converting the dopamine monomer into PDA to form Ni2(NDISA)–PDA. It was hypothesized that the polymerization process, which inhibits the collapse of the mesoporous MOF channels33 during solvent removal, might also lead to a carbon having higher surface areas. Finally, melamine was pyrolyzed in a separate boat, placed downstream from the Ni2(NDISA)–PDA sample. Nitrogen-rich precursors, like melamine, evolve NH3 gas during pyrolysis and hence can be used to provide extra nitrogen species to the MOF sample during high temperature treatment.46,47 In light of the low nitrogen content in free-base dopamine (4.54 atom%) and the NDISA ligand (3.70 atom%), melamine (40.0 atom%) was introduced as an extra nitrogen source to help further stabilize the nickel species during heat treatment. Next, all three samples including Ni2(NDISA), Ni2(NDISA)–PDA, and Ni2(NDISA)–PDA with melamine underwent pyrolysis. The resulting materials are etched with 3 mol L−1 HCl aqueous solution for 12 h to remove the Ni particles on the surface,48 and the final nickel-containing nitrogen-doped-carbon catalysts are denoted as Ni/NC, Ni/NC-D, and Ni/N-CNTs, respectively. It is noted that the decomposition of melamine produces reducing gases, including NH3 and H2, that facilitated the growth of carbon nanotubes (CNTs) during the pyrolysis of Ni2(NDISA)–PDA.49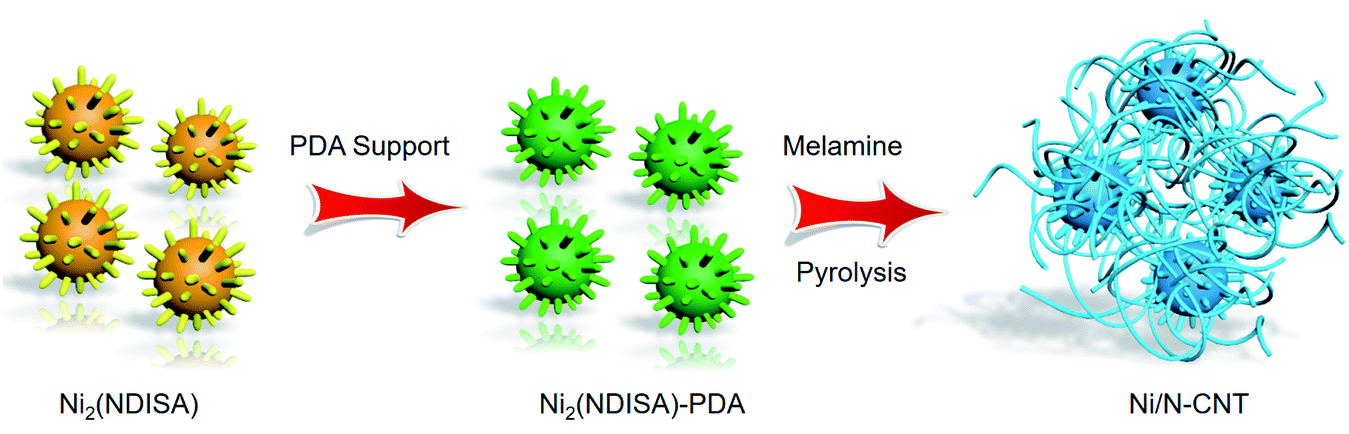 | ||
| Scheme 1 The preparation process used to prepare Ni/N-CNTs composites. | ||
Before pyrolysis, the MOF and MOF composite were characterized. Analysis of the X-ray diffraction (XRD) pattern of the as-prepared Ni2(NDISA) matches well with the simulated one, confirming successful preparation of the MOF (Fig. S1†). Scanning electron microscopy (SEM) images of the Ni2(NDISA) show a spherical chestnut-like morphology with lots of needle-like nanorods assembled on the surface (Fig. S2†). Due to the collapse of mesoporous Ni2(NDISA), its surface area, determined via N2 adsorption measurements at 77 K, is only 246 m2 g−1 after solvent removal. This value is well-below the theoretical surface area of 2872 m2 g−1 determined by Zeo++ using a probe radii of 1.860 Å (Fig. S3a†).50 During the polymer insertion, Ni2(NDISA) turned from an earthy yellow color to black, which is indicative of the presence of the highly conjugated PDA (Fig. S4†). The XRD pattern of the Ni2(NDISA)–PDA is also consistent with the simulated powder pattern obtained from the crystal structure of Ni2(NDISA), implying that the material was well preserved during the polymerization reaction (Fig. S5†). FT-IR spectra also imply there is no structural difference between the parent MOF and MOF/polymer composite (Fig. S6†). Further, Raman spectroscopy, Fig. S7,† reveals that the composite spectrum is the sum of the features observed for PDA and Ni2(NDISA), and SEM images also show no change in crystal morphology (Fig. S8†). The BET surface area of Ni2(NDISA)–PDA, 1298 m2 g−1, is over five times that of the parent MOF, 246 m2 g−1 (Fig. S3a†). This is accompanied by an increase of pore volume from 0.152 cm3 g−1 to 0.816 cm3 g−1 after polymer addition. In addition, pore size distribution of Ni2(NDISA)–PDA indicates a peak centered at 3.02 nm, which is close to the theoretical value of 3.3 nm determined from the crystal structure (Fig. S3b†). The increased porosity is due to the polymer support, which inhibits deformation of the metal helices that is caused by π–π stacking interactions between the adjacent naphthalene groups of the NDISA ligands.33 In addition to an increase in surface area, the CO2 capacity is doubled, from 0.80 mmol g−1 to 1.50 mmol g−1 at approximately 1.2 bar, after the polymer loading (Fig. S9†). This observation is likely due to the presence of amine groups on the polydopamine backbone combined with increased accessible surface area.51 TGA analysis of the Ni2(NDISA)–PDA in air revealed that the polymer loading is ∼20.5 wt% (Fig. S10†).
