
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltra-small cobalt nanoparticles from molecularly-defined Co–salen complexes for catalytic synthesis of amines†
Thirusangumurugan
Senthamarai
a,
Vishwas G.
Chandrashekhar
a,
Manoj B.
Gawande
 b,
Narayana V.
Kalevaru
a,
Radek
Zbořil
b,
Paul C. J.
Kamer
a,
Rajenahally V.
Jagadeesh
*a and
Matthias
Beller
*a
b,
Narayana V.
Kalevaru
a,
Radek
Zbořil
b,
Paul C. J.
Kamer
a,
Rajenahally V.
Jagadeesh
*a and
Matthias
Beller
*a
aLeibniz-Institut für Katalyse e. V. an der Universität Rostock, Albert-Einstein-Str. 29a, 18059 Rostock, Germany. E-mail: jagadeesh.rajenahally@catalysis.de; matthias.beller@catalysis.de
bRegional Centre of Advanced Technologies and Materials, Department of Physical Chemistry, Faculty of Science, Palacký University, Olomouc, Šlechtitelů 27, Olomouc, 78371, Czech Republic
First published on 21st February 2020
Abstract
We report the synthesis of in situ generated cobalt nanoparticles from molecularly defined complexes as efficient and selective catalysts for reductive amination reactions. In the presence of ammonia and hydrogen, cobalt–salen complexes such as cobalt(II)–N,N′-bis(salicylidene)-1,2-phenylenediamine produce ultra-small (2–4 nm) cobalt-nanoparticles embedded in a carbon–nitrogen framework. The resulting materials constitute stable, reusable and magnetically separable catalysts, which enable the synthesis of linear and branched benzylic, heterocyclic and aliphatic primary amines from carbonyl compounds and ammonia. The isolated nanoparticles also represent excellent catalysts for the synthesis of primary, secondary as well as tertiary amines including biologically relevant N-methyl amines.
Introduction
In recent years, 3d metal-based nanoparticles (NPs) emerged as promising catalysts for the synthesis of functionalized and complex organic molecules for advanced applications in life and material sciences.1 Traditionally, such syntheses are performed using homogeneous organometallic complexes,2 which are often sensitive and more difficult to recycle compared to heterogeneous materials.1,2 For the preparation of stable but at the same time active and selective NPs, the use of suitable precursors and optimal methods is crucial.1 Commonly, nanoparticles are prepared by chemical reduction processes, calcination or pyrolysis in the presence of suitable supports and metal precursors. The resulting materials are applied particularly in industrially-relevant bench mark reactions of less functionalized molecules.3 However, in recent years there is an increasing interest to use such catalysts for advanced organic synthesis, specifically for the preparation of life science products.1 In this respect, the preparation of specific NPs by immobilization and pyrolysis of organometallic complexes or metal organic frameworks (MOFs) on heterogeneous supports attracted also attention.1,4 These supported NPs show high activity and selectivity for the preparation of functionalized amines,1d–i nitriles,1k,4c carboxylic acid derivatives,1f,k and cycloaliphatic compounds.1j Although this preparation represents a highly useful tool to produce novel nano-structured catalysts on lab-scale, the upscaling can be difficult and requires specialized equipment.1,4 Thus, the use of alternative, more convenient methods is highly desired. One possibility is the practical in situ generation of active heterogeneous NPs.5 Based on this idea, herein we report a straightforward approach for the generation of cobalt-based NPs in situ from molecularly-defined metal complexes and their application in reductive amination reactions using ammonia and molecular hydrogen (Fig. 1). | ||
| Fig. 1 In situ generation of Co-NPs for reductive aminations. | ||
