
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing enzymatic activity – a radical approach†
Neil C.
Taylor
a,
Gary
Hessman
 a,
Holger B.
Kramer‡
b and
Joanna F.
McGouran
*a
a,
Holger B.
Kramer‡
b and
Joanna F.
McGouran
*a
aSchool of Chemistry, Trinity College Dublin, Trinity Biomedical Sciences Institute, 152-160 Pearse St., Dublin 2, Ireland. E-mail: jmcgoura@tcd.ie
bDepartment of Physiology, Anatomy and Genetics, University of Oxford, Parks Road, Oxford, OX1 3PT, UK
First published on 6th February 2020
Abstract
Deubiquitinating enzymes (DUBs) are known to have numerous important interactions with the ubiquitin cascade and their dysregulation is associated with several diseases, including cancer and neurodegeneration. They are an important class of enzyme, and activity-based probes have been developed as an effective strategy to study them. Existing activity-based probes that target the active site of these enzymes work via nucleophilic mechanisms. We present the development of latent ubiquitin-based probes that target DUBs via a site selective, photoinitiated radical mechanism. This approach differs from existing photocrosslinking probes as it requires a free active site cysteine. In contrast to existing cysteine reactive probes, control over the timing of the enzyme–probe reaction is possible as the alkene warhead is completely inert under ambient conditions, even upon probe binding. The probe's reactivity has been demonstrated against recombinant DUBs and to capture endogenous DUB activity in cell lysate. This allows more finely resolved investigations of DUBs.
Introduction
Ubiquitin is a 76 amino acid protein which is added post-translationally to protein substrates to modulate their activity and interactions.1 Ubiquitination is orchestrated by the widely conserved E1, E2 and E3 enzymes that sequentially activate, conjugate and ligate the ubiquitin monomers to substrate proteins.2 Deubiquitinating enzymes (DUBs) possess ubiquitin C-terminal hydrolytic activity and are responsible for removal of ubiquitin from its conjugates.3 The human genome encodes for approximately 100 DUBs split into eight classes, seven of which are cysteine proteases.4,5 The repertoire of conjugation machinery and DUBs in eukaryotes, along with the differences in their relative promiscuity result in highly complex enzymatic networks that regulate numerous critical cellular events. Disruptions within these networks is associated with disorders including cancer, neurodegeneration and inflammation.4 Therefore, there is a demand to expand the molecular toolbox to study these enzymes, and activity-based probes (ABPs) continue to play a central role in this area.ABPs have been successful in identifying new DUB family members,6 DUB inhibitor assessment,7,8 characterisation of linkage selectivity9–14 and they have aided DUB crystallisation.15 Recent advances include cell permeable probes,16 methodologies to precisely identify labelling sites17 and probes resembling ubiquitin–substrate conjugates.18–22 In existing probes, a reactive C-terminal moiety is aligned with the active site cysteine upon binding and a covalent bond is immediately formed via nucleophilic attack. Therefore, there is no external control over the timing of adduct formation, even in cases of probes that are inert prior to enzyme binding.23
We present the development of a ubiquitin-based ABP that is inert under ambient conditions and can be activated upon exposure to UV light. The probe works via a radical mechanism within the enzyme active site. Initiators act as the primary radical source and abstract a hydrogen from the active site cysteine. The resulting thiyl radical reacts with the aligned alkene moiety of the probe affording a carbon-centred radical which likely abstracts a hydrogen from within the active-site. The result is a covalent bond between the C-terminus of the probe and the active site cysteine. The active site is expected to remain accessible even post-binding as DUBs typically act upon ubiquitinated substrates, leaving significant space proximal to our monoubiquitin probe. Selected crystal structures of monoubiquitin bound DUBs OTUB1, OTUB2 and UCHL3 were analysed to visualise this (ESI Fig. 1†).24–26 All structures analysed showed a cavity at the C-terminus of the bound ubiquitin. This mechanism allows for the establishment of a binding equilibrium between the probe and target enzymes followed by a short activation period, allowing for temporal control which is not achievable using existing cysteine reactive probes (Scheme 1). DUBs operate in several complex enzymatic networks and are known to be post translationally modified.4 Therefore, the ability to induce the formation of a probe–enzyme complex in vitro at specific times or after different external inputs could help offer a greater understanding of how these enzymes act within cells. This coupling reaction represents a promising and novel strategy to target the active form of these enzymes. It differs from existing photo crosslinking strategies as it is mechanism based with high selectivity.27 Although it has been used to form ubiquitin dimers and trimers,28 this is to our knowledge the first example of a thiol–ene reaction successfully employed in activity-based protein profiling. Furthermore, this approach is readily applicable to a wide variety of other enzyme classes beyond the ubiquitin-proteasome system and cysteine proteases.
