
Industrial synthesis of reactive silicones: reaction mechanisms and processes
Theresia
Köhler
a,
Andrea
Gutacker
b and
Esteban
Mejía
 *c
*c
aHenkel Belgium N.V., Nijverheidstraat 7, B-2260 Westerlo, Belgium
bHenkel AG & Co. KGaA, Henkelstr. 67, 40191 Düsseldorf, Germany
cLeibniz Institute for Catalysis, Albert-Einstein-Str. 29a, 18059 Rostock, Germany. E-mail: Esteban.mejia@catalysis.de
First published on 19th October 2020
Abstract
Silicones and silicone rubbers are ubiquitous in our daily lives, being used in a broad range of applications ranging from consumer products and adhesives to medicinal and electronic devices. The chemistry and synthesis of these versatile materials have been reviewed so many times that it is difficult to have a comprehensive overview. Hence, with this tutorial review we aim to provide a concise account of the most important reactions and industrial processes to furnish silicones, focusing specially on reactive silicones, specifically in telechelic OH-terminated polysiloxanes, which represent one of the most relevant members of this important materials family.
 Theresia Köhler | Theresia Köhler obtained her doctoral degree in 2018 at the Leibniz Institute for Catalysis (LIKAT)/Rostock University in the group of Polymer Chemistry and Catalysis under the supervision of Esteban Mejia and Udo Kragl. After a postdoctoral stay in the same group she joined Henkel's R&D team in Westerlo, Belgium in 2019. She is currently working as a product development chemist for adhesives used in electronics. |
 Andrea Gutacker | Andrea Gutacker obtained her doctoral grade in 2011 at the University of Wuppertal in the group of Prof. Ullrich Scherf. She joined Henkel's adhesives research department in the same year. After several R&D projects at Henkel, mainly dealing with technology development and raw materials for sealants and adhesives, she is now responsible for product development on silicones in Henkel's Consumers & Craftsmen Adhesive Technology Division. |
 Esteban Mejía | Esteban Mejía studied chemistry at the National University of Colombia in Bogota, where he also obtained his master's degree in polymer chemistry. In 2008 he moved to Switzerland to pursue his PhD in homogeneous catalysis at the ETH Zurich under the supervision of Antonio Togni. In 2012 he joined the group of Matthias Beller at the Leibniz Institute for Catalysis (LIKAT) in Rostock (Germany) as a postdoc. In 2014 he started his independent career at LIKAT where he is currently leader of the group of Polymer Chemistry and Catalysis. |
1. Introduction
Silicones or polyorganosiloxanes are a class of hybrid organic/inorganic polymers whose chains are characterized by a backbone of alternating silicon and oxygen atoms which might be linear or branched (Fig. 1). They represent an important materials class with special properties compared to common organic polymers.1 They find multiple applications in the construction sector, the automotive industry, electronics, medicine, personal care as well as many consumer goods. Due to their special characteristics they are widely used as insulating materials, spreading agents, defoamers, parting agents, implants, matrices in drug delivery systems, adhesives and sealants.2–4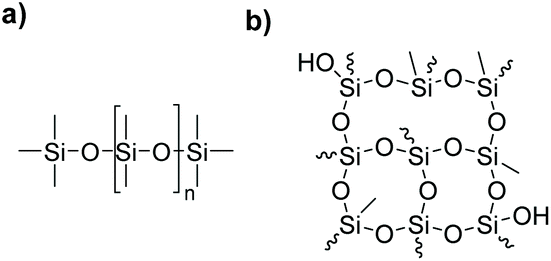 | ||
| Fig. 1 Exemplary structures of silicone materials: (a) linear polydimethylsiloxane (PDMS) and (b) highly branched hydroxy functional silicone resin. | ||
The term “silicone” was coined by Frederick S. Kipping who studied silicon and its compounds at the beginning of the 20th century.5 He called the resin-like materials which he eventually obtained “silicon ketones” or “silicones” as he regarded them as aggregates of discrete molecules rather than as polymers. Because of their limited variety and their virtual inertness, Kipping did not foresee any application.6,7 Later on, James F. Hyde produced silicone resins for impregnating and coating glass in the 1930s building on Kipping's work.8,9 Before the development of the direct synthesis of organohalosilanes10,11 – also known as the Müller–Rochow process – in the 1940s, the availability of the raw materials for its preparation was limited to silica and mineral silicates, leading to a very slow development of silicone chemistry.6 After a “dark period” of intensive research owing to World War II (keeping most of the advances in silicone chemistry unknown to the public for decades), the silicone field flourished dramatically.12 In 2008, an estimated 480![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 t per a of silicone products were produced by the biggest European silicone companies.13 In 2011 the worldwide production of silicones was around 7.3 million t per a and it was increased to 7.4 million t per a in 2017.14 Other market studies state an increase in sales of approximately 4% per a until 2024.15 This indicates a growing demand for basic siloxane products and a constant innovation potential in this area.
000 t per a of silicone products were produced by the biggest European silicone companies.13 In 2011 the worldwide production of silicones was around 7.3 million t per a and it was increased to 7.4 million t per a in 2017.14 Other market studies state an increase in sales of approximately 4% per a until 2024.15 This indicates a growing demand for basic siloxane products and a constant innovation potential in this area.
The unique properties of silicones are mostly attributed to the special characteristics of the Si–O bond.1 For instance, silicones show only slight changes of their physical properties over broad temperature ranges as well as low glass transition temperatures and high gas permeability. This results from the high flexibility of the silicone chains.16,17 The Si–O bond is longer than the C–O bond and the Si–O–Si bond angle is wider than that in its carbon analogues.18,19 The angle might change with variations in the organic moieties attached to the silicon atoms. Both the longer bonds and the wider angles allow easier rotation of the substituents, leading to the said high flexibility of the polysiloxane chains.19
Silicones show good dielectric behavior due to the partial ionic character of the Si–O bond (∼50%) resulting from the high difference in electronegativity (ΔENSi–O = 1.76).20 Thus, silicones are widely used as insulator materials.21 Moreover, the partial ionic character is a very effective tool for the synthesis and modification of silicone polymers through so-called “equilibration” or “redistribution” reactions in the presence of strong acids or bases, allowing the preparation of organofunctional terminated silicones.19 Furthermore, silicones show high thermal and oxidative stability which results from the high bond dissociation energy (BDE) of the Si–O bond (460 kJ mol−1), much higher than that for the C–O bond (318 kJ mol−1).18,19
Linear polyorganosiloxanes, mainly polydimethylsiloxanes (PDMS), are among the most important silicone products (besides cyclic siloxanes). There are many processes to obtain these linear polymers and new processes are being developed constantly.22–35 A key element of these transformations is controlling the nature of the functional groups that will end up at the ends of the chains, since, depending of their reactivity, these end-groups determine the subsequent use of these materials. Alkyl-terminated PDMS are used as lubricants, plasticizers, and heating oils due to their inertness, while hydroxy, alkoxy, hydrido and vinyl terminated telechelic PDMS are employed as the base of household and structural adhesives and curable formulations for sealings and coatings. These groups are especially important for curing these polymers to obtain solid silicone rubbers.21 From these reactive silicones, OH-terminated PDMS are arguably the most relevant in industry.
The term “telechelic” refers to polymers with reactive chain-ends which can undergo further polymerization reactions or polymer analogue reactions.36,37 Polymer analogue reactions are defined as those in which the structure of the polymer is changed by the reaction of its reactive moieties. The chain length, and therefore its degree of polymerization, is not changed.38 It is important to mention that the reactive end-groups in telechelic polymers derive from the initiator, or terminating or chain-transfer agents used in the polymerization, but never from the monomer. Polymers having the same reactive moieties at the chain-ends are commonly referred to as homotelechelic polymers, whereas polymers with two different reactive chain ends are called heterotelechelic.37
Many reviews and book chapters are devoted to the preparation of linear polyorganosiloxanes and their corresponding mechanisms from a purely academic viewpoint.21,39–43 However, there very few (if any) collecting the synthesis of linear polysiloxanes from both academic and industrial perspectives, and importantly, there are no reviews focusing on OH-terminated reactive silicones. With this contribution, and without aiming to be comprehensive, we want to fill this gap and provide an overview of the academic and industrial processes to produce these important materials.
