
Radical reactions promoted by trivalent tertiary phosphines
Dingwu
Pan
,
Guihua
Nie
,
Shichun
Jiang
,
Tingting
Li
and
Zhichao
Jin
 *
*
Laboratory Breeding Base of Green Pesticide and Agricultural Bioengineering, Key Laboratory of Green Pesticide and Agricultural Bioengineering Ministry of Education, Guizhou University, Huaxi District, Guiyang 550025, China. E-mail: zcjin@gzu.edu.cn
First published on 22nd June 2020
Abstract
Tertiary phosphines have been extensively developed as effective organic catalysts/reagents to promote various modern organic transformations in both racemic and enantioselective fashions. However, their applications in radical generation and reactions remain relatively less explored. Phosphine-centered radical species, generated from various tertiary phosphines via phosphine–oxidant charge transfer processes, photoredox catalysis and electrochemical oxidations, can give rise to many unprecedented activation modes and reactions. Single-electron-transfer (SET) reactions associated with tertiary phosphines in particular have recently gained popularity, affording novel and promising synthetic approaches to challenging molecular structures from readily available starting materials upon simple operation. Summarized herein are the historical background and recent breakthroughs in this dynamic field of phosphine-mediated radical organic reactions.
 Dingwu Pan | Dingwu Pan received his BS and MSc degrees from Guizhou University in 2012 and 2016, respectively. He is now pursuing his PhD degree under the guidance of Prof. Yonggui Robin Chi and Prof. Zhichao Jin at Guizhou University. |
 Guihua Nie | Guihua Nie received her BS degree from Central South University of Forestry and Technology in 2018. She then entered Guizhou University in 2018 to pursue her PhD degree under the guidance of Prof. Yonggui Robin Chi and Prof. Zhichao Jin. |
 Shichun Jiang | Shichun Jiang received her BS degree from Anshan Normal University in 2017. She then entered Guizhou University in 2017 to pursue her PhD degree under the guidance of Prof. Yonggui Robin Chi and Prof. Zhichao Jin. |
 Tingting Li | Tingting Li received his BS degree from Qingdao Agricultural University in 2014 and MSc degree from Anhui Normal University in 2017. He then entered Guizhou University in 2017 to pursue his PhD degree under the guidance of Prof. Yonggui Robin Chi and Prof. Zhichao Jin. |
 Zhichao Jin | Prof. Zhichao Jin received his MSc degree from East China University of Science and Technology in 2011 under the guidance of Prof. Jinxing Ye, and PhD degree in chemistry from Nanyang Technological University in 2015 under the guidance of Prof. Yonggui Robin Chi. After postdoctoral training with Prof. Yixin Lu at National University of Singapore, he started his independent academic career at Guizhou University in 2017. His current research interests include the development of novel catalytic activation modes and functional molecules for applications in pesticides. |
1. Introduction
Phosphine organocatalysis has seen prosperous growth in the past 20 years.1 The use of tertiary phosphines as promoters for organic reactions could date back to 1963, when Rauhut and Currier described their pioneering work of the dimerization of electron-deficient alkenes in their patent.2 To date, countless efficient methods have been established with tertiary phosphines being used as the reaction catalysts for chemo-, regio- and stereoselective transformations. However, most of the well-developed methodologies are limited to electron-pair-transfer reactions. The development of the single-electron-transfer (SET) variants is relatively rare (Fig. 1). Numerous elegant reviews focusing on phosphine organocatalysis have been well-documented,1 but a systematic review concentrating on tertiary phosphine-associated radical reactions has not been presented.3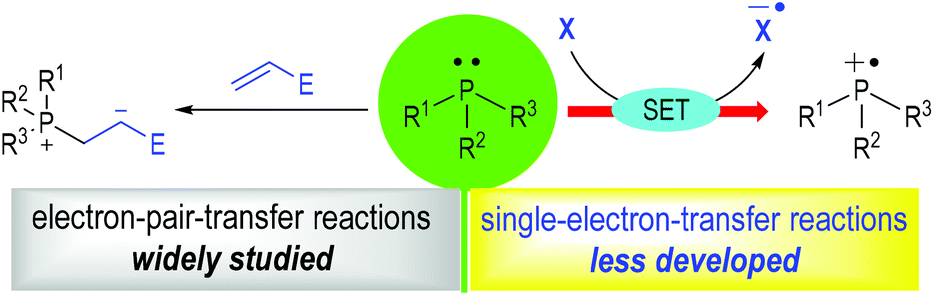 | ||
| Fig. 1 Phosphine-catalyzed electron-pair-transfer reactions and SET reactions. | ||
The discovery of radical ions derived from trivalent amines has proven to be a milestone in organic synthesis.4 The applications of such radical species have attracted considerable interest with numerous efficient synthetic methods established in the past 20 years.5 In contrast, the applications of trivalent tertiary phosphine-derived radical species in organic synthesis are relatively less developed, although they have been established for more than half a century.6 Given the rapid development in radical chemistry and phosphine organocatalysis in recent years, it has become more and more attractive to develop novel synthetic methods using phosphine-derived radical intermediates. The concept of a tertiary phosphinium radical ion as a reaction intermediate has also proven to be significant in modern organic synthesis.
Therefore, we would like to provide an overview on the radical reactions promoted by trivalent tertiary phosphine molecules. The postulated reaction mechanisms involved in these SET processes will also be discussed in detail. This review article will be organized into four sections: (i) radical reactions of tertiary phosphines with oxidative reagents, (ii) radical reactions of tertiary phosphines in irradiative reactions, (iii) radical reactions of tertiary phosphines in electrochemical reactions, and (iv) the summary and outlook in tertiary phosphine-associated radical reactions. Although several excellent examples might inevitably be missed out in this review, the aim is to provide a systematic summarization of the tertiary phosphine-associated radical transformations and reaction mechanisms. Radical reactions promoted by phosphorus molecules other than trivalent tertiary phosphines will be excluded in this review, since they have already been summarized in the corresponding review articles.3 We hope this review will provide researchers with a scientific auxiliary tool in phosphine/radical chemistry, inspiring more strategic synthetic applications, and stimulating future mechanistic research within this dynamic and exciting field.
2. Radical reactions of tertiary phosphines with oxidative reagents
2.1. Phosphine-associated radical reactions with quinone oxidants
The generation of radical ion species from tertiary phosphines was first evidenced by Ramirez and co-workers in 1956.6 The authors observed two different types of interactions between triphenylphosphine and p-quinones (Fig. 2). Simple p-quinone 1 reacted with triphenylphosphine 2 through a conjugate addition to form intermediate 3, which finally afforded the zwitterion 4 through a proton transfer process (Fig. 2a). The sterically hindered p-quinone 5, however, reacted with 2 at the oxygen atoms and constructed O–P bonds to give the ion pair I consisting of the anion 6 and cation 8 as the final product (Fig. 2b). This reaction was supposed to go through SET processes. The triphenylphosphine 2 was oxidized by p-chloranil 5 through two steps of SET reactions which gave the dianion 6 and the radical cation 7 (Fig. 2b, eqn (1)). The radical cation 7 then reacted with another molecule of 5 through SET processes to afford the dication 8 (Fig. 2b, eqn (2)).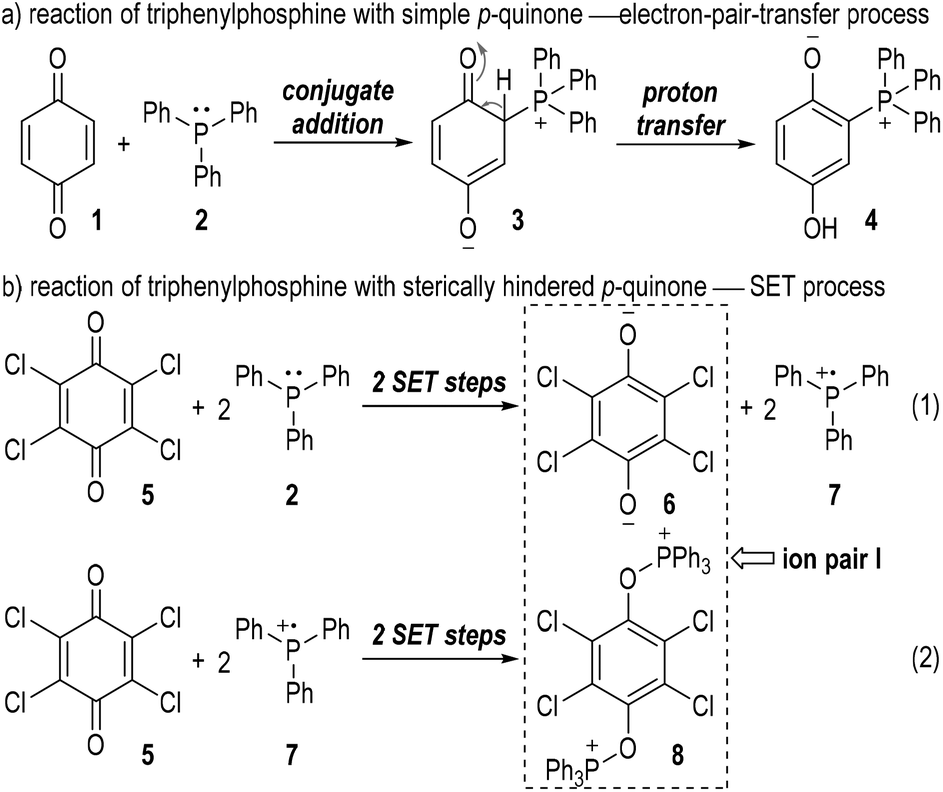 | ||
| Fig. 2 The first evidence of the radical ion species generated from tertiary phosphines. | ||
Although supportive demonstration for the formation of the radical cation 7 could be observed from paramagnetic resonance absorption measurements, there was no direct experimental evidence for the existence of such a species. In their following work the authors further studied the reaction mechanism by means of electron spin resonance (ESR) and UV absorption spectroscopy.7 A charge transfer complex 9 was expected to be formed (Fig. 3, eqn (1)) which rapidly underwent oxidations by chloranil 5 to give a chloranil semiquinone radical anion 10 together with a phenoxyradical phosphonium cation 11 (Fig. 3, eqn (2)).
