
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConsensus virtual screening of dark chemical matter and food chemicals uncover potential inhibitors of SARS-CoV-2 main protease†
Marisa G. Santibáñez-Morán a,
Edgar López-Lópezb,
Fernando D. Prieto-Martíneza,
Norberto Sánchez-Cruza and
José L. Medina-Franco*a
a,
Edgar López-Lópezb,
Fernando D. Prieto-Martíneza,
Norberto Sánchez-Cruza and
José L. Medina-Franco*a
aDIFACQUIM Research Group, Department of Pharmacy, School of Chemistry, Universidad Nacional Autónoma de México, Mexico City, Mexico. E-mail: medinajl@unam.mx; jose.medina.franco@gmail.com; Tel: +52 (55) 5622-3899, ext. 44458
bDepartment of Pharmacology, Center of Research and Advanced Studies of the National Polytechnic Institute (CINVESTAV), Mexico City, Mexico
First published on 1st July 2020
Abstract
The pandemic caused by SARS-CoV-2 (COVID-19 disease) has claimed more than 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 lives worldwide, and more than nine million people are infected. Unfortunately, an effective drug or vaccine for its treatment is yet to be found. The increasing information available on critical molecular targets of SARS-CoV-2 and active compounds against related coronaviruses facilitates the proposal (or repurposing) of drug candidates for the treatment of COVID-19, with the aid of in silico methods. As part of a global effort to fight the COVID-19 pandemic, herein we report a consensus virtual screening of extensive collections of food chemicals and compounds known as dark chemical matter. The rationale is to contribute to global efforts with a description of currently underexplored chemical space regions. The consensus approach included combining similarity searching with various queries and fingerprints, molecular docking with two docking protocols, and ADMETox profiling. We propose compounds commercially available for experimental testing. The full list of virtual screening hits is disclosed.
000 lives worldwide, and more than nine million people are infected. Unfortunately, an effective drug or vaccine for its treatment is yet to be found. The increasing information available on critical molecular targets of SARS-CoV-2 and active compounds against related coronaviruses facilitates the proposal (or repurposing) of drug candidates for the treatment of COVID-19, with the aid of in silico methods. As part of a global effort to fight the COVID-19 pandemic, herein we report a consensus virtual screening of extensive collections of food chemicals and compounds known as dark chemical matter. The rationale is to contribute to global efforts with a description of currently underexplored chemical space regions. The consensus approach included combining similarity searching with various queries and fingerprints, molecular docking with two docking protocols, and ADMETox profiling. We propose compounds commercially available for experimental testing. The full list of virtual screening hits is disclosed.
1. Introduction
Coronaviruses (COVs) per se can infect humans and other animal species. Some of them cause a variety of previously studied diseases such as Severe Acute Respiratory Syndrome (SARS) and Middle East Respiratory Syndrome (MERS). SARS-CoV-2 is an emergent virus that generates the COVID-19 disease1 which is currently considered a “pandemic” according to the World Health Organization (WHO), with more than ten million confirmed cases and more than 500000 deaths worldwide (as per June 30th, 2020).2
SARS-CoV-2 has a complex architecture, and as happens with different viruses, there are several proteins involved in viral internalization and replication. The life cycle of SARS-CoV-2 starts with the viral recognition of its spike protein by a cellular receptor (ACE receptor and TMPRSS2). After that, the internalization and uncoating process is mediated by membrane proteins. Once into the host cell, RNA replication, and biosynthesis of viral polypeptides are carried out (RdRp – ribosomes). Finally, the processing of precursors proteins by the main protease (3CLpro or Mpro) and the assembly of these, contributes to the generation of new viruses.3–5 These main targets offer a venue for the development of new treatments via rational drug design. Examples include spike protein, RNA polymerase, and chymotrypsin-like cysteine protease (3CLpro or Mpro) which are presented in Fig. 1.3–5 Of these, the main protease (Mpro) is a promising target for the design and proposal of new therapies due to the lack of homologous proteins in humans.6 Also, its selective inhibition would take advantage of the natural life cycle of SARS-CoV-2, avoiding its replication and dissemination. Several research groups are actively pursuing Mpro as a molecular target to identify drug candidates for the treatment of COVID-19.
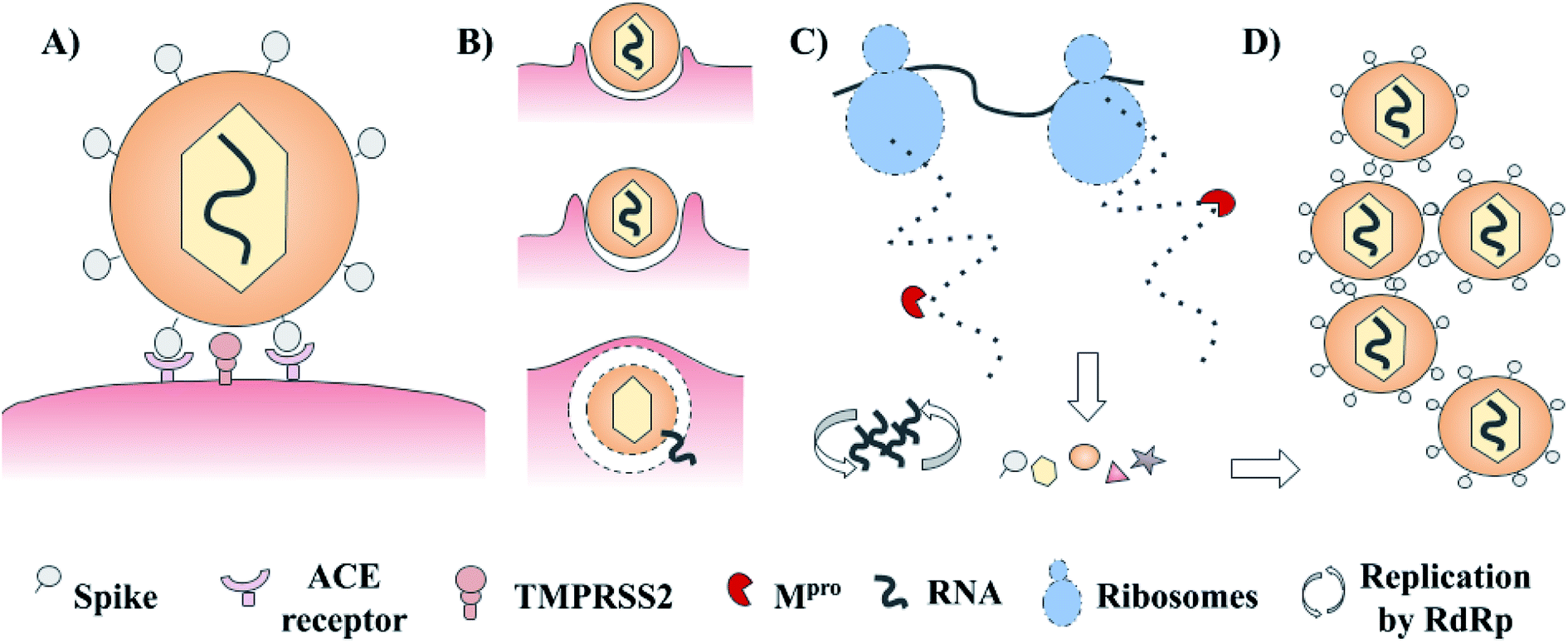 | ||
| Fig. 1 Schematic life cycle and main studied targets of SARS-CoV-2. (A) Cellular recognition; (B) internalization and uncoating process; (C) biosynthesis of viral proteins and RNA replication; and (D) assembly of new virions. | ||
Computational methods represent an approach with the power of efficiently filter large and diverse compound libraries to select potential candidates for drug development.7,8 Recently published works show a tendency towards drug repurposing and to search structurally different libraries (e.g., with broad scaffold diversity), and natural products.9–13 Moreover, the search for novel compounds commercially available or with the possibility of being synthesized has had a vital rebound (e.g., screening part or the entire ZINC database).9,14–16 Table 1 summarizes representative examples of virtual screening (VS) studies directed to different molecular targets, including SARS-CoV-2 Mpro. Most of these efforts relied on structure-based drug design (SBDD). Few others include similarity searching and quantitative structure–activity relationship (QSAR) modeling.17 In this sense, there are many compounds suggested by computational methods that could be evaluated quickly with in vitro techniques. However, the use of computational consensus methodologies could improve the performance of each technique.