2. Methods for the synthesis of OH-terminated PDMS
OH-terminated PDMS are commonly synthesized by hydrolysis and condensation reactions of organochlorosilanes or by the ROP of cyclic polyorganosiloxanes (Scheme 1). The synthesis via condensation is not well-controlled and leads to broad polydispersities of the obtained polymers. In contrast, by the ROP of cyclic siloxanes, which also represents the main pathway in industry, silicones with tailored molecular weights can be obtained in a highly controlled fashion. The variety of suitable initiators, end-capping agents, additives, reactors etc. is broad.39,44 In particular, for the anionic ring-opening polymerization (vide infra) many new initiators have been reported in the past few years. Metal hydroxides (like KOH, which is one of the earliest examples)27 can be replaced with metal-free initiators such as tetraethylammonium or tetraethylphosphonium salts and phosphazene bases.39 The most recent reports have dealt with nitrogen-containing bases such as N-heterocyclic carbenes,45 bycyclic guanidines46 and phosphatranes.47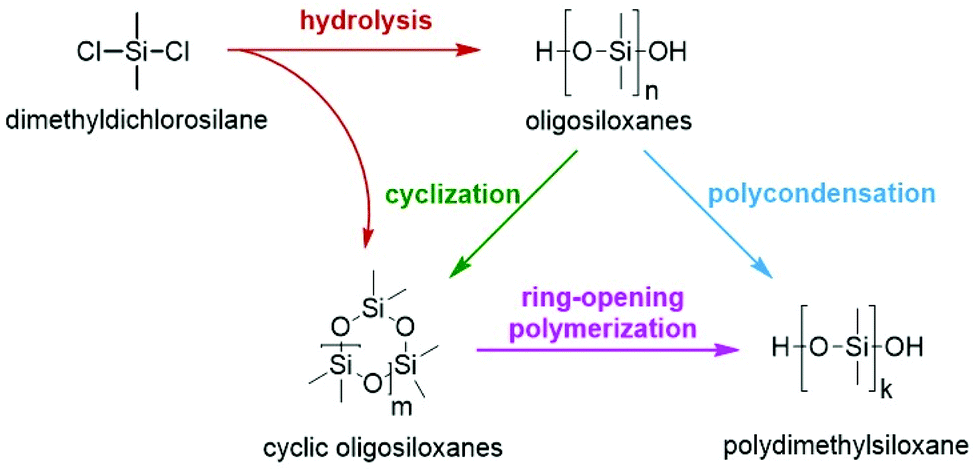 | ||
| Scheme 1 Possible synthesis routes for OH-terminated polydimethylsiloxanes starting from dimethyldichlorosilane. | ||
In general, producing a silicone comprises at least three essential steps: first, synthesis of oligomeric silicone prepolymers from dimethyldichlorosilane followed by condensation polymerization or ring-opening polymerization (ROP); second, neutralizing or quenching the catalyst; and third, stripping the silicone polymer from volatiles.33 The neutralization of the catalyst is especially crucial for the success of the overall process since basic or acidic catalyst residues lead to degradation of the polymer.21
Depending on the chosen polymerization method, the technical requirements must be adjusted. Considering these requirements, the following reactor setups have performed remarkably well at an industrial scale: (a) polymerization vessel; (b) stirred-tank cascade; (c) screw extruder reactor; (d) cell reactor/plug-flow-reactor; and (e) fixed bed reactor.21,41
2.1. From silicon to silicone
The main reactions for the synthesis of methylchlorosilanes by the Müller–Rochow process are presented in Scheme 2.49 The main reaction (a) leads to dimethyldichlorosilane:48 other methylchlorosilanes are products of side reactions (b and c) leading to a broad mixture of silanes (also referred to as “crude silanes”). This mixture also includes hydride-containing silanes such as (CH3)SiHCl2 (e), which are crucial raw materials for hydrosilation reactions.50 The mixture is normally separated via laborious distillation. However, through adjustment of reaction temperature and the usage of promoters, the composition of the different silanes can be influenced. Anyhow, the main product always is dimethyldichlorosilane. An exemplary composition is given in Table 1.48
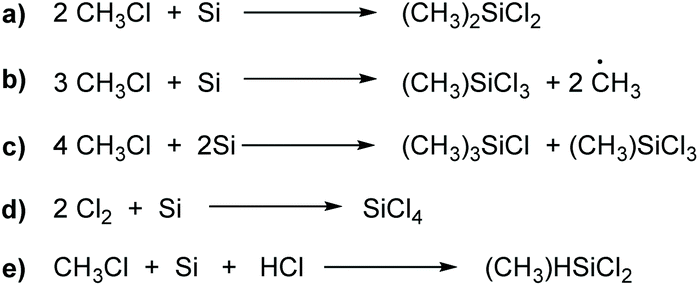 | ||
| Scheme 2 Reactions for the synthesis of methylchlorosilanes by the Müller–Rochow process. | ||
| Compound | Boiling point [°C] | Proportion in the “crude silane” [wt%] |
|---|---|---|
| Typical reaction conditions: 0.5–3 wt% copper catalyst; zinc or zinc oxide as a promoter; 250–320 °C.54 | ||
| (CH3)2SiCl2 | 70 | 70–90 |
| (CH3)SiCl3 | 66 | 5–15 |
| (CH3)3SiCl | 57 | 2–4 |
| (CH3)HSiCl2 | 41 | 1–4 |
| (CH3)2HSiCl | 35 | 0.1–0.5 |
Organochlorosilanes are very sensitive to decomposition in the presence of protic compounds such as water or alcohols under the release of hydrochloric acid or chloroalkanes. This feature is exploited for the synthesis of polymeric silicone materials, by which the purified organochlorosilanes are converted to cyclic, linear21,40,41 or branched polyorganosiloxanes or highly branched silicone resins51via methanolysis or hydrolysis reactions.3,4,52,53
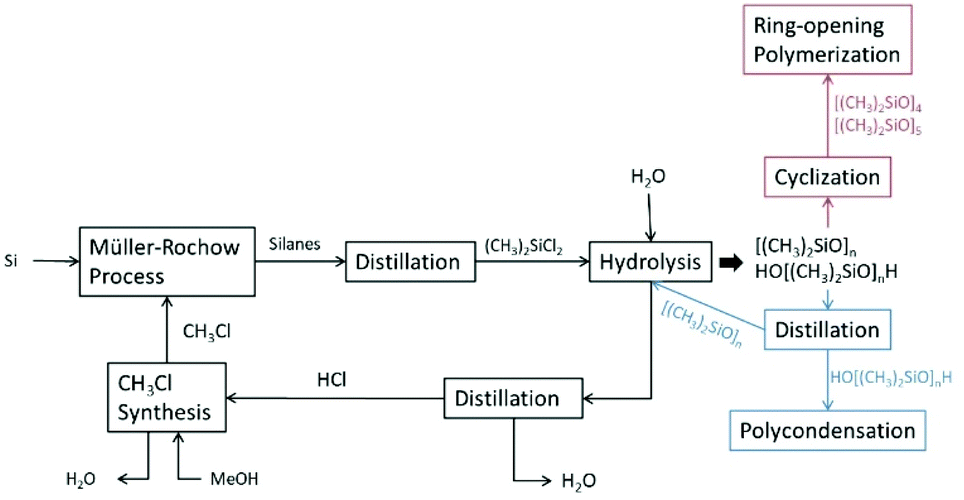 | ||
| Scheme 3 Simplified flow diagram of the two-step process for the synthesis of OH-terminated PDMS via hydrolysis. | ||
Commonly, the silicone synthesis process is directly connected to the Müller–Rochow process. For this procedure chloromethane is needed as the starting material to furnish the chlorosilanes. Therefore, aqueous hydrochloric acid which accumulates in the hydrolysis reaction is first distilled to yield pure HCl, which is then reacted with methanol. The prepared chloromethane is then used for the Müller–Rochow process. Instead of hydrolysis, methanolysis of dichlorosilanes can be performed. This bears the advantage that chloromethane is recovered directly instead of aqueous hydrochloric acid, reducing the overall cost of the process.21,40,41
In patent US 496085035 they describe a batch process for the polycondensation of oligomeric siloxanediols in order to obtain a silanol (Si–OH) end-capped polymer. Therein, the oligomeric siloxane diols are first stirred under reduced pressure to remove volatile compounds out of the substrate. Then they return to atmospheric pressure by purging with nitrogen. The reaction mixture is heated to a certain temperature (50–130 °C) and triflic acid is added to the reaction mixture. Again, the pressure is reduced, and the mixture can react until the desired viscosity of the product is obtained. The reaction is quenched by adding hexamethylcyclotrisilazane. The obtained polymer is then stripped to remove volatile contents.35
2.2. Ring-opening polymerization of cyclic siloxanes
The ROP of cyclic siloxanes is commonly not the method of choice in industrial processes due to its higher cost compared to hydrolysis and polycondensation approaches. However, it is still highly relevant for special applications in which a careful control of the molecular weight and polydispersity of the produced polysiloxanes is required. The cyclic monomers used are mainly organocyclotetrasiloxanes (like octamethylcyclotetrasiloxane, also called D4) or organocyclotrisiloxanes (like hexamethylcyclotrisiloxane, also called D3). Other monomers include cyclic carbosiloxanes such as 2,2,5,5-tetramethyl-2,5-disila-1-oxacyclopentane. Known ROP techniques are: ionic, radical, and peroxide- or plasma-initiated ROP.17 The ionic ROP can be initiated either by strong bases or acids. If the polymerization is initiated by strong bases an anionic mechanism (AROP) takes place. Initiation with strong acids leads to a cationic (CROP) mechanism.17Anionic or cationic ROP of both strained (D3) and unstrained (D4, D5 and D6) cyclic siloxanes leads to the same equilibrium in which the polymer and cyclic byproducts co-exist. Nevertheless, the mechanism leading to this equilibrium is different, which also results in two possible pathways for ROP: kinetically or thermodynamically controlled (Scheme 4).57,58 The thermodynamically controlled ROP leads to the parallel formation of cyclic oligosiloxanes and linear polymers, whereas the kinetically controlled ROP firstly leads to linear polymers and, in a second step, equilibration processes take place.57
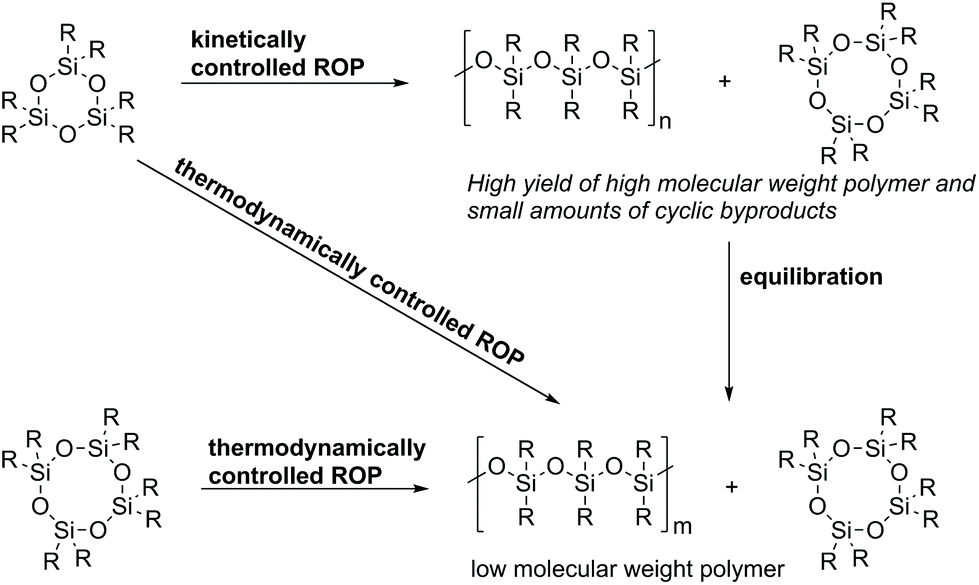 | ||
| Scheme 4 Differences between thermodynamically and kinetically controlled ROP of cyclic siloxanes. | ||