Next, samples including Ni2(NDISA), Ni2(NDISA)–PDA, and Ni2(NDISA)–PDA/melamine, underwent pyrolysis at 1000 °C for 2 hours and acid etching, forming Ni/NC, Ni/NC-D, and Ni/N-CNTs, respectively. ICP-OES analysis shows the Ni loadings in the Ni/NC, Ni/NC-D and Ni/N-CNTs are 1.67, 0.80 and 2.36 wt%, respectively. XRD patterns of the three samples reveal low-intensity diffraction peaks associated with metallic nickel, which results from Ni NPs embedded inside the carbon layer (Fig. 1a).52 This was further confirmed by the TEM characterization (Fig. S11†). It is expected that because the Ni nanoparticles are protected from the etching acid (HCl) by the graphitic layers, that the NPs also cannot come into contact with the electrolyte/gas during catalytic reactions. Despite their unlikely role in the catalytic reaction, the Ni particles are conductive and thus can provide a pathway for electrons, enhancing the conductivity as observed in previous studies.53,54 Moreover, residual nickel nanoparticles in the carbon matrix can potentially act as a source for the formation of more single atom nickel sites during catalyst preparation if extra polymer and the second thermal treatment was adopted.55,56 In addition to residual NPs, high-angle annular dark-field (HAADF) STEM images and EDXS elemental maps clearly confirm the presence of single Ni atom sites in all three carbons (Fig. S12–S16† and Fig. 2). The BET surface areas are 164, 287, and 50 m2 g−1 for Ni/NC, Ni/NC-D and Ni/N-CNTs, respectively, and the materials have an irregular pore size distribution (Fig. S17†). It is noted that the highest surface area carbon is obtained with the help of the PDA support; this phenomenon is expected given that PDA inhibits the MOF collapse. It is likely that the decreased surface area of the Ni/N-CNT sample is due to the introduction of melamine, which promotes the formation of CNTs that are denser than the MOF-derived porous carbon. Raman spectra show the presence