The resulting amines represent privileged molecules widely used in chemistry, medicine, biology, and material science.6 For their synthesis, catalytic reductive amination of carbonyl compounds using molecular hydrogen is widely applied as cost-effective methodology in both academic research and industry.1e,h,7–10 Among reductive aminations, the synthesis of primary amines, which can be easily functionalized to high value products, continues to be especially important.1e,h,j,8–10 Regarding catalysts for this reaction, precious metals-based ones are known to a large extent.8,9 However, in recent years Co-1e and Ni1h,i-based nanocatalysts have successfully been developed in addition to RANEY® nickel.10
Results and discussion
In situ generation of Co-NPs and their activities
Following our concept, we initially investigated the reaction of cobalt salen complexes to obtain NPs. For example, using the cobalt–N,N′-bis(salicylidene)-1,2-phenylenediamine (complex I) in water–THF as solvent in the presence of ammonia and molecular hydrogen at 120 °C a black precipitate of Co NPs is formed, which can be magnetically separated (Fig. 1 and S3†). To explore their reactivity, preliminary catalytic experiments were carried out for the reductive amination of 4-bromobenzaldehyde 1 to 4-bromobenzylamine 2 in presence of ammonia and molecular hydrogen (Fig. 2). Indeed, using a mixture of cobalt(II) acetate and N,N′-bis(salicylidene)-1,2-phenylenediamine (L1) led to the formation of 15% of 2. In contrast, testing simple cobalt(II) acetate under the same conditions produced no desired product. Remarkably, the defined complex Co-L1 (complex I) exhibited excellent activity as well as selectivity in the bench mark reaction (98% of 4-bromobenzylamine). In addition, other molecularly-defined Co–salen complexes have also been tested (Fig. S1†) and complexes II–IV showed good activity (85–90% yield), while complex V resulted in lower product yield (50%). In all cases of active complexes, the reaction mixtures turned black after some hours. Hence, we assumed the in situ formed cobalt-NPs are the “real” active species for the reductive amination reaction. To confirm this, we performed a standard mercury test in the presence of complex I and after addition of 15 mg Hg the reductive amination reaction did not occur. Hot filtration of NPs and testing the filtrate for the reaction showed that Co-NPs did not go into solution as soluble particles. Studying the course of the benchmark reaction at different intervals of time showed a prolonged catalyst preformation time and only after 10 h 4-bromobenzylamine started to form (Fig. S3†). Apparently, complex I generated nanoparticles slowly, which then catalyze the desired amination process. For comparison, we also prepared cobalt nanoparticles separately by mixing complex I, ammonia and hydrogen (see S7a†). After isolation, they were tested under similar conditions and exhibited comparable activity and selectivity to that of in situ generated ones. Due to their physical properties, the Co NPs could be magnetically separated and were conveniently re-used up to three times (Fig. 2). However, after the third cycle we observed a significant decrease in activity and selectivity. In addition, the stability of the catalyst system was also confirmed by recycling the NPs after reduced reaction time (Fig. S4†). Next, we compared the reactivity of these active NPs with related supported NPs. However, addition of carbon or silica support to the reaction led to completely inactive materials (Fig. 2). On the other hand, materials prepared by immobilization of complex I on carbon or silica and subsequent pyrolysis produced catalysts with moderate activity (Fig. 2; 40–50% yield of 2). In addition, specific cobalt nanoparticles have been prepared by using chemical reduction of cobalt salts11 and tested for their activities. However, none of these cobalt nanoparticles formed the desired product, 4-bromobenzylamine (Table S1,† entries 5–6). All these results reveal the superiority of the simply in situ generated Co NPs (Fig. 3).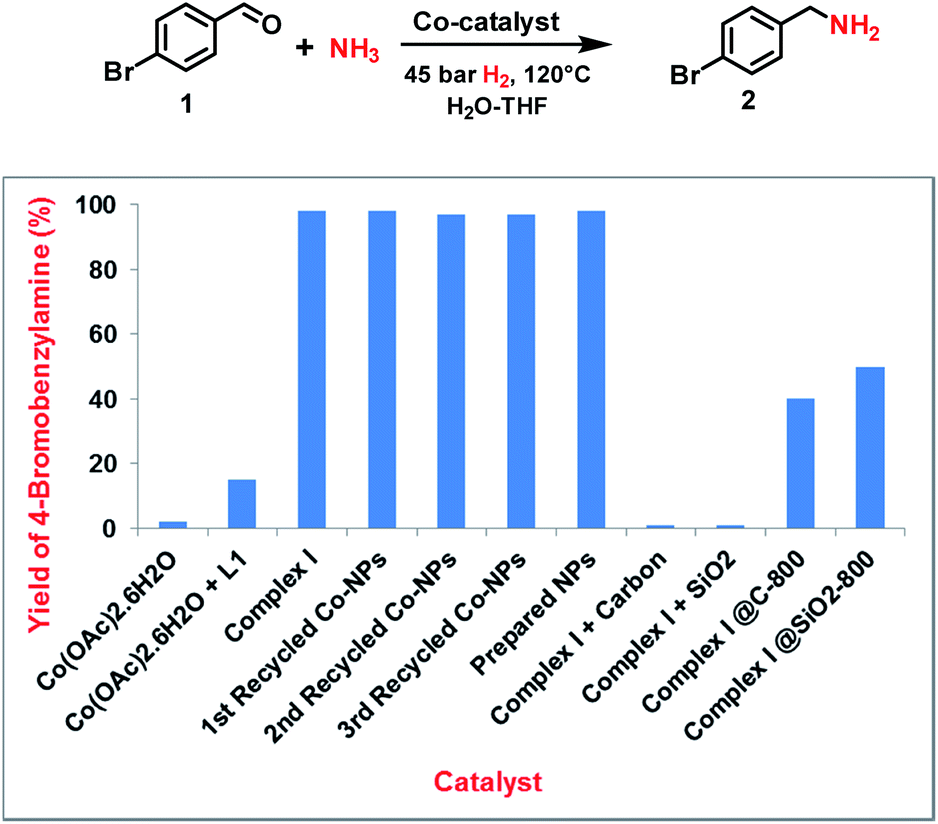 | ||