| Target | Experimental methods | Libraries | Compounds screened/outcome | Ref. |
|---|---|---|---|---|
| a Computational hits.b Active hits. | ||||
| Mpro | Deep docking | ZINC 15 | 1.3 billion/1,000a | 9 |
| Mpro | Pharmacophore model, molecular docking, and dynamics | Marine natural products | 14064/17a |
10 |
| Mpro | Pharmacophore screening and molecular docking | ZINC | 50000/10a |
15 |
| Spike protein | Homology modeling and molecular docking | FDA | 3300/12a | 18 |
| Mpro, PLpro and RdRp | Homology modeling, molecular docking, and dynamics | DrugBank and traditional Chinese medicine | 1973/57a | 11 |
| ACE2 | Molecular docking | Literature compilation (natural products) | —/5a | 12 |
| Mpro | Molecular docking | Literature compilation (natural products) | 80/8a | 13 |
| Mpro | Molecular docking | FDA | 486/20a | 19 |
| Mpro | Molecular docking, and dynamics | ZINC | 606 million/12a | 20 |
| Mpro | Similarity search and QSAR modeling | DrugBank (marketed, withdrawn, experimental, and investigational) | 9615/41a | 17 |
| Mpro | Molecular docking and dynamics | DrugBank (approved and drug candidates in clinical trials) | 2201/5a | 21 |
| Mpro and TMPRSS2 | Homology modeling and molecular docking | ZINC | 34500/8a |
14 |
| Mpro | Induced fit docking | In-house | 10000/6b |
22 |
The goal of this work is to propose active compounds against Mpro from SARS-CoV-2 and related coronaviruses. One of the novelties of the present study relies on the probed chemical space: food chemicals and molecules in the Dark Chemical Matter (DCM), which to the best of our knowledge, have been explored for SARS-CoV-2 on a limited basis. Thus, the rationale was to expand the search of chemical space and suggest molecules for experimental screening. Active compounds could be later optimized to increase activity. As a screening strategy, we started with similarity searching using different fingerprints to pre-select compounds using data fusion strategies. Selected compounds from similarity searching were screened with molecular docking with two different software. The final selection of computational hits was based on consensus scoring, information of protein–ligand contacts, and the ADMETox (absorption, distribution, metabolism, excretion, and toxicity) profile of compounds. Additional criteria used to guide the selection of hit candidates for testing included predictions by machine learning (ML) models for SARS-CoV-2 activity developed by Collaborations Pharmaceuticals, Inc and freely available.23
2. Materials and methods
Herein we combined ligand- and structure-based methods to virtually screen compounds from two primary molecular databases and select hit candidates for testing. Ligand-based methods were based on similarity searching using the principles of data fusion.24,25 Structure-based approaches were based on molecular docking and consensus scoring.26 The selection of hit compounds was also made considering the predicted ADMETox profile as well as prediction by ML models made freely available by Collaborations Pharmaceuticals. Fig. 2 outlines the main VS strategy and hit selection. Overall, two main general approaches were considered that are distinguished by the type of reference compounds used in the similarity searching. In one method (left-hand side of Fig. 2), three HIV-1 protease inhibitors approved for clinical use were used as queries. As elaborated below in Section 3.1, the three compounds have shown in vitro activity against SARS-CoV or SARS-CoV-2. In the second approach, (right-hand side of Fig. 2) 1052 compounds with potential affinity for SARS-CoV-2 Mpro or SARS-CoV Mpro were used as queries. The workflow in Fig. 2 is described in more detail in the next subsections.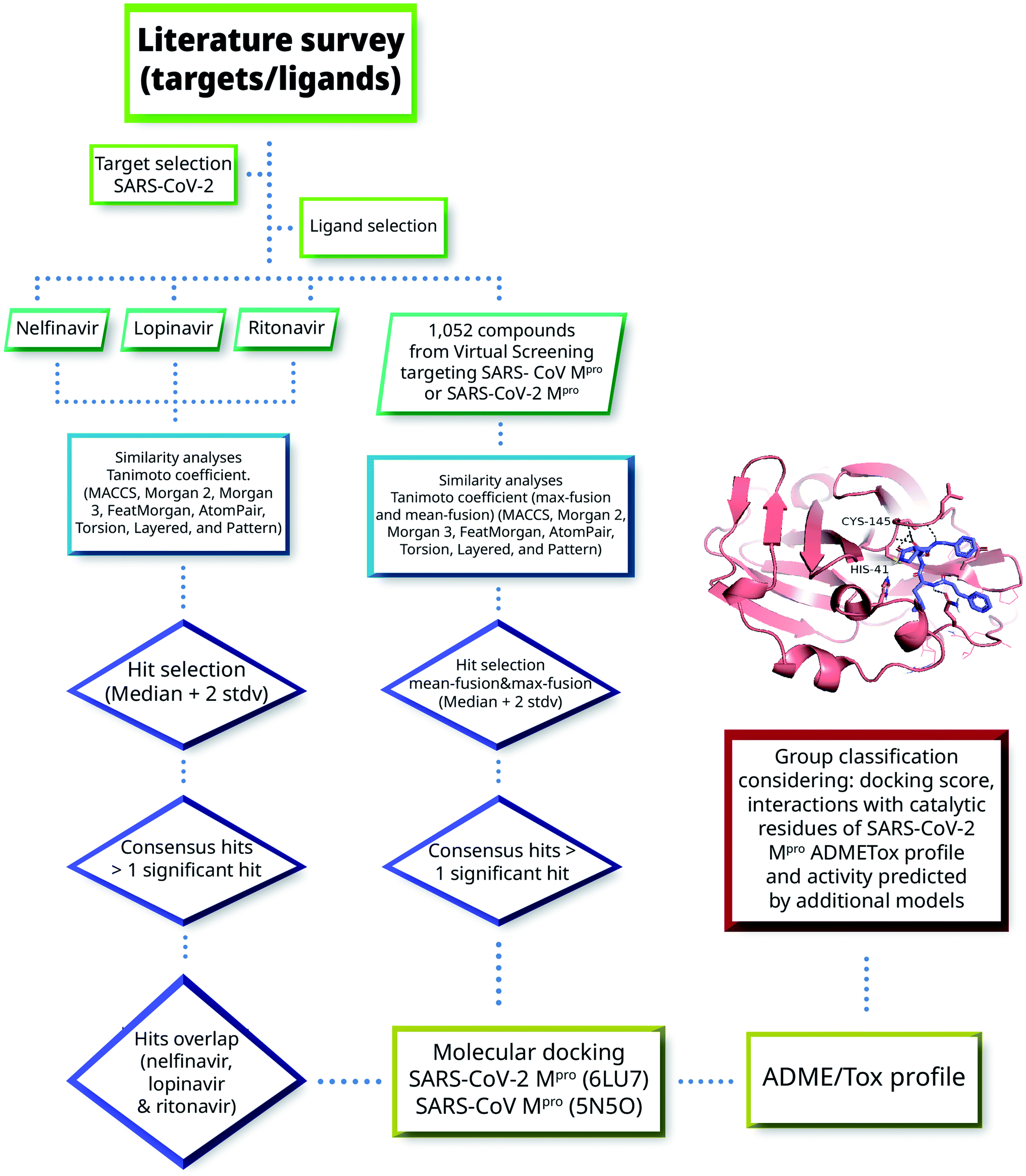 | ||
| Fig. 2 General workflow of the virtual screening approach used in this work. | ||
2.1 Screening and reference databases
Table 2 summarizes the four major types of data sets considered in this study.| Dataset | Content overview and sizea | Rationale | Ref. |
|---|---|---|---|
| a After data curation. | |||
| Actives | N3, alpha-ketoamides 11a, 11r, and 11s, carmofur, cinaserin, disulfiram, ebselen, PX12, shikonin, and tideglusib | Reference compounds used in docking to compare docking scores and predicted binding modes | 22 and 27 |
| FooDB | 22880 compounds |
Large library of food chemicals. Smaller food chemical data sets have been screened | 28 |
| Dark chemical matter (DCM) | 139329 compounds |
Large screening library underexplored. Likelihood to shade light into the darkness of the COVID-19 pandemic | 29 |
| ZINC (top-ranked hits) | 10 top-ranked virtual screening hits of ZINC using deep docking/Glide and SARS-CoV-2 Mpro (PDB ID: 6LU7) | Further consensus of published computational hits with other docking programs (Vina and MOE) | 9 |
One of the screening databases was the public food chemical database (FooDB) with 23883 compounds.28 The chemical diversity and coverage of chemical space of FooDB have been reported revealing that food chemicals are structurally diverse and have, in general, large molecular complexity.30 DCM was the other screening database. DCM is a collection of 139352 compounds that had shown no activity when tested in at least 100 screening assays.29 Even though DCM has a low activity profile against common targets, the rationale of screening this collection was to explore regions in chemical space currently overlooked. Moreover, DCM has yielded active molecules in other assays31,32 probing the value of screening this region of the chemical space. The structures of FooDB and DCM were curated and standardized, employing RDKit, CDK (Chemistry Development Kit), and ChemAxon tools. The largest component of molecules with more than one fragment was retained, compounds containing an atom type other than H, C, O, N, S, P, F, Cl, Br, I, B, Si, and Se were removed. The tautomer with the lowest energy for each remaining compound was generated.