Thermodynamically controlled ROP: The thermodynamically controlled ROP is entropy-driven and is mainly applied for the polymerization of cyclotetrasiloxanes or higher cyclosiloxanes due to the fact that all bonds in both monomers and polymers are thermodynamically equivalent.59 This leads to the parallel formation of linear poly- and oligocyclosiloxanes. Thereby, the polymer concentration increases until equilibrium is achieved. This kind of polymerization is also known as equilibrium polymerization or equilibration because it stops after reaching the equilibrium state.57
An advantage of this process is that it is not dependent on the nature of the used initiators. Therefore, initiators which achieve equilibrium fast and under mild conditions can be chosen; they do not interact with the monomer and can be separated easily from the product. Furthermore, the polymerization does not have to be quenched at a certain moment.57 A limitation of this approach is the formation of oligocycles. Therefore, the thermodynamically controlled ROP should be used for polymerizations which give a high yield of the polymer in the equilibrium state. The ratio of linear polymers and cyclic oligomers in equilibrium is especially influenced by the polarity and bulkiness of the substituents at the silicon atoms. This method is also not recommended for polymers with very low polydispersities and for copolymers which should have a specific structure (e.g. alternating).57
Kinetically controlled ROP: The kinetically controlled ROP is characterized by the fact that the chain propagation proceeds much faster than the back-biting or chain transfer reactions. This applies only for strained cyclotrisiloxanes where the ROP is not entropy-but enthalpy-driven, with the ring strain release being the driving force of the reaction.57 The reaction has to be quenched directly after reaching the highest monomer conversion in order to avoid equilibration reactions. If the reaction is stopped before equilibrium is attained, a high molecular weight polymer with only small amounts of cyclic byproducts is obtained. An advantage of this process is that polymers with controlled structures and higher yields can be obtained. Conversely, the processes require more expensive monomers and very pure reactants and only selected initiators can be used.57
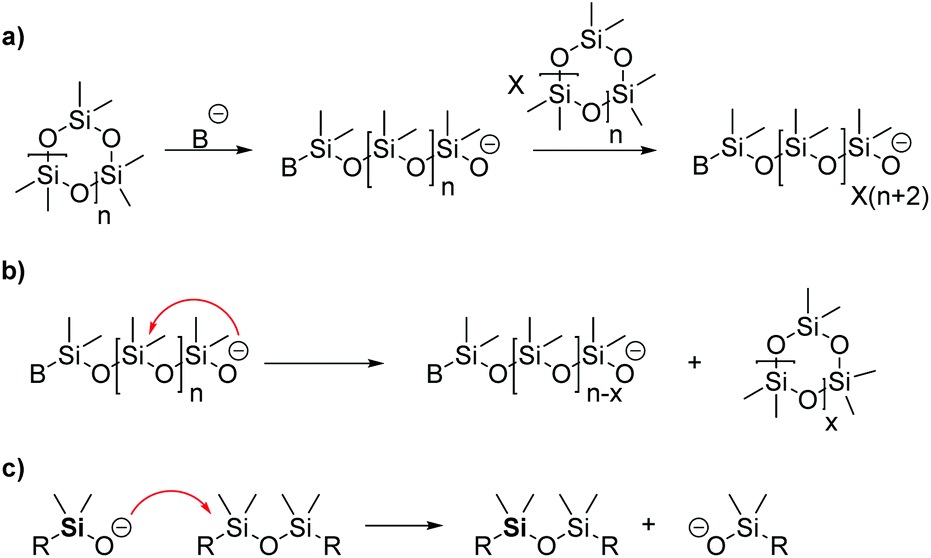 | ||
| Scheme 5 Anionic ring-opening polymerization of cyclosiloxanes. (a) Initiation reaction by the formation of silanolate anions and propagation. (b) Back-biting reaction on a growing PDMS chain. (c) Chain-transfer reaction. | ||
One problem which occurs with the AROP is the so-called back-biting reaction. It represents the reversed reaction of propagation and leads to breakdown of the polymeric chain. Herein, the active center reacts with the growing chain by cleaving the siloxane bond under the formation of cyclic siloxanes. As a consequence, smaller and bigger cyclic siloxanes are generated as by-products when equilibrium is achieved (Scheme 5b).39 This can be avoided under certain conditions by stopping the reaction directly before equilibrium is achieved and it is commonly called non-equilibrium polymerization.57 Chain transfer is another side reaction which can occur (Scheme 5c). Herein, the active center attacks a silicon atom at another polymer chain, breaking the siloxane bond and leading to chain randomization which is also called equilibration. As long as the active center is not deactivated, the polymerization proceeds without termination which also enhances the described side reactions.
Chain termination occurs always as side reaction in AROP due to traces of protonic impurities (i.e. water) in the reaction. It can also be forced by using the initiator in an aqueous solution or by quenching the reaction with water or alcohols.40 Importantly, the molecular weight can be controlled by the amount of water used.61 An alternative is the usage of end-capping agents, typically disiloxanes including tetramethyldisiloxane (TMDS), hexamethyldisiloxane (HMDS) or divinyltetramethyldisiloxane (DVS).33 These end-capping agents may also be used to regulate the molecular weight of the polymer and/or to add a functionality different from OH, for instance a vinyl group.62
The polymerization rate depends on many factors such as the nature of the initiator, the polymerization medium and the chosen monomer. However, the key factor governing the kinetics of the ring-opening polymerization is the silanolate–counterion interaction (K+ and Na+ being the most common in industry) leading to the formation of aggregates which are inactive in AROP.63,64
Initiators: Many different initiators are well known for this route and those widely described are alkali metal or alkali earth metal hydroxides (e.g. KOH and NaOH),27 alcoholic complexes thereof, alkali metal or alkali earth metal silanolates,65 tetraalkylammonium hydroxide, phosphazene bases66 and, recently, N-heterocyclic carbenes45 or bicyclic guanidines (Scheme 6).39,67 It has been observed that the polymerization rate increases with the size of the counterion: Li+ < Na+ < K+ < Rb+ < Cs+ ∼ Et4N+ ∼ Et4P+. With increasing size, the cations become less electrophilic and therefore the concentration of free anions increases. Therefore, the most active initiators are phosphazenes.63
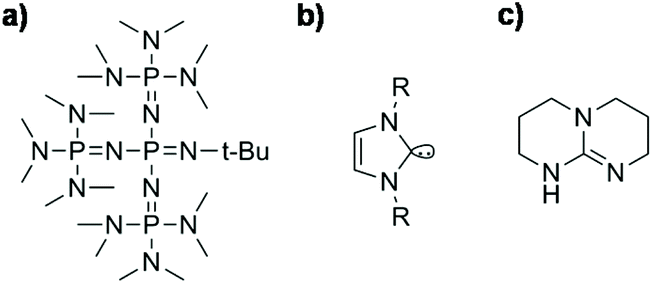 | ||
| Scheme 6 Modern initiators for ROP of cyclosiloxanes: (a) an exemplary molecular structure of a phosphazene: (1-tert-butyl-4,4,4-tris(dimethylamino)-2,2-bis[tris(dimethylamino)-phosphoranyliden-amino]-2λ5,4λ5-catenadi(phosphazene)) (P4-tBu phosphazene), (b) the general molecular structure of a N-heterocyclic carbene (NHC), and (c) the molecular structure of 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD). | ||
Potassium hydroxide is one of the earliest initiators used for the ROP of cyclic siloxanes. Its usage dates back to 1948, when the ring-opening polymerization with alkali metals was first patented.27 Potassium hydroxide is neither soluble in the monomer nor in the polysiloxane. Therefore, it needs to be dispersed as a fine powder in the reaction system and high temperatures (>150 °C) are necessary to start the polymerization.30 Nowadays, potassium silanolates are often used as initiators for the synthesis of silicones. They represent a KOH-surrogate which is soluble in siloxanes68,69 and may also be used for polycondensation.69 They can be obtained by reacting potassium hydroxide with siloxanes.65
Phosphazene bases (Scheme 6a), which belong to the class of so-called superbases, are a relatively new class of initiators in the ROP of cyclic siloxanes. They are very efficient for the ring-opening polymerization, especially in the presence of proton donors such as methanol or traces of water.66 The interaction with proton donors leads to the formation of silanolates with a very bulky and soft counterion in which the positive charge is delocalized. Phosphazene bases can also catalyze condensation reactions when equilibrium is achieved.29,32,70 For example, Möller et al. showed that P4-tBu phosphazene is very active at room temperature at very low concentrations (10−3 mol L−1). Polymerization of D4 starts immediately and equilibrium is achieved after 1 min.66,71,72
