
The present and future synthetic strategies of structural modifications of sinomenine
Jerome P. L.
Ng
a,
Paolo
Coghi
b,
Betty Yuen Kwan
Law
a,
Liang
Liu
*a and
Vincent Kam Wai
Wong
 *a
*a
aState Key Laboratory of Quality Research in Chinese Medicine, Macau University of Science and Technology, Avenida Wai Long, Taipa, Macau, China. E-mail: lliu@must.edu.mo; bowaiwong@gmail.com
bSchool of Pharmacy, Macau University of Science and Technology, Avenida Wai Long, Taipa, Macau, China
First published on 14th October 2020
Abstract
Sinomenine is a tetracyclic alkaloid that is extracted from the traditional Chinese medicine sinomenium acutum. It has been reported to possess low cytotoxicity and a variety of biological activities, such as anti-tumor, anti-inflammatory and anti-arthritic effects. However, the relatively short biological half-life of sinomenine hinders its extensive clinical application. Hence, extensive research on its structural modifications has been carried out in recent decades. The reaction sites can be classified into four categories based on functional groups—aromatic ring (A-ring), benzylic position (B-ring), enone (C-ring) and trialkylamine (D-ring). This review summarizes the representative examples of each modification to date and discusses the synthetic difficulties, especially the chemoselectivity and stereoselectivity of reactions. The future prospects of synthesis of sinomenine derivatives from sinomenine-like derivatives and by some novel late-stage functionalizations, which can be potentially applicable to sinomenine, of structurally similar derivatives are also discussed.
 Jerome P. L. Ng | Jerome P. L. Ng received his Ph.D. from the University of Hong Kong in 2019 under the supervision of Prof. Pauline Chiu. He is currently a postdoctoral fellow of Dr Vincent Wong's group at the State Key Laboratory in Macau University of Science and Technology since 2019. His research focuses on the organic synthesis of derivatives of natural products derived from traditional Chinese medicine. |
 Paolo Coghi | Paolo Coghi received his PhD (2008) from the University of Milan (Italy) guided by Dr Diego Monti. After a postdoctoral stay (2008–2011) at the National Research Council of Italy and working in the group of Prof. Antonio Papagni (2011–2014), he joined the group of Dr Vincent Wong working on the synthesis of triterpenoid derivatives. Since 2017 he has been a lecturer in the School of Pharmacy at Macau University of Science and Technology. His research interests focus on infective disease and cancer. |
 Betty Yuen Kwan Law | Betty Yuen Kwan Law obtained her Ph.D. degree from the University of Hong Kong in 2009. She is currently working as an associate professor in the Macau University of Science and Technology (State Key Laboratory of Quality Research in Chinese Medicines). Her research interest includes the identification of novel autophagic enhancers from Chinese medicinal herbs and therapeutic applications of the identified autophagic modulators. |
 Liang Liu | Liang Liu received his Ph.D. in Medicine from Guangzhou University of Traditional Chinese Medicine in 1990. He is currently the President of Macau University of Science and Technology and Director of State Key Laboratory of Quality Research in Chinese Medicine. His research in the integrative approach of Western and traditional Chinese medicine has been recognized by many awards and honorary titles. |
 Vincent Kam Wai Wong | Vincent Kam Wai Wong received his Ph.D. from the University of Hong Kong in 2006 and then worked as a post-doctoral research fellow at Hong Kong Baptist University for three years. In 2009, he was promoted to a research assistant professor. He started working as an assistant professor in the State Key Laboratory of Macau University of Science and Technology in 2011 and was promoted to an associate professor in 2015. His research focuses on the relationship between autophagy and multidrug-resistant, apoptosis–resistant anti-tumor effects via SERCA inhibition and AMPK activation. |
1. Introduction
Sinomenine 1 is an alkaloid and is a biologically active component extracted from the root of an herbal plant sinomenium acutum. Traditionally, sinomenium acutum has been used for treating arthritis patients in China.1 Recent studies showed that its primary ingredient, sinomenine 1, was responsible for the anti-arthritic effect.2–4 Sinomenine has also exhibited a wide spectrum of biological activities, such as anti-tumor, anti-arrhythmic, immunosuppressive, anti-hypertensive, and anti-inflammatory effects.1,5,6 In particular, sinomenine was used as a combinational therapy with methotrexate to treat rheumatoid arthritis effectively.7 Despite the promising biological properties of sinomenine, some potential adverse clinical effects and relatively short biological half-life of sinomenine were reported.8,9 Structural modification thus became a hot topic for optimizing the properties of sinomenine.Sinomenine is a morphine-type compound consisting of a tetracyclic framework (Fig. 1). Structurally, it is similar to codeine except for having an oxa-bridge between C4 and C5, opposite stereochemistry of the D-ring and different substituents at C6 and C7 positions. Other natural products like dextromethorphan 2c also possess a similar tetracyclic framework without having the oxa-bridge that is found in morphine 2a and codeine 2b. Sinomenine is composed of three labile functional groups—tetrasubstituted aromatic ring, α,β-unsaturated ketone and trialkylamine. Therefore, four major reaction sites are identified as aromatic ring (A-ring), benzylic position (B-ring), enone (C-ring) and amine (D-ring). The multifunctional groups of sinomenine also give rise to difficulties in the chemoselectivity of reactions upon modifications. In some cases, one single step generated several products that pure compounds were inseparable10 or one useful synthetic intermediate was obtained in a low yield.11 In view of these, developing high selectivity synthetic protocols is of the utmost importance for advanced research in the future drug design of sinomenine.
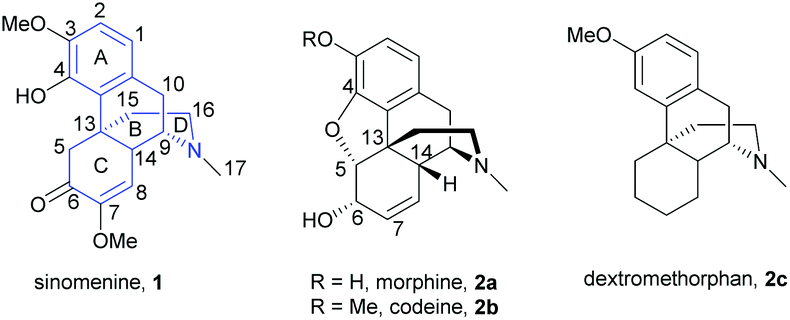 | ||
| Fig. 1 Structures of sinomenine 1, morphine 2a, codeine 2b and dextromethorphan 2c. | ||
The pharmacological effects, mechanisms and clinical applications of sinomenine and its derivatives have been summarized in numerous reviews.1,5,6,12 Although some reviews briefly summarized the synthesis of some important derivatives, the systematic analysis of selectivity and reactivity issues in synthetic protocols was not made.12–14 Thus, we would like to reveal some representative examples of each modification and discuss the synthetic issues found in the synthesis. To provide new insight into the development of novel sinomenine derivatives, synthetic strategies of sinomenine derivatives from functionalizations of other naturally or commercially available sinomenine-like derivatives are also reviewed.
2. Modifications of the aromatic ring (A-ring)
Among different chemically active functional groups of sinomenine 1, the tetrasubstituted aromatic ring of sinomenine is the most considerable site for modifications due to its broad scope of chemical reactivity and less sterically hindered environment.2.1 Electrophilic aromatic substitution
Electrophilic aromatic substitution is a typical reaction for benzene. The A-ring of sinomenine 1 is highly prone to electrophilic attack due to the high electron density and extra stabilization of cationic intermediates by electron-rich substituents. Electrophilic aromatic substitution of the A-ring of 1 and its derivatives has been reported with high regioselectivity at the C1 position by halogenation, nitration and Friedel–Crafts alkylation.Halogenation of 1 with N-halosuccinimide (NXS, X = Cl, Br or I) resulted in aryl halides 3a–c with moderate to high yields (55–85%, Scheme 1).15,16 In the absence of the enone moiety, the iodination of C-ring non-functionalized derivatives also provided iodide 4 with a comparable yield (84%).17 Halogenations using NXS also worked well in other substrates with 1,2- and 1,4-reduced enone of the C-ring.16 Only mono-halogenated products were observed when using NXS as the halogenating reagent. Similarly, bromination of 1 with bromine in dichloromethane only generated mono-halogenated derivatives. However, the reaction of 1 with bromine in acetonitrile afforded intermediate 3b′′ with dibromide at C1 and C5 positions, and subsequently basic treatment caused ring closure between the C4-hydroxy group and C5-bromide to afford morphine derivatives.18 The α-bromination of ketone at C5 involves the formation of enol (Scheme 2). A more polar solvent stabilizes the formation of enol and consequently facilitates the α-bromination. Hence, bromination using a relatively more polar solvent, MeCN (dielectric constant of MeCN vs. CH2Cl2 = 37.5 vs. 8.93), occurs at both C1 and C5.
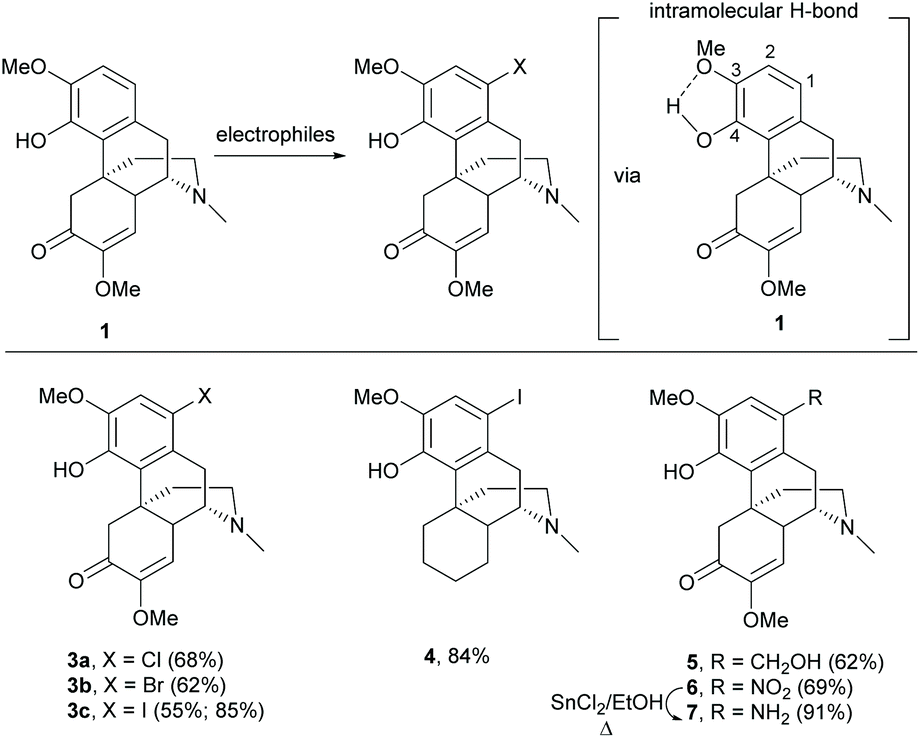 | ||
| Scheme 1 Electrophilic aromatic substitutions of sinomenine 1. | ||
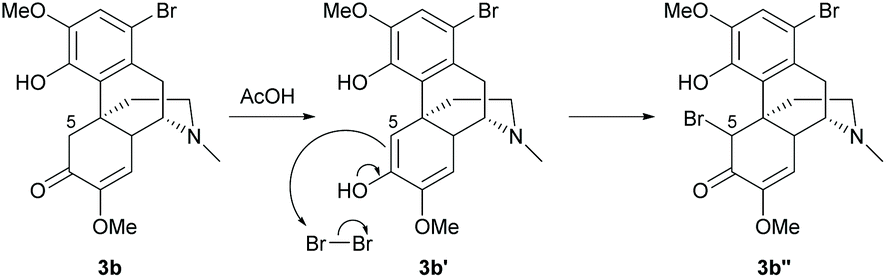 | ||
| Scheme 2 Proposed reaction mechanism of α-bromination of 3b. | ||
Other 1-substituted electron-withdrawing groups were synthesized from halides 3a/3c but not directly from 1 (Scheme 3).19 Fluoride 3d was generated by fluorine–halogen exchange using potassium fluoride as the fluorinating reagent. Similarly, nucleophilic trifluoromethylation of iodide 3c afforded 1-trifluoromethyl derivative 8 (Scheme 3).
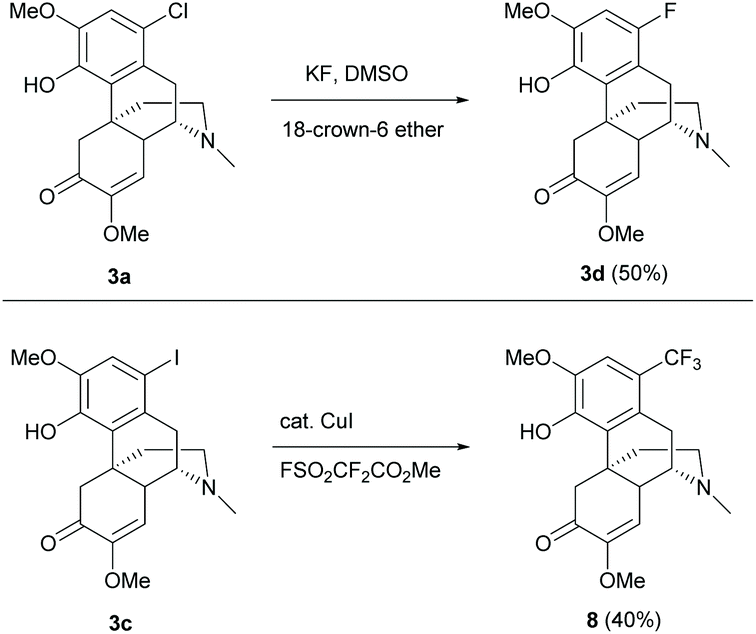 | ||
| Scheme 3 Fluorine–halogen exchange of 3a and nucleophilic trifluoromethylation of 3c. | ||
The Friedel–Crafts type reaction with formaldehyde and nitration with sodium nitrite generated 1-hydroxymethyl derivative 5 and 1-nitro derivative 6 respectively (Scheme 1), which were important synthetic intermediates for further functionalization, such as alkylation and acylation.20–22 Furthermore, the reduction of the nitro group with SnCl2/EtOH under reflux conditions was performed selectively to yield amine 7 without over-reduction in the C-ring. Otherwise, typical reduction conditions using a palladium catalyst and hydrogen afforded the 1,4-reduced product as mentioned in Scheme 25.
Interestingly, the electrophilic aromatic substitution of 1 is highly regioselective at the C1-position. The intramolecular H-bond between C3-methoxy and C4-hydroxy was proposed to increase the mesomeric and inductive effects of the C4-hydroxy group toward the para-position (C1) (Scheme 1).23 Meanwhile, the intramolecular H-bond diminished both effects of the C3-methoxy group toward the ortho-position (C2). The steric strains for substitutions at C1- and C2-positions are similar. Therefore, the regioselectivity of 1 was governed by the electronic factor.
2.2 C(sp2)–C(sp2) coupling reaction
The C(sp2) functionalization of the A-ring of 1 can be achieved by palladium-catalyzed C(sp2)–C(sp2) coupling reaction via the key intermediates aryl halides derived from halogenation as mentioned in section 2.1.Catalytic cross-coupling reactions of aryl iodide 4 with various acrylates and Pd(OAc)2 by the Heck reaction generated cinnamate derivatives 9a–e with high yields (84–93%, Scheme 4).17 Using a microreactor for the Heck reaction provided the same products 9f–j with higher yields by 46–68% in a short reaction time (20 min).24 Generally, palladium-catalyzed cross-coupling reactions afforded (E)-isomers of adducts as the major products because the syn-β-hydride elimination of the reaction occurred via the less steric congestion to afford the thermodynamically favourable (E)-olefin. Milder reaction conditions using Suzuki coupling with aryl boronates also produced the corresponding aryl–aryl coupling products 10a–b with comparable yields (86–93%, Scheme 4).25 Further extension of the substrate scope of coupled products is potentially performed by using the Heck reaction in conjunction with CO insertion.