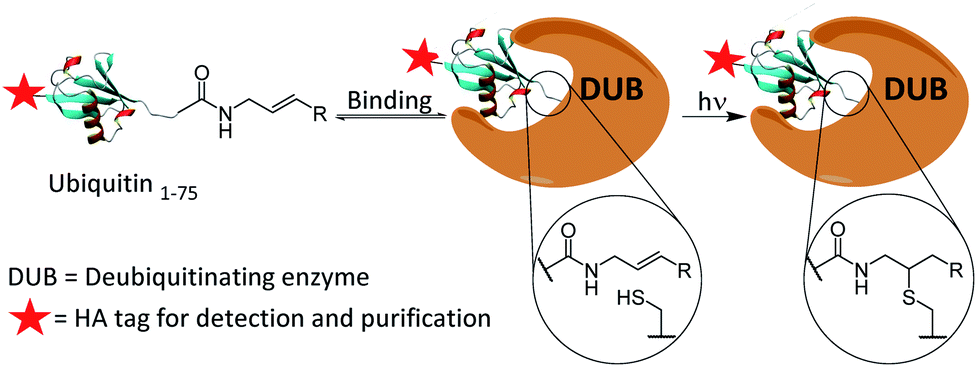 | ||
| Scheme 1 Proposed covalent bond formation of probes to DUBs via a thiol–ene reaction following the establishment of a binding equilibrium. | ||
Results and discussion
Firstly, the probe was generated using semi-synthetic techniques, employing an activated thioester intermediate as previously described.29 Its structure is comprised of a human influenza hemagglutinin (HA) tag and ubiquitin 1-75 (Ub75). The C-terminal glycine 76 residue that is present in wild-type ubiquitin is replaced with an alkene moiety, initial studies focused on a terminal alkene (Scheme 1, R = H).The intact probe was characterised using MALDI mass spectrometry and silver staining, with both showing a single protein of the expected molecular weight (Fig. 1A and C). LC-MS/MS located the allyl amide functionality to the C-terminus of the protein, confirming the structure of our probe (Fig. 1B).
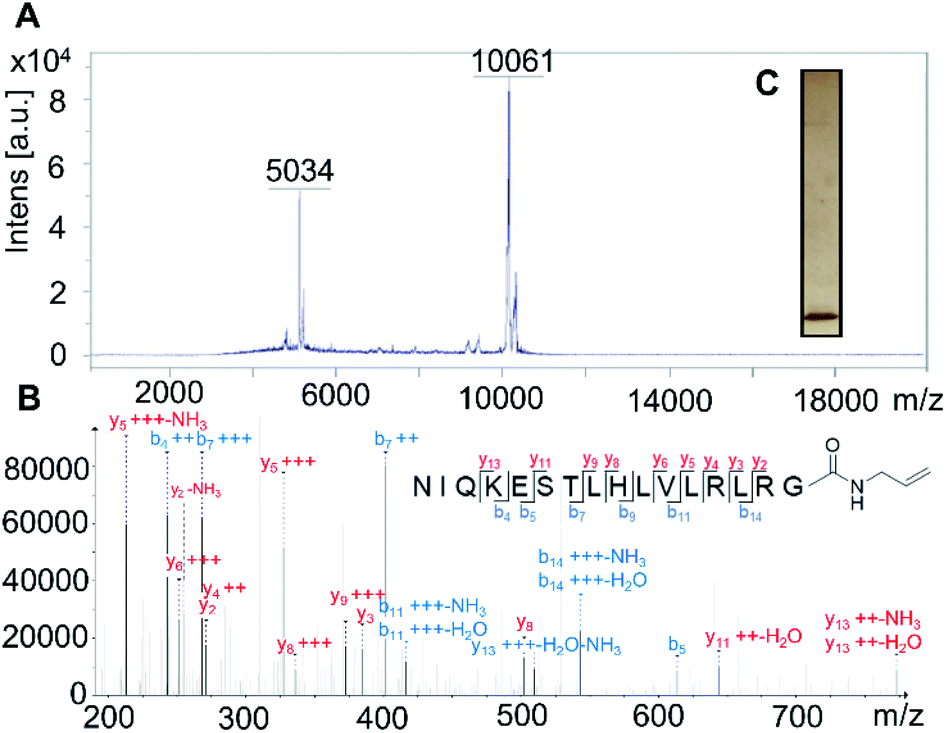 | ||
Fig. 1 (A) MS analysis of the intact probe 1 using MALDI, calculated m/z 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 062 (M + H)+ and 5032 (M + 2H)2+, found m/z 10061 (M + H)+ and 5034 (M + 2H)2+. (B) LC-MS/MS identification of Ub75 C-terminal peptide of probe 1. (C) Purified probe 1 was resolved using 12% SDS-PAGE and visualised using silver staining. 062 (M + H)+ and 5032 (M + 2H)2+, found m/z 10061 (M + H)+ and 5034 (M + 2H)2+. (B) LC-MS/MS identification of Ub75 C-terminal peptide of probe 1. (C) Purified probe 1 was resolved using 12% SDS-PAGE and visualised using silver staining. | ||
We next validated that probe 1 could react with DUBs. We tested with His-tagged, recombinant DUB OTUB1 (Fig. 2). Following a 60 minute preincubation at 37 °C, the radical initiator 2,2-dimethoxy-2-phenylacetophenone (DPAP) was added along with radical stabiliser 4′-methoyacetophenone (MAP). Samples were then degassed with N2 for 2 min prior to exposure to UV light (365 nm). The reaction was analysed by gel electrophoresis and visualised by silver staining, anti-HA and anti-His western blots (Fig. 2A, B and ESI 2†). After labelling, a new band corresponding to the expected molecular weight of the probe–enzyme adduct is observed in lane 5 of both gels, as indicated by the blue arrow. When OTUB1 was denatured using heat or SDS no new band is visible. This validates the need for a specific binding interaction between the probe and the active form of an enzyme for the reaction to occur. In-gel digestion of the new band confirmed the presence of both the probe and OTUB1 (ESI Fig. 2†). Further to this, we also demonstrated elucidation the Kd of our probe using OTUB1 as a model system. We derived a Kd of 7.8 μM and applied in combination with reversible inhibitors, this system could be a simple technique to evaluate their potency (ESI Fig. 3†).