of D, G, and 2D bands at 1342, 1573, 2682 cm−1 and with ID/IG values of 0.75, 0.55, 0.58 for Ni/NC, Ni/NC-D and Ni/N-CNTs, respectively (Fig. 1b). The reduced ID/IG values indicate the degree of graphitization is enhanced in Ni/NC-D and Ni/N-CNTs; this structural feature can improve the electrical conductivity of carbon matrix facilitating electron transport for electrochemical applications.57
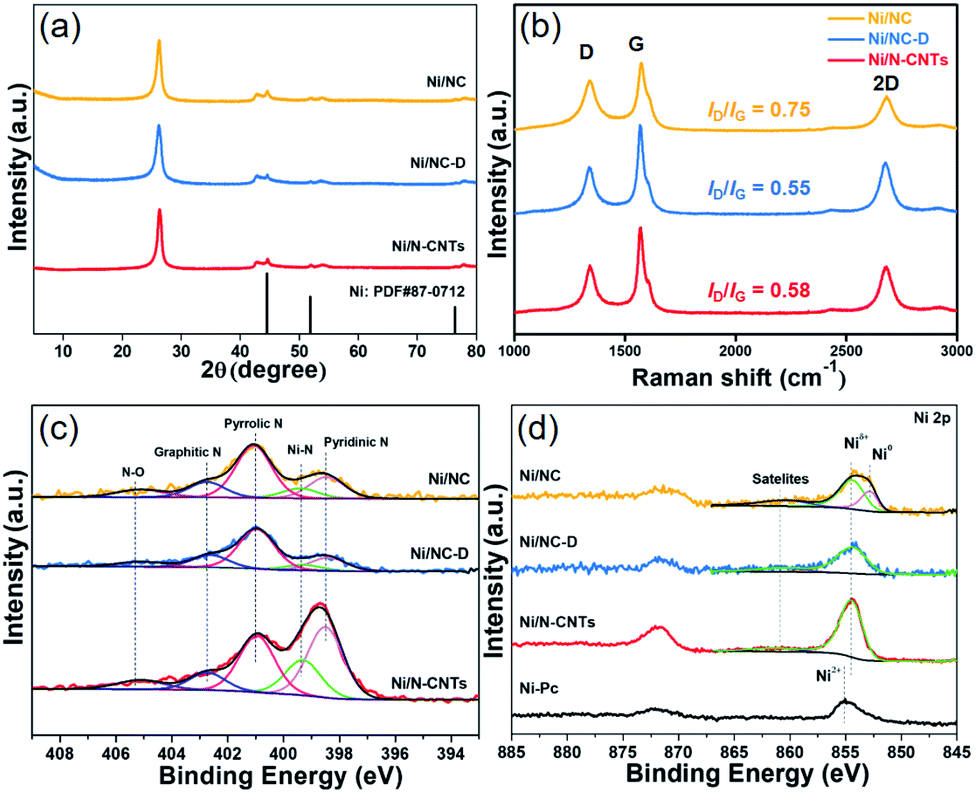 | ||
| Fig. 1 XRD patterns (a), Raman spectra (b), high resolution XPS N 1s (c), and high resolution XPS Ni 2p spectra (d) of the Ni/NC (orange), Ni/NC-D (blue) and the Ni/N-CNTs (red). | ||
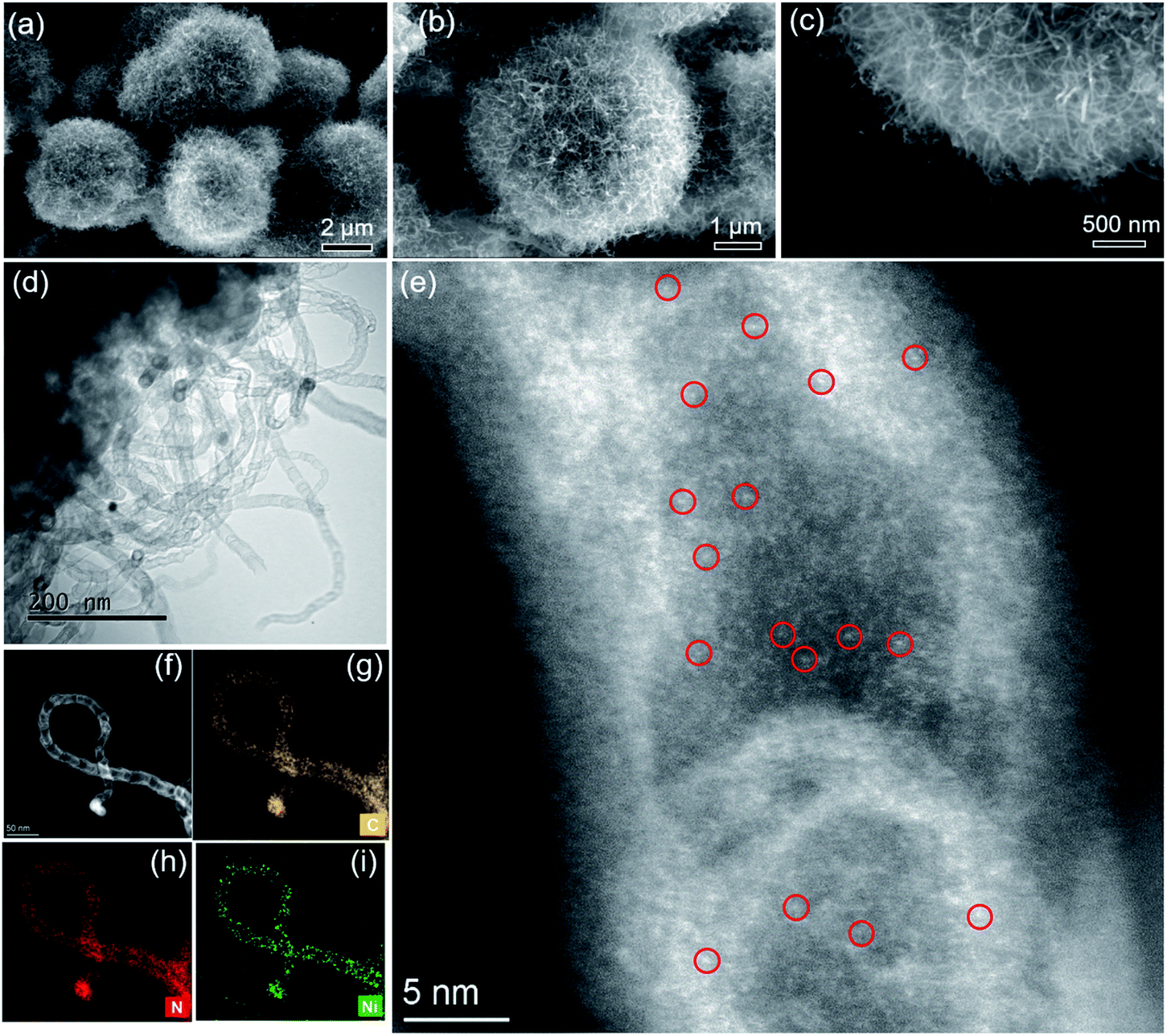 | ||
| Fig. 2 SEM (a–c), bright-field TEM (d) and HAADF-STEM images of Ni/N-CNTs (e and f). STEM-EDXS elemental maps (g–i) of C, N and Ni of Ni/N-CNTs sample shown in (f). Due to the high density of single atoms inside the N-CNTs, only a few metal single atoms are indicated by red circles in (e). | ||
As shown in Fig. 2, after the pyrolysis of Ni2(NDISA)–PDA/melamine and a further acid etching step meant to remove Ni NPs, the spherical structure of Ni2(NDISA)–PDA was maintained meanwhile large quantities of carbon nanotubes grow on the material's surface (Fig. 2a–c). Upon a closer look, the CNTs are found to be entangled, forming a three-dimensional network. As expected, no CNT growth was observed for the other two samples that exclude melamine (Fig. S12 and S14†).49 Further, after acid etching, a small amount of nickel nanoparticles (NPs) still remain in the Ni/N-CNTs (Fig. 2d and S18†); this is likely due to the dense graphitic carbon layer that protects the NPs.55,58,59 Upon closer inspection of the CNTs, a considerable amount of single atom nickel species are found distributed throughout the CNTs (Fig. 2e). Further, energy dispersive X-ray spectroscopy (EDXS) in scanning TEM mode (STEM) indicates that C, N and Ni are also well-dispersed throughout the Ni/N-CNTs (Fig. 2g–i and S16†).