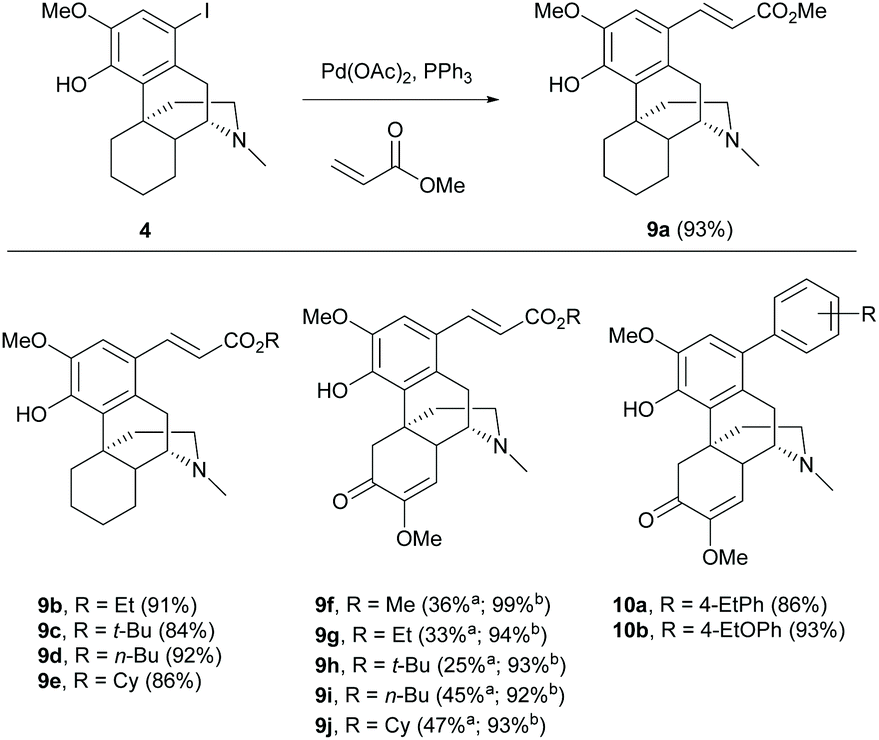 | ||
Scheme 4 Cross-coupling reactions of aryl halides with aryl and alkenyl coupling partners catalyzed by palladium. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reactions were performed without a microreactor. bReactions were performed with a microreactor. Reactions were performed without a microreactor. bReactions were performed with a microreactor. | ||
2.3 O-Alkylations and esterifications
Sinomenine possesses the C4-hydroxy group which is prone to further chemical modifications and is one of the major nucleophilic sites of sinomenine. To date, the O-alkylations and esterifications have been reported leading to ether and ester derivatives respectively.Alkylations of the labile C4-hydroxy group proceeded either via SN2 reactions with alkyl halides21 or the Mitsunobu reaction with alcohols.26 Alkylation of 1 with ethyl bromide using K2CO3 as the base resulted in a low yield of 11a (15%, Scheme 5) because of the formation of a N-alkylated product (81% yield).26 Generally, N-alkylation is more favorable than O-alkylation. When Cs2CO3 was used as a base, the yield of 11a was improved up to 83% (Scheme 5).21 High yield and chemoselective alkylation of the C4-hydroxy group was also observed in derivative 11b probably due to the cesium effect. The cesium effect resulted in a weak coordination of the cesium cation with the phenolic anion. The phenolic anion became more nucleophilic and the O-alkylation was predominant over N-alkylation.27 Moreover, the cesium cation was proposed to coordinate with trialkylamine to form a quaternary ammonium compound.28 The lone pair of N atom was no longer available for the alkylation. Alternatively, the alkylation of the C4-hydroxy group by the Mitsunobu reaction provided chemoselective alkylated products 12a–c in high yields (50–81%, Scheme 5).26
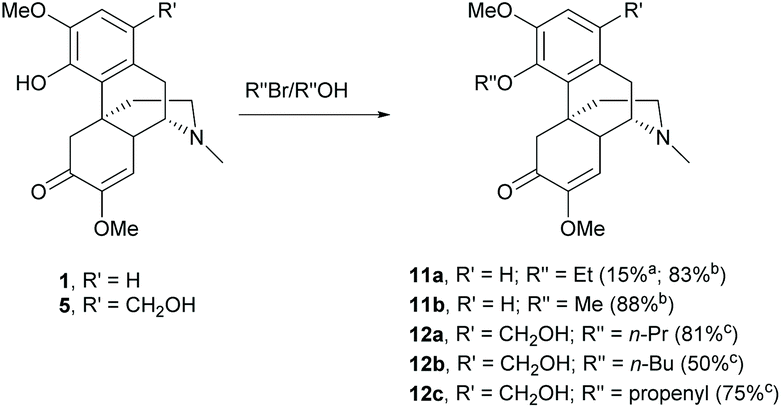 | ||
| Scheme 5 The 4-O-alkylations of sinomenine derivatives. aK2CO3 was used as the base. bCs2CO3 was used as the base. cReactions were performed under Mitsunobu reaction conditions. | ||
Esterification of the C4-hydroxy group of 1 afforded ester derivatives 13a–c with saturated and unsaturated alkyl chains in similar yields (∼70%, Scheme 6).29,30 Moreover, the phenolic hydroxy group at the C4 position reacted chemoselectively over the secondary hydroxy group at the C6-position, resulting in esters 14–15 due to the higher acidity of the C4-hydroxy group.30 Notably, the primary alcohol at the C1-position of derivative 5 was also susceptible to esterification, resulting in di-esters 16a–b.29 Incorporation of the PEG moiety at the C4-substituent by esterification with EDC was also reported.31 Despite using EDC as the coupling reagent, the acylation of 5 with acid anhydrides generated di-esters 16c–d with higher yields (75–85%, Scheme 6).21 However, the installation of longer alkyl chains by acid anhydrides is not as good as the methodology using EDC with carboxylic acids due to expensive or commercially unavailable acid anhydrides with long alkyl chains and the poor atom economy of the reaction.
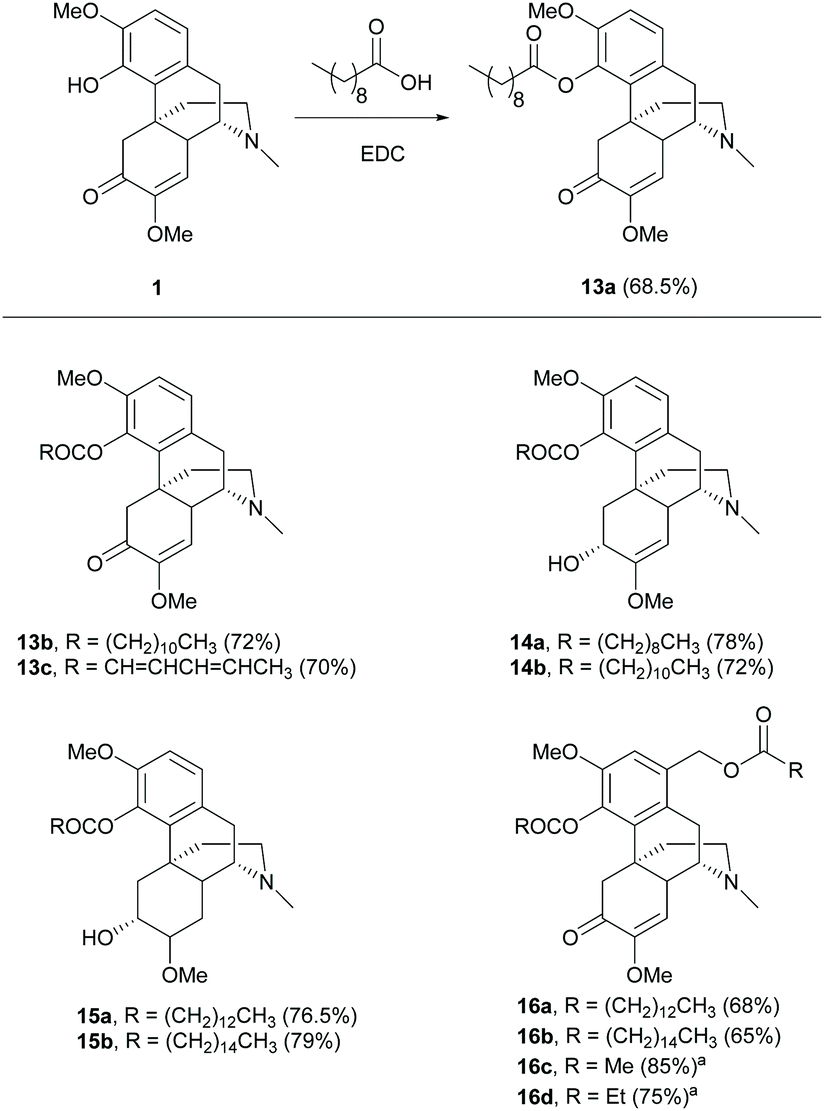 | ||
| Scheme 6 The 4-O-acylations of sinomenine derivatives. aReactions were performed with acid anhydrides. | ||
Derivative 17 with terminal alkyne was obtained using KOH as the base in an exceptionally high yield (96%, Scheme 7). The tolerance of sinomenine derivative 17 towards 1,3-dipolar cycloadditions was reported.32 The 1,3-dipole was not prepared from sinomenine due to the incompatibility of the C6-carbonyl group towards its formation. Therefore, the alkyne component was incorporated into the C4-position of 1 by an SN2 reaction with 3-chloropropyne (Scheme 7). The subsequent cycloaddition of 17 worked well with both electron-rich and electron-deficient 1,3-dipoles derived from the corresponding aromatic aldehydes, to afford cycloadducts 18a–e with moderate to high yields (76–91%, Scheme 7). Under similar reaction conditions, 1,3-dipolar cycloaddition of C1-cinnamate derivative 9f (Scheme 4) with 1,3-dipoles resulted in similar yields (81–93%).24 The cycloaddition of the enone of the C-ring was not observed probably due to the steric effect of the α-substituent.
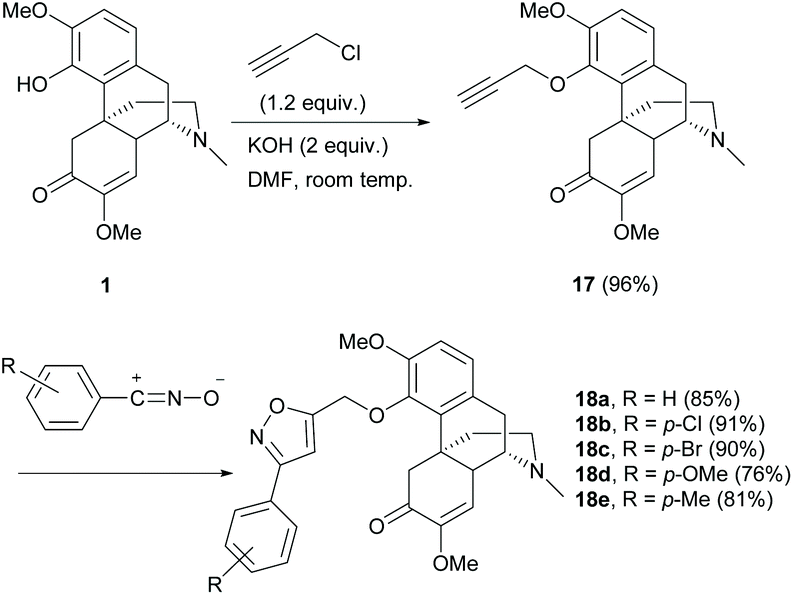 | ||
| Scheme 7 The 4-O-alkylations of 1 and the subsequent 1,3-dipolar cycloaddition of alkyne derivative 17. | ||
2.4 Dimerizations
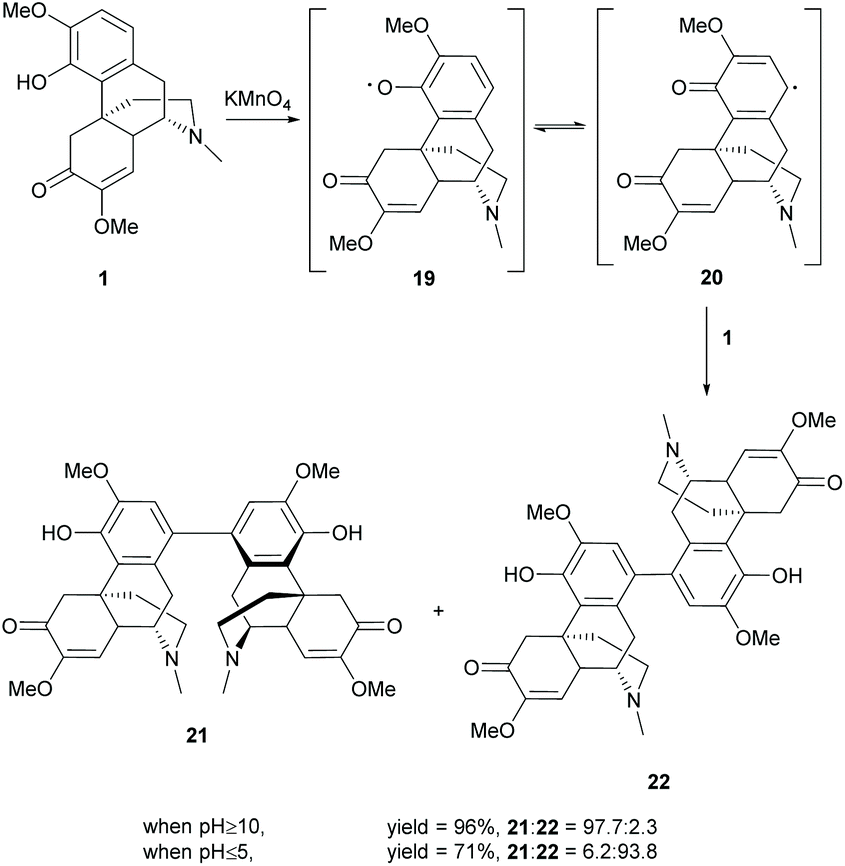
Synthesis of dimers of sinomenine was reported by using oxidative coupling reactions and the 4-O-alkylation/esterification conditions as mentioned in section 2.3. Dimers generated by 4-O-alkylation/esterification used dibromoalkanes, diacyl chlorides or diols as linkers. The full substrate scope can be referred to the literature.33Another strategy for coupling of 1 at the C1-position by oxidation with KMnO4 synthesized stereoisomers 21 and 22 stereoselectively at different pH values (Scheme 8).34 The oxidation of 1 under basic conditions (pH ≥ 10) provided 21 as the major stereoisomer (21:22 = 97.7:2.3) while that under acidic conditions (pH ≤ 5) provided 22 as the major stereoisomer (21:22 = 6.2:93.8). The total yield of both stereoisomers diminished at low pH probably due to the strong oxidative effect of KMnO4 under acidic conditions.
 | ||
| Scheme 8 Dimerization of 1 by oxidative coupling. | ||
The reaction was proposed to undergo a radical oxidative aromatic coupling via aryl radical 20, similar to the synthesis of BINOL.35 The stereoselectivity was probably controlled by the reaction rate of radical formation. Under acidic conditions, the stronger oxidizing power of KMnO4 allows the fast formation of radicals, leading to kinetically stable product 22 with less steric hindrance. While the alkaline conditions diminished the oxidizing power of KMnO4, the slower formation of radicals allowed the re-organization of the conformations of radicals 20 and 1. Finally, thermodynamically stable product 21 was formed probably due to the extra stability arising from intermolecular π–π stacking of the A-ring.
For faster screening of the bioactivity of both stereoisomers, other oxidants, such as MnO2 and FeCl3/H2O2, afforded both stereoisomers in a ratio of approximately 1:1. More examples of dimers with a modified C-ring of 1 were found in the literature.36
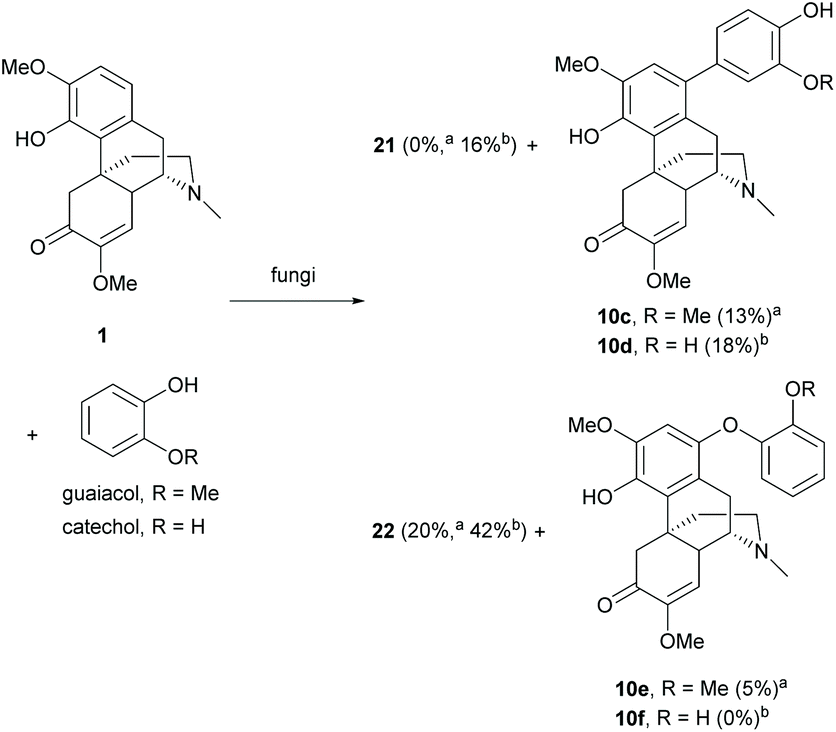
Other than the chemical methods, dimerization of 1 can also be achieved by biotransformation.37,38 Biotransformation of 1 with Antrodiella semisupina generated dimer 22 in a low yield (20%).38 The reaction was capable of coupling on multiple substrates. The biotransformation of 1 with guaiacol generated dimer 22 (20%) together with C–C coupled derivative 10c (13%) and C–O coupled derivative 10e (5%, Scheme 9).
 | ||
| Scheme 9 Dimerization and cross-coupling of 1 by biotransformation. aReaction was performed with Antrodiella semisupina. bReaction was performed Coriolus unicolor. | ||
However, no cross-coupled product was obtained when the reaction of 1 and catechol was performed with the same fungus Antrodiella semisupina.37 Another screened fungus Coriolus unicolor catalyzed the reaction to synthesize both dimers 21 and 22 (58% combined yield) and C–C coupled derivative 10d (18%) as a minor product (Scheme 9).
As proposed by the authors, the biotransformation proceeded via a mechanism similar to the oxidative radical pathway as depicted in Scheme 8. The oxidation of 1 and guaiacol/catechol generated oxygen radicals at hydroxy groups, which were delocalized to form carbon radicals for subsequent homo- and cross-couplings. The proposed mechanism was verified by acetylation of the 4-hydroxy group of 1.38 Once the 4-hydroxy group was protected, no coupled product was observed. In general, the homo-coupling of 1 was predominant over cross-coupling probably due to the formation of a more stable radical derived from 1.
