DOI:
10.1039/D0QO00764A
(Review Article)
Org. Chem. Front., 2020,
7, 3284-3311
The preparation of (4H)-imidazol-4-ones and their application in the total synthesis of natural products
Received
30th June 2020
, Accepted 22nd August 2020
First published on 26th August 2020
Abstract
(4H)-Imidazol-4-ones are an important scaffold for a variety of applications, including natural products, medicine, agriculture, and other applications. Over the years, there have been a number of preparations published for the synthesis of imidazol-4-ones. This review discusses the progress made on the synthesis of imidazol-4-ones, and their application towards the total synthesis of a range of imidazol-4-one containing natural products. Emphasis is made on areas of the field that still need progress.
 Katarina L. Keel | Katarina Keel is currently a PhD student in Dr Jetze Tepe's lab at Michigan State University. Her research interests focus on the synthesis of heterocycles for their application as a new class of novel proteasome modulators as well as the total synthesis of natural products. |
 Jetze Tepe | Jetze J. Tepe is a professor of chemistry at the Department of Chemistry at Michigan State University. His research program is focused on the synthesis and biological evaluation of small heterocyclic scaffolds and natural products, as part of an academic drug discovery program. |
1. Introduction
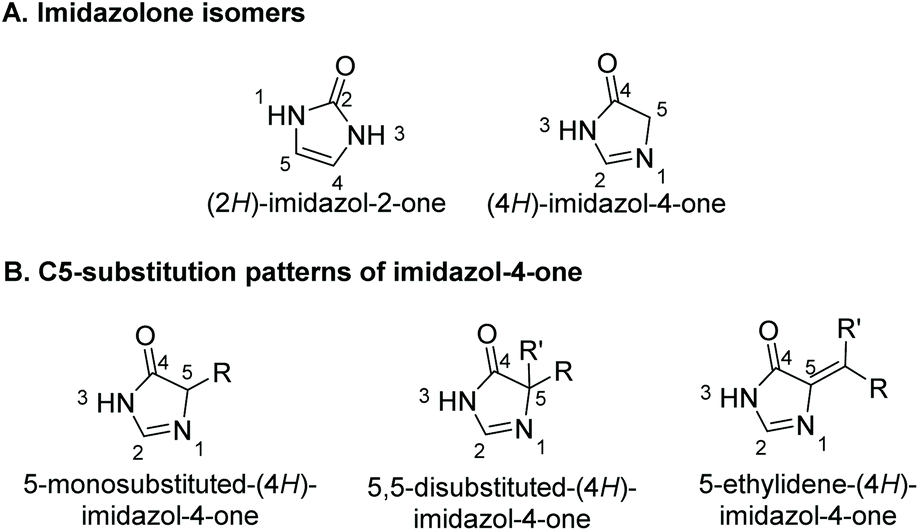
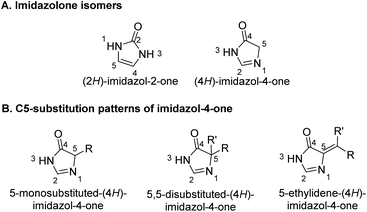
Heterocycles represent an important core in many isolated natural products. Imidazolones are five-membered heterocyclic rings containing two non-adjacent nitrogens and a carbonyl group. There are two isomers of imidazolones, depending on the placement of the carbonyl: imidazol-2-ones and imidazol-4-ones (Fig. 1A).
 |
| | Fig. 1 Structures of imidazolones. | |
Imidazol-4-ones are an important heterocycle utilized for a large range of applications, including medicinal chemistry,1–5 fluorescent protein chromophores,6–8 agrochemicals,9 and natural products. This heterocyclic structural motif is also found naturally occurring in the body. Imidazol-4-ones are found as advanced glycation end products (AGE),10–12 post-translational modifications of several amino acids-aka 3,5-dihydro-5-methylidene-4H-imidazol-4-one (MIO),13 and creatinine, a waste product used to indicate kidney health.14 Despite being found in a vast assortment of fields, there has never been a review on the preparative methods of imidazol-4-ones.
Preparation of (4H)-imidazol-4-ones goes back as far as 1907, when H. Finger first reported the synthesis of a (4H)-imidazol-4-one.15 Since then, a number of unique methodologies have been developed for the production of imidazol-4-ones. These methodologies can be used to produce three C5-substitution patterns, as shown in Fig. 1B. In an effort to centralize this information, section 2 of this review summarizes the methodologies developed for the synthesis of imidazol-4-ones. Details of each reaction are discussed, as well as any advantages or disadvantages to the method. Section 3 of this review will highlight the application of these preparative methods in the total synthesis of known imidazol-4-one containing natural products. Additionally, this section of the review presents several natural products that have yet to be synthesized. Overall, this review provides the first ever analysis of the known preparative methods of imidazol-4-ones, and the only review summarizing imidazol-4-one containing natural products.
2. Preparation of (4H)-imidazol-4-ones
Since the first report of the synthesis of imidazol-4-ones, there have been numerous reported methods of preparation. In this section, many preparative methods of imidazol-4-ones will be discussed. For the purpose of this review, the methods have been categorized into three main transformations: condensation reactions, aza-Wittig reactions, and heterocyclic rearrangements.
2.1 Condensation reactions
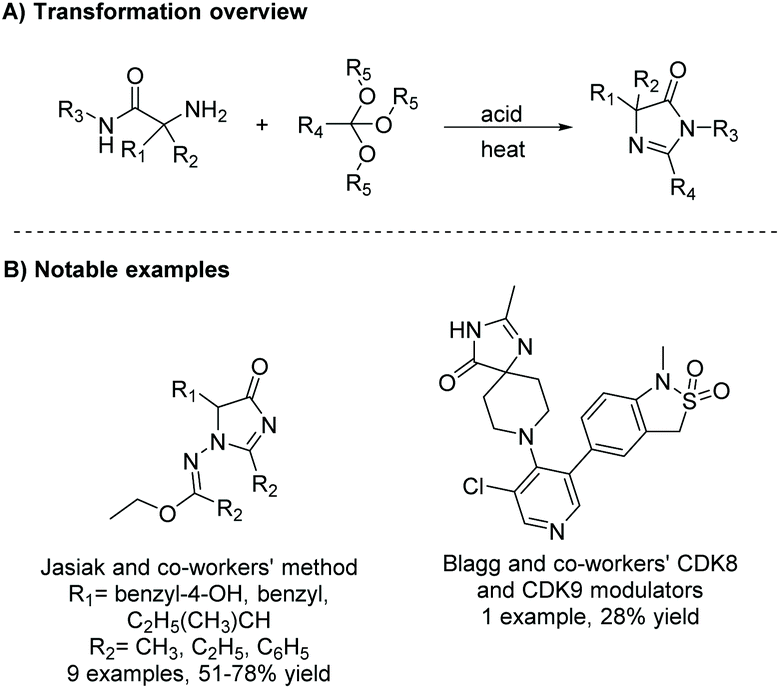
2.1.1 Cyclization of diamides.
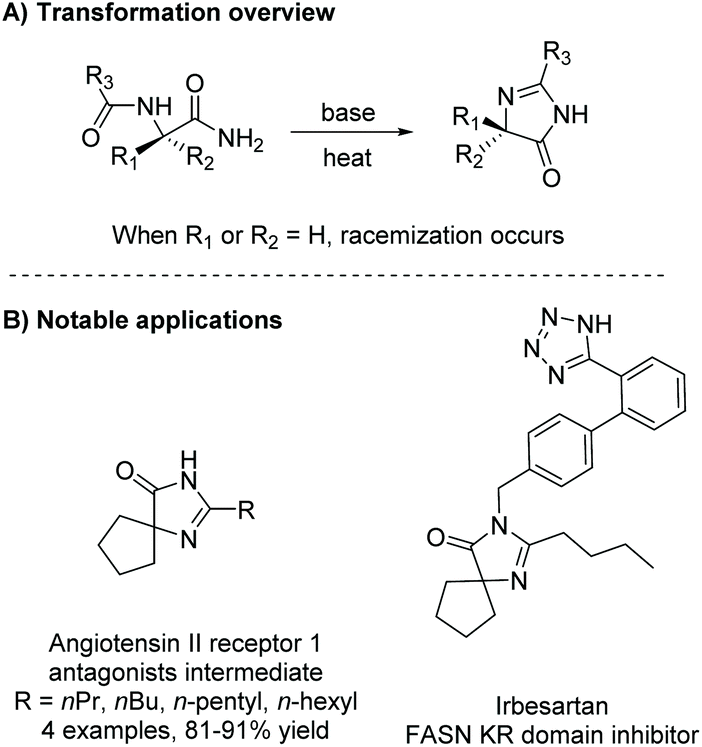
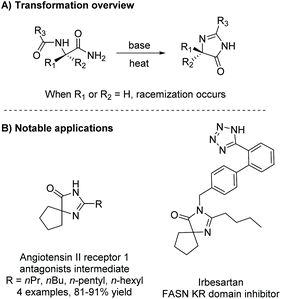
Under basic conditions, with or without heat, a diamide will cyclize to produce a range of substituted imidazol-4-ones. Some notable examples of this reaction being used within the last 5 years include the synthesis of derivatives of irbesartan for fatty acid synthase (FASN) KR domain inhibition5,16 and angiotensin II receptor 1 antagonists (Scheme 1).1 This method has also been used to produce enantiopure 5,5-disubstituted-4-imidazolones from enantiopure diamides, but does not work well for the production of enantiopure 5-monosubstituted-4-imidazolones, as heat and base can cause the stereocenter to tautomerize.17 Additionally, this reaction has been used to produce a number of 5-ethylidene-4-imidazolones, with the alkene's conformation being driven by steric interactions with the imidazolone's carbonyl.
 |
| | Scheme 1 General transformation from diamide to imidazol-4-one. | |
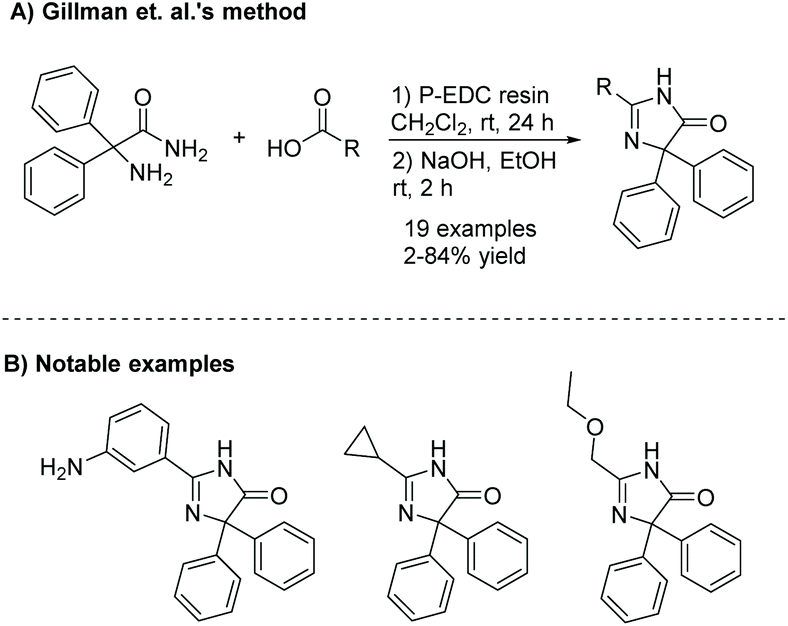
The diamide is a common intermediate in several preparative methods of imidazol-4-ones. One example is the condensation of an α-amino amide and acid derivative. Gillman et al. synthesized a number of imidazol-4-ones using an α-amino amide and carboxylic acid (see Scheme 2).18 Here, the amine and carboxylic acid were coupled together using a polymer-supported carbodiimide reagent, producing diamides in relatively low yields (10–50%), due to the steric bulk surrounding the amine. The diamide was then cyclized upon addition of sodium hydroxide in ethanol.
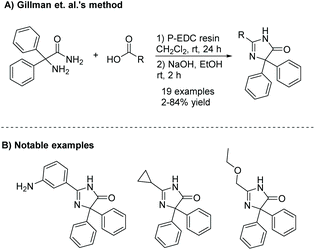 |
| | Scheme 2 Gillman et al.'s synthesis of imidazolones as neuropeptide Y5 receptor antagonists. | |
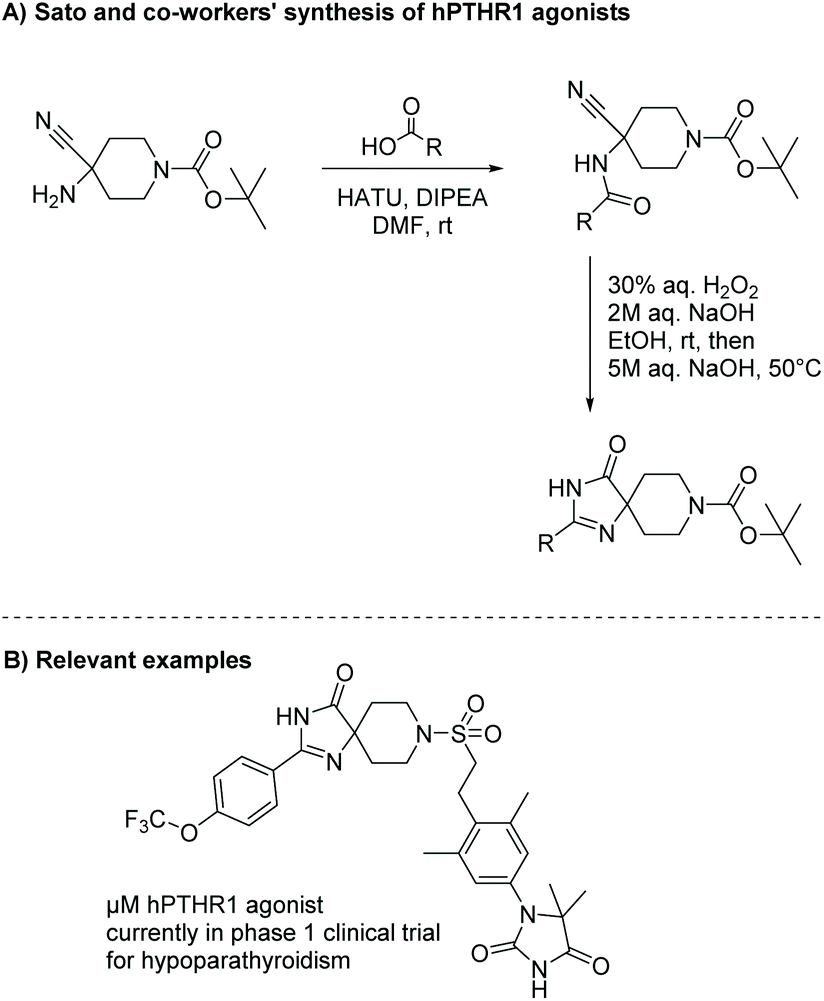
Diamides can also be produced through the oxidation of α-amino nitriles using hydrogen peroxide. α-Amino nitriles were first used to synthesize imidazol-4-ones in 1981 by Marinus Los.19 In the first step of this synthesis, an amine reacts with a carboxylic acid or acid chloride to produce an amide. The second step involves oxidation of the nitrile to an amide using hydrogen peroxide, which leads to the formation of an imidazol-4-one through cyclization of the diamide intermediate. Nishimura et al. used this methodology to produce a range of imidazol-4-ones as human parathyroid hormone receptor 1 (hPTHR1) agonists for treatment of hypoparathyroidism, shown in Scheme 3.20,21 One of their analogues, shown in Scheme 3B, is currently in a phase 1 clinical trial for the treatment of hypoparathyroidism.
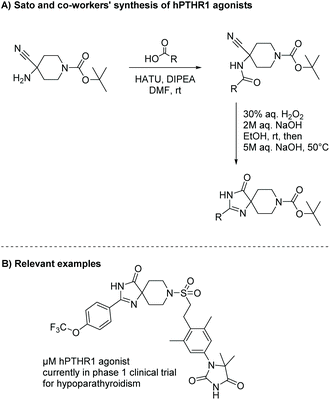 |
| | Scheme 3 The synthesis of piroimidazol-4-ones for treatment of hypoparathyroidism. | |
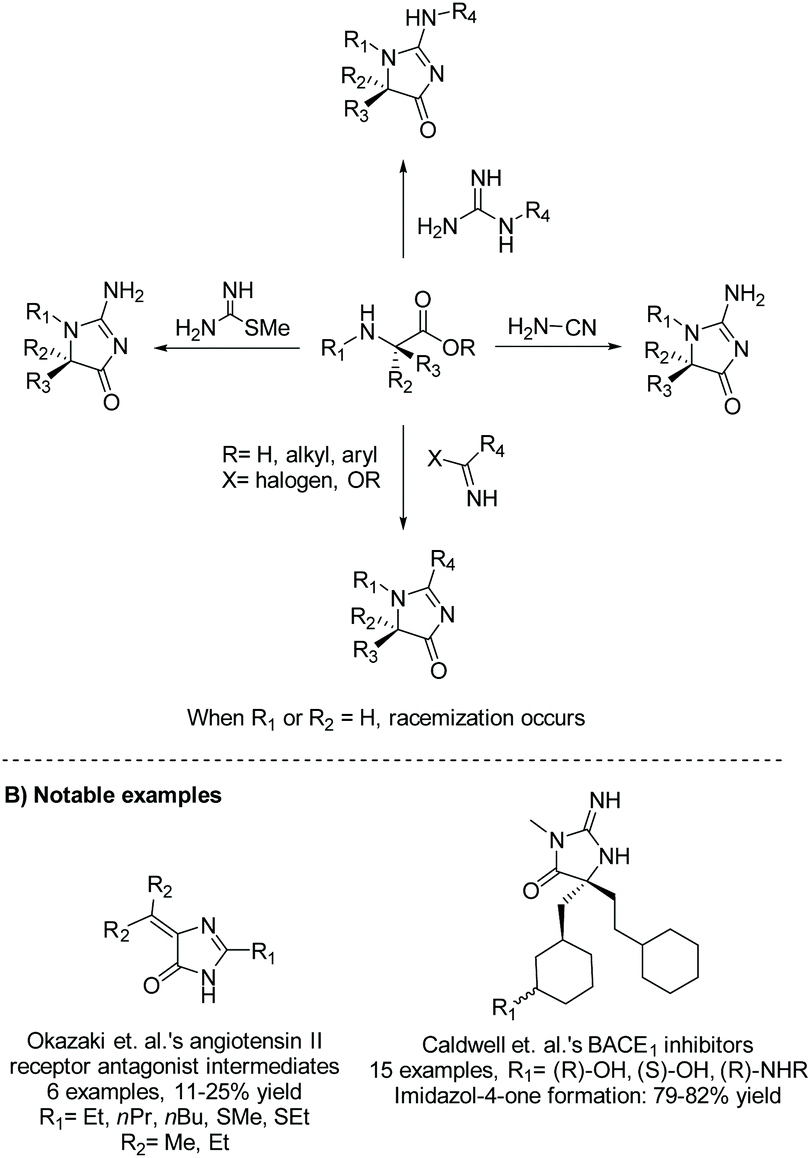
2.1.2 Amino ester and cyanamide/guanidine.
Amino esters are versatile building blocks used to synthesize a range of heterocycles, including imidazol-4-ones. The condensation of amino esters with cyanamides, guanidines, imino halides, imidates, or methyl carbamimidothioates has been used to produce a range of 5-monosubstituted, 5,5-disubstituted, and 5-ethylidene imidazol-4-ones for biological applications and natural product syntheses.22–25Scheme 4 summarizes the different nucleophiles that react with amino esters to produce imidazol-4-ones. Also demonstrated in Scheme 4, this method can be used to produce enantiopure 5,5-disubstituted imidazol-4-ones, as this reaction is considered stereospecific when the 5-position is disubstituted, similar to the cyclization of diamides.24
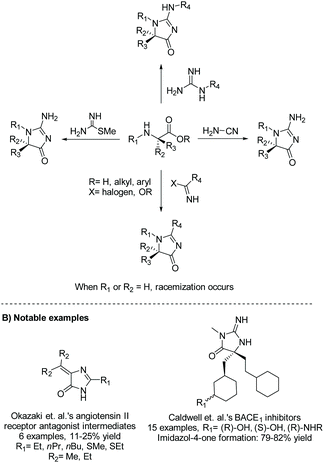 |
| | Scheme 4 Transformation of amino ester to imidazolone using a variety of nucleophiles. | |
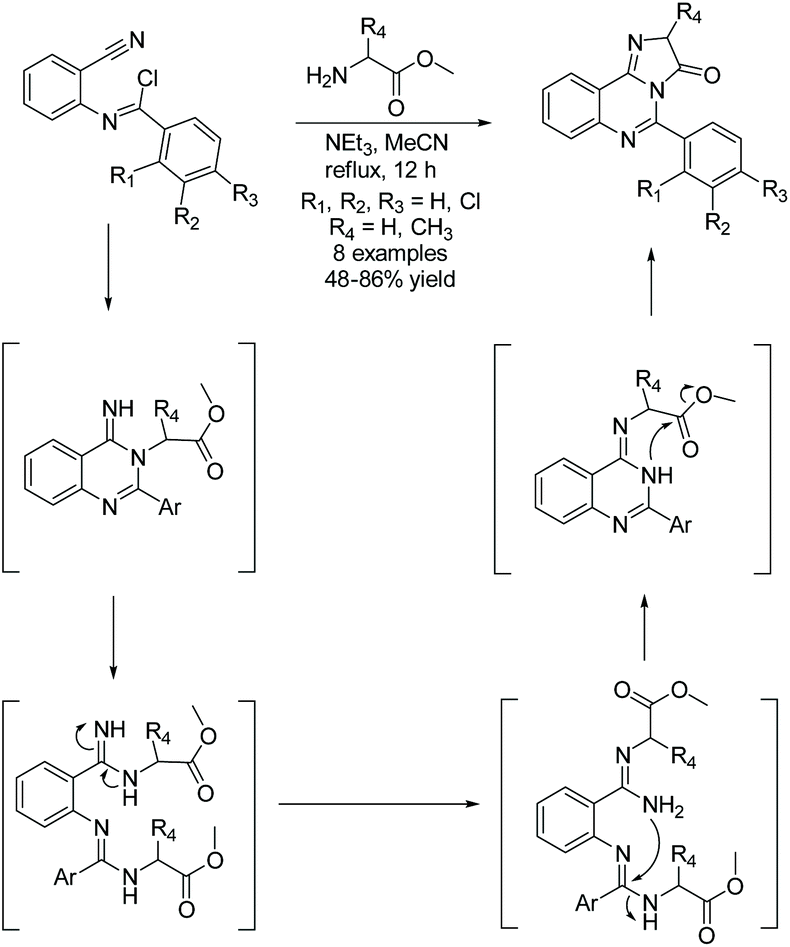
In 2020, Fathalla et al. utilized an intramolecular cyclization of amidine and amino ester to produce imidazoquinazolinones through a domino synthesis (Scheme 5).26 Here, a range of benzyimidoyl chlorides were reacted with glycine or L-alanine methyl ester hydrochloride to produce a few quinazolin-4(3H)-imines. The quinazolin-4(3H)-imine then undergoes a series of ring deconstructions and formations, ultimately producing some complex imidazoquinazolinones in 48–86% yield. This one-pot methodology was also used to construct pyrimidoquinazolinones by reacting benzymidoyl chloride with a β-alanine methyl ester.26
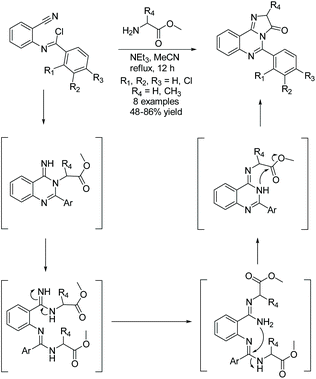 |
| | Scheme 5 Domino synthesis of imidazoquinazolinones. | |
2.1.3 Imidate and α-imino ester.
In section 2.1.2 imidates were used to produce a range of imidazol-4-ones upon reaction with α-amino esters. Moreover, imidates and thioimidates can be used to produce 5-ethylidene-4H-imidazol-4-ones when reacted with α-imino esters (Scheme 6).27,28 The first report of the cyclization of thioimidate and α-imino ester was by Ikejiri et al. in 2012, where they synthesized five fused-ring 5-ethylidene-4H-imidazol-4-ones in 43–85% yield.28Scheme 7 displays the proposed mechanism for this reaction. The key step to this mechanism is the formation of an aziridine, which then ring opens to the imidazol-4-one product. In the past two years alone, this reaction has been reported for the production of a number of fluorescent protein chromophores, which require the ethylidene moiety for their function.29–32 While this method is used for the production of 5-ethylidene-4H-imidazol-4-ones, there are no reported cases of it being used to produce 5-monsubstituted or disubstituted imidazol-4-ones.
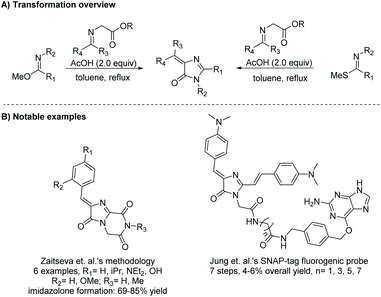 |
| | Scheme 6 General condensation of iminoester and α-imino or α-amino ester. | |
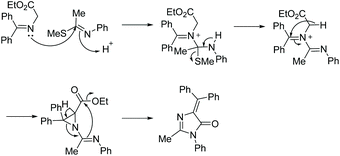 |
| | Scheme 7 Miyashita and co-worker's proposed mechanism for the formation of 5-ethylidene-4H-imidazol-4-ones. | |
2.1.4 Orthoester and α-amino amide.
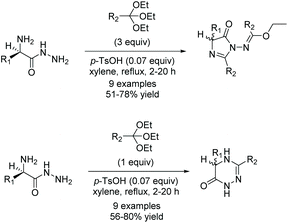
Another way of synthesizing (4H)-imidazol-4-ones is through the condensation of orthoesters and amino amides (Scheme 8). One of the first reports of this reaction was in 1956 by Brunken and Bach.33,34 In this reaction, the amine from the amino amide reacts with an orthoester, creating an α-imino amide in situ which then cyclizes to form an imidazol-4-one. Typically, this reaction needs to be activated by acid and/or heat to get good conversion to cyclized product. Since the first report of this reaction, it has been used a number of times in the production of di- and tri-substituted imidazolones.35–37
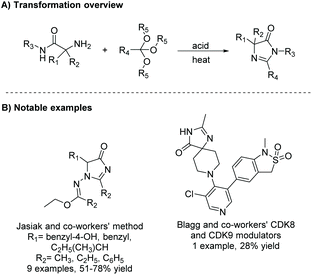 |
| | Scheme 8 General condensation of orthoester and α-amino amide. | |
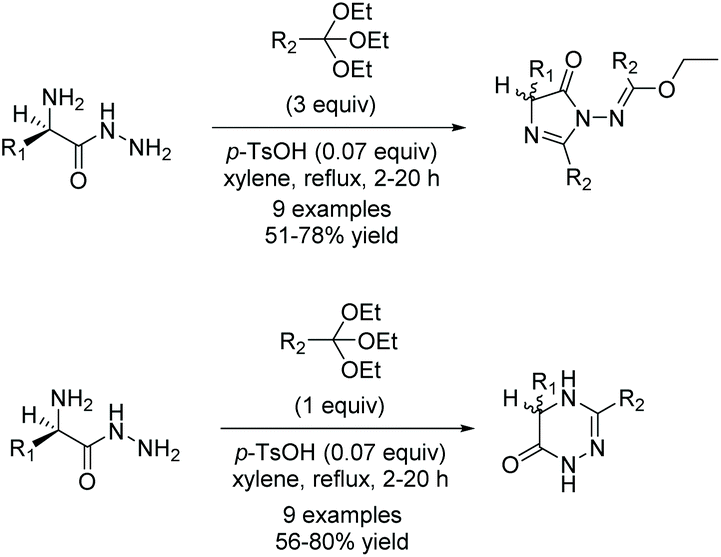
A study done by Jasiak and co-workers in 2013 demonstrates the synthesis of imidazol-4-ones from optically active α-amino carboxylic acid hydrazides (Scheme 9).35 Nine examples were synthesized in good yields (51–78%). Additionally, several triazines were produced using almost the exact same conditions. It is known that excess orthoester is required to produce an imidazol-4-one as the major product. When equimolar amounts of orthoester and amino amide are used, the major product isolated is the triazine (Scheme 8). Interestingly, even though optically pure amino amides were used in this reaction, racemates were produced. While the chiral carbon is not directly affected by the transformation to imidazol-4-one, the carbon can tautomerize through hydrogen migration from the stereogenic carbon to the imino or carbonyl groups, causing a shift of the double bond, leading to loss of optical activity.
 |
| | Scheme 9 Jasiak and co-workers’ synthesis of imidazol-4-ones from optically active α-aminocarboxylic acid hydrazides. | |
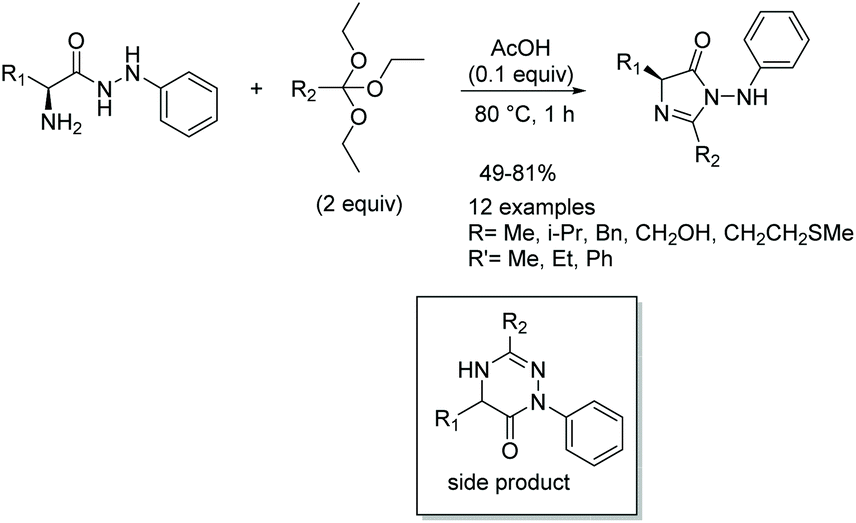
In 2014, Kacem and Hassine published an interesting modification to this reaction, leading to the synthesis of enantioselective, 5-monosubstituted imidazol-4-ones.9 This methodology involved a solvent-free condensation between chiral α-amino acid phenylhydrazides and triethyl orthoesters with catalytic dry acetic acid (Scheme 10). While solvent-free conditions produced the highest yields in the shortest amount of time, this reaction was also performed under a variety of solvents. Under solvent free conditions, all the compounds were prepared enantioselectively within an hour. While they do not mention why this reaction was enantioselective when previous reports were not, it may have to do with the milder conditions and shortened reaction time the solvent-free synthesis allowed for.