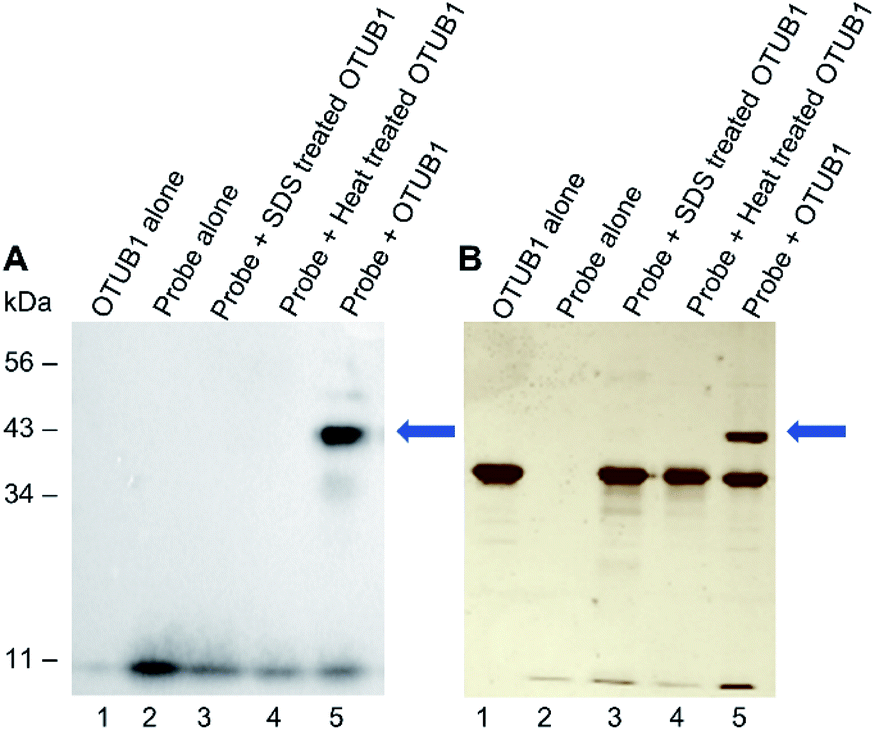 | ||
| Fig. 2 Labelling of recombinant OTUB1 (2 μg) with probe 1 (4 μg). Visualised using (A) anti-HA western blot and (B) silver stain. Recombinant OTUB1 (2 μg) was denatured using heat or SDS prior to incubation with the probe 1 (4 μg) in lanes 3 and 4. Probe 1 was pre-incubated for 90 min prior to addition of initiators and exposure to UV light (365 nm) for 5 min. | ||
After confirming reactivity with a recombinant DUB, probe 1 was tested in HEK 293T cell lysate using conditions analogous to those described for the recombinant enzyme labelling. (Fig. 3). The absence of detectable labelling in lane 1 confirms the requirement of radical initiation for covalent labelling (Fig. 3A). Exposure time to UV light was optimised, with conditions tested from 0 to 360 minutes (Fig. 3A, lanes 2–7). No labelling is observed at the 0 minute time point (Fig. 3A, lane 2). There is no discernible difference between the 1 minute and 10 minute timepoints (Fig. 3A, lanes 3 and 4). One minute of UV light exposure is sufficient for complete labelling under these conditions. This suggests the binding interaction between the enzyme and probe 1 has pre-ordered the reaction by aligning the C-terminal alkene such that the active site cysteine is primed to attack. Prolonged exposure to UV light showed degradation of the sample at longer timepoints (Fig. 3A, lanes 5–7). Two further control experiments were performed to validate the need for free thiols and to compare to an existing probe. Comparison between lanes 8 and 4 reveals that the addition of thiol alkylator N-ethylmaleimide (NEM) inhibited labelling (Fig. 3A). Covalent bond formation therefore requires free cysteine residues. A labelling reaction with the known HA-Ub75CH2CH2Br6 probe was subjected to the same conditions as the thiol–ene labelling to confirm there was no degradation of the lysate under these conditions (Fig. 3A, lanes 9 and 10). Interestingly, radical labelling conditions appear to enhance labelling with this probe. This data suggests that this probe can react via both electrophilic and radical mechanisms under our radical labelling conditions.