2.5 Future prospects of synthesis of A-ring modified sinomenine derivatives from late-stage functionalizations of other derivatives with similar core structures
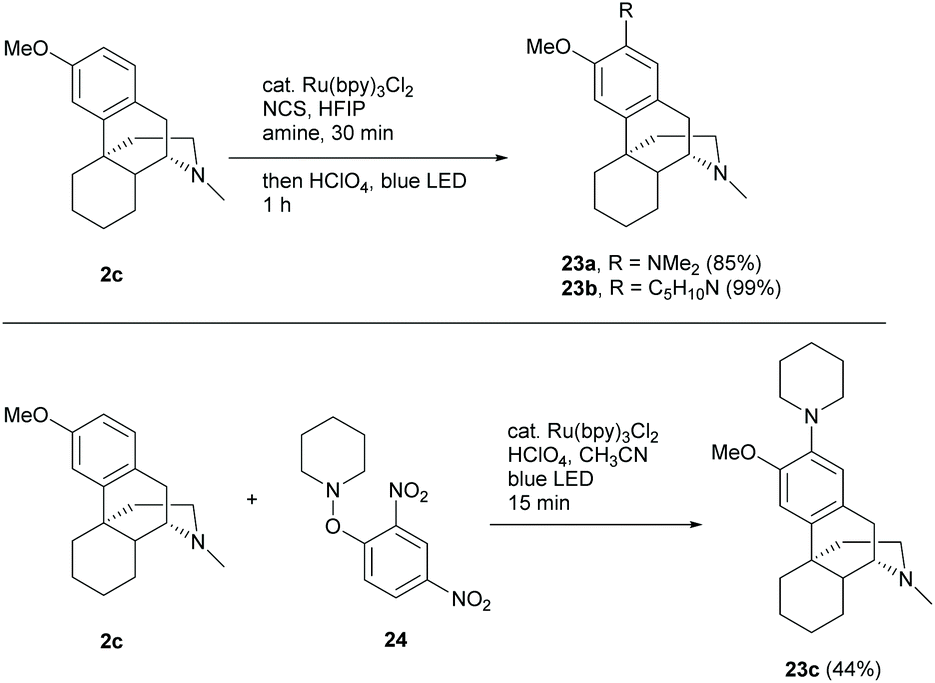
Sinomenine and the related structures possessed an absolute configuration of the D-ring opposite to that of codeine derivatives and this was confirmed by X-ray crystallography of the cyclization product of 1,4-reduced derivative 61 (Scheme 25).39 Despite the direct modification of sinomenine 1, dextromethorphan 2c or other structurally similar derivatives became an alternative option of the starting material due to the similarity of the tetracyclic framework and low cost. For example, 2c was shown to be a neuroprotective and anti-inflammatory agent, similar to 1.40 Moreover, the novel functionalizations of 2c and other similar derivatives may be possibly applicable to sinomenine, especially C-ring non-functionalized derivative 58 (Scheme 24).Recent developments of novel chemical reactions showed their compatibility towards complex biomolecules by using dextromethorphan as one of the examples. Ruffoni et al. showed the regioselective radical amination of arenes with aliphatic amines by photocatalysis.41 The amination of dextromethorphan 2c with 24 different aliphatic amines afforded 2-aminated products with >50% yields in most cases of which two representative examples are shown in Scheme 10.
 | ||
| Scheme 10 Regioselective amination of 2c by alkyl amines. | ||
The reaction showed high tolerance to the hydroxy group, halide, ester and alkene. This may allow the reaction to be applied in the late-stage modifications of sinomenine and its derivatives with a protecting-group-free strategy. Notably, radical amination of 2c with O-aryl hydroxylamine 24 afforded 23c with a diminished yield (44%) but a faster conversion within 15 min (Scheme 10).42
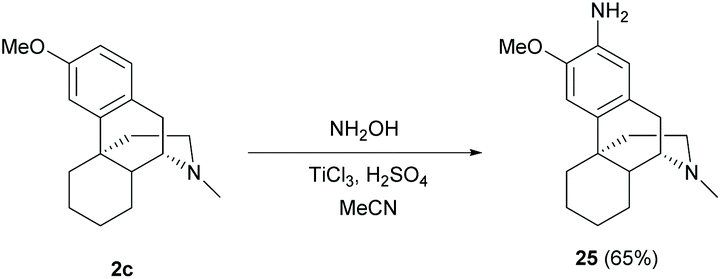
The direct C–H amination of arenes with hydroxylamine mediated by titanium(III) chloride was reported to be applicable in complex natural products including sinomenine-like derivative 2c to provide a shorter synthetic step than typical amination steps (nitration and subsequent nitro reduction).43 The reaction of 2c with hydroxylamine proceeded via the formation of the aminyl radical (˙NH2), which was derived from TiCl3-mediated N–O bond cleavage, to afford aniline 25 in one step (65%, Scheme 11). Notably, labile functional groups such as hydroxy and enone have to be protected prior to utilizing this protocol.
 | ||
| Scheme 11 The TiCl3-mediated C–H amination of arene 2c with hydroxylamine. | ||
Similar to the reaction mechanism of the C(sp2)–C(sp2) coupling reaction as described in section 2.2, Ichii et al. demonstrated the Hiyama cross-coupling of aryl bromides with aryl(trialkoxy)silanes at a ppm level loading of Pd catalyst 27.44 Only one example of the cross-coupling reaction of silylated dextromethorphan 26 was demonstrated in a high yield (90%, Scheme 12). Notably, the synthesis of silylated 26 involved the use of n-BuLi which was incompatible with the enone of the C-ring of 1. However, using C-ring non-functionalized derivative 58 or switching the role of 1 from a silylating reagent to a bromide reagent will be feasible for the reaction.
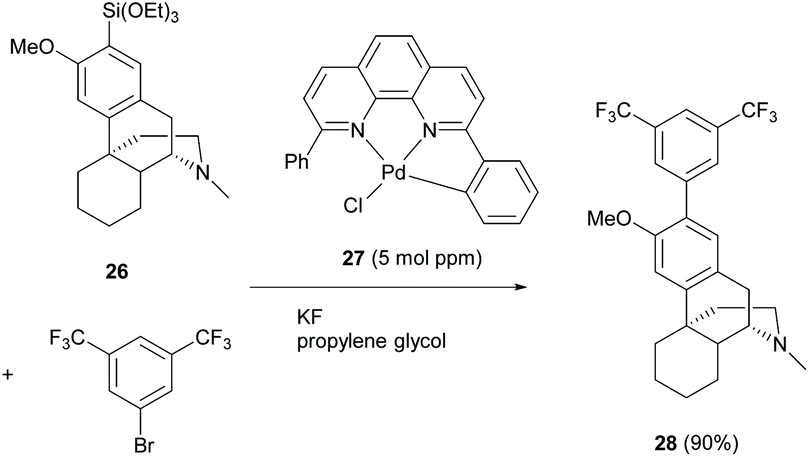 | ||
| Scheme 12 Hiyama cross-coupling of dextromethorphan derivative 26 with aryl bromide. | ||
The synthesis of unsymmetrical diaryl sulfide 29 by direct C(sp2)–H thioarylation using phenyl methyl sulfoxide activated by triflic anhydride resulted in a high yield (70%, Scheme 13).45 The reactions demonstrated a high functional group tolerance, such as aldehyde and enone, which showed its suitability in the late-stage modification of sinomenine derivatives.
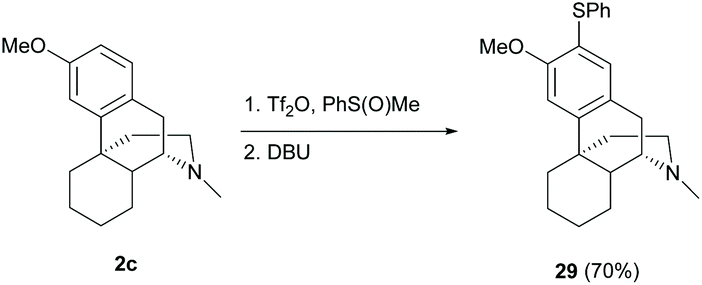 | ||
| Scheme 13 The C(sp2)–H thioarylation of 2c with sulfoxide. | ||
Direct modification of the C3-methoxy group of 2c with Grignard reagents by nickel catalysis underwent C–O cleavage and subsequent C–C bond formation to afford derivatives 30a–d in moderate to high yields (52–74%, Scheme 14).46,47 Similarly, C-ring non-functionalized derivative 58 will work well for the reaction preventing 1 from the 1,2-addition with Grignard reagents.
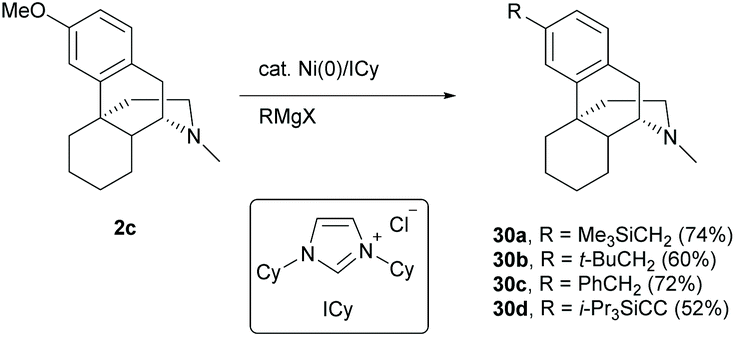 | ||
| Scheme 14 Nickel-catalyzed cross-couplings of 2c with Grignard reagents. | ||
The amination of the aryl ring of aryl triflate 31, an enantiomeric derivative of 2c, was also reported by Pd-catalyzed cross-coupling of aryl triflate with aromatic amine. A nearly quantitative yield of coupled product 33 was isolated (98%, Scheme 15).48 The opposite enantiomer of 31 also underwent the reaction analogously to yield the corresponding enantiomer of 33 in 89% yield. However, another Pd-catalyzed amination of triflate derivatives with amines was reported with low yields (<20%) probably due to improper choice of ligands.49
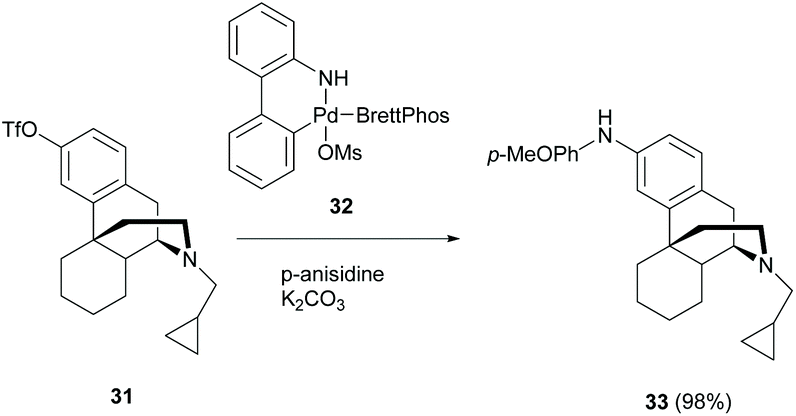 | ||
| Scheme 15 Buchwald–Hartwig amination of aryl triflate 31 with p-anisidine. | ||
A novel synthetic protocol for allylic C(sp2)–H amination was developed. One example of Pd catalysis of C(sp3)–N fragment coupling reaction of olefin 34 derived from 2c was reported to give 35 in a high yield (91%) and E-selectivity (Scheme 16).50 The reaction is well-tolerant to tertiary amine (D-ring) by protonation with dichloroacetic acid.
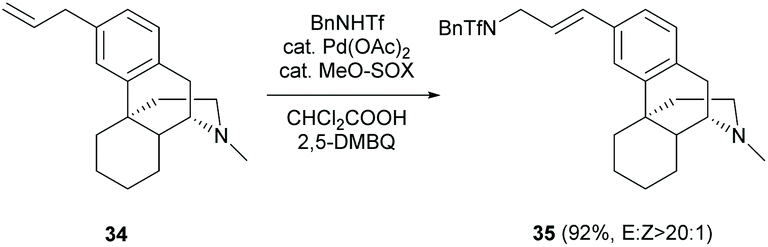 | ||
| Scheme 16 The C(sp3)–N fragment coupling reaction of olefin 34 with BnNHTf. | ||
Other than C(sp2)–H functionalization at the A-ring, C(sp3)–H amination at the benzylic position of 3-alkyl substituted 36 by manganese catalysis was reported in a moderate yield (44%, Scheme 17).51 The amination occurred chemoselectively at the remote benzylic position but not the benzylic position at C-10 resulting from the electron-deficiency induced by the protonation of 3° amine with HBF4. Changing the catalyst from manganese to rhodium provided amination at 3° amine to furnish hydrazine as the major product.52
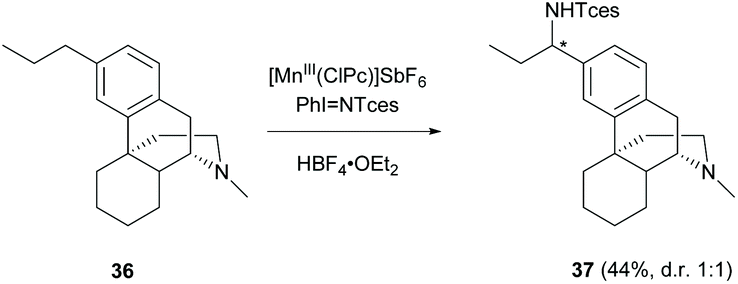 | ||
| Scheme 17 The C(sp3)–H amination of 36 by manganese catalysis. | ||
Alkyne, an electron-rich functional group, has demonstrated one of its reactivities towards 1,3-dipolar cycloaddition in the modifications of sinomenine (Scheme 7). Recently, diprenorphine 38 with similar core structures to sinomenine possessed terminal alkyne which underwent double hydroboration.53 Unlike the typical hydroboration using transition metals, the double hydroboration of diprenorphine was catalyzed by 9-borabicyclo[3.3.1]nonane (H-B-9BBN) to afford gem-diboryl product 39 (67%, Scheme 18).
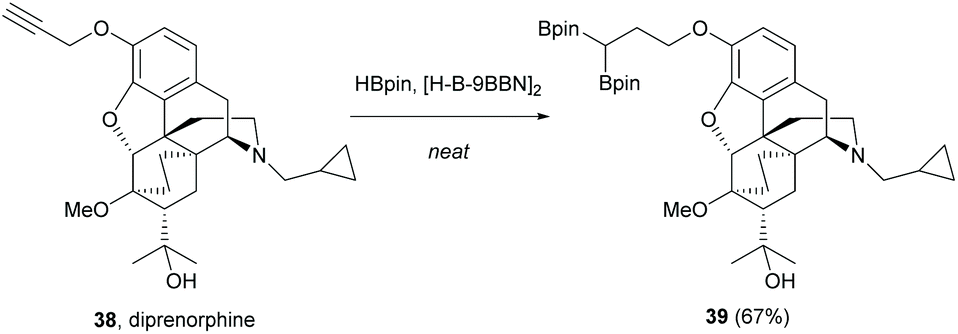 | ||
| Scheme 18 Borane-catalyzed alkyne double hydroboration of diprenorphine 38. | ||
3. Modifications of the B-ring
3.1 The modifications of the benzylic position of sinomenine (B-ring)
The B-ring of sinomenine is one of the most difficult positions for chemical modifications due to the steric effects of A-, C- and D-rings.One strategy is to functionalize the benzylic position by oxidizing the A-ring of 1 into a quinone methide 40 with PhI(OAc)2 (DIB).54,55 The products obtained were solvent-dependent (Scheme 19). The oxidative dearomatization in H2O provided quinone methide 40 while that in methanol generated re-aromatized products 41a and 41b. The formation of 41a and 41b was postulated to result from 1,6-conjugated addition of 40 prepared in situ from 1, with methanol from solvent or acetate from DIB. The results showed that activated 40 was highly susceptible to the attack of nucleophiles, even as weak as acetate. Similar nucleophilic addition was also observed when 41a was oxidized with DIB in methanol. After the first oxidation of 41a, 1,2-addition of oxidized product 44 proceeded with methanol to afford 43 as the major product (Scheme 19).
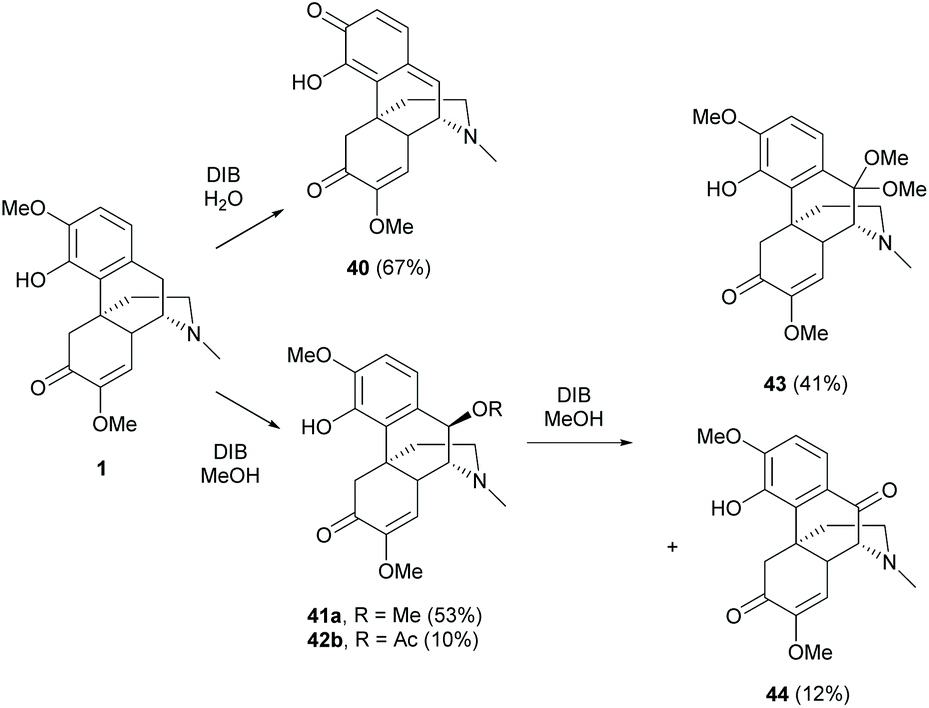 | ||
| Scheme 19 Oxidation of 1 by DIB in different solvents. | ||
After the formation of quinone methide 40, the subsequent conjugated addition with thiophenol occurred selectively to afford adduct 45a with a good yield (81%, Scheme 20). Aliphatic and electron-rich aromatic thiols also worked well in the reaction (≥79% yields). Moreover, the reaction proceeded with hard nucleophiles, alcohols and amines, to afford 42 and 46 respectively, but with lower yields (44–67%, Scheme 20) compared with soft nucleophiles (thiols). Generally, the chemoselectivity of the quinone (A-ring) over the enone in the C-ring resulted from the formation of a more energetically feasible product by rearomatization of the A-ring upon the addition. Surprisingly, the conjugated addition occurred stereospecifically at the Si-face of 40 due to the sterically hindered D-ring.
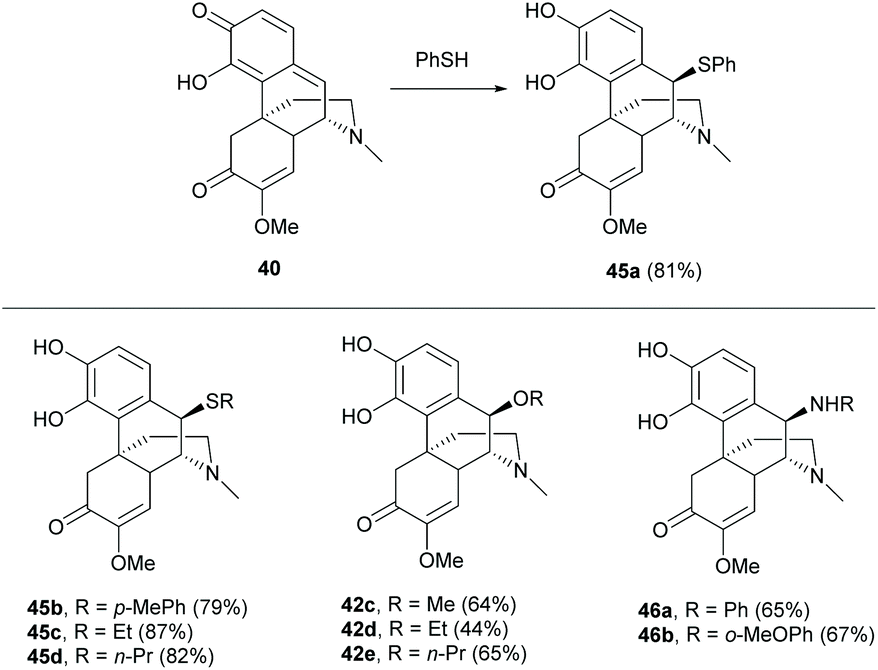 | ||
| Scheme 20 The 1,6-conjugated addition of 40 with thiols, alcohols and amines. | ||
3.2 Future approaches of functionalizations of ring B of sinomenine
As described in section 3.1, the present synthetic strategy on the benzylic position of ring B is limited to quinone methide 40 and its 1,6-conjugated adducts. Future prospects of modifications of ring B are possible to be developed by modifying the synthetic protocols of sinomenine-like derivatives.The C10-H oxygenation of sinomenine-like derivative 2c generated mesylate 47via proton coupled electron transfer between the mesyloxyl radical and 2c (Scheme 21).56 Subsequent SN1 reaction of mesylate with acetate and hydrolysis provided C10-hydroxy derivative 48 (53%). A minor over-oxidation product with C10-carbonyl was also observed (10%).