In the past few years, initiation via Lewis bases has been of special interest in silicone research. In particular, the use of metal-free organocatalysts such as N-heterocyclic carbenes (NHCs, Scheme 6b) or guanidines has been reported,73 even for the ROP of other cyclic monomers such as caprolactones.74 In the literature, NHCs with cyclohexyl or tert-butyl moieties have been reported as active initiators for the ROP of cyclic oligosiloxanes using primary alcohols such as methanol or benzyl alcohol as co-initiators.45 NHCs act as nucleophilic reagents, where the electron rich carbon of the NHC attacks a silicon atom of the cyclic monomer and binds to it while the monomer undergoes ring-opening. The formed zwitterionic intermediate contains the silanolate center which is needed for the chain propagation described above. The alcohols described as co-initiators are necessary for chain termination. By releasing the initiator they form a heterotelechelic silicone which is alkoxy end-capped on one side and silanol-terminated on the other (Scheme 7).75,76 NHCs have the great disadvantage of being very sensitive to moisture. In the literature, CO2-protected NHCs are also described, which can be thermally deprotected and allow a premixing of the reactants under normal conditions. This is especially advantageous for industrial applications.77
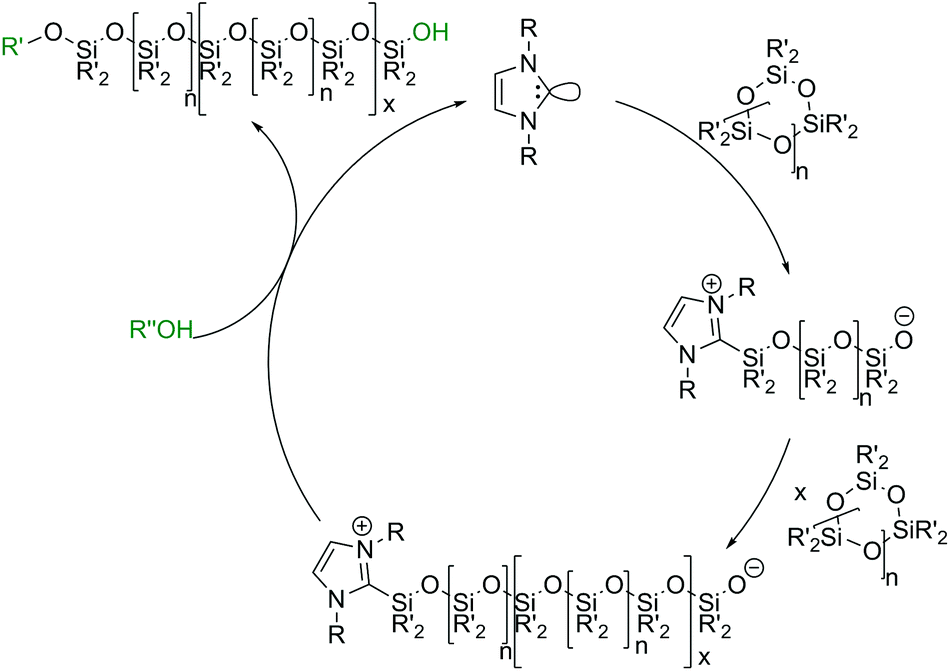 | ||
| Scheme 7 Proposed reaction mechanism for the NHC initiated ring-opening polymerization of cyclic siloxanes by monomer activation. | ||
Molecules such as 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) (Scheme 6c) have been successfully used for the ROP of cyclic carbosiloxanes such as 2,2,5,5-tetramethyl-1-oxa-2,5-disilacyclopentane or hexamethylcyclotrisiloxane. Hedrick et al. used TBD at a concentration of 1.2 mol% in combination with an alcohol and could confirm via NMR that they obtained heterotelechelic polymers with an alkoxy and silanol terminated chain-end. TBD acts in this polymerization as a co-initiator by deprotonation of the added alcohol. The formed alcoholate acts as an initiator for the ring opening polymerization.67 TBD is less active than NHCs but the obtained polymers show a smaller polydispersity which indicates a more controlled process.78 In Patent US 0215097 a process for preparing silicones by ROP of cyclosiloxanes in the presence of bicyclic guanidines (especially TBD) is described. Therein, alcohols are not necessarily used and additionally disiloxanes are used as end-capping agents.46
Influence of solvents and activators: The polymerization itself can be carried out neatly in solvents or in emulsion. However, the solubility of the initiator in the reaction medium plays an important role in the reaction kinetics. Suitable solvents are liquid hydrocarbons such as hexane or silicone fluids.33 In some examples THF is used as the solvent in combination with a hard counter-ion, namely Li+, in the ROP of D3.79–83 Reaction conditions were found under which back-biting and chain-transfer reactions have been avoided and polymers with very narrow polydispersities (<1.1) have been obtained. Advantageous to this process is the absence of any additives which could contaminate the polymer. The reason for these results is the stronger interaction of the counter-ion with the solvent than with the active center at the chain-ends.81 Morton and Bostick reported a solution polymerization of D4 in THF, showing 71% conversion after 24 h at 60 °C. Under neat conditions, a reaction temperature of 140 °C was necessary, demonstrating that the choice of solvent has an accelerating effect.84,85
Various additives and coinitiators which improve the polymerization rate have been described and these include hexamethylphosphoric triamide,86–88 dimethyl sulfoxide,86,89–92 2,5,8-trioxanonane (diglyme),93 2,5,8,11-tetraoxadodecane (triglyme),94 1,2-dimethoxyethane,86 dimethylformamide,95 and N-methylpyrrolidone.96 The success of these approaches lies in the fact that the counter-ion shows a stronger interaction with these activators compared to that with the active center of the polymer chain.88,91,97–99 For instance, when crown ethers are used, they form a complex with the counter-ion and thereby reduce the silanolate–cation interaction and the formation of aggregates resulting in an increase in the polymerization rate.100 Some of these additives and some other polar aprotic substances such as acetonitrile and acetone have been studied in the ROP of 1,3,5-tri(3,3,3-trifluoropropyl-)-1,3,5-trimethylcyclotrisiloxane (DF3) with sodium siloxanolate as an initiator. The reaction is of first order with the monomer if no additive is used. Some of these results are summarized in Table 2.95,101
| Solvent | c Additive [mol L−1] | T [°C] | k/k0 |
|---|---|---|---|
| [Sodium siloxanolate] = 2 × 10−2 mol L−1. k0 is the observed first-order rate constant in the absence of an activator. | |||
| Nitrobenzene | 0.1 | 40 | 2.1 |
| Tetrahydrofurane | 0.1 | 40 | 10.5 |
| Acetonitrile | 0.1 | 40 | 34 |
| Acetone | 0.1 | 40 | 95 |
| Dimethylformamide | 0.01 | 40 | 150 |
| Dimethylsulfoxide | 0.01 | 40 | 155 |
| Hexamethylphosphortriamide | 0.001 | 40 | 28 |
| Diglyme | 0.001 | 40 | 28 |
Effect of the monomer structure: In general, the reactivity of the monomers decreases with increasing ring size: D3 ≫ D4 > D5 > D6. Hexamethyltrisiloxane (D3) has the largest ring strain among cyclic siloxanes and shows very high reactivity in ROP and therefore, is often used in non-equilibrium polymerization processes.58 This has the advantage that minimal back-biting and chain randomization take place.39 Nevertheless, the preparation of D3 is complex and therefore its commercial availability is limited.41 The reactivity order of cyclosiloxanes is reversed if the polymerization is performed under neat conditions or in a non-polar solvent in combination with an alkali metal silanolate.40,102 This might be explained by the formation of crown ether-like complexes due to a multidentate interaction of D6 or D7 rings with the cation.103
The polymerization mechanism using triflic acid as an initiator has been studied in depth, and will serve as an example for the following mechanistic considerations.105,109 In general, it is accepted that the Si–O-bond is cleaved by strong protic acids during the initiation reaction. Thereby, the corresponding silyl ester silanol is formed which starts the chain propagation.104 It is proposed that homocomplexes of the acid (TfOH·TfO−; [(TfO2)H]−) or the hydrate of the acid ([TfO·H2O]−) are formed and they act as initiators since the ester group is inactive.110 Toskas et al. proposed that chain propagation is based on the formation of trisiloxonium ions (Scheme 8a).107 The most relevant side reactions during CROP are chain-transfer (Scheme 8b)111 and back-biting. The latter, unlike in the AROP, takes place via intramolecular condensation processes leading to a product mixture of macrocyclic rings and linear polymers.104,107
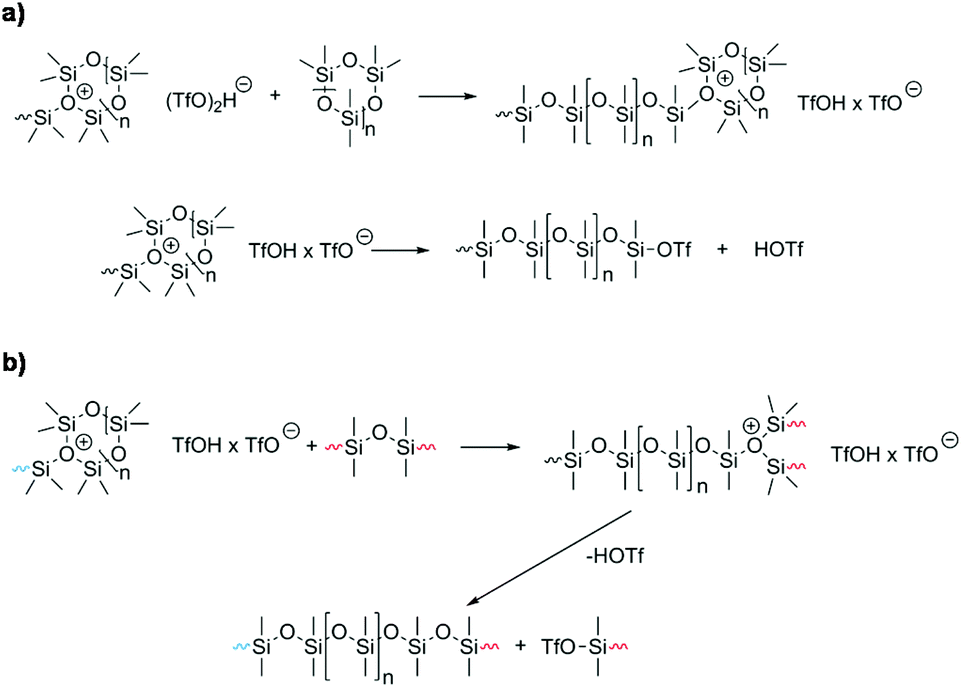 | ||
| Scheme 8 Mechanistic details of the CROP of cyclosiloxanes. (a) Chain propagation (as postulated by Toskas et al.) and (b) chain transfer reaction. | ||
Strong protic acids such as sulfuric,25,112 sulfonic31,107,109,113 and perchloric acid initiators are used in the CROP.101 Sulfuric acid is the earliest reported initiator for the CROP of cyclic siloxanes.112 Sulfonic acids which are described in the literature as successful initiators include triflic acid (HOTf)107 and bis(trifluoromethyl)sulfonimide (TFSI-H).114 Heterogeneous initiators such as ion exchange resins, acid-treated graphite115 or acid treated clays116 can also be used in CROP.101 They bear the advantage that they can be easily separated from the product after the reaction.