The surface chemical composition and elemental valence states of Ni/NC, Ni/NC-D and Ni/N-CNTs were further investigated by XPS. For all three samples, high-resolution N 1s spectra revealed the presence of the porphyrin-like Ni–Nx moieties at 399.5 eV, as well as pyridinic (398.5 eV), pyrrolic (401.0 eV), graphitic (402.7 eV), and N–Ox (405.2 eV) species (Fig. 1c).60,61 The Ni–Nx fraction relative to total nitrogen content for Ni/N-CNTs (18.3%) was higher than that for Ni/NC (9.0%) and Ni/NC-D (7.0%). Further, Ni/N-CNTs exhibited much higher Ni and N content (0.9 at% and 6.2 at%, respectively) on the surface compared with Ni/NC and Ni/NC-D (Table S1†). While XPS has limitations with regard to probe depth, these results indicate that the nitrogen content in Ni/N-CNTs is indeed enhanced, a structural feature that can help stabilize Ni species during the pyrolysis and acid etching. Further, the binding energies of Ni 2p3/2 in Ni/NC-D and Ni/N-CNTs are approximately 854.5 eV, which is higher than metallic Ni0 (853.0 eV) but lower than Ni2+ in nickel phthalocyanine (NiPc, 855.0 eV); this suggests that low-valence state Ni species are present on the surface of these two samples (Fig. 1d).62 For the Ni/NC sample, a weak Ni0 peak can also be observed at 853.0 eV, which can be ascribed to the existence of non-coordinated Ni NPs (Fig. S11a†). These results indicate that the addition of polymer can efficiently improve the dispersion and the stabilization of Ni species.
Due to the high surface sensitivity of XPS (detection depth often less than 10 nm), we collected ex situ X-ray absorption near-edge spectroscopy (XANES) and extended X-ray absorption fine structure (EXAFS) data, using synchrotron radiation. Such techniques can provide a complete determination of the electronic structures and the coordination environment of the Ni atoms in the three samples. The XANES spectra (Ni K-edge measurements) of Ni/NC, Ni/NC-D, and Ni/N-CNTs and two references samples including Ni foil and NiPc are shown in Fig. 3a. The samples have a shift in the absorption edge to higher energies and an increase in the white-line intensity (denoted by the purple arrows) with the following trend: Ni/NC < Ni/NC-D < Ni/N-CNTs. The shift indicates a stepwise increase in the oxidation state of the Ni atoms from left to right and an intermediate oxidation state between 0 (metallic) and +2 (NiPc) for all three samples. It is known that higher oxidations states can be stabilized by the presence of electronegative nitrogen species,56 and hence it is no surprise that the shifting absorption edge correlates well with the increase N-content in the samples, Ni/NC < Ni/NC-D < Ni/N-CNTs. To further confirm this hypothesis, EXAFS spectra were obtained. Fig. 3b and Table S2† displays the Fourier transform of the phase uncorrected EXAFS for the three Ni-containing materials. According to the standards, the peaks at ca. 1.45 and 2.19 Å can be assigned to Ni–N and Ni–Ni bonds, respectively. While Ni/NC and Ni/NC-D display similar features as the metallic Ni foil, showing predominantly a Ni–Ni contribution and only a minor contribution from the Ni–N scattering path, the Ni/N-CNTs have a highly pronounced contribution from Ni–N. Fittings of the EXAFS data indicate an increase in the coordination number (C.N.) of the Ni–N shell from 0.44 (±0.29) < 1.38 (±0.52) < 3.13 (±0.25), and a reduction in the C.N. of the Ni–Ni shell from 8.63 (±0.29) > 6.55 (±0.32) > 1.74 (±0.19) for Ni/NC, Ni/NC-D, and Ni/N-CNTs, respectively. Note that X-ray absorption spectroscopy is a more bulk sensitive technique when compared to XPS, thus the former technique is able to resolve more metallic Ni signals from the Ni particles coated in the carbon layers and provides the coordination environment of both Ni particles and Ni single atoms.63 As XPS results show that no Ni0 can be detected on the surface of Ni/NC-D and Ni/N-CNTs and EXAFS results show that there is no clear Ni–C coordination, we anticipate that the single atom Ni is with a Ni–Nx configuration.64 While in case of Ni/NC, both Niδ+ and Ni0 were detected with XPS, we propose that the coordinate environment of surface Ni atoms for this sample is Ni–Nix–Ny. Overall, from the XPS, XANES and EXAFS results we can infer that the added polymer, which provides extra nitrogen to the MOF precursor, results in more single atom Ni species coordinated to N.