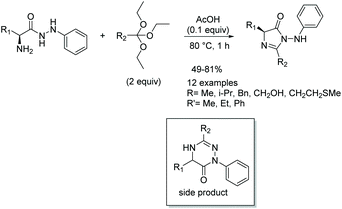 |
| | Scheme 10 Kacem and Hassine's solvent-free synthesis of imidazolones. | |
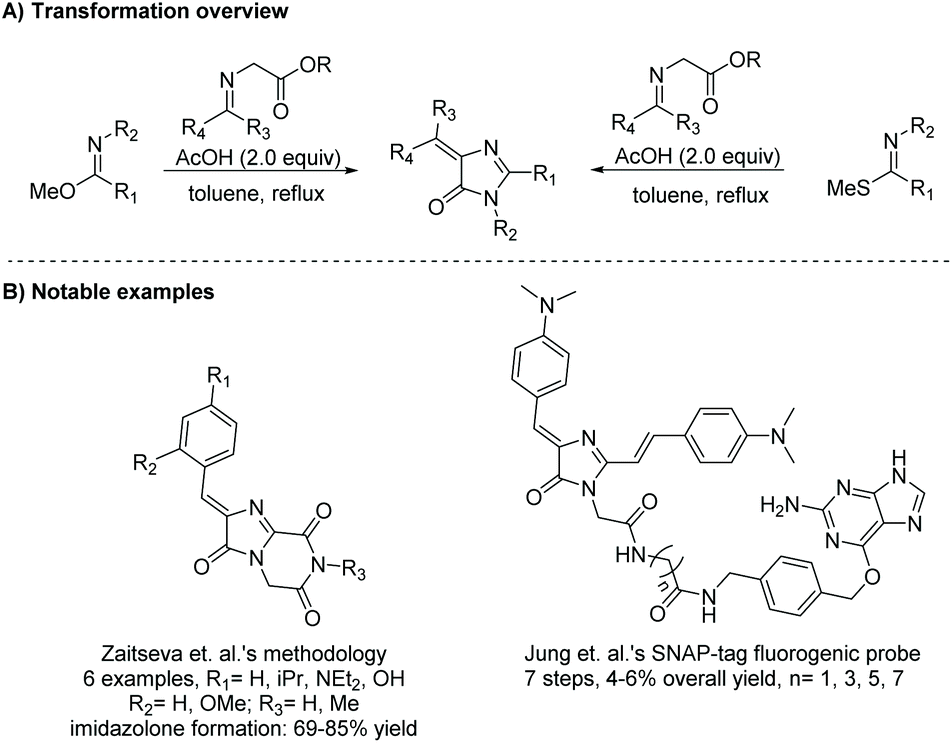
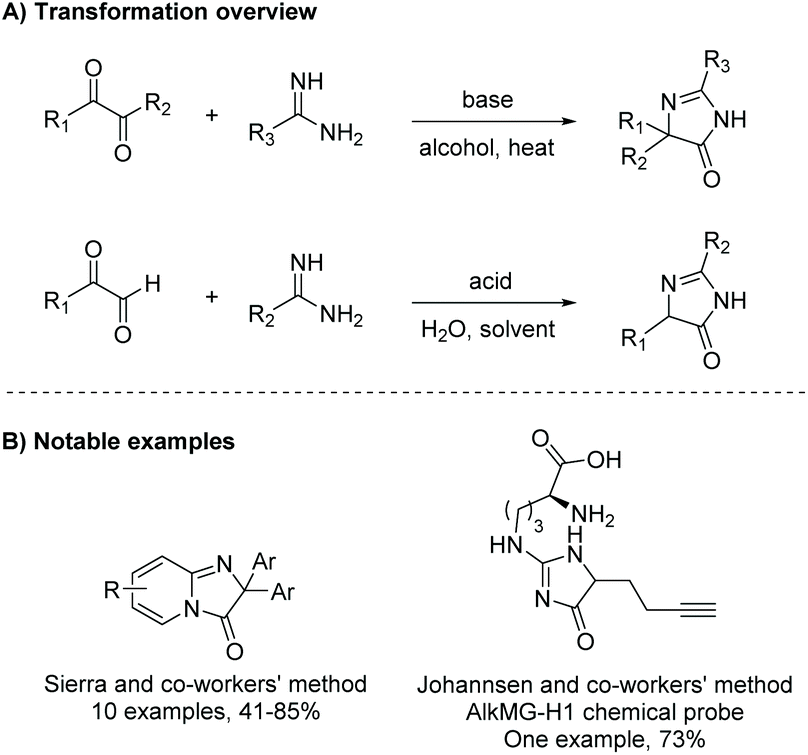
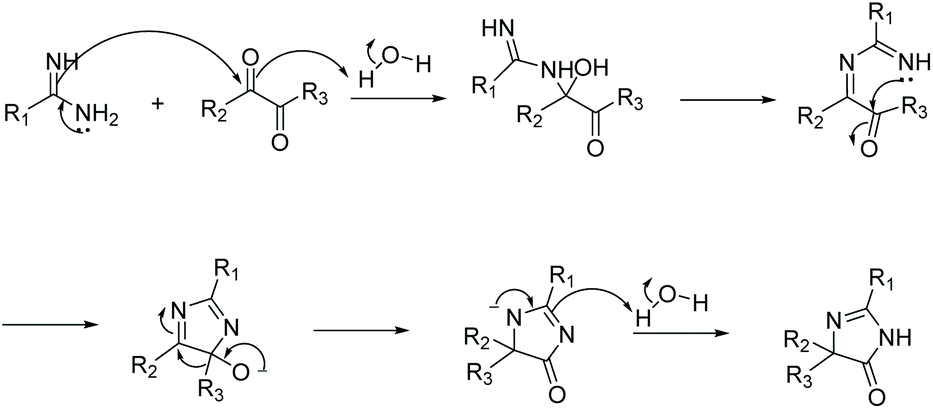
2.1.5 Diketone and amidine/guanidine.
Condensation of a diketone and an amidine or guanidine is a common way to produce 5,5-disubstituted imidazol-4-ones (Scheme 11). One of the first reports of this reaction was in 1950.38 In this reaction, the diketone and amidine react under basic conditions, creating a di-imine intermediate, which then cyclizes to a 5-hydroimidazole. The 5-hydroimidazole then undergoes a 1,5-dialkyl migration to produce the desired imidazol-4-one product. This mechanism is displayed in Scheme 12.39 Furthermore, 5-monosubstituted imidazol-4-ones can be produced starting with an α-keto aldehyde under acidic conditions (Scheme 11). Over the past 10 years, this reaction has been used to produce imidazol-4-ones and 2-aminoimidazol-4-ones for an assortment of medicinal applications.40–42 Additionally, this cyclization can be used to produce bicyclic imidazolones through the condensation of a diketone and 2-aminopyridine or 2-aminopyrimidine.43,44
 |
| | Scheme 11 General condensation of diketone and amidine to produce di- and tri-substituted imidazol-4-ones. | |
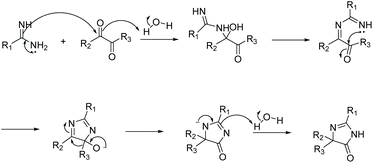 |
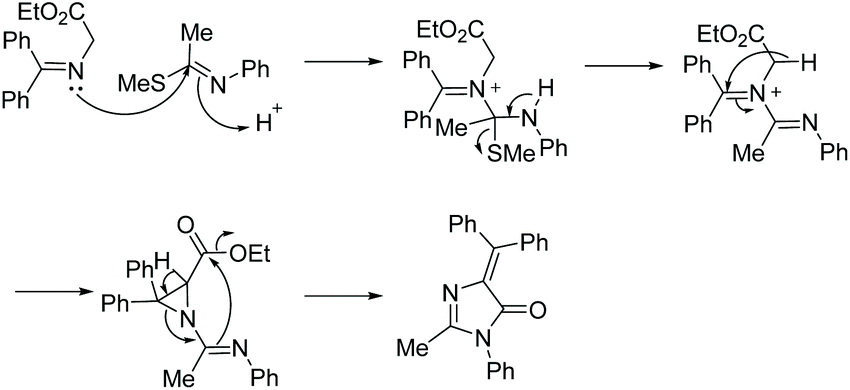
| | Scheme 12 Proposed mechanism for the cyclization of amidine and diketone to produce 5,5-disubstituted imidazol-4-ones. | |
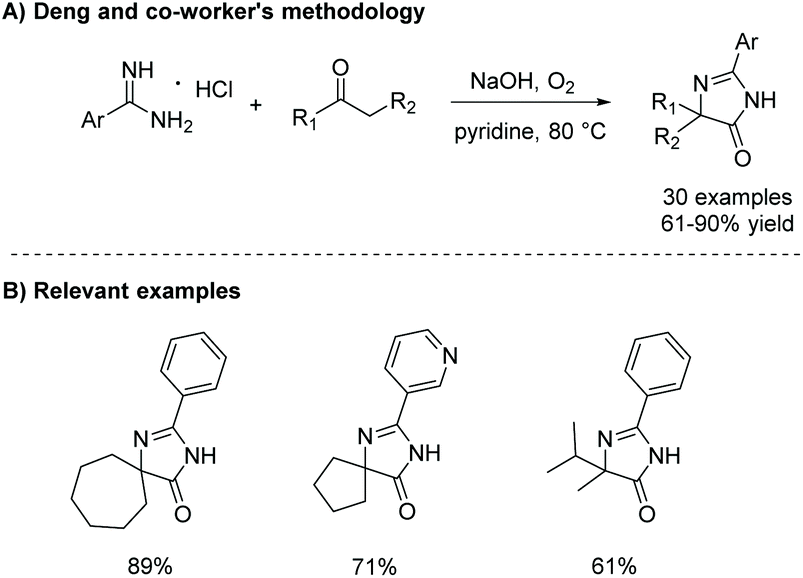
In 2015, Xie et al. reported a one-pot oxidative condensation of ketones and amidines (Scheme 13).39 Here, molecular oxygen is used to oxidize the α-keto carbon to a diketone in situ, which then cyclizes under basic conditions to produce tri-substituted imidazol-4-ones, including spiroimidazol-4-ones, in good yields (30 examples, 61–90% yield). This reaction has not been used to produce any enantioselective imidazolones thus far, mainly due to racemization which occurs during the 1,5-dialkyl migration.
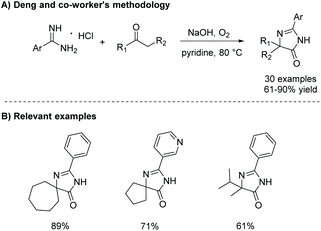 |
| | Scheme 13 Deng and co-workers’ synthesis of imidazol-4-ones from ketone and amidine containing starting materials. | |
2.2 Aza-Wittig reaction
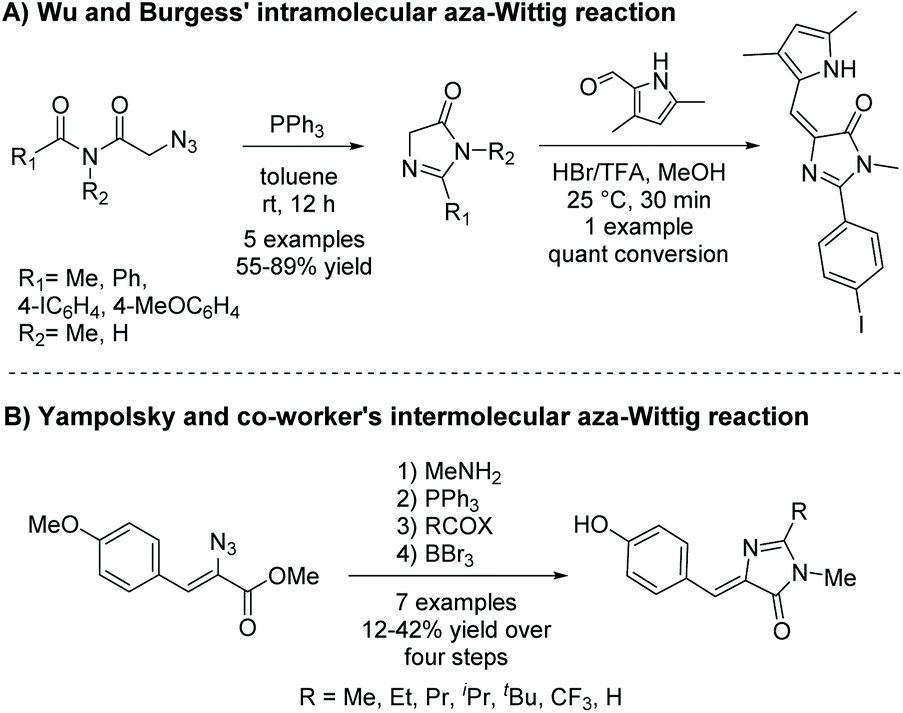
2.2.1 Intramolecular aza-Wittig reaction.
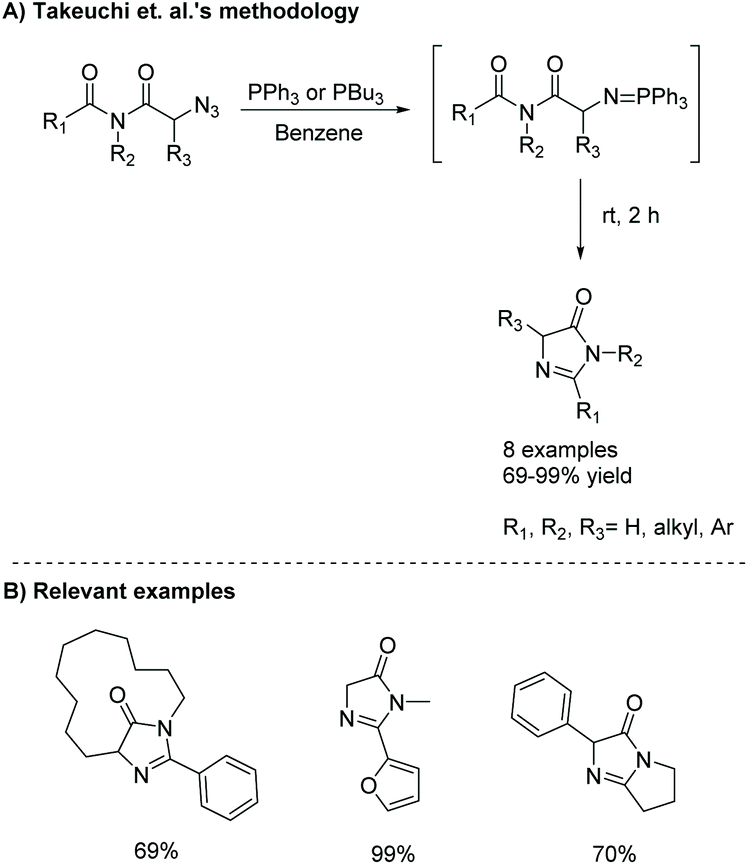
The intramolecular aza-Wittig reaction is another common way to produce imidazol-4-ones. Takeuchi et al. was one of the first to report this reaction in 1989, using azido-substituted imides.45 These imides reacted with triphenylphosphine to afford some 5-monosubstituted-4-imidazolones in good yields (69–99%) via a Staudinger reaction, followed by an intramolecular aza-Wittig reaction (Scheme 14).
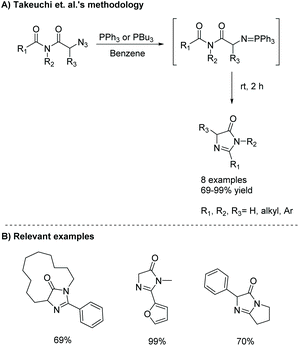 |
| | Scheme 14 Takeuchi et al.'s intramolecular aza-Wittig reaction to produce imidazolones. | |
Thus far, this reaction appears to be used mainly to produce a range of 5-ethylidene-4-imidazolones.46–49 There are two different pathways this reaction can be implemented to produce 5-ethylidene-4-imidazolones; both are shown in Scheme 15. In method A, the more popular method, the imidazol-4-one ring is formed from a terminal azidoimide.50 Then, the 5-ethylidene substituent is added via a Knoevenagel condensation reaction. Method B first condenses the azidoamide and aldehyde to produce an internal azide.49 This then reacts with triphenylphosphine, followed by an acid halide to produce an iminoyl halide, which will cyclize upon condensation with the present amide. Alkene conformation is driven by steric interactions as well as the potential to hydrogen bond. Variations of this reaction have been used a couple times to produce 5,5-disubstituted-4-imidazolones.51,52
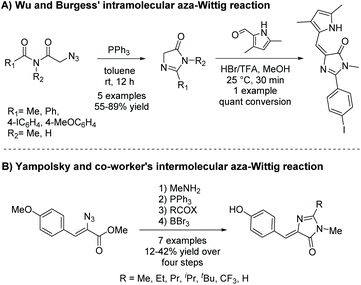 |
| | Scheme 15 Aza-Wittig reaction to produce 5-ethylidene-4-imidazolones. | |
2.2.2 Tandem aza-Wittig/heterocumulene-mediated annulation.
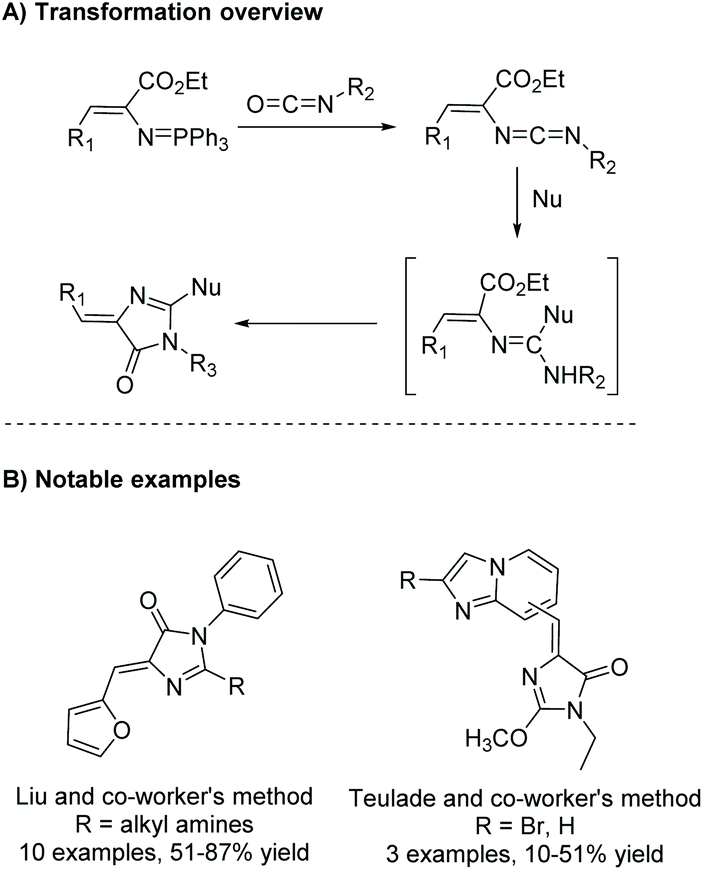
The aza-Wittig reaction can also be used as part of a heterocumulene-mediated annulation reaction to produce imidazol-4-ones. The first report of this reaction was in 1992 by Molina et al. for the synthesis of aplisinopsin-like alkaloids.53 In this reaction, an aza-Wittig reagent reacts with isocyanate to produce a heterocumulene intermediate. Upon addition of a nucleophile, the intermediate will cyclize to produce a variety of 5-ethylidene imidazol-4-ones. The mechanism and notable examples of this reaction are shown in Scheme 16. Some examples of nucleophiles (Nu) include alcohols, amines, thiols, and heterocycles.4,54–56
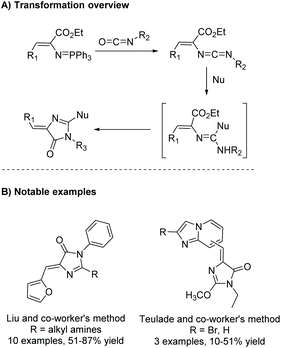 |
| | Scheme 16 General heterocumulene-mediated annulation initiated by an aza-Wittig reagent. | |
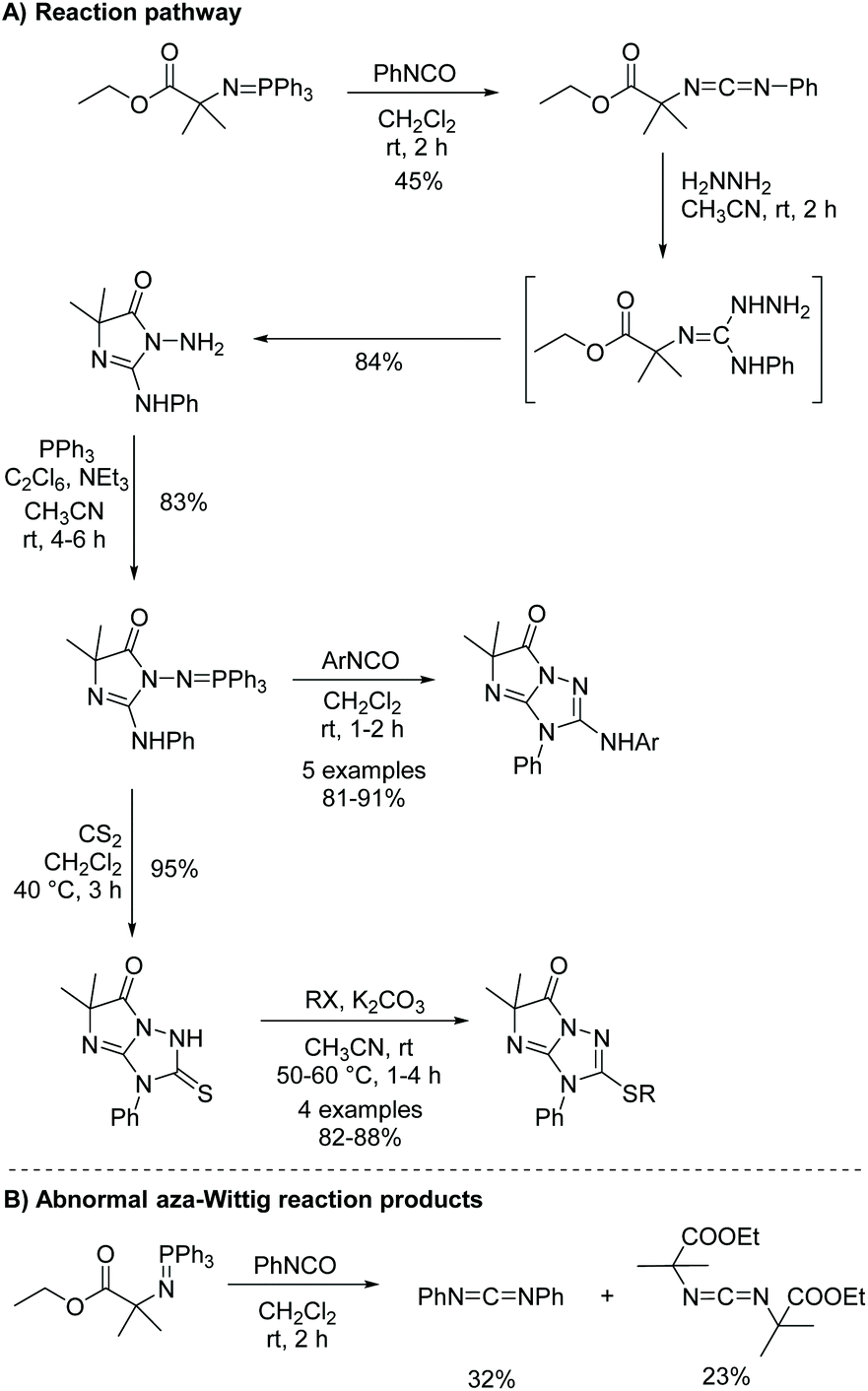
While more uncommon, the aza-Wittig/heterocumulene-mediated annulation can also be used to produce 5,5-disubstituted imidazol-4-ones. Yang and co-workers reported an interesting cascade of reactions to produce 3,5-dihydro-6H-imidazo[1,2-b]-1,2,4-triazol-6-ones (Scheme 17).57 In this report they highlighted some abnormal aza-Wittig reactions, which led to unanticipated side products, reducing the yield of the isocyanate intermediate (Scheme 17B). It was noted that only the main aza-Wittig reaction product was produced when a similar but less sterically hindered iminophosphorane was used. Therefore, one limitation to this method is sterics, which can hinder formation of the isocyanate intermediate. This may explain why more 5,5-disubstituted imidazol-4-ones are not made via the aza-Wittig/heterocumulene mediated annulation methodology. There is also one report of this reaction being used to synthesize 5-monosubstituted imidazol-4-ones.58 Interestingly, this reaction was performed stereospecifically, using enantiopure amino acids to produce enantiopure 5-monosubstituted imidazolones.
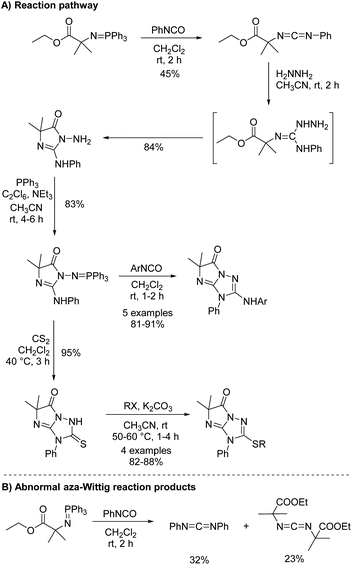 |
| | Scheme 17 Yang and co-worker's synthesis of 3,5-dihydro-6H-imidazo[1,2-b]-1,2,4-triazol-6-ones. | |
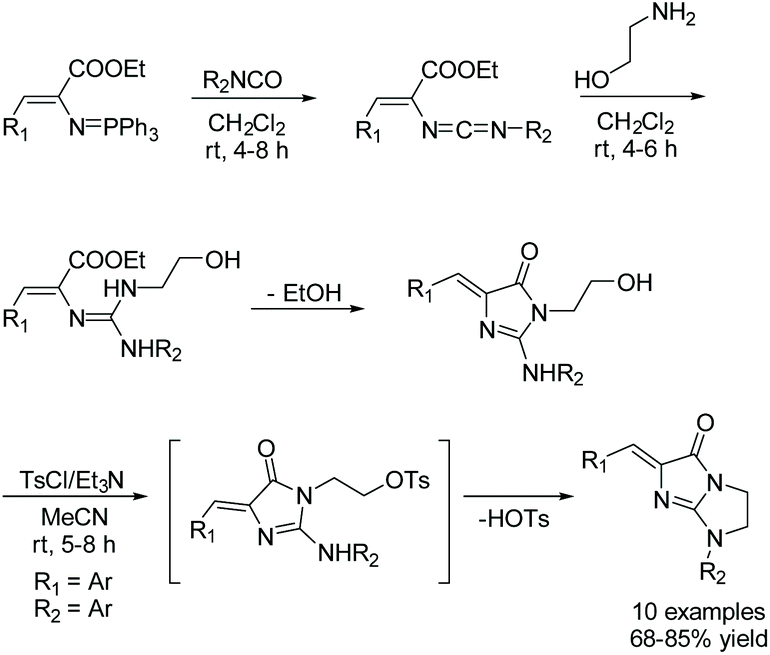
In 2019, a one-pot, three step reaction was reported by Ding and co-workers using an aza-Wittig/heterocumulene mediated annulation to produce a variety of bicycloimidazol-4-ones.59Scheme 18 describes their one-pot, three-step synthesis of a variety of bicycloimidazol-4-ones in good yields (68–85%). In this reaction, a (vinylimino)phosphorane is treated with a variety of aromatic isocyanates to produce a carbodiimide. 2-aminoethanol is then added to the reaction, which produces a guanidine intermediate that cyclizes upon the loss of ethanol. Then, upon addition of tosyl chloride (TsCl) and triethylamine (NEt3), the primary alcohol is converted to a tosyl ether, which leaves upon the formation of the second ring. These three steps were performed in sequence, without isolation.59
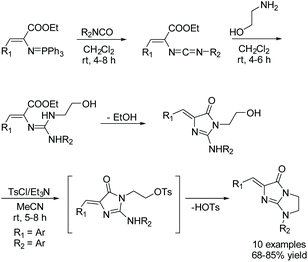 |
| | Scheme 18 One pot, multicomponent synthesis of bicycloimidazolones reported by Ding and co-workers. | |
2.3 Heterocyclic conversion/rearrangements
2.3.1 Thiohydantoin conversion to imidazolone.
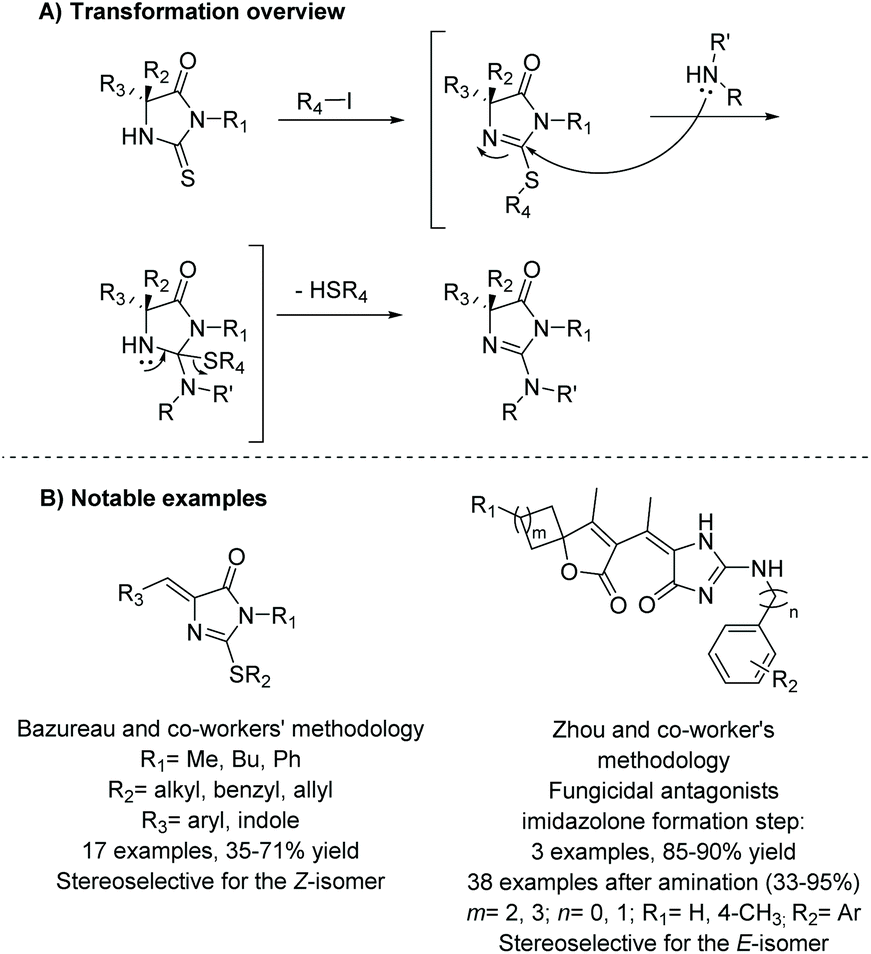
One of the most commonly used methodologies for the production of imidazol-4-ones is the conversion of thiohydantoins using alkyl halides. This was first reported by Daboun and Ibrahim in 1981.60 Since then, there have been numerous reports of this transformation being used to synthesize various imidazol-4-ones for medicinal applications61,62 and the total synthesis of natural products.63–67Scheme 19 describes the basic transformation from thiohydantoin to 2-aminoimidazol-4-one. This reaction usually takes two steps. In the first step, the thiohydantoin is converted to 2-(alkylthio)imidazol-4-one by reacting the thiohydantoin with an alkyl halide and some base. The second step is then addition of a nucleophile (anime, hydrazine, boronic acid, ether, etc.) with heat, which converts the 2-thioether to a variety of functional groups. Substitution at the 5 position of the ring is also versatile, withstanding mono- and di-substitution as well as an ethylidene functional group. Moreover, the 5-position substitution is not affected during transformation from thiohydantoin to imidazol-4-one, which allows this method to be used for the production of stereoselective compounds.68 As with many of these methods, the stereochemistry of the 5-ethylidene group is driven by sterics, to avoid interactions with the imidazolone's carbonyl. This typically leads to the Z-isomer being favored.62 However, in some instances, the E-isomer may be favored, like in Zhou and coworker's work, mentioned in Scheme 19B.61 The E-isomer was identified from a X-ray crystal structure.