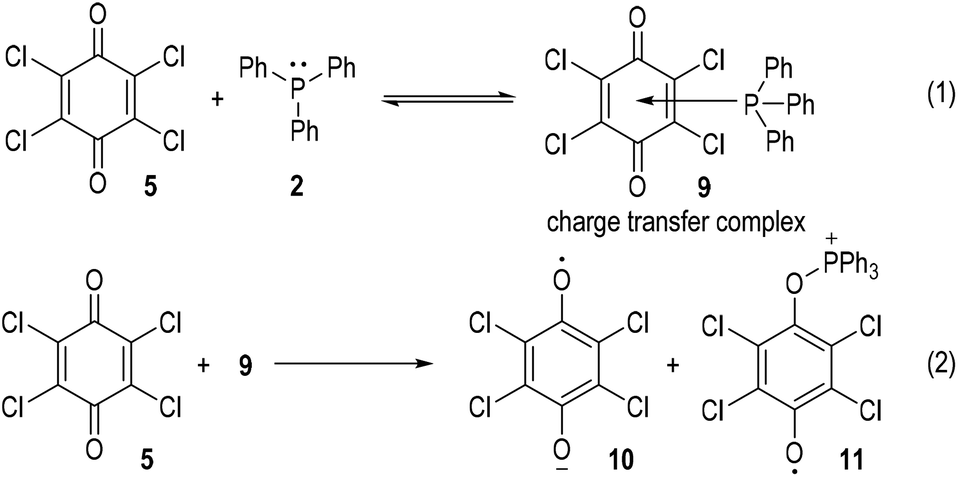 | ||
| Fig. 3 Another possible reaction pathway for the SET reaction between triphenylphosphine 2 and chloranil 5. | ||
Bartlett and co-workers also observed unusual catalytic activities of triphenylphosphine in the reactions with mercaptans, where the phosphinium radical cation 7 had been suggested as the reaction intermediate.8 But there had, again, been no definitive proof to support the generation of such phosphinium radical cations until in 1969 when Powell and Hall reported their observations on the racemization of an optically active triarylphosphine 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9 under the oxidation reaction of 7,7,8,8-tetracyanoquinodimethane (TCNQ) 13 (Fig. 4).10
9 under the oxidation reaction of 7,7,8,8-tetracyanoquinodimethane (TCNQ) 13 (Fig. 4).10
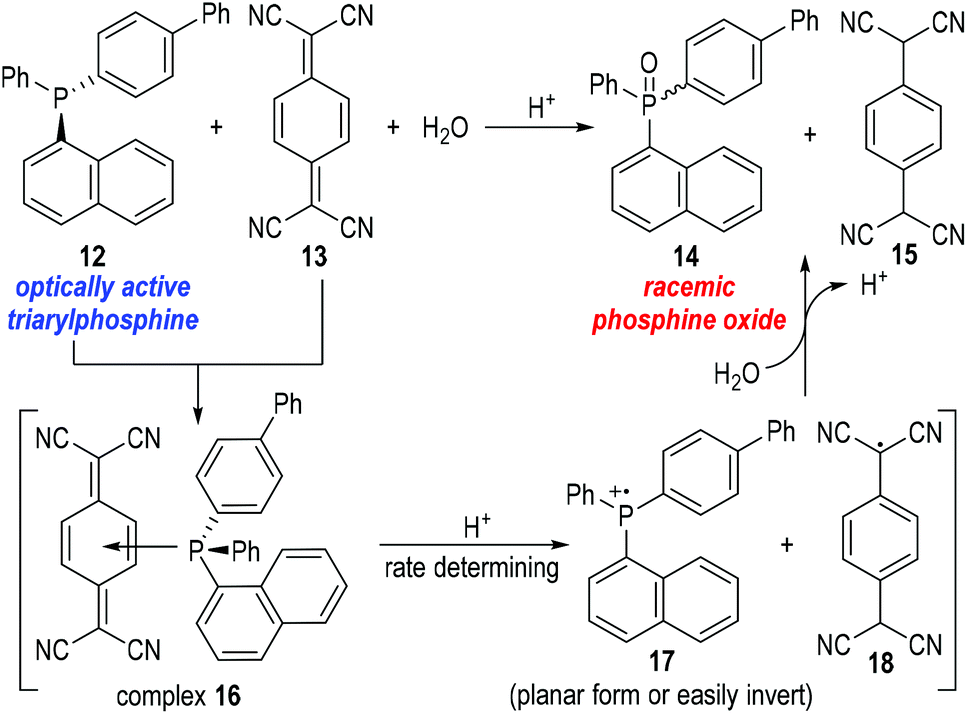 | ||
| Fig. 4 Oxidation of the chiral triarylphosphine 12 by TCNQ 13. | ||
Powell and Hall used optically active 4-biphenyl-α-naphthylphenylphosphine 12 to react with the oxidative TCNQ 13 in the presence of water and a catalytic amount of HCl. Racemic phosphine oxide 14 and TCNQH215 were afforded in almost quantitative yields. In a control experiment, optically active phosphine oxide (the chiral form of 14) could be obtained through oxidation of 12 with H2O2 in acetic acid and the afforded enantiomerically enriched product did not racemize in the presence of HCl or 15. The results of the control experiment indicated that the phosphine oxide 14 was racemized upon its formation from the reaction between 12 and 13. Therefore, a planar or easily inverted phosphinium radical cation 17 was postulated as the key intermediate for this oxidative process. Kinetic studies on this oxidation reaction suggested that complex 16 was quickly formed from 12 and 13, and underwent a rate-determining fragmentation with acids to give the radical cation 17 and the neutral radical 18. The radical species 17 and 18 then reacted with H2O to give the final product 14 and 15 and released one equiv. of proton for additional catalytic cycles. The radical cation 17 was believed to bear a planar structure or a low inversion energy barrier and thus gave the phosphine oxide product 14 in racemic forms.
2.2. Phosphine-associated radical reactions with iodine-containing oxidants
Perfluoroalkyl iodides can react with trivalent phosphines through SET processes and generate phosphinium radical cations together with free perfluoroalkyl carbon radicals. Various radical chain reactions can therefore be initiated through this protocol.In 1990, Huang and Zhang reported the first organophosphine-initiated radical addition reaction of perfluoroalkyl iodides 19 with alkenes 20 (Fig. 5).11 Both carbons of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of the alkenes 20 were functionalized by the perfluoroalkyl iodides 19 and the adducts 21 were afforded in good to excellent yields (Fig. 5a).
C double bond of the alkenes 20 were functionalized by the perfluoroalkyl iodides 19 and the adducts 21 were afforded in good to excellent yields (Fig. 5a).
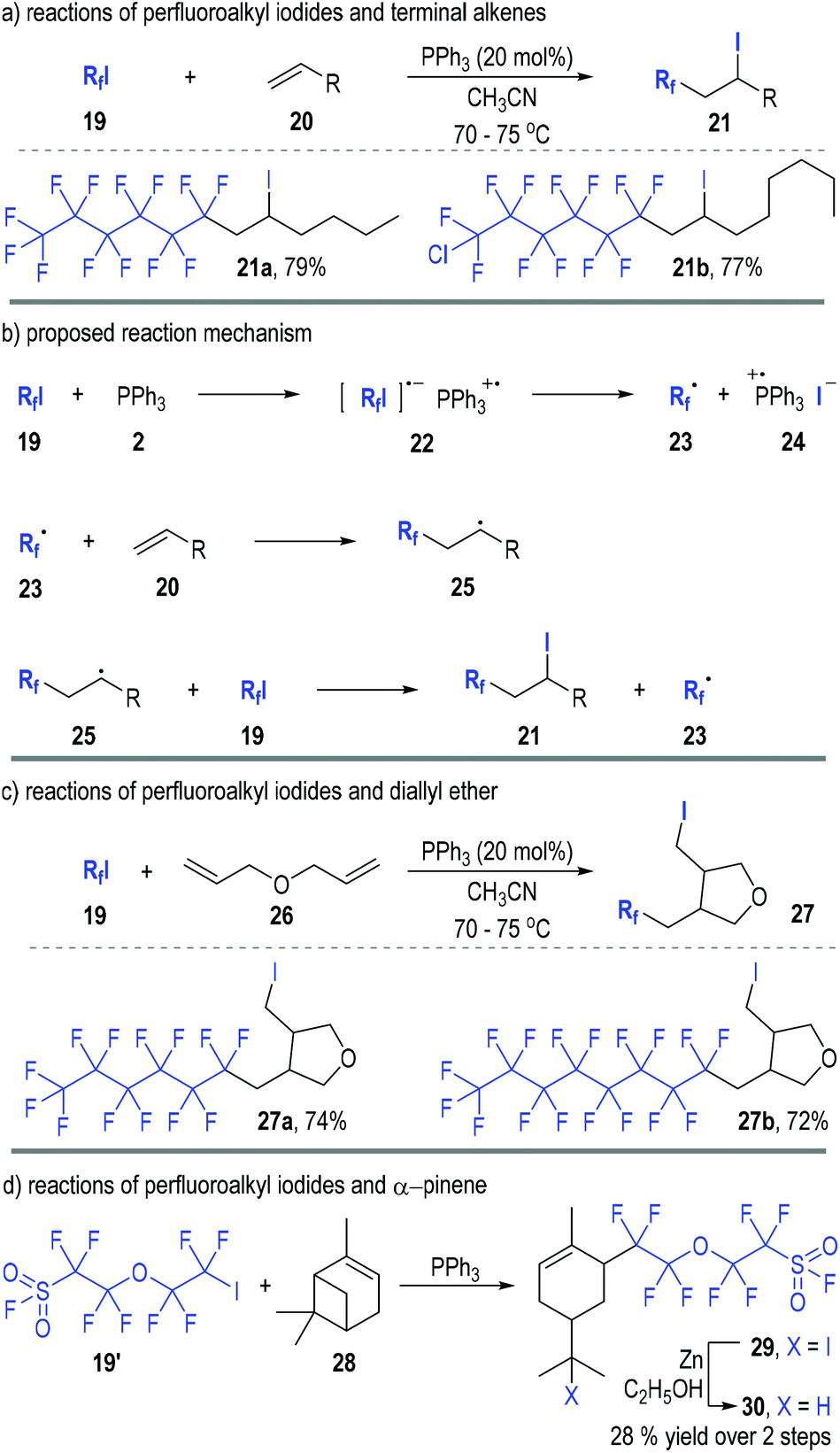 | ||
| Fig. 5 Phosphine initiated radical addition reaction of perfluoroiodides and alkenes. | ||
The reaction was believed to go through radical addition pathways (Fig. 5b). Perfluoroalkyl iodides 19 first reacted with triphenyl phosphine 2 to give a radical ion complex 22, which would quickly crack to the free carbon radical 23 and the phosphinium radical ion pair 24. The free radical 23 could add to alkene 20 and gave the radical adduct 25, which then reacted with another perfluoroalkyl iodide 19 to afford the final product 21 and release the free radical 23 for additional radical chain propagation processes.