An alternative method for initiating the CROP is the generation of trisiloxonium ions by an in situ hydride transfer. It has the advantage that a simple addition polymerization takes place since the chain-propagation takes place only in one direction.108 Another alternative is the CROP of D3 with trimethylsilyl triflate in the absence of strong protic acids.117
A continuous process for the synthesis of silanol-terminated polymers via AROP has been described in patent US 4250290 (Scheme 9).24 It is a cascade of plug-flow reactors where the monomer is fed continuously. The reaction is conducted at 145 °C. Potassium silanolate is used as an initiator and the reaction is quenched with water at an early stage. This is necessary to control the rapidly increasing viscosity of the reaction mixture, hence avoiding mixing problems.24
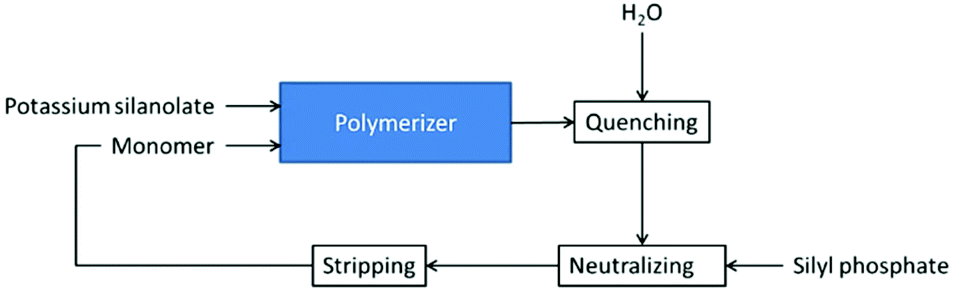 | ||
| Scheme 9 Simplified flow diagram of the process for the production of OH-terminated silicones via AROP.24 | ||
Another patent describes the synthesis of silanol-terminated PDMS via CROP. The polymerization of a mixture of cyclic siloxanes with either aqueous H2SO4 or a combination of aqueous HCl with tetramethylammonium tetrachloroferrate is performed in a loop-type reactor (Scheme 10). Both the time of residence and the reaction temperature depend on the chosen initiator. The unstripped polymer is obtained via phase separation. In the case of sulfuric acid as an initiator the yield is about 55% and the polymer contains less than 1 ppm of H2SO4.25
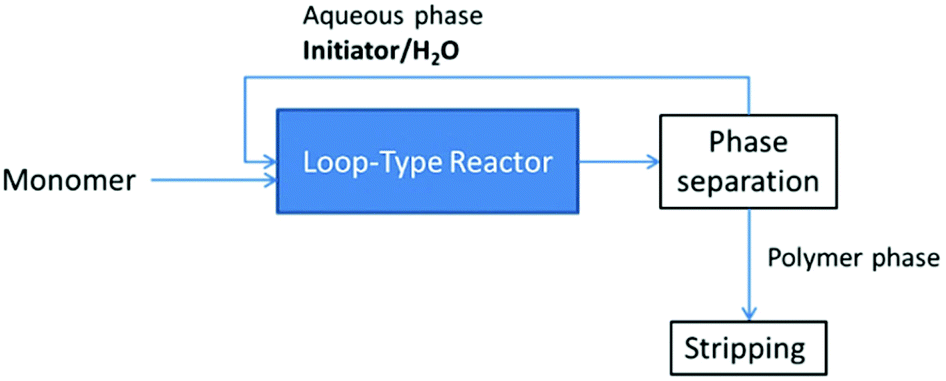 | ||
| Scheme 10 Simplified flow diagram of the process for the synthesis of silanol terminated PDMS via CROP.25 | ||
Phosphazene bases can also catalyze condensation reactions.29,32,70 For example patent EP 1008611 claims the synthesis of silicone polymers via ring-opening polymerization and condensation polymerization at the same time in an equilibrium state.70 In the reaction mixture, the siloxanes and the catalyst are mixed with water along with a OH-terminated polysiloxane or an alcohol to yield a polymer. Additionally, an end-capping agent may be used to produce a polymer with a targeted molecular weight. In general, a silanol-terminated siloxane or an alkoxy- and silanol-terminated siloxane is used as a starting material.70 With this method also hydroxyl-terminated polymers can be obtained.70
2.3. Emulsion polymerization
First patented in 1959,118 the polymerization of organosiloxanes in aqueous emulsions either by ROP31,113 or polycondensation,119,120 is of great importance regarding sustainability.39 Silicone emulsions of polyorganosilanols in water are obtained as a product, which can be used in disperse dyes,121 disperse adhesives,122 and in personal-123 or home-care applications.124Regarding the emulsion polymerization of cyclic siloxanes, the process itself and the mechanism behind it are relatively complex. The process involves a delicate balance among chemical reactions, physico-chemical phenomena such as diffusion processes, phase equilibria and the nature of the interface, commonly modified by the use of surfactants.40 Favored surfactants used in the emulsion polymerization of cyclic siloxanes are fatty sulfonic acids (anionic) such as dodecylbenzenesulfonic acid or quaternary ammonium surfactants (cationic) such as dodecylbenzyldimethylammonium hydroxide. These surfactants can initiate the polymerization themselves. Nevertheless, they can be used in combination with classic initiators. Cationic emulsifiers may be used in addition to basic initiators, whereas anionic emulsifiers are used in combination with acidic initiators.40
Besides the basic reactions of the ROP (initiation, chain growth and termination), condensation reactions of the OH-terminated oligomers and polymers can occur. Other side reactions are back-biting and redistribution as well as depolymerization. Recent reports propose that the initiation and propagation of the ROP of cyclic siloxanes take place via homogeneous nucleation in the continuous phase.125,126
The reaction mechanism of the emulsion polymerization of cyclic siloxanes can be divided into three stages (Scheme 11).125,126 In Stage I ROP is initiated by the emulsifier itself, leading to small oligomeric silanolates which undergo propagation until the chain becomes too hydrophobic and forms a coil, leading to precursor particles becoming polymer particles by coagulation and further stabilization from surfactant molecules diffusing from the micelles. When the growing polymer chain is terminated by water the resulting disilanols can undergo condensation reactions at the polymer particle surface. Stage II starts in the absence of micelles. Then, no new polymer particles can be formed, and newly formed chains diffuse into the polymer particles or monomer droplets, whereby the particles grow. Stage III is characterized by a constant number and size of particles. Only the molecular weight of the polymer changes to an equilibrium value via equilibration. The possibility of micelle nucleation is neglected, since step-growth polymerization occurs via condensation of small oligomers which subsequently form macrocyclic siloxanes by intramolecular condensation.125,126
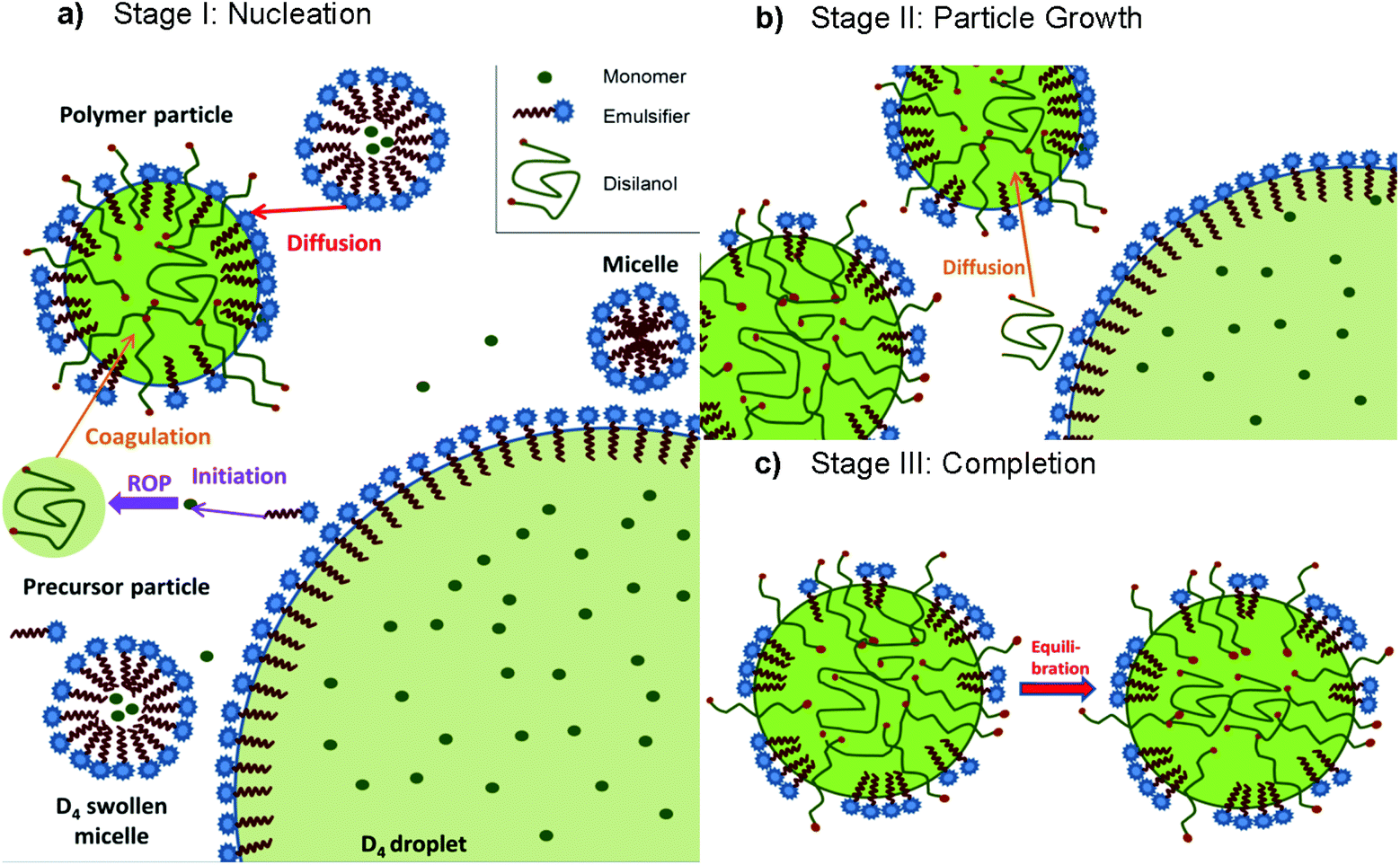 | ||
| Scheme 11 Schematic presentation of the three stages of emulsion polymerization of cyclic siloxanes (exemplary for D4).125,126 | ||
As already mentioned, emulsifiers can initiate the polymerization process themselves. In patent US 4999398 the inventors describe that dodecylbenzenesulfonic acid is capable of forming a stable microemulsion with D4 in aqueous media as well as initiating CROP.113 In patent US 3294725 the same phenomena are observed by using a combination of dodecylbenzene sulfonic acid and its sodium salt.31 Polycondensation in emulsions is also possible. In patent US 6232396 they use dodecylbenzenesulfonic acid as an emulsifier, which is neutralized with Na2SO4 at the beginning of the reaction. Then, an emulsion is prepared with water and a low molecular weight silanol-terminated PDMS. The condensation is then initiated by the addition of concentrated sulfuric acid.119 In patent US 9156954 they describe a similar process to yield silanol terminating groups in the final polymer.120 All the described processes have an occurrence in common that in the end a stable aqueous polysiloxane emulsion is obtained which is not further treated. Interestingly, the polymerization reaction can be terminated by simply adding a salt such as NaCl.
3. Conclusions
There is broad palette of available synthetic methodologies to prepare reactive silicones, specially telechelic OH-terminated polysiloxanes, making use of many catalytic technologies. These methods include the classic anionic and cationic ring-opening polymerizations and more “sophisticated” modern approaches such as aqueous emulsion polymerization and polycondensation reactions. Interestingly, even though these reactions are currently a subject of multi-ton industrial processes, their underlying reaction mechanisms are still a subject of debate.Consequently, as it is common practice in industry, the control of these polymerization processes and the tailoring of the physico-chemical properties of the produced materials are done mostly empirically, hampering the further advancement of the field. Hence, it is necessary to deepen the chemical knowledge available on silicon(e) chemistry by applying modern analysis and modeling techniques for mechanistic investigations.
Moreover, considering the current society's drive towards a “greener” and more sustainable chemical industry, it is foreseeable that novel reactive silicone materials will emerge, displaying novel properties and reactivities (including biodegradability, hydrolysability, bio-compatibility, etc.) which will enable their use in even more applications in tune with the current human needs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr Johann Klein and Prof. Udo Kragl for their constant help and enlightening discussions. The financial support (T. K.) from Henkel AG & Co. KGaA is gratefully acknowledged.Notes and references
- J. E. Mark, R. West and H. R. Allcock, in Inorganic Polymers, Oxford University Press, New York, 2nd edn, 2005, pp. 154–199 Search PubMed.
- R. N. Meals, Silicones, Ann. N. Y. Acad. Sci., 1965, 125, 137–146 CrossRef CAS.
- S. Geier, H. Schmitz, U. Göschel, P. Eyerer, A. Ostrowicki, N. Woicke, C. Ulrich, W. Lutz, J. Eschl, G. Rüb, M. Keuerleber, J. Diemert, J. Hauk, A. Stieneker, J. Woidasky, I. Fischer, K. Kretschmer, L. Ober, C. Kohlert, S. Ganslmeier, C. Schlade, H. Schüle, K. Kurz, K. U. Tönnes, P. Elsner, R. Protte, D. Liebing, A. Rodríguez, S. R. Raisch, H.-J. Dern, S. Schlünken, R. Bräuning and A. König, in Kunststoffe: Eigenschaften und Anwendungen, ed. P. Elsner, P. Eyerer and T. Hirth, Springer, Berlin, Heidelberg, 2012, ch. 2, pp. 115–1201, DOI:10.1007/978-3-642-16173-5_2.
- A. Tomanek, Silicones & Industry: A Compendium for Practical Use, Instruction and Reference, Wacker-Chemie, 1991 Search PubMed.
- F. S. Kipping, XXII.-Organic derivatives of silicon. Part II. The synthesis of benzylethylpropylsilicol, its sulphonation, and the resolution of the dl-sulphonic derivative into its optically active components, J. Chem. Soc., Trans., 1907, 91, 209–240 RSC.
- F. S. Kipping, The bakerian lecture organic derivatives of silicon, Proc. R. Soc. London, Ser. A, 1937, 159, 139–148 CAS.
- J. F. Hyde, Chemical Background of Silicones, Science, 1965, 147, 829–836 CrossRef CAS.
- J. F. Hyde, Fibrous glass textile material for electrical insulation, Corning Glassworks, US Pat, 2209850, 1938 Search PubMed.
- J. F. Hyde, Glass building unit joining method, Corning Glassworks, CA Patent, 355761, 1936 Search PubMed.
- R. Müller, Verfahren zur Herstellung von Kohlenstoff-Silicium-Halogenverbindungen, VEB Silikonchemie, DD Pat, 5348, 1953 (patent application D.R.P. C 57411 was filed on 6 June 1942) Search PubMed.
- E. G. Rochow, Preparation of organosilicon halides General Electric, US Pat, 2380995, 1945 (patent application was filed on 26 September 1941) Search PubMed.
- E. G. Rochow, The chemistry of silicones, Sci. Am., 1948, 179, 50–53 CrossRef CAS.
- http://www.chemanager-online.com/news-opinions/nachrichten/europas-silikonindustrie-schafft-werte , (accessed 18.01.2016).
- https://www.statista.com/statistics/573585/global-silicon-production/ , (accessed 15.03.2018).
- https://www.ceresana.com/de/marktstudien/kunststoffe/silikone/ , (accessed 23.03.2018).
- J. E. Mark, Some Interesting Things about Polysiloxanes, Acc. Chem. Res., 2004, 37, 946–953 CrossRef CAS.
- M. Butts, J. Cella, C. D. Wood, G. Gillette, R. Kerboua, J. Leman, L. Lewis, S. Rajaraman, S. Rubinsztajn, F. Schattenmann, J. Stein, J. Wengrovius and D. Wicht, in Encyclopedia of Polymer Science and Technology, John Wiley & Sons, Inc., 2002, DOI:10.1002/0471440264.pst338.
- M. G. Voronkov, V. P. Mileshkevich and I. A. Iuzhelevskii, Siloxane Bond: Physical Properties and Chemical Transformations, Springer US, New York, 1978 Search PubMed.
- E. Yilgör and I. Yilgör, Silicone containing copolymers: Synthesis, properties and applications, Prog. Polym. Sci., 2014, 39, 1165–1195 CrossRef.
- L. Pauling, The Nature of the Chemical Bond and the Structure of Molecules and Crystals: An Introduction to Modern Structural Chemistry, Cornell University Press, 1960 Search PubMed.
- H.-H. Moretto, M. Schulze and G. Wagner, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000, DOI:10.1002/14356007.a24_057.
- M. Sato, M. Furuya and M. Maruyama, Method for the preparation of diorganopolysiloxane end-blocked with silanolic hydroxy groups, Shin-Etsu Chemical Co. Ltd., US Pat, 5475077, 1995 Search PubMed.
- S. Rubinsztajn, Catalysts for polycondensation and redistribution of organosiloxane polymers, General Electric Company, US Pat, 5403909, 1995 Search PubMed.
- L. P. Petersen, Process for the continuous manufacture of siloxane polymers, General Electric, US Pat, 4250290, 1981 Search PubMed.
- R. Ottlinger and R. Reitmeier, Method for preparing linear organopolysiloxanediols, Wacker-Chemie GmbH, US Pat, 4762937, 1988 Search PubMed.
- K. Itoh, T. Shinohara, H. Kizaki, S. Tanaka, Y. Satou and K. Umemura, Method of producing dimethylpolysiloxanes, Shin-Etsu Chemical Co. Ltd., US Pat, 5473037, 1995 Search PubMed.
- J. F. Hyde and W. H. Daudt, Production of organosiloxanes, Corning Glass Works, US Pat, 2443353, 1948 Search PubMed.
- P. Hupfield, A. Surgenor and R. Taylor, Polymerisation of siloxanes, Dow Corning Corporation, EP Pat, 1008611, 2001 Search PubMed.
- P. Hupfield, A. Surgenor and R. Taylor, Polymerisation of siloxanes, Dow Corning Corporation, EP Pat, 1008610, 2000 Search PubMed.
- E. Fleury, J. M. Mas and K. Ramdani, Method for the preparation of organopolysiloxane by polymerization and rearrangement of cyclic siloxanes, Rhodia Chimie, US Pat, 7776988, 2010 Search PubMed.
- D. E. Findley and D. R. Weyenberg, Method of polymerizing siloxanes and silcarbanes in emulsion by using a surface active sulfonic acid catalyst, Dow Corning, US Pat, 3294725, 1966 Search PubMed.
- D. Eglin, J. De La Cro Habimana, P. Hupfield, A. Surgenor and R. Taylor, Polymerisation catalyst and polymerisation process, Dow Corning Corporation, EP Pat, 1008598, 2004 Search PubMed.
- J. Currie, P. Griffith, W. Herron and R. Taylor, Process for producing a silicone polymer, Dow Corning Limited, US Pat, 6054548, 2000 Search PubMed.
- J. Burkhardt, Verfahren zur Herstellung von linearen Organopolysiloxanen mit Triorganosiloxygruppen als endständige Einheiten, Wacker-Chemie GmbH, EP Pat, 0208285, 1993 Search PubMed.
- L. Billet and J. J. Lebrun, Production of diorganopolysiloxanes having silanol end groups, Rhone-Poulenc Chimie, US Pat, 4960850, 1990 Search PubMed.
- V. Percec, C. Pugh, O. Nuyken and S. D. Pask, in Comprehensive Polymer Science and Supplements, ed. G. Allen and J. C. Bevington, Pergamon, Amsterdam, 1989, vol. 6, ch. 9, pp. 281–357 Search PubMed.
- K. Horie, M. Barón, R. B. Fox, J. He, M. Hess, J. Kahovec, T. Kitayama, P. Kubisa, E. Maréchal, W. Mormann, R. F. T. Stepto, D. Tabak, J. Vohlídal, E. S. Wilks and W. J. Work, Definitions of terms relating to reactions of polymers and to functional polymeric materials (IUPAC Recommendations 2003), Pure Appl. Chem., 2004, 76, 889–906 CAS.
- P. J. Roth and P. Theato, in Reference Module in Materials Science and Materials Engineering, Elsevier, 2016, DOI:10.1016/B978-0-12-803581-8.01420-X.
- F. Ganachaud and S. Boileau, in Handbook of Ring-Opening Polymerization, ed. P. Dubois, O. Coulembier and J. M. Raquez, Wiley-VCH, 2009, ch. 3, pp. 65–95 Search PubMed.
- J. Chojnowski and M. Cypryk, in Silicon-Containing Polymers: The Science and Technology of Their Synthesis and Applications, ed. R. G. Jones, W. Ando and J. Chojnowski, Springer Netherlands, Dordrecht, 2000, ch. 1, pp. 3–41, DOI:10.1007/978-94-011-3939-7_1.
- J. Ackermann and V. Damrath, Chemie und Technologie der Silicone II. Herstellung und Verwendung von Siliconpolymeren, Chem. Unserer Zeit, 1989, 23, 86–99 CrossRef CAS.
- R. Schliebs and J. Ackermann, Chemie und Technologie der Silicone I, Chem. Unserer Zeit, 1987, 21, 121–127 CrossRef CAS.
- J. Chojnowski, in Siloxane Polymers, ed. S. J. Clarson and J. A. Semlyen, Prentice Hall, 1993, pp. 1–71 Search PubMed.
- W. Noll, Chemistry and Technology of Silicones, Academic Press Inc., New York, 1968 Search PubMed.
- M. Rodriguez, S. Marrot, T. Kato, S. Stérin, E. Fleury and A. Baceiredo, Catalytic activity of N-heterocyclic carbenes in ring opening polymerization of cyclic siloxanes, J. Organomet. Chem., 2007, 692, 705–708 CrossRef CAS.
- K. Sivasubramanian, P. P. Anderson and V. Khare, Catalyst for synthesis of siloxanes, Momentive Performance Materials Inc., US Pat, 20160215097, 2016 Search PubMed.
- K. Sivasubramanian and P. Anderson, Catalyst for synthesis of siloxanes Momentive Performance Materials Inc., US Pat, 9663620, 2017 Search PubMed.
- L. Rösch, P. John and R. Reitmeier, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000. DOI:10.1002/14356007.a24_021.
- M. P. Clarke, The direct synthesis of methylchlorosilanes, J. Organomet. Chem., 1989, 376, 165–222 CrossRef CAS.
- B. Marciniec, in Comprehensive Handbook on Hydrosilylation, ed. B. Marciniec, Pergamon, Amsterdam, 1992, pp. 8–98. DOI:10.1016/B978-0-08-040272-7.50007-7.
- H. M. Bank, M. E. Cifuentes and T. E. Martin, Process for the synthesis of soluble, condensed hydridosilicone resins containing low levels of silanol, Dow Corning Corporation, US Pat, 5010159, 1991 Search PubMed.
- S. C. Burgess, M. M. Crisanti, L. Li, S. Majeti, S. Mitra, E. A. Reno and J. Yue, Compositions Comprising organosiloxane resins for delivering oral care substances, Procter & Gamble, EP Pat, 1196135, 2002 Search PubMed.
- A. K. Serobian, Compositions and Methods for Treating Automotive Surfaces, The Armor All/STP Products Company, US Pat, 9399722, 2016 Search PubMed.
- A. F. Hollemann and E. Wiberg, Lehrbuch der anorganischen Chemie, Walter de Gruyter GmbH, Berlin/Boston, 101th edn, 1995 Search PubMed.
- J. N. Kostas, Process for preparing cyclic polysiloxanes from linear polysiloxanes, Hercules Incorporated, US Pat, 5491249, 1996 Search PubMed.
- G. R. Haines, D. E. Puckett and L. H. Wood, Process for preparing cyclic organohydrogensiloxanes, Dow Corning Corporation, US Pat, 5395956, 1995 Search PubMed.
- M. Cypryk, in Polymer Science: A Comprehensive Reference, ed. M. Möller, Elsevier, Amsterdam, 2012, vol. 4, ch. 4.17, pp. 451–476 Search PubMed.
- J. Beckmann, D. Dakternieks, A. Lim, K. Lim and K. Jurkschat, Understanding ring strain and ring flexibility in six- and eight-membered cyclic organometallic group 14 oxides, J. Mol. Struct., 2006, 761, 177–193 CrossRef CAS.
- T. C. Kendrick, B. Parbhoo and J. W. White, in The Silicon–Heteroatom Bond, ed. D. A. Armitage, R. J. P. Corriu, T. C. Kendrick, B. Parbhoo, T. D. Tilley, J. W. White and J. C. Young, 1991, pp. 67–140. DOI:10.1002/9780470772447.ch3.
- S. Penczek, M. Cypryk, A. Duda, P. Kubisa and S. Słomkowski, Living ring-opening polymerizations of heterocyclic monomers, Prog. Polym. Sci., 2007, 32, 247–282 CrossRef CAS.
- M. Cazacu and M. Marcu, Silicone Rubbers. Ix. Contributions to Polydimethylsiloxane-α,ω-Diols Synthesis by Heterogeneous Catalysis, J. Macromol. Sci., Part A: Pure Appl. Chem., 1995, 32, 1019–1029 CrossRef.
- L. E. Geipel, Molecular weight control and polysiloxanes, Stauffer Wacker Silicone Corp., US Pat, 3661962, 1972 Search PubMed.
- P. V. Wright, in Ring Opening Polymerization, ed. K. J. Ivin and T. Saegusa, Elsevier, London, 1984, vol. 2, ch. 14, p. 1055 Search PubMed.
- L. Wilczek and J. P. Kennedy, Aggregation in the Anionic Polymerization of Hexamethylcyclotrisiloxane with Lithium Counterion, Polym. J., 1987, 19, 531–538 CrossRef CAS.
- D. Graiver, A. W. Lomas, E. T. Rasmussen and K. J. Wall, Method for the preparation of potassium silanolates, Dow Corning Corporation, US Pat, 5856546, 1999 Search PubMed.
- A. Molenberg and M. Möller, A fast catalyst system for the ring-opening polymerization of cyclosiloxanes, Macromol. Rapid Commun., 1995, 16, 449–453 CrossRef CAS.
- B. G. G. Lohmeijer, G. Dubois, F. Leibfarth, R. C. Pratt, F. Nederberg, A. Nelson, R. M. Waymouth, C. Wade and J. L. Hedrick, Organocatalytic Living Ring-Opening Polymerization of Cyclic Carbosiloxanes, Org. Lett., 2006, 8, 4683–4686 CrossRef CAS.
- E. R. Evans, Process for the polymerization of cyclic diorganopolysiloxanes with cation-complex catalysts, General Electric Company, US Pat, 4122247, 1978 Search PubMed.
- D. Graiver, L. A. Wade, J. G. Matisons and A. Provatas, Preparation of polyorganosiloxanes by interfacial polymerization, Dow Corning Corporation/University of South Australia, EP Pat, 0799852, 1997 Search PubMed.
- P. Hupfield, A. Surgenor and R. Taylor, Polymerisation von Siloxanen, Dow Corning Corporation, EP Pat, 1008611, 2001 Search PubMed.
- B. Eßwein, A. Molenberg and M. Möller, Use of polyiminophosphazene bases for ring-opening polymerizations, Macromol. Symp., 1996, 107, 331–340 CrossRef.
- A. Molenberg and M. Möller, Polymerization of cyclotrisiloxanes by organolithium compounds and P2-Et base, Macromol. Chem. Phys., 1997, 198, 717–726 CrossRef CAS.
- K. Fuchise, M. Igarashi, K. Sato and S. Shimada, Organocatalytic controlled/living ring-opening polymerization of cyclotrisiloxanes initiated by water with strong organic base catalysts, Chem. Sci., 2018, 9, 2879–2891 RSC.
- M. Fevre, J. Pinaud, Y. Gnanou, J. Vignolle and D. Taton, N-Heterocyclic carbenes (NHCs) as organocatalysts and structural components in metal-free polymer synthesis, Chem. Soc. Rev., 2013, 42, 2142–2172 RSC.
- H. A. Brown, Y. A. Chang and R. M. Waymouth, Zwitterionic Polymerization to Generate High Molecular Weight Cyclic Poly(Carbosiloxane)s, J. Am. Chem. Soc., 2013, 135, 18738–18741 CrossRef CAS.
- V. Katiyar and H. Nanavati, Ring-opening polymerization of L-lactide using N-heterocyclic molecules: mechanistic, kinetics and DFT studies, Polym. Chem., 2010, 1, 1491–1500 RSC.
- S. Naumann, F. G. Schmidt, R. Schowner, W. Frey and M. R. Buchmeiser, Polymerization of methyl methacrylate by latent pre-catalysts based on CO2-protected N-heterocyclic carbenes, Polym. Chem., 2013, 4, 2731–2740 RSC.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Organocatalytic Ring-Opening Polymerization, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS.
- H. Kazama, Y. Tezuka and K. Imai, Syntheses and Reactions of Uniform Size Poly(dimethylsiloxane) with Various Reactive End Groups, Polym. J., 1987, 19, 1091–1100 CrossRef CAS.
- H. Kazama, Y. Tezuka and K. Imai, Synthesis and reactions of uniform-size poly(dimethylsiloxanes) having carboxylic acid as a single end group and both end groups, Macromolecules, 1991, 24, 122–125 CrossRef CAS.
- V. Bellas, H. Iatrou and N. Hadjichristidis, Controlled Anionic Polymerization of Hexamethylcyclotrisiloxane. Model Linear and Miktoarm Star Co- and Terpolymers of Dimethylsiloxane with Styrene and Isoprene, Macromolecules, 2000, 33, 6993–6997 CrossRef CAS.
- Y. Kawakami, Y. Miki, T. Tsuda, R. A. N. Murthy and Y. Yamashita, Silicone Macromers for Graft Polymer Synthesis, Polym. J., 1982, 14, 913–917 CrossRef CAS.
- W. Li and B. Huang, Synthesis of a polydimethylsiloxane macromonomer and its copolymerization with ethylene, Makromol. Chem., 1989, 190, 2373–2380 CrossRef CAS.
- M. Morton and E. E. Bostick, Anionic polymerization of octamethylcyclotetrasiloxane in tetrahydrofuran solution, J. Polym. Sci., Part A: Gen. Pap., 1964, 2, 523–538 CrossRef CAS.
- M. Morton, M. A. Deisz and E. E. Bostick, Base-catalyzed solution polymerization of octamethylcyclotetrasiloxane, J. Polym. Sci., Part A: Gen. Pap., 1964, 2, 513–522 CrossRef.
- H. J. Hölle and B. R. Lehnen, Preparation and characterization of polydimethylsiloxanes with narrow molecular weight distribution, Eur. Polym. J., 1975, 11, 663–667 CrossRef.
- B. G. Zavin, A. A. Zhdanov, M. Ścibiorek and J. Chojnowski, The anionic oligomerization of hexamethylcyclotrisiloxane with methylmethoxysilanes, Eur. Polym. J., 1985, 21, 135–140 CrossRef CAS.
- W. H. Dickstein and C. P. Lillya, Telechelic star poly (dimethylsiloxane) s of highly defined structure, Macromolecules, 1989, 22, 3886–3888 CrossRef CAS.
- C. L. Lee and O. K. Johannson, Polymerization of hexamethylcyclotrisiloxane with a biscatecholsiliconate. I. Nature of polymerization, J. Polym. Sci., Polym. Chem. Ed., 1976, 14, 729–742 CrossRef CAS.
- C. L. Lee, O. W. Marko and O. K. Johannson, Polymerization of hexamethylcyclotrisiloxane with a biscatecholsiliconate. II. Kinetics of polymerization, J. Polym. Sci., Polym. Chem. Ed., 1976, 14, 743–758 CrossRef CAS.
- T. Suzuki, Preparation of poly(dimethylsiloxane) macromonomers by the ‘initiator method’: 2. Polymerization mechanism, Polymer, 1989, 30, 333–337 CrossRef CAS.
- T. Suzuki and T. Okawa, Poly(dimethylsiloxane) macromonomers having both alkenyl and polymerizable groups. Application to crosslinkable copolymers, Polymer, 1988, 29, 2095–2099 CrossRef CAS.
- J. C. Saam, D. J. Gordon and S. Lindsey, Block Copolymers of Polydimethylsiloxane and Polystyrene, Macromolecules, 1970, 3, 1–4 CrossRef CAS.
- S. Boileau, in Ring-Opening Polymerization, ed. J. E. McGrath, American Chemical Society, Washington, D. C., 1985, vol. 286, ch. 2, pp. 23–35 Search PubMed.
- Y. A. Yushlevskii, E. G. Kagan and N. N. Fedoseeva, Polymerization of cyclosiloxanes by bases in the presence of activators, Dokl. Akad. Nauk SSSR, 1970, 190, 647–650 CAS.
- H. Kazama, Y. Tezuka and K. Imai, A new bifunctional initiator for the living polymerization of hexamethylcyclotrisiloxane, Polym. Bull., 1989, 21, 31–37 CrossRef CAS.
- Y. Zhang, Z. Zhang, Q. Wang and Z. Xie, Synthesis of well-defined difunctional polydimethylsiloxane with an efficient dianionic initiator for ABA triblock copolymer, J. Appl. Polym. Sci., 2007, 103, 153–159 CrossRef CAS.
- C. A. Veith and R. E. Cohen, Kinetic modelling and optimization of the trifluoropropylmethylsiloxane polymerization, J. Polym. Sci., Part A: Polym. Chem., 1989, 27, 1241–1258 CrossRef CAS.
- B. Momper, T. Wagner, U. Maschke, M. Ballauff and E. Fischer, Preparation and characterization of narrowly distributed poly (methylphenylsiloxane) by anionic polymerization of the cyclic trimers, Polym. Commun., 1990, 31, 186–189 CAS.
- R. C. Hedden and C. Cohen, Preparation of poly(diethylsiloxane) with the NaOH/12-crown-4 catalyst, Polymer, 2000, 41, 6975–6979 CrossRef CAS.
- T. C. Kendrick, B. M. Parbhoo and J. W. White, in Comprehensive Polymer Science and Supplements, ed. J. C. Bevington, Pergamon, Amsterdam, 1989, vol. 4, ch. 25, pp. 459–523 Search PubMed.
- M. Mazurek and J. Chojnowski, Anionic polymerization of siloxanes, 2. Internal multifunctional assistance of siloxane system to the siloxane bond cleavage by alcali metal silanolates, Makromol. Chem., 1977, 178, 1005–1017 CrossRef CAS.
- J. S. Ritch and T. Chivers, Silicon Analogues of Crown Ethers and Cryptands: A New Chapter in Host–Guest Chemistry?, Angew. Chem., Int. Ed., 2007, 46, 4610–4613 CrossRef CAS.
- M. Cypryk, Polymerization of Cyclic Siloxanes, Silanes, and Related Monomers, Elsevier, 2012 Search PubMed.
- J. Chojnowski, M. Mazurek, M. Šcibiorek and L. Wilczek, Cationic polymerization of siloxanes. Approach to the mechanistic studies, Makromol. Chem., 1974, 175, 3299–3303 CrossRef CAS.
- L. Wilczek and J. Chojnowski, Acidolytic ring opening of cyclic siloxane and acetal monomers. Role of hydrogen bonding in cationic polymerization initiated with protonic acids, Macromolecules, 1981, 14, 9–17 CrossRef CAS.
- G. Toskas, G. Besztercey, M. Moreau, M. Masure and P. Sigwalt, Cationic polymerization of hexamethylcyclotrisiloxane by trifluoromethanesulfonic acid and its derivatives, 2. Reaction involving activated trifluoromethylsulfonates, Macromol. Chem. Phys., 1995, 196, 2715–2735 CrossRef CAS.
- G. K. S. Prakash, C. Bae, Q. Wang, G. Rasul and G. A. Olah, Tris(trimethylsilyl)sulfonium and Methylbis(trimethylsilyl)sulfonium Ions: Preparation, NMR Spectroscopy, and Theoretical Studies1, J. Org. Chem., 2000, 65, 7646–7649 CrossRef CAS.
- J. Chojnowski and L. Wilczek, Mechanism of the polymerization of hexamethylcyclotrisiloxane (D3) in the presence of a strong protonic acid, Makromol. Chem., 1979, 180, 117–130 CrossRef CAS.
- F. Ganachaud, S. Boileau and B. Boury, Silicon Based Polymers: Advances in Synthesis and Supramolecular Organization, Springer Netherlands, 2008 Search PubMed.
- J. Chojnowski, M. Cypryk and K. Kaźmierski, Cationic Polymerization of a Model Cyclotrisiloxane with Mixed Siloxane Units Initiated by a Protic Acid. Mechanism of Polymer Chain Formation, Macromolecules, 2002, 35, 9904–9912 CrossRef CAS.
- W. Patnode and D. F. Wilcock, Methylpolysiloxanes, J. Am. Chem. Soc., 1946, 68, 358–363 CrossRef CAS.
- D. Graiver and O. Tanaka, Methods for making polydiorganosiloxane microemulsions, Dow Corning Corporation, US Pat, 4999398, 1991 Search PubMed.
- J.-R. Desmurs, L. Ghosez, J. Martins, T. Deforth and G. Mignani, Bis(trifluoromethane)sulfonimide initiated ring-opening polymerization of octamethylcyclotetrasiloxane, J. Organomet. Chem., 2002, 646, 171–178 CrossRef CAS.
- G. Siciliano, Continuous process for producing polysiloxanes oils utilizing a carbon black catalyst, General Electric, US Pat, 3853933A, 1974 Search PubMed.
- G. Siciliano and N. Holdstock, Continuous process for producing polysiloxane oils, General Electric, US Pat, 3853934, 1974 Search PubMed.
- A. Jallouli and J. Saam, Silyl Triflate-Initiated Ring-Opening Polymerizations of Cyclosiloxanes, J. Inorg. Organomet. Polym., 1998, 8, 179–203 CrossRef CAS.
- J. F. Hyde and J. R. Wehrley, Polymerization of organopolysiloxanes in aqueous emulsion, Dow Corning Corporation, US Pat, 2891920A, 1959 Search PubMed.
- S. Dong, F. J. Traver and J. F. Warrenchak, Emulsion polymerization process, General Electric Company, US Pat, 6232396, 2001 Search PubMed.
- S. Cauvin, S. Hanssens, A. Petrosino, B. Rassart and B. Van Roy, Emulsion polymerisation method, Dow Corning Corporation, US Pat, 9156954, 2015 Search PubMed.
- I. G. McMullen, H. I. Haden and R. Broadhurst, Dyeing of polyester and cellulose fiber blends with a silicone-containing solution of a reactive dye and a disperse dye and an aminoplast precursor treatment thereof, Imperial Chemical Industries Limited, US Pat, 3504996, 1970 Search PubMed.
- J. A. Kosal, Silicone pressure sensitive adhesive compositions, Dow Corning, US Pat, 6545086, 2003 Search PubMed.
- R. P. Gee, Silicone emulsion for personal care application, Dow Corning, US Pat, 5300286, 1994 Search PubMed.
- A. R. Avery, D. Charmot, J. M. Frechet, D. Hajduk, E. Khoshdel and M. Liu, Home and personal care compositions comprising silicon based lubricants, Conopco, Inc., US Pat, 2006100126, 2006 Search PubMed.
- R. P. Gee, Emulsion polymerization of dimethylcyclosiloxane in cationic emulsion: mechanism study utilizing two phase liquid–liquid reaction kinetics, Colloids Surf., A, 2015, 481, 297–306 CrossRef CAS.
- R. P. Gee and B. L. Zimmerman, Cationic emulsion polymerization of dimethylcyclosiloxane utilizing anionic surfactant: A mechanism study, Colloids Surf., A, 2016, 508, 101–109 CrossRef CAS.
| This journal is © the Partner Organisations 2020 |