
Industrial synthesis of reactive silicones: reaction mechanisms and processes

Abstract
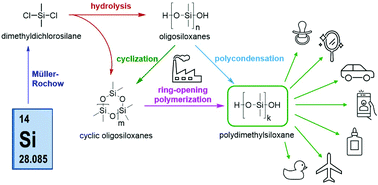
Silicones and silicone rubbers are ubiquitous in our daily lives, being used in a broad range of applications ranging from consumer products and adhesives to medicinal and electronic devices. The chemistry and synthesis of these versatile materials have been reviewed so many times that it is difficult to have a comprehensive overview. Hence, with this tutorial review we aim to provide a concise account of the most important reactions and industrial processes to furnish silicones, focusing specially on reactive silicones, specifically in telechelic OH-terminated polysiloxanes, which represent one of the most relevant members of this important materials family.
 Please wait while we load your content...
Please wait while we load your content...