Active compounds from the study of Jin et al.22 were used as a reference. These were the peptide-like inhibitor N3, carmofur, cinaserin, disulfiram, ebselen, PX12, shikonin, tideglusib, and alpha-keto amides (11a, 11r, 11s).27 Lopinavir, nelfinavir, and ritonavir were other reference compounds for the molecular docking performed in AutoDock Vina.
To identify additional potential hit compounds, we included the top 10 ranked virtual screening hits from the study of Ton et al.9 Authors of that work screened the ZINC database against the SARS-CoV-2 Mpro (PDB ID 6LU7) using the docking program Glide. The rationale of using this set was to explore further the predicted profile of top-ranked compounds using different docking programs (i.e., Vina and MOE, vide infra).
2.2 Similarity searching
Eight two-dimensional molecular fingerprints (Molecular ACCess System-MACCS-keys (166-bits), Morgan 2 [ECFP4-like], Morgan 3 [ECFP6-like], FeatMorgan, AtomPair, Torsion, Layered, and Pattern) were generated for all the queries, the 22880 compounds in FooDB, and 139329 molecules in DCM.
In the first virtual screening approach (Fig. 2), nelfinavir, lopinavir, and ritonavir were used as independent queries (vide infra). The molecular similarity between each of the queries and each of the molecules in FooDB and DCM was estimated with the Tanimoto coefficient.33 The compounds with a Tanimoto coefficient higher than the median plus two standard deviations were considered as a hit. The molecules labeled as hits according to more than one molecular fingerprint (consensus hits), were selected. The consensus hits for the three queries were additionally analyzed by molecular docking.
In the second approach (Fig. 2), 1052 compounds with potential affinity for SARS-CoV-2 Mpro or SARS-CoV Mpro were selected from published molecular docking studies9,19,27,34,35,59 and used as queries. The structure file with the chemical structures of the 1052 compounds is available in the ESI.† Mean-fusion similarity scores and max-fusion similarity scores were determined using the eight molecular fingerprints and the Tanimoto coefficient.36 Compounds with max-fusion similarity scores and mean-fusion similarity scores higher than the median plus two standard deviations for more than one fingerprint were selected as consensus hits and evaluated by molecular docking.
The molecular similarity analyses were generated in KNIME employing the RDKit node for molecular fingerprints generation and the CDK node for the similarity calculation.37,38
2.3 Molecular docking
To enhance the likelihood of finding active compounds, two docking programs with different algorithms were used, namely; Autodock Vina, version 1.1.2,39 and Molecular Operating Environment (MOE) v.2019.40 As explained hereunder, the docking protocols for each program were validated with experimental information available.Docking with Autodock Vina was conducted with two crystallographic structures obtained from the Protein Data Bank (PDB),41 namely, SARS-CoV-2 Mpro (PDB ID 6LU7)22 and the structurally related SARS-CoV Mpro (PDB ID 5N5O).42 Both structures are co-crystallized with a peptide-like (N3) and an alpha-ketoamide (11s) inhibitor, respectively. The crystal structures were prepared in Autodock Tools. The grid-box was constructed based on the binding site of the alpha-ketoamide inhibitors 11a and 11s. The ligands were normalized, their clean 3D form was generated, hydrogens were added, and molecules were optimized using the Universal Force Field (UFF) in KNIME. The results were visualized in PyMol (version 2.3).
Induced fit docking protocol for the Mpro (PDB ID 6LU7) of SARS-CoV-2 was carried out with MOE software v.2019. The protein was prepared with the “Quick prepare” tool using the parameters assigned by the PFROSST force field. The peptide-like inhibitor N3 was removed, and their binding site was used to direct the docking. Triangle matcher method was refined with the induced fit protocol, and the other parameters were established by default. This protocol was validated using experimental information recently published by Jin et al.22 The binding poses were successfully reproduced. The binding scores showed a correlation of 0.703 with the in vitro inhibition values of the data set.
2.4 ADME/Tox profiling
Early consideration of ADMET/Tox properties is fundamental in current drug discovery efforts. Due to the availability of several free chemoinformatic resources,43 herein we employed SwissADME44 to calculate more than 40-related properties including descriptors associated with drug-likeness, solubility, blood-brain barrier (BBB) permeability, Pgp substrate, inhibition of CYPs, Bioavailability Score, PAINS alerts, and the number of violations to empirical rules (Lipinski, Veber, Egan, Brenk). The full list of ADME/Tox related properties calculated with SwissADME is in the ESI.† We have used SwissADME to profile other compound databases of pharmaceutical relevance.453. Results and discussion
We describe the results of similarity searching, molecular docking, and ADMETox followed by the combined analysis to select hit compounds for experimental testing.3.1 Similarity searching
As previously stated, Mpro is a promising drug target due to its importance in COVs life cycle (Fig. 1, vide supra). The recent publication of the SARS-CoV-2 Mpro crystal structure showed a 96% similarity with the SARS-CoV Mpro and the conservation of the active binding site. To search for SARS-CoV-2 Mpro inhibitors in underexplored regions of the chemical space, we assessed the molecular similarity of FooDB and DCM databases with compounds that potentially inhibit SARS-CoV Mpro or SARS-CoV-2 Mpro. As a first approach, three HIV-1 protease inhibitors approved for clinical use, namely; lopinavir, ritonavir, and nelfinavir were used as queries or reference compounds. Lopinavir and ritonavir have shown activity against SARS-CoV46,47 and are currently under clinical trials for the treatment of COVID-19. In addition, molecular dynamics predicted binding affinity of both molecules for the active site of SARS-CoV Mpro48 and there is recent evidence of in vitro activity of lopinavir against SARS-CoV-2.49 Another protease inhibitor with in vitro activity against SARS-CoV,50 nelfinavir, has been predicted to have high binding affinity to the SARS-CoV-2 Mpro by molecular dynamics.51,52 Thus, nelfinavir was also included as a reference for the similarity search. Despite those observations, there is still no conclusive evidence of the effectiveness of these drugs in the treatment of COVID-19 (ref. 53–57) which encourages the identification of other existing molecules that target SARS-CoV-2.After the ligands were prepared (as described in the Methods Section 2.3), 143 consensus hits from FooDB were found to be highly similar to nelfinavir, lopinavir, and ritonavir (i.e., with similarity values above than the median plus two standard deviations). From the 143 consensus hits, 40 compounds with drug-like properties were selected for more analyses. Five hundred compounds were selected from the DCM database with significantly high Tanimoto similarity values to nelfinavir, lopinavir, and ritonavir. DCM compounds are constantly tested in HTS assays, and therefore, they were considered to have suitable physicochemical properties for drug development. In this sense, it is not surprising that a more significant number of consensus hits for the three drugs were found in DCM, considering that the molecular and physicochemical properties of DCM do not significantly differ from approved drugs. In contrast, FooDB was not assembled to be “drug-like.”