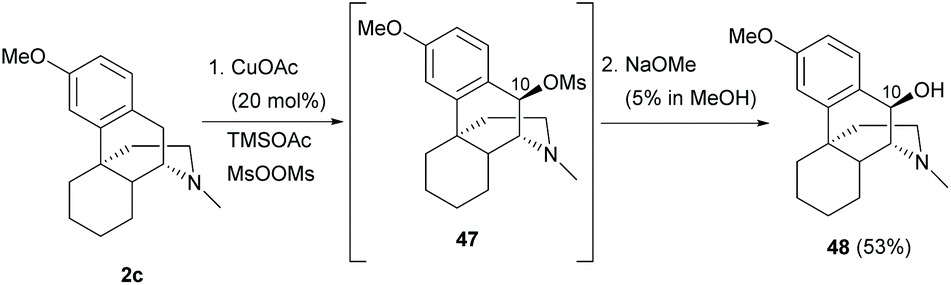 | ||
| Scheme 21 The C10-H oxidation of 2c with bis(methanesulfonyl) peroxide. | ||
Despite the functionalization at the C10-benzylic position of sinomenine, oxidation of 4-O-benzyl protected derivative 49 with MnO2 yielded the C14-hydroxy derivative 50 (15%, Scheme 22a).57 Notably, the protection of the C4-hydroxy group is the key to C14 oxidation. Otherwise, the dimerization of sinomenine occurred as shown in Scheme 8.
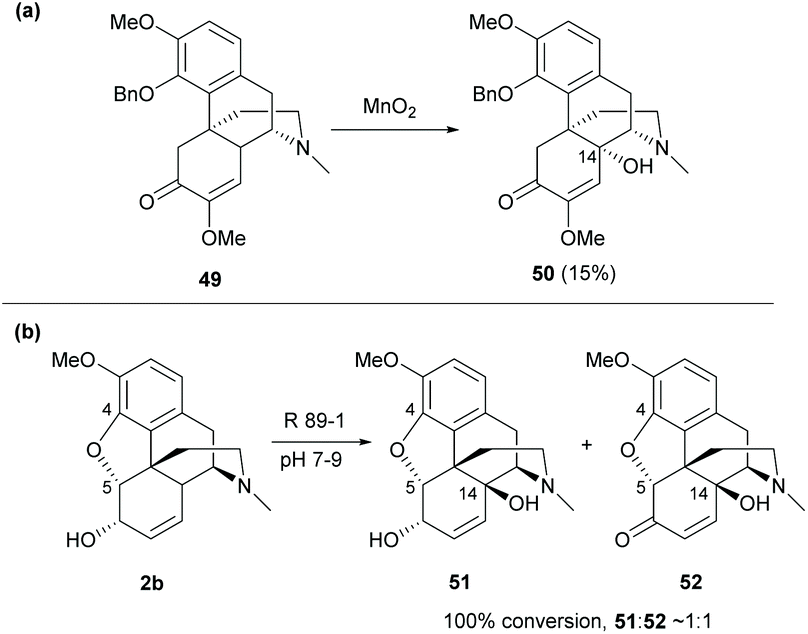 | ||
| Scheme 22 (a) The chemical oxidation of 4-O-benzyl protected derivative 49 by MnO2; (b) the oxidation of codeine 2b by the biocatalyst Rhizobium radiobacter R89-1. | ||
Other than the chemical method for C14 oxidation, it is also possible to synthesize the C14-hydroxy derivatives from codeine by using a biocatalyst. The biotransformation of 2b with the biocatalyst Rhizobium radiobacter R89-1 afforded C14-hydroxy derivatives 51–52 in nearly 1:1 (Scheme 22b).56 The tetracyclic framework of sinomenine can be obtained by C4,5-ether cleavage of codeine derivatives which will be further discussed in section 4.2.
Recently, some authors58 discovered that a novel fused-ring between B- and D-rings was constructed by the Burgess reagent59 (Et3NSO2NCO2Me) in a one-pot sequence from a sinomenine-like derivative, oxymorphone. Based on the protocol, C14-hydroxy sinomenine derivative 53 was proposed to form intermediate 54 with C4-acetylation and N-oxide formation (Scheme 23). Notably, the N-oxidation of sinomenine has been reported in the literature (Scheme 43a).60 Iminium 55 was postulated to be obtained by demethylation of N-oxide using the Burgess reagent, and then trapped by 14-hydroxy to form a novel oxazolidone ring in 56. Future strategies could involve the functionalization between C14- and N-positions to increase the structural complexity of sinomenine.
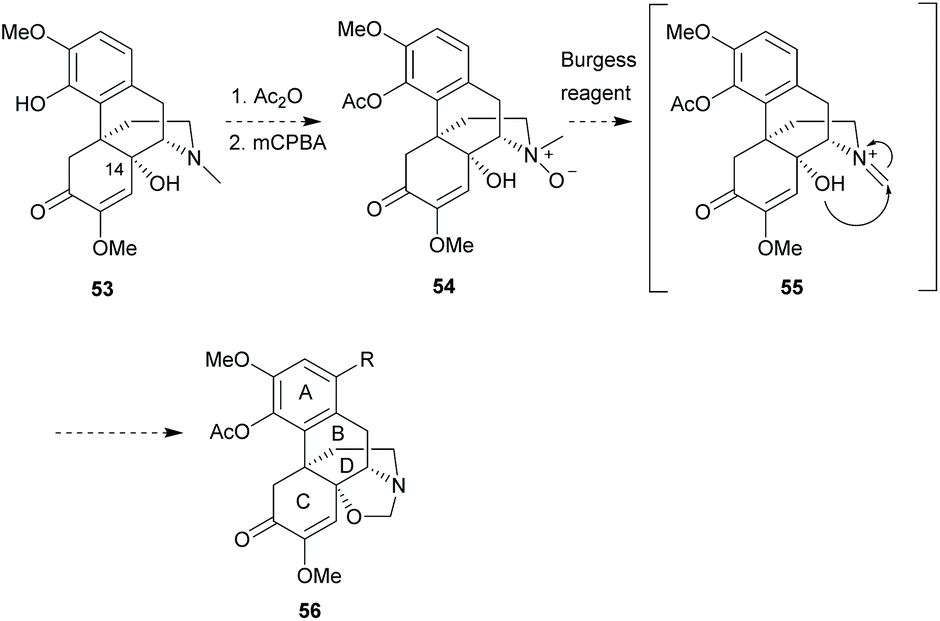 | ||
| Scheme 23 Proposed synthetic plan for forming a new fused-ring between rings B and D. | ||
4. Modifications of the C-ring
4.1 The modifications of enone of sinomenine (C-ring)
The C-ring of sinomenine possesses the major functional group, enone, which is reactive towards nucleophilic attack, amination, reduction, etc.Interestingly, simple acidic treatment of sinomenine 1 yielded several different products. The enol ether of 1 was hydrolyzed to a diketone 57 by using HCl in H2O (86% yield, Scheme 24).61 Further reduction of the C-ring yielded 58 in one-pot from 1 with a high yield (81%) via the Clemmensen reduction of the in situ generated diketone 57 from hydrolysis (Scheme 24).17 A small amount of two incomplete reduction products was also reported with either one ketone or one hydroxy group left in the C-ring, but the actual structural configurations were not determined.
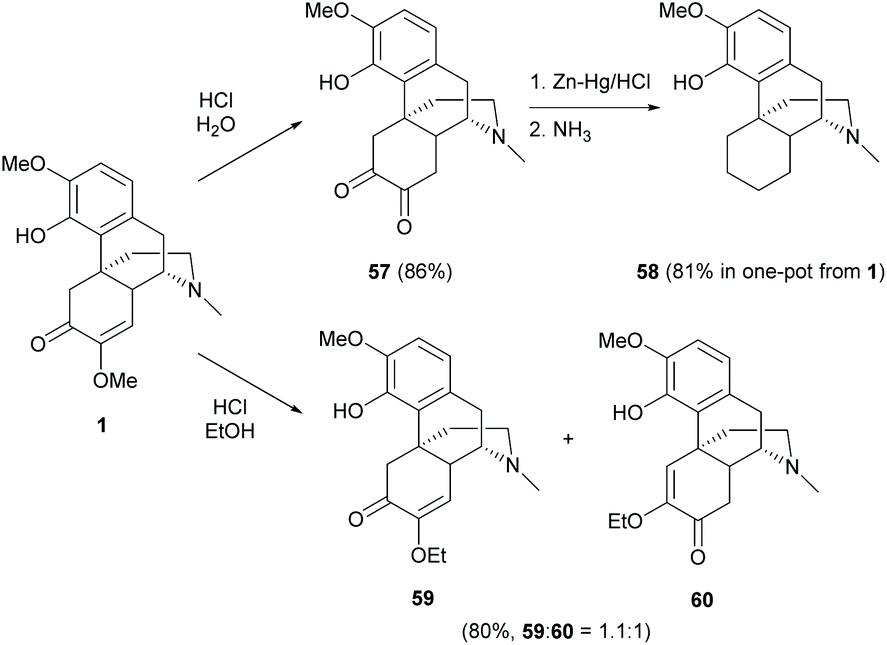 | ||
| Scheme 24 Hydrolysis and/or hydrogenation of the C-ring of sinomenine 1. | ||
When treating 1 with HCl in ethanol, the transformation provided the displacement of OMe with the OEt group and/or rearrangement of enone 59 into 60 in a ratio of nearly 1:1 (Scheme 24).61 Various chain lengths of saturated and unsaturated alcohols also worked smoothly as reported in the literature.62
Hydrogenation of enone 1 was performed chemoselectively and stereoselectively with different catalysts (Scheme 25). Treatment of enone 1 with Pd/C as the catalyst generated 1,4-reduction product 61 in high yields (83%;29 79%61), while that with PtO2 afforded over-reduction product 62 in a comparable yield (70%).61 Under hydrogenation conditions, Pt catalyst reacted with the carbonyl group faster than Pd catalyst.63 Therefore, reduction of 1 with PtO2 afforded 62via the reduction of the C6-carbonyl group followed by C7,8-alkene. As shown in the half-chair conformation of 1, the sterically hindered A-ring prevents the catalyst from approaching the top side; thus, the hydrogenation occurs at the less hindered bottom side, resulting in stereoselective products 61 and 62 (Scheme 25).
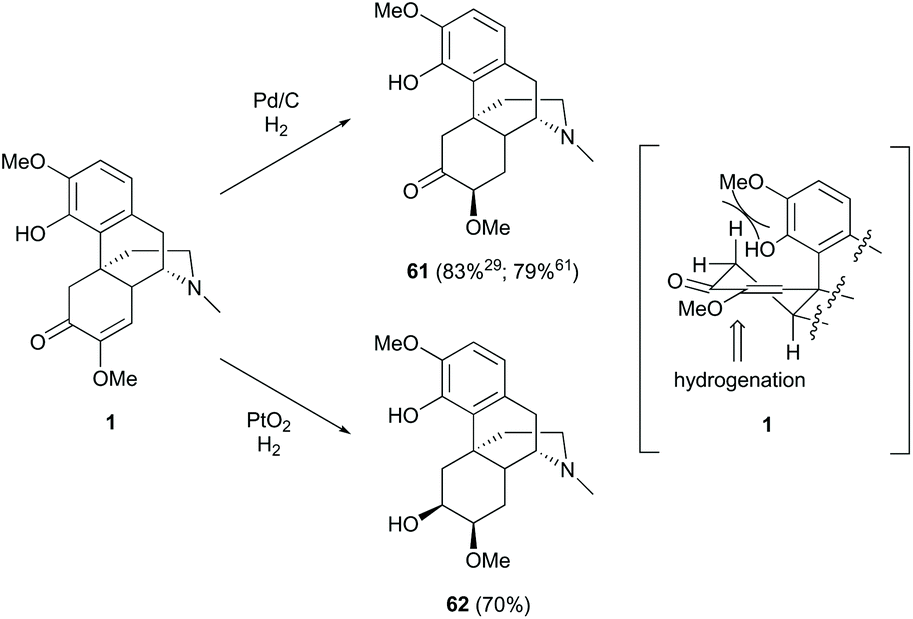 | ||
| Scheme 25 Catalytic reduction of 1 with hydrogen. | ||
To avoid unnecessary side reactions like nucleophilic addition in the modifications of other reaction sites, protection of enone with ethylene glycol under acidic conditions yielded vicinal diacetal 63a in a moderate yield (59%, Scheme 26).61 Inconsistently, higher yields of diketals 63a–b (87–91%) were reported probably due to the driving force from removing water by the Dean-Stark technique.62
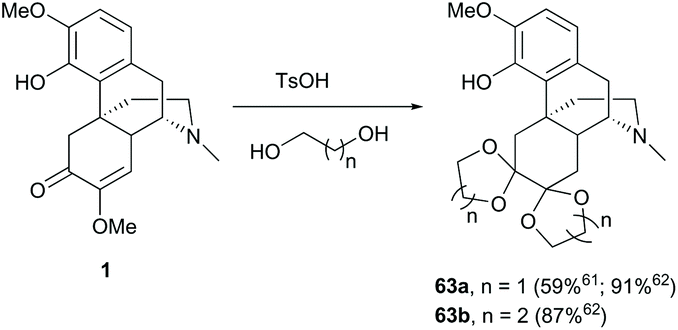 | ||
| Scheme 26 Protection of 1 with glycols under acidic conditions. | ||
Alternatively, hydrolysis of 1 in the presence of HCl and ammonium hydroxide produced keto-enamine 64 with a high yield (78%, Scheme 27).11 The regioselectivity of the nucleophilic attack of NH3 on the C6-carbonyl group of 57 was probably facilitated by the intramolecular H-bond of phenol in 57′ (Scheme 28a). Treating 64 with lead tetraacetate caused oxidative cleavage of the C-ring and intramolecular ring-closure between the C4-hydroxy group and C5 position respectively to afford nitrile ester 65 as a single diastereomer (Scheme 27).
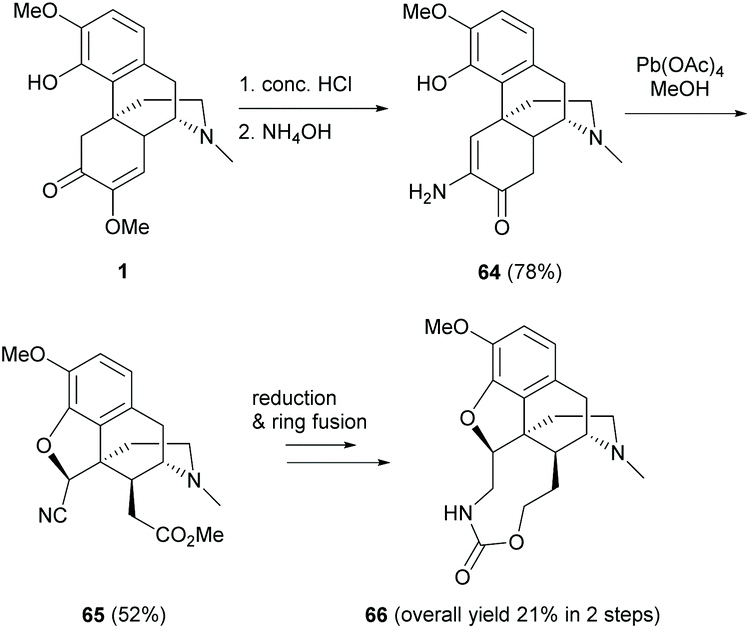 | ||
| Scheme 27 Hydrolysis and subsequent ring distortion of sinomenine 1. | ||
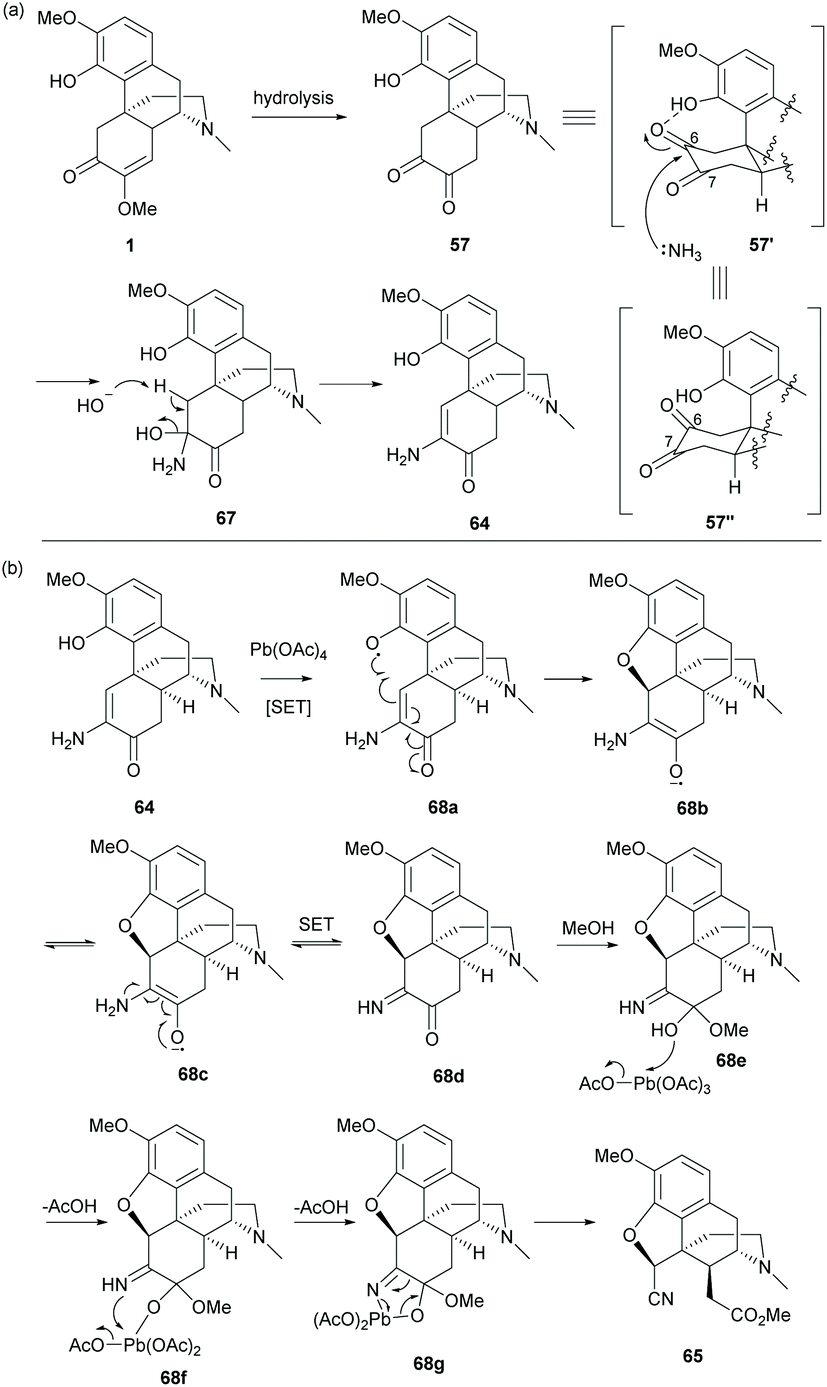 | ||
| Scheme 28 Proposed reaction mechanisms of (a) the formation of enamine; (b) ring closure and subsequent ring cleavage. | ||
As depicted in the proposed mechanism in Scheme 28b, the ring closure of 64 proceeded via a radical oxidation with Pb(OAc)4, resulting in keto-imine 68d. The stereochemistry of C5 is in the (S)-configuration due to the ring closure at the less hindered β-face. The consequent oxidative cleavage of 68d generated 65 as a single diastereomer without altering the stereochemistry at C14. Further modification of 65 to 66 was performed by reduction with LiAlH4 followed by ring fusion with triphosgene (Scheme 27).