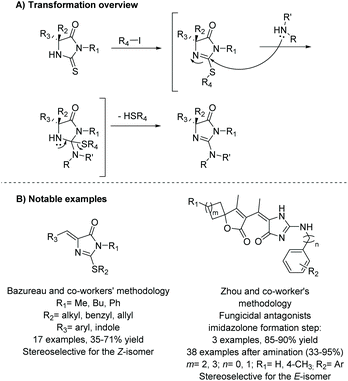 |
| | Scheme 19 Conversion of thiohydantoin into 2-aminoimidazolone. | |
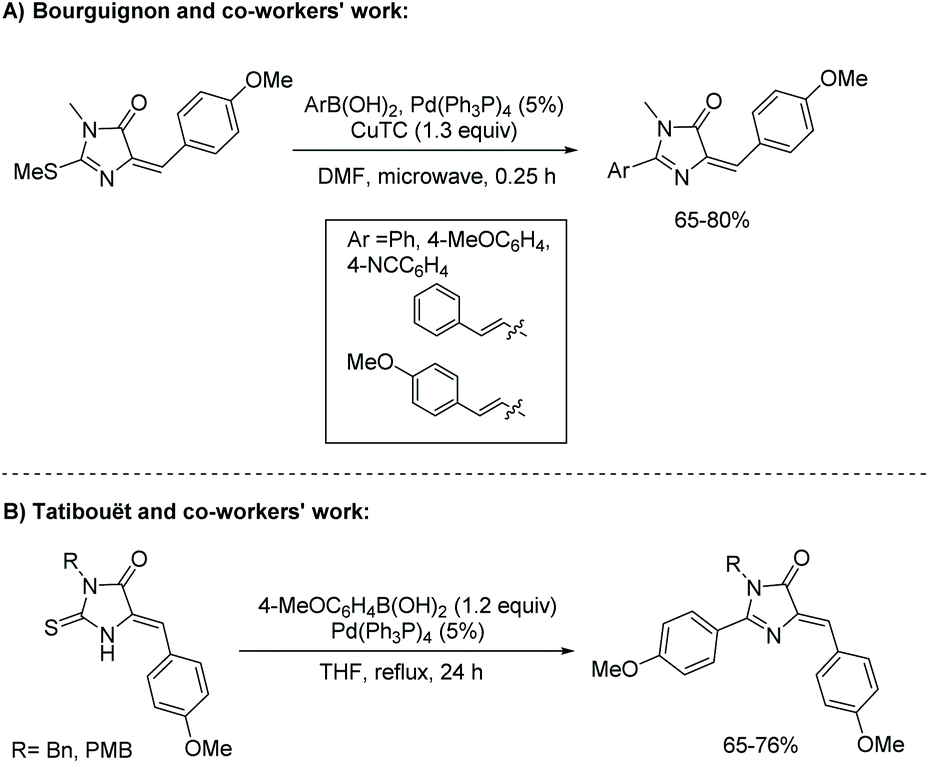
Ease of alteration of the 2-position of the thiohydantoin ring can lead to a multitude of unique imidazol-4-ones. Amination of the 2-position is performed with a desired amine and heat; this is the same procedure for substitution to a hydrazine. Etherfication can be accomplished using an alcohol and some base, along with heat.69 Arylation of the 2-position of the imidazol-4-one is also possible, following the Liebeskind-Srogl reaction. This method was first reported by Bourguignon and co-workers in 2004 (Scheme 20).70 The Liebeskind-Srogl reaction creates a C–C bond through the cross coupling of a boronic acid and thioether in the presence of copper(I) thiophene-2-carboxylate (CuTC) and tetrakis(triphenylphosphine)palladium (Pd(Ph3P)4).71 Surprisingly, the authors report the E-isomer was isolated from the Knoevenagel condensation of 4-methoxy benzaldehyde and thiohydantoin. This led to isolation of E-imidazol-4-one. In another study, Tatibouët and co-workers reported the use of boronic acids and Pd(PPh3)4 to directly convert thiohydantoins into 2-arylimidazolones (see Scheme 20).71 In their work, the Knoevenagel condensation of 4-methoxy benzaldehyde and thiohydantoin led to the isolation of the Z-isomer of thiohydantoin and subsequentially, Z-imidazol-4-one.
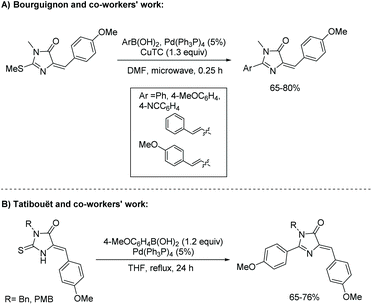 |
| | Scheme 20 Conversion of thiohydantoin to 2-aryl-imidazol-4-one. | |
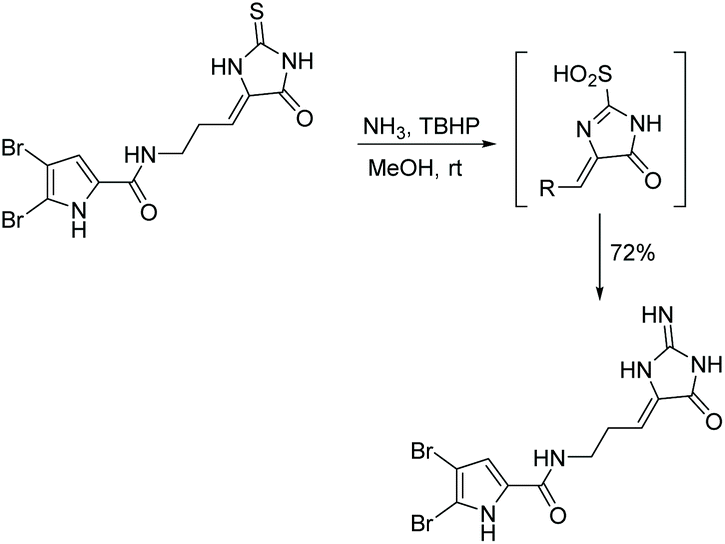
The conversion of thiohydantoin to 2-aminoimidazol-4-one can be performed using a one-pot, two step procedure with tert-butylhydroperoxide (TBHP) and aqueous ammonia in methanol at room temperature. This reaction was first mentioned in the total synthesis of dispacamide, reported by Lindel and Hoffmann in 1997 (Scheme 21).63 In this reaction, TBHP is used to oxidize the sulfur to a sulfinic acid in situ, which is then easily removed by nucleophilic attack of ammonia. Since this reaction was first reported, it has been used to synthesize a range of (4H)-imidazol-4-ones.67,72,73
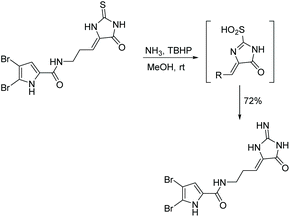 |
| | Scheme 21 First reported conversion of thiohydantoin to imidazolone using TBHP. | |
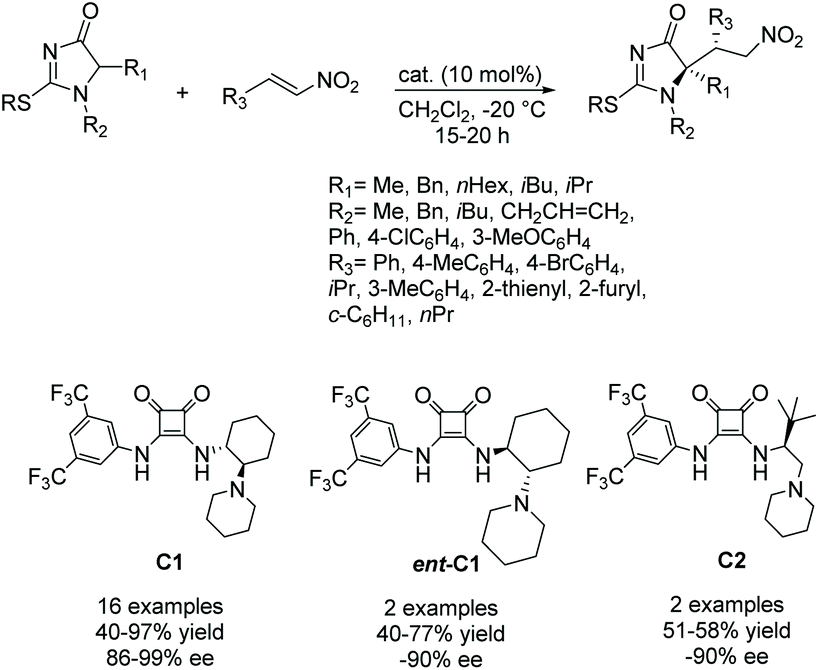
Palomo and co-workers have been working on a method to produce enantiopure 5,5-disubstituted imidazolones from 5-monosubstituted imidazolones.74–76 In their reaction, a 5-monosubstituted imidazolone reacts with an electrophile under the influence of a bifunctional Brønsted base/H-bond catalyst to create a stereoselective 5,5-disubstituted product (Scheme 22). Additionally, they used this method to produce some enantioselective bi- or tricyclic imidazol-4-ones. Since this first report, they have elaborated on the variety of groups used to enantioselectively alkylate the 5-position of the ring, including aldol products and enols. A variety of catalysts were also used, with varying success.74–76
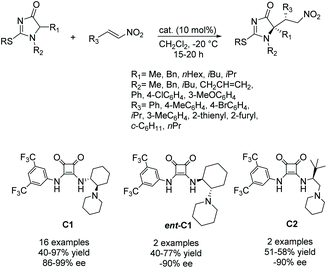 |
| | Scheme 22 Enantioselective reaction between imidazol-4-ones and nitroolefins. | |
2.3.2 Oxazolone rearrangement.
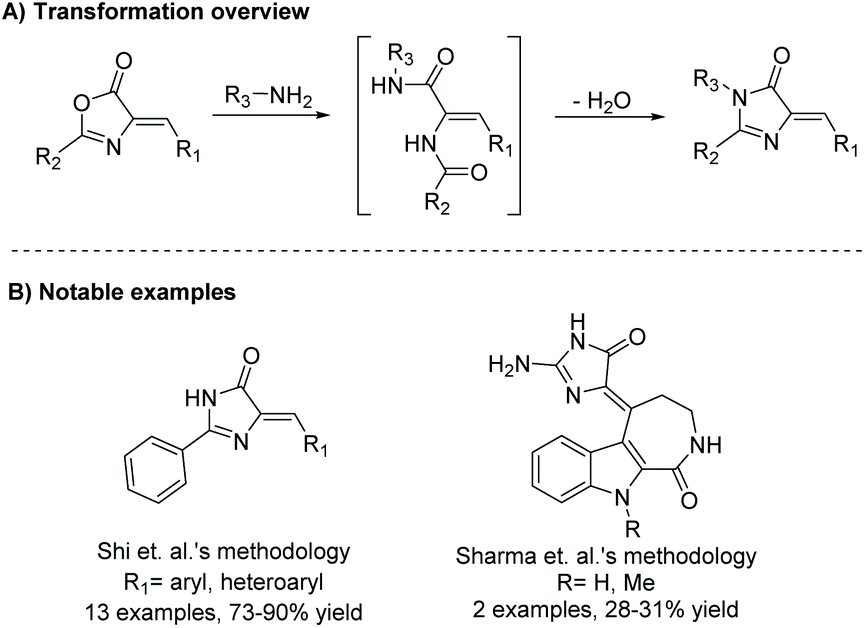
Rearrangement of an oxazolone ring upon addition of an amine can be used to produce di-substituted and tri-substituted imidazol-4-ones for a wide range of biological applications,2,77–80 total syntheses,81 and other applications.82Scheme 23 displays the basic mechanism for this reaction. The reaction is initiated by nucleophilic attack of a primary amine, which opens the oxazolone ring into a diamide. The diamide will then cyclize to form an imidazol-4-one upon loss of water. Reagents used to promote this reaction can vary, and the reaction can be run under acidic, neutral, or basic conditions; however, most reactions require heat. Some popular conditions include refluxing the amine in pyridine or ethanol or refluxing with sodium acetate in acetic acid. A wide range of amines can be used as the nucleophile for the ring opening amination, including primary aryl, alkyl, benzyl, silyl and alkenyl amines, amino acids, ammonium acetate, and hydrazines.2,3,77 Additionally, the use of benzyl carbamimidothioate has been reported to produce 2-amino-4H-imidazol-4-ones.83,84 This reaction is mainly used to produce 5-ethylidene-4-imidazolones, but has been used several times to produce 5,5-disubstituted-4-imidazolones.85,86 There have been no reports of 5-monosubstituted imidazol-4-ones being produced via this method. A slight variation of the oxazolone rearrangement is the use of a thiazol-4(5H)-one, which ring opens upon addition of a secondary amine. The intermediate will then recyclize, releasing sulfur in the process. This reaction has been reported only a few times, beginning in 2007.87–89
 |
| | Scheme 23 General transformation of oxazolone to imidazolone. | |
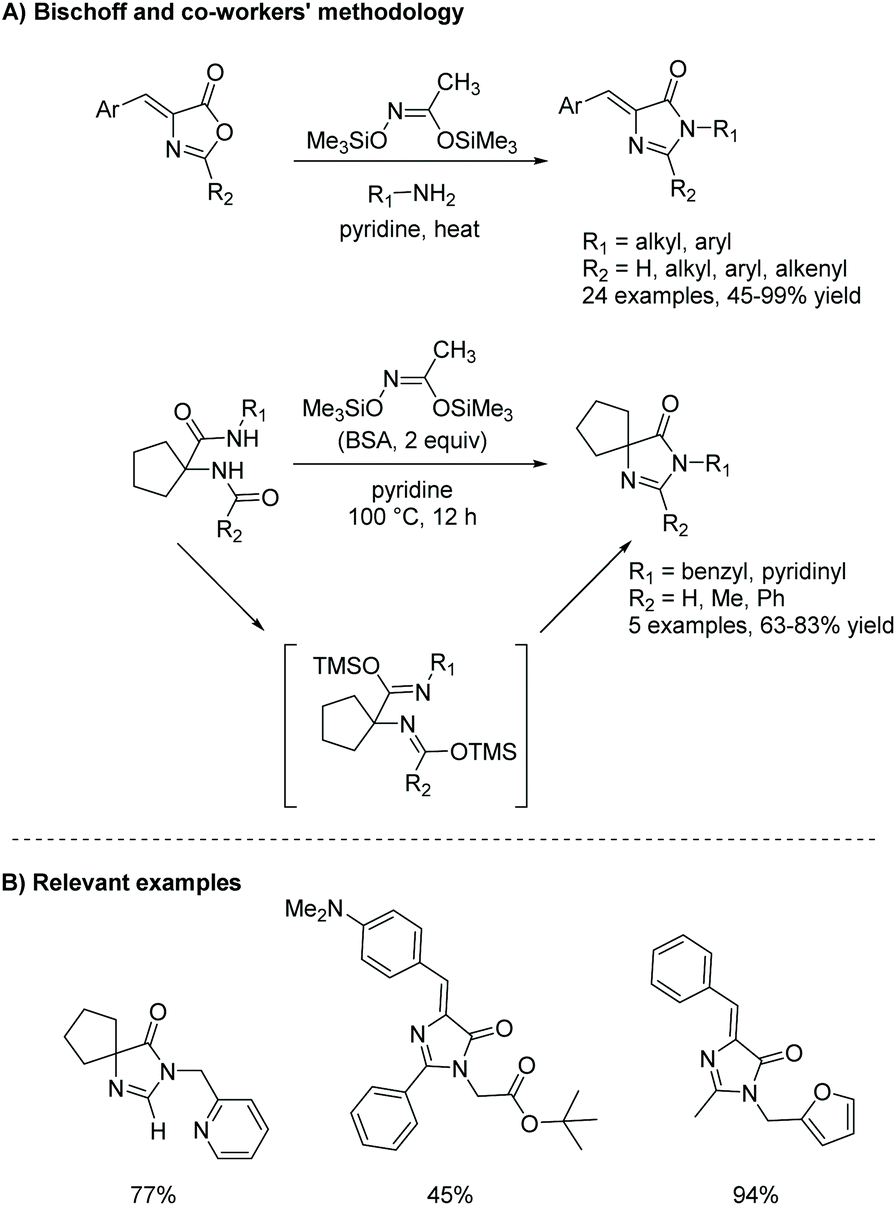
One of the downfalls of this reaction is the harsh conditions needed to convert from diamide to imidazol-4-ones, which limits this reaction, avoiding the use of sterically bulky amines. An interesting alteration to this reaction is described in an article by Bischoff and co-workers, where they used N,O-bistrimethylsilylacetamide (BSA) to promote the transformation of oxazolone to imidazol-4-one.90 In this reaction, BSA is a dehydration reagent, used to speed up the dehydration of diamide under mild conditions. This reaction proved to be a mild, one-pot method to produce a range of imidazolones (Scheme 24). BSA provided them with a large tolerance towards a variety of functional groups, including 5,5-dialkyl and 5-benzylidene imidazolones as well as 2H- and 2-substituted imidazolones, in mild to good yields (45–99%).90 Some of the highlights from this procedure include its compatibility with tert-butyl groups, formamides, activated double bonds, and N-arylamides, which are poorly reactive.
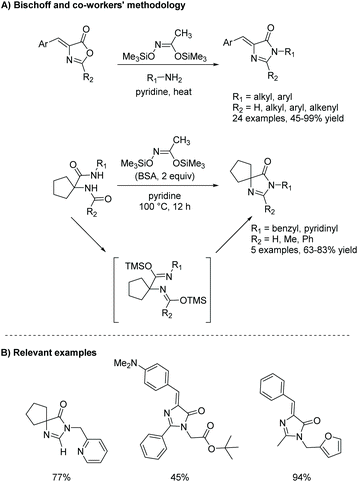 |
| | Scheme 24 Bischoff and co-workers’ work on a BSA-mediated formation of imidazolones from oxazolones. | |
2.3.3 Oxidative pinacol-like rearrangement of imidazole.
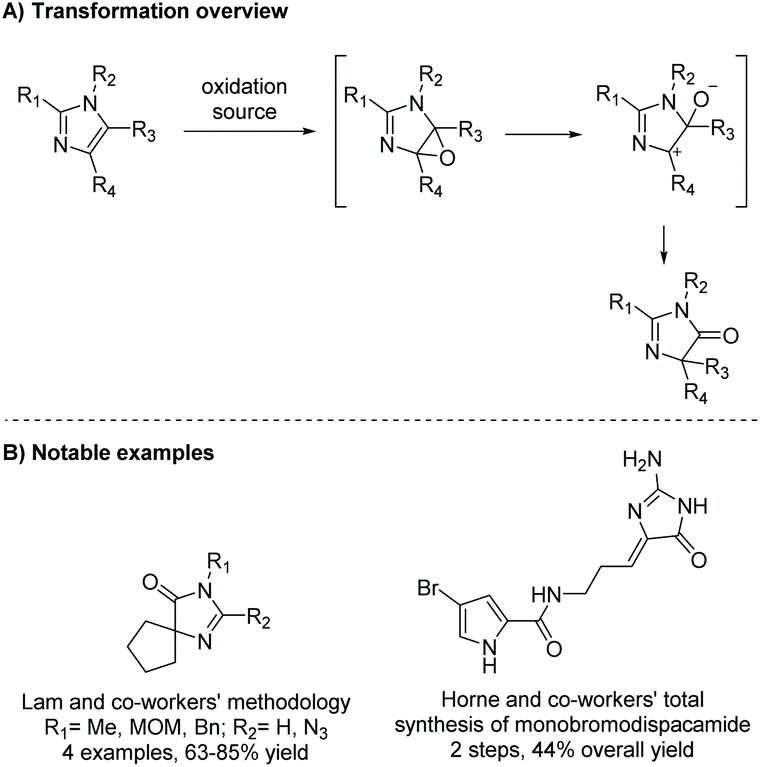
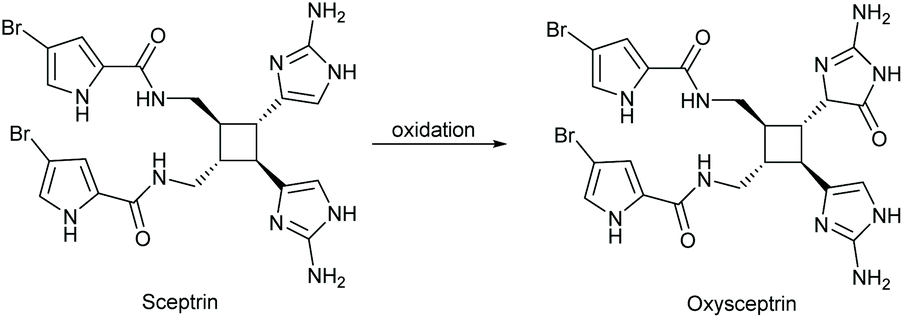
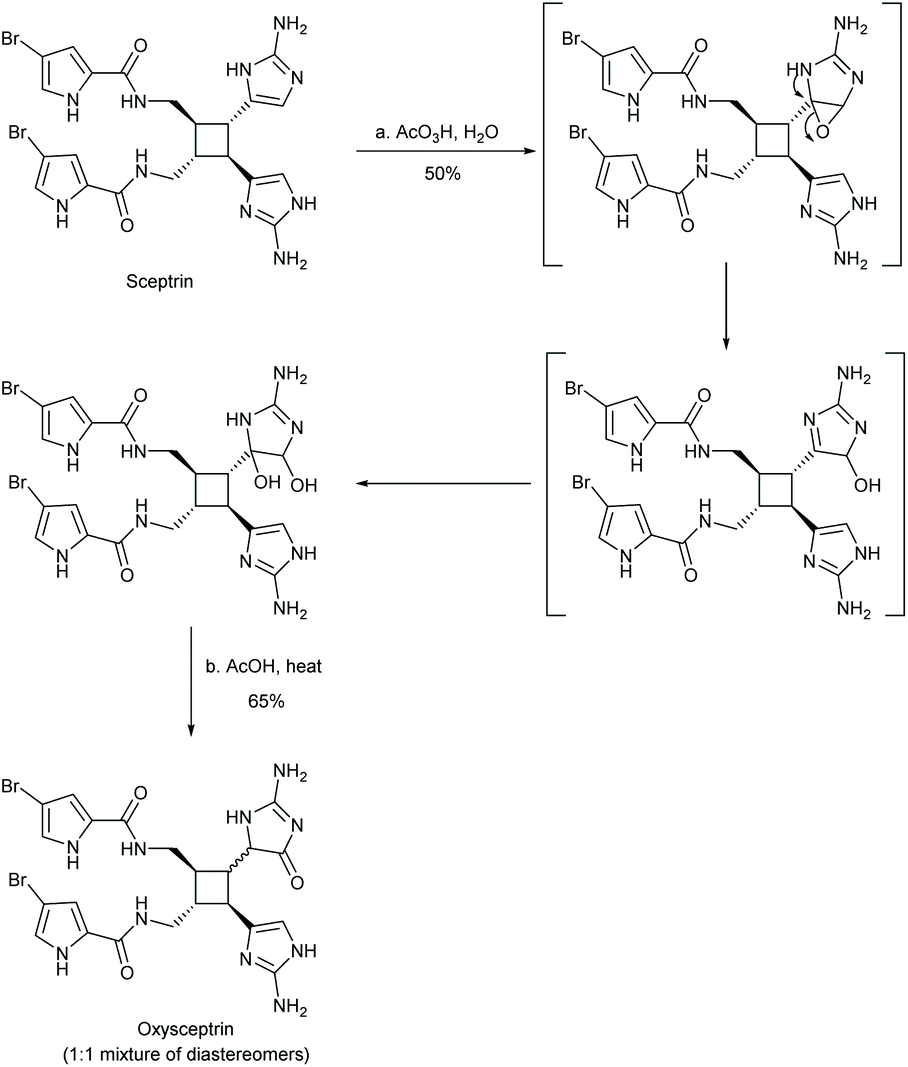
The oxidative rearrangement of imidazoles is an alternative method of producing imidazol-4-ones. In this reaction, an oxidation source, like singlet oxygen or dimethyldioxirane (DMDO), is used to epoxidate the imidazole, which can subsequently undergo a pinacol-like rearrangement to form an imidazol-4-one (Scheme 25). One of the first reports of this reaction was by Guy Rio and Bernard Serkiz in 1975, where they used molecular oxygen as an oxygen radical source.91 Since then, this reaction has been performed using a variety of oxidants, such as DMDO,92 Davis reagent,93,94 m-chloroperbenzoic acid (mCPBA),95 and other peroxides.96 This reaction has been used to produce a wide range of imidazolones for natural product total synthesis, including calcaridine A, hymenialdisine, oxysceptrin and monobromodispacamide.97–101 Additionally, there is some evidence to support the formation of enantioselective products from the pinacol-like rearrangement. Kimura and co-workers report a stereoselective 1,5-phenyl migration on several imidazolols upon addition of base in DMSO, producing 5,5-diarylated imidazol-4-ones (ee > 90%).102 In nature, the pinacol rearrangement is used to convert 2′-deoxyguanosine to a spiroimidazolone.103 This reaction was found to occur naturally through single-electron oxidation under basic conditions (pH > 8) and can also take place in acetic acid with m-chloroperbenzoic acid (mCPBA) or dimethyldioxirane (DMDO).104
 |
| | Scheme 25 General mechanism for the pinacol-like rearrangement of imidazole to imidazolone. | |
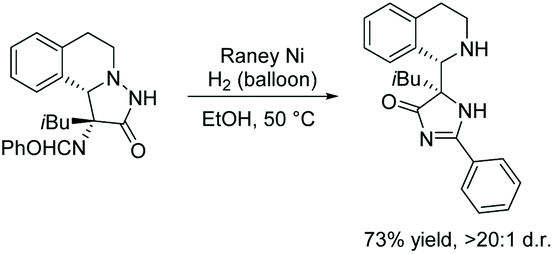
2.3.4 Rearrangement of pyrazolidin-3-one.
In 2016, Su and co-workers reported the rearrangement of pyrazolidin-3-one using RANEY® nickel and hydrogen (Scheme 26).105 RANEY® nickel and atmospheric hydrogen converts the pyrazolidine-3-one to imidazol-4-one through a reductive cyclization via cleavage of the nitrogen–nitrogen bond. Not much is reported on this reaction, as it has only been used once for the purpose of synthesizing an imidazolone, however, it has been used previously to produce an indolizidine from N-amino-3,4-dihydroisoquinoline.106,107
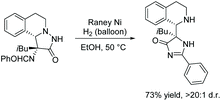 |
| | Scheme 26 Reductive rearrangement of pyrazolidine-3-one to imidazol-4-one. | |
2.3.5 Ring expansion of amino-β-lactam.
In 2015, Dražić et al. reported a base-promoted amino-β-lactam ring expansion that produced 2-amino-4-imidazolones (Scheme 27).108 In this reaction, the amino-β-lactam reacts first with thiourea. Then, upon treatment with potassium carbonate, there is an amidolysis via the N1–C2 bond, which leads to rearrangement to a 5-membered imidazol-4-one ring. It was found that they could not isolate 2-guanidine β-lactams that contained an electron withdrawing group on the guanidine nitrogen. Instead, those compounds rearranged to a 5-membered ring without addition of potassium carbonate. Presumably, the electron withdrawing groups (i.e. NO2, CN) allow for easier deprotonation, which makes triethylamine a strong enough base to deprotonate the guanidine, allowing it to react with the C2 carbon.