Fig. 2 Reductive amination of 4-bromobenzaldehyde: Activity of cobalt catalystsa. aReaction conditions: 0.5 mmol 4-bromobenzaldehyde, 6 mol% Co-complex, (Co NPs), 5 bar NH3, 45 bar H2, 2.5 mL H2O–THF (1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 120 °C, 24 h, GC yields using n-hexadecane as standard. :1), 120 °C, 24 h, GC yields using n-hexadecane as standard. | ||
 | ||
| Fig. 3 Reductive amination of carbonyl compounds in presence of NH3 and H2 using different Co NPs produced from cobalt–salen complex. | ||
Characterization of in situ Co-NPs
To understand the reactivity and to know the structural features of the most active cobalt nanoparticles, we performed detailed characterizations using transmission electron microscopy (TEM), energy dispersive X-ray spectroscopy (EDX), X-ray diffraction (XRD), and X-ray photoelectron spectroscopy (XPS).TEM analysis of cobalt-particles at different magnification showed sheets and at some places thread bundles like morphology where cobalt nanoparticles are embedded in carbon and nitrogen framework (Fig. 4). Further detailed morphological investigations were performed by HRTEM-STEM analysis. A close inspection of HRTEM images at 20 nm, revealed the presence of ultra-small (range 2–4 nm) cobalt nanoparticles (Fig. 4 and S6†) supported on graphitic carbon. The HAADF-elemental mapping displayed a homogeneous distribution of the cobalt nanoparticles (Fig. 4). In case of the recycled catalyst, we observed that these particles were still intact and there are no noticeable changes in the morphology (Fig. S8†).
 | ||
| Fig. 4 TEM images of in situ generated Co-NPs from complex I. (a and b) HRTEM images of cobalt catalyst, (C) magnified STEM image, (d–g) elemental mapping images where C, N, O and Co are in blue, yellow, green and red colours. | ||
XRD patterns of in situ generated and reused Co-nanoparticles do not show variations on the phase composition (Fig. S9†). Two allotropes of metallic cobalt have been identified, one with face centered cubic arrangement (Co-fcc, space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, PDF card 01-089-7093), and the other one with hexagonal closed packing (Co-hcp, space group P63/mmc, PDF card 01-089-7373). Elemental analysis of the bulk material showed 96.8 wt% of Co, 0.15 wt% of C and only 0.5 wt% of N. Complementary, XPS analysis displayed the presence of larger amounts of C, N, and O (C = 34.4, N = 1.2, O = 47.24 and Co = 16.8 at%) on the surface (Fig. S10†). The high resolution XP spectra of NPs in C1s region can be deconvoluted into five peak components with binding energies of 284.6, 285.6, 286.5, 288.5 and 289.4 eV corresponding to C–C sp2, C–C sp3, C–O/C–N, and C
m, PDF card 01-089-7093), and the other one with hexagonal closed packing (Co-hcp, space group P63/mmc, PDF card 01-089-7373). Elemental analysis of the bulk material showed 96.8 wt% of Co, 0.15 wt% of C and only 0.5 wt% of N. Complementary, XPS analysis displayed the presence of larger amounts of C, N, and O (C = 34.4, N = 1.2, O = 47.24 and Co = 16.8 at%) on the surface (Fig. S10†). The high resolution XP spectra of NPs in C1s region can be deconvoluted into five peak components with binding energies of 284.6, 285.6, 286.5, 288.5 and 289.4 eV corresponding to C–C sp2, C–C sp3, C–O/C–N, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and OC–O type bonds with individual atomic% of 64.09, 19.30, 8.59, 3.20 and 4.82 respectively, showing the graphitic nature of the carbon material (Fig. 5a). The presence of a specific N1s peak at 399.7 confirms pyrrolic nitrogen (Fig. 5b). The three peak components at 529.5, 531.4, and 532.9 eV in O1s spectra originate from the presence of Co(OH)2 (6.99%), CO (73.09%), and O–C on the surface of cobalt (19.92%). This reveals partial oxidation at the surface of the optimal material (Fig. 5c). In agreement, the two main component peaks having binding energy at 780.7 eV (50.81%) and 782.5 eV (22.54%) confirm the presence of Co2+ (Co(OH)2) (Fig. 5d).12a Three small peaks having binding energies at 778.09 (2.45%), 781.09 (0.59%) and 783.09 (0.35%) eV indicate the presence of metallic cobalt.12b