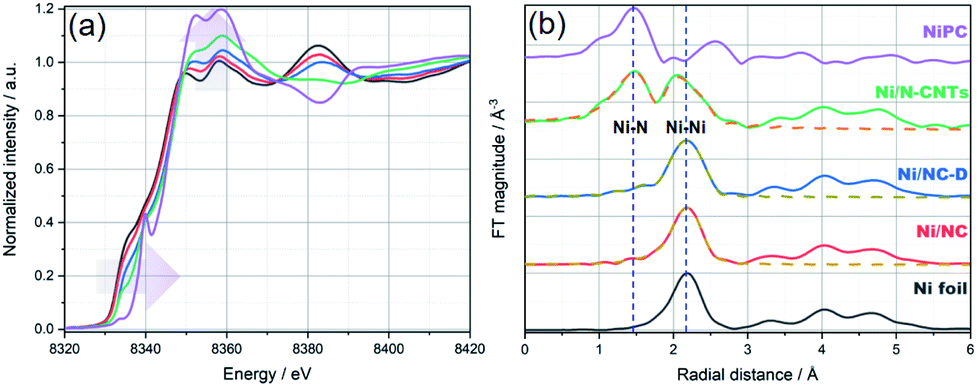 | ||
| Fig. 3 (a) Normalized XANES spectra and (b) Fourier transform (FT) of k2-weighted EXAFS (solid lines) and the fit results (dashed lines) for Ni foil (black), Ni/NC (red), Ni/NC-D (blue), Ni/N-CNTs (green), and Ni-PC (purple), respectively. The blue vertical dashed lines at ca. 1.45 and 2.19 Å in (b) are to mark the position of Ni–N and Ni–Ni bonds, respectively. Details on the EXAFS analysis are provided in the ESI.† | ||
During the last few years, single-atom nickel species coordinated to nitrogen doped carbon materials have been identified as highly selective catalysts for CO formation in the CO2RR.14,56,65 However, more often than not, sophisticated approaches are required for the formation of the single-atom catalysts.48 Considering that the Ni/N-CNT offers both a 3D conductive network and highly dispersed nickel species, CO2RR was chosen as a proof-of-concept application to demonstrate that polymers can lend to enhanced performance of MOF precursors employed in pyrolysis.
The CO2RR performance was evaluated in a batch reactor (H-cell) using 0.5 M KHCO3 as the electrolyte. The gaseous and liquid products were quantified using gas chromatography (GC) and high performance liquid chromatography (HPLC).66 Similar to previously reported single-atom Ni catalysts, only H2 and CO were detected as products in the examined potential range (Fig. 4a and S19†) for all three samples. At a low overpotential range (−0.4 to −0.8 V vs. RHE), the CO faradaic efficiency (FE) follows a trend of Ni/N-CNTs > Ni/NC-D > Ni/NC, which correlates with the average valency of the Ni (as well as the Ni–Nx content), indicating that the Ni single atom sites play the dominant role for electroreduction of CO2 to CO. Specifically, both Ni/NC-D and Ni/N-CNTs showed over 96% FE for CO at −0.67 V vs. RHE. In addition, we find that the CO selectivity of the Ni/N-CNTs sample is also higher than that of the sample without acid-etching (Fig. S20 and S21†), which further confirms the necessity of removing metallic Ni particles to expose only Ni–Nx sites for CO2RR.
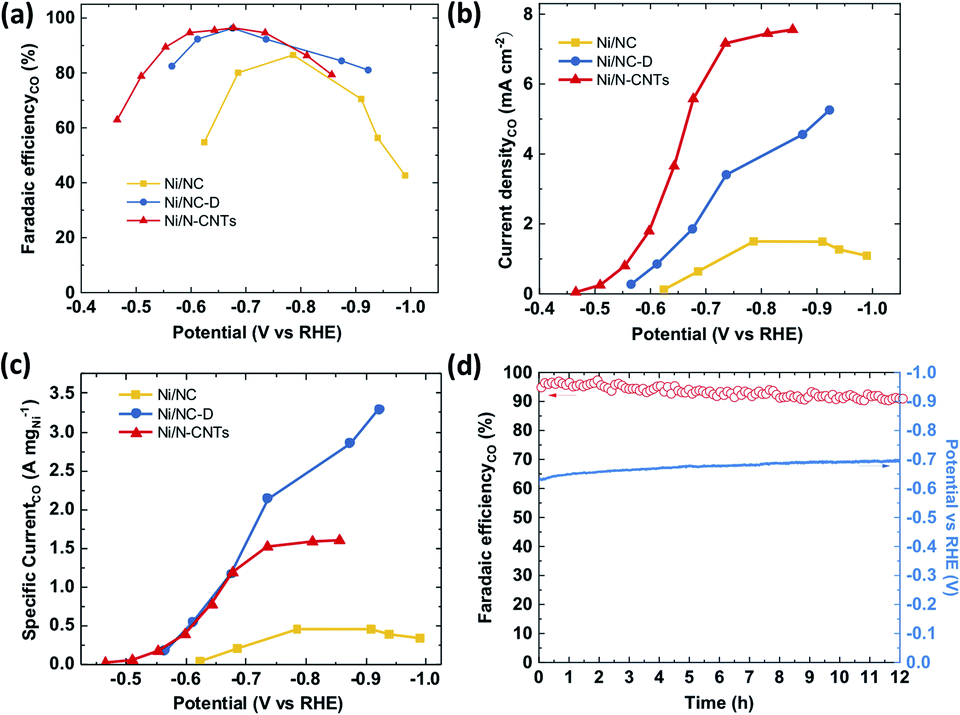 | ||
| Fig. 4 The performance of the electrochemical CO2 reduction on the as-prepared Ni/NC, Ni/NC-D, Ni/N-CNTs catalysts. (a) Faradaic efficiency, (b) geometric partial current density and (c) mass partial current density of CO as a function of potential; (d) faradaic efficiency and potential as a function of testing time for Ni/N-CNTs. | ||
For all three samples, both the CO partial current density and the mass activity increased with the decrease of applied potential (Fig. 4c and d). Ni/N-CNTs exhibited the highest CO partial current density compared with those of Ni/NC and Ni/NC-D (Fig. 4b), due to its high concentration of Ni–Nx sites. On the other hand, the Ni/NC sample derived from pure MOF had the lowest Ni–Nx content as well as the lowest Ni–N coordination number compared with the other two samples; thus this sample showed the worst CO selectivity and activity.63 Interestingly, at high overpotentials, Ni/NC-D exhibited an even better mass activity than the Ni/N-CNTs, likely because the high geometric activity of the Ni/N-CNTs leads to a more severe CO2 mass transfer limitation.67 During stability tests, Ni/N-CNTs maintained over 90% CO FE for more than 12 h and the Ni particles remained under the protection of graphite layers (Fig. S22†). In comparison, the CO FE for Ni/NC-D decreased slowly from 90% to 80% in 2 hours and Ni/NC showed rapid deterioration in 0.5 hour (Fig. S23†). These results unambiguously demonstrate that using MOF-polymer as precursor instead of pure MOF can dramatically improve the CO2RR performance, including activity, CO selectivity and stability. These properties might be further improved by adding extra N in the pyrolysis process to generate more stabilized Ni–Nx species.