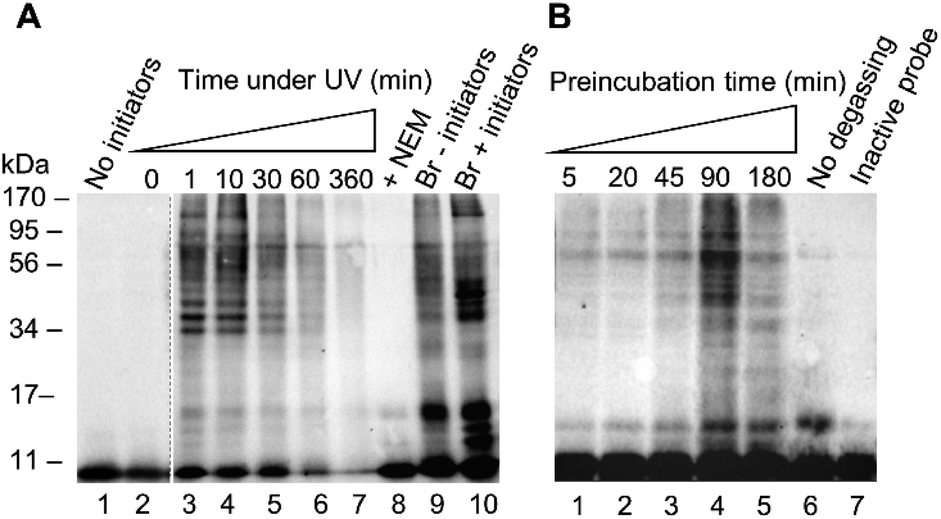 | ||
| Fig. 3 (A) HEK 293T cell lysate (50 μg) was incubated with probe 1 (1 μg) for 60 min before exposure to UV light (365 nm) for a range of times. Controls included using NEM (10 mM), the Ub75CH2CH2Br probe (1 μg) and a labelling excluding initiators. (B) HEK 293T cell lysate (50 μg) was pre-incubated for varying periods of time with probe 1 (1 μg) before exposure to UV light (365 nm) for 2 min. Controls included no degassing and an inactive probe (1 μg) incubated for 45 min. | ||
We next examined how the preincubation period affected the labelling pattern (Fig. 3B, lanes 1–5). We reasoned that prior to activation, a binding equilibrium must be established between the probe and target enzymes to yield the rapid labelling observed. The conditions of the labelling were altered slightly from the recombinant enzyme labelling with a shortened UV light exposure time of 2 min and variations in the pre-incubation time. After a 5 minute incubation there was little visible labelling. Increasing timepoints from 20 to 90 minutes correlated to an increase in the intensity of the labelling pattern. A drop-off is observed by the 180 minute timepoint, indicating that the binding equilibrium is fully established by 90 minutes. This drop-off can likely be attributed to a reduction in DUB activity after an extended incubation time in cell lysate. These results were consistent with our hypothesis that a pre-organised reaction is occurring following a specific binding interaction. Two further experiments were carried out to confirm the thiol–ene reaction is responsible for the covalent labelling. Samples were degassed prior to exposure to UV light to reduce the influence of reactive oxygen species which have been demonstrated to be inhibitory to certain DUBs.30 Omitting this step reduced labelling (Fig. 3B, lane 6 vs. lane 5). The absence of labelling when HA-Ub75 is used confirms that the alkene moiety is necessary for covalent bond formation (Fig. 3B, lane 7). These results, in combination with our findings that the reaction does not proceed when cysteine residues are alkylated and when initiators are absent, provide compelling evidence that the thiol–ene reaction is responsible for the observed labelling. Optimisation was also carried out for initiator concentration along with more refined optimisation of time under UV (ESI Fig. 4†).
We next set out to test the modulation of labelling using the known pan DUB inhibitor, PR-619.31 PR-619 was incubated with cell lysate for 30 min prior to the addition of probe 1 and the labelling was then carried out under optimised conditions with a 90 min pre-incubation time. A clear concentration-dependent reduction in labelling intensity was observed when the lysate was preincubated with PR-619 (Fig. 4A, lanes 1–6). This implies specificity of probe 1 towards DUBs. To demonstrate how the thiol–ene mediated labelling mechanism can be used advantageously in inhibitor studies, an additional experiment was performed whereby the binding equilibrium was established between the probe 1 and DUBs in lysate during a 90 min incubation. PR-619 was titrated into the mixture at increasing concentrations and incubated for a further 30 min before the addition of initiators and exposure to UV light (Fig. 4B). A parallel experiment was set up with the Ub75CH2CH2Br probe for comparison (Fig. 4C). A similar trend is observed to the previous experiment where PR-619 abrogates the labelling in a concentration dependent manner when using our probe 1 (Fig. 4B). When the conditions of lanes 2 and 6, 0 μM and 100 μM PR-619 respectively, are replicated with the Ub75CH2CH2Br probe, only minor difference is observed. (Fig. 4C, lanes 2 and 3). To check the integrity of the probe and cell lysate under these conditions a silver stain of the samples was also performed (ESI Fig. 5†). This demonstrates how DUB inhibitors can disrupt the binding equilibrium, thus allowing the study of these inhibitors in a novel way with greater control. Similarly, we observed analogous effects using wild-type ubiquitin in place of PR-619, illustrating how probe 1 can be utilised study reversible binding events (ESI Fig. 6†).