O, and OC–O type bonds with individual atomic% of 64.09, 19.30, 8.59, 3.20 and 4.82 respectively, showing the graphitic nature of the carbon material (Fig. 5a). The presence of a specific N1s peak at 399.7 confirms pyrrolic nitrogen (Fig. 5b). The three peak components at 529.5, 531.4, and 532.9 eV in O1s spectra originate from the presence of Co(OH)2 (6.99%), CO (73.09%), and O–C on the surface of cobalt (19.92%). This reveals partial oxidation at the surface of the optimal material (Fig. 5c). In agreement, the two main component peaks having binding energy at 780.7 eV (50.81%) and 782.5 eV (22.54%) confirm the presence of Co2+ (Co(OH)2) (Fig. 5d).12a Three small peaks having binding energies at 778.09 (2.45%), 781.09 (0.59%) and 783.09 (0.35%) eV indicate the presence of metallic cobalt.12b
 | ||
| Fig. 5 HR-XPS spectra of in situ generated Co NPs. | ||
The HR-XPS of reused catalysts revealed that there is no shifting of binding energy in Co 2p3/2 peak but the ratio of metallic cobalt vs. cobalt hydroxide was slightly changed (Fig. S11d†). On the other hand, no perceptible change in the binding energies of C1s and N1s was discerned except for the slight shifting of the pyrrolic nitrogen peak from 399.7 eV to 400.2 eV, thus reiterating no apparent alteration in the chemical nature of the carbon shell of the catalyst (Fig. S11a and b†).
It is interesting to note that these cobalt-particles exhibit ferromagnetic behaviour with distinct values of coercivity field and remanent magnetization (Fig. S12†). Ferromagnetic behaviour at room temperature is due to the stronger effect of the magnetic dipole interaction compared with thermal fluctuations. We do not observe any blocking temperature suggesting the size of nanoparticles above 10–15 nm (e.g. the system is not superparamagnetic at room temperature).
Synthesis of linear primary amines
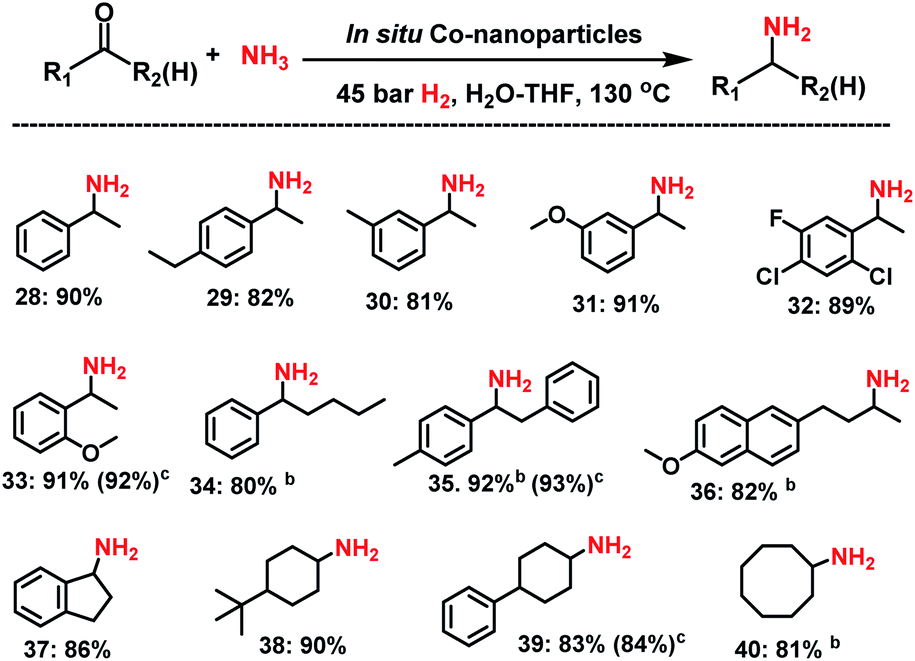
Then, we tested the general applicability of our in situ generated nanoparticles for the synthesis of primary amines. As shown in Schemes 1 and 2, a variety of structurally diverse and functionalized benzylic, heterocyclic and aliphatic linear and branched primary amines can be prepared in good to excellent yields. Simple and substituted aldehydes underwent smooth reaction to give primary benzylic amines in up to 92% yield (Scheme 1, products 3–7). For example, fluoro-, chloro-, and bromo-substituted benzaldehydes produced corresponding amines without significant dehalogenations in 86–92% yields (Scheme 1, entries 8–12). Different functionalized benzylic amines containing methoxy, trifluoromethoxy, dimethylamino, and ester groups as well as C–C double bonds were synthesized in up to 95% yield (Scheme 1, products 14–23). In addition to benzylic amines, primary aliphatic ones were also prepared under similar conditions (Scheme 1, products 25–27). Interestingly, the natural product perillaldehyde was successfully aminated to produce the corresponding amine in 87% yield (product 27). | ||
| Scheme 1
In situ generated Co-nanoparticles catalyzed synthesis of linear primary amines from aldehydesa. aReaction conditions: 0.5 mmol aldehyde, 6 mol% complex I (22 mg), 5–7 bar NH3, 45 bar H2, 2.5 mL H2O–THF (1.5:1), 120 °C, 24h, isolated yields. b Same as ‘a’ at 130 °C. c Same as ‘a’ in 2.5 mL H2O. d same as ‘a’ using prepared and isolated Co-NPs from complex I (2 mg; 6.5 mol% Co). | ||
 | ||
| Scheme 2 Synthesis of branched primary amines from ketones using in situ generated Co-nanoparticlesa. aReaction conditions: 0.5 mmol ketone, 6 mol% complex I (22 mg) 5–7 bar NH3, 45 bar H2, 2.5 mL H2O, 130 °C, 24 h, isolated yields. b Same as ‘a’ in H2O–THF (1.5:1 ratio). cUsing prepared and isolated Co-NPs from complex I (2 mg; 6.5 mol% Co). | ||
Synthesis of branched primary amines