The generation and participation of the radical intermediates in this reaction was supported by the cyclization reaction of perfluoroalkyl iodides 19 with diallyl ether 26 (Fig. 5c). The free radical intermediate 25 would preferentially react with an intramolecular CC double bond when the R group contained an alkene moiety (e.g., 26). Hence, the tetrahydrofuran derivatives 27 were obtained as the final products from the radical addition reactions between the perfluoroalkyl iodides 19 and the diallyl ether 26.
Interestingly, pinenes were also effective substrates for this radical addition reaction (Fig. 5d). For example, α-pinene 28 reacted with the perfluoroalkyl iodide 19′ in the presence of a catalytic amount of triphenylphosphine and created the rearrangement product 29via a radical-associated ring opening process. The iodide 29 was further reduced to 30 in a 28% overall yield after purification.
Moreno-Mañas and co-workers further studied this reaction in 2002.12 They monitored the reaction process with 31P NMR and found that triphenylphosphine had been the only significant phosphorus compound detectable at the beginning of the reaction. The signal for triphenylphosphine oxide appeared slowly during the reaction with no other phosphorus species observed. This indicated that the SET process from triphenylphosphine 2 to perfluoroalkyl iodides 19 was a rate determining step in this radical chain reaction.
It is worth noting that neither the work reported by Huang and Zhang nor the one by Moreno-Mañas and co-workers had commented on the potential roles of light in the phosphine-promoted alkene iodo perfluoroalkylation reactions.
In 2019, Czekelius and co-workers reported their discovery on the key roles of light in this transformation (Fig. 6).13 They have repeated some of the previously reported protocols12 for alkene perfluoroalkylations in the dark and found no conversions could be detected after 48 h. This is strong evidence supporting that the phosphine-associated perfluoroalkylation of alkenes is indeed a photo-induced radical reaction. The authors had systematically studied the reaction conditions for this alkene perfluoroalkylation reaction and found that the tri-tert-butylphosphine could efficiently promote this transformation under irradiation by a blue LED (461 nm). An electron donor–acceptor (EDA) complex was likely formed from the perfluoroalkyliodide and the trivalent phosphine. The EDA complex could be activated by blue light and fragment into the perfluoroalkyl radical 23. The radical 23 could react with the alkene 20 through a radical addition process and gave the radical adduct 25, which could react with another iodide 19 to afford the final product 21 and regenerate the perfluoroalkyl radical 23 for additional radical propagation reactions.
 | ||
| Fig. 6 The key role of light in trivalent phosphine associated alkene perfluoroalkylation. | ||
In addition, Zhao, He and co-workers further developed this chemistry and used the iododifluoroacetate 31 as the alkylation reagent in the phosphine-initiated difluoroalkylation reaction of alkenes (Fig. 7).14 In their report, they observed optical absorption by the reactants between 350 nm and 370 nm, which indicated that visible light might play unclarified roles in this transformation. Bis(diphenylphosphino)methane 34 was used as the radical chain initiator and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone 35 was used as an additive. The additive 35 could reduce the level of formation of the main by-product HCF2CO2C2H5 and thus enhance the yields of the target products. Two types of reaction products were obtained in this transformation. Alkyl substituted terminal alkenes gave the 1,2-addition products 32 in good to excellent yields with a broad range of substitution groups being tolerated. Substituted styrenes and internal aryl alkenes led to the Heck-type products 33 in moderate to excellent yields. The authors also studied the reaction mechanism through control experiments and provided substantial evidence for the involvement of radical intermediates in such transformations. The 1,2-addition products 32 were supposed to be afforded through a similar radical addition reaction pathway as depicted in Fig. 5b. However, when the R group of the radical intermediate 25 was a phenyl group (36, generated from styrene), it would be more stable than the alkyl radical species, thus giving a cation 37via a SET process. Deprotonation of the cation intermediate 37 finally led to the Heck-type product 33 in generally moderate to good yields.
 | ||
| Fig. 7 Phosphine-promoted difluoroalkylation of alkenes. | ||
The deoxygenation of alcohols (38) has always been a valuable strategy in organic synthesis.15 Koreeda and Zhang have proved that such important transformations could be efficiently achieved through a trivalent phosphine-associated radical reaction process (Fig. 8).16 The phosphite derivative 39 bearing an iodobenzene unit was used as the key reaction promoter in this strategy. Alcohol 38 was readily converted to the phosphite 40 by 39 under classical conditions. The iodophenyl-containing phosphite 40 could fragment homolytically into the radical intermediate 41 under standard radical initiation conditions. An intramolecular radical addition process led to the formation of the phosphoranyl radical 42, which could undergo a β-scission process to give the phosphinate 43 and the free carbon radical 44. The carbon radical could take away a hydrogen atom from the reducing reagent to afford the deoxygenated product 45 (Fig. 8a). Of note, a number of biologically interesting molecules containing hydroxyl groups17 could be effectively deoxygenated through this protocol (Fig. 8b).
 | ||
| Fig. 8 Phosphite-associated deoxygenation of alcohols. | ||
This phosphite-associated radical deoxygenation strategy was further developed by Miller and Jordan in 2012.18 They adapted this method to a catalytic phosphoramidite transfer process that they had previously developed (Fig. 9). Phosphoramidite 46 was used as the trivalent phosphite precursor and phenyltetrazole 47 was used as the reaction catalyst. The iodophenyl-containing phosphite 40 was efficiently afforded from 38 and 46 under the catalysis of 47. The released amine by-product could be scavenged by the phenyl isocyanate that had been added into the reaction system. Then the afforded crude phosphite intermediate (40) was directly subjected to a standard radical reaction condition without purification and gave the desired deoxygenated product 45 in good yields with retention of the optical purities. Highly oxygenated molecules such as 48 worked smoothly in this transformation, with the deoxygenated product 49 being secured in 84% yield.
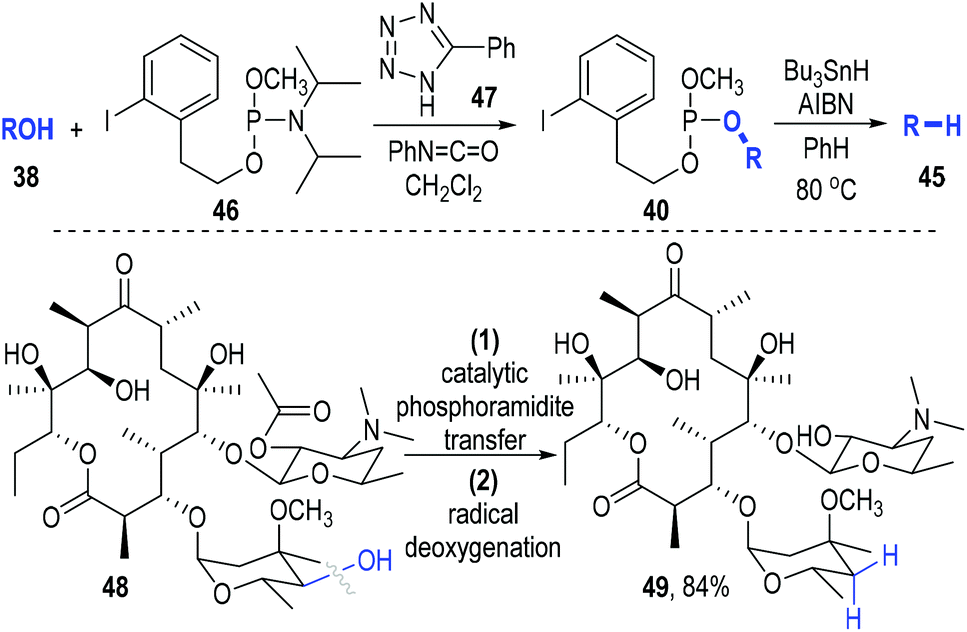 | ||
| Fig. 9 Combination of catalytic phosphoramidite transfer with radical deoxygenation of alcohols. | ||
In 2015, Liu, Tan and co-workers reported a phosphine-catalyzed radical reaction in the one-pot synthesis of bistrifluoromethylated enamindes (Fig. 10).19 Both the amide β-C–H and the alkene group of the N-(1-(2-allylphenyl)ethyl)benzamide 50 were functionalized by Togni's reagent 51 under the catalysis of the phosphine 52. The bistrifluoromethylated product 53 was afforded through multiple SET processes.