A small dataset of 1052 compounds with predicted affinity to SARS-CoV-2 Mpro was assembled to broaden the search of potential Mpro inhibitors. Although these alternative reference compounds are potentially (but not confirmed) active, it has been suggested that they can increase the likelihood to identify active molecules. Such an approach is reminiscent of what has been described as “turbo-similarity searching”.58 As more data becomes available, a more chemically diverse and larger set could be integrated. Meanwhile, the top hits reported in six peer-reviewed molecular docking studies were included.9,19,27,34,35,59 After ligand preparation, 178 and 174 consensus hits from FooDB and DCM were recovered, respectively. Significant hits were found for five of the eight molecular fingerprints, highlighting the advantages of using multiple molecular fingerprints.60
Four compounds were overlapping consensus hits from both similarities searching methods. DBB13044 and DBB18117 from FooDB, and DCM33835 and DCM97265 from DCM database.
The total number of consensus hits further analyzed by molecular docking and ADMETox in silico profiling was 888 compounds (including stereoisomers).
3.2 Molecular docking
Molecular docking of SARS-CoV Mpro was performed with Autodock Vina (PDB ID 5N5O). The docking scores for the reference compounds ranged from −8.5 to −4.1 kcal mol−1, with a mean value of −6.8 kcal mol−1. Of note, lopinavir, ritonavir, and nelfinavir were included as references. A total of 393 compounds, from the hits selected by molecular similarity, fell above (less favorable) the mean docking score. However, reference compounds with docking scores above the mean value, such as ebselen (−6.2 kcal mol−1) bound to the active site of SARS-CoV Mpro by four hydrogen bonds with residues Lys141, Gly143, Ser144, and Cys145. Hence, a hard cut-off value purely based on docking scores was not established. The docking scores for the reference compounds docked to SARS-CoV-2 Mpro carried out in MOE ranged from −9.4 to −5.16 kcal mol−1.Fig. 3 shows the predicted binding mode of representative hits compounds with Mpro. As discussed hereunder in the Section 3.4 Hit Selection (vide infra), the selected hit compounds shown in Fig. 3 had favorable docking scores with Vina and MOE and had at least one interaction with the catalytic residues His41, Cys145 and/or Glu166 (key interactions reported).22 According to the docking models, other important key interactions were observed. DBB2790 makes Pi–H interactions with sidechain of His 41, H-bond interaction with the sidechain of Cys 145 and H-bonds interactions with the sidechain and backbone of Glu 166; DCM78683 makes H-bond interactions with the sidechain of Asn 142 and Cys 145, and DCM111769 makes Pi–H interactions with Glu 166. These proposed compounds are predicted to preferentially bind to the P1, P2, and P3 regions.
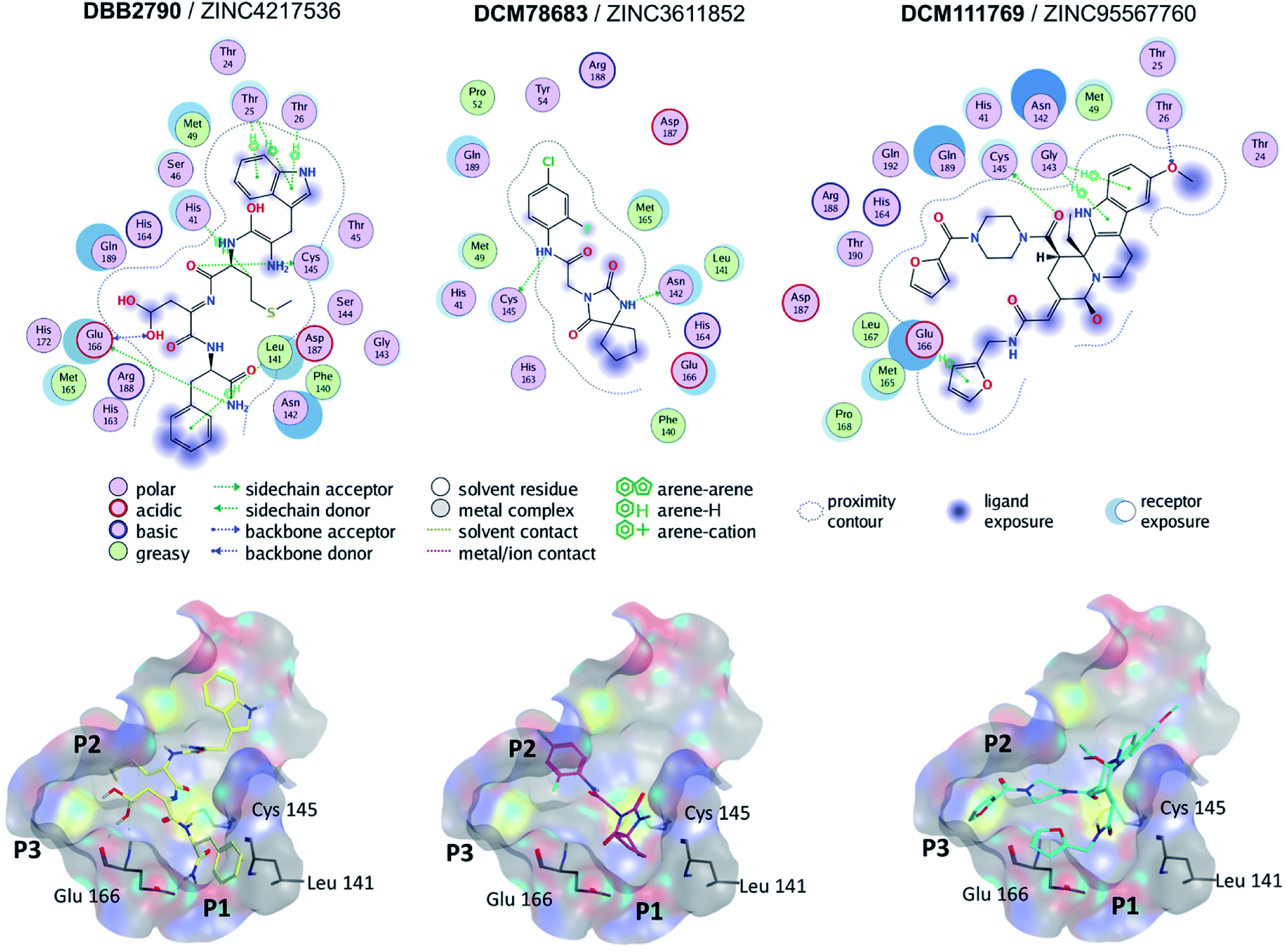 | ||
| Fig. 3 Binding modes of three selected hits within SARS-CoV-2 Mpro (PDB ID 6LU7) as predicted by Molecular Operating Environment. | ||
A literature survey revealed that the VS hit DBB2790 (Fig. 3) has a high structural similarity to compound GC373 (a molecule with nanomolar activity against Mpro from SARS-CoV-2).61 In 2013 Kim et al. reported GC373 as an inhibitor of Mpro from feline coronavirus.62 Moreover, the protein–ligand interactions of both compounds and Mpro are similar. These observations support the potential antiviral activity of DBB2790.