Conclusions
In summary, using a Ni–MOF–polymer composite as a precursor to pyrolysis, we have prepared a catalyst that consists of highly dispersed Ni–Nx species. The nitrogen-containing polymer, polydopamine, improves the surface area of the resulting carbon and helps to minimize the formation of Ni–Ni bonds.It is noted, that carbons prepared only from MOF precursors, more often than not, offer minimal nitrogen content and hence low metal dispersion.59 We believe, using composite precursors is an easy and effective strategy to enhance the performance of MOF-derived carbons in various electrochemical applications. As a demonstration, we designed a Ni/N-CNT catalyst that was derived from a MOF/polymer composite; the resulting carbon offers high activity, selectivity and stability during electrochemical CO2 reduction to CO. This performance is dramatically better than the carbon derived only from the parent Ni–MOF, denoted as Ni/NC. Considering the vast number of MOFs and polymers that can be employed as composite building blocks, we believe this strategy can provide a platform for the future design of high performing catalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Swiss National Science Foundation under grant PYAPP2_160581 and PZ00P2_179989. This research was also part of the activities SCCER HeE, which is financially supported by Innosuisse-Swiss Innovation Agency. L. P. is supported by the National Natural Science Foundation of China (NSFC grant number 21903066). M. A. is financially supported by the Swiss Commission for Technology and Innovation (CTI). O. T. is financially supported by SNSF grant CRSK-2_190823. We also acknowledge the SuperXAS beamline at Paul Scherrer Institute (PSI) for beamtime allocation and Dr Maarten Nachtegaal and Dr Olga Safonova for their assistance on the beamline.Notes and references
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef.
- H. Furukawa, U. Müller and O. M. Yaghi, Angew. Chem., Int. Ed., 2015, 54, 3417–3430 CrossRef CAS.
- X. Han, S. Yang and M. Schröder, Nat. Rev. Chem., 2019, 3, 108–118 CrossRef CAS.
- J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1213–1214 RSC.
- M. Li, D. Li, M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2014, 114, 1343–1370 CrossRef CAS.
- Z. Chen, P. Li, R. Anderson, X. Wang, X. Zhang, L. Robison, L. R. Redfern, S. Moribe, T. Islamoglu, D. A. Gómez-Gualdrón, T. Yildirim, J. F. Stoddart and O. K. Farha, Science, 2020, 368, 297–303 CrossRef CAS.
- X. Xiao, L. Zou, H. Pang and Q. Xu, Chem. Soc. Rev., 2020, 49, 301–331 RSC.
- J. W. Maina, C. Pozo-Gonzalo, L. Kong, J. Schutz, M. Hill and L. F. Dumee, Mater. Horiz., 2017, 4, 345–361 RSC.
- H. Zhang, J. Li, Q. Tan, L. Lu, Z. Wang and G. Wu, Chem.–Eur. J., 2018, 24, 18137–18157 CrossRef CAS.
- N. C. Burtch, H. Jasuja and K. S. Walton, Chem. Rev., 2014, 114, 10575–10612 CrossRef CAS.
- Q. Zhu, D. Yang, H. Liu, X. Sun, C. Chen, J. Bi, J. Liu, H. Wu and B. Han, Angew. Chem., Int. Ed., 2020, 59, 8896–8901 CrossRef CAS.
- C. Wang, J. Kim, J. Tang, M. Kim, H. Lim, V. Malgras, J. You, Q. Xu, J. Li and Y. Yamauchi, Chem, 2020, 6, 19–40 CAS.
- W.-H. Lai, B.-W. Zhang, Z. Hu, X.-M. Qu, Y.-X. Jiang, Y.-X. Wang, J.-Z. Wang, H. K. Liu and S.-L. Chou, Adv. Funct. Mater., 2019, 29, 1807340 CrossRef.
- T. Sun, L. Xu, D. Wang and Y. Li, Nano Res., 2019, 12, 2067–2080 CrossRef CAS.
- S. Dang, Q.-L. Zhu and Q. Xu, Nat. Rev. Mater., 2017, 3, 17075 CrossRef.