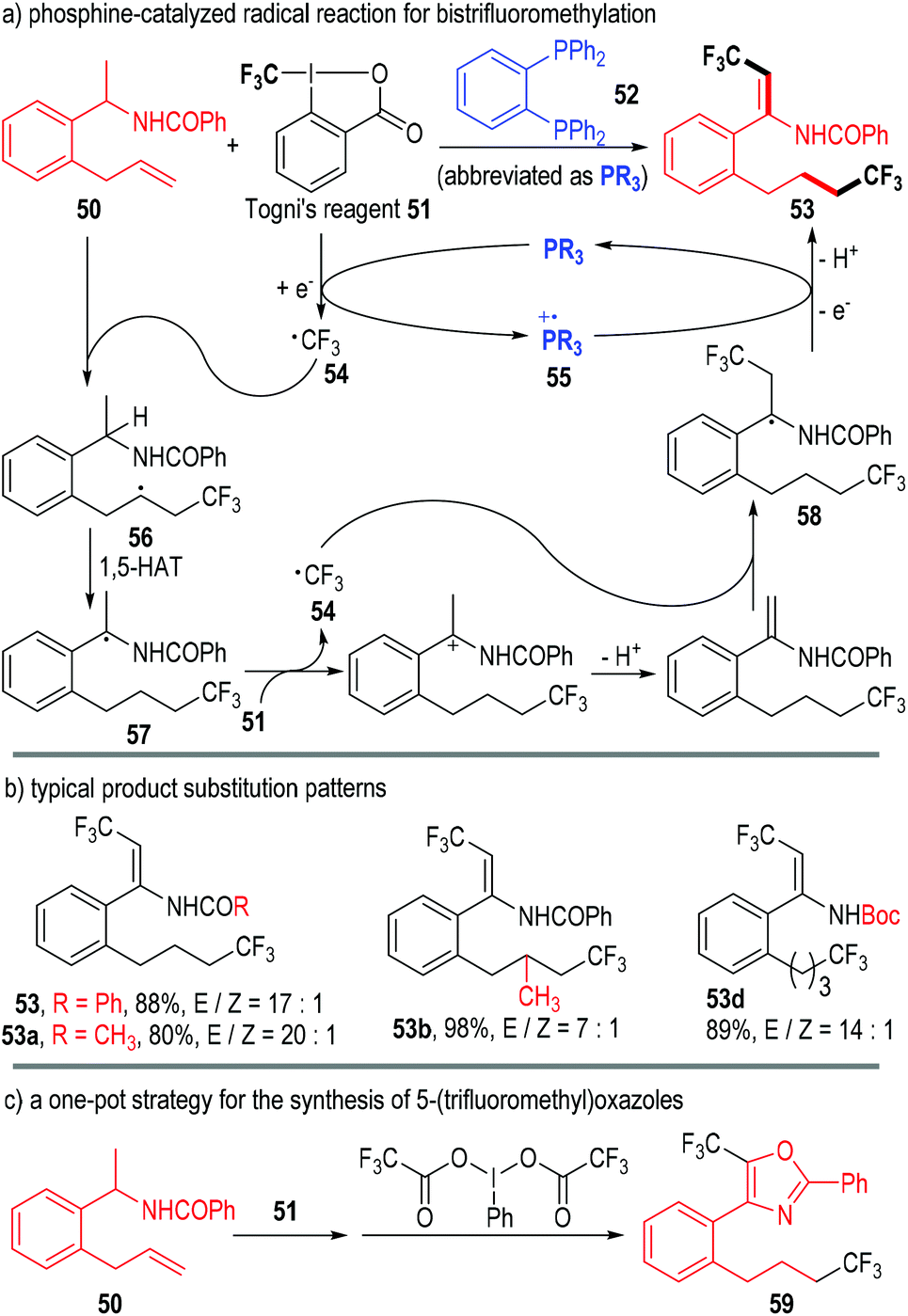 | ||
| Fig. 10 Phosphine-catalyzed bistrifluoromethylation of allyphenyl amide. | ||
The trivalent phosphine 52 first reacted with the oxidative Togni's reagent 51 through SET processes and gave the free trifluoromethyl radical 54 and the phosphinium radical cation 55. The carbon radical 54 next added onto the alkene unit of the substrate 50 and afforded the radical adduct 56. An intramolecular 1,5-hydrogen atom transfer (1,5-HAT) process in 56 led to the new radical intermediate 57, which then reacted with another Togni's reagent 51 through a SET/deprotonation process, generating the radical intermediate 58. One electron could be transferred from 58 to the phosphinium radical cation 55, and the product 53 was finally afforded via a further deprotonation step; meanwhile, the trivalent phosphine 52 was regenerated for subsequent catalytic cycles (Fig. 10a).
Various substituents and substitution patterns were well tolerated in this protocol and the corresponding products could be obtained in moderate to excellent yields and stereoselectivities (Fig. 10b).
The authors also successfully combined this transformation with an iodane-associated oxidative enamide cyclization reaction in one-pot. The bioactive molecule 59 bearing a 5-(trifluoromethyl)oxazole fragment was achieved in a good yield through this protocol (Fig. 10c).
2.3. Phosphine-associated radical reactions with electron-deficient nitrogen-containing oxidants
Aryldiazonium salts are also suitable oxidative reactants that can accept one electron from trivalent phosphine molecules. Yasui and co-workers reported in 1991 that the 4-nitrophenyldiazonium salt 60 could react with triphenylphosphine 2 in alcoholic solvents 61 and give nitrobenzene 62, triphenylphosphine oxide 63 and ketones 64 as the final products (Fig. 11a).20 A radical chain reaction mechanism was proposed for the generation of all the above products. Mechanism-wise, the 4-nitroarene 60 first reacted with triphenylphosphine 2 through a SET process and gave the radical intermediates 65 and 7. The aryl radical 65 could then react with the alcoholic solvent molecule 61 through a hydrogen-atom-transfer (HAT) process to afford the arene product 62 and the radical 66. A reaction between the radical intermediate 66 and another diazonium salt 60 gave the ketone product 64, with the aryl radical 65 being regenerated for further chain propagation reactions. In fact, the phosphinium radical cation intermediate 7 could also react with the alcohol 61 and give the deprotonated radical adduct 67, which would be quickly decomposed to give the phosphine oxide 63 and a carbon radical 68.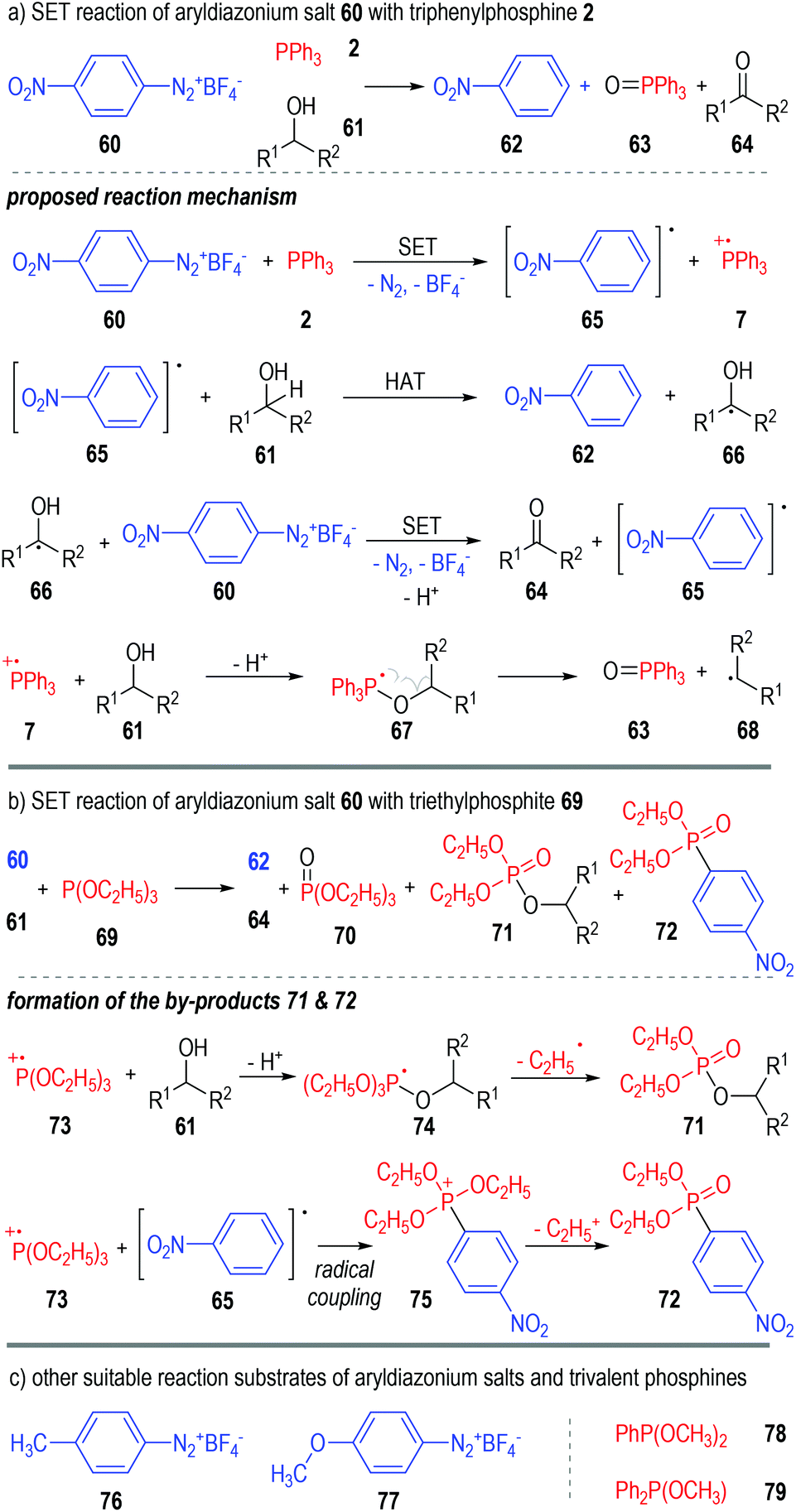 | ||
| Fig. 11 SET reactions of aryldiazonium salts with trivalent phosphines. | ||
The triphenylphosphine 2 used in this reaction can also be replaced with the triethylphosphite 69 (Fig. 11b). The formation of additional by-products 71 and 72 was observed in this case, which further supported the SET reaction mechanism as proposed. The transesterificated phosphate 71 was supposed to be formed through fragmentation of the deprotonated radical adduct 74. The radical coupling reaction between 73 and 65 gave the phosphonium intermediate 75, which then released one equiv. of the ethyl carbocation and afforded 72 as the stable product.