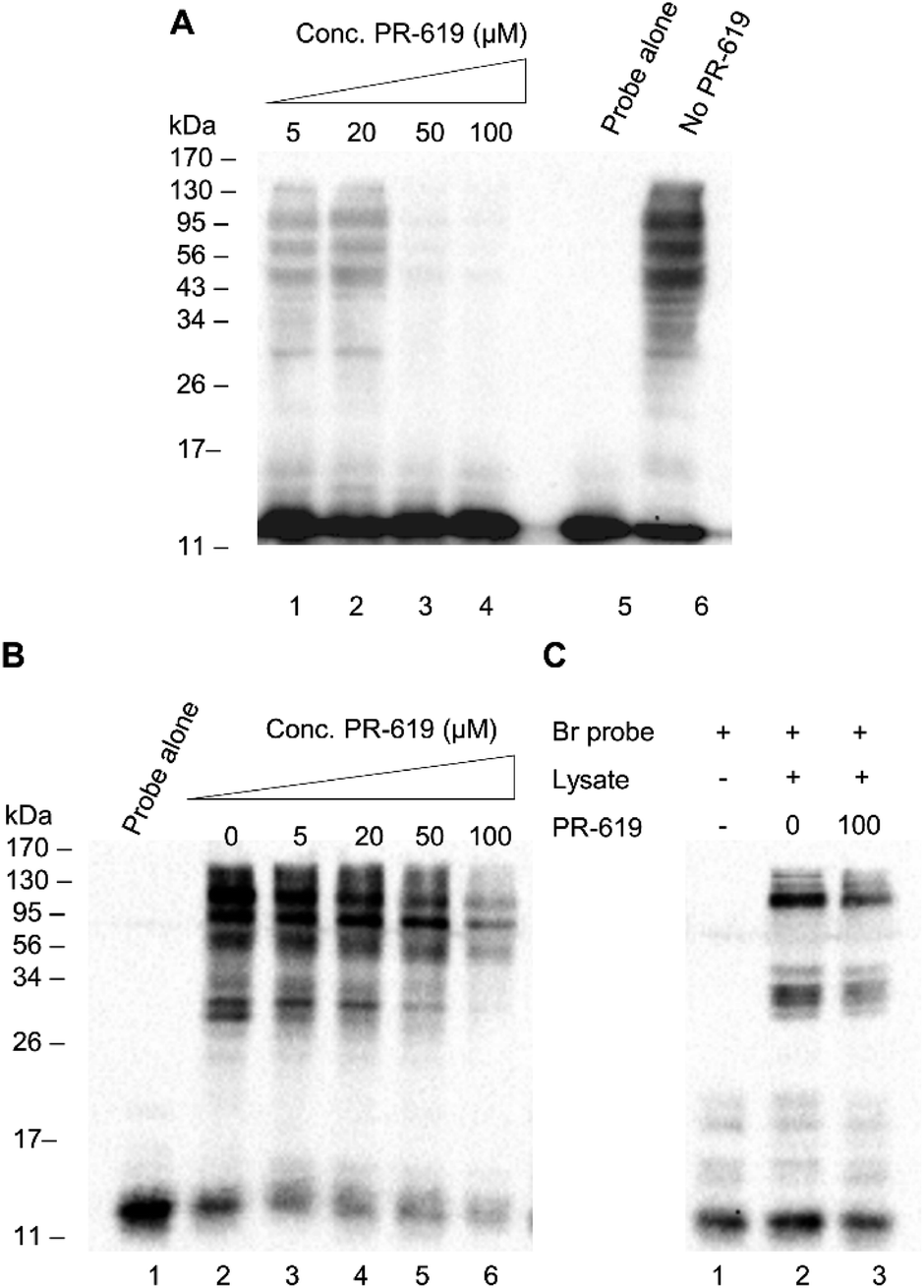 | ||
| Fig. 4 (A) HEK 293T cell lysate (50 μg) was preincubated for 90 min with increasing concentrations of PR-619 before being labelled with the probe 1 (1 μg). (B) HEK 293T cell lysate (50 μg) was preincubated with probe 1 (1 μg) for 90 min before PR-619 was added at increasing concentrations. (C) HEK 293T cell lysate (50 μg) was incubated with Ub75CH2CH2Br and PR-619 (100 μM) was added to one sample. All samples were exposed to UV light (365 nm) for 2 min. | ||
In order to extend the scope of thiol–ene mediated DUB capture and explore its structural requirements, a phenyl-substituted alkene probe 2 (Scheme 1, R = Ph) was synthesised to examine if a more substituted alkene would alter reactivity and selectivity. Probe 2 was tested in HEK 293T lysate alongside the terminal alkene probe 1 which showed a different, generally reduced, labelling pattern by western blot (ESI†). Despite the more modest labelling observed with phenyl-substituted probe 2, it was taken forward for further comparative analysis with terminal alkene probe 1.
Finally, to unambiguously characterise the specificity and reactivity of our probes in complex cellular environments, an immunoprecipitation (IP) was performed on material captured by a thiol–ene labelling. Tagged proteins were enriched and analysed by anti-HA western blot and LC-MS/MS following tryptic digest (Fig. 5). Both the terminal alkene probe 1 and the phenyl-substituted alkene probe 2 were used along with a probe-free negative control condition, each carried out in triplicate. The inputs and eluates for this experiment were visualised using anti-HA western blot (ESI Fig. 7†).