Based on the synthesis of C10-substituted product 42c as mentioned in section 3.1, treatment of diol-protected 69 under Schmidt reaction conditions (NaN3/H2SO4) proceeded via substitution at the C10-position and/or ring expansion to afford compounds 70 and 71 respectively (Scheme 29a).11 Both C-10 substituted derivatives 70 and 71 were obtained with retention of stereochemistry. The azide approached from the less hindered upper face of secondary carbocation which was formed and stabilized by delocalization of the A-ring. Similar ring-expansion product 72 was also observed (51% yield) when 44 was treated under Schmidt reaction conditions (Scheme 29a). The proposed mechanism of the ring-expansion reaction under Schmidt conditions is exemplified by 44 (Scheme 29b). Under acidic conditions, azide attacks the activated and less hindered C6-carbonyl group, leading to intermediate 73b. The azide easily extrudes nitrogen gas in many reactions such as the Staudinger ligation. The formation of nitrogen gas as the leaving group becomes the driving force for generating intermediate 73e by a Baeyer–Villiger-like reaction. Finally, derivative 72 was obtained.
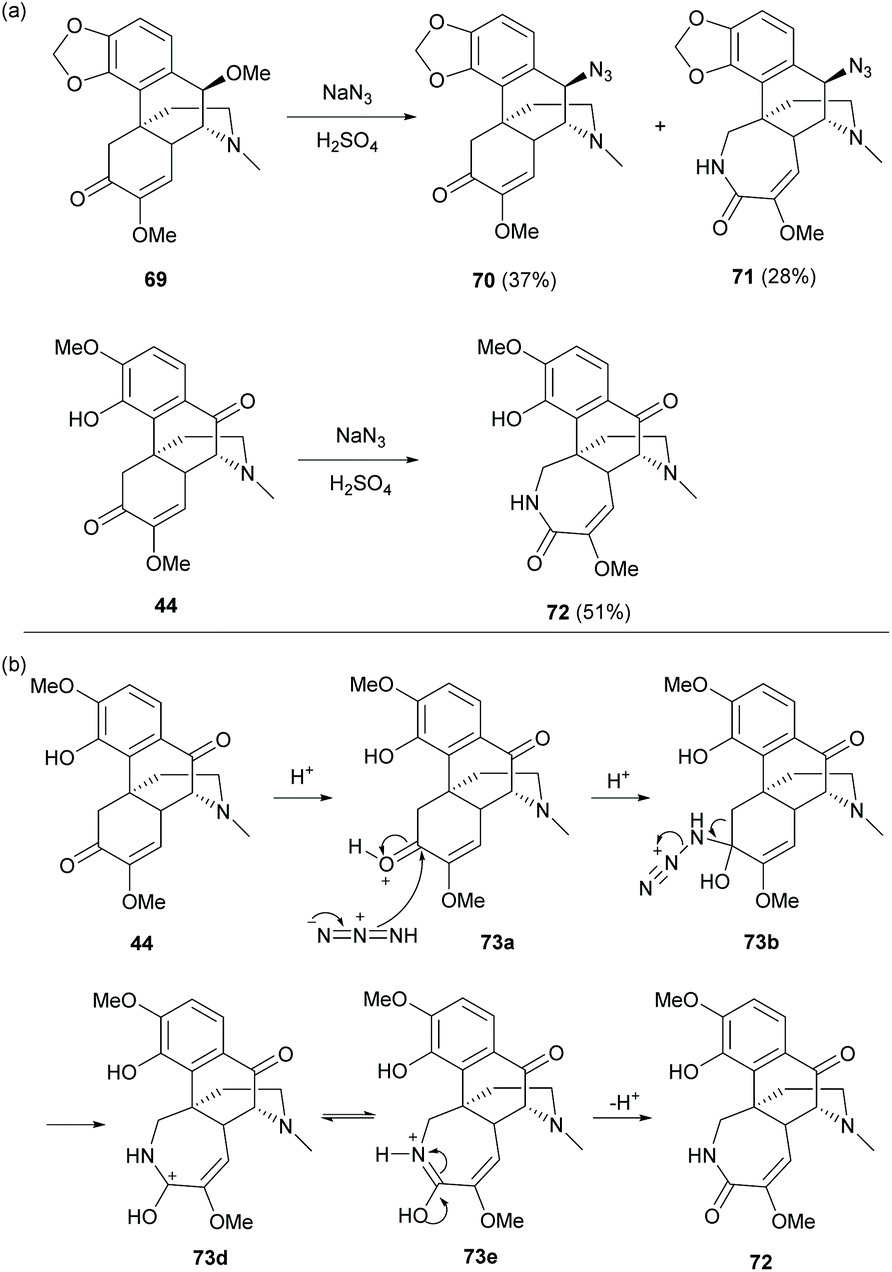 | ||
| Scheme 29 (a) Substitution and/or ring expansion of 69 and 44 under Schmidt reaction conditions; (b) proposed reaction mechanism of ring expansion of 44 under Schmidt reaction conditions. | ||
A series of sinomenine-pyrazine derivatives were prepared from the condensation of diketone 57, which was derived from the HCl hydrolysis of sinomenine 1, with diamines (Scheme 30).10 Condensation reactions of 57 with symmetrical diamines provided 74a–c as single products in high yields (≥75%). The reactions with unsymmetrical diamines also afforded 74d–e in high yields (70–72%) but poor regioselectivity (ranging from 3.7:1 to 6:1). Unfortunately, the regioisomers of 74d and 74e were inseparable and it was difficult to determine their structures due to the spectral similarity. One solution was to synthesize separable regioisomers by increasing the polarity difference of regioisomers by switching either the 1′ or 2′-position with a more polar hydroxy group.64 Therefore, condensation of 57 with phenylglycinamide generated two separable regioisomers 75 and 76 in a high combined yield (68%), where 75 was the major regioisomer (Scheme 30). The phenolic hydroxy groups of 75 and 76 at 1′ and 2′-positions were further diversified by triflylation and Suzuki couplings with various boronic acids for biological evaluation. Over 30 examples of condensation products were synthesized, including imidazole-fused derivatives.10,61,64–66
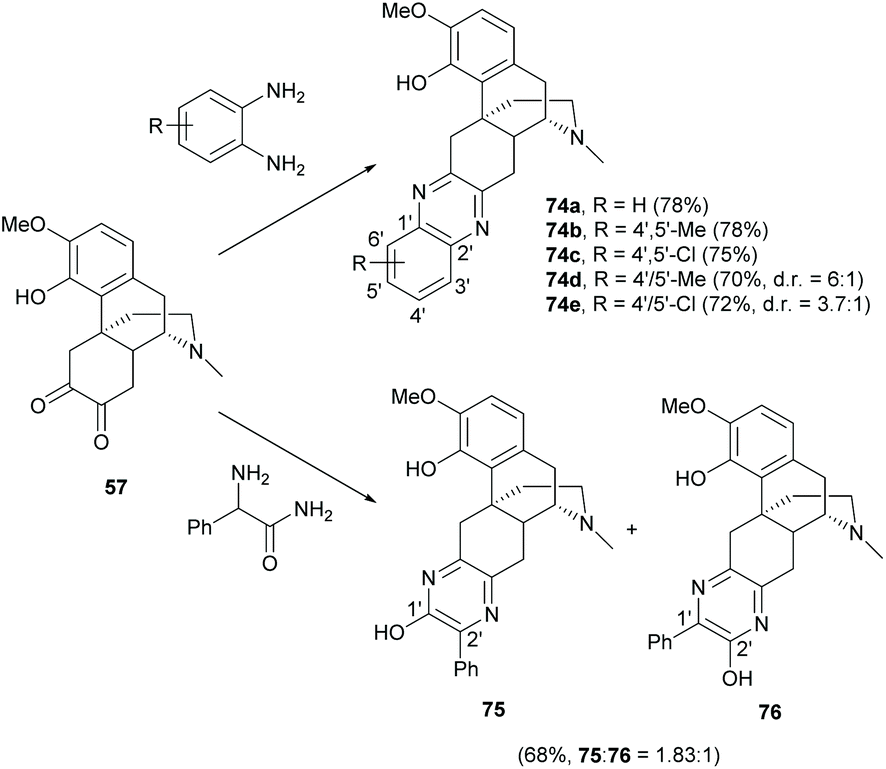 | ||
| Scheme 30 Synthesis of sinomenine-pyrazine derivatives by condensation reactions of diketone 57 with diamines. | ||
Similarly, C6-ketone of 1 was also reactive towards the condensation reaction without any pre-treatment. The reaction proceeded smoothly with hydroxylamine and phenyl hydrazine respectively to afford 77 and 78 in nearly quantitative yields (Scheme 31).36,67 Condensations of 1 with other aromatic amines and hydrazines produced imine and hydrazone derivatives with yields of ≥81%.62 Further transformation of oxime 77 yielded sulfonamides 79a–b by the stereoselective reductions of imine and enol ether with Pd/C and subsequent reactions of amines with sulfonyl chlorides (Scheme 31).36 Alternatively, tosylated oxime was also obtained by direct tosylation of 77 with TsCl.62
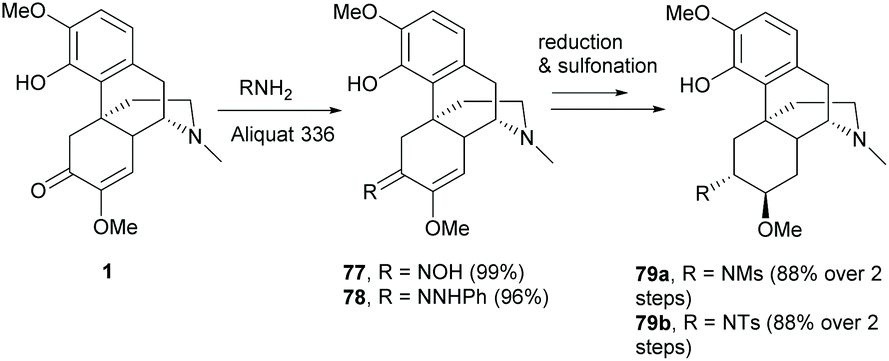 | ||
| Scheme 31 Condensation reactions of 1 with hydroxylamine and phenyl hydrazine and further transformation of 77 to 79 by reduction and sulfonation. | ||
Despite the 1,4-reduction of enone 1 as depicted in Scheme 25, 1,2-reduction of enone proceeded stereoselectively with LiAlH4 to afford alcohol derivatives 81a–d with high yields (88–95%) and in the (S)-configuration at the C6-position (Scheme 32a).68 Reductions of other 1- or 4-substituted derivatives were also reported with high yields and in the (S)-configuration.16,68 The C6-hydroxy group with the (R)-configuration was prepared by using another reducing reagent, NaBH4, in a lower yield even in large excess (19 equiv., Scheme 32a).69 The different stereochemical outcomes between LiAlH4 and NaBH4 were probably due to different intermediates formed prior to reduction (Scheme 32b). The reaction of 1 with LiAlH4 first generated lithiated intermediate 84. The hydride was then proposed to approach C6-carbonyl from the less sterically hindered side of 84. In contrast, the reaction of 1 with NaBH4 generated a boron complex 85 by alcoholysis.70 The hydride was delivered to C6-carbonyl from C4-borohydride of 85, resulting in a different stereochemical outcome.
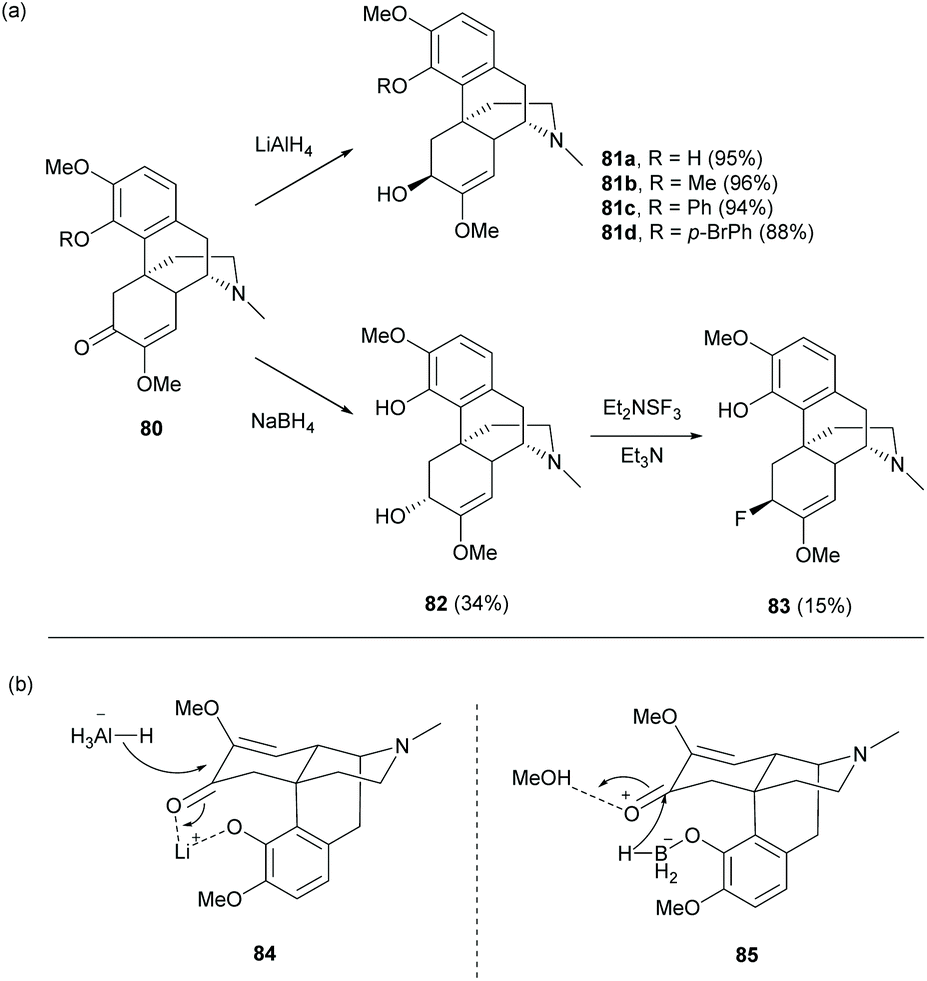 | ||
| Scheme 32 (a) 1,2-Reduction of enone 80 and fluorination of alcohol 82; (b) proposed intermediates for different stereoselectivity between LiAlH4 and NaBH4. | ||
Treatment of the C6-hydroxy group of 82 with DAST (Et2NSF3) underwent an SN2-like mechanism to provide C6-fluorinated derivative 83 with inversion of stereochemistry.
4.2 Future prospects of synthesizing sinomenine derivatives from other naturally and commercially available products (C-ring)
Apart from direct modification of sinomenine, other morphine derivatives with the C4,5-ether bridge, such as codeine, would be a readily available source for synthesizing opposite enantiomers of sinomenine by breaking the C4,5-ether bridge.The ring-opening of 2b with excess n-BuLi resulted in sinomenine-like tetracyclic derivative 87 in a high yield (74%, Scheme 33).71 The reaction was proposed to proceed via an E1cB (Elimination Unimolecular conjugate Base) mechanistic pathway with the formation of dilithiated intermediate 86.
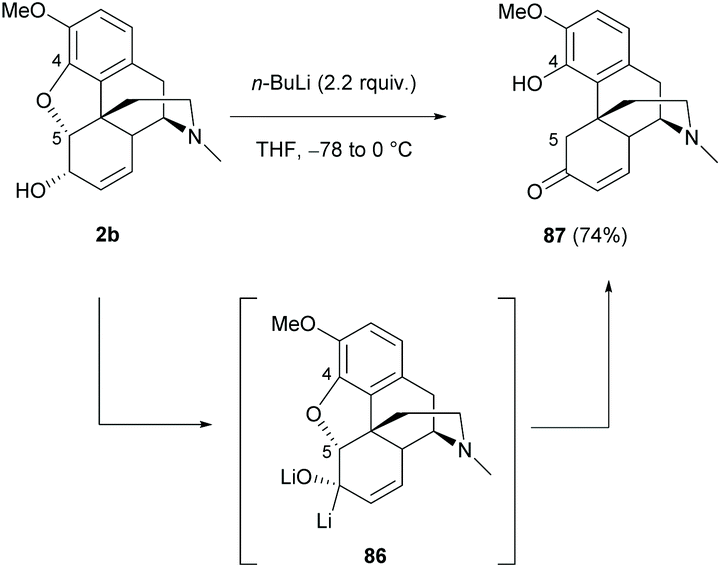 | ||
| Scheme 33 Ring-opening of the 4,5-ether bridge of 2b by n-BuLi. | ||
The breaking of the oxa-bridge of thebaine derivative 88 with zinc under acidic conditions also yielded sinomenine derivative 90 (90%, Scheme 34).72 The reductive cleavage of C4,5-ether bridge was postulated to proceed via zinc-activated complex 89.