Switching the electron-withdrawing 4-NO2 group on the nitrophenyldiazonium salt 60 into electron-donating groups (e.g., 4-CH3, 76 & 4-OCH3, 77) was found to have little effect on the target product yields (Fig. 11c, left part).21
Similarly, several trivalent phosphines such as dimethyl phenylphosphonite 78 and methyl diphenylphosphinite 79 were also suitable single-electron donors for this radical chain reaction (Fig. 11c, right part).22
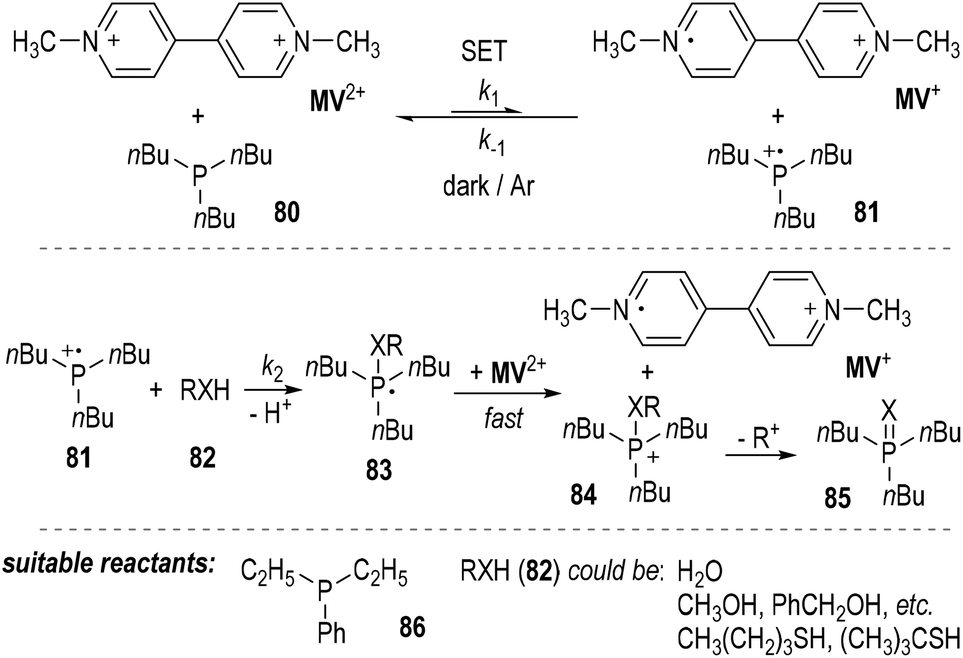
The authors also found that 1,1′-dimethyl-4,4′-bipyridinium dication (methylviologen, abbreviated as MV2+) could be used as a suitable one-electron acceptor for the SET reactions employing trivalent phosphorus compounds (Fig. 12).23MV2+ was readily reduced by tributylphosphine 80 in the presence of water, alcohols or thiols. The one-electron-reduced form MV+ was gradually accumulated without the formation of the two-electron-reduced product of MV0. The generation of the MV+ from MV2+ was believed to result from a SET process between tributylphosphine 80 and MV2+ (Fig. 12, top). The afforded phosphinium radical cation 81 then reacted with the nucleophilic compound 82 and gave a phosphoranyl radical 83, which could be quickly oxidized by MV2+ to become a phosphonium ion 84 and another one-electron-reduced product of MV+. Elimination of an alkyl carbocation (R+) from 84 led to the formation of the oxidized product 85 (Fig. 11, middle). Diethylphenylphosphine 86 could also be used instead of the tributylphosphine 80 as the reducing reagent in this reaction. Water and various alcohols and thiols could be used as the oxidative quenching reagents (Fig. 12, bottom).
 | ||
| Fig. 12 One electron reduction of MV2+ by trivalent phosphines. | ||
Kinetic studies revealed that the reaction was not a simple first-order reaction.24 The reaction rate of the backward SET process from MV+ and 81 (k−1) is comparable with that of the ionic reaction between 81 and 82 (k2). Since the oxidation potential of the phosphoranyl radical 83 was estimated to be much lower than that of MV2+, the SET oxidation of 83 by MV2+ was believed to be very fast and irreversible. When benzyl alcohol was used as the oxidative quenching reagent 82 (RXH), benzyl ether was detected as a by-product. This observation further supported the radical reaction mechanism, since benzyl ether would be generated during the decomposition process of the phosphonium ion 84 to the oxidized product 85.
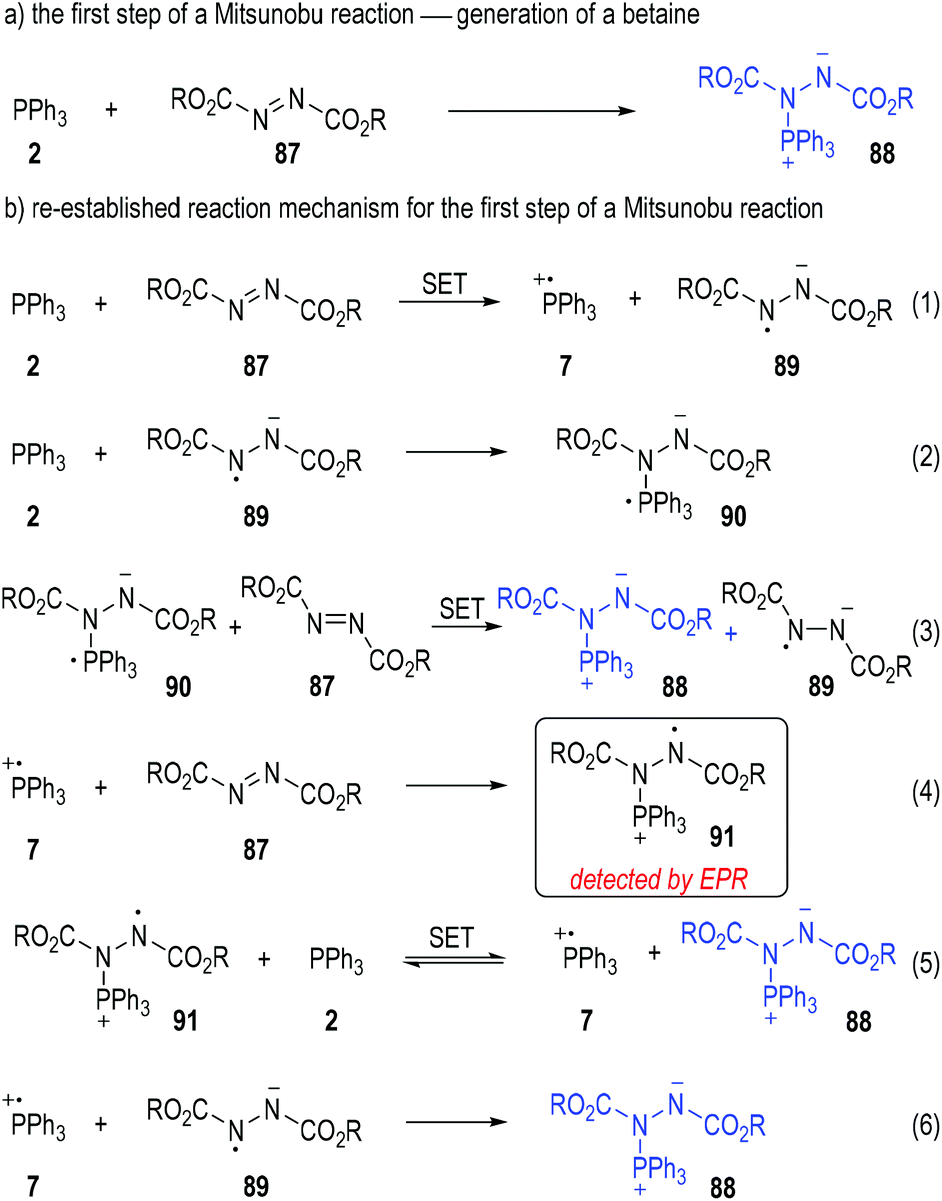
Disubstituted azo molecules are significant nitrogen-containing oxidants in organic synthesis. They are widely employed in the Mitsunobu reaction25 to convert the hydroxyl group existing in an organic compound into other nucleophilic groups. It is widely accepted that the first step of a Mitsunobu reaction involves the formation of a betaine (88) through a Michael-type nucleophilic addition of triphenylphosphine (2) to the dialkyl azodicarboxylate (87) (Fig. 13a).26 However, the yellow to deep red coloured solution generated from mixing the triphenylphosphine and dialkyl azodicarboxylates prompted scientists to re-evaluate the reaction mechanism of the beginning step.27
 | ||
| Fig. 13 Re-investigation of the mechanism of Mitsunobu reaction. | ||
Mitsunobu has first noted the colour changes in these reactions and suggested that radical species might be generated during these processes.28 Jenkins and co-workers further studied the reaction mixtures with electron paramagnetic resonance (EPR) spectroscopy in 1995 and observed the formation of radicals in these processes.29 With this in mind, Jenkins and co-workers claimed that it was also possible to form the betaine intermediate 88via a radical chain mechanism (Fig. 13b).
Triphenylphosphine 2 could first react with the azodicarboxylate 87 through a SET process to give the radical cation 7 and the radical anion 89 to initiate the radical chain processes (Fig. 13b, eqn (1)).
When triphenylphosphine 2 was present in an excess amount in the reaction mixture, the radical anion 89 would react with another molecule of 2 and afforded the new radical anion 90 (eqn (2)). The azodicarboxylate 87 could then receive one electron from 90 to form the target betaine 88, with the radical anion 89 regenerated (eqn (3)) for more chain propagation reactions (eqn. (2) and (3)).
In the case that the azodicarboxylate 87 was present in an excess amount, the radical cation 7 generated from eqn (1) would react with 87 to give the new radical cation 91, which then took one electron from 2 to afford the betaine 88 with the phosphinium radical cation 7 regenerated as radical chain carriers (eqn (4) and (5)).
The radical chain would be finally closed when both the radical cation 7 and the radical anion 89 existed in large amounts in the reaction mixture, since they would collide into the betaine 88 through radical coupling reactions (eqn (6)).
As a technical note, the formation of the radical cation 91 was detected by EPR analysis during the reactions (eqn (4)). None of the other radical species was clearly observed in this study, probably because of the very short lifetime of these highly reactive transient structures.