Next, we tested the reductive amination of ketones (Scheme 2), which is more challenging compared to aldehydes.Nevertheless, at higher temperature (130 °C) nine aromatic and six aliphatic branched primary amines were prepared in up to 92% yield. In addition, separately prepared Co NPs from complex I, gave similar yields of amines to those obtained by in situ generated nanoparticles.
Synthesis of secondary and tertiary amines
Apart from primary amines synthesis, we explored the applicability of Co-NPs for the synthesis of secondary and tertiary amines. Interestingly, testing complex I which generates the active NPs vide supra for the reaction of benzaldehyde and aniline at 120 °C in presence of molecular hydrogen (40 bar) led to the formation of imine (N-benzylideneaniline) as the sole product. Under these conditions no nanoparticles could be isolated after the reaction.Apparently, the presence of both ammonia and hydrogen are required for the generation of the active NPs! Indeed, using isolated Co NPs, which were prepared from complex I, ammonia and hydrogen, led to excellent activity and selectivity for the synthesis of secondary and tertiary amines including N-methyl amines (Scheme 3). As representative examples different benzaldehydes were reacted with substituted anilines and the corresponding N-benzylanilines were obtained in 87–98% yields (Scheme 1; products 41–45). Similarly, reactions of different benzaldehydes with benzylic and aliphatic amines produced selectively the corresponding secondary and tertiary amines (Scheme 3; products 46–55). In addition, aliphatic aldehydes and 4-fluoroaniline underwent reductive amination and gave the corresponding secondary amines (Scheme 3, products 56–57). Finally, N,N′-dimethylamines were also prepared from three different aldehydes and aqueous N,N′-dimethyl amine (Scheme 3, products 58–60).
 | ||
| Scheme 3 Synthesis of secondary, tertiary and N-methyl amines using Co-nanoparticles prepared from complex Ia. aReaction conditions: 0.6 mmol aldehyde, 0.5 mmol amine, 2 mg Co-NPS (6.5 mol% Co), 45 bar H2, 2.5 mL H2O, 130 °C, 20 h, isolated yields. bSame as ‘a’ using 1 mL aq. N,N′-dimethylamine instead of amine. | ||
Reaction upscaling
In order to demonstrate the synthetic utility of this novel reductive amination protocol, we performed the amination of 5 carbonyl compounds in 5–10 g scale (Scheme 4). As expected, all the tested reactions could be successfully upscaled and the yields (92–96%) of the corresponding primary amines were comparable to that of small scale (0.5 mmol) reactions. | ||
| Scheme 4 Gram-scale synthesis of selected primary amines using in situ generated Co-NPs a. aReaction conditions: 5–10 g of carbonyl compound, weight of complex I corresponds to 6 mol%, 5–7 bar NH3, 45 bar H2, 75–150 mL H2O–THF (1:1), 120 °C, 24h, isolated yields. b Same as ‘a’ at 130 °C in 75 mL H2O. | ||
Experimental
General considerations
All substrates were obtained commercially from various chemical companies and their purity has been checked before use. Cobalt(II) acetate tetrahydrate (cat no. 208396–50G), salicylaldehyde, phenylenediamine and other ligand precursors were purchased from Sigma-Aldrich. Silica (silicon(IV) oxide, amorphous fumed, S.A. 300–350 m2 g−1) was obtained from Alfa Aesar. Carbon powder, VULCAN® XC72R with Code XVC72R and CAS No. 1333-86-4 was obtained from Cabot Corporation Prod. The pyrolysis experiments were carried out in a Nytech-Qex oven.X-ray diffraction patterns were recorded with an Empyrean (PANalytical, The Netherlands) diffractometer in the Bragg–Brentano geometry, Co-Kα radiation (40 kV, 30 mA, λ = 0.1789 nm) equipped with a PIXcel3D detector (1D mode) and programmable divergence and diffracted beam anti-scatter slits. The measurement range was 2θ: 5–105°, with a step size of 0.026°. The identification of crystalline phases was performed using the High Score Plus software (PANalytical) that includes the PDF-4+ database.