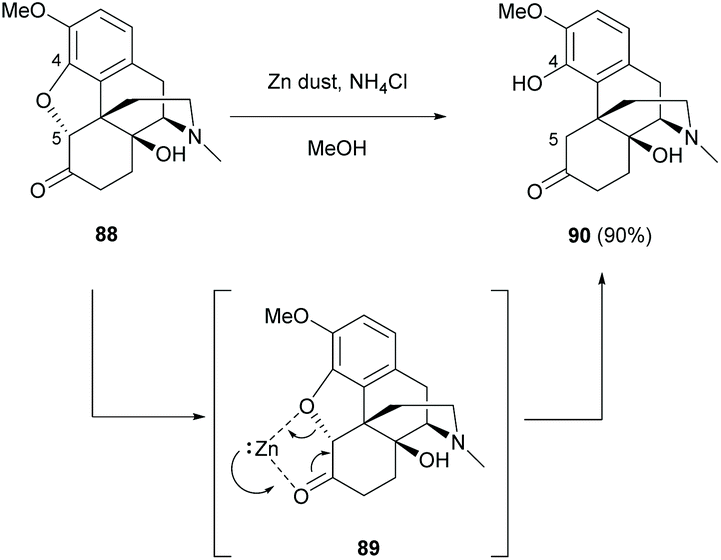 | ||
| Scheme 34 Reductive ring-opening of thebaine derivative 88 with Zn dust under acidic conditions. | ||
Alternatively, the cleavage of the C4,5-ether bridge of the morphine derivative naloxone was reported by a modified Wolff–Kishner reduction (Scheme 35).73 The reaction proceeded via a one-pot, two-step synthesis. The C6-carbonyl of 91 was first converted to N-tert-butyldimethylsilylhydrazone derivative 92 with 1,2-bis(tert-butyldimethylsilyl)hydrazine. Subsequent modified Wolff–Kishner reduction at room temperature generated the desired tetracyclic sinomenine derivative 93 (81%).
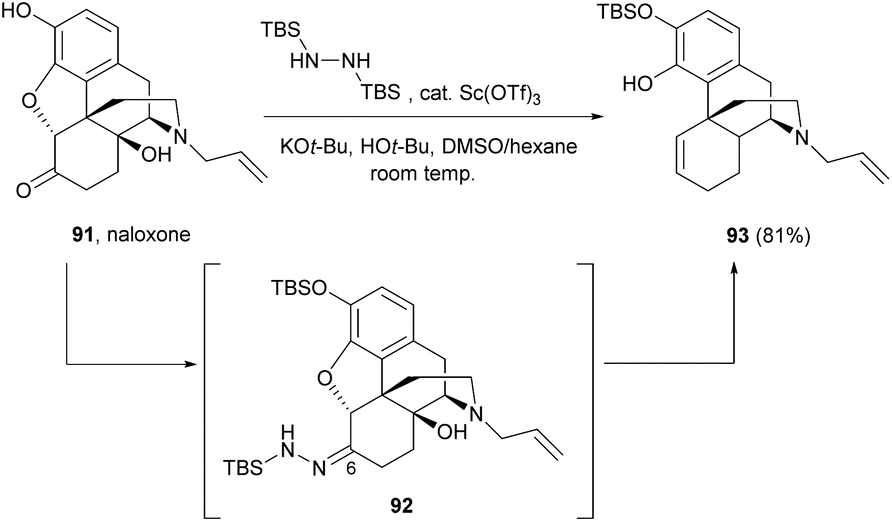 | ||
| Scheme 35 Ring-opening of naloxone 91 by Wolff–Kishner reduction. | ||
As illustrated by the above examples, breaking the C4,5-ether bridge of morphine derivatives provides a future prospect for diverse C-ring derivatization of the enantiomeric isomer of sinomenine, which is naturally unavailable.
5. Modifications of the D-ring
5.1 The modifications of trialkylamine of sinomenine (D-ring)
The D-ring of sinomenine comprises electron-rich trialkylamine which is a very reactive nucleophile. As mentioned in section 2.3, the nucleophilic amine competed with other nucleophiles in the substitution reactions. Efforts have been made on modifications of the D-ring by deactivating the reactivity of amine and switching the N-methyl substituent to various biologically active moieties.In order to replace the N-methyl substituent with other functionalities, several demethylation methods were reported to synthesize precursors 97–99 for further alkylation.74–77N-Demethylation of 4-O-unprotected 1 was performed directly in one step using 1-chloroethyl chloroformate (Scheme 36). However, demethylated product 97 was obtained albeit in low yield (13%). In contrast, 4-O-protected 94–95 underwent demethylations smoothly in quantitative yields (Scheme 36).75–77 An additional step of 4-O-deprotection was required after the functionalization of amine groups of 98–99. The 4-O-BzOBz group of 98 was removed by using trifluoroacetic acid (TFA), while the 4-O-benzyl group of 99 was removed by Pd/C in ethanol with cyclohexane (1–10 equiv.). Notably, the debenzylation of 99 under similar reductive conditions as shown in Scheme 25 did not cause 1,4-reduction of enone in the absence of hydrogen.
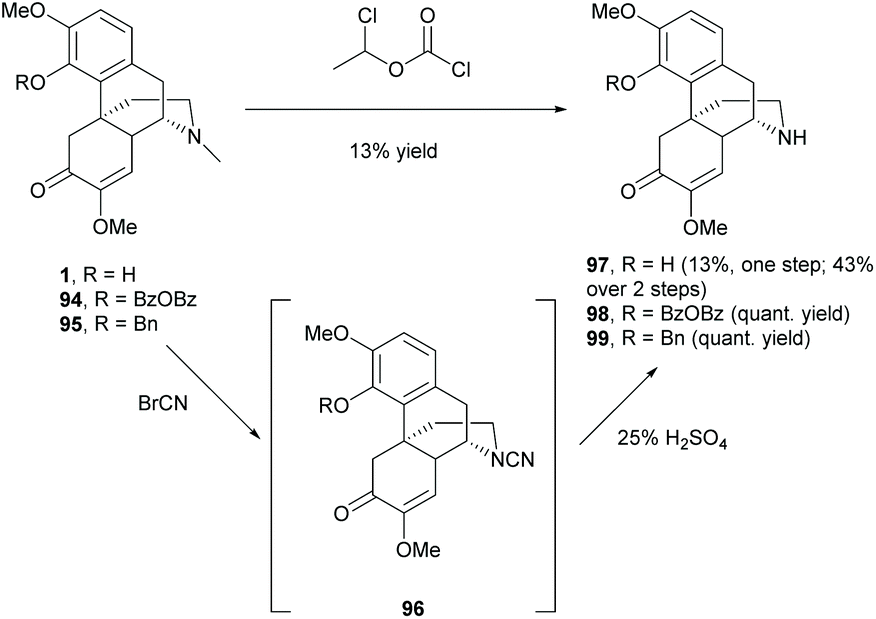 | ||
| Scheme 36 N-Demethylations of 1, 94 and 95. | ||
Alternatively, 97 was also obtained from a 2-step synthesis with an improved yield (43%) compared with the one-step demethylation of 1 (13%, Scheme 36). The N-methyl substituent of 1 was first converted to cyanamide 96, and then subjected to hydrolysis using sulfuric acid without further purification to afford 97 with a yield up to 43%. Finally, the obtained dialkylamines 97–99 were available for further functionalizations like alkylation, protection, etc.
After N-demethylation, the N-alkylation of dialkylamine 97 was reported to proceed by either an SN2 reaction with alkyl halide or a reductive amination with aldehyde (Scheme 37).74,75 Generally, the latter method provided higher yields by avoiding side reactions, such as 4-O-alkylation or N-dialkylation. More examples of N-alkylations of 97–99 were reported in the literature.75 Further transformation of 100 to triazole derivatives by 1,3-dipolar cycloaddition with azides was found to be similar to those shown in Scheme 7.74
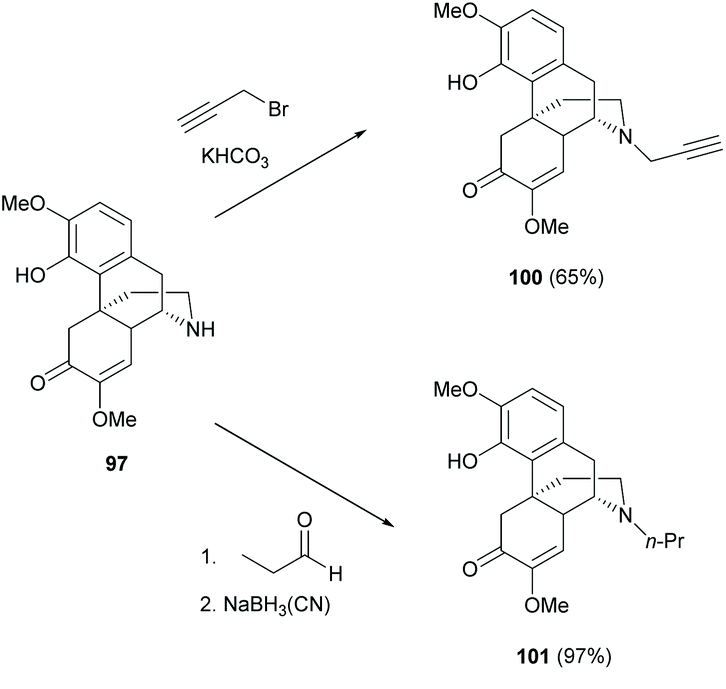 | ||
| Scheme 37 N-Alkylations of dialkylamine 97. | ||
Similar to the reaction conditions of 4-O-acylation as mentioned in section 2.3, N-acylation proceeded via either a substitution reaction with acyl chlorides or a DCC coupling reaction with carboxylic acids (Scheme 38).77 Both strategies generated N-acylated derivatives 102 and 104 with high yields (89% and 80% respectively) and the 4-O-BzOBz groups were selectively removed by treatment with TFA. Sulfonation of dialkylamine 98 was also reported by replacing acyl chloride with sulfonyl chloride.76 Detailed substrate scopes of N-acylation and N-sulfonation were found in the literature.76,77
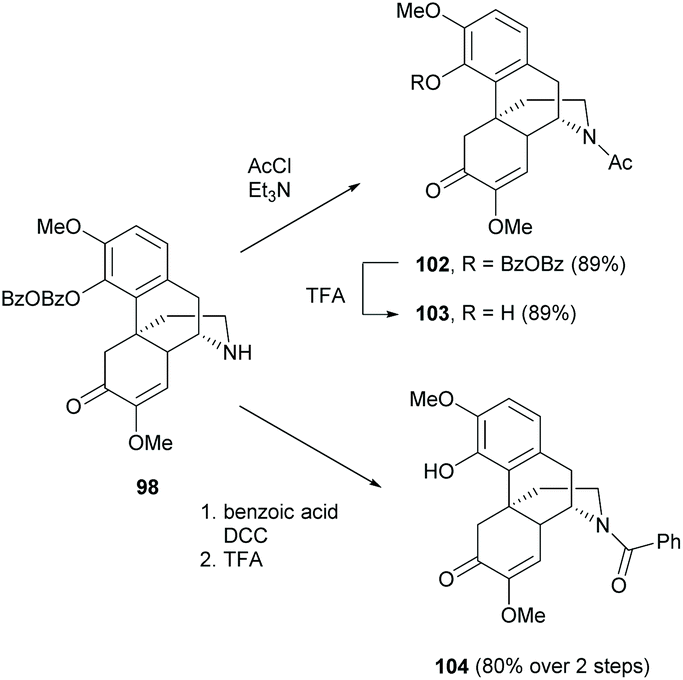 | ||
| Scheme 38 N-Acylations of 98 and its subsequent 4-O-deprotection. | ||
A few examples of complex ring distortions of the D-ring of sinomenine 1 were proposed.11 One distinguished example was the D-ring opening of sinomenine derivatives by Hofmann-type elimination (Scheme 39) while the others, such as ring rearrangements and contractions, were reported albeit in low yields (≤11%). Sequential additions of iodomethane and potassium carbonate to sinomenine derivatives cleaved the C–N bond, leading to ring-opened derivatives 105 and 107–108 in moderate yields (38–58%) except for 106 (3%). Only 1 gave alkyl-shifted product 105.
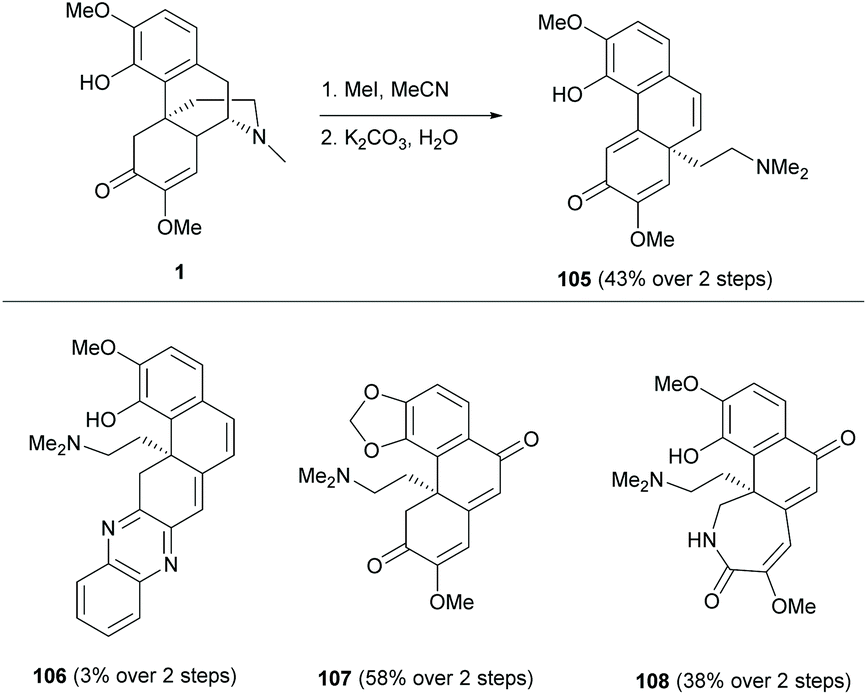 | ||
| Scheme 39 D-Ring distortions of sinomenine derivatives. | ||
The mechanism of the cleavage of the C–N bond by Hofmann elimination is exemplified in the formation of derivative 107 (Scheme 40a).
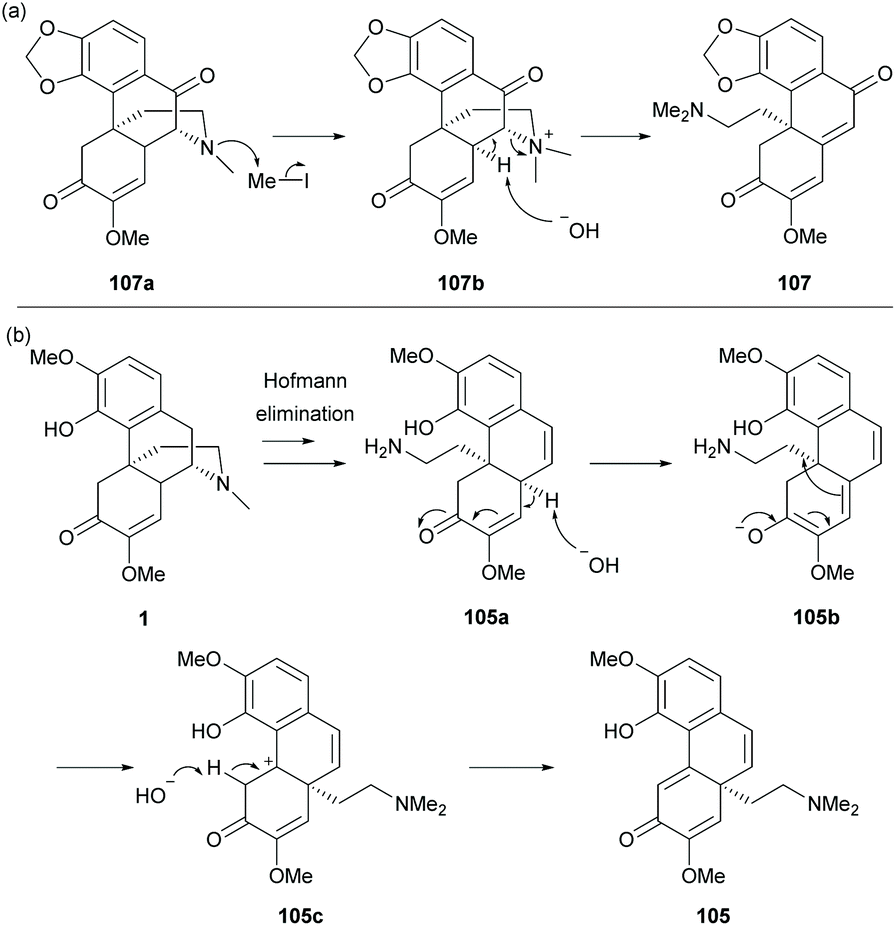 | ||
| Scheme 40 Proposed reaction mechanisms of (a) Hofmann elimination; (b) Hofmann elimination and rearrangement reactions. | ||
The methylation of trialkylamine provided quaternary ammonium derivative 107b which became a labile leaving group for the subsequent C–N bond cleavage by deprotonation at the C14-position. For the reaction of sinomenine, derivative 105a was formed, similar to 107b, but the deprotonation occurred at less hindered C10-H (Scheme 40b). Under basic conditions, the γ-proton of enone 105a was deprotonated, followed by a rearrangement reaction to generate carbocation 105c. The alkyl shift product 105 was obtained by the deprotonation of the C5-proton. The rearrangement reaction was not observed in other substrates probably due to the absence of C14-γ-H of enone.
Based on the C–N bond cleavage, a cascade reaction involving hexadehydro-Diels–Alder (HDDA) and Hofmann-type elimination was developed.78 Under thermal conditions, aryne 110 was in situ generated from tetrayne 109 and then trapped by 1 regioselectively at the 3′-position owing to a strong electronic effect of the N-Ms group directing at the meta-position (Scheme 41).79 The subsequent Hofmann-type elimination of intermediates 111a and 111b afforded derivatives 112a and 112b (74% combined yield) by trapping the protons at C10 and C14 respectively. The formation of 112b was less favourable probably due to the 1,3-allylic strain of the bulky phenyl group of 111b.