- S. Zheng, X. Li, B. Yan, Q. Hu, Y. Xu, X. Xiao, H. Xue and H. Pang, Adv. Energy Mater., 2017, 7, 1602733 CrossRef.
- S. Fu, C. Zhu, J. Song, D. Du and Y. Lin, Adv. Energy Mater., 2017, 7, 1700363 CrossRef.
- X. Li, X. Yang, H. Xue, H. Pang and Q. Xu, EnergyChem, 2020, 2, 100027 CrossRef.
- X. Li and Q.-L. Zhu, EnergyChem, 2020, 2, 100033 CrossRef.
- K. Chen, J.-L. Ling and C.-D. Wu, Angew. Chem., Int. Ed., 2020, 59, 1925–1931 CrossRef CAS.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang, W. Huang, J. Yang, X. Hong, S. Wei, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 CrossRef CAS.
- S. Yang, L. Peng, S. Bulut and W. L. Queen, Chem.–Eur. J., 2019, 25, 2161–2178 CrossRef CAS.
- S. Wei, A. Li, J.-C. Liu, Z. Li, W. Chen, Y. Gong, Q. Zhang, W.-C. Cheong, Y. Wang, L. Zheng, H. Xiao, C. Chen, D. Wang, Q. Peng, L. Gu, X. Han, J. Li and Y. Li, Nat. Nanotechnol., 2018, 13, 856–861 CrossRef CAS.
- Y. Xiong, J. Dong, Z.-Q. Huang, P. Xin, W. Chen, Y. Wang, Z. Li, Z. Jin, W. Xing, Z. Zhuang, J. Ye, X. Wei, R. Cao, L. Gu, S. Sun, L. Zhuang, X. Chen, H. Yang, C. Chen, Q. Peng, C.-R. Chang, D. Wang and Y. Li, Nat. Nanotechnol., 2020, 15, 390–397 CrossRef CAS.
- X.-C. Huang, Y.-Y. Lin, J.-P. Zhang and X.-M. Chen, Angew. Chem., Int. Ed., 2006, 45, 1557–1559 CrossRef CAS.
- W. Xu, H. Chen, K. Jie, Z. Yang, T. Li and S. Dai, Angew. Chem., Int. Ed., 2019, 58, 5018–5022 CrossRef CAS.
- A. Kirchon, L. Feng, H. F. Drake, E. A. Joseph and H.-C. Zhou, Chem. Soc. Rev., 2018, 47, 8611–8638 RSC.
- Y. Lin, P. Liu, E. Velasco, G. Yao, Z. Tian, L. Zhang and L. Chen, Adv. Mater., 2019, 31, 1808193 CrossRef.
- M. Kalaj, K. C. Bentz, S. Ayala, J. M. Palomba, K. S. Barcus, Y. Katayama and S. M. Cohen, Chem. Rev., 2020, 120, 8267–8302 CrossRef CAS.
- T. Kitao, Y. Zhang, S. Kitagawa, B. Wang and T. Uemura, Chem. Soc. Rev., 2017, 46, 3108–3133 RSC.
- S. Li and F. Huo, Nanoscale, 2015, 7, 7482–7501 RSC.
- H.-L. Jiang, Y.-N. Gong, L. Jiao, Y. Qian, C.-Y. Pan, L. Zheng, X. Cai, B. Liu and S.-H. Yu, Angew. Chem., Int. Ed., 2020, 59, 2705–2709 CrossRef.
- L. Peng, S. Yang, S. Jawahery, S. M. Moosavi, A. J. Huckaba, M. Asgari, E. Oveisi, M. K. Nazeeruddin, B. Smit and W. L. Queen, J. Am. Chem. Soc., 2019, 141, 12397–12405 CrossRef CAS.
- D. T. Sun, L. Peng, W. S. Reeder, S. M. Moosavi, D. Tiana, D. K. Britt, E. Oveisi and W. L. Queen, ACS Cent. Sci., 2018, 4, 349–356 CrossRef CAS.
- D. T. Sun, N. Gasilova, S. Yang, E. Oveisi and W. L. Queen, J. Am. Chem. Soc., 2018, 140, 16697–16703 CrossRef CAS.
- S. Yang, L. Peng, O. A. Syzgantseva, O. Trukhina, I. Kochetygov, A. Justin, D. T. Sun, H. Abedini, M. A. Syzgantseva, E. Oveisi, G. Lu and W. L. Queen, J. Am. Chem. Soc., 2020, 142, 13415–13425 CrossRef CAS.
- S. Yang, V. V. Karve, A. Justin, I. Kochetygov, J. Espín, M. Asgari, O. Trukhina, D. T. Sun, L. Peng and W. L. Queen, Coord. Chem. Rev., 2021, 427, 213525 CrossRef CAS.
- V. V. Karve, D. T. Sun, O. Trukhina, S. Yang, E. Oveisi, J. Luterbacher and W. L. Queen, Green Chem., 2020, 22, 368–378 RSC.
- S. Yang, L. Peng, P. Huang, X. Wang, Y. Sun, C. Cao and W. Song, Angew. Chem., Int. Ed., 2016, 55, 4016–4020 CrossRef CAS.
- Y. Chen, S. Ji, S. Zhao, W. Chen, J. Dong, W.-C. Cheong, R. Shen, X. Wen, L. Zheng, A. I. Rykov, S. Cai, H. Tang, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Nat. Commun., 2018, 9, 5422 CrossRef CAS.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS.