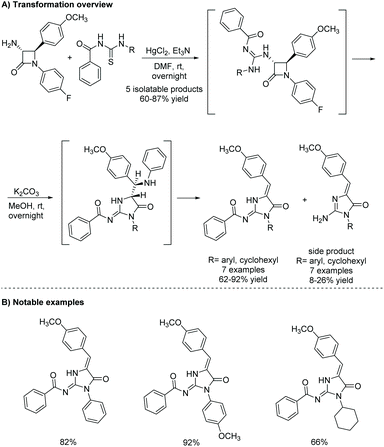 |
| | Scheme 27 Habuš and co-workers’ beta-lactam ring expansion to produce 2-amino-4-imidazolones. | |
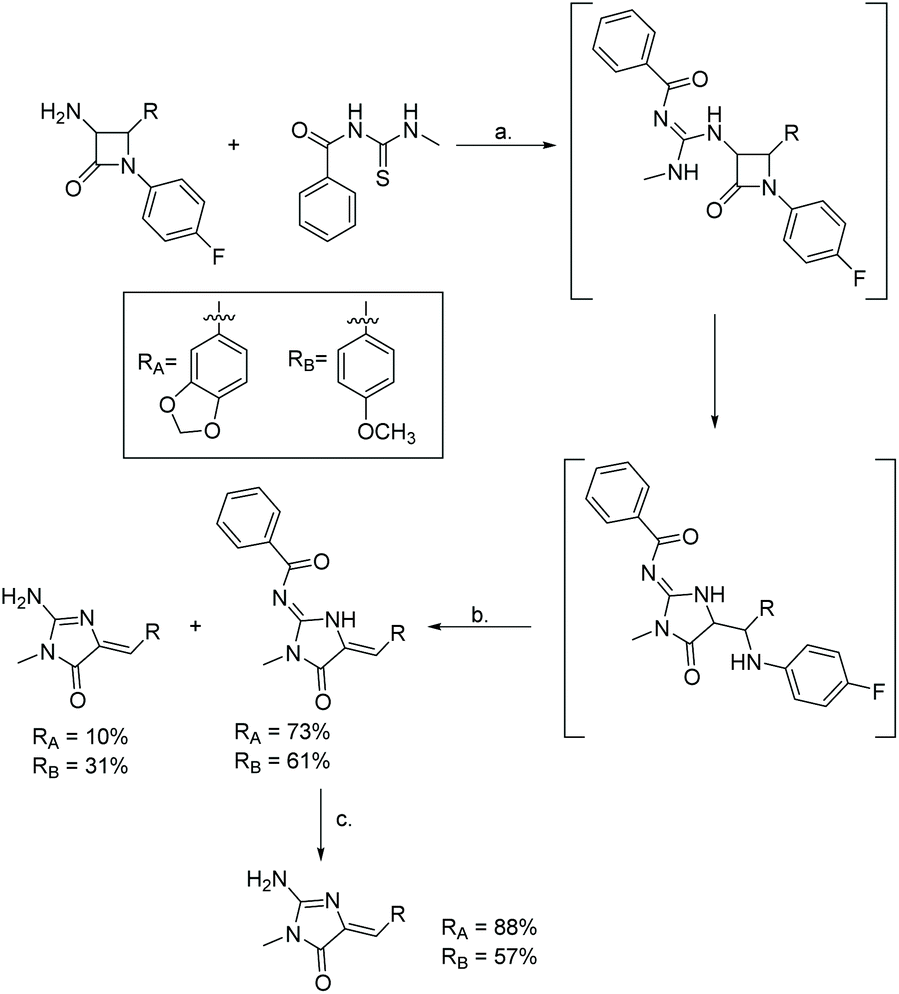
This rearrangement has been used previously to produce other heterocycles, like thiohydantoins, hydantoins, and imidazolines.109,110 In 2017, this methodology was used to produce leucettamine B and C.111
2.3.6 Boulton-Katritzky rearrangement of isoxazole.
In 2019, Cai and co-workers reported a stereoselective synthesis of (E)-5-tetrasubstituted-ylidene-3,5-dihydro-4H-imidazol-4-ones starting from isoxazoles.112 This reaction is a combination of a Michael addition reaction followed by a Boulton-Katritzky rearrangement, using a nucleophilic carbon during the rearrangement process. Several conditions were screened, including changes in the base, base equivalents, solvent, temperature, and time. Ultimately, the best conditions were reported as using potassium carbonate (0.5 equivalents) in DMSO at 120 °C for 45 minutes (81% yield). These conditions were used to produce a range of imidazol-4-ones, varying at three different positions (see Scheme 28A) in moderate to good yields (35–85%). Additionally, this reaction produced some 2-amino-4-imidazolones in good yields (71–91%) by altering the starting material (see Schemes 28B and C). The mechanism of this reaction is displayed in Scheme 28D. The E stereoselectivity is due to a hydrogen bond formation between the NH of the alkene and oxygen atom of the carbonyl group. Notably, this reaction has only been used to produce 5-ethylidene-4-imidazolones.
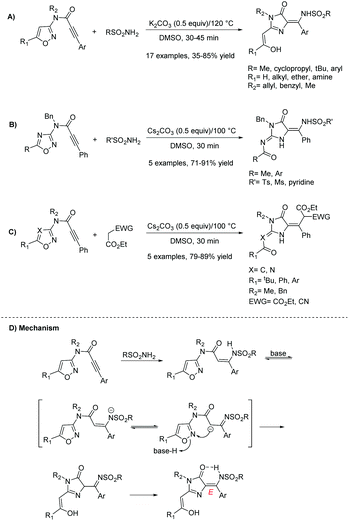 |
| | Scheme 28 Cai and co-workers’ synthesis of imidazol-4-ones via rearrangement of isoxazoles. | |
In summary, section 2 of this review aimed to highlight the known efforts towards the development of novel methodologies for the preparation of imidazol-4-ones. A number of methods were described, with examples of their usability for medicinal and other applications. Advantages and disadvantages of each method were highlighted, including the scope of the reaction, limitations to substitution patterns of the imidazol-4-ones, and abilities to produce enantioselective products.
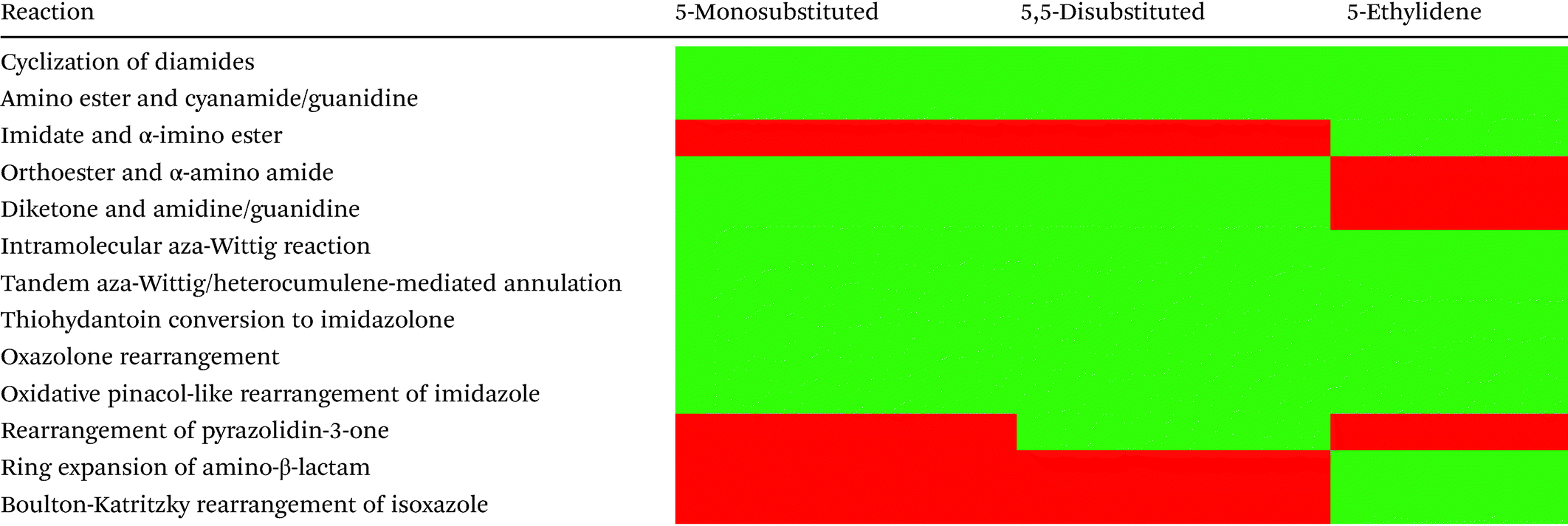
Table 1 highlights the methods discussed in this section and summarizes the substitution patterns which can be produced from each method. There are six methods which can be used to produce all three substitution patterns: cyclization of diamides, cyclization of amino ester and cyanamide/guanidine, tandem aza-Wittig/heterocumulene-mediated annulation, thiohydantoin conversion to imidazolone, oxazolone rearrangement, and oxidative pinacol-like rearrangement of imidazole. Several of the other methods were specifically designed for the synthesis of 5-ethylidene-4-imidazolones, including the cyclization of imidates and α-imino esters, ring expansion of amino-β-lactam, and the Boulton-Katritzky rearrangement of isoxazole. There are plenty of methods for the production of 5-monosubstituted and 5,5-disubstituted imidazol-4-ones, however, there is a lack of methods that produce enantiopure 5-substituted imidazol-4-ones. In fact, there is only one reaction reported that produces enantiopure 5,5-disubstitued imidazol-4-ones stereoselectively from a starting material where the stereocenter was not already in place.74–76
Table 1 Substitution patterns of imidazol-4-ones produced from referenced reactions, where green symbolizes produced scaffolds and red symbolizes scaffolds which have not been produced from that methodology
In the next section of this review, the preparation of imidazol-4-ones will be applied to a series of natural product total syntheses.
3. (4H)-Imidazol-4-one containing natural products and their total syntheses
There are many isolated (4H)-imidazol-4-one containing nat-Ural products known to date, however, they have never been summarized in one report. These natural products can be separated into three main categories: indole alkaloids, pyrrole alkaloids, and other 2-aminoimidazol-4-one alkaloids. Section 3 of this review covers all known (4H)-imidazol-4-one containing natural products, their biological activities, and efforts towards their total syntheses. Some of the methods highlighted in section 2 of this review will be used to produce these natural products.
3.1 Indole alkaloids
3.1.1 Aplysinopsin.
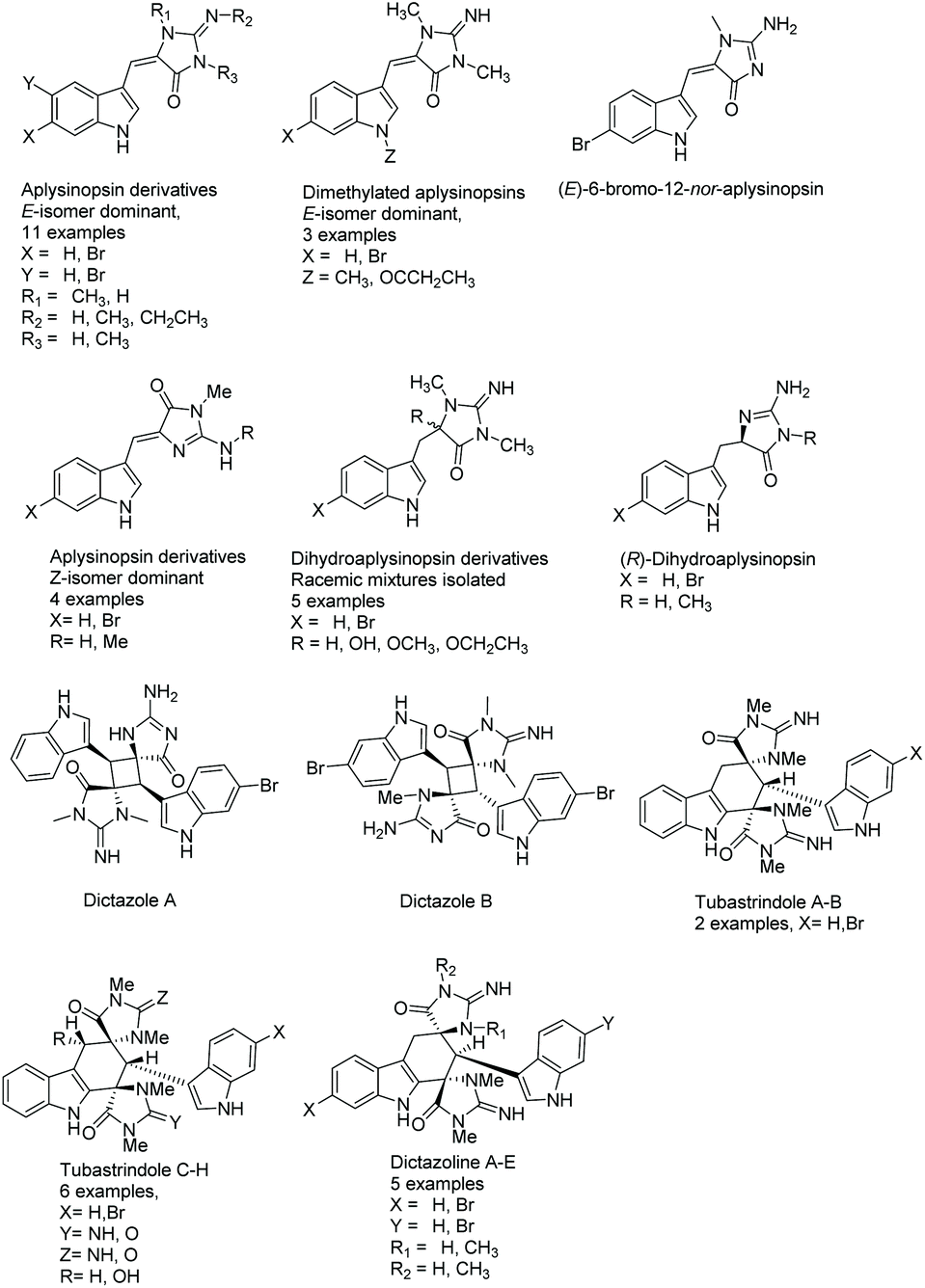
The first aplysinopsin derivatives were isolated by Kazlauskas, Rymantas, and co-workers in 1977 from the dictoyocceratid sponge Aplysinopsis.113 This family of natural products is derived from tryptophan and has been isolated from a number of different sources, including sponges and scleractinian corals.114 There are over 30 known variations of aplysinopsin, of which the imidazolone containing derivatives are displayed in Fig. 2.115–119 There are a number of hydantoin-containing aplysinopsin analogues as well, but for the purpose of this review we will only be discussing the (4H)-imidazolon-4-one containing scaffolds. The natural derivatives of aplysinopsin differ by variations in the structure of the imidazolone ring (oxidation state, position and number of N-methylations), bromination pattern of the indole, presence and absence of a double bond on the 5 position of the imidazol-4-one ring, dimerization, and the stereochemistry.
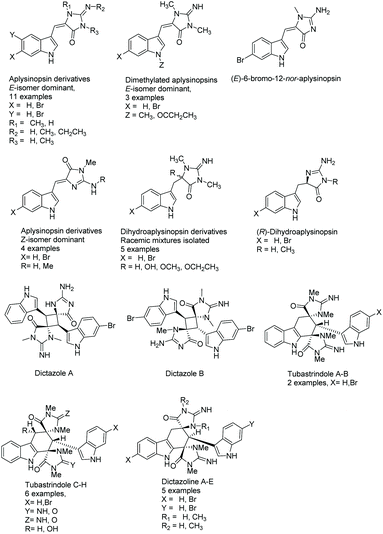 |
| | Fig. 2 (4H)-Imidazol-4-one containing derivatives of aplysinopsin. | |
Aplysinopsins are known for their range of biological activities, including anticancer, antimicrobial, and antiplasmodial activities.115 However, the most significant biological activity is modulation of neurotransmission through regulation of monoaminooxidase (MAO), serotonin receptors, and nitric oxide synthase (NOS) activities.120 Methylaplysinopsin is a potent reversible MAO inhibitor, found to decrease MAO concentrations below 1 ng mL−1 over 4–8 hours in vivo.121 6-Bromoaplysinopsin was found to have an affinity for human serotonin 5-HT2 receptors with a similar Ki as serotonin.115 5,6-dibromo-2′-demethylaplysinopsin (Z) showed 100% inhibition of nNOS at 125 μg mL−1 and 32% inhibition of iNOS.120 Most recently, tubastrindole B was found to be a glycine-gated chloride channel receptor α1 selective GlyR antagonist (IC50 = 25.9 μM with a IC50 > 300 μM for α3).122 Aplysinopsin was not found to have any inhibitory activity in comparison. GlyRs play a pivotal role in running inhibitory neurotransmission in the spinal cord, brain stem and retina. This could be beneficial in the treatment of inflammatory pain, opioid-induced breathing disorders, epilepsy, and movement disorders.122
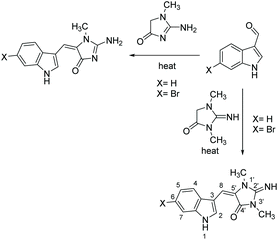
Two of the original total syntheses for (E)-aplysinopsin and its 6-bromo derivative were reported by Guella et al. (1988) and Fattorusso et al. (1985).123,124 Their syntheses are summarized in Scheme 29 and include a method to produce exo and endo products. The key step is a Knoevenagel condensation of methyl creatinine with indol-3-carboxaldehyde and its 6-bromo derivative. Additionally, Guella et al. studied the photoisomerization of the (E) isomer and found they were able to increase the amount of (Z) isomer to establish H–C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O heteronuclear coupling constants for both.124 Knowing that the (E) isomer has an appreciably larger coupling constant (10.5 Hz) than (Z) (5.1 Hz) has allowed researchers to quickly determine whether they synthesized the (E) or (Z) isomer in subsequent studies. This synthesis has since been used to make a variety of analogues,117,125 including 5,6-dibromo-2′-demethylaplysinopsin126 and several series of N-alkylated aplysinopsin derivatives used for biological testing.127–129 The conditions for this reaction vary but typically require heat and addition of a base, such as piperidine. A microwave-assisted method has been reported as well, where the reaction was microwaved for 30–60 seconds with creatinine and NaOAc.129
O heteronuclear coupling constants for both.124 Knowing that the (E) isomer has an appreciably larger coupling constant (10.5 Hz) than (Z) (5.1 Hz) has allowed researchers to quickly determine whether they synthesized the (E) or (Z) isomer in subsequent studies. This synthesis has since been used to make a variety of analogues,117,125 including 5,6-dibromo-2′-demethylaplysinopsin126 and several series of N-alkylated aplysinopsin derivatives used for biological testing.127–129 The conditions for this reaction vary but typically require heat and addition of a base, such as piperidine. A microwave-assisted method has been reported as well, where the reaction was microwaved for 30–60 seconds with creatinine and NaOAc.129
 |
| | Scheme 29 First reported total synthesis of (E)-aplysinopsin derivatives. | |
Since this first synthesis of aplysinopsin was published, many other groups have reported variations of the total synthesis of aplysinopsin and its derivatives.81,122,125,130 A particularly interesting synthesis was reported by Molina et al.53,131 The authors reported synthesizing several aplysinopsin derivatives through a tandem aza-Wittig/heterocumulene-mediated annulation (Scheme 30). Using this method, four different analogues of aplysinopsin were synthesized in moderate yields, all containing Z-isomerization. This paper was one of the first reports of synthesizing (Z)-aplysinopsin derivatives.131
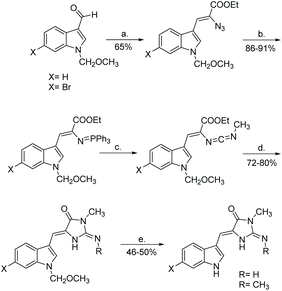 |
| | Scheme 30 Iminophosphorane-mediated ring formation to produce (Z)-aplysinopsin derivatives. Reagents: (a) N3CH2COOC2H5, NaOEt, EtOH, −15 °C, (b) PPh3, CH2Cl2, rt, (c) CH3-NCO, toluene, rt, (d) NH2R, toluene, 45 °C, (e) HCOOH, reflux. | |
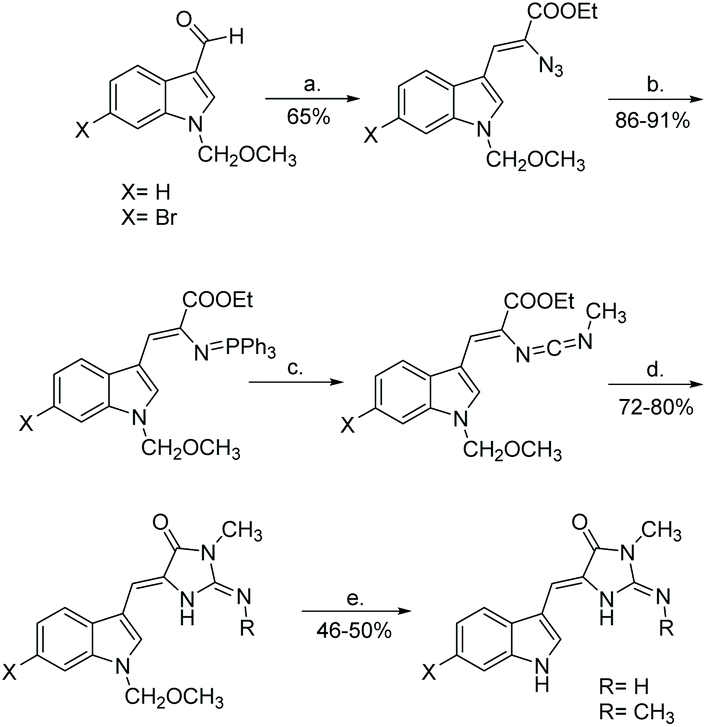
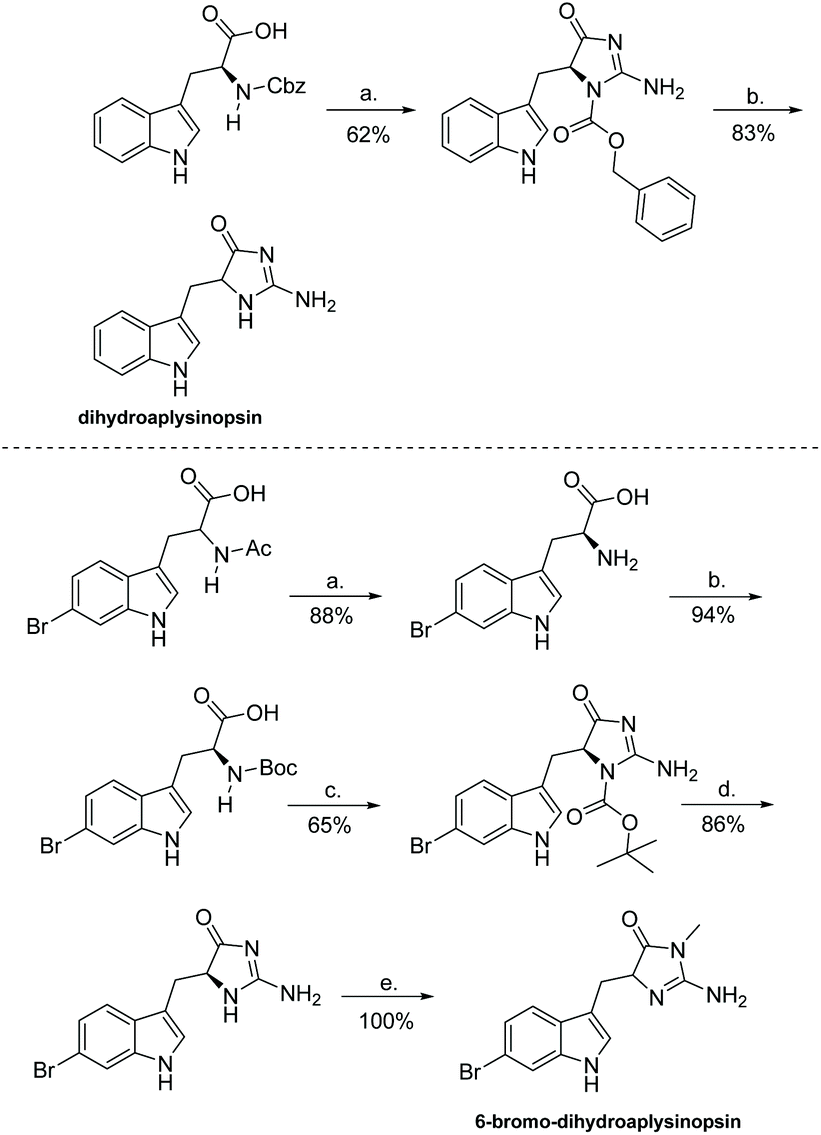
There are other, unique derivatives within the aplysinopsin family that cannot be synthesized via the same means as reported above. Examples include dihydroaplysinopsin and 6-bromo-dihydroaplysinopsin, which were isolated and synthesized in 2015 by Shaker et al. (Scheme 31).119 The formation of (4H)-imidazol-4-one involved L-tryptophan being treated with N-hydroxysuccinimide and N,N-dicyclohexylcarbodiimide to form an activated NHS-ester. This was then introduced to aqueous sodium cyanamide, which produced the protected imidazolone in good yields. Because they started with optically pure amino acids (L-tryptophan), Shaker used the configuration of the final synthesized products, which were determined as (S) even after partial racemization occurred, to determine the configuration of the natural products as (R) based on measured optical rotation.119
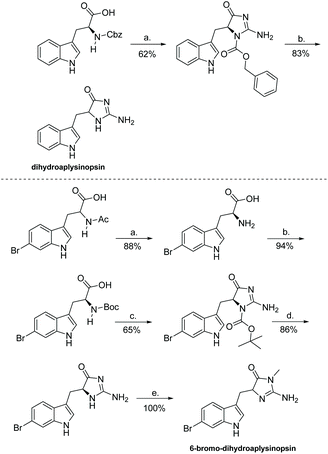 |
| | Scheme 31 Total synthesis of dihydroaplysinopsin and its 6-bromo derivative. Reagents: (a) 1. THF, DCC, NHS, 0 °C, 3 h, 2. NaHNCN, H2O, rt, 16 h, (b) MeOH, Pd/C, H2, rt, 1 h. Reagents: (a) Borate buffer, CoCl2*6H2O, pH = 8.5, L-aminoacyclase, 38 °C, 48 h, (b) 1,4-dioxane, H2O, KOH, Boc2O, rt, 16 h, (c) 1. THF, DCC, NHS, 2 h 2. NaHNCN, H2O, rt, 16 h, (d) MeCN, SnCl4, 0 °C, under Ar, 45 min, (e) MeCN, EtOH, MeI, rt, 24 h. | |
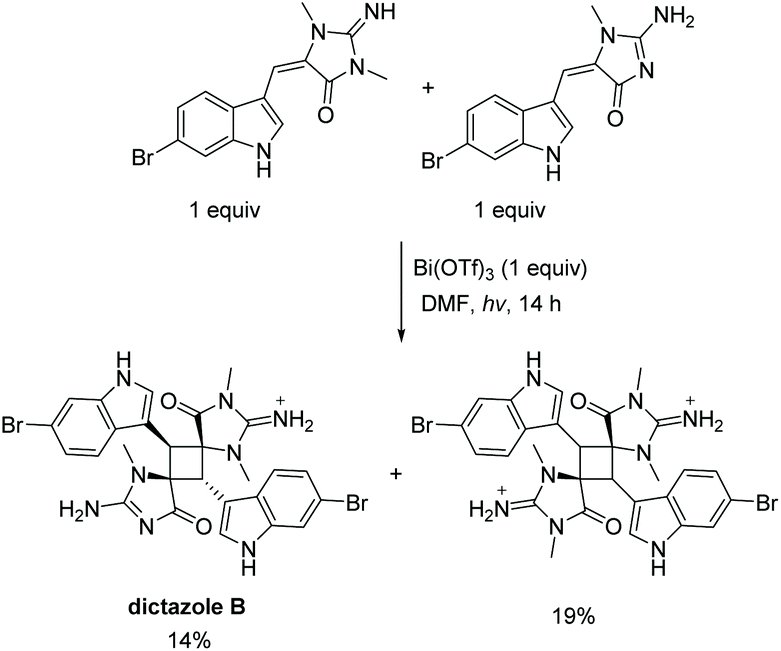
Another unique scaffold from the aplysinopsin family is dictazole. Dictazole A and B are distinctive aplysinopsins, formed through the dimerization of two aplysinopsin monomers. Several papers report on the synthesis of dictazole A and B through a [2 + 2] cycloaddition of aplysinopsin monomers. The first report of the formation of dictazole B was in 2014 by Skiredj et al.116 The authors were able to isolate 14% of dictazole B through a heterodimerization of an endo and exo monomer of 6-bromoaplysinopsin (Scheme 32). Additionally, they isolated 19% yield of the anti, head-to-tail homodimer of the exo starting material. The same group has published several other variations to this synthesis in similar yields.132,133
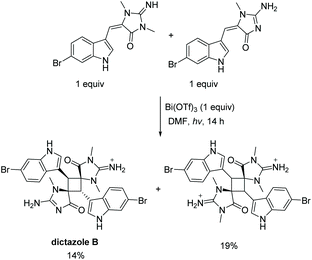 |
| | Scheme 32 The first total synthesis of dictazole B. | |
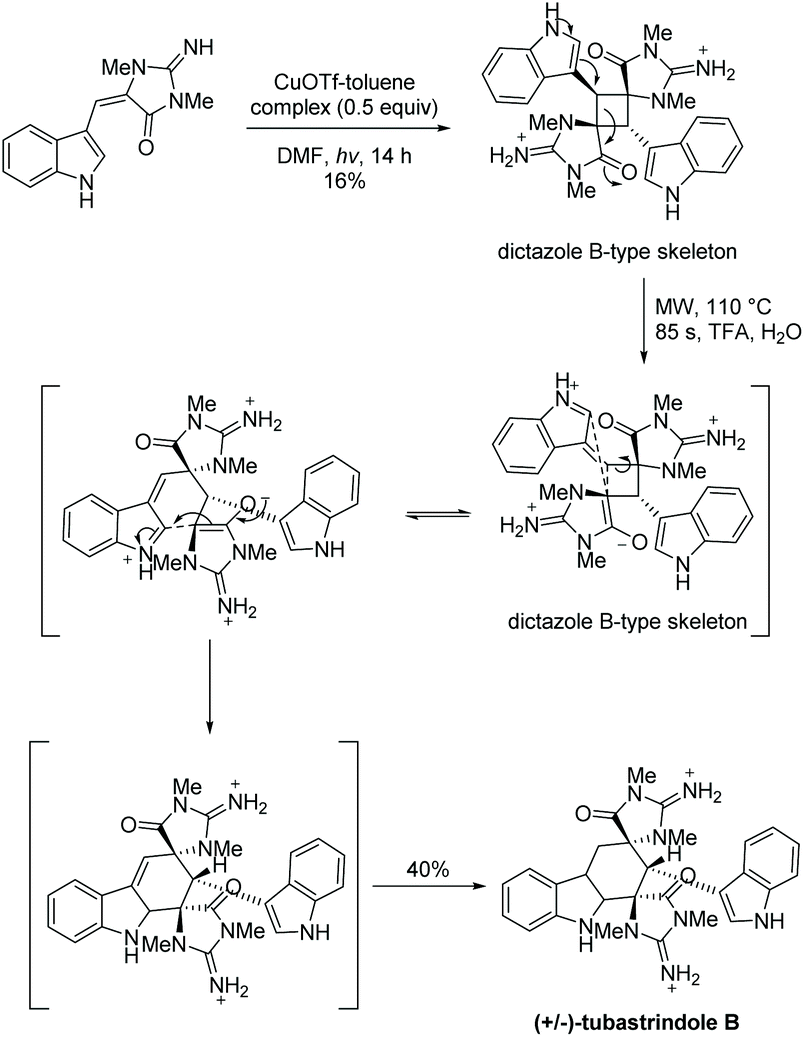
Skiredj et al. also worked on the total synthesis of tubastrindole B, the ring expanded product of dictazole B.134Scheme 33 illustrates their reported aplysinopsin cascade, where ring expansion of dictazole B occurred upon addition of heat and trifluoroacetic acid. This mimics the proposed biomimetic formation of tubastrindole A–H and dictazoline A–E.134