3.3 ADMETox
For 888 selected hits, the ADMETox-related descriptors were computed with SwissADME. As described hereunder, some of these descriptors were used as a guide for the classification of hit compounds in different priority groups. The main types of ADMETox descriptors considered were those associated with drug-likeness, solubility, and cytochromes' inhibition.3.4 Hit selection
Instead of establishing stringent (and arguably heuristic and hard) cut-off values, the compounds selected by molecular similarity were classified into four groups considering their interactions with the catalytic residues of the SARS-CoV-2 Mpro (H41 and C145), their commercial availability, ADMETox characteristics, and their predicted activity by ML. Thereby, most compounds with suitable profiles were classified into one of the groups. The number of the group is associated with the priority for acquisition and testing. Table 3 summarizes the group classification strategy and the number of compounds that were classified into each group. A further description of each group is presented below.| Group | Number of compounds | Commercial availabilitya | In silico safety criteriab | Hydrogen bonds with H41 or C145 | Active according to machine learning |
|---|---|---|---|---|---|
| a Compounds reported as “in-stock” in the ZINC database were considered commercially available.b Compounds that do not have PAINS alerts, do not pass through the BBB, and are predicted to not inhibit CYP1A2, CYP2C19, CYP2C9, CYP2D6 or CYP3A4. | |||||
| 1 | 41 | Available | Safe | Present | Active/inactive |
| 2 | 10 | Available | Safe | Not present | Active |
| Available | Not safe | Present | Active | ||
| 3 | 34 | Not available | Safe | Present | Active/inactive |
| 4 | 20 | Not available | Safe | Not present | Active |
| Not available | Not safe | Present | Active | ||
Group 1 includes commercially available compounds that meet our safety criteria (based on the predictions of SwissADME), i.e., they do not have PAINS alerts, do not pass through the BBB, and do not inhibit CYP1A2, CYP2C19, CYP2C9, CYP2D6 or CYP3A4. The molecules in this group are predicted to form hydrogen bonds with at least one of the catalytic residues of PDB ID 6LU7. Table 4 summarizes the 41 molecules that fell into this top priority group.
| Set | ID | ZINC ID | Vina's score 5N5O kcal mol−1 | MOE's score 6LU7 kcal mol−1 | GIa absorption | Pgpb substrate | Alic log_S | Ali class | Lipinski violations | Brenk violations | Bioavailabilityd |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a GI gastrointestinal.b Pgp P-glycoprotein.c Ali topological method implemented from Ali J. et al. 2012.63d Probability that the compound will have F > 10%.e Compounds that do not violate any of the following rules: Lipinski, Ghose, Veber, Egan, and Muegge.f Compounds predicted to be active by the ML model. | |||||||||||
| foodb_mfsm | DBB9450 | 169676920 | −6.6 | −10.9 | Low | Yes | −8.39 | Poorly soluble | 3 | 4 | 0.17 |
| foodb_mfsm | DBB5554 | 85545908 | −7.9 | −10.9 | Low | Yes | −6.76 | Poorly soluble | 3 | 2 | 0.17 |
| foodb_mfsm | DBB2790 | 4217536 | −7.8 | −9.3 | Low | Yes | −6.4 | Poorly soluble | 3 | 3 | 0.17 |
| dcm_ch | DCM110214 | 34805301 | −7.4 | −9.2 | Low | Yes | −2.7 | Soluble | 1 | 1 | 0.55 |
| dcm_ch | DCM122034 | 15990331 | −7 | −8.9 | High | Yes | −3.55 | Soluble | 0 | 1 | 0.55 |
| dcm_ch | DCM73598 | 8918473 | −7.2 | −8.7 | Low | Yes | −4.01 | Moderately soluble | 1 | 1 | 0.55 |
| foodb_mfsm | DBB2455 | 53057130 | −7.6 | −8.6 | Low | Yes | −4.76 | Moderately soluble | 1 | 1 | 0.55 |
| dcm_ch | DCM2279 | 38144961 | −6.8 | −8.5 | Low | Yes | −3.84 | Soluble | 1 | 1 | 0.55 |
| dcm_ch | DCM82216 | 4270581 | −7.1 | −8.3 | High | Yes | −2.36 | Soluble | 0 | 1 | 0.55 |
| dcm_ch | DCM55533 | 8917865 | −6.4 | −8.3 | High | Yes | −1.82 | Very soluble | 0 | 1 | 0.55 |
| dcm_ch | DCM119353 | 9409555 | −7.8 | −8.2 | Low | Yes | −4.18 | Moderately soluble | 0 | 0 | 0.56 |
| dcm_ch | DCM65267 | 100771995 | −6.2 | −8.2 | Low | Yes | −1.75 | Very soluble | 0 | 0 | 0.55 |
| foodb_mfsm | DBB13825 | 4228235 | −7.4 | −8.1 | Low | No | 0.85 | Highly soluble | 2 | 4 | 0.17 |
| dcm_ch | DCM131779 | 9159501 | −6.4 | −8 | Low | Yes | −3.37 | Soluble | 1 | 1 | 0.55 |
| dcm_ch | DCM65270 | 100778159 | −7.2 | −7.9 | High | Yes | −0.63 | Very soluble | 0 | 0 | 0.55 |
| dcm_ch | DCM82831 | 9109751 | −7.8 | −7.8 | Low | No | −2.37 | Soluble | 0 | 1 | 0.55 |
| foodb_mfsm | DBB13483 | 5283951 | −6.3 | −7.8 | High | No | −3.7 | Soluble | 0 | 2 | 0.55 |
| foodb_mfsm | DBB13002 | 2005305 | −7.3 | −7.8 | Low | No | −3.27 | Soluble | 2 | 4 | 0.11 |
| foodb_mfsm | DBB14163 | 8577218 | −7.4 | −7.7 | Low | No | −2.11 | Soluble | 2 | 2 | 0.11 |
| dcm_ch | DCM131783 | 15954557 | −6.9 | −7.7 | High | Yes | −2.24 | Soluble | 0 | 1 | 0.55 |
| dcm_ch | DCM93255e | 32980237 | −7.2 | −7.7 | High | No | −3.22 | Soluble | 0 | 1 | 0.55 |
| foodb_mfsm | DBB13917 | 2036915 | −7.5 | −7.7 | Low | No | −2.74 | Soluble | 2 | 2 | 0.11 |
| dcm_mfsm | DCM116923e | 2970717 | −6.5 | −7.7 | High | Yes | −2.54 | Soluble | 0 | 1 | 0.55 |
| dcm_ch | DCM10478 | 4083870 | −6.6 | −7.6 | Low | No | 0.02 | Highly soluble | 1 | 3 | 0.55 |
| dcm_ch | DCM28770 | 100778693 | −6.7 | −7.6 | High | Yes | −2.52 | Soluble | 0 | 0 | 0.55 |
| dcm_ch | DCM33486e | 1181094 | −6.6 | −7.5 | High | Yes | −3.62 | Soluble | 0 | 1 | 0.55 |
| dcm_ch | DCM30682e | 1577795 | −6.4 | −7.5 | High | No | −3.76 | Soluble | 0 | 2 | 0.55 |
| dcm_ch | DCM110206e | 12652624 | −7.2 | −7.5 | High | Yes | −4.15 | Moderately soluble | 0 | 2 | 0.55 |
| dcm_mfsm | DCM91011e | 6754750 | −7.7 | −7.4 | High | No | −1.49 | Very soluble | 0 | 1 | 0.55 |
| foodb_mfsm | DBB13919 | 4228265 | −7.7 | −7.4 | Low | No | −1.97 | Very soluble | 2 | 2 | 0.17 |
| foodb_mfsm | DBB17132e | 20431033 | −6.2 | −7.1 | High | No | −1.7 | Very soluble | 0 | 2 | 0.55 |
| dcm_ch | DCM131782e | 2126038 | −7.1 | −7.1 | High | No | −0.08 | Very soluble | 0 | 1 | 0.55 |
| dcm_mfsm | DCM71724 | 18056800 | −6.2 | −7.1 | Low | Yes | −4.37 | Moderately soluble | 0 | 2 | 0.55 |
| dcm_mfsm | DCM94188e | 18143600 | −7.1 | −6.9 | High | No | −2.63 | Soluble | 0 | 0 | 0.55 |
| foodb_mfsm | DBB20185 | 2242693 | −6.1 | −6.6 | Low | No | 0.98 | Highly soluble | 0 | 2 | 0.55 |
| foodb_mfsm | DBB17114 | 4090721 | −7 | −6.5 | High | No | −1.38 | Very soluble | 0 | 2 | 0.55 |
| foodb_mfsm | DBB18961 | 4321512 | −6.8 | −6.5 | Low | No | −0.96 | Very soluble | 0 | 0 | 0.55 |
| foodb_mfsm | DBB18947 | 1303441 | −6.1 | −6.1 | High | No | −0.42 | Very soluble | 0 | 0 | 0.55 |
| foodb_mfsm | DBB19736 | 2040854 | −5.4 | −6.1 | High | No | 2.05 | Highly soluble | 0 | 0 | 0.55 |
| foodb_mfsm | DBB19719 | 1532770 | −5.6 | −5.9 | High | No | 1.67 | Highly soluble | 0 | 0 | 0.55 |
| foodb_mfsm | DBB21857e,f | 895813 | −5.8 | −5.6 | High | No | −1.75 | Very soluble | 0 | 0 | 0.56 |
Group 2 comprises ten commercially available compounds that are predicted to be active by ML, but they violate one of the other two criteria. They can meet our safety criteria and do not form hydrogen bonds with the catalytic residues. Else, they can form hydrogen bonds with the catalytic residues but do not meet our safety criteria.