- K. Zhao, Y. Liu, X. Quan, S. Chen and H. Yu, ACS Appl. Mater. Interfaces, 2017, 9, 5302–5311 CrossRef CAS.
- I. Hod, M. D. Sampson, P. Deria, C. P. Kubiak, O. K. Farha and J. T. Hupp, ACS Catal., 2015, 5, 6302–6309 CrossRef CAS.
- K. AlKaabi, C. R. Wade and M. Dincă, Chem, 2016, 1, 264–272 CAS.
- H. B. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- L. Zhao, Y. Zhang, L.-B. Huang, X.-Z. Liu, Q.-H. Zhang, C. He, Z.-Y. Wu, L.-J. Zhang, J. Wu, W. Yang, L. Gu, J.-S. Hu and L.-J. Wan, Nat. Commun., 2019, 10, 1278 CrossRef.
- C. Zhao, Y. Wang, Z. Li, W. Chen, Q. Xu, D. He, D. Xi, Q. Zhang, T. Yuan, Y. Qu, J. Yang, F. Zhou, Z. Yang, X. Wang, J. Wang, J. Luo, Y. Li, H. Duan, Y. Wu and Y. Li, Joule, 2018, 3, 584–594 CrossRef.
- M. Du, D. Song, A. Huang, R. Chen, D. Jin, K. Rui, C. Zhang, J. Zhu and W. Huang, Angew. Chem., Int. Ed., 2019, 58, 5307–5311 CrossRef CAS.
- T. F. Willems, C. H. Rycroft, M. Kazi, J. C. Meza and M. Haranczyk, Microporous Mesoporous Mater., 2012, 149, 134–141 CrossRef CAS.
- L. Peng, S. Yang, D. T. Sun, M. Asgari and W. L. Queen, Chem. Commun., 2018, 54, 10602–10605 RSC.
- R. V. Jagadeesh, K. Murugesan, A. S. Alshammari, H. Neumann, M.-M. Pohl, J. Radnik and M. Beller, Science, 2017, 358, 326–332 CrossRef CAS.
- S. Xin, Y.-G. Guo and L.-J. Wan, Acc. Chem. Res., 2012, 45, 1759–1769 CrossRef CAS.
- J. Wang, X. Yan, Z. Zhang, H. Ying, R. Guo, W. Yang and W. Q. Han, Adv. Funct. Mater., 2019, 29, 1904819 CrossRef.
- Q. Fan, P. Hou, C. Choi, T. S. Wu, S. Hong, F. Li, Y. L. Soo, P. Kang, Y. Jung and Z. Sun, Adv. Energy Mater., 2020, 10, 1903068 CrossRef CAS.
- J. Yang, Z. Qiu, C. Zhao, W. Wei, W. Chen, Z. Li, Y. Qu, J. Dong, J. Luo, Z. Li and Y. Wu, Angew. Chem., Int. Ed., 2018, 57, 14095–14100 CrossRef CAS.
- C. C. Hou, L. Zou and Q. Xu, Adv. Mater., 2019, 31, 1904689 CrossRef CAS.
- M. Jia, C. Choi, T.-S. Wu, C. Ma, P. Kang, H. Tao, Q. Fan, S. Hong, S. Liu, Y.-L. Soo, Y. Jung, J. Qiu and Z. Sun, Chem. Sci., 2018, 9, 8775–8780 RSC.
- L. Fan, P. F. Liu, X. Yan, L. Gu, Z. Z. Yang, H. G. Yang, S. Qiu and X. Yao, Nat. Commun., 2016, 7, 10667 CrossRef CAS.
- K. Artyushkova, B. Kiefer, B. Halevi, A. Knop-Gericke, R. Schlogl and P. Atanassov, Chem. Commun., 2013, 49, 2539–2541 RSC.
- W. Ju, A. Bagger, G.-P. Hao, A. S. Varela, I. Sinev, V. Bon, B. Roldan Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Nat. Commun., 2017, 8, 944 CrossRef.
- C. Yan, H. Li, Y. Ye, H. Wu, F. Cai, R. Si, J. Xiao, S. Miao, S. Xie, F. Yang, Y. Li, G. Wang and X. Bao, Energy Environ. Sci., 2018, 11, 1204–1210 RSC.
- X. Li, H. Rong, J. Zhang, D. Wang and Y. Li, Nano Res., 2020, 13, 1842–1855 CrossRef CAS.
- Y. J. Sa, H. Jung, D. Shin, H. Y. Jeong, S. Ringe, H. Kim, Y. J. Hwang and S. H. Joo, ACS Catal., 2020, 10, 10920–10931 CrossRef.
- Y. Cheng, S. Zhao, B. Johannessen, J.-P. Veder, M. Saunders, M. R. Rowles, M. Cheng, C. Liu, M. F. Chisholm, R. De Marco, H.-M. Cheng, S.-Z. Yang and S. P. Jiang, Adv. Mater., 2018, 30, 1706287 CrossRef.
- J. Zhang, W. Luo and A. Züttel, J. Mater. Chem. A, 2019, 7, 26285–26292 RSC.
- W. Luo, J. Zhang, M. Li and A. Züttel, ACS Catal., 2019, 9, 3783–3791 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed information for the synthesis and characterizations of all the materials. See DOI: 10.1039/d0sc04512h |
| ‡ These authors have contributed equally to this study. |
| This journal is © The Royal Society of Chemistry 2020 |