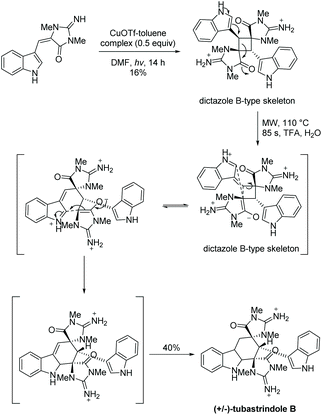 |
| | Scheme 33 The first total synthesis of tubastrindole B. | |
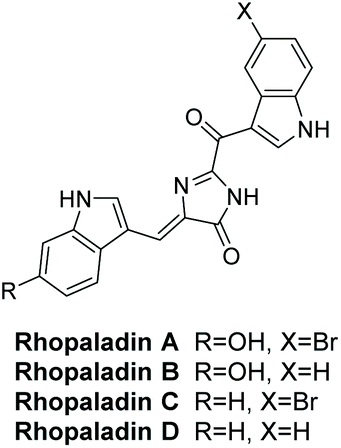
3.1.2 Rhopaladins A–D.
Rhopaladins A–D were first isolated by Sato et al. in 1998 from the marine tunicate Rhopalaea sp.135 Their structures are shown in Fig. 3. Their geometry was determined as (Z) by running a NOESY experiment on rhopaladin C. Sato et al. reported that rhopaladin C showed some antibacterial activity and rhopaladin B exhibited inhibitory activity against c-erb-2 kinase and cyclin dependent kinase 4 (IC50 = 7.4 and 12.5 μg mL−1, respectively).135
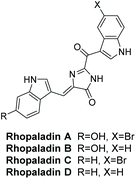 |
| | Fig. 3 Rhopaladin A–D. | |
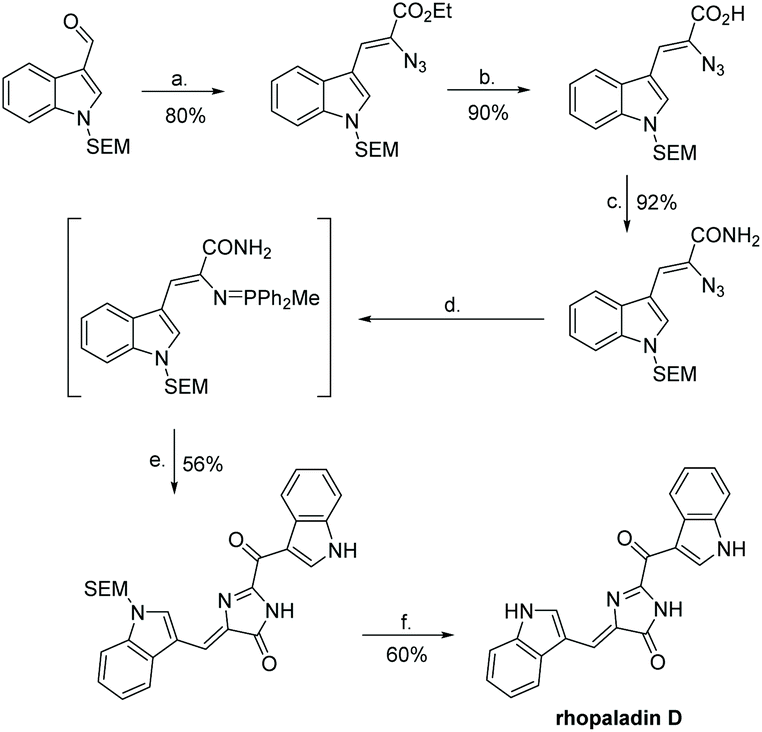
The first total synthesis of rhopaladin D was reported by Fresneda et al. in 2000 (Scheme 34).136 The key step to imidazol-4-one formation was an intermolecular aza-Wittig reaction, where the intermediate reacted with indolyl-3-glyoxylyl chloride to produce an imidoyl chloride, which cyclized to form the central imidazolone ring. This total synthesis was completed in 6 steps, with an overall yield of 19%. However, after step (e.), the product was isolated as a 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 mixture of E/Z isomers and, even after chromatographic separation, the Z isomer underwent isomerisation to E isomer upon sunlight irradiation. Thus, the rhopaladin D they synthesized was a mixture of E/Z isomers as well. The other three rhopaladin analogues have yet to succumb to a total synthesis.
:4 mixture of E/Z isomers and, even after chromatographic separation, the Z isomer underwent isomerisation to E isomer upon sunlight irradiation. Thus, the rhopaladin D they synthesized was a mixture of E/Z isomers as well. The other three rhopaladin analogues have yet to succumb to a total synthesis.
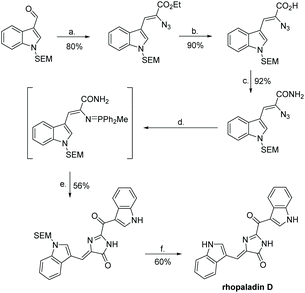 |
| | Scheme 34 First total synthesis of rhopaladin D. Reagents (a) N3CH2COOEt, NaEtO, EtOH, −15 °C; (b) LiOH, THF, H2O; (c) carbonyldiimidazole (CDI), NH3, DMF; (d) PPh2Me, THF; (e) indolyl-3-glyoxylyl chloride, polymer-bound BEMP, THF; (f) TBAF, THF, reflux. | |
3.1.3 Nortopsentine D.
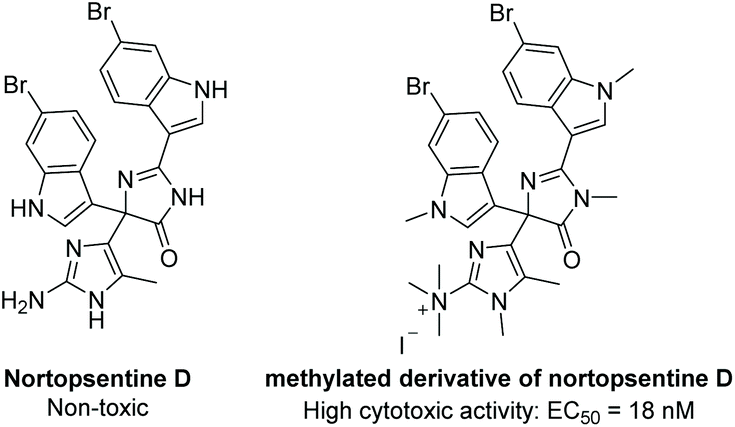
Nortopsentine D was isolated in 1996 from the axinellid sponge Dragmacidon sp.137 It is part of a family of imidazolediylbis(indole) alkaloids. While most of this family is comprised of imidazole linkers, nortopsentine D contains a (4H)-imidazol-4-one as its core scaffold (Fig. 4). This natural product was tested for its cytotoxicity against KB tumor cells, antibacterial activity (S. Aureus), and antifungal activity (Candida albicans), but it proved inactive for all three. However, when methylated (Fig. 4), nortopsentine D proved to be highly cytotoxic, with an EC50 = 0.014 μg mL−1, or 18 nM.137 The total synthesis of nortopsentine D has yet to be reported.
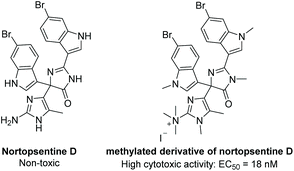 |
| | Fig. 4 Structures of nortopsentine D and its methylated derivative. | |
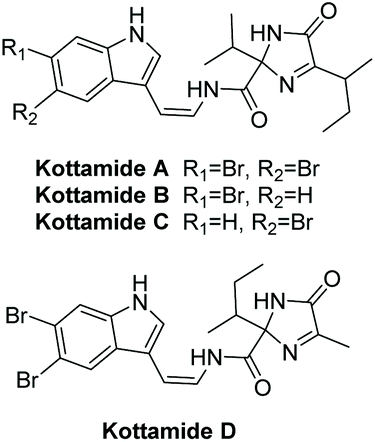
3.1.4 Kottamides A–D.
Kottamides A–D are 2,2,5-trisubstituted (4H)-imidazol-4-one alkaloids isolated in 2002 from the New Zealand ascidian Pycnoclavella kottae (Fig. 5).138 The stereochemistry of C5 was never deduced for kottamides A–D, however they were able to confirm the Z-configuration of the enamide using JHH measurements. Kottamides A–D were found to have a range of biological activities, including anti-inflammatory and anti-metabolic, as well as cytotoxicity towards several tumor cell lines. Kottamide D was found to have potent anti-metabolic activity in cells using MTT assays (IC50 = 6–10 μM).138 Kottamides A–D were also assayed for cytotoxic and antimicrobial properties. All four were found to have moderate activity against P388 cells. Kottamide A was tested for cytotoxicity/antiviral activity against the African Green Monkey kidney cell line (BSC-1) infected with the RNA virus PV110. It was found to have some antiviral activity (zone size 1–2 mm) and moderate cytotoxicity (zone size > 4.5 mm, 240 μg loading).138 The total synthesis of kottamide A–D has yet to be elucidated.
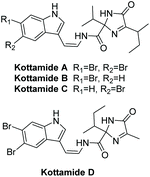 |
| | Fig. 5 Kottamides A–D. | |
3.1.5 Other indole alkaloids.
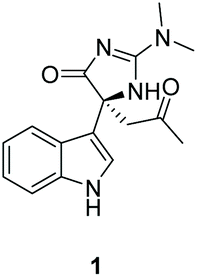
An unnamed (4H)-imidazol-4-one containing indole alkaloid natural product was isolated from the marine tunicate Dendrodoa grossularia in 1998 by Riche and coworkers (Fig. 6).139 They were able to identify the relative stereochemistry of compound 1 by measuring the optical rotation and isolating a single-crystal X-ray structure of the compound.
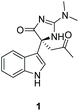 |
| | Fig. 6 Indole alkaloid isolated from D. grossularia. | |
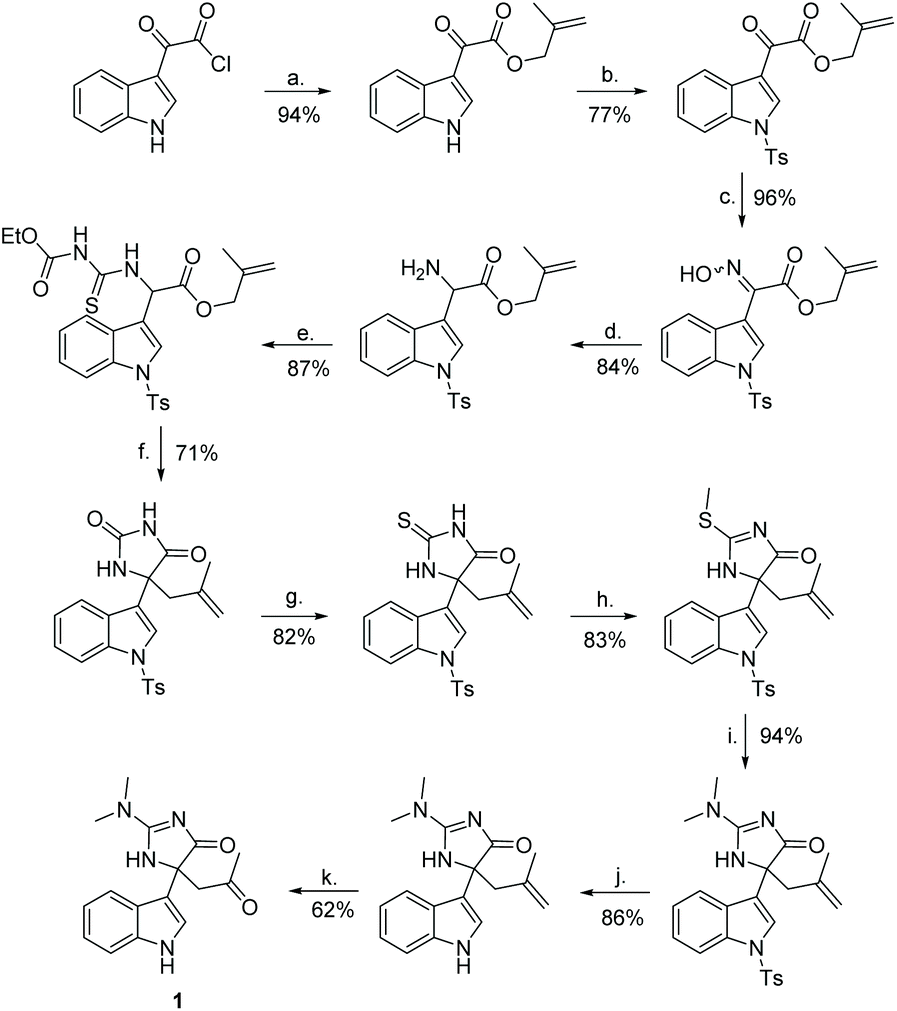
The first total synthesis of compound 1 was completed by Hupp and Tepe in 2008 (Scheme 35).140,141 In this synthesis, the quaternary carbon was formed via a oxazole rearrangement reaction, which produced an hydantoin product. The hydantoin was then converted into a thiohydantoin, which was reacted to form a 2-aminoimidazolone through standard conditions. In total, this total synthesis took 14 steps and had an overall yield of 12%. One thing to note with this total synthesis is the product was a racemate of compound 1.
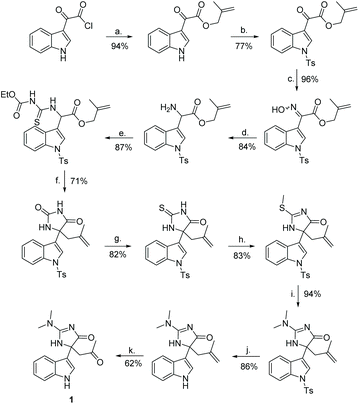 |
| | Scheme 35 The total synthesis of indole alkaloid 1. Reagents (a) 2-methylprop-2en-1-ol, ethyl acetate, rt, 16 h; (b) TsCl, DMAP, DIPEA, Ch2Cl2, rt, 16 h; (c) NH2OH*HCl, dioxane/H2O, pyridine, reflux, 16 h; (d) Zn, AOH, 0 °C, 2 h; (e) O-ethyl carbonisothiocyanatidate, CH2Cl2, rt, 16 h; (f) 1. EDCl, Et3N, CH2Cl2, 0 °C to reflux, 9 h, 2. NaOMe, MeOH, rt, 4 h, 3. HCl (aq.), 5 min; (g) Lawesson's reagent, toluene, reflux, 24 h; (h) MeI, DMAP, DIPEA, CH2Cl2, rt, 2 h; (i) dimethylamine in THF, sealed tube, 75 °C, 4 h; (j) KOEt, EtOH, reflux, 24 h; (k) 1. OsO4, NMO, rt, 4 h then 2. NalO4, 0 °C, 2 h. | |
3.2 Pyrrole alkaloids
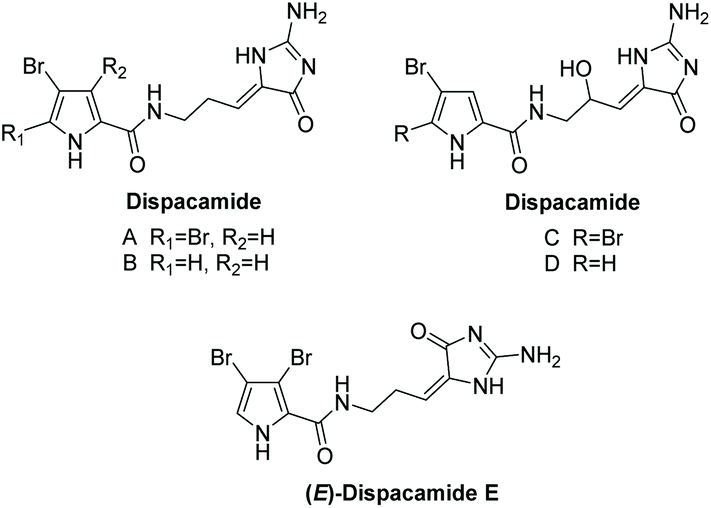
3.2.1 Dispacamide.
Dispacamide and its mono-brominated derivative were first isolated by Cafieri et al. in 1996 from four Caribbean Agelas sponges.142 Cafieri et al. later isolated dispacamides C and D, which are racemic mixtures of the 9-hydroxyl derivatives of dispacamides A and B, in 1997 from the same sponges.143 In 2014, dispacamide E was isolated by Ebada et al. from two Indonesian Stylissa sponges.144 These natural products are known precursors to the oroidin family and their cyclized derivatives, like hymenialdisine.145 Their structures are displayed in Fig. 7. Dispacamides have proven to have a range of useful biological activities, including antihistamine and antimalarial activity. Dispacamides C and D have been found to have impressive antihistamine activity when tested on isolated Guinea pig ileum; with just 1 μM, the response to histamine was almost completely abolished.143 Moreover, dispacamide B was found to have potent antiplasmodial activity (IC50 = 1.34 μg mL−1) against the multiple-drug resistant strain of P. falciparum, while being devoid of any cytotoxicity towards rat myoblast cells at 90 μg mL−1.146
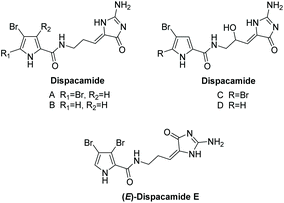 |
| | Fig. 7 Dispacamide derivatives. | |
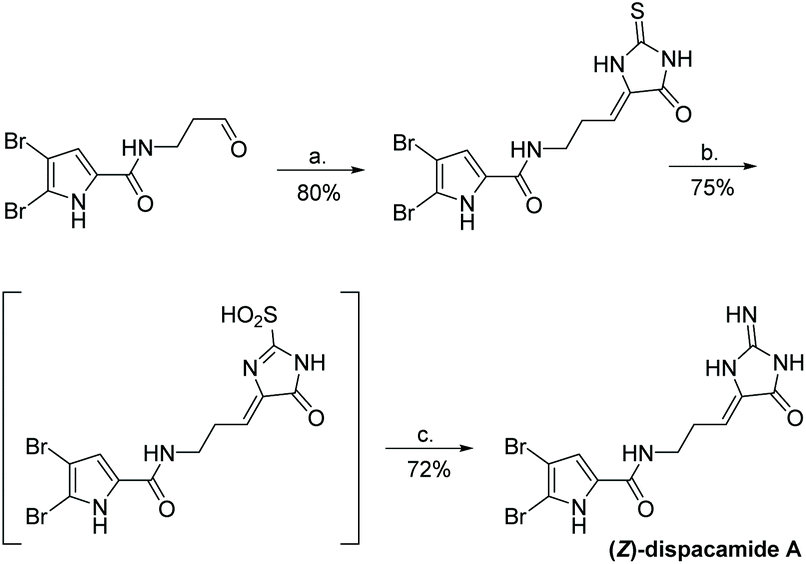
The first total synthesis of dispacamide A was reported in 1997 by Lindel et al.63 In this synthesis, the authors coupled an aliphatic aldehyde with thiohydantoin using piperidine to provide stereochemically pure (Z)-alkylidene thiohydantoin. This was then S-methylated and converted to a 2-amino-4-imidazolone upon treatment with heat and NH3/NH4Cl in methanol. They also reported an attempt to directly coupling creatinine with an aliphatic aldehyde, which was unsuccessful. The full synthesis is shown in Scheme 36.
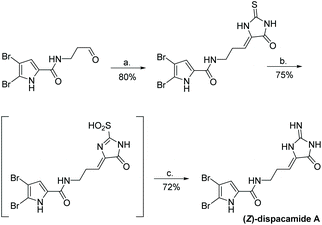 |
| | Scheme 36 The first total synthesis of dispacamide A. Reagents (a) piperidine, EtOH/H2O (8:2), rt, 4 h, (b) aq. 25% NH3, aq. 70% TBHP, MeOH, rt, 12 h. | |
Dispacamide B was first synthesized a year later, in 1998, by Olofson et al. (Scheme 37).99 In this synthesis, the key step is the oxidation of imidazole to imidazol-4-one using bromine and DMSO. The full synthesis, shown in Scheme 37, was performed in two steps and had an overall yield of 44%. Ando et al. has reported similar syntheses, using tetra-n-butylammonium tribromide-DMSO to convert the 2-aminoimidazole to 2-amino-4-imidazolone.147,148
 |
| | Scheme 37 The first total synthesis of monobromo-dispacamide. Reagents: (a) Br2, DMSO, rt, (b) 4-bromo-2-(trichloroacetyl)pyrrole, DMF, rt. | |
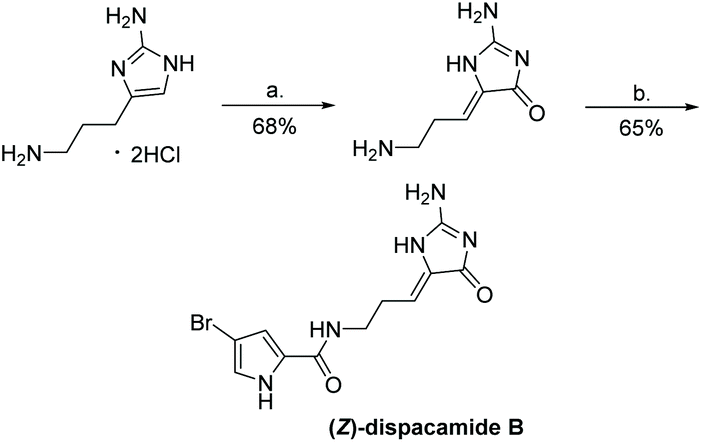
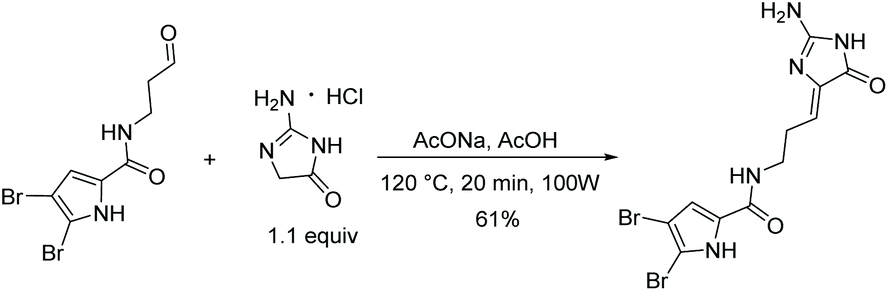
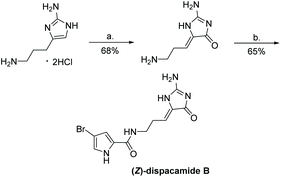
The most recently reported total synthesis of dispacamide A was in 2012, where the authors directly condensed creatinine with an alkyl aldehyde under acidic conditions with heat to complete their total synthesis in 61% yield.149 This reaction accomplished what Lindel et al. unsuccessfully attempted in 1997 by using a different set of conditions.63 This one step, microwave-assisted procedure is shown in Scheme 38.
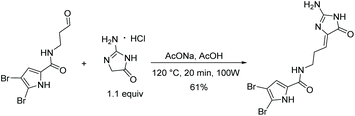 |
| | Scheme 38 Microwave-assisted total synthesis of dispacamide A. | |
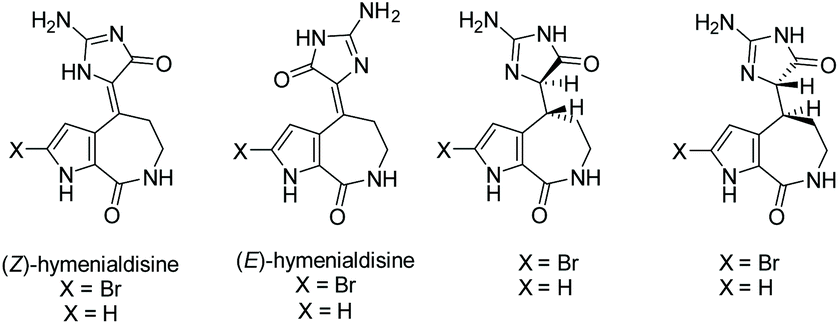
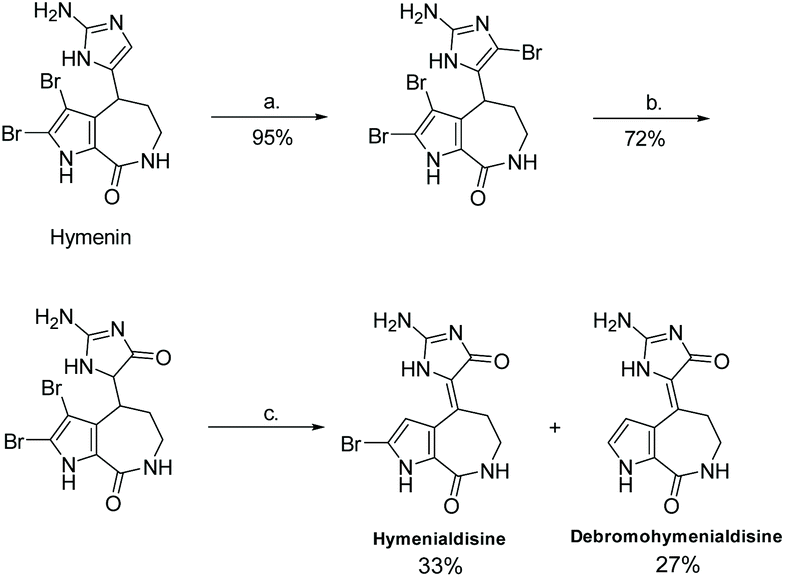
3.2.2 Hymenialdisine.
Hymenialdisine was first isolated in 1981 by C. A. Mattia et al. from the sponge Acanthella aurantiaca.150 Since then, a few other derivatives of hymenialdisine have been isolated from various marine sponges (see Fig. 8).151–153 Hymenialdisine has been found to have a number of different biological activities, most notable as a nM kinase inhibitor (by competing with ATP for binding the kinases), resulting in inhibition of the pro-inflammatory transcription factor, NF-κB.84,154–156 Additionally, derivatives of hymenialdisine were found to be highly potent inhibitors of checkpoint kinase II (IC50 = 8 nM).83,157–159 Moreover, it has been found to have some antitumor and anti-inflammatory activity.160
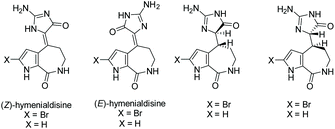 |
| | Fig. 8 Isolated derivatives of hymenialdisine. | |
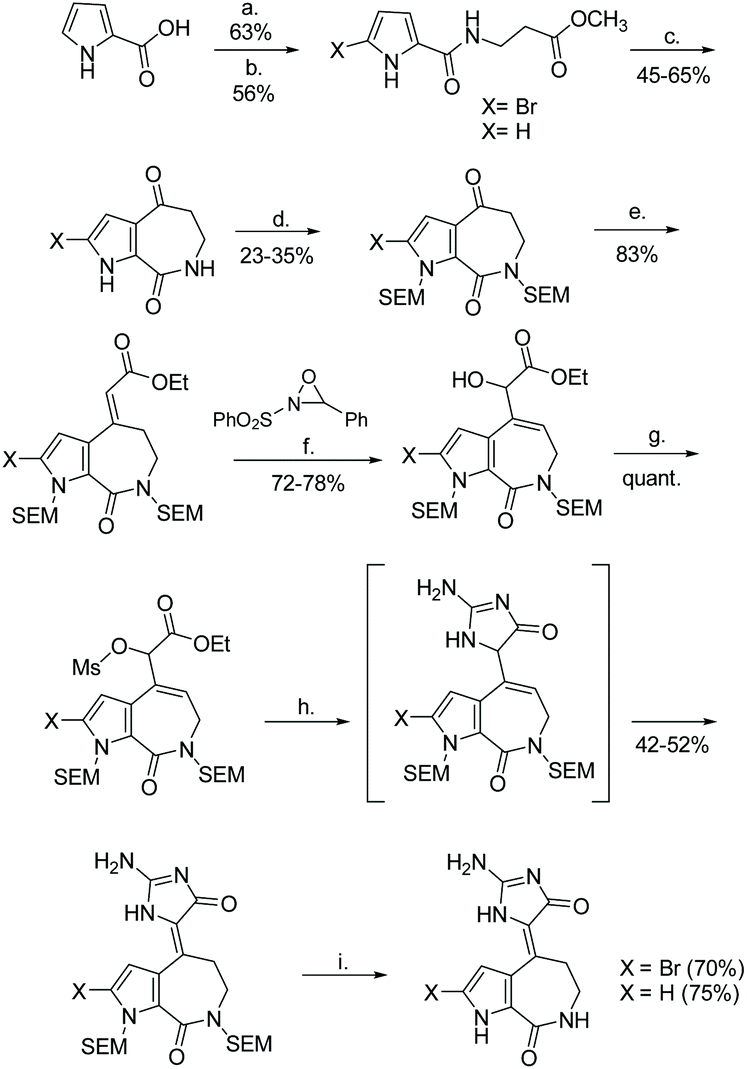
The first total synthesis of (Z)-hymenialdisine and (Z)-debromo-hymenialdisine was reported in 1995 by Annoura and Tatsuoka.23 This synthesis required 9 steps, starting from a 2-carboxyl-pyrolle, and had an overall yield of 1.5%. The imidazolone formation step took a α-ether-ester and cyclized it with guanidine to produce an imidazol-4-one (Scheme 39).