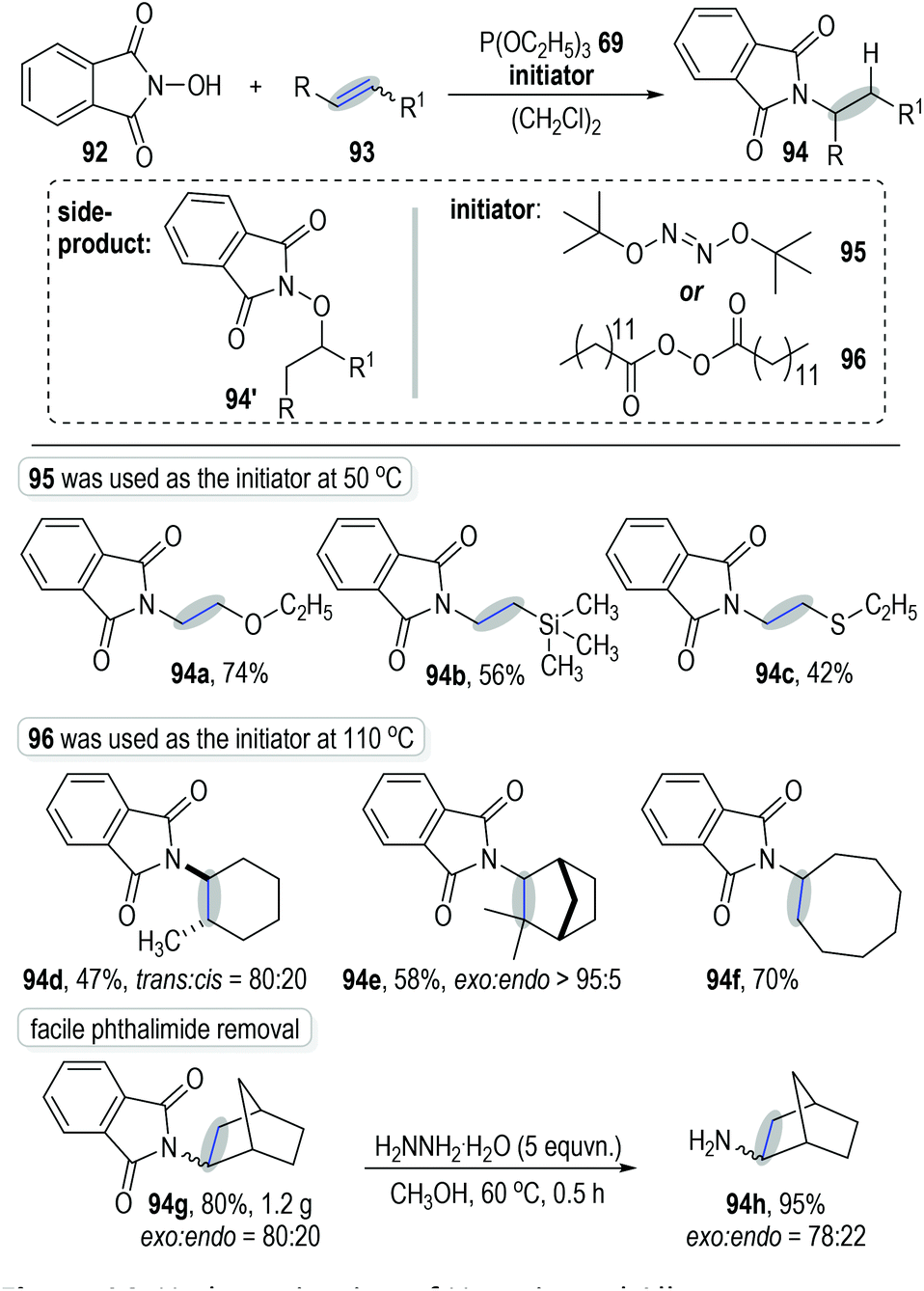
Schmidt and Lardy developed a novel method for the anti-Markovnikov alkene hydroamination through a trivalent phosphite-associated radical addition reaction (Fig. 14).30N-Hydroxyphthalimide 92 was used as the amination reagent to react with alkene 93 under the catalysis of phosphite 69 and gave the hydroaminated product 94 with moderate to excellent isolated yields. The polar addition product 94′ was observed as the main side-product at high reaction temperatures. Therefore, tert-butyl hyponitrite 95, which displayed a lower fragmentation temperature, was applied as the initiator for this radical chain reaction. Excellent anti-Markovnikov regio-selectivities were observed when using electron-rich alkenes as the reaction substrates (e.g., Fig. 14, 94a to 94c). However, when electron-unbiased alkenes were used as reaction substrates, dilauroyl peroxide 96 was needed as the radical initiator at a higher temperature in order to reach satisfactory product yields (e.g., 94d to 94f). The radical addition reactions could be carried out at gram scales without obvious erosion on the product yields (e.g., 94g). Of note, the phthalimide protecting group existing in the final products could be effectively removed through simple protocols to give the corresponding free primary amines in good yields (e.g., 94h).
 | ||
| Fig. 14 Hydroamination of unactivated alkenes. | ||
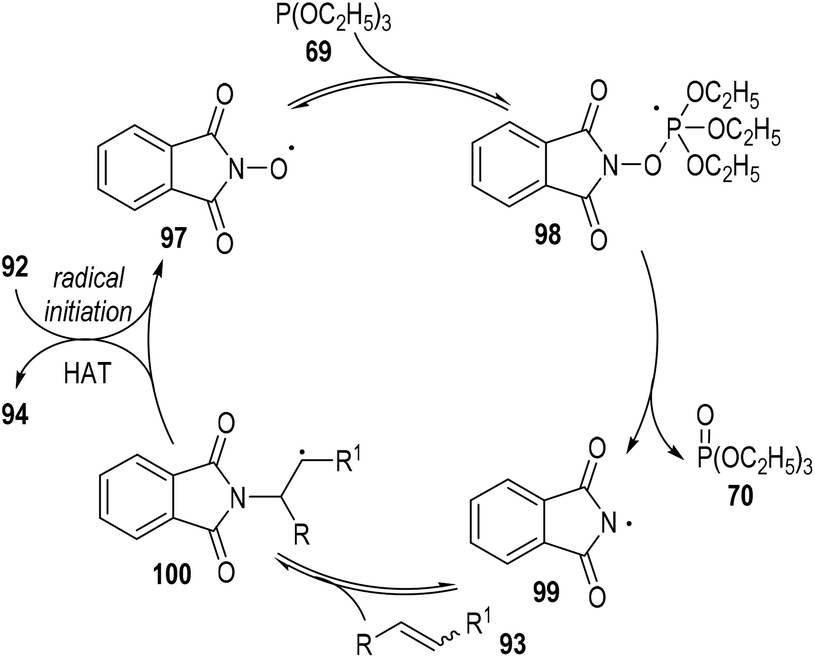
A radical chain reaction mechanism was proposed for this phosphite-associated alkene hydroamination reaction (Fig. 15). The active radical precursor 95 or 96 could fragment into free radicals under heating conditions and abstracted a hydrogen atom from the N-hydroxyphthalimide 92 to give the O-centred radical 97. The radical 97 then added to the phosphite 69 and gave the phosphoranyl radical intermediate 98, which could then go through a β-scission step to generate the N-centred radical 99 with one equiv. of phosphate 70 released. The phthalimidyl radical 99 could react with the alkene substrate 93 and give the radical adduct intermediate 100. Finally, the free carbon radical 100 took away one hydrogen atom from the N-hydroxyphthalimide 92 to afford the target product 94, with the O-centred radical 97 regenerated for additional propagation reactions.
 | ||
| Fig. 15 Proposed reaction mechanism for the hydroamination of unactivated alkenes. | ||
3. Radical reactions of tertiary phosphines in irradiative reactions
3.1. Generation and detection of radical species from tertiary phosphines under irradiation
The characterization of phosphine-centered radical species has been well established in the early 1970s.31 Meanwhile, ESR spectroscopy has played a key role throughout the 1970s. A variety of tertiary phosphines were exposed to 60Co γ-rays at 77 K and the reaction mixtures were subjected to ESR analyses.32 To cite the trialkylphosphite 101 as an example (Fig. 16),32b the phosphine-centered radical cation (e.g., 102), dimeric radical cation (e.g., 103), and neutral radical (e.g., 105) can all be detected through this method. The reaction mechanisms for the generations of these radical species have been postulated based on the evidence of the detected radicals and the afforded reaction products. The radical anion 104 generated from an electron addition to the trialkylphosphite 101 was detected. It might quickly fragment into the neutral radical 105 and the alkoxyl anion 106. The free O-centered radical 107 and the C-centered radical 110 could also be sometimes observed due to fragmentations of the dimeric radical cation 103 and the phosphoranyl radical 108. The trialkyl phosphate 109 was always noticed as the final product in this phosphite irradiative transformation.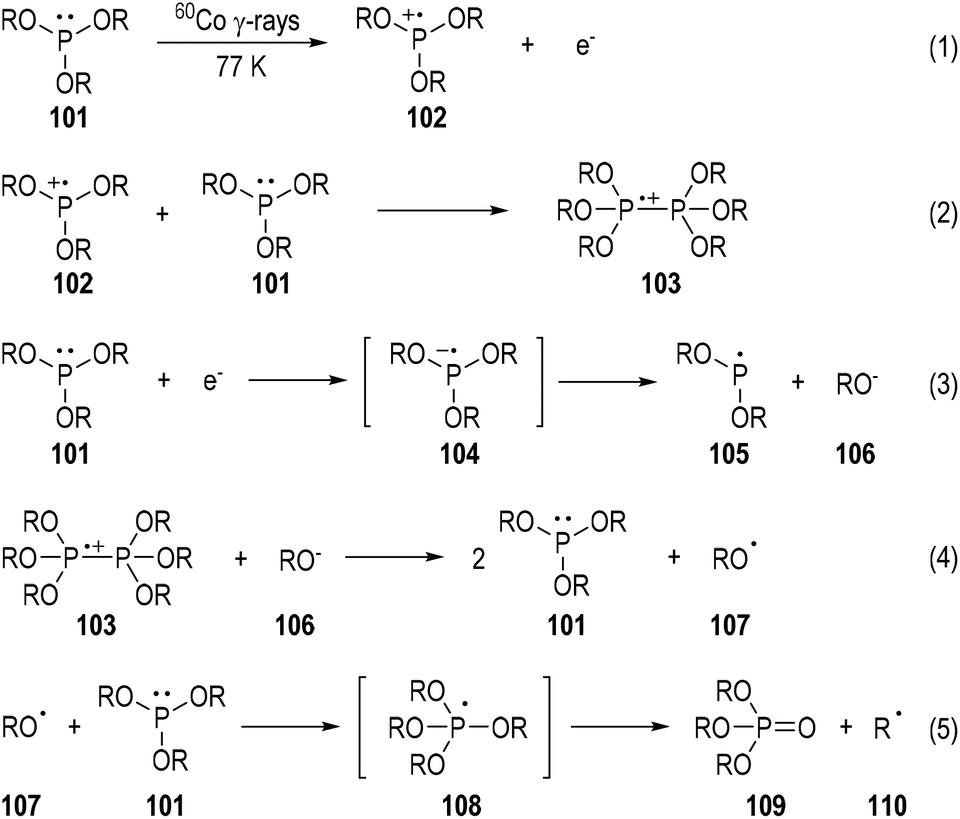 | ||
| Fig. 16 ESR studies on the radiation mechanisms for the generations of phosphine radical species. | ||
3.2. Photo-induced radical reactions of tertiary phosphines with electron-deficient arenes