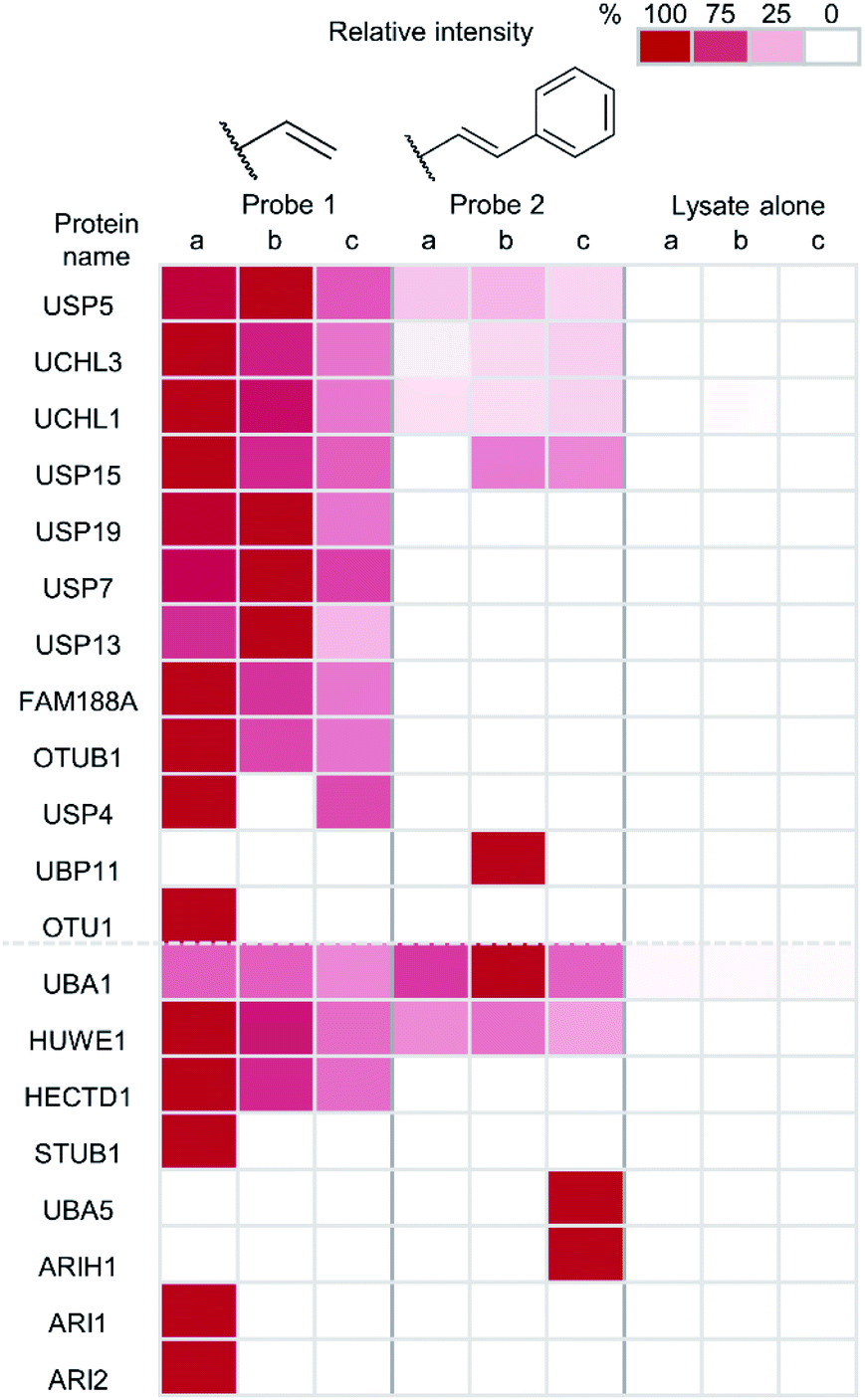 | ||
| Fig. 5 Heatmap showing the enrichment of DUBs and ubiquitin conjugation machinery after immunoprecipitation with anti-HA coupled agarose beads using probe 1, probe 2 and lysate alone. Intensities calculated using label-free quantification following LC-MS/MS analysis. The replicate that had the highest intensity for each individual enzyme was taken as 100% and the values of other replicates for the enzyme are expressed as a percentage of this value. | ||
Protein intensities were calculated using label-free quantification with the replicate that had the highest intensity for each individual enzyme being taken as 100%. The values of other replicates are expressed as a percentage of this. Twelve DUBs were identified from the IP as well as eight components of the ubiquitin conjugation machinery. Enrichment of all but one of the identified DUBs was observed in samples treated with the terminal alkene probe 1 (Fig. 5). Enrichment of certain DUBs was also observed for the phenyl-substituted probe 2, although this is less pronounced. It appears the terminal alkene probe 1 yields more efficient capture in this case. Additionally, enrichment of ubiquitin conjugation machinery was also observed in both cases. Our probe design is therefore also applicable to labelling the less nucleophilic active site cysteines of these enzymes.