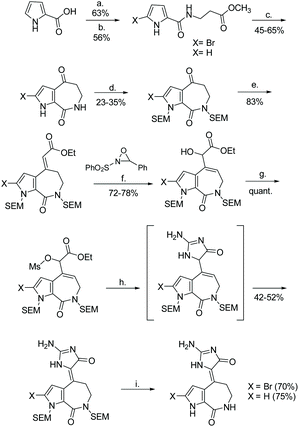 |
| | Scheme 39 The first total synthesis of hymenialdisine and debromohymenialdisine by Annoura and Tatsuoka. Reagents (a) SOCl2, cat. DMF, toluene, 60 °C, 1 h, then H2NCH2CH2COOMe, Et3N, CH2Cl2, rt, 3 h; (b) NBS, THF, rt, 2 h; (c) 10% aq. NaOH-MeOH (2:1), rt, 5 h, then PPA-P2O5, 100 °C, 1 h; (d) NaH (2 eq.), SEMCl (2 eq.), DMF, rt, 2 h; (e) (EtO)2POCH2COOEt, NaH, DME, 50 °C, 24 h; (f) KHMDS, THF, −78 °C, 2 h (g) MsCl, Et3N, CH2Cl2, 0 °C; (h) guanidine, DMF, 50 °C, 5 h; (i) 5% aq. HCl-MeOH (1:1), 80 °C, 2 h. | |
In 1997, Horne and co-workers reported a different synthesis of (Z)-hymenialdisine and (Z)-debromo-hymenialdisine, where the formation of the imidazolone ring came from the oxidation of 2-amino-4-bromoimidazole under acidic conditions with heat (Scheme 40).100 This reaction took only three steps and had an overall yield of 23% and 18%, respectively.
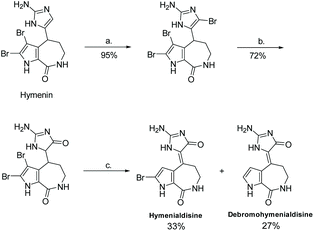 |
| | Scheme 40 Synthesis of hymenialdisine and debromohymenialdisine through the oxidation of 2-amino-4-bromoimidazole. Reagents (a) Br2, TFA, rt; (b) AcOH/H2O, reflux (c) CH3SO3H, HBr (cat.), 90 °C, sealedtube, 12 h. | |
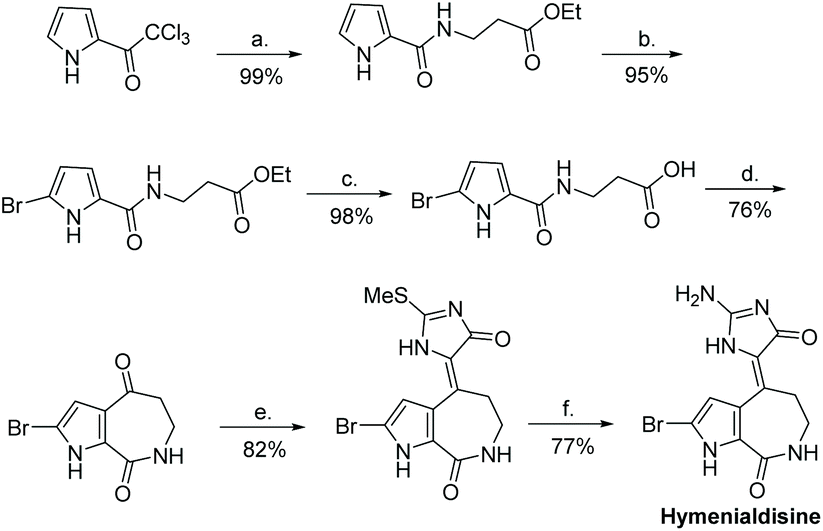
In recent years, Saleem and Tepe reported a new total synthesis for hymenialdisine and its de-brominated derivative, illustrated in Scheme 41.161 In this synthesis, 2-(methylthio)-1,5-dihydro-4H-imidazol-4-one was condensed with 2-bromoaldisine. The methylthio group was converted to an amino substituent via reaction with ammonium hydroxide. This total synthesis took 6 steps and had an overall yield of 44%. Another paper used a very similar condensation reaction between 2-methylthiol-imidazol-4-one and 2-bromoaldisine, at somewhat lower yields.162 The (E)-hymenialdisine and dihydrohymenialdisine derivatives have yet to be synthesized.
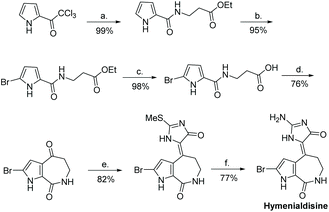 |
| | Scheme 41 An efficient total synthesis of hymenialdisine. (a) H2NCH2CH2COOEt-HCl, Et3N, DCM, rt; (b) DBDMH, MeOH/THF, −78 °C-rt; (c) LiOH or KOH, EtOH, H2O, rt, 18 h; (d) P2O5, MeSO3H, 110 °C, 2 h; (e) 2-(methylthio)-1,5-dihydro-4H-imodazol-4-one, TiCl4, py., THF, −10 °C-rt; (f) NH4OH, THF, sealed tube, 110 °C. | |
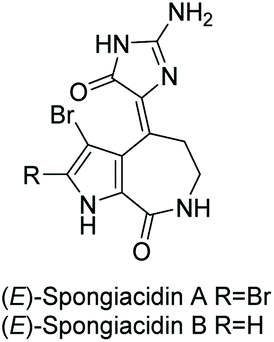
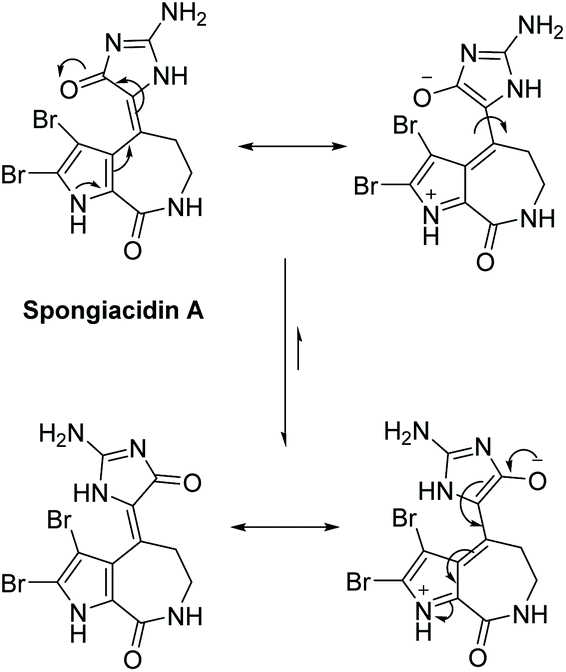
3.2.3 Spongiacidins A and B.
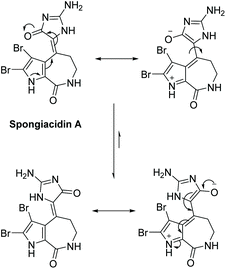
Spongiacidins A and B were isolated by Inaba et al. in 1998 from the sponge Hymeniacidon.163 Their structures are shown in Fig. 9. This class of natural products is part of the hymenialdisine family, differing by their bromo substitution pattern and alkene stereochemistry. Spongiacidin A was identified as being cytotoxic to L5178Y and HCT116 cancer cell lines. It was also found to be a protein kinase inhibitor.164 Unlike hymenialdisine, there have been no reports of the total synthesis of spongiacidins A or B. This may be in part due to the spontaneous conversion of spongiacidin's (E)-isomer to the (Z)-isomer, driven by the reduction of steric strain upon conversion.165 The proposed isomerization is shown in Scheme 42. The steric strain exerted between the C-3 bromine and oxygen on C-15 prevents a straightforward synthesis of the (E)-isomer required to produce spongiacidin.
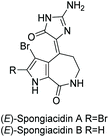 |
| | Fig. 9 Spongiacidin A and B. | |
 |
| | Scheme 42 Isomerization of (E)-spongiacidin A to the (Z)-isomer. | |
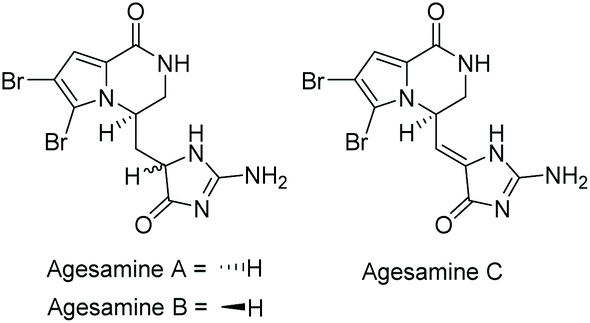
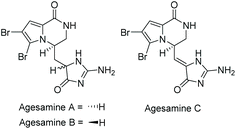
3.2.4 Agesamines A–C.
Agesamine A and B were isolated by Katsuki et al. in 2019 from the sponge Agelas sp.166 Agesamine C was isolated by Kovalerchik et al. in 2020 from the sponge Agelas oriodes.167 Their structures are illustrated in Fig. 10. Agesamine A and B were found to have some cytotoxicity towards HeLa cells, and although the synthesis of the related hydantoin analogue has been reported,168,169 the total syntheses of agesamines A–C have yet to be completed.
 |
| | Fig. 10 Agesamine derivatives. | |
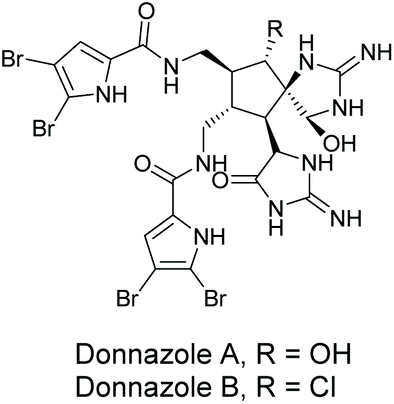
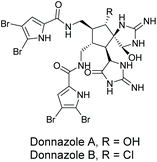
3.2.5 Donnazoles A and B.
Donnazoles A and B were isolated by Al-Mourabit and co-workers in 2012 from the marine sponge Axinella donnani (Fig. 11).170 These natural products are dimeric members of the same class of pyrrole-aminoimidazole (PAI) alkaloids as oroidin, sceptrin, massadine, and palau'amine. The structure's absolute configuration was determined by comparison of their circular dichroism (CD) with sceptrin. To date, there has been no biological activity reported for donnazoles A and B. While some efforts have been made towards developing the methodology needed to produce donnazoles A and B,171 they have yet to be synthesized.
 |
| | Fig. 11 Donnazoles A and B. | |
3.3 Other 2-amino-(4H)-imidazol-4-one alkaloids
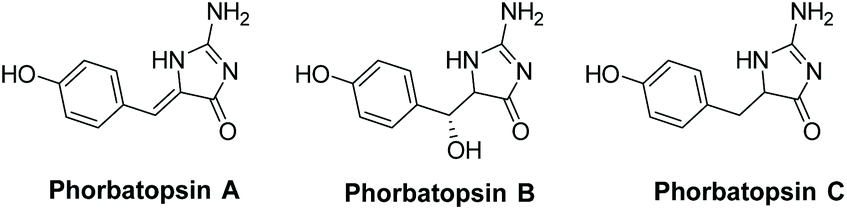
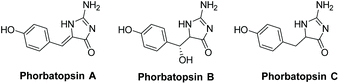
3.3.1 Phorbatopsins A–C.
Phorbatopsins A–C were isolated by Nguyen et al. in 2012 from the Mediterranean sponge Phorbas topsenti (Fig. 13).174 One biological application for these compounds is their antioxidant activity, which was tested using the Oxygen Radical Absorbance Capacity (ORAC) assay. All were shown to have some activity, with phorbatopsin A being most active, having an ORAC value of 0.88 (which is comparable to the positive control Trolox's ORAC value of 1). Derivatives of phorbatopsin A were also tested for antitumor activity, with several derivatives having over 90% inhibition at a concentration of 50 μM.72
 |
| | Fig. 13 Isolated phorbatopsin analogues. | |
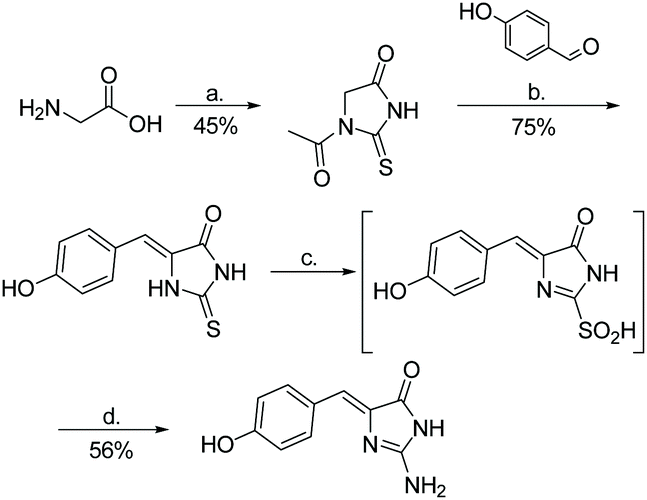
The first total synthesis of phorbatopsin A was reported in 2013 (Scheme 44).72 In this synthesis, glycine was treated with NH4SCN under acidic conditions. The formed thiohydantoin was then condensed with 4-hydroxybenzaldehyde and converted to a 2-aminoimidazolone using TBHP and ammonia. Phorbatopsin A was produced in four steps with an overall yield of 19%. Phorbatopsins B and C have yet to be synthesized.
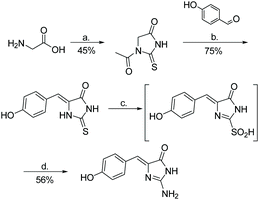 |
| | Scheme 44 The first total synthesis of phorbatopsin A. Reagents (a) NH4SCN, Ac2O, AcOH, reflux, 2 h; (b) NaOAc, AcOH, reflux, 5 h; (c) TBHP, MeOH, rt, 2 h; (d) NH3, MeOH, rt, 10 h. | |
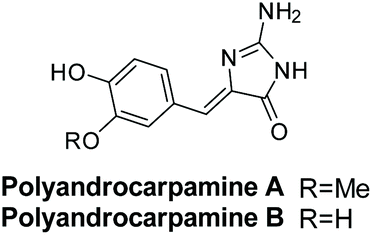
3.3.2 Polyandrocarpamines A and B.
Polyandrocarpamines A and B were isolated by Davis et al. in 2002 from the Fijian ascidian Polyandrocarpa sp.175 Their structures are shown in Fig. 14. Polyandrocarpamine A was found to have selective cytotoxicity against the CNS cell line SF 268 with a GI value of 65 μM.176 Then, in 2017, both derivatives were found to inhibit mammalian and protozoan DYRK and CLK kinases.177
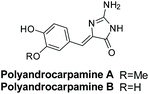 |
| | Fig. 14 Polyandrocarpamine A and B. | |
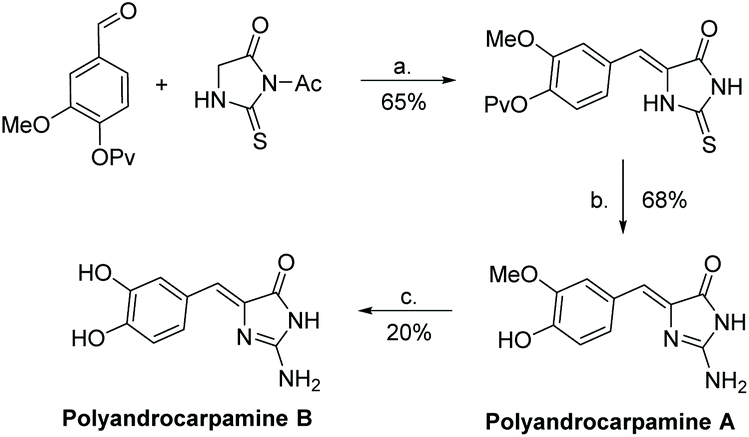
The first total synthesis of polyandrocarpamine A and B was reported by Davis et al. after their isolation in 2002.175 This synthesis contains only three steps: condensation of an aryl aldehyde and thiohydantoin, then conversion to 2-aminoimidazolone using tetrabutyl hydrogen peroxide (TBHP) and ammonia. Polyandrocarpamine A was converted to polyandrocarpamine B through a demethylation using a boron tribromide dimethyl sulfide complex. The full synthesis of polyandrocarpamines A and B were completed in 44% and 9% overall yield, respectively (Scheme 45). In 2009, Davis et al. reported a microwave assisted synthesis, producing polyandrocarpamine A and B in one step with yields of 56% and 80%, respectively.178
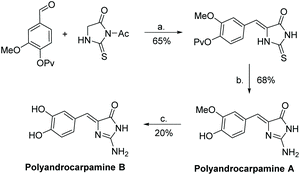 |
| | Scheme 45 The first total synthesis of polyandrocarpamine A and B. Reagents (a) NaOAc, AcOH, reflux, 2 h; (b) TBHP (15 equiv.), aq. NH4OH, MeOH, rt, 72 h; (c) BBr3-SMe2, DCE, reflux, 15 min. | |
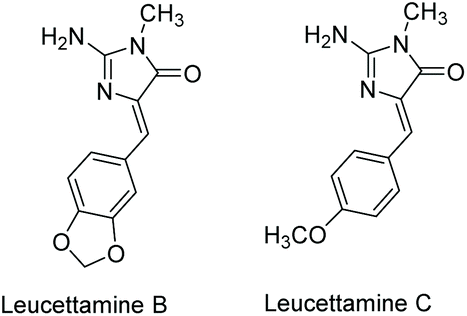
3.3.3 Leucettamine B and C.
Leucettamine B was first isolated by Chan et al. in 1993 from the marine sponge Leucetta microraphis,179 and leucettamine C was isolated from Leucetta sponges in 2003.180 These compounds have since been isolated from several other natural sources.181,182 Their structures are shown in Fig. 15. While leucettamine B and C have limited biological activity, derivatives of leucettamine B have been found to inhibit protein kinase activity.66,177,183
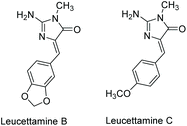 |
| | Fig. 15 Imidazolone-containing leucettamine natural products. | |
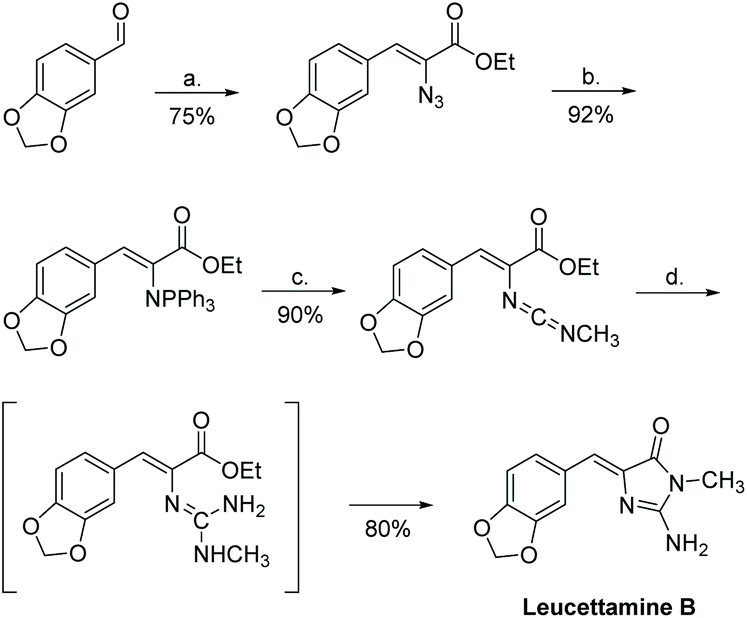
Leucettamine B was first synthesized in 1994 by Molina et al.184 The synthesis was completed in four steps, with an overall yield of 50% (Scheme 46). In this synthesis, the key transformation is an aza-Wittig/heterocumulene-mediated annulation to build the 2-aminoimidazolone ring.
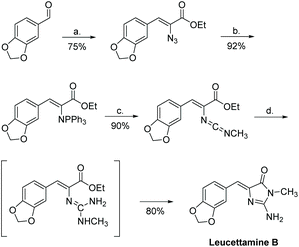 |
| | Scheme 46 The first total synthesis of leucettamine B. Reagents (a) N3CH2COOEt, NaOET, −15 °C; (b) Ph3P, CH2Cl2, rt; (c) CH3NCO, toluene, rt; (d) NH3, sealed tube, 45 °C. | |
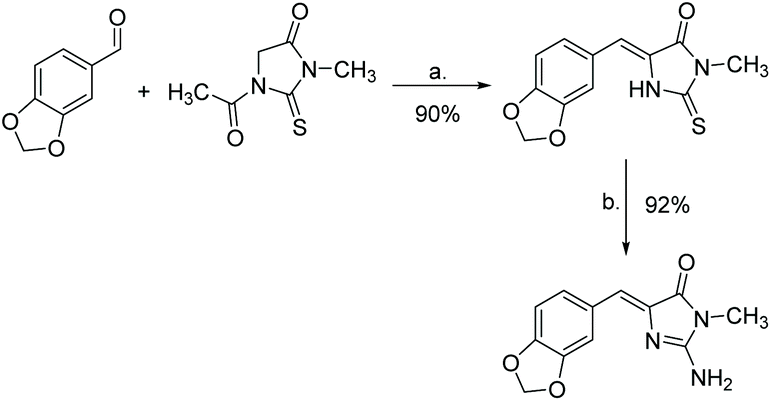
Another popular method of synthesizing leucettamine B is through the condensation of thiohydantoin and an aryl aldehyde or imine. This is then converted to a 2-amino-imidazole through varying conditions. The first report of this synthesis was in 1999 by Roué and Bergman, which produced leucettamine B in 83% yield.185 Their synthesis is shown in Scheme 47. Since then, several other similar methods have been reported.64,186,187
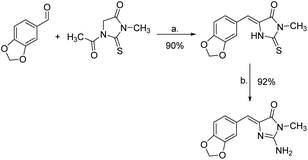 |
| | Scheme 47 Synthesis of leucettamine B via a thiohydantoin intermediate. Reagents (a) CH3CO2H, CH3COONa, heat; (b) NH3, TBHP. | |
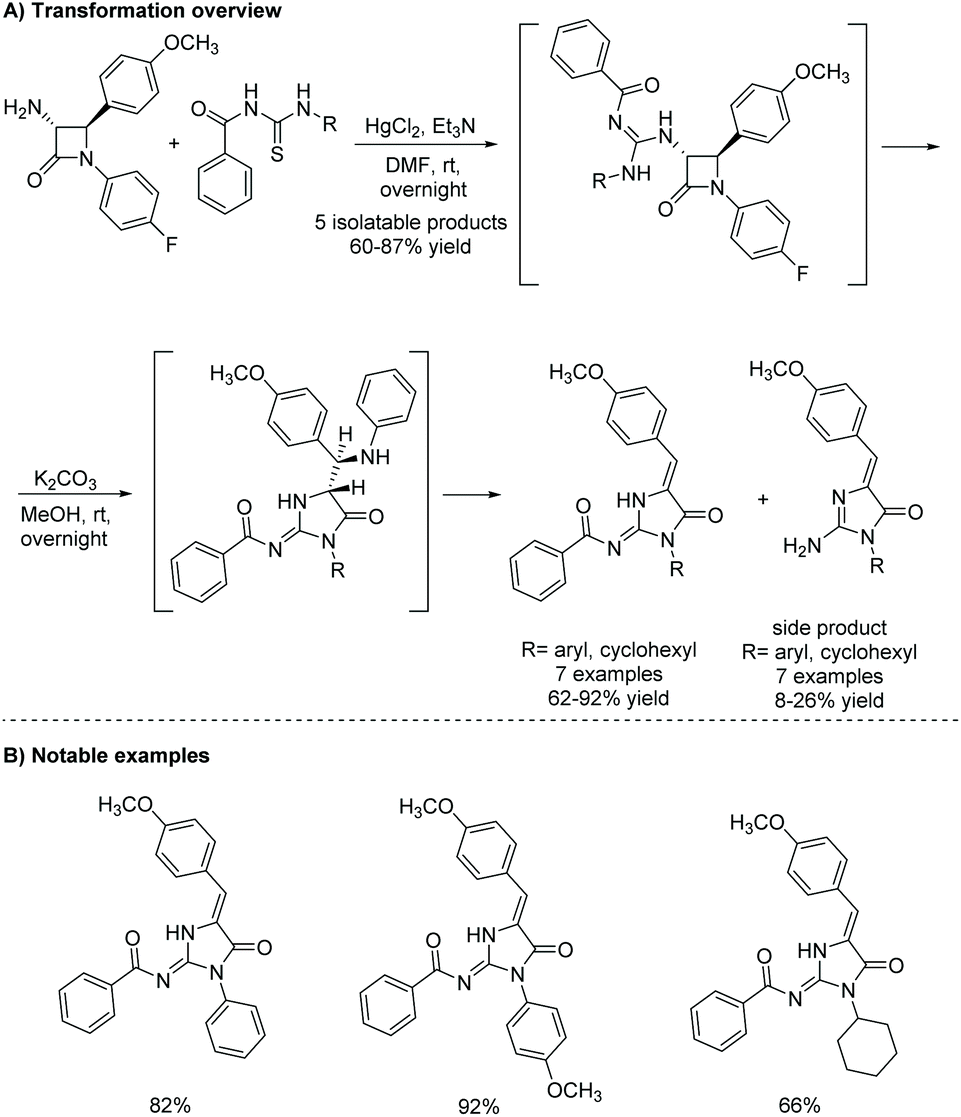
The most recently reported total synthesis of both leucettamines B and C was in 2017 by Dražić et al.111 Their synthesis is shown in Scheme 48 and starts with a β-lactam, which first reacts with N-(methylcarbamothioyl)benzamide and then undergoes a ring expansion under basic conditions to produce the desired imidazol-4-one. This was the first report of the total synthesis of leucettamine C.