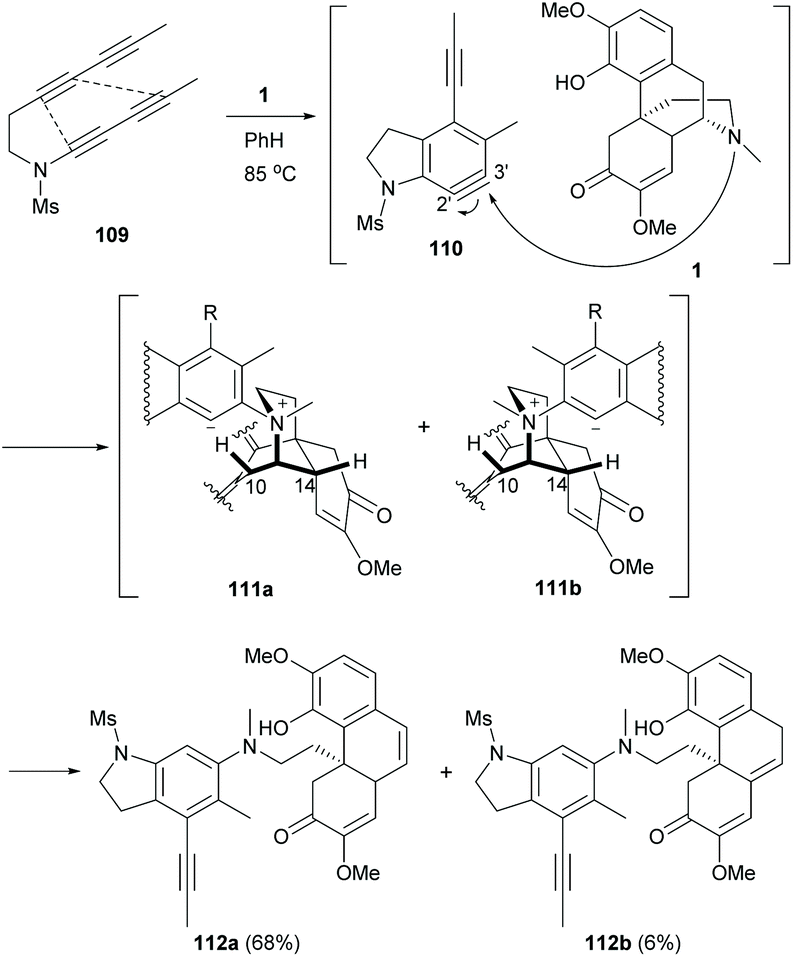 | ||
| Scheme 41 The two-stage HDDA cascade reaction of 1 with tetrayne 109. | ||
In addition, the biotransformation of 4-O-protected derivatives at the D-ring was reported using two screened microorganisms, Mucor plumbeus and Absidia corymbifera (Scheme 42).80 The reaction of 95 with M. plumbeus afforded N-demethylated derivative 99 (31%) and N-oxide derivative 113 (24%). Both derivatives were the metabolites which resulted from a detoxifying action of microorganisms. They have been identified in the herbal plant Sinomenium acutum81 and the metabolites in rats.82
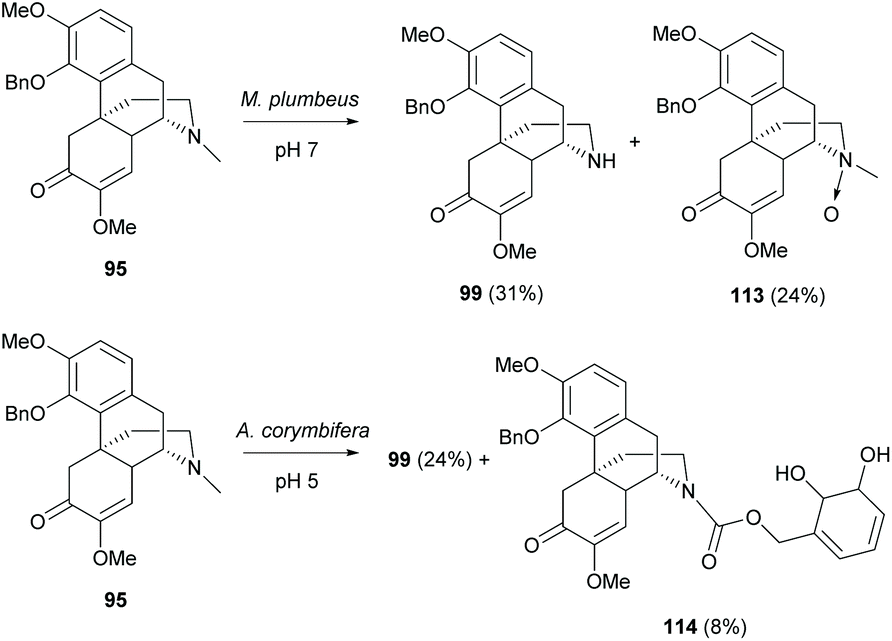 | ||
| Scheme 42 The biotransformations of 4-O-protected derivatives 95 and 114 by microorganisms. | ||
Similarly, the reaction of 95 with A. corymbifera also provided N-demethylated derivative 99 (24%) as the major product (Scheme 42). A minor dihydroxylated derivative 114 (8%) was obtained. Further experiments suggested only one oxygen atom of 114 originating from oxygen by using 18O2 labelling. This illustrated that the mechanistic pathway possibly proceeds via the formation of an epoxide followed by its ring-opening by hydrolysis.
Direct N-functionalization of 1 was also reported by either N-oxidation or N-amination (Scheme 43).60 The typical N-oxidation of 1 with mCBPA proceeded smoothly to afford N-oxide 115 in a high yield (76%, Scheme 43a).
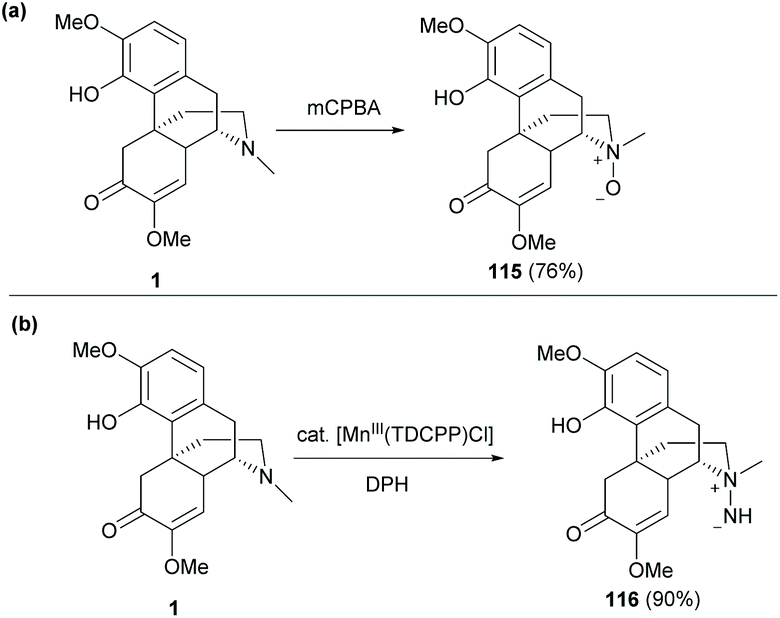 | ||
| Scheme 43 (a) N-Oxidation of 1 with mCPBA; (b) N-amination of 1 with hydroxylamine catalyzed by Mn(III). | ||
Another novel strategy was the N-amination of sinomenine with O-(2,4-dinitrophenyl) hydroxylamine (DPH) catalyzed by [Mn(TDCPP)Cl] (Scheme 43b). The reaction was first initiated by the formation of active species, Mn![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH, which was trapped by the trialkylamine of 1 by nucleophilic addition to afford unprotected aminimide 116 (90%).
NH, which was trapped by the trialkylamine of 1 by nucleophilic addition to afford unprotected aminimide 116 (90%).
5.2 Future approaches of functionalizations of ring D of sinomenine
As mentioned in section 5.1, modifications of the D-ring mainly focused on a stepwise manner—N-demethylation and subsequent N-substitution. A few examples of direct functionalization of the N-methyl group of sinomenine have been shown to be an effective method for D-ring diversification. In view of these, late-stage functionalizations in sinomenine-like derivatives become a fast track for diversifying the D-ring of sinomenine without N-demethylation.Direct C–H functionalization of the N-methyl substituent of 2c with dehydroalanine 117 (Dha) was reported to yield amine-Dha adduct 118via the formation of the α-amino radical (64%, Scheme 44).83 The novel N-methyl functionalization with unnatural amino acids and peptides would be useful for increasing the complexity of sinomenine derivatives and exploring the corresponding biological activities.
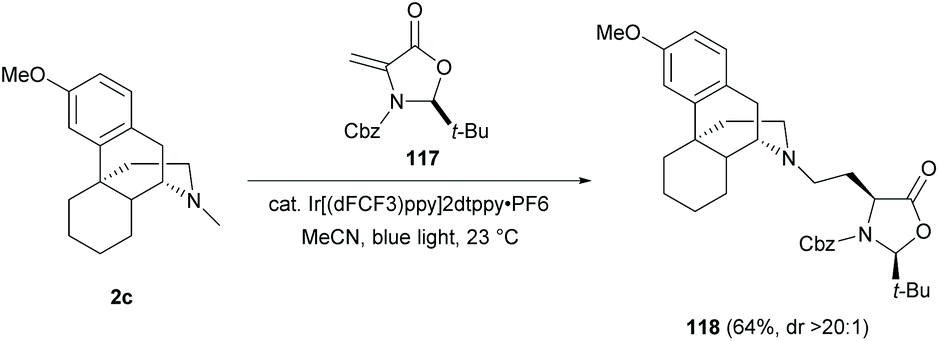 | ||
| Scheme 44 Amine conjugated addition of 2c to dehydroalanine 117 by photoredox C–H functionalization. | ||
Similarly, another direct N-methyl functionalization was reported to proceed with the in situ generated α-amino radical by radical hydrogen atom abstraction with the DABCO radical cation (Scheme 45).84 The subsequent addition of Grignard reagents provided sinomenine-like derivatives 119 and 121 in low to high yields (78% and 38% respectively).
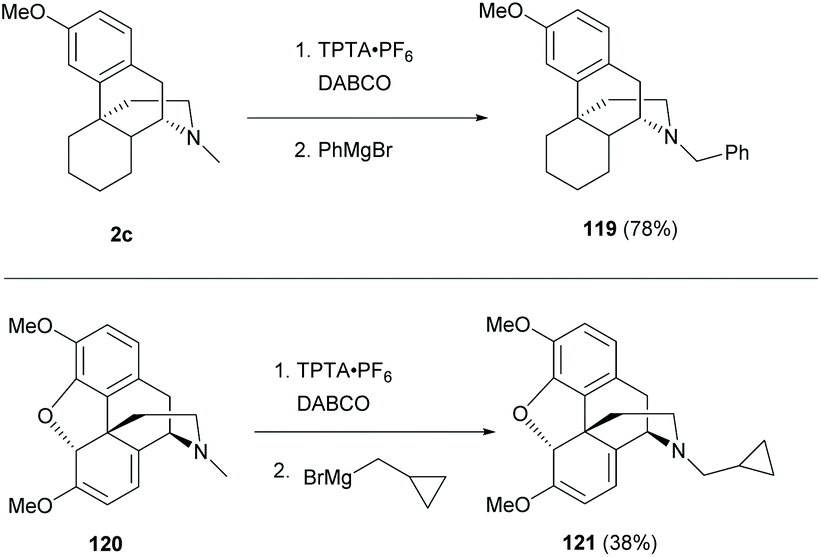 | ||
| Scheme 45 N-Methyl C–H functionalization of trialkylamines 2c and 120via hydrogen atom abstraction. | ||
Late-stage C–H functionalizations of N-methyl sinomenine-like derivatives 2c and 120via intermolecular rhodium-carbenoid insertion were reported with high regioselectivities toward the N-methyl C–H bond due to sterically demanding carbenoid (Scheme 46).85 The donor/acceptor carbenoid derived from diazo 122 inserted selectively to the N-methyl of 2c and 120 to generate derivatives 123–124 (87% and 52% respectively).
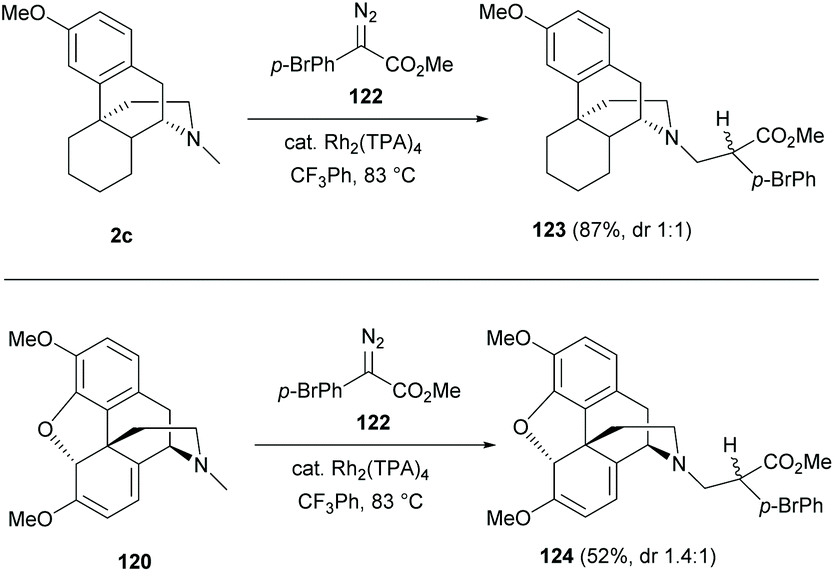 | ||
| Scheme 46 Dirhodium(II)-catalyzed intermolecular C17-H insertion of sinomenine-like derivatives 2c and 120 with diazo 122. | ||
6. Conclusions
This review summarizes the chemical modifications of sinomenine by demonstrating selected examples from each type of chemical reaction, in order to show the applicability of organic reactions in this particular chemical structure and analyze the synthetic issues that have been found. Besides chemical modifications, only a few examples of biotransformations have been reported to date. The full substrate scope of each type of reaction was not shown but could be found in detail in the literature. Based on the structure of sinomenine, the reaction sites are generally categorized into substituted aromatic ring (A-ring), benzylic position (B-ring), enone (C-ring) and trialkylamine (D-ring).Sinomenine benefits from its sterically hindered polycyclic structure. The stereoselectivity of the reactions was not a major concern in the synthesis because the formations of stereoisomers were mostly controlled by steric factors.
Further modifications of sinomenine may consider the latest novel reactions that have been applied in structurally similar natural products and their semi-synthetic products. The enantiomeric isomers of sinomenine derivatives were easily obtained by C4,5-ether cleavage of morphine derivatives. Furthermore, the C3-methoxy group and C2-position of sinomenine have rarely been modified to date. Extensive studies on those positions will help in a deeper understanding of the structure–activity relationships of sinomenine.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by an FDCT grant from the Macao Science and Technology Development Fund (Project code: 0048/2018/A2; 0003/2019/AKP) and Foshan Medicine Dengfeng Project of China (2019–2021). We thank Chunsong Cheng from the Faculty of Chinese Medicine of Macau University of Science and Technology for the photos of herbal plants.Notes and references
- X.-X. Zhao, C. Peng, H. Zhang and L.-P. Qin, Sinomenium acutum: A review of chemistry, pharmacology, pharmacokinetics, and clinical use, Pharm. Biol., 2012, 50, 1053–1061 CrossRef.
- H.-D. Pan, Y. Xiao, W.-Y. Wang, R.-T. Ren, E. L.-H. Leung and L. Liu, Traditional Chinese Medicine as a Treatment for Rheumatoid Arthritis: From Empirical Practice to Evidence-Based Therapy, Engineering, 2019, 5, 895–906 CrossRef CAS.
- B. D. Huang, A. S. He, M. Fu, P. Sheng and W. M. Liao, Sinomenine suppresses expression of interleukin-1beta-induced matrix metalloproteinases in human osteoarthritic chondrocytes, J. Med. Plants Res., 2010, 4, 1830–1836 CAS.
- L. Liu, E. Buchner, D. Beitze, C. B. Schmidt-Weber, V. Kaever, F. Emmrich and R. W. Kinne, Amelioration of rat experimental arthritides by treatment with the alkaloid sinomenine, Int. J. Immunopharmacol., 1996, 18, 529–543 CrossRef CAS.
- Q. Wang and X.-K. Li, Immunosuppressive and anti-inflammatory activities of sinomenine, Int. J. Immunopharmacol., 2011, 11, 373–376 CrossRef CAS.
- Y.-S. Zhang, J.-Y. Han, O. Iqbal and A.-H. Liang, Research Advances and Prospects on Mechanism of Sinomenin on Histamine Release and the Binding to Histamine Receptors, Int. J. Mol. Sci., 2019, 20, 70 CrossRef.
- R.-Y. Huang, H.-D. Pan, J.-Q. Wu, H. Zhou, Z.-G. Li, P. Qiu, Y.-Y. Zhou, X.-M. Chen, Z.-X. Xie, Y. Xiao, Q.-C. Huang and L. Liu, Comparison of combination therapy with methotrexate and sinomenine or leflunomide for active rheumatoid arthritis: A randomized controlled clinical trial, Phytomedicine, 2019, 57, 403–410 CrossRef CAS.
- L. Li, C. L. Zhang and B. W. Song, Pharmacological research and clinical application of sinomenine, Tradit. Chin. Drug Res. Clin. Pharmacol., 2006, 17, 310–313 Search PubMed.
- J. Wu and Y.-H. Li, The Side Effects and Prevention of Drug Treatment by Zhengqingfengtongning, Chin. J. Med. Guide, 2012, 14, 250–251 Search PubMed.
- Z.-J. Yao and H.-B. Zhou, China Pat, CN1687065A, 2005 Search PubMed.
- A. Garcia, B. S. Drown and P. J. Hergenrother, Access to a Structurally Complex Compound Collection via Ring Distortion of the Alkaloid Sinomenine, Org. Lett., 2016, 18, 4852–4855 CrossRef CAS.
- J. Tang, A. Raza, J. Chen and H. Xu, A Systematic Review on the Sinomenine Derivatives, Mini-Rev. Med. Chem., 2018, 18, 906–917 CrossRef CAS.
- T.-L. Wang, H.-Q. Liu, Y.-Q. Chen, N.-G. Li and W. Li, Recent advance of sinomenine derivatives, Prog. Mod. Biomed., 2016, 16, 1167–1171 CrossRef CAS.
- X. Li, Q. Zhao, J. Dong, Y. Jiang and G. Ye, Research progress in sinomenine structural modification, J. Pharm. Pract., 2018, 36, 204–214 Search PubMed.
- Y.-H. Zhang, Z.-X. Gao, C.-L. Zhong, H.-B. Zhou, L. Chen, W.-M. Wu, X.-J. Peng and Z.-J. Yao, An inexpensive fluorescent labeling protocol for bioactive natural products utilizing Cu(I)-catalyzed Huisgen reaction, Tetrahedron, 2007, 63, 6813–6821 CrossRef CAS.