Group 3 consists of 34 molecules that are not commercially available but meet the safety criteria and form hydrogen bonds with at least one of the catalytic residues. These compounds would be suited for synthesis and testing.
Group 4 contains 20 molecules that are not commercially available and are predicted to be active by ML. However, they do not meet the safety criteria or do not form hydrogen bonds with the catalytic residues. According to our classification, compounds in this group would have the lowest priority for acquiring (synthesizing since they are not commercially available) and testing. Compounds that do not fall into any of these four groups were considered as non-priority for acquisition.
Table 4 summarizes the in silico profile of representative hit compounds selected for experimental validation.
Table 5 summarizes the information of 18 compounds listed in group 1 from FooDB with their corresponding IDs and annotated sources. Interestingly, some of the selected hits that were structurally similar to potential Mpro inhibitors were from endogenous sources. For instance, angiotensin II (DBB9450) and angiotensin IV (DBB5554) (a degradation product) were predicted as binders of the active site of SARS-CoV-2 Mpro. Key interactions predicted were hydrogen-bonds with His41, Ser46, Cys145, Gln189 (DBB9450) and Thr26, Met49, Cys145, and Glu166 (DBB5554). Angiotensin II (ANG-II) is an octapeptide hormone product of angiotensin I's cleavage by the angiotensin-converting enzyme (ACE). ANG-II binds to AT1 and AT2 receptors; the activation of AT1 receptors by ANG-II induces vasoconstriction, vasopressin and aldosterone release, thirst, renal sodium reabsorption, angiogenesis, vascular aging, and inflammation. ANG-II can be converted to angiotensin 1–7 by the angiotensin-converting enzyme II (ACE2). The action of aminopeptidase A and aminopeptidase N produces angiotensin III and angiotensin IV, respectively.
| IDs | FooDB annotation |
|---|---|
| DBB9450/FDB022383 | Angiotensin II, endogenous |
| DBB5554/FDB022385 | Angiotensin IV |
| DBB2790/FDB023765 | Tetragastrin, endogenous |
| DBB2455/FDB023767 | Morphiceptin, endogenous |
| DBB13825/FDB031192 | Tetrahydrofolate |
| DBB13483/FDB013079 | Neotame, artificial sweetener |
| DBB13002/FDB022600 | 5-Methyltetrahydrofolic acid (5-MTHF) |
| DBB14163/FDB014504 | Folic acid |
| DBB13917/FDB022702 | Aminopterin |
| DBB13919/FDB022395 | Dihydrofolic acid |
| DBB17132/FDB028374 | Phenylbutyrylglutamine, metabolite of phenylbutyrate |
| DBB20185/FDB003618 | Gamma-L-glutamyl-L-phenylalanine, soft-necked garlic |
| DBB17114/FDB029352 | Indole acetyl glutamine, endogenous |
| DBB18961/FDB023789 | N4-Acetylcytidine, endogenous |
| DBB18947/FDB022917 | 5-Methyldeoxycytidine (5-mdc) |
| DBB19736/FDB012937 | Carnosine 44A |
| DBB19719/FDB022217 | Homocarnosine, metabolite |
| DBB21857/FDB022212 | Hydroxyphenylacetylglycine, endogenous human metabolite |
Angiotensin 1–7 has opposite actions to ANG-II. Because ACE2 mediates the entry of SARS-CoV-2 to the host cells and ACE2 activity may be downregulated after virus infection, the accumulation of ANG-II could be linked to the development of severe symptoms of COVID-19 disease. If Mpro inhibitors are structurally similar to ANG-II, their potential binding affinity for the active site of ACE2 should be evaluated. Some studies have assessed the ability of ACE2 inhibitors to prevent SARS-CoV from entering into the cells.64 However, the inhibition of the ACE2 function could cause overaccumulation of ANG-II and promote its undesired effects.
Nonetheless, probably, DCM compounds may not elicit a dual inhibition of SARS-CoV-2 Mpro and ACE2, considering that these molecules had shown no activity against common targets evaluated in HTS assays.
Food folates like 5-MTHF, folic acid, dihydrofolic acid, and tetrahydrofolate (Table 5) were also among the compounds in the top priority group with observed hydrogen bonds to the catalytic residues of the SARS-CoV-2 Mpro, and favorable docking scores (below −7.4 kcal mol−1). Folates are cofactors in many one-carbon transfer reactions, including nucleotide synthesis for DNA and RNA synthesis, interconversion of serine and glycine, methionine generation and methylation of histones, DNA, proteins, phospholipids, and neurotransmitters. Folate deficiency has been linked to neural tube defects, brain dysfunction, coronary heart disease, and increased risk of colorectal and breast cancer.65 Since mammalian cells cannot synthesize de novo folate, naturally occurring food folates and synthetic folic acid are used in dietary supplements and fortified food.
Nevertheless, recent studies showed that a high intake of folic acid might be associated with a risk of developing leukemia and other conditions such as cancer, arthritis, insulin resistance, and masking deficiency of vitamin B12.66 Thus, the implications of low and high plasma levels of folates in COVID-19 patients must be evaluated. Our results suggest that folates could inhibit SARS-CoV-2 Mpro, but their activity in in vitro and in vivo assays remains to be confirmed. To broaden our knowledge of the impact of a healthy diet, and the specific mechanisms through which food chemicals participate in the progression of COVID-19 disease could be a simple approach for the prevention and combat of the current pandemic.
Intriguingly, aminopterin (DBB13917), a folic acid analog that inhibits the dihydrofolate reductase enzyme was also a potential Mpro inhibitor. Aminopterin is one of the so-called antifolates that interfere with folate metabolism and in turn nucleotide synthesis. Currently, an aminopterin analog with less toxic effects, methotrexate, is under clinical trials for the treatment of COVID-19 disease (NCT04352465). Methotrexate is an immunosuppressant used in the treatment of cancer and inflammatory conditions; it is often concurrently administered with folic acid.
3.5 Top-ranked hits from deep docking of ZINC
The ten top-ranked compounds from the analysis conducted by Ton et al. were included in this study (vide supra).9 Even though the ML model did not predict activity against the main protease for these molecules, they represent new hits selected from billions of compounds in the ZINC database. They had good docking scores in our analyses, and three of them ZINC1218583693, ZINC1186058814, and ZINC1655436520 met our safety criteria and had interactions with the catalytic residues of SARS-CoV-2 Mpro. Furthermore, ZINC1655436520 also formed hydrogen bonds with residues Phe140, Leu141, Gly143, Ser144, Cys145, and Glu166 of SARS-CoV Mpro, it is predicted to have good water solubility and high GI absorption, and it does not violate Lipinski's, Ghose, Veber, Egan or Muegge rules.4. Conclusions
Herein we report a consensus structure- and ligand-based virtual screening of two large chemical databases, namely, 22880 food chemicals and 139329 compounds classified as dark chemical matter to identify potential drug candidates for the treatment of COVID-19 targeting the SARS-CoV-2 Mpro. This work is part of our continued effort to identify systematically bioactive food chemicals.67 We also screened top-ranked hits identified in a previous VS of 1.6 billion molecules from ZINC using Glide.9 The similarity searching was done following two approaches. The first approach yielded 40 drug-like food chemicals and 500 DCM molecules with high similarity to nelfinavir, lopinavir, and ritonavir. The data fusion approach returned 178 food chemicals and 174 DCM compounds. In total, 888 hit compounds were subject to molecular docking with two docking programs. The hit compounds were selected considering docking score, predicted interactions with key residues, and ADMETox profiling. An additional criterion used as a guide was a prediction by ML models developed by collaborators in North Carolina, USA.68 After the selection criteria, 105 hits in total were identified, of which several are commercially available (and with reasonable prices) and ready for experimental testing. The full list of hit compounds annotated with the in silico profile is available in the ESI.† We disclose that a preliminary version of this work is available as a preprint.69 This work contributes to a global effort to screen compound databases from different sources aimed at identifying candidate drugs for the treatment of COVID-19. To the best of our knowledge, this is one of the first reports to systematically screen a large food chemical database and one of the first to explore the molecules in DCM for COVID-19.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
E. López-López thanks CONACyT (Consejo Nacional de Ciencia y Tecnología, Mexico) for the scholarship granted: 762342. F. D. Prieto-Martínez and N. Sánchez-Cruz are also grateful to CONACYT for the PhD scholarship granted, No. 660465/576637, and 335997, respectively. We thank the financial support of the NUATEI (Nuevas Alternativas para el Tratamiento de Enfermedades Infecciosas) program IBT-UNAM to purchase MOE license. Valuable discussions with Artem Cherkasov and members of the DIFACQUIM research group, in particular Noemi Angeles Durán-Iturbide and Alejandro Gómez-García, are greatly acknowledged. Authors would like to thank Kenia Morales-Bermeo for help designing Fig. 2.References
- E. de Wit, N. van Doremalen, D. Falzarano and V. J. Munster, Nat. Rev. Microbiol., 2016, 14, 523–534 CrossRef CAS PubMed.