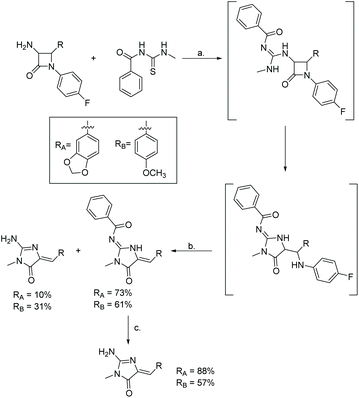 |
| | Scheme 48 Total synthesis of leucettamines B and C via β-lactam ring expansion. Reagents (a) HgCl2, Et3N, DMF, rt, overnight; (b) K2CO3, MeOH, rt, overnight; (c) K2CO3, MeOH, 50 °C, overnight. | |
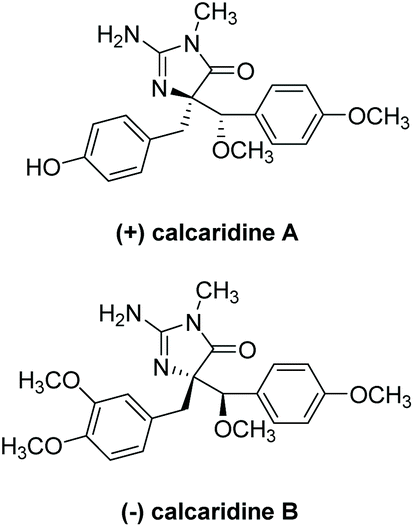
3.3.4 Calcaridines A and B.
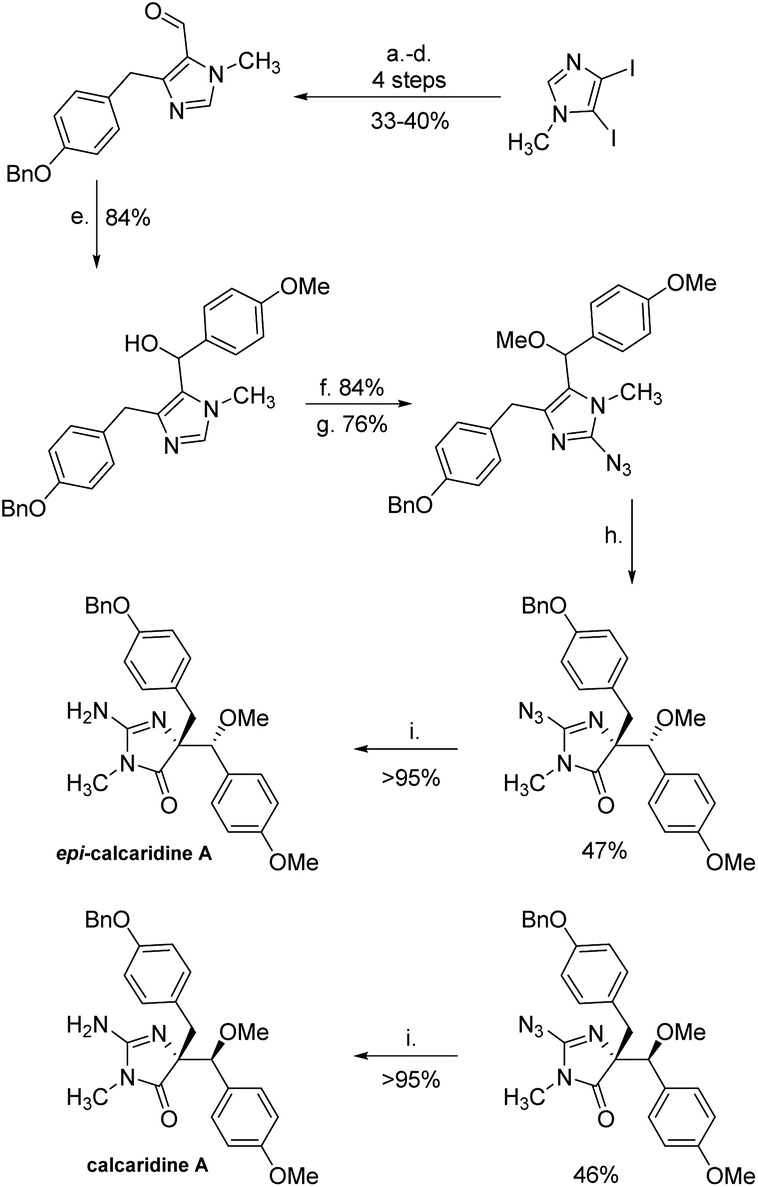
(+)-Calcaridine A was first isolated by Edrada et al. in 2003 from the sponge Leucetta.188 (−)-Calcaridine B was isolated by Tang et al. in 2019 from the marine sponge Leucetta chagosensis.189 Their structures are shown in Fig. 16. (−)-Calcaridine B was found to exhibit mild cytotoxicity toward the MCF-7 cancer cell line with an IC50 value of 25.3 μM, whereas (+)-calcaridine A has no known biological activity to date. The first total synthesis of calcaridine A was reported by Koswatta et al.97,190 In this synthesis (Scheme 49), a 2-azidoimidazole is converted to a 2-amino-4-imidazolone through sequential oxidation and reduction after a number of alkylation steps. This total synthesis was inspired by the proposed biomimetic synthesis. In this proposed pathway, calcaridine A is said to be derived from the rearrangement and/or oxidation of naamine A, an imidazole-containing natural product, also isolated from Leucetta sponges. The downside to this synthesis is the isolation of both (+)-calcaridine A and its epimer. They attempted to convert the individual diastereomers into each other upon reaction with catalytic HCl in methanol, but found no discernable epimerization, even at a range of different temperatures. (−)-Calcaridine B has yet to be synthesized.
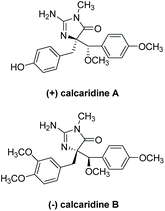 |
| | Fig. 16 Calcaridine A and B. | |
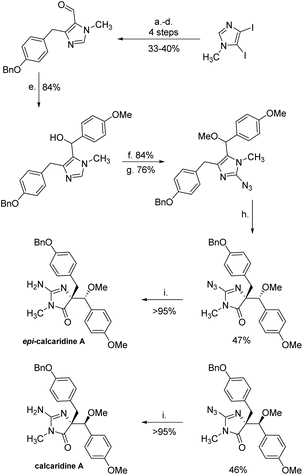 |
| | Scheme 49 The first total synthesis of calcaridine A. Reagents (a) EtMgBr, CH2Cl2, rt, then N-methyl-formanilide; (b) ethylene glycol, p-TsOH, PhH, reflux; (c) EtMgBr, THF, rt, then aryl aldehyde; (d) HCl (aq.), THF, reflux, then Et3SiH, TFA, CH2Cl2, rt; (e) 4-MeOC6H4MgBr, THF, reflux; (f) NaH, THF, 0 °C-rt to 0 °C, then MeI; (g) BuLi, THF, −78 °C, then TsN3; (h) N-sulfonylaziridine. CHCl3, rt; (i) Pd(OH)2/C, H2, EtOH. | |
4. Conclusions
In section three of this review, the known imidazol-4-one containing natural products and their total syntheses were discussed in detail. Table 2 summarizes the main imidazolone formation steps found in the mentioned total syntheses. Interestingly, most of the total syntheses followed one of a few common methods for imidazol-4-one formation. Seven of the natural products were formed via a conversion of thiohydantoin to imidazol-4-one, and the majority of these total syntheses also employed a Knoevenagel condensation to substitute the 5-position of the ring. All the natural products synthesized by this combination of reactions were 5-ethylidene-4-imidazolones. The aza-Wittig/heterocumulene-mediated annulation was also used to produce a couple 5-ethylidene-4-imidazolone containing natural products, namely aplysinopsin and leucettamine B. Two other approaches to synthesizing 5-ethylidene-4-imidazolones utilized in the total synthesis of natural products were an intermolecular aza-Wittig reaction, which was used to synthesize rhopaladin D, and a β-lactam ring expansion, used to synthesize leucettamines B and C.
Table 2 Summary of common transformations used in the synthesis of (4H)-imidazol-4-one containing natural products
| Thiohydantoin conversion |
Knoevenagel condensation |
Aza-Wittig/Heteroxumulene mediated annulation |
Oxidative pinacol-like rearrangement of imidazole |
Condensation of ester and guanidine/cyanamide |
| Dispacamide |
Dispacamide |
Aplysinopsin |
Hymenialdisine |
Hymenialdisine |
| Hymenialdisine |
Hymenialdisine |
Leucettamine B |
Monobromodispacamide |
Dihydroaplysinopsin |
| Polyandrocarpamine A |
Polyandrocarpamine A |
|
Calcaridine A |
|
| Polyandrocarpamine B |
Polyandrocarpamine A |
|
Oxysceptrin |
|
| Leucettamine B |
Leucettamine B |
|
|
|
| Indole alkaloid 1 |
Aplysinopsin |
|
|
|
| Phorbatopsin A |
|
|
|
|
Moreover, a number of imidazol-4-one containing natural products have yet to be synthesized, namely rhopaladins A–C, nortopsentine D, kottamides A–D, dispacamides C–E, dihydro-hymenialdisine derivatives, spongiacidins A–B, agesamines A–C, donnazoles A–B, phorbatopsin B and C, and (−)-calcaridine B. Analyzing the natural products that have yet to be synthesized, most of them are 5-monosubstituted or 5,5-disubstituted imidazol-4-ones. While there are many preparative methods highlighted in section 2 to produce these imidazolones, the majority of them have not been utilized in any total syntheses. Two reactions used to produce 5-mono or 5-disubstituted-4-imidazolone containing natural products highlighted in section 3 are the oxidative pinacol-like rearrangement of imidazole to produce oxysceptrin and calcaridine A and the condensation of ester and guanidine or cyanamide, used to produce dihydroaplysinopsin.
In summary, this review has covered a large range of preparative methods of imidazol-4-ones. Within section 2 of the review, advantages and disadvantages of each method were discussed, as well as their versatility upon substituting the 2 and 5 positions of the ring. Section 3 applied those preparative methods to a range of total syntheses of natural products. This section also highlighted several natural products that have yet to succumb to total synthesis. Here, the disparity between total syntheses of 5-ethylidene-4-imidazolones and other imidazol-4-ones became apparent. Looking towards the future, it is evident more effort is needed in the realm of 5-monosubstituted and 5,5-disubstituted imidazol-4-ones, specifically highlighting enantioselective methods which can be used for the total synthesis of natural products. Overall, this review highlighted the importance of imidazol-4-ones in a variety of applications, and the preparative methods explored to date.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge financial support for this work from the National Institutes of Health 1R01 AG066223-01A1.
Notes and references
- X. Bao,
et al., Design, Synthesis and Evaluation of Novel Potent Angiotensin II Receptor 1 Antagonists, Eur. J. Med. Chem., 2016, 123, 115–127 CrossRef CAS.
- M. El-Araby, A. Omar, H. H. Hassanein, A.-G. H. El-Helby and A. A. Abdel-Rahman, Design, Synthesis and in Vivo Anti-Inflammatory Activities of 2,4-Diaryl-5-4H-Imidazolone Derivatives, Molecules, 2012, 17, 12262–12275 CrossRef CAS.
- B. B. Baldaniya, Synthesis, Characterization and Biological Activity of 5-Arylidene-3-(6,7-Dicloro-1,3-Benzothiazol-2-Yl)-Phenyl-3,5-Dihydro-4 H -Imidazol-4-Ones, Eur. J. Chem., 2010, 7, 81–84 CAS.
- Y. Sun, B.-Q. Fu and M.-W. Ding, One-Pot Synthesis and Fungicidal Activities of Derivatives of Imidazo [2,1-b]-1,3,4-Thiadiazol-5(6 H)-One, Phosphorus, Sulfur Silicon Relat. Elem., 2006, 181, 1437–1443 CrossRef CAS.
- T. Lu,
et al., Design and Synthesis of a Series of Bioavailable Fatty Acid Synthase (FASN) KR Domain Inhibitors for Cancer Therapy, Bioorg. Med. Chem. Lett., 2018, 28, 2159–2164 CrossRef CAS.
- K. Santra, I. Geraskin, M. Nilsen-Hamilton, G. A. Kraus and J. W. Petrich, Characterization of the Photophysical Behavior of DFHBI Derivatives: Fluorogenic Molecules That Illuminate the Spinach RNA Aptamer, J. Phys. Chem. B, 2019, 123, 2536–2545 CrossRef CAS.
- E. A. Dolgopolova,
et al., Confinement-Driven Photophysics in Cages, Covalent−Organic Frameworks, Metal–Organic Frameworks, and DNA, J. Am. Chem. Soc., 2020, 142, 4769–4783 CrossRef CAS.
- S. Gutiérrez,
et al., Highly Fluorescent Green Fluorescent Protein Chromophore Analogues Made by Decorating the Imidazolone Ring, Chem. – Eur. J., 2015, 21, 18758–18763 CrossRef.
- B. B. Hassine and Y. Kacem, Solvent-Free Synthesis of New Chiral 3-Phenylamino-3,5-Dihydro-4H-Imidazol-4-One Derivatives from α-Amino Acid Phenylhydrazides, Heterocycles, 2014, 89, 197 CrossRef.
- P. Matafome, C. Sena and R. Seiça, Methylglyoxal, Obesity, and Diabetes, Endocrine, 2013, 43, 472–484 CrossRef CAS.
- S. L. Fishman, H. Sonmez, C. Basman, V. Singh and L. Poretsky, The Role of Advanced Glycation End-Products in the Development of Coronary Artery Disease in Patients with and without Diabetes Mellitus: A Review, Mol. Med., 2018, 24, 1–12 CAS.
- V. Srikanth,
et al., Methylglyoxal, Cognitive Function and Cerebral Atrophy in Older People, J. Gerontol., Ser. A, 2013, 68, 68–73 CrossRef.
- P. A. Sánchez-Murcia, J. A. Bueren-Calabuig, M. Camacho-Artacho, Á. Cortés-Cabrera and F. Gago, Stepwise Simulation of 3,5-Dihydro-5-Methylidene-4 H- Imidazol-4-One (MIO) Biogenesis in Histidine Ammonia-Lyase, Biochemistry, 2016, 55, 5854–5864 CrossRef.
- H. Krawczyk, 1H And13C NMR Spectra and Solution Structures of Novelderivatives of 5-Substituted Creatinines, Spectrochim. Acta, Part A, 2006, 66, 9–16 CrossRef.
- H. Finger, Einwirkung von Imidoäthern Auf Aminoester, J. Prakt. Chem., 1907, 76, 93–97 CrossRef CAS.
- M. Hoffmann,
et al., 18 F–Labeled Derivatives of Irbesartan for Angiotensin II Receptor PET Imaging, ChemMedChem, 2018, 13, 2546–2557 CrossRef CAS.
- P. Drabina, P. Funk, A. Růžička, J. Moncol and M. Sedlák, The Structures of Cobalt(II) and Copper(II) Complexes Derived from 6-(4,5-Dihydro-1H-Imidazol-5-on-2-Yl)Pyridine-2-Carboxylic Acid, Polyhedron, 2012, 34, 31–40 CrossRef CAS.
- K. W. Gillman,
et al., Synthesis and Evaluation of 5,5-Diphenylimidazolones as Potent Human Neuropeptide Y5 Receptor Antagonists, Bioorg. Med. Chem., 2006, 14, 5517–5526 CrossRef CAS.
-
M. Los, Substituted Imidazolinyl Nicotine Acids, Esters and Salts and Use Thereof as Herbicidal Agents, American Cyanamid Co., EP0041623, 1981 Search PubMed.
- T. Tamura,
et al., Identification of an Orally Active Small-Molecule PTHR1 Agonist for the Treatment of Hypoparathyroidism, Nat. Commun., 2016, 7, 1–14 Search PubMed.
- Y. Nishimura, T. Esaki, Y. Isshiki, Y. Furuta, A. Mizutani, T. Kotake, T. Emura, Y. Watanabe, M. Ohta, T. Nakagawa, K. Ogawa, S. Arai, H. Noda, H. Kitamura, M. Shimizu, T. Tamura and H. Sato, Lead Optimization and Avoidance of Reactive Metabolite Leading to PCO371, a Potent, Selective, and Orally Available Human Parathyroid Hormone Receptor 1 (hPTHR1) Agonist, J. Med. Chem., 2020, 63, 5089–5099 CrossRef CAS.
- T. Okazaki,
et al., Studies on Nonpeptide Angiotensin II Receptor Antagonists. III. Synthesis and Biological Evaluation of 5-Alkylidene-3, 5-Dihydro-4H-Imidazol-4-One-Derivatives, Chem. Pharm. Bull., 1998, 46, 777–781 CrossRef CAS.
- H. Annoura and T. Tatsuoka, Total Syntheses of Hymenialdisine and Debromohymenialdisine: Stereospecific Construction of the 2-Amino-4-Oxo-2-Imidazolin-5(Z)-Disubstituted y Ylidene Ring System, Tetrahedron Lett., 1995, 36, 413–416 CrossRef CAS.
- J. P. Caldwell,
et al., Discovery of Potent Iminoheterocycle BACE1 Inhibitors, Bioorg. Med. Chem. Lett., 2014, 24, 5455–5459 CrossRef CAS.
- X. Wang, S. Dixon, N. Yao, M. J. Kurth and K. S. Lam, Microwave Assisted DIC-Promoted Intramolecular Cyclization for Solid Phase Synthesis of Trisubstituted Imidazolidinones and Pyrimidinones, Tetrahedron Lett., 2005, 46, 5747–5750 CrossRef CAS.
- W. Fathalla, E. Y. Nofal and M. A. El-Moneim, Domino Synthesis of Pyrimido and Imidazoquinazolinones, J. Heterocycl. Chem., 2020, 57, 1266–1274 CrossRef CAS.
- T. Okuda,
et al., Synthesis and Properties of 4-(Diarylmethylene)Imidazolinone-Conjugated Fluorescent Nucleic Acids, Tetrahedron Lett., 2016, 57, 3129–3132 CrossRef CAS.
- M. Ikejiri,
et al., Design and Concise Synthesis of a Novel Type of Green Fluorescent Protein Chromophore Analogue, Org. Lett., 2012, 14, 4406–4409 CrossRef CAS.
- E. R. Zaitseva, A. Yu. Smirnov, I. N. Myasnyanko, A. I. Sokolov and M. S. Baranov, Synthesis of 2-Arylideneimidazo[1,2-a]Pyrazine-3,6,8(2H,5H,7H)-Triones as a Result of Oxidation of 4-Arylidene-2-Methyl-1H-Imidazol-5(4H)-Ones with Selenium Dioxide, Chem. Heterocycl. Compd., 2020, 56, 116–119 CrossRef CAS.
- N. V. Povarova,
et al., Red–Shifted Substrates for FAST Fluorogen–Activating Protein Based on the GFP–Like Chromophores, Chem. – Eur. J., 2019, 25, 9592–9596 CrossRef CAS.
- C. Chen,
et al., Designing Redder and Brighter Fluorophores by Synergistic Tuning of Ground and Excited States, Chem. Commun., 2019, 55, 2537–2540 RSC.
- K. H. Jung,
et al., A SNAP-Tag Fluorogenic Probe Mimicking the Chromophore of the Red Fluorescent Protein Kaede, Org. Biomol. Chem., 2019, 17, 1906–1915 RSC.
- J. Brunken and G. Bach, Synthesen in der Imidazolon-Reihe, Chem. Ber., 1956, 89, 1363–1373 CrossRef CAS.
- S. Ginsburg, 1,2,4-Substituted 5(4H)-Imidazolones 1, J. Org. Chem., 1962, 27, 4062–4063 CrossRef CAS.
- A. Kudelko, W. Zieliński and K. Jasiak, Synthesis of Novel 1-[(1-Ethoxymethylene)Amino]Imidazol-5(4H)-Ones and 1,2,4-Triazin-6(5H)-Ones from Optically Active α-Aminocarboxylic Acid Hydrazides, Tetrahedron Lett., 2013, 54, 4637–4640 CrossRef CAS.
- A. Mallinger,
et al., Discovery of Potent, Selective, and Orally Bioavailable Small-Molecule Modulators of the Mediator Complex-Associated Kinases CDK8 and CDK19, J. Med. Chem., 2016, 59, 1078–1101 CrossRef CAS.
- L. H. Jones, T. Dupont, C. E. Mowbray and S. D. Newman, A Concise and Selective Synthesis of Novel 5-Aryloxyimidazole NNRTIs, Org. Lett., 2006, 8, 1725–1727 CrossRef CAS.
- C. Hoffmann, New Derivatives of 5,5-Diphenylhydantoin. II, Bull. Soc. Chim. Fr., 1950, 659–660 CAS.
- Y. Xie,
et al., Efficient 4,5-Dihydro-1H-Imidazol-5-One Formation from Amidines and Ketones under Transition-Metal Free Conditions, Green Chem., 2015, 17, 209–213 RSC.
- S. Karch,
et al., A New Fluorogenic Small-Molecule Labeling Tool for Surface Diffusion Analysis and Advanced Fluorescence Imaging of β-Site Amyloid Precursor Protein-Cleaving Enzyme 1 Based on Silicone Rhodamine: SiR-BACE1, J. Med. Chem., 2018, 61, 6121–6139 CrossRef CAS.
- J. D. Scott,
et al., Discovery of the 3-Imino-1,2,4-Thiadiazinane 1,1-Dioxide Derivative Verubecestat (MK-8931)–A β-Site Amyloid Precursor Protein Cleaving Enzyme 1 Inhibitor for the Treatment of Alzheimer's Disease, J. Med. Chem., 2016, 59, 10435–10450 CrossRef CAS.
- C. Sibbersen, J. Palmfeldt, J. Hansen, N. Gregersen, K. A. Jørgensen and M. Johannsen, Development of a Chemical Probe for Identifying Protein Targets of α-Oxoaldehydes, Chem. Commun., 2013, 49, 4012 RSC.
- B. Alcaide, J. Plumet and M. A. Sierra, The Reaction of α-Diketones with Primary Heteroaromatic Amines.Synthesis and Reactions of Imidazo[1, 2-a]Pyridin-3(2H)-Ones and N-Heteroaryl α-Iminoketones, Tetrahedron, 1989, 45, 6841–6856 CrossRef CAS.
- M. Adib, M. Mahdavi, A. Abbasi, A. H. Jahromi and H. R. Bijanzadeh, Efficient Synthesis of Imidazo[1,2-a]Pyridin-3(2H)-Ones, Tetrahedron Lett., 2007, 48, 3217–3220 CrossRef CAS.
- H. Takeuchi, S. Hagiwara and S. Eguchi, A New Efficient Synthesis of Imidazolinones and Quinazolinone by Intramolecular Aza-Wittig Reaction, Tetrahedron, 1989, 45, 6375–6386 CrossRef CAS.
- Y.-A. Chen,
et al., N−H-Type Excited-State Proton Transfer in Compounds Possessing a Seven-Membered-Ring Intramolecular Hydrogen Bond, Chem. – Eur. J., 2016, 22, 14688–14695 CrossRef CAS.
- Y.-H. Hsu,
et al., Locked Ortho - and Para -Core Chromophores of Green Fluorescent Protein; Dramatic Emission Enhancement via Structural Constraint, J. Am. Chem. Soc., 2014, 136, 11805–11812 CrossRef CAS.
- J. Wang,
et al., New Alternating Electron Donor–Acceptor Conjugated Polymers Entailing (E)-[4,4′-Biimidazolylidene]-5,5′(1H,1′H)-Dione Moieties, Polym. Chem., 2013, 4, 5283 RSC.
- M. S. Baranov, K. M. Solntsev, K. A. Lukyanov and I. V. Yampolsky, A Synthetic Approach to GFP Chromophore Analogs from 3-Azidocinnamates. Role of Methyl Rotors in Chromophore Photophysics, Chem. Commun., 2013, 49, 5778 RSC.
- L. Wu and K. Burgess, Syntheses of Highly Fluorescent GFP-Chromophore Analogues, J. Am. Chem. Soc., 2008, 130, 4089–4096 CrossRef CAS.
- K. E. Szabó,
et al., Glucopyranosylidene-Spiro-Imidazolinones, a New Ring System: Synthesis and Evaluation as Glycogen Phosphorylase Inhibitors by Enzyme Kinetics and X-Ray Crystallography, J. Med. Chem., 2019, 62, 6116–6136 CrossRef.
- S. O. Zaitseva,
et al., Azidoacetic Acid Amides in the Synthesis of Substituted Arylidene-1-H-Imidazol-5-(4H)-Ones, ChemistrySelect, 2018, 3, 8593–8596 CrossRef CAS.
- P. Molina, P. M. Fresneda and P. Almendros, A Simple and General Entry to Aplysinopsine- Type Alkaloids by Tandem Aza-Wittig/Heterocumulene-Mediated Annelation., Tetrahedron Lett., 1992, 33, 4491–4494 CrossRef CAS.
- J. M. Chezal,
et al., Heterocyclization of Functionalized Vinylic Derivatives of Imidazo[1,2- a ]Pyridines, J. Org. Chem., 2001, 66, 6576–6584 CrossRef CAS.
- M.-W. Ding, G.-P. Zeng and Z.-J. Liu, A Regioselective Synthesis of 1-Heterocyclic Substituted 1,2,4-Triazoles via Addition Reaction of 1,2,4-Triazole with Functionalized Carbodiimide, Synth. Commun., 2002, 32, 3057–3062 CrossRef CAS.
- M.-W. Ding, S.-J. Yang, Y. Sun, Z.-J. Liu and X.-P. Liu, A Solution-Phase Parallel Synthesis of 2-Amino-5-Furfurylidene-4H-Imidazolin-4-Ones, Heterocycl. Commun., 2002, 8, 493–496 CAS.
- J.-Z. Yuan, B.-Q. Fu, M.-W. Ding and G.-F. Yang, New Facile Synthesis of 3,5-Dihydro-6H-Imidazo[1,2-b]-1,2,4-Triazol-6-Ones by an Iminophosphorane-Mediated Annulation, Eur. J. Org. Chem., 2006, 4170–4176 CrossRef CAS.
-
J. H. Kang, J. T. Moon, J. Kim, D. J. Joo and J. Y. Lee, Synthesis of 2-Arylsubstituted Imidazolone Derivatives, 2.
- F. Tan, Z.-Z. Meng, X.-Q. Xiong, G.-P. Zeng and M.-W. Ding, One-Pot Regioselective Synthesis of 2,5,6,7-Tetrahydroimidazo [1,2-a]Imidazol-3-Ones Starting from (Vinylimino)Phosphoranes, Synlett, 2019, 30, 857–859 CrossRef CAS.
- H. A. Daboun and Y. A. Ibrahim, Rearrangement of 3-Arylhydantoins into 3-Aminoglycocyamidines, J. Heterocycl. Chem., 1982, 19, 41–43 CrossRef CAS.
- B. Tang, A. Guan, Y. Zhao, J. Jiang, M. Wang and L. Zhou, Synthesis and Fungicidal Activity of (E)-5-[1-(2-Oxo-1-Oxaspiro[4,5]Dec/Non-3-En-3-Yl)Ethylidene]-2-Aminoimidazolin-4-One Derivatives, Chin. J. Chem., 2017, 35, 1133–1140 CrossRef CAS.
- S. Renault, S. Bertrand, F. Carreaux and J. P. Bazureau, Parallel Solution-Phase Synthesis of 2-Alkylthio-5-Arylidene-3,5-Dihydro-4 H -Imidazol-4-One by One-Pot Three-Component Domino Reaction, J. Comb. Chem., 2007, 9, 935–942 CrossRef CAS.
- T. Lindel and H. Hoffmann, Synthesis of Dispacamide from the Marine Sponge Agelas Dispar, Tetrahedron Lett., 1997, 38, 8935–8938 CrossRef CAS.
- J.-R. Chérouvrier, F. Carreaux and J. P. Bazureau, Microwave-Mediated Solventless Synthesis of New Derivatives of Marine Alkaloid Leucettamine B, Tetrahedron Lett., 2002, 43, 3581–3584 CrossRef.
- B. Portevin, R. M. Golsteyn, A. Pierré and G. De Nanteuil, An Expeditious Multigram Preparation of the Marine Protein Kinase Inhibitor Debromohymenialdisine, Tetrahedron Lett., 2003, 44, 9263–9265 CrossRef CAS.
- G. Burgy,
et al., Chemical Synthesis and Biological Validation of Immobilized Protein Kinase Inhibitory Leucettines, Eur. J. Med. Chem., 2013, 62, 728–737 CrossRef CAS.
- M. Debdab,
et al., Leucettines, a Class of Potent Inhibitors of Cdc2-Like Kinases and Dual Specificity, Tyrosine Phosphorylation Regulated Kinases Derived from the Marine Sponge Leucettamine B: Modulation of Alternative Pre-RNA Splicing, J. Med. Chem., 2011, 54, 4172–4186 CrossRef CAS.
- L. E. Overman, B. N. Rogers, J. E. Tellew and W. C. Trenkle, Stereocontrolled Synthesis of the Tetracyclic Core of the Bisguanidine Alkaloids Palau'amine and Styloguanidine, J. Am. Chem. Soc., 1997, 119, 7159–7160 CrossRef CAS.
-
S. Khan, R. Mahar, V. Bajpai, B. Kumar and P. M. S. Chauhan, Expedient Base-Mediated Desulfitative Dimethylamination, Oxidation, or Etherification of 2-(Methylsulfanyl)-3,5-Dihydro-4H-Imidazol-4-One Scaffolds, N. Y., 2013, pp. 8 Search PubMed.
- S. Oumouch, M. Bourotte, M. Schmitt and J.-J. Bourguignon, An Expeditious Synthesis of 2,4-Disubstituted 2-Imidazolin-5-Ones, Synthesis, 2005, 25–27 CAS.
- S. Gosling, P. Rollin and A. Tatibouët, Thiohydantoins: Selective N- and S-Functionalization for Liebeskind-Srogl Reaction Study, Synthesis, 2011, 3649–3660 CAS.
- Y. Ling,
et al., Benzylidene 2-Aminoimidazolones Derivatives: Synthesis and in Vitro Evaluation of Anti-Tumor Carcinoma Activity, Chem. Pharm. Bull., 2013, 61, 1081–1084 CrossRef CAS.
- S. Porwal, S. S. Chauhan, P. M. S. Chauhan, N. Shakya, A. Verma and S. Gupta, Discovery of Novel Antileishmanial Agents in an Attempt to Synthesize Pentamidine−Aplysinopsin Hybrid Molecule, J. Med. Chem., 2009, 52, 5793–5802 CrossRef CAS.
- J. Etxabe, J. Izquierdo, A. Landa, M. Oiarbide and C. Palomo, Catalytic Enantioselective Synthesis of N,Cα,Cα-Trisubstituted α-Amino Acid Derivatives Using 1H-Imidazol-4(5H)-Ones as Key Templates, Angew. Chem., Int. Ed., 2015, 54, 6883–6886 CrossRef CAS.
- J. Izquierdo, J. Etxabe, E. Duñabeitia, A. Landa, M. Oiarbide and C. Palomo, Enantioselective Synthesis of 5,5-Disubstituted Hydantoins by Brønsted Base/H-Bond Catalyst Assisted Michael Reactions of a Design Template, Chem. – Eur. J., 2018, 24, 7217–7227 CrossRef CAS.
- J. Izquierdo,
et al., Asymmetric Synthesis of Adjacent Tri– and Tetrasubstituted Carbon Stereocenters: Organocatalytic Aldol Reaction of an Hydantoin Surrogate with Azaarene 2−Carbaldehydes, Chem. – Eur. J., 2019, 25, 12431–12438 CrossRef CAS.
- D. Kumar, G. Mariappan, A. Husain, J. Monga and S. Kumar, Design, Synthesis and Cytotoxic Evaluation of Novel Imidazolone Fused Quinazolinone Derivatives, Arabian J. Chem., 2017, 10, 344–350 CrossRef CAS.
- A. A. Beloglazkina,
et al., Synthesis and Biological Testing of (5Z)-2-Aryl-5-Arylmethylidene-3,5-Dihydro-4H-Imidazol-4-Ones as Antimitotic Agents, Med. Chem. Res., 2016, 25, 1239–1249 CrossRef CAS.
- N. S. Hari Narayana Moorthy, V. Saxena, C. Karthikeyan and P. Trivedi, Synthesis, in Silico Metabolic and Toxicity Prediction of Some Novel Imidazolinones Derivatives as Potent Anticonvulsant Agents, J. Enzyme Inhib. Med. Chem., 2012, 27, 201–207 CrossRef CAS.