- J. Tang, R. Zhang, X.-Q. Xu, C.-W. Wen, Y.-S. Jin, Q.-Y. Wu and H.-S. Chen, Synthesis, characterization, and NF-kappaB pathway inhibition of 1-halogenated sinomenine derivatives, Chem. Nat. Compd., 2013, 48, 1031–1034 CrossRef CAS.
- H. Pan, T. Lu, X. Wu, C. Jiang, C. Gu, K. Zhang and J. Jin, Synthesis of C-ring hydrogenated sinomenine cinnamate derivatives via Heck reactions, J. Chem. Res., 2019, 43, 469–473 CrossRef CAS.
- P. X. Wang, T. Jiang, G. L. Cantrell, D. W. Berberich, B. N. Trawick, S. Liao and J. Brandt, US Pat, US8614224B2, 2013 Search PubMed.
- J. Tang, R.-J. Liu, X.-Q. Xu and X.-F. Yu, China Pat, CN101899004A, 2010 Search PubMed.
- J.-X. Meng and J.-L. Xu, China Pat, CN106986826A, 2017 Search PubMed.
- Z.-J. Zhao, C. Zhao, J. Xiao and J.-C. Wang, Transdermal permeation and anti-inflammation activities of novel sinomenine derivatives, Molecules, 2016, 21, 1520 CrossRef.
- F.-C Wu, X.-Z. Feng, K.-M. Wu, G.-F. Cheng, Y.-M. Huang, X.-R. Ye, P. Qiu and X.-L. Zheng, China Pat, CN1876634A, 2006 Search PubMed.
- N. Barroeta, G. Chuchani and J. Zabicky, Kinetics and Substituent Effects in Electrophilic Aromatic Substitution. II.1 Tritylation of Catechol and its Monoethers, J. Org. Chem., 1966, 31, 2330–2333 CrossRef CAS.
- J. Jin, P. Teng, H.-L. Liu, J. Wu, Y.-M. Liu, Q. Xu and J.-X. Li, Microfluidics assisted synthesis and bioevaluation of sinomenine derivatives as antiinflammatory agents, Eur. J. Med. Chem., 2013, 62, 280–288 CrossRef CAS.
- Y.-Y. Jiang, X.-Z. Li, G.-M. Ye and Q.-J. Zhao, China Pat, CN108863932A, 2018 Search PubMed.
- X. Zheng, D. Luo, H. Gao, N. Jiang and A. Ding, Highly regioselective synthesis of C-4 (ring A) sinomenine ether derivatives, J. Chem. Res., 2012, 36, 315–317 CrossRef CAS.
- J. P. Parrish, B. Sudaresan and K. W. Jung, Improved Cs2CO3 Promoted O-Alkylation of Phenols, Synth. Commun., 1999, 29, 4423–4431 CrossRef CAS.
- J.-C. Castillo, J. Orrego-Hernández and J. Portilla, Cs2CO3-Promoted Direct N-Alkylation: Highly Chemoselective Synthesis of N-Alkylated Benzylamines and Anilines, Eur. J. Org. Chem., 2016, 3824–3835 CrossRef CAS.
- C.-J. Wei, F. Xu, M.-J. Shi, J.-W. Hu, J.-J. Wang, B. Zhen, X. Wang, T.-F. Ji, J.-H. Wang and G.-H. Du, Synthesis and antitumor activities of sinomenine derivatives on rings A and C, J. Asian Nat. Prod. Res., 2018, 20, 277–291 CrossRef CAS.
- D. Wang, R. Zhang, C. Jiang, A. Raza, J. Tang, Z. Ouyang, Z. Su and H. Xu, Synthesis and Anti-Inflammatory Effect of Sinomenine 4-Hydroxy Esters, Chem. Nat. Compd., 2018, 54, 131–136 CrossRef CAS.
- S.-C. Li, Z.-B. Zhang, Q.-S. Zhang, Q. Yang, W.-M. Zhong, A. Li and X. Bao, WO Pat, WO2018068696A1, 2018 Search PubMed.
- H. Pan, T. Lu, X. Wu, C. Gu, N. Tao, B. Zhang, A. Wang, G. Chen, K. Zhang, J. Cheng and J. Jin, Design and synthesis of sinomenine isoxazole derivatives via 1,3-dipolar cycloaddition reaction, Nat. Prod. Res., 2019, 1–5, DOI:10.1080/14786419.2019.1677649.
- P. Teng, H.-L. Liu, L. Zhang, L.-L. Feng, Y. Huai, Z.-S. Deng, Y. Sun, Q. Xu and J.-X. Li, Synthesis and biological evaluation of novel sinomenine derivatives as anti-inflammatory agents, Eur. J. Med. Chem., 2012, 50, 63–74 CrossRef CAS.
- Z.-S. Deng, Y. Zhao, C.-C. He, J. Jin, Y.-M. He and J.-X. Li, pH-Dependent, Stereoselective Dimerization of Sinomenine, Org. Lett., 2008, 10, 3879–3882 CrossRef CAS.
- J. M. Brunel, BINOL: A Versatile Chiral Reagent, Chem. Rev., 2005, 105, 4233–4233 CrossRef CAS.
- P. Teng, H.-L. Liu, Z.-S. Deng, Z.-B. Shi, Y.-M. He, L.-L. Feng, Q. Xu and J.-X. Li, Synthesis and biological evaluation of unique stereodimers of sinomenine analogues as potential inhibitors of NO production, Bioorg. Med. Chem., 2011, 19, 3096–3104 CrossRef CAS.
- Z. S. Deng, D. Zhao, Y. Hu, J. X. Li, K. Zou and J. Z. Wang, Biocatalyzed cross-coupling of sinomenine and 1,2-dihydroxybenzene by Coriolus unicolor, Chin. Chem. Lett., 2012, 23, 321–324 CrossRef CAS.
- Z.-S. Deng, J.-X. Li, P. Teng, P. Li and X.-R. Sun, Biocatalyzed Cross-Coupling of Sinomenine and Guaiacol by Antrodiella semisupina, Org. Lett., 2008, 10, 1119–1122 CrossRef CAS.
- J. Minamikawa, K. C. Rice, A. E. Jacobson, A. Brossi, T. H. Williams and J. V. Silverton, Studies in the (+)-morphinan series. 7. Unusual crystallographic and tautomeric properties of (+)-4-hydroxy-7-oxo-3-methoxy-17-methyl-5,6-dehydromorphinan: An interlacing double helix, J. Org. Chem., 1980, 45, 1901–1905 CrossRef CAS.
- Y. Liu, L. Qin, G. Li, W. Zhang, L. An, B. Liu and J.-S. Hong, Dextromethorphan Protects Dopaminergic Neurons against Inflammation-Mediated Degeneration through Inhibition of Microglial Activation, J. Pharmacol. Exp. Ther., 2003, 305, 212–218 CrossRef CAS.
- A. Ruffoni, F. Juliá, T. D. Svejstrup, A. J. McMillan, J. J. Douglas and D. Leonori, Practical and regioselective amination of arenes using alkyl amines, Nat. Chem., 2019, 11, 426–433 CrossRef CAS.
- T. D. Svejstrup, A. Ruffoni, F. Juliá, V. M. Aubert and D. Leonori, Synthesis of Arylamines via Aminium Radicals, Angew. Chem., Int. Ed., 2017, 56, 14948–14952 CrossRef CAS.
- Y. Y. See and M. S. Sanford, C–H Amination of Arenes with Hydroxylamine, Org. Lett., 2020, 22, 2931–2934 CrossRef CAS.
- S. Ichii, G. Hamasaka and Y. Uozumi, The Hiyama Cross-Coupling Reaction at Parts Per Million Levels of Pd: In Situ Formation of Highly Active Spirosilicates in Glycol Solvents, Chem. – Asian J., 2019, 14, 3850–3854 CrossRef CAS.
- J. A. Fernández-Salas, A. P. Pulis and D. J. Procter, Metal-free C-H thioarylation of arenes using sulfoxides: A direct, general diaryl sulfide synthesis, Chem. Commun., 2016, 52, 12364–12367 RSC.
- M. Tobisu, T. Takahira, A. Ohtsuki and N. Chatani, Nickel-catalyzed alkynylation of anisoles via C-O bond cleavage, Org. Lett., 2015, 17, 680–683 CrossRef CAS.
- M. Tobisu, T. Takahira and N. Chatani, Nickel-Catalyzed Cross-Coupling of Anisoles with Alkyl Grignard Reagents via C-O Bond Cleavage, Org. Lett., 2015, 17, 4352–4355 CrossRef CAS.
- A. W. Sromek, B. A. Provencher, S. Russell, E. Chartoff, B. I. Knapp, J. M. Bidlack and J. L. Neumeyer, Preliminary pharmacological evaluation of enantiomeric morphinans, ACS Chem. Neurosci., 2014, 5, 93–99 CrossRef CAS.
- M. Decker, Y.-G. Si, B. I. Knapp, J. M. Bidlack and J. L. Neumeyer, Synthesis and Opioid Receptor Binding Affinities of 2-Substituted and 3-Aminomorphinans: Ligands for μ, κ, and δ Opioid Receptors, J. Med. Chem., 2010, 53, 402–418 CrossRef CAS.
- R. Ma and M. C. White, C-H to C-N Cross-Coupling of Sulfonamides with Olefins, J. Am. Chem. Soc., 2018, 140, 3202–3205 CrossRef CAS.
- J. R. Clark, K. Feng, A. Sookezian and M. C. White, Manganese-catalysed benzylic C(sp3)-H amination for late-stage functionalization, Nat. Chem., 2018, 10, 583–591 CrossRef CAS.
- J. Li, J. S. Cisar, C.-Y. Zhou, B. Vera, H. Williams, A. D. Rodríguez, B. F. Cravatt and D. Romo, Simultaneous structure–activity studies and arming of natural products by C–H amination reveal cellular targets of eupalmerin acetate, Nat. Chem., 2013, 5, 510–517 CrossRef CAS.
- J. H. Docherty, K. Nicholson, A. P. Dominey and S. P. Thomas, A Boron–Boron Double Transborylation Strategy for the Synthesis of gem-Diborylalkanes, ACS Catal., 2020, 10, 4686–4691 CrossRef CAS.
- Y. Wang, L. Wu, H. Cai, H. Lei, C.-M. Ma, L. Yang, H. Xu, Q. Zhu, Z. Yao and Y. Wu, YL064 directly inhibits STAT3 activity to induce apoptosis of multiple myeloma cells, Cell Death Discovery, 2018, 4, 1–10 Search PubMed.
- Y.-T. Lou, L.-Y. Ma, M. Wang, D. Yin, T.-T. Zhou, A.-Z. Chen, Z. Ma, C. Bian, S. Wang, Z.-Y. Yang, B. Sun and Z.-J. Yao, Regio- and stereoselective C10β-H functionalization of sinomenine: an access to more potent immunomodulating derivatives, Tetrahedron, 2012, 68, 2172–2178 CrossRef CAS.
- L. Tanwar, J. Börgel and T. Ritter, Synthesis of Benzylic Alcohols by C–H Oxidation, J. Am. Chem. Soc., 2019, 141, 17983–17988 CrossRef CAS.
- B. N. Trawick, D. W. Berberich and C. W. Grote, U.S. Pat, US20150335638, 2015 Search PubMed.
- M. A. A. Endoma-Arias, D. P. Cox and T. Hudlicky, General Method of Synthesis for Naloxone, Naltrexone, Nalbuphone, and Nalbuphine by the Reaction of Grignard Reagents with an Oxazolidine Derived from Oxymorphone, Adv. Synth. Catal., 2013, 355, 1869–1873 CrossRef CAS.
- S. Santra, Burgess Reagent: From Oblivion to Renaissance in Organic Synthesis, Synlett, 2009, 328–329 CrossRef CAS.
- S. Zhang, Y. Liu, F. Xing and C.-M. Che, Direct preparation of unprotected aminimides (R3N+–NH−) from natural aliphatic tertiary alkaloids (R3N) by [Mn(TDCPP)Cl]-catalysed N-amination reaction, Chem. Commun., 2020, 56, 9102–9105 RSC.
- M. Wang, L. Ma, Y. Lou, C. Bian, T. Zhou, H. Zhou, H. Liao, Z. Ma, D. Yin, A. Chen, S. Wang, Z. Yang, B. Sun and Z. Yao, Sinomenine derivatives with embedment of nitrogen-containing heterocycles exhibiting potent TNF-alphainhibitory activity, Sci. China: Chem., 2012, 55, 2537–2547 CrossRef CAS.
- Q. Tang, J. Luo, Q. Zhu, Y. Li and S. Yin, Synthesis and anti-inflammatory activities investigation of sinomenine derivatives on ring C, Nat. Prod. Res., 2006, 20, 1015–1023 CrossRef CAS.
- S. Choi and T. Kazunori, Concentration Dependence of Ketone Hydrogenation Catalyzed by Ru, Pd, and Pt. Evidence for Weak Ketone Adsorption on Pd Surface, Bull. Chem. Soc. Jpn., 1982, 55, 2275–2276 CrossRef.
- T. Zhou, J. Hou, M. Wang, L. Ma, L. Wu, S. Wang, B. Sun and Z.-J. Yao, Regio-controlled synthesis of unsymmetrical pyrazine-fused sinomenine derivatives and discriminate substitution effects on TNF-α inhibitory activity, Tetrahedron, 2014, 70, 5475–5482 CrossRef CAS.
- Z.-J. Yao and H.-B. Zhou, China Pat, CN1687070A, 2005 Search PubMed.
- Y.-T. Lou, H.-B. Zhou, J. Zou, L.-C. Yan, E.-G. Bi, B. Sun and Z.-J. Yao, Modification of poorly bioactive sinomenine into more potent immunosuppressive agents by embedding of drug-like fragments, Tetrahedron Lett., 2010, 51, 485–488 CrossRef CAS.
- X. Cui, B. Li, T. Liu and C. Li, A Practical solution for aqueous reactions of water-insoluble high-melting-point organic substrates, Green Chem., 2012, 14, 668–672 RSC.
- X. Zheng, W. Zhu, L. Han, K. Zhang and T. Pan, Highly stereoselective synthesis of functionalized sinomenine derivatives by reducing the C-6 (ring C), J. Chem. Res., 2014, 38, 734–736 CrossRef CAS.
- X.-R. Ye, K.-X. Yan, K.-M. Wu, X.-Z. Feng, Y.-M. Huang and P. Qiu, Synthesis and anti-inflammatory analgesic activities of sinomenine derivatives, Acta Pharm. Sin., 2004, 39, 180–183 CAS.
- M. G. Constantino, L. G. d. O. Matias, G. V. J. d. Silva, E. Barbieri and M. T. d. P. Gambardella, Stereoselective sodium borohydride reductions of cyclopentanones: influence of ceric chloride on the stereochemistry of reaction, Quim. Nova, 1998, 21, 719–721 CrossRef CAS.
- A. Coop and K. C. Rice, Direct and Simple Conversion of Codeine to Thebainone-A and Dihydrothebainone, Heterocycles, 1999, 50, 39–42 CrossRef CAS.
- F. Li, L. Gaob, C. Yin, J. Chen, J. Liu, X. Xie and A. Zhang, Synthesis and opioid receptor activity of indolopropellanes, Bioorg. Med. Chem. Lett., 2009, 19, 4603–4606 CrossRef CAS.
- M. E. Furrow and A. G. Myers, Practical Procedures for the Preparation of N-tert-Butyldimethylsilylhydrazones and Their Use in Modified Wolff–Kishner Reductions and in the Synthesis of Vinyl Halides and gem-Dihalides, J. Am. Chem. Soc., 2004, 126, 5436–5445 CrossRef CAS.
- F. Yang, Z.-J. Guan, X.-Y. Chai, Q.-Y. Wu and Q.-G. Meng, Synthesis of N-substituted sinomenine derivatives and its inhibitory effect against NF-κB transcriptional activity, Acad. J. Second Mil. Med. Univ., 2015, 36, 413–417 CrossRef CAS.
- Y. Pan, Y.-F. Li, Q.-M. Bu, L.-Q. Huang, J. Wang and J.-X. Li, China Pat, CN1785976A, 2006 Search PubMed.
- Y. Pan, Y.-F. Li, Q.-M. Bu, L.-Q. Huang, J. Wang and J.-X. Li, China Pat, CN1785977A, 2006 Search PubMed.
- Y. Pan, Y.-F. Li, Q.-M. Bu, L.-Q. Huang, J. Wang and J.-X. Li, China Pat, CN1821244A, 2006 Search PubMed.
- S. P. Ross and T. R. Hoye, Reactions of hexadehydro-Diels–Alder benzynes with structurally complex multifunctional natural products, Nat. Chem., 2017, 9, 523–530 CrossRef CAS.
- R. Karmakar, S. Y. Yun, K.-P. Wang and D. Lee, Regioselectivity in the Nucleophile Trapping of Arynes: The Electronic and Steric Effects of Nucleophiles and Substituents, Org. Lett., 2014, 16, 6–9 CrossRef CAS.
- R. Joyeau, M. Planchon, J. Abessolo, K. Aissa, C. Bance and D. Buisson, Combinatorial approach to the selection of active microorganisms in biotransformation: Application to sinomenine, J. Mol. Catal. B: Enzym., 2013, 85–86, 65–70 CrossRef CAS.
- G.-H. Bao, G.-W. Qin, R. Wang and X.-C. Tang, Morphinane Alkaloids with Cell Protective Effects from Sinomenium acutum, J. Nat. Prod., 2005, 68, 1128–1130 CrossRef CAS.
- W.-M. Cheng, F. Qiu and X.-S. Yao, Three major urinary metabolites of sinomenine in rats, J. Asian Nat. Prod. Res., 2007, 9, 13–18 CrossRef CAS.
- R. A. Aycock, C. J. Pratt and N. T. Jui, Aminoalkyl Radicals as Powerful Intermediates for the Synthesis of Unnatural Amino Acids and Peptides, ACS Catal., 2018, 8, 9115–9119 CrossRef CAS.
- J. P. Barham, M. P. John and J. A. Murphy, Contra-thermodynamic Hydrogen Atom Abstraction in the Selective C–H Functionalization of Trialkylamine N-CH3 Groups, J. Am. Chem. Soc., 2016, 138, 15482–15487 CrossRef CAS.
- J. He, L. G. Hamann, H. M. L. Davies and R. E. J. Beckwith, Late-stage C–H functionalization of complex alkaloids and drug molecules via intermolecular rhodium-carbenoid insertion, Nat. Commun., 2015, 6, 5943 CrossRef.
| This journal is © the Partner Organisations 2020 |