Electron-deficient aromatic nitriles have long been used as good electron acceptors in photo-induced electron-transfer reactions.33 The solution of 1,4-dicyanonaphthalene (abbreviated as DCN, 111) could give fluorescence emission at 395 nm when it was exposed to photoirradiation at 320 nm (Fig. 17, eqn (1)).34 The fluorescence could be effectively quenched by trivalent phosphines via SET processes (eqn (2) to (5)).35 The photo-excited 111* could accept one electron from the electron-rich triphenylphosphine 2 and give the radical ionic pairs 7 and 112 (eqn (2)). The radical cation 7 then reacted with the nucleophilic H2O to give the radical cation intermediate 113 (eqn (3)), which subsequently transferred one electron to 111 and afforded the phosphoranyl cation 114 after a deprotonative process (eqn (4)). A further deprotonation of 114 led to the formation of the triphenylphosphine oxide 63 as the final product (eqn (5)). It is worth noting that no intermolecular reaction product from 2 and 111 was observed in this process. The DCN 111 was recovered in almost quantitative yield after this photolysis reaction. A further redox process between the radical anion 112 and the released protons might happen in the reaction system.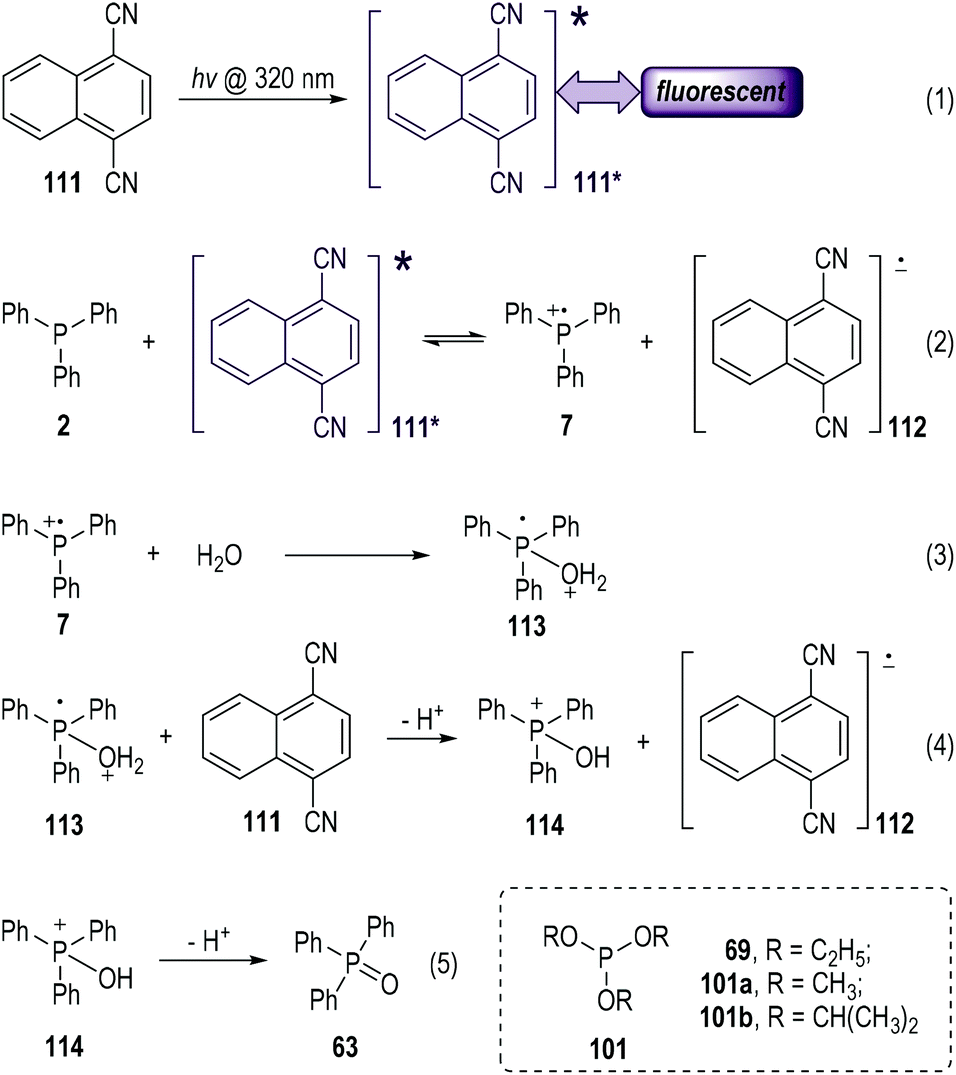 | ||
| Fig. 17 Quenching of DCN fluorescence by trivalent phosphines. | ||
As a technical note, the electron transfer process depicted in eqn (2) would undergo a backward process to re-generate 2 and 111 (111*) in the absence of H2O that was used for the fast deactivation of the radical cation 7. Moreover, various trivalent phosphites (e.g., 69, 101a, 101b) also worked well in this process and gave the corresponding trialkyl phosphates as the radical reaction products.
9,10-Dicyanoanthracene 115 can also be used as the reaction promoter for this photo-induced SET process.36 Visible light can activate 115 to generate the first excited singlet state 115*, which is an effective SET reagent for the intramolecular rearrangement of the 2-phenylallyl phosphite 116 (Fig. 18).37 One electron was readily transferred from the trivalent phosphite 116 to the excited intermediate 115* and the phosphinium radical cations 117 (117a & 117b) were afforded with elimination of the radical anion 115˙−. Since the oxidation potential of the trivalent phosphite is comparable to that of styrene, both of the radical cations 117a and 117b might be formed in this SET oxidative step. However, the radical cations 117a and 117b gave the same phosphoranyl radical cation intermediate 118 through intramolecular cyclization processes. β-Scission of the phosphoranyl radical cation 118 led to the phosphonate-containing carbon radical cation 119, which absorbed one electron from the radical anion 115˙− to give the final rearrangement phosphonate product 120 with regeneration of the 9,10-dicyanoanthracene 115 for further photo-induced SET processes. Alternatively, the cyclized radical cation 118 could absorb one electron from 115˙− to give the diradical intermediate 121, which then went through a radical coupling process to break the ring structure and afford the target rearrangement product 120.
 | ||
| Fig. 18 Rearrangement of 2-phenylallyl phosphite. | ||
This photo-induced radical reaction showed good functional group tolerance (e.g., 116a, 116b) with the corresponding phosphite rearrangement products being obtained in moderate to good yields.
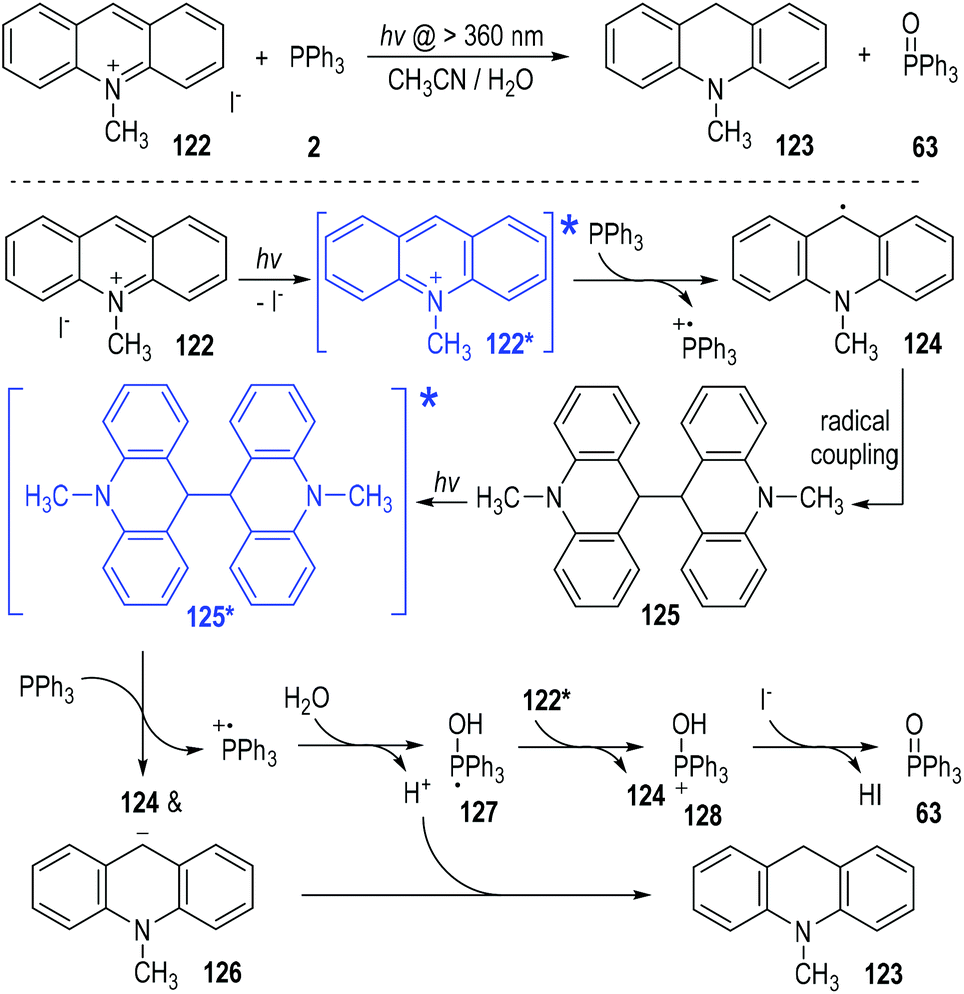
In 1991, Yasui and Ohno reported a photo-induced SET reduction of 10-methylacridinium iodide 122 by triphenylphosphine 2 in aqueous solutions (Fig. 19).38 The hydrogenated 10-methylacridan 123 and the triphenylphosphine oxide 63 were afforded as the final products. The 10-methylacridinium ion of 122 was activated under visible light irradiation to give the excited state 122*, which was ready to accept one electron from triphenylphosphine to give the free radical 124. Homo radical coupling of the radical 124 gave the dimer intermediate 125. The intermediate 125 was stable in aqueous methanol and could be isolated in good yield at the early stage of the reaction process. However, in acetonitrile the dimer 125 was further reduced since it was slightly soluble into the reaction system. The dimer 125 could also absorb visible light to form the excited state and get one electron from triphenylphosphine to crack into the free radical 124 and the carbanion 126. The carbanion 126 could be protonated by the aqueous system to give the final product 123. The triphenylphosphinium generated during the SET processes could react with H2O or the alcoholic solvents to form the phosphoranyl radical 127. Transferring one electron from 127 to the electron-deficient methylacridinium ion of 122* led to the phosphoranyl cation 128, which was finally deprotonated to afford the phosphine oxide 63 as the product. The counter anion of I− existing in 122 was considered as the base to facilitate the deprotonation step of the intermediate 128.