- F. Shi, X.-N. Zeng, F.-Y. Wu, S. Yan, W.-F. Zheng and S.-J. Tu, Efficient Microwave-Assisted Synthesis and Antioxidant Activity of 4-Arylidene-2-Phenyl-1H-Imidazol-5(4H)-Ones, J. Heterocycl. Chem., 2012, 49, 59–63 CrossRef CAS.
- K. F. Suzdalev and M. N. Babakova, Synthesis of Analogues of Indole Alkaloids from Sea Sponges – Aplysinopsins by the Reaction of Amines with (4Z)-4-[(1H-Indol-3-Yl)-Methylene]-1,3-Oxazol-5(4H)-Ones, J. Heterocycl. Chem., 2016, 53, 1200–1206 CrossRef CAS.
- M. A. Sofan, T. M. Abou Elmaaty, A.-K. M. Elkafafy and A. E. M. Abdel Mageed, Synthesis of Novel 5-Substituted Imidazolinones as Insecticides against Cotton Leaf Worm (Spodoptera Littoralis), J. Heterocycl. Chem., 2020, 57, 377–389 CrossRef CAS.
- V. Sharma and J. J. Tepe, Potent Inhibition of Checkpoint Kinase Activity by a Hymenialdisine-Derived Indoloazepine, Bioorg. Med. Chem. Lett., 2004, 14, 4319–4321 CrossRef CAS.
- V. Sharma, T. A. Lansdell, G. Jin and J. J. Tepe, Inhibition of Cytokine Production by Hymenialdisine Derivatives, J. Med. Chem., 2004, 47, 3700–3703 CrossRef CAS.
-
J. Bessa Belmunt, Process for Preparing an Angiotensin II Receptor Antagonist, CA02617289, 2007.
-
Y. Chen, et al., Substituted 3,5-Dihydro-4h-Imidazol-4-Ones for the Treatment of Obesity, WO2004058727, July 15, 2004 Search PubMed.
- S. M. Ramsh and N. L. Medvedskii, Aminolysis of 2-Amino-5-Benzylidene-1,3-Thiazol-4(5H)-One in Cyclic Amines: Recyclization to 2-Amino-5-Benzylidene-1,5-Dihydro-4H-Imidazol-4-One Derivatives, Russ. J. Gen. Chem., 2007, 77, 971–972 CrossRef CAS.
- S. M. Ramsh, A. V. Smirnova, S. Yu. Solovyova and S. Minyan, Mechanism of the Recyclization of 2-Amino-5-Benzylidene-1,3-Thiazol-4(5H)-One into Derivatives of 2-Amino-5-Benzylidene-1,5-Dihydro-4H-Imidazol-4-One, Chem. Heterocycl. Compd., 2008, 44, 92–95 CrossRef CAS.
- M. R. Mahmoud, H. M. F. Madkour, E. A. E.-F. El-Bordany and E.-S. A. Soliman, Synthesis and Reactions of (Z)-2-Imino-5-(3,4,5-Trimethoxy Benzylidene)Thiazolidin-4(H)One, Eur. J. Chem., 2011, 2, 475–479 CrossRef CAS.
- M. Muselli, L. Colombeau, J. Hédouin, C. Hoarau and L. Bischoff, Mild, Efficient, One-Pot Synthesis of Imidazolones Promoted by N,O-Bistrimethylsilylacetamide (BSA), Synlett, 2016, 27, 2819–2825 CrossRef CAS.
- G. Rio and B. Serkiz, A Photo-Oxide of the 1,2′ Dimer of the 2,4,5-Triphenylimidazolyl (Lophyl) Radical, J. Chem. Soc., Chem. Commun., 1975, 849–850 RSC.
- C. J. Lovely, H. Du, Y. He and H. V. Rasika Dias, Oxidative Rearrangement of Imidazoles with Dimethyldioxirane, Org. Lett., 2005, 7, 749–749 CrossRef CAS.
- R. Sivappa, P. Koswatta and C. J. Lovely, Oxidative Reactions of Tetrahydrobenzimidazole Derivatives with N-Sulfonyloxaziridines, Tetrahedron Lett., 2007, 48, 5771–5775 CrossRef.
- W. Susanto and Y. Lam, Oxidation Reactions Using Polymer-Supported 2-Benzenesulfonyl-3-(4-Nitrophenyl)Oxaziridine, Tetrahedron, 2011, 67, 8353–8359 CrossRef CAS.
- X. Wang,
et al., A Biomimetic Route for Construction of the [4+2] and [3+2] Core Skeletons of Dimeric Pyrrole–Imidazole Alkaloids and Asymmetric Synthesis of Ageliferins, J. Am. Chem. Soc., 2012, 134, 18834–18842 CrossRef CAS.
- Z. Ma, X. Wang, Y. Ma and C. Chen, Asymmetric Synthesis of Axinellamines A and B, Angew. Chem., Int. Ed., 2016, 55, 4763–4766 CrossRef CAS.
- P. B. Koswatta, R. Sivappa, H. V. R. Dias and C. J. Lovely, Total Synthesis of (±)-Calcaridine A and (±)- Epi -Calcaridine A, Org. Lett., 2008, 10, 5055–5058 CrossRef CAS.
- P. Koswatta, R. Sivappa, H. Dias and C. Lovely, Total Synthesis of the Leucetta-Derived Alkaloid Calcaridine A, Synthesis, 2009, 2970–2982 CAS.
- A. Olofson, K. Yakushijin and D. A. Horne, Synthesis of Marine Sponge Alkaloids Oroidin, Clathrodin, and Dispacamides. Preparation and Transformation of 2-Amino-4,5-Dialkoxy-4,5-Dihydroimidazolines from 2-Aminoimidazoles, J. Org. Chem., 1998, 63, 1248–1253 CrossRef CAS.
- Y. Xu, K. Yakushijin and D. A. Horne, Synthesis of C11N5 Marine Sponge Alkaloids: (±)-Hymenin, Stevensine, Hymenialdisine, and Debromohymenialdisine, J. Org. Chem., 1997, 62, 456–464 CrossRef CAS.
-
D. P. O'Malley, K. Li, M. Maue, A. L. Zografos and P. S. Baran, Total Synthesis of Dimeric Pyrrole-Imidazole Alkaloids: Sceptrin, Ageliferin, Nagelamide E, Oxysceptrin, Nakamuric Acid, and the Axinellamine Carbon Skeleton, 14.
- G. Lu, A. Katoh, Z. Zhang, Z. Hu, P. Lei and M. Kimura, Preparation of 2,4,5-Triarylimidazol-4-Ols and Their Stereoselective Rearrangement by 1,5-Phenyl Migration, J. Heterocycl. Chem., 2010, 47, 932–938 CrossRef CAS.
- A. M. Fleming, E. I. Armentrout, J. Zhu, J. G. Muller and C. J. Burrows, Spirodi(Iminohydantoin) Products from Oxidation of 2′-Deoxyguanosine in the Presence of NH4Cl in Nucleoside and Oligodeoxynucleotide Contexts, J. Org. Chem., 2015, 80, 711–721 CrossRef CAS.
- W. Ye,
et al., Iminohydantoin Lesion Induced in DNA by Peracids and Other Epoxidizing Oxidants, J. Am. Chem. Soc., 2009, 131, 6114–6123 CrossRef CAS.
- X. Liu, Y. Wang, D. Yang, J. Zhang, D. Liu and W. Su, Catalytic Asymmetric Inverse-Electron-Demand 1,3-Dipolar Cycloaddition of C,N-Cyclic Azomethine Imines with Azlactones: Access to Chiral Tricyclic Tetrahydroisoquinolines, Angew. Chem., Int. Ed., 2016, 55, 8100–8103 CrossRef CAS.
- C. Guo, M. Fleige, D. Janssen-Müller, C. G. Daniliuc and F. Glorius, Switchable Selectivity in an NHC-Catalysed Dearomatizing Annulation Reaction, Nat. Chem., 2015, 7, 842–847 CrossRef CAS.
- H. D. S. Guerrand, H. Adams and I. Coldham, Cascade Cyclization, Dipolar Cycloaddition of Azomethine Imines for the Synthesis of Pyrazolidines, Org. Biomol. Chem., 2011, 9, 7921 RSC.
- T. Dražić,
et al., Synthesis of New 2-Aminoimidazolones with Antiproliferative Activity via Base Promoted Amino-β-Lactam Rearrangement, Tetrahedron, 2015, 71, 9202–9215 CrossRef.
- V. Mehra and V. Kumar, Facile Diastereoselective Synthesis of Functionally Enriched Hydantoins via Base-Promoted Intramolecular Amidolysis of C-3 Functionalized Azetidin-2-Ones, Tetrahedron Lett., 2013, 54, 6041–6044 CrossRef CAS.
- R. Raj,
et al., Discovery of Highly Selective 7-Chloroquinoline-Thiohydantoins with Potent Antimalarial Activity, Eur. J. Med. Chem., 2014, 84, 425–432 CrossRef CAS.
- T. Dražić, K. Molčanov, M. Jurin and M. Roje, Synthesis of Marine Alkaloids Leucettamines B and C by β-Lactam Ring Rearrangement, Synth. Commun., 2017, 47, 764–770 CrossRef.
- R. Kotagiri, Z. Deng, W. Xu and Q. Cai, Stereospecific Synthesis of (E)-5-Tetrasubstituted-Ylidene-3,5-Dihydro-4H-Imidazol-4-Ones, Org. Lett., 2019, 21, 3946–3949 CrossRef CAS.
- R. Kazlauskas, P. T. Murphy, R. J. Quinn and R. J. Wells, Aplysinopsin, a New Tryptophan Derivative from a Sponge, Tetrahedron Lett., 1977, 18, 61–64 CrossRef.
- N. Cachet, L. Loffredo, O. O. Vicente and O. P. Thomas, Chemical Diversity in the Scleractinian Coral Astroides Calycularis, Phytochem. Lett., 2013, 6, 205–208 CrossRef CAS.
- D. Bialonska and J. K. Zjawiony, Aplysinopsins - Marine Indole Alkaloids: Chemistry, Bioactivity and Ecological Significance, Mar. Drugs, 2009, 7, 166–183 CrossRef CAS.
- A. Skiredj,
et al., Spontaneous Biomimetic Formation of (±)-Dictazole B under Irradiation with Artificial Sunlight, Angew. Chem., Int. Ed., 2014, 53, 6419–6424 CrossRef CAS.
- G. Guella, I. Mancini, H. Zibrowius and F. Pietra, Aplysinopsin-Type Alkaloids from Dendrophyllia Sp., a Scleractinian Coral of the Family Dendrophylliidae of the Philippines, Facile Photochemical (Z/E) Photoisomerization and Thermal Reversal, Helv. Chim. Acta, 1989, 72, 1444–1450 CrossRef CAS.
- T. Iwagawa,
et al., Aplysinopsin Dimers from a Stony Coral. Tubastraea Aurea, Heterocycles, 2008, 75, 2023 CrossRef CAS.
- K. H. Shaker, M. Göhl, T. Müller and K. Seifert, Indole Alkaloids from the Sea Anemone Heteractis Aurora and Homarine from Octopus Cyanea, Chem. Biodivers., 2015, 12, 1746–1755 CrossRef CAS.
- P. M. Pauletti,
et al., Halogenated Indole Alkaloids from Marine Invertebrates, Mar. Drugs, 2010, 8, 1526–1549 CrossRef CAS.
- J. Baird-Lambert, P. A. Davis and K. M. Taylor, Methylaplysinopsin: A Natural Product of Marine Origin with Effects on Serotonergic Neurotransmission, Clin. Exp. Pharmacol. Physiol., 1982, 9, 203–212 CrossRef CAS.
- W. Balansa,
et al., Australian Marine Sponge Alkaloids as a New Class of Glycine-Gated
Chloride Channel Receptor Modulator, Bioorg. Med. Chem., 2013, 21, 4420–4425 CrossRef CAS.
- E. Fattorusso, V. Lanzotti, S. Magno and E. Novellino, Tryptophan Derivatives from a Mediterranean Anthozoan, Astroides Calycularis, J. Nat. Prod., 1985, 48, 924–927 CrossRef CAS.
- G. Guella, I. Mancini, H. Zibrowius and F. Pietra, Novel Aplysinopsin-Type Alkaloids from Scleractinian Corals of the Family Dendrophylliidae of the Mediterranean and the Philippines. Configurational-Assignment Criteria, Stereospecific Synthesis, and Photoisomerization, Helv. Chim. Acta, 1988, 71, 773–782 CrossRef CAS.
- J. E. Johnson, D. C. Canseco, D. D. Dolliver, J. A. Schetz and F. R. Fronczek, Synthesis and Characterization of Aplysinopsin Analogs, J. Chem. Crystallogr., 2009, 39, 329–336 CrossRef CAS.
- E. Boyd and J. Sperry, Synthesis of the Selective Neuronal Nitric Oxide Synthase (NNOS) Inhibitor 5,6-Dibromo-2′-Demethylaplysinopsin, Synlett, 2011, 826–830 CAS.
- D. F. Cummings, D. C. Canseco, P. Sheth, J. E. Johnson and J. A. Schetz, Synthesis and Structure–Affinity Relationships of Novel Small Molecule Natural Product Derivatives Capable of Discriminating between Serotonin 5-HT1A, 5-HT2A, 5-HT2C Receptor Subtypes, Bioorg. Med. Chem., 2010, 18, 4783–4792 CrossRef CAS.
- K. Lewellyn, D. Bialonska, M. J. Loria, S. W. White, K. J. Sufka and J. K. Zjawiony, In Vitro Structure–Activity Relationships of Aplysinopsin Analogs and Their in Vivo Evaluation in the Chick Anxiety–Depression Model, Bioorg. Med. Chem., 2013, 21, 7083–7090 CrossRef CAS.
- N. R. Penthala, T. R. Yerramreddy and P. A. Crooks, Microwave Assisted Synthesis and in Vitro Cytotoxicities of Substituted (Z)-2-Amino-5-(1-Benzyl-1H-Indol-3-Yl)Methylene-1-Methyl-1H-Imidazol-4(5H)-Ones against Human Tumor Cell Lines, Bioorg. Med. Chem. Lett., 2010, 20, 591–593 CrossRef CAS.
- K. Lewellyn, D. Bialonska, N. D. Chaurasiya, B. L. Tekwani and J. K. Zjawiony, Synthesis and Evaluation of Aplysinopsin Analogs as Inhibitors of Human Monoamine Oxidase A and B, Bioorg. Med. Chem. Lett., 2012, 22, 4926–4929 CrossRef CAS.
- P. Molina, P. Almendros and P. M. Fresneda, Iminophosphorane-Mediated Imidazole Ring Formation: A New and General Entry to Aplysinopsin-Type Alkaloids of Marine Origin., Tetrahedron, 1994, 50, 2241–2254 CrossRef CAS.
- A. Skiredj, M. Beniddir, D. Joseph, G. Bernadat, L. Evanno and E. Poupon, Harnessing the Intrinsic Reactivity within the Aplysinopsin Series for the Synthesis of Intricate Dimers: Natural from Start to Finish, Synthesis, 2015, 47, 2367–2376 CrossRef CAS.
- N. Duchemin,
et al., DNA–Templated [2+2] Photocycloaddition: A Straightforward Entry into the Aplysinopsin Family of Natural Products, Angew. Chem., Int. Ed., 2018, 57, 11786–11791 CrossRef CAS.
- A. Skiredj,
et al., A Unified Bioinspired “Aplysinopsin Cascade”: Total Synthesis of (±)-Tubastrindole B and Related Biosynthetic Congeners, Org. Lett., 2014, 16, 4980–4983 CrossRef CAS.
- H. Sato, M. Tsuda, K. Watanabe and J. Kobayashi, Rhopaladins A ∼ D, New Indole Alkaloids from Marine Tunicate Rhopalaea Sp, Tetrahedron, 1998, 54, 8687–8690 CrossRef CAS.
- P. M. Fresneda, P. Molina and M. A. Sanz, The First Synthesis of the Bis(Indole) Marine Alkaloid Rhopaladin D, Synlett, 2000, 1190–1192 CAS.
- I. Mancini, G. Guella, C. Debitus, J. Waikedre and F. Pietra, From Inactive Nortopsentin D, a Novel Bis(Indole) Alkaloid Isolated from the Axinellid Sponge Dragmacidon Sp. from Deep Waters South of New Caledonia, to a Strongly Cytotoxic Derivative, Helv. Chim. Acta, 1996, 79, 2075–2081 CrossRef CAS.
- D. R. Appleton, M. J. Page, G. Lambert, M. V. Berridge and B. R. Copp, Kottamides A−D: Novel Bioactive Imidazolone-Containing Alkaloids from the New Zealand Ascidian Pycnoclavella k Ottae, J. Org. Chem., 2002, 67, 5402–5404 CrossRef CAS.
- A. Loukaci, M. Guyot, A. Chiaroni and C. Riche, A New Indole Alkaloid from the Marine Tunicate Dendrodoa g Rossularia, J. Nat. Prod., 1998, 61, 519–522 CrossRef CAS.
- C. D. Hupp and J. J. Tepe, Total Synthesis of a Marine Alkaloid from the Tunicate Dendrodoa Grossularia, Org. Lett., 2008, 10, 3737–3739 CrossRef CAS.
- C. D. Hupp and J. J. Tepe, 1-Ethyl-3-(3-Dimethylaminopropyl)Carbodiimide Hydrochloride-Mediated Oxazole Rearrangement: Gaining Access to a Unique Marine Alkaloid Scaffold, J. Org. Chem., 2009, 74, 3406–3413 CrossRef CAS.
- F. Cafieri, E. Fattorusso, A. Mangoni and O. Taglialatela-Scafati, Dispacamides, Anti-Histamine Alkaloids from Caribbean Agelas Sponges, Tetrahedron Lett., 1996, 37, 3587–3590 CrossRef CAS.
- F. Cafieri, R. Carnuccio, E. Fattorusso, O. Taglialatela-Scafati and T. Vallefuoco, Anti-Histaminic Activity of Bromopyrrole Alkaloids Isolated from Caribbean Agelas Sponges, Bioorg. Med. Chem. Lett., 1997, 7, 2283–2288 CrossRef CAS.
- S. S. Ebada,
et al., Dispacamide E and Other Bioactive Bromopyrrole Alkaloids from Two Indonesian Marine Sponges of the Genus Stylissa, Nat. Prod. Res., 2015, 29, 231–238 CrossRef CAS.
- B. Forte, B. Malgesini, C. Piutti, F. Quartieri, A. Scolaro and G. Papeo, A Submarine Journey: The Pyrrole-Imidazole Alkaloids, Mar. Drugs, 2009, 7, 705–753 CrossRef CAS.
- F. Scala,
et al., Bromopyrrole Alkaloids as Lead Compounds against Protozoan Parasites, Mar. Drugs, 2010, 8, 2162–2174 CrossRef CAS.
- N. Ando and S. Terashima, A Novel Synthesis of the 2-Aminoimidazol-4-Carbaldehyde Derivatives, Versatile Synthetic Intermediates for 2-Aminoimidazole Alkaloids, Synlett, 2006, 2836–2843 CrossRef CAS.
- N. Ando and S. Terashima, A Novel Synthesis of the 2-Amino-1H-Imidazol-4-Carbaldehyde Derivatives and Its Application to the Efficient Synthesis of 2-Aminoimidazole Alkaloids, Oroidin, Hymenidin, Dispacamide, Monobromodispacamide, and Ageladine A, Tetrahedron, 2010, 66, 6224–6237 CrossRef CAS.
- S. Guihéneuf, L. Paquin, F. Carreaux, E. Durieu, L. Meijer and J. P. Bazureau, An Efficient Approach to Dispacamide A and Its Derivatives, Org. Biomol. Chem., 2012, 10, 978–987 RSC.
- C. A. Mattia and L. M. R. Puliti, 4-(2-Amino-4-Oxo-2-Imidazolin-5-Ylidene)-2-Bromo-4,5,6,7-Tetrahydropyrrolo- [2,3-c]Azepin-8-One Methanol Solvate: A New Bromo Compound from the Sponge, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, 38, 2513–2515 CrossRef.
- I. Kitagawa, M. Kobayashi, K. Kitanaka, M. Kido and Y. Kyogoku, Marine Natural Products XII On the Chemical Constituents of the Okinawan Marine Sponge Hymeniacidon Aldis, Chem. Pharm. Bull., 1983, 31, 2321–2328 CrossRef CAS.
- D. H. Williams and D. J. Faulkner, Isomers and Tautomers of Hymenialdisine and Debromohymenialdisine, Nat. Prod. Lett., 1996, 9, 57–64 CrossRef CAS.
- P. Sauleau,
et al., Dihydrohymenialdisines, New Pyrrole-2-Aminoimidazole Alkaloids from the Marine Sponge Cymbastela Cantharella, Tetrahedron Lett., 2011, 52, 2676–2678 CrossRef CAS.
- N. Ruocco, S. Costantini, F. Palumbo and M. Costantini, Marine Sponges and Bacteria as Challenging Sources of Enzyme Inhibitors for Pharmacological Applications, Mar. Drugs, 2017, 15, 173 CrossRef.
- N. Bailon-Moscoso, G. Cevallos-Solorzano, J. C. Romero-Benavides and M. I. R. Orellana, Natural Compounds as Modulators of Cell Cycle Arrest: Application for Anticancer Chemotherapies, Curr. Genomics, 2017, 18, 106–131 CrossRef CAS.
- D. Skropeta, N. Pastro and A. Zivanovic, Kinase Inhibitors from Marine Sponges, Mar. Drugs, 2011, 9, 2131–2154 CrossRef CAS.
- R. S. Z. Saleem, T. A. Lansdell and J. J. Tepe, Synthesis and Evaluation of Debromohymenialdisine-Derived Chk2 Inhibitors, Bioorg. Med. Chem., 2012, 20, 1475–1481 CrossRef CAS.
- T. N. T. Nguyen, R. S. Z. Saleem, M. J. Luderer, S. Hovde, R. W. Henry and J. J. Tepe, Radioprotection by Hymenialdisine-Derived Checkpoint Kinase 2 Inhibitors, ACS Chem. Biol., 2012, 7, 172–184 CrossRef CAS.
- T. N. T. Nguyen and J. J. Tepe, Preparation of Hymenialdisine, Analogues and Their Evaluation as Kinase Inhibitors, Curr. Med. Chem., 2009, 16, 3122–3143 CrossRef CAS.
- N. D'Orazio, M. A. Gammone, E. Gemello, M. De Girolamo, S. Cusenza and G. Riccioni, Marine Bioactives: Pharmacological Properties and Potential Applications against Inflammatory Diseases, Mar. Drugs, 2012, 10, 812–833 CrossRef.
- R. S. Z. Saleem and J. J. Tepe, A Concise Total Synthesis of Hymenialdisine, Tetrahedron Lett., 2015, 56, 3011–3013 CrossRef CAS.
- G. Papeo, H. Posteri, D. Borghi and M. Varasi, A New Glycociamidine Ring Precursor: Syntheses of (Z)-Hymenialdisine, (Z,)-2-Debromohymenialdisine, and (±)- Endo -2-Debromohymenialdisine †, Org. Lett., 2005, 7, 5641–5644 CrossRef CAS.
- K. Inaba, H. Sato, M. Tsuda and J. Kobayashi, Spongiacidins A−D, New Bromopyrrole Alkaloids from Hymeniacidon Sponge, J. Nat. Prod., 1998, 61, 693–695 CrossRef CAS.
- A. N. E. Hamed,
et al., Bioactive Pyrrole Alkaloids Isolated from the Red Sea: Marine Sponge Stylissa Carteri, Z. Naturforsch. C., 2018, 73, 199–210 CAS.
- C. Eder,
et al., New Alkaloids from the Indopacific Sponge Stylissa Carteri, J. Nat. Prod., 1999, 62, 184–187 CrossRef CAS.
- S. Tsukamoto, A. Katsuki, H. Kato, Y. Ise, F. Losung and R. E. P. M, angindaan, Agesamines A and B, New Dibromopyrrole Alkaloids from the Sponge Agelas Sp., Heterocycles, 2019, 98, 558 CrossRef.
- D. Kovalerchik,
et al., Bromopyrrole Alkaloids of the Sponge Agelas Oroides Collected Near the Israeli Mediterranean Coastline, J. Nat. Prod., 2020, 83, 374–384 CrossRef CAS.
- B. M. Trost, M. Osipov and G. Dong, Palladium-Catalyzed Dynamic Kinetic Asymmetric Transformations of Vinyl Aziridines with Nitrogen Heterocycles: Rapid Access to Biologically Active Pyrroles and Indoles, J. Am. Chem. Soc., 2010, 132, 15800–15807 CrossRef CAS.
- B. M. Trost and G. Dong, Asymmetric Annulation toward Pyrrolopiperazinones: Concise Enantioselective Syntheses of Pyrrole Alkaloid Natural Products, Org. Lett., 2007, 9, 2357–2359 CrossRef CAS.
- J. Muñoz, C. Moriou, J.-F. Gallard, P. D. Marie and A. Al-Mourabit, Donnazoles A and B from Axinella Donnani Sponge: Very Close Derivatives from the Postulated Intermediate ‘Pre-Axinellamine’, Tetrahedron Lett., 2012, 53, 5828–5832 CrossRef.
- R. A. Rodriguez,
et al., Axinellamines as Broad-Spectrum Antibacterial Agents: Scalable Synthesis and Biology, J. Am. Chem. Soc., 2014, 136, 15403–15413 CrossRef CAS.
- P. A. Keifer, R. E. Schwartz, M. E. S. Koker, R. G. Hughes, D. Rittschof and K. L. Rinehart, Bioactive Bromopyrrole Metabolites from the Caribbean Sponge Agelas Conifera, J. Org. Chem., 1991, 56, 2965–2975 CrossRef CAS.
- J. Kobayashi, M. Tsuda and Y. Ohizumi, A Potent Actomyosin ATPase Activator from the Okinawan Marine SpongeAgelas Cf.Nemoechinata, Experientia, 1991, 47, 301–304 CrossRef CAS.
- T. D. Nguyen,
et al., Antioxidant Benzylidene 2-Aminoimidazolones from the Mediterranean Sponge Phorbas Topsenti, Tetrahedron, 2012, 68, 9256–9259 CrossRef CAS.
- R. A. Davis, W. Aalbersberg, S. Meo, R. M. da Rocha and C. M. Ireland, The Isolation and Synthesis of Polyandrocarpamines A and B. Two New 2-Aminoimidazolone Compounds from the Fijian Ascidian, Polyandrocarpa Sp., Tetrahedron, 2002, 58, 3263–3269 CrossRef CAS.
- M. Sugumaran and W. E. Robinson, Bioactive Dehydrotyrosyl and Dehydrodopyl Compounds of Marine Origin, Mar. Drugs, 2010, 8, 2906–2935 CrossRef CAS.
- N. Loaëc,
et al., Marine-Derived 2-Aminoimidazolone Alkaloids. Leucettamine B-Related Polyandrocarpamines Inhibit Mammalian and Protozoan DYRK & CLK Kinases, Mar. Drugs, 2017, 15, 316 CrossRef.
- R. A. Davis, P. S. Baron, J. E. Neve and C. Cullinane, A Microwave-Assisted Stereoselective Synthesis of Polyandrocarpamines A and B, Tetrahedron Lett., 2009, 50, 880–882 CrossRef CAS.
- G. W. Chan,
et al., New Leukotriene B4 Receptor Antagonist: Leucettamine A and Related Imidazole Alkaloids from the Marine Sponge Leucetta Microraphis, J. Nat. Prod., 1993, 56, 116–121 CrossRef CAS.
- P. Crews, D. P. Clark and K. Tenney, Variation in the Alkaloids among Indo-Pacific Leucetta Sponges, J. Nat. Prod., 2003, 66, 177–182 CrossRef CAS.
- K. Watanabe, Y. Tsuda, M. Iwashima and K. Iguchi, A New Bioactive Triene Aldehyde from the Marine Sponge Leucetta Microraphis, J. Nat. Prod., 2000, 63, 258–260 CrossRef CAS.
- W. H. B. Hassan, A. M. Al-Taweel and P. Proksch, Two New Imidazole Alkaloids from Leucetta Chagosensis Sponge, Saudi Pharm. J., 2009, 17, 295–298 CrossRef.
- M. Debdab,
et al., Synthesis and Preliminary Biological Evaluation of New Derivatives of the Marine Alkaloid Leucettamine B as Kinase Inhibitors, Eur. J. Med. Chem., 2010, 45, 805–810 CrossRef CAS.
- P. Molina, P. Almendros and P. M. Fresneda, An Iminophosphorane-Mediated Efficient Synthesis of the Alkaloid Leucettamine B of Marine Origin, Tetrahedron Lett., 1994, 35, 2235–2236 CrossRef CAS.
- N. Roué and J. Bergman, Synthesis of the Marine Alkaloid Leucettamine B, Tetrahedron, 1999, 55, 14729–14738 CrossRef.
- F. Carreaux,
et al., An Efficient Method for the Preparation of New Analogs of Leucettamine B under Solvent-Free Microwave Irradiation, Heterocycles, 2009, 78, 1191 CrossRef.
- M. Selvaraju and C.-M. Sun, Unprecedented One-Pot Chemocontrolled Entry to Thioxoimidazolidinones and Aminoimidazolones: Synthesis of Kinase Inhibitor Leucettamine B, ACS Comb. Sci., 2015, 17, 182–189 CrossRef CAS.
- R. A. Edrada, C. C. Stessman and P. Crews, Uniquely Modified Imidazole Alkaloids from a Calcareous Leucetta Sponge, J. Nat. Prod., 2003, 66, 939–942 CrossRef CAS.
- W.-Z. Tang,
et al., (-)-Calcaridine B, a New Chiral Aminoimidazole-Containing Alkaloid from the Marine Sponge Leucetta Chagosensis, J. Asian Nat. Prod. Res., 2019, 21, 1123–1128 CrossRef CAS.
- P. Koswatta, R. Sivappa, H. Dias and C. Lovely, Total Synthesis of the Leucetta-Derived Alkaloid Calcaridine A, Synthesis, 2009, 2970–2982 CAS.
|
| This journal is © the Partner Organisations 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*