TEM images were obtained using a HRTEM TITAN 60-300 with X-FEG type emission gun, operating at 80 kV. This microscope is equipped with a Cs image corrector and a STEM high-angle annular dark-field detector (HAADF). The point resolution is 0.06 nm in TEM mode. The elemental mappings were obtained by STEM-Energy Dispersive X-ray Spectroscopy (EDS) with an acquisition time of 20 min. For HRTEM analysis, the powder samples were dispersed in ethanol and ultrasonicated for 5 min. One drop of this solution was placed on a copper grid with holey carbon film.
XPS surface investigation has been performed on the PHI 5000 VersaProbe II XPS system (Physical Electronics) with monochromatic Al-Kα source (15 kV, 50 W) and photon energy of 1486.7 eV. Dual beam charge compensation was used for all measurements. All the spectra were measured in the vacuum of 1.3 × 10−7 Pa and at room temperature of 21 °C. The analyzed area on each sample was a spot of 200 μm in diameter. The survey spectra were measured with pass energy of 187.850 eV and electronvolt step of 0.8 eV while for the high resolution spectra was used pass energy of 23.500 eV and electronvolt step of 0.2 eV. The spectra were evaluated with the MultiPak (Ulvac – PHI, Inc.) software. All binding energy (BE) values were referenced to the carbon peak C 1s at 284.80 eV.
Magnetic properties of cobalt-nanoparticles were analyzed using a Quantum Design Physical Properties Measurement System (PPMS Dynacool system) with the vibrating sample magnetometer (VSM) option. The experimental data were corrected for the diamagnetism and signal of the sample holder. The temperature dependence of the magnetization was recorded in a sweep mode of 1 K min−1 in the zero-field-cooled (ZFC) and field-cooled (FC) measuring regimes. To get the ZFC magnetization curve, the sample was firstly cooled down from 300 to 5 K in a presence of zero magnetic field and the measurement was carried out on warming from 5 to 300 K under the external magnetic field (1000 Oe). In the case of the FC magnetization measurements, the sample was cooled from 300 to 5 K in an external magnetic field (1000 Oe) and the measurement was carried out on warming from 5 to 300 K at the same value of the external magnetic field (1000 Oe). Hysteresis loops were measured at room temperature (300 K) and at low temperature (5 K).
GC conversion and yields were determined by GC-FID, HP6890 with FID detector, column HP530 m × 250 mm × 0.25 μm. 1H, 13C, NMR data were recorded on a Bruker ARX 300 and Bruker ARX 400 spectrometers using DMSO-d6 and CDCl3 solvents.
All catalytic reactions were carried out in 300 mL and 100 mL autoclaves (PARR Instrument Company). In order to avoid unspecific reactions, catalytic reactions were carried out either in glass vials, which were placed inside the autoclave, or glass/Teflon vessel fitted autoclaves.
Preparation of Co–salen complexes (see scheme S1†)
Procedure for reductive amination
:1)) was added and the vial was fitted with septum, cap and needle. The reaction vials (8 vials with different substrates at a time) were placed into a 300 mL autoclave. The autoclave was flushed with hydrogen twice at 40 bar pressure and then it was pressurized with 5–7 bar ammonia and 45 bar hydrogen. The autoclave was placed into an aluminium block preheated at 135 °C in case of aldehydes and 145 °C in case of ketones and the reactions were stirred for the required time. During the reaction, the inside temperature of the autoclave was measured to be 120 °C in case of aldehydes and 130 °C in case of ketones and this temperature was used as the reaction temperature. After completion of the reactions, the autoclave was cooled to room temperature. The remaining ammonia and hydrogen were discharged and the vials containing reaction products were removed from the autoclave. The reaction mixtures containing the products were filtered off and washed thoroughly with ethanol. The reaction products were analyzed by GC-MS. The crude product was purified by column chromatography using ethyl acetate and n-heptane as the eluent. The corresponding primary amines were converted to their respective hydrochloride salt and characterized by NMR and GC-MS analysis. For converting into hydrochloride salt of amine, 0.3–0.5 mL 7 M HCl in dioxane or 1.5 M HCl in methanol was added to the dioxane solution of respective amine and stirred at room temperature for 4–5 h. Then, the solvent was removed and the resulted hydrochloride salt of amine was dried under high vacuum. The yields were determined by GC for the selected amines: after completing the reaction, n-hexadecane (100 μL) as standard was added to the reaction vials and the reaction products were diluted with ethyl acetate followed by filtration using plug of silica and then analyzed by GC.
Isolation of in situ generated cobalt nanoparticles
After the completion of the reductive amination reaction of carbonyl compound in presence of ammonia and hydrogen as described in Section S3a,† the in situ generated cobalt nanoparticles from the solution containing products were separated using the magnetic stir bar. Then, they were separated from the magnetic stir bar and washed with water and ethanol. Finally the recycled Co NPs were dried under vacuum and stored in a glass vial.Elemental analysis (wt%): Co = 96.8% C = 0.15% and N = 0.5%.
Recycling of in situ generated cobalt-nanoparticles
A magnetic stirring bar and 5 mmol 4-bromobenzaldehyde were transferred to a glass fitted 100 mL autoclave and then 20 mL THF–water (1.5:1) was added. Subsequently, 20 mg isolated in situ generated Co NPs were added. The autoclave was flushed with 40 bar hydrogen and then it was pressurized with 5–7 bar ammonia gas and 45 bar hydrogen. The autoclave was placed into the heating system and the reaction was allowed to progress at 120 °C (temperature inside the autoclave) by stirring for 24 h. After completion of the reaction, the autoclave was cooled and the remaining ammonia and hydrogen pressure was discharged. To the reaction products, 200 μL n-hexadecane as standard was added. The catalyst was then separated by centrifugation and the supernatant containing the reaction products was subjected to GC analysis for determining conversion and yield. The separated catalyst was then washed with ethanol, dried under vacuum and used without further purification or reactivation for the next run.
Gram-scale reactions
The Teflon or glass fitted 300 mL autoclave was charged with a magnetic stirring bar and 5–10 g of carbonyl compound (aldehyde or ketone) and complex I (weight of complex I corresponds 6 mol%). Then, 75–150 mL of solvent (THF/H2O (1.5:1) in case of aldehyde and H2O in case of ketones) was added and the autoclave was flushed with hydrogen twice at 40 bar pressure. Afterwards, it was pressurized with 5–7 bar ammonia and 45 bar hydrogen. The autoclave was placed into an aluminium block preheated at 135 °C in case of aldehydes and 145 °C in case of ketones and the reactions were stirred for the required time. During the reaction, the inside temperature of the autoclave was measured to be 120 °C in case of aldehydes and 130 °C in case of ketones and this temperature was used as the reaction temperature. After completion of the reaction, the autoclave was cooled to room temperature and the remaining ammonia and hydrogen were discharged. The reaction mixtures containing the products were filtered off and washed thoroughly with ethanol. The reaction products were analysed by GC-MS and the crude primary amine product was purified by column chromatography using ethyl acetate and n-heptane as the eluent.
Preparation of cobalt-nanoparticles
:1) was added. The autoclave was flushed with 40 bar hydrogen and then it was pressurized with 5–7 bar ammonia and 45 bar hydrogen. The autoclave was placed into the heating system and the reaction was allowed to progress at 120 °C (temperature inside the autoclave) by stirring for 24 h. After 24 h of reaction time, the autoclave was removed from the heating system and cooled to room temperature. The remaining ammonia and hydrogen pressure was discharged. The cobalt nanoparticles formed were separated from solution by using the magnetic stir bar. Then, the nanoparticles were separated from the magnetic stir bar and washed with water and ethanol. Finally obtained nanoparticles were dried under vacuum and stored in a glass vial.
g of cobalt acetate and 1.5mL of oleic acid were mixed in 40mL of diphenyl ether (DPE) and the reaction mixture was heated to 200 °C under N2 atmosphere. Then, 1.0mL of TOP (trioctylphosphine) was added and the mixture was again heated to 250 °C. Subsequently, the reducing solvent such as 4.0g of 1,2-dodecanediol dissolved in 10mL DPE at 80 °C, was injected into the reaction mixture. Then whole reaction mixture was held at 250 °C for 30min until the completion of the reduction. The reaction products were cooled to room temperature and ethanol was added to precipitate nanoparticles. The formed cobalt nanoparticles were separated by centrifugation and were finally dried and stored in glass vial.
Method-II: 2 mmol of cobalt acetate and 0.4 mmol of oleic acid were mixed in 1 mL ethanol. Then, 3 mmol of NaBH4 dissolved in 1 mL of ethanol, was slowly added to the above mixture at room temperature under stirred condition. The whole mixture was stirred at room temperature for 4 h. The formed cobalt nanoparticles were separated by centrifugation and finally dried and stored in glass vial.
Conclusions
In conclusion, we demonstrated that the in situ formation of cobalt nanoparticles from molecularly defined precursors is straightforward and convenient. Such approach can be used as a versatile tool to prepare selective and active heterogeneous catalysts. In our specific case, Co NPs are formed from cobalt–salen complexes (e.g. cobalt(II)–N,N′-bis(salicylidene)-1,2-phenylenediamine) in the presence of ammonia and hydrogen. Thereby, well-defined ultra-small metallic cobalt and cobalt hydroxide nanoparticles embedded in a cobalt–nitrogen framework are formed. The resulting NPs are stable in the presence of air and water and allow for the preparation of various functionalized and structurally diverse linear and branched benzylic, heterocyclic and aliphatic primary amines as well as secondary and tertiary amines. Moreover, they can be easily magnetically separated enabling easy catalyst recycling and product purification.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the European Research Council (EU project 670986-NoNaCat), and the State of Mecklenburg-Vorpommern for financial and general support. We thank the analytical team of the Leibniz-Institute for Catalysis, Rostock for their excellent service. We gratefully thank Ondrej Tomanec, and Martin Petr (both from Palacky University) for HRTEM elemental mapping and XPS data respectively. The authors gratefully acknowledge the support by the Operational Programme Research, Development and Education – European Regional Development Fund, projects no. CZ.02.1.01/0.0/0.0/16_019/0000754 and CZ.02.1.01/0.0/0.0/15_003/0000416 of the Ministry of Education, Youth and Sports of the Czech Republic.Notes and references
-
(a) D. Wang and D. Astruc, Chem. Soc. Rev., 2017, 46, 816–854 RSC
; (b) L. Liu and A. Corma, Chem. Rev., 2018, 8, 4981–5079 CrossRef PubMed
-
(a)
G. W. Parshall and S. D. Ittel, Homogeneous Catalysis: The Applications and Chemistry of Catalysis by Soluble Transition Metal Complexes, Weily, 1992 Search PubMed
-
(a)
L. Filipponi and D. Sutherland, Nanotechnologies: Principles, Applications, Implications and Hands-on Activities, European Commission, European Union, 2012 Search PubMed;
(b) M. B. Gawande, S. P. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 3371–3393 RSC
-
(a) S. Dang, Q.-L. Zhu and Q. Xu, Nat. Rev. Mater., 2017, 3, 17075 CrossRef
-
(a) N. Yan, Y. Yuan and P. J. Dyson, Dalton Trans., 2013, 42, 13294–13304 RSC
-
(a)
A. Ricci, Amino group chemistry: From synthesis to the life sciences, Wiley-VCH, 2008 Search PubMed
-
(a) S. Gomez, J. A. Peters and T. Maschmeyer, Adv. Synth. Catal., 2002, 344, 1037–1057 CrossRef CAS
-
(a) Y. Nakamura, K. Kon, A. S. Touchy, K.-I. Shimizu and W. Ueda, ChemCatChem, 2015, 7, 921–924 CrossRef CAS
-
(a) T. Gross, A. M. Seayad, M. Ahmad and M. Beller, Org. Lett., 2002, 4, 2055–2058 CrossRef CAS PubMed
- (a) Z. Wang, “Mignonac reaction” in comprehensive organic name reactions and reagents, Wiley, 2010 CrossRef; (b) https://erowid.org/archive/rhodium/chemistry/reductive.amination.html, 2004.
- A. S. Zola, R. U. Ribeiro, J. M. C. Bueno, D. Zanchet and P. A. Arroyo, J. Exp. Nanosci., 2014, 9, 398–405 CrossRef CAS
-
(a) J. Yang, H. Liu, W. N. Martens and R. L. Frost, J. Phys. Chem., 2010, 114, 111–119 CAS
-
(a) A. A. Khandar, B. Shaabani, F. Belaj and A. Bakhtiari, Inorg. Chim. Acta, 2007, 360, 3255–3264 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04963k |
| This journal is © The Royal Society of Chemistry 2020 |