Conclusions
In conclusion, we have successfully developed a novel activity-based monoubiquitin probe that is completely inactive under ambient conditions and can be selectively activated to label DUBs and enzymes of the ubiquitin conjugation machinery through the active site cysteine. We have demonstrated its reactivity against recombinant, purified DUB OTUB1 and HEK 293T lysate. Experiments using heat and SDS treated recombinant enzyme showed the probe is selective for the active form of the DUB, requiring a specific binding interaction. We have shown the ability of our probe to study the effects of inhibitors displacing bound probe, potentially accelerating development of novel inhibitors. Moreover, we found that selectivity can be influenced by the addition of groups proximal to the alkene moiety opening the potential to develop probes more specific to particular DUB classes. Finally, we have demonstrated for the first time the successful application of thiol–ene chemistry in activity-based protein profiling.The short activation period of the probe following specific binding provides a snapshot of the DUBs bound at that time. The consequence of capturing the equilibrium in such a way results in a small proportion of DUBs captured relative to similar existing probes, however, it does provide an opportunity to study binding affinity and inhibitor potency in new ways for both reversable and irreversible binding interactions. Unlike existing photocrosslinking probes this technique offers a residue specific, mechanism-based method to provide a time-resolved readout of enzyme activity rather than protein binding. The current requirement for radical initiators and a degassing step of cell lysate prior to exposure to UV light, may limit the applicability of this method in living cells. However, with careful optimisation this approach could be explored for its applicability to intact cells. This radical labelling approach is broadly applicable and provides further opportunities in chemical proteomics beyond the study of reactivity and selectivity in the ubiquitin system as any enzyme bearing an active site cysteine could be targeted in this way. The alkene moiety is chemically inert and sterically undemanding to introduce to other biological entities, such as carbohydrates, DNA or inhibitor scaffolds to access a wide variety of enzyme classes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Dr Edel Durack for her assistance with the MALDI spectra. This work was supported by Trinity College Dublin School of Chemistry.Notes and references
- G. Goldstein, M. Scheid, U. Hammerling, D. H. Schlesinger, H. D. Niall and E. A. Boyse, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 11–15 CrossRef CAS PubMed.
- A. Hershko, H. Heller, S. Elias and A. Ciechanover, J. Biol. Chem., 1983, 258, 8206–8214 CAS.
- R. Hough and M. Rechsteiner, J. Biol. Chem., 1986, 261, 2391–2399 CAS.
- P. Hanpude, S. Bhattacharya, A. K. Dey and T. K. Maiti, IUBMB Life, 2015, 67, 544–555 CrossRef CAS PubMed.
- S. A. Abdul Rehman, Y. A. Kristariyanto, S. Y. Choi, P. J. Nkosi, S. Weidlich, K. Labib, K. Hofmann and Y. Kulathu, Mol. Cell, 2016, 63, 146–155 CrossRef CAS PubMed.
- A. Borodovsky, H. Ovaa, N. Kolli, T. Gan-Erdene, K. D. Wilkinson, H. L. Ploegh and B. M. Kessler, Chem. Biol., 2002, 9, 1149–1159 CrossRef CAS PubMed.
- H. B. Kramer, B. Nicholson, B. M. Kessler and M. Altun, Biochim. Biophys. Acta, 2012, 1823, 2029–2037 CrossRef CAS PubMed.
- A. P. Turnbull, S. Ioannidis, W. W. Krajewski, A. Pinto-Fernandez, C. Heride, A. C. L. Martin, L. M. Tonkin, E. C. Townsend, S. M. Buker, D. R. Lancia, J. A. Caravella, A. V. Toms, T. M. Charlton, J. Lahdenranta, E. Wilker, B. C. Follows, N. J. Evans, L. Stead, C. Alli, V. V. Zarayskiy, A. C. Talbot, A. J. Buckmelter, M. Wang, C. L. McKinnon, F. Saab, J. F. McGouran, H. Century, M. Gersch, M. S. Pittman, C. G. Marshall, T. M. Raynham, M. Simcox, L. M. D. Stewart, S. B. McLoughlin, J. A. Escobedo, K. W. Bair, C. J. Dinsmore, T. R. Hammonds, S. Kim, S. Urbé, M. J. Clague, B. M. Kessler and D. Komander, Nature, 2017, 550, 481–486 CrossRef CAS PubMed.
- J. F. McGouran, S. R. Gaertner, M. Altun, H. B. Kramer and B. M. Kessler, Chem. Biol., 2013, 20, 1447–1455 CrossRef CAS PubMed.
- D. Flierman, G. J. van der Heden van Noort, R. Ekkebus, P. P. Geurink, T. E. Mevissen, M. K. Hospenthal, D. Komander and H. Ovaa, Cell Chem. Biol., 2016, 23, 472–482 CrossRef CAS PubMed.
- G. Li, Q. Liang, P. Gong, A. H. Tencer and Z. Zhuang, Chem. Commun., 2014, 50, 216–218 RSC.
- N. Haj-Yahya, H. P. Hemantha, R. Meledin, S. Bondalapati, M. Seenaiah and A. Brik, Org. Lett., 2014, 16, 540–543 CrossRef CAS PubMed.
- A. Weber, P. R. Elliott, A. Pinto-Fernandez, S. Bonham, B. M. Kessler, D. Komander, F. El Oualid and D. Krappmann, Cell Chem. Biol., 2017, 24, 1299–1313 CrossRef CAS PubMed.