- Johns Hopkins Coronavirus Resource Center, https://coronavirus.jhu.edu/ Search PubMed.
- B. W. Neuman and M. J. Buchmeier, Adv. Virus Res., 2016, 96, 1–27 CrossRef CAS PubMed.
- J. S. Morse, T. Lalonde, S. Xu and W. R. Liu, ChemBioChem, 2020, 21, 730–738 CrossRef CAS PubMed.
- H. Lee, A. Mittal, K. Patel, J. L. Gatuz, L. Truong, J. Torres, D. C. Mulhearn and M. E. Johnson, Bioorg. Med. Chem., 2014, 22, 167–177 CrossRef CAS PubMed.
- T. Pillaiyar, M. Manickam, V. Namasivayam, Y. Hayashi and S.-H. Jung, J. Med. Chem., 2016, 59, 6595–6628 CrossRef CAS PubMed.
- F. D. Prieto-Martínez, E. López-López, K. Eurídice Juárez-Mercado, and J. L. Medina-Franco, in In Silico Drug Design, Elsevier, 2019, pp. 19–44 Search PubMed.
- E. López-López, C. Barrientos-Salcedo, F. D. Prieto-Martínez and J. L. Medina-Franco, Adv. Protein Chem. Struct. Biol., 2020 DOI:10.1016/bs.apcsb.2020.04.001.
- A.-T. Ton, F. Gentile, M. Hsing, F. Ban and A. Cherkasov, Mol. Inf., 2020 DOI:10.1002/minf.202000028.
- D. Gentile, V. Patamia, A. Scala, M. T. Sciortino, A. Piperno and A. Rescifina, Mar. Drugs, 2020, 18, 225 CrossRef PubMed.
- A. Huang, X. Tang, H. Wu, J. Zhang, W. Wang, Z. Wang, L. Song, M. Zhai, L. Zhao, H. Yang, X. Ma, S. Zhou, J. Cai, Preprints 2020, http://www.preprints.org/manuscript/202003.0239/v1.
- H. Chen and Q. Du, Preprints, 2020 DOI:10.20944/preprints202001.0358.v3.
- S. Adem, V. Eyupoglu, I. Sarfraz, A. Rasul and M. Ali, Preprints, 2020 DOI:10.20944/preprints202003.0333.v1.
- A. D. Elmezayen, A. Al-Obaidi, A. T. Şahin and K. Yelekçi, J. Biomol. Struct. Dyn., 2020 DOI:10.1080/07391102.2020.1758791.
- B. Andrade, P. Ghosh, D. Barth, S. Tiwari, R. José Santana Silva, W. Rodrigues de Assis Soares, T. Silva Melo, A. dos Santos Freitas, P. González-Grande, L. Sousa Palmeira, L. Carlos Junior Alcantara, M. Giovanetti, A. Góes-Neto and V. Ariston de Carvalho Azevedo, Preprints, 2020 DOI:10.20944/preprints202004.0003.v1.
- A. Fischer, M. Sellner, S. Neranjan, M. A. Lill and M. Smieško, ChemRxiv, 2020 DOI:10.26434/chemrxiv.11923239.v1.
- T. Bobrowski, V. Alves, C. C. Melo-Filho, D. Korn, S. S. Auerbach, C. Schmitt, E. Muratov and A. Tropsha, ChemRxiv, 2020 DOI:10.26434/chemrxiv.12153594.v1.
- K. Senathilake, S. Samarakoon and K. Tennekoon, Preprints, 2020 DOI:10.20944/preprints202003.0042.v2.
- M. Kandeel and M. Al-Nazawi, Life Sci., 2020, 251, 117627 CrossRef CAS PubMed.
- A. Fischer, M. Sellner, S. Neranjan, M. Smieško and M. A. Lill, Int. J. Mol. Sci., 2020, 21, 3626 CrossRef PubMed.
- J. Wang, J. Chem. Inf. Model., 2020, 60, 3277–3286 CrossRef CAS PubMed.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang, C. Peng, Y. Duan, J. Yu, L. Wang, K. Yang, F. Liu, R. Jiang, X. Yang, T. You, X. Liu, X. Yang and H. Yang, Nature, 2020, 582, 289–293 CrossRef CAS PubMed.
- Molecular Materials Informatics, Inc., Assay Central Predictions, available online, accessed on May 19, 2020, https://assaycentral.github.io/ Search PubMed.
- P. Willett, J. Chem. Inf. Model., 2013, 53, 1–10 CrossRef CAS PubMed.
- B. Chen, C. Mueller and P. Willett, Mol. Inf., 2010, 29, 533–541 CrossRef CAS PubMed.
- M. Feher, Drug Discov. Today, 2006, 11, 421–428 CrossRef CAS PubMed.
- L. Zhang, D. Lin, Y. Kusov, Y. Nian, Q. Ma, J. Wang, A. von Brunn, P. Leyssen, K. Lanko, J. Neyts, A. de Wilde, E. J. Snijder, H. Liu and R. Hilgenfeld, J. Med. Chem., 2020, 63, 4562–4578 CrossRef CAS PubMed.
- Computer software, Canada: The Metabolomics Innovation Centre. The Metabolomics Innovation Centre: FooDB (Version 1), available online, accessed on May 19, 2020, https://foodb.ca/ Search PubMed.
- A. M. Wassermann, E. Lounkine, D. Hoepfner, G. Le Goff, F. J. King, C. Studer, J. M. Peltier, M. L. Grippo, V. Prindle, J. Tao, A. Schuffenhauer, I. M. Wallace, S. Chen, P. Krastel, A. Cobos-Correa, C. N. Parker, J. W. Davies and M. Glick, Nat. Chem. Biol., 2015, 11, 958–966 CrossRef CAS PubMed.
- J. J. Naveja, M. P. Rico-Hidalgo and J. L. Medina-Franco, F1000Research, 2018, 7, DOI:10.12688/f1000research.15440.2.
- F. Ballante, A. Rudling, A. Zeifman, A. Luttens, D. D. Vo, J. J. Irwin, J. Kihlberg, J. Brea, M. I. Loza and J. Carlsson, J. Med. Chem., 2020, 63, 613–620 CrossRef CAS.
- A. M. Wassermann, M. Tudor and M. Glick, Drug Discov. Today Technol., 2017, 23, 69–74 CrossRef PubMed.
- D. Bajusz, A. Rácz and K. Héberger, J. Cheminf., 2015, 7, 20 Search PubMed.
- Y. W. Chen, C.-P. B. Yiu and K.-Y. Wong, F1000Research, 2020, 9, 129 Search PubMed.
- S. Liu, Q. Zheng and Z. Wang, Bioinformatics, 2020, 36, 3295–3298 CrossRef PubMed.
- J. L. Medina-Franco, G. M. Maggiora, M. A. Giulianotti, C. Pinilla and R. A. Houghten, Chem. Biol. Drug Des., 2007, 70, 393–412 CrossRef CAS PubMed.
- S. Beisken, T. Meinl, B. Wiswedel, L. F. de Figueiredo, M. Berthold and C. Steinbeck, BMC Bioinf., 2013, 14, 257 CrossRef PubMed.
- M. R. Berthold, N. Cebron, F. Dill, T. R. Gabriel, T. Kötter, T. Meinl, P. Ohl, C. Sieb, K. Thiel and B. Wiswedel, in Data Analysis, Machine Learning and Applications, ed. C. Preisach, H. Burkhardt, L. Schmidt-Thieme and R. Decker, Springer Berlin Heidelberg, Berlin, Heidelberg, 2008, pp. 319–326 Search PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- F. D. Prieto-Martínez, M. Arciniega and J. L. Medina-Franco, Tip. Rev. Espec. Ciencias Químico-Biol., 2018, 21, 65–87 Search PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- L. Zhang and R. Hilgenfeld, 2020, available online, accessed on, May 18, 2020, DOI:10.2210/pdb5N5O/pdb.
- M. González-Medina, J. J. Naveja, N. Sánchez-Cruz and J. L. Medina-Franco, RSC Adv., 2017, 7, 54153–54163 RSC.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- N. A. Durán-Iturbide, B. I. Díaz-Eufracio and J. L. Medina-Franco, ACS Omega, 2020 DOI:10.1021/acsomega.0c01581.
- K. S. Chan, S. T. Lai, C. M. Chu, E. Tsui, C. Y. Tam, M. M. L. Wong, M. W. Tse, T. L. Que, J. S. M. Peiris, J. Sung, V. C. W. Wong and K. Y. Yuen, Hong Kong Med. J., 2003, 9, 399–406 CAS.
- C. M. Chu, V. C. C. Cheng, I. F. N. Hung, M. M. L. Wong, K. H. Chan, K. S. Chan, R. Y. T. Kao, L. L. M. Poon, C. L. P. Wong, Y. Guan, J. S. M. Peiris, K. Y. Yuen and HKU/UCH SARS Study Group, Thorax, 2004, 59, 252–256 CrossRef CAS PubMed.
- V. Nukoolkarn, V. S. Lee, M. Malaisree, O. Aruksakulwong and S. Hannongbua, J. Theor. Biol., 2008, 254, 861–867 CrossRef CAS.
- K.-T. Choy, A. Y.-L. Wong, P. Kaewpreedee, S. F. Sia, D. Chen, K. P. Y. Hui, D. K. W. Chu, M. C. W. Chan, P. P.-H. Cheung, X. Huang, M. Peiris and H.-L. Yen, Antiviral Res., 2020, 178, 104786 CrossRef CAS PubMed.
- N. Yamamoto, R. Yang, Y. Yoshinaka, S. Amari, T. Nakano, J. Cinatl, H. Rabenau, H. W. Doerr, G. Hunsmann, A. Otaka, H. Tamamura, N. Fujii and N. Yamamoto, Biochem. Biophys. Res. Commun., 2004, 318, 719–725 CrossRef CAS.
- S. Khaerunnisa, H. Kurniawan, R. Awaluddin, S. Suhartati and S. Soetjipto, Preprints, 2020 DOI:10.20944/preprints202003.0226.v1.
- Z. Xu, C. Peng, Y. Shi, Z. Zhu, K. Mu, X. Wang and W. Zhu, bioRxiv, 2020 DOI:10.1101/2020.01.27.921627.
- Q. Cai, D. Huang, P. Ou, H. Yu, Z. Zhu, Z. Xia, Y. Su, Z. Ma, Y. Zhang, Z. Li, Q. He, Y. Fu, L. Liu and J. Chen, medRxiv, 2020 DOI:10.1101/2020.02.17.20024018.
- B. Cao, Y. Wang, D. Wen, W. Liu, J. Wang, G. Fan, L. Ruan, B. Song, Y. Cai, M. Wei, X. Li, J. Xia, N. Chen, J. Xiang, T. Yu, T. Bai, X. Xie, L. Zhang, C. Li, Y. Yuan and C. Wang, N. Engl. J. Med., 2020, 382, 1787–1799 CrossRef PubMed.
- L. Hu, S. Chen, Y. Fu, Z. Gao, H. Long, H. Ren, Y. Zuo, H. Li, J. Wang, Q. Xv, W. Yu, J. Liu, C. Shao, J. Hao, C. Wang, Y. Ma, Z. Wang, R. Yanagihara, J. Wang and Y. Deng, medRxiv, 2020 DOI:10.1101/2020.03.25.20037721.
- Y. Li, Z. Xie, W. Lin, W. Cai, C. Wen, Y. Guan, X. Mo, J. Wang, Y. Wang, P. Peng, X. Chen, W. Hong, G. Xiao, J. Liu, L. Zhang, F. Hu, F. Li, F. Li, F. Zhang, X. Deng and L. Li, medRxiv, 2020 DOI:10.1101/2020.03.19.20038984.
- D. Yan, X. Liu, Y. Zhu, L. Huang, B. Dan, G. Zhang and Y. Gao, medRxiv, 2020 DOI:10.1101/2020.03.22.20040832.
- E. J. Gardiner, V. J. Gillet, M. Haranczyk, J. Hert, J. D. Holliday, N. Malim, Y. Patel and P. Willett, Stat. Anal. Data Min., 2009, 2, 103–114 CrossRef.
- X. Liu and X.-J. Wang, J. Genet. Genomics, 2020, 47, 119–121 CrossRef.
- J. L. Medina-Franco, K. Martínez-Mayorga, A. Bender, R. M. Marín, M. A. Giulianotti, C. Pinilla and R. A. Houghten, J. Chem. Inf. Model., 2009, 49, 477–491 CrossRef CAS.
- W. Vuong, M. B. Khan, C. Fischer, E. Arutyunova, T. Lamer, J. Shields, H. A. Saffran, R. T. McKay, M. J. van Belkum, M. Joyce, H. S. Young, D. L. Tyrrell, J. C. Vederas and M. J. Lemieux, bioRxiv, 2020 DOI:10.1101/2020.05.03.073080.
- Y. Kim, S. R. Mandadapu, W. C. Groutas and K.-O. Chang, Antiviral Res., 2013, 97, 161–168 CrossRef CAS PubMed.
- J. Ali, P. Camilleri, M. B. Brown, A. J. Hutt and S. B. Kirton, J. Chem. Inf. Model., 2012, 52, 420–428 CrossRef CAS PubMed.
- M. J. Huentelman, J. Zubcevic, J. A. Hernández Prada, X. Xiao, D. S. Dimitrov, M. K. Raizada and D. A. Ostrov, Hypertension, 2004, 44, 903–906 CrossRef CAS PubMed.
- B. N. Ames, Ann. N. Y. Acad. Sci., 1999, 889, 87–106 CrossRef CAS PubMed.
- J. Selhub and I. H. Rosenberg, Biochimie, 2016, 126, 71–78 CrossRef CAS PubMed.
- A. Peña-Castillo, O. Méndez-Lucio, J. R. Owen, K. Martínez-Mayorga and J. L. Medina-Franco, in Applied chemoinformatics: achievements and future opportunities, ed. T. Engel and J. Gasteiger, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2018, pp. 501–525 Search PubMed.
- S. Ekins, M. Mottin, P. R. P. S. Ramos, B. K. P. Sousa, B. J. Neves, D. H. Foil, K. M. Zorn, R. C. Braga, M. Coffee, C. Southan, A. C. Puhl and C. H. Andrade, Drug Discov. Today, 2020, 25, 928–941 CrossRef CAS.
- M. S. Santibáñez-Morán, E. López-López, F. D. Prieto-Martínez, N. Sánchez-Cruz and J. L. Medina-Franco, ChemRxiv, 2020 DOI:10.26434/chemrxiv.12420860.v1.
Footnote |
| † Electronic supplementary information (ESI) available: Excel file with ten worksheets that report all similarity values, docking scores, and ADMETox profile of the hit compounds outlined in Fig. 2. Structure file of the 1052 queries used for the similarity searching. See DOI: 10.1039/d0ra04922k. |
| This journal is © The Royal Society of Chemistry 2020 |