 | ||
| Fig. 19 Reduction of 10-methylacridinium ion by triphenylphosphine. | ||
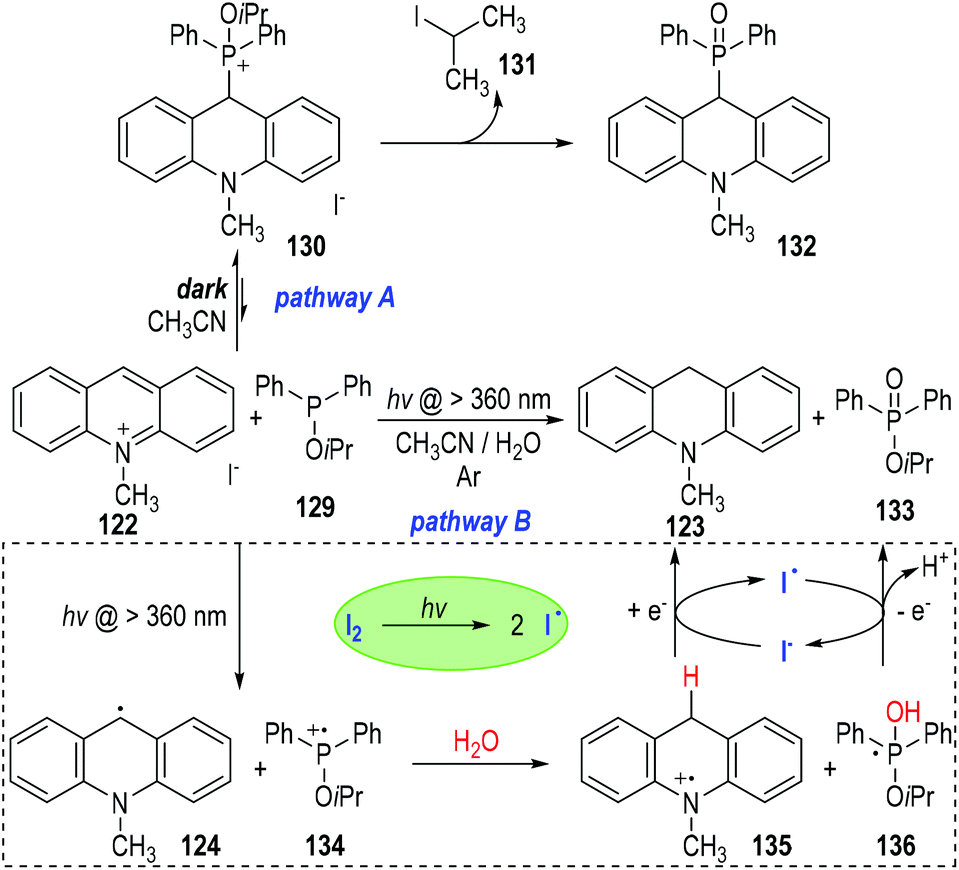
The authors also observed two different reaction pathways between the 10-methylacridinium iodide 122 and the isopropyl diphenylphosphinite 129 (Fig. 20).39 An intermolecular nucleophilic addition of 129 to 122 happened in the dark to give a phosphonium ion 130, which gradually decomposed to give the phosphine oxide 132 as the final product (Fig. 20, pathway A). However, when the reaction system was irradiated with visible light under an argon atmosphere, 10-methylacridan 123 and isopropyl diphenylphosphinate 133 were formed as the final products (pathway B).
 | ||
| Fig. 20 Photo-induced reduction of 10-methylacridinium iodide by isopropyl diphenylphosphite. | ||
The formation of 10-methylacridan 123 and isopropyl diphenylphosphinate 133 was believed to go through a photo-induced radical reaction pathway.40 It has been mentioned that 122 could be activated by visible light to form the excited state 122* (Fig. 19). The excited methylacridinium 122* was readily to accept one electron from the trivalent phosphine species 129 and afforded the radical 124 and the phosphinium cation 134. Reactions of these radical intermediates with water gave the new radical cation 135 and the phosphoranyl radical 136 as the reaction intermediates. An electron transfer process between 135 and 136 finally produced the desired product of 123 and 133, respectively. It should be noted that, the electron transfer process between 135 and 136 could be promoted by a catalytic amount of I2, which could crack into the free iodo radicals I˙ to carry on the single electrons between 135 and 136.
3.3. Photo-induced radical reactions of tertiary phosphines with iodonium salts and iodides
Disubstituted iodonium salts41 are reactive reagents and can be used as effective single-electron oxidants in the radical reactions with tertiary phosphines. Particularly, alkynyliodonium tetrafluoroborate 137 could react with triphenylphosphine 2 at −78 °C in dichloromethane under sunlight irradiation (Fig. 21).42 The synthetically useful (alkylethynyl)triphenylphosphonium salt 138 was afforded as the final product in almost quantitative yield. The reaction was believed to go through a visible light-induced SET process. The hypervalent iodine(III) was activated by sunlight irradiation and obtained one electron from triphenylphosphine 2 to give the radical cation 7 and the iodo radical 139. Radical 139 added onto the triphenylphosphine 2 to afford the phosphoranyl radical 140, which underwent an intramolecular ligand coupling process to give the new phosphoranyl radical 141 with elimination of a phenyl iodide. The phosphoranyl radical 141 transferred one electron to the iodonium salt 137 and gave the target product 138 with the regeneration of the iodo radical 139 for additional radical propagation reactions. In our opinion, the SET process between the phosphoranyl radical 141 and the phosphinium radical cation 7 could also happen and facilitate the formation of the final product 138 with the regeneration of triphenylphosphine 2.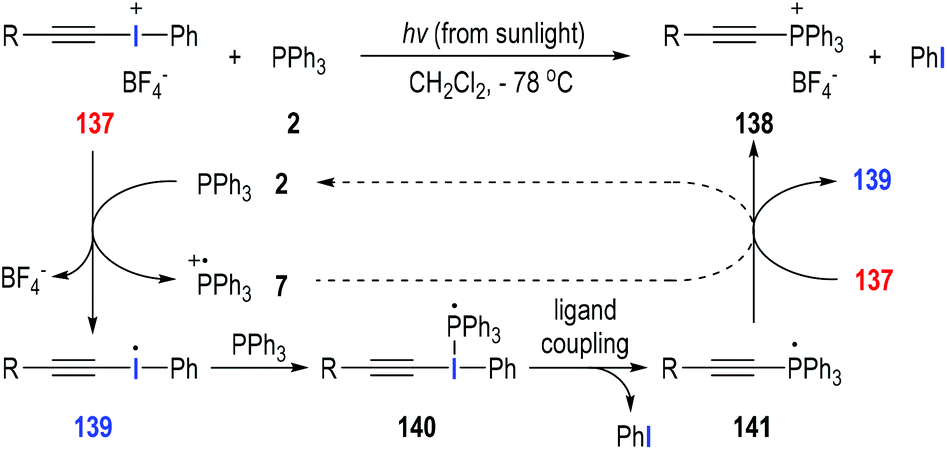 | ||
| Fig. 21 Preparation of (alkylethynyl)triphenylphosphonium salts through photo-induced radical reactions. | ||
It has also been well-known that trivalent phosphines can react with various alkyl iodides through SET processes and generate reactive radical intermediates for versatile reactions (for details, see section 2.2). Visible light has also proven to be crucial in some of these transformations (section 2.2, Fig. 6).13 Moreover, the formation of a phosphine-iodine complex radical (e.g., Ph3P-I˙) have been observed via EPR spectroscopy in the early 1970s.43
However, in the field of photoredox catalysis,44 the most applied reaction catalysts were expensive transition-metal complexes45 and organic dyes.46 To address the shortcomings of existing catalysts, the application of inexpensive and readily available phosphine-iodine complexes as catalysts in photoredox reactions became attractive.
Very recently, Shang, Fu and co-workers disclosed a photo-induced, triphenylphosphine and sodium iodide co-catalyzed decarboxylative alkylation reaction (Fig. 22).47 The aliphatic redox-active ester 142 was used as the alkyl radical precursor to react with silyl enol ether 143 through photocatalytic SET processes (Fig. 22a). The alkylated ketones 144 were afforded as the final products in moderate to excellent yields. The reaction could tolerate a broad range of substituents and substitution patterns. N-Heteroarenes 145 could also be used as the alkyl radical acceptors in this protocol, with the Minisci-type reaction products 146 being afforded in good yields with excellent regio-selectivities (Fig. 22b). Gratifyingly, this photocatalytic Minisci-type decarboxylative alkylation reaction could also be carried out in enantioselective fashion (Fig. 22c). In this case, chiral phosphoric acid 150 was used in a catalytic amount instead of trifluoroacetic acid in the photo-induced radical coupling reaction between 147 and 148. The corresponding Minisci-type product 149 was afforded in an excellent yield and optical purity, only under slightly adjusted catalytic conditions.