- M. P. Mulder, F. El Oualid, J. ter Beek and H. Ovaa, Chembiochem, 2014, 15, 946–949 CrossRef CAS PubMed.
- S. Misaghi, P. J. Galardy, W. J. Meester, H. Ovaa, H. L. Ploegh and R. Gaudet, J. Biol. Chem., 2005, 280, 1512–1520 CrossRef CAS PubMed.
- W. Gui, C. A. Ott, K. Yang, J. S. Chung, S. Shen and Z. Zhuang, J. Am. Chem. Soc., 2018, 140, 12424–12433 CrossRef CAS PubMed.
- D. S. Hewings, J. Heideker, T. P. Ma, A. P. AhYoung, F. El Oualid, A. Amore, G. T. Costakes, D. Kirchhofer, B. Brasher, T. Pillow, N. Popovych, T. Maurer, C. Schwerdtfeger, W. F. Forrest, K. Yu, J. Flygare, M. Bogyo and I. E. Wertz, Nat. Commun., 2018, 9, 1162 CrossRef PubMed.
- S. M. Mali, S. K. Singh, E. Eid and A. Brik, J. Am. Chem. Soc., 2017, 139, 4971–4986 CrossRef CAS PubMed.
- R. Meledin, S. M. Mali, S. K. Singh and A. Brik, Org. Biomol. Chem., 2016, 14, 4817–4823 RSC.
- P. Gong, G. A. Davidson, W. Gui, K. Yang, W. P. Bozza and Z. Zhuang, Chem. Sci., 2018, 9, 7859–7865 RSC.
- M. Jbara, S. Laps, M. Morgan, G. Kamnesky, G. Mann, C. Wolberger and A. Brik, Nat. Commun., 2018, 9, 3154 CrossRef PubMed.
- K. C. Pao, M. Stanley, C. Han, Y. C. Lai, P. Murphy, K. Balk, N. T. Wood, O. Corti, J. C. Corvol, M. M. Muqit and S. Virdee, Nat. Chem. Biol., 2016, 12, 324–331 CrossRef CAS PubMed.
- R. Ekkebus, S. I. van Kasteren, Y. Kulathu, A. Scholten, I. Berlin, P. P. Geurink, A. de Jong, S. Goerdayal, J. Neefjes, A. J. Heck, D. Komander and H. Ovaa, J. Am. Chem. Soc., 2013, 135, 2867–2870 CrossRef CAS PubMed.
- W. Tian, C. Chen, X. Lei, J. Zhao and J. Liang, Nucleic Acids Res., 2018, 46, W363–W367 CrossRef CAS PubMed.
- A. Ernst, G. Avvakumov, J. Tong, Y. Fan, Y. Zhao, P. Alberts, A. Persaud, J. R. Walker, A. M. Neculai, D. Neculai, A. Vorobyov, P. Garg, L. Beatty, P. K. Chan, Y. C. Juang, M. C. Landry, C. Yeh, E. Zeqiraj, K. Karamboulas, A. Allali-Hassani, M. Vedadi, M. Tyers, J. Moffat, F. Sicheri, L. Pelletier, D. Durocher, B. Raught, D. Rotin, J. Yang, M. F. Moran, S. Dhe-Paganon and S. S. Sidhu, Science, 2013, 339, 590–595 CrossRef CAS PubMed.
- M. Altun, T. S. Walter, H. B. Kramer, P. Herr, A. Iphöfer, J. Boström, Y. David, A. Komsany, N. Ternette, A. Navon, D. I. Stuart, J. Ren and B. M. Kessler, PLoS One, 2015, 10, e0115344 CrossRef PubMed.
- C. N. Braxton, E. Quartner, W. Pawloski, D. Fushman and T. A. Cropp, Biochemistry, 2019, 58, 883–886 CrossRef CAS PubMed.
- E. M. Valkevich, R. G. Guenette, N. A. Sanchez, Y. C. Chen, Y. Ge and E. R. Strieter, J. Am. Chem. Soc., 2012, 134, 6916–6919 CrossRef CAS PubMed.
- A. Borodovsky, B. M. Kessler, R. Casagrande, H. S. Overkleeft, K. D. Wilkinson and H. L. Ploegh, EMBO J., 2001, 20, 5187–5196 CrossRef CAS PubMed.
- P. Gopinath, A. Mahammed, S. Ohayon, Z. Gross and A. Brik, Chem. Sci., 2016, 7, 7079–7086 RSC.
- X. Tian, N. S. Isamiddinova, R. J. Peroutka, S. J. Goldenberg, M. R. Mattern, B. Nicholson and C. Leach, Assay Drug Dev. Technol., 2011, 9, 165–173 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: For additional recombinant enzyme and cell lysate experiments as well as experimental details on the probe synthesis, in vitro labelling conditions, immunoprecipitation, mass spectrometry methods and chemical synthesis. See DOI: 10.1039/c9sc05258e |
| ‡ Present address: MRC London Institute of Medical Sciences, Hammersmith Hospital Campus, Du Cane Road, London, W12 0NN, UK. |
| This journal is © The Royal Society of Chemistry 2020 |