
The present and future synthetic strategies of structural modifications of sinomenine

Abstract
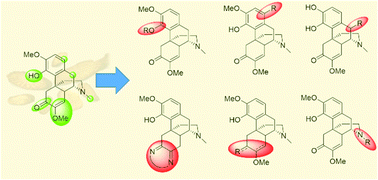
Sinomenine is a tetracyclic alkaloid that is extracted from the traditional Chinese medicine sinomenium acutum. It has been reported to possess low cytotoxicity and a variety of biological activities, such as anti-tumor, anti-inflammatory and anti-arthritic effects. However, the relatively short biological half-life of sinomenine hinders its extensive clinical application. Hence, extensive research on its structural modifications has been carried out in recent decades. The reaction sites can be classified into four categories based on functional groups—aromatic ring (A-ring), benzylic position (B-ring), enone (C-ring) and trialkylamine (D-ring). This review summarizes the representative examples of each modification to date and discusses the synthetic difficulties, especially the chemoselectivity and stereoselectivity of reactions. The future prospects of synthesis of sinomenine derivatives from sinomenine-like derivatives and by some novel late-stage functionalizations, which can be potentially applicable to sinomenine, of structurally similar derivatives are also discussed.
 Please wait while we load your content...
Please wait while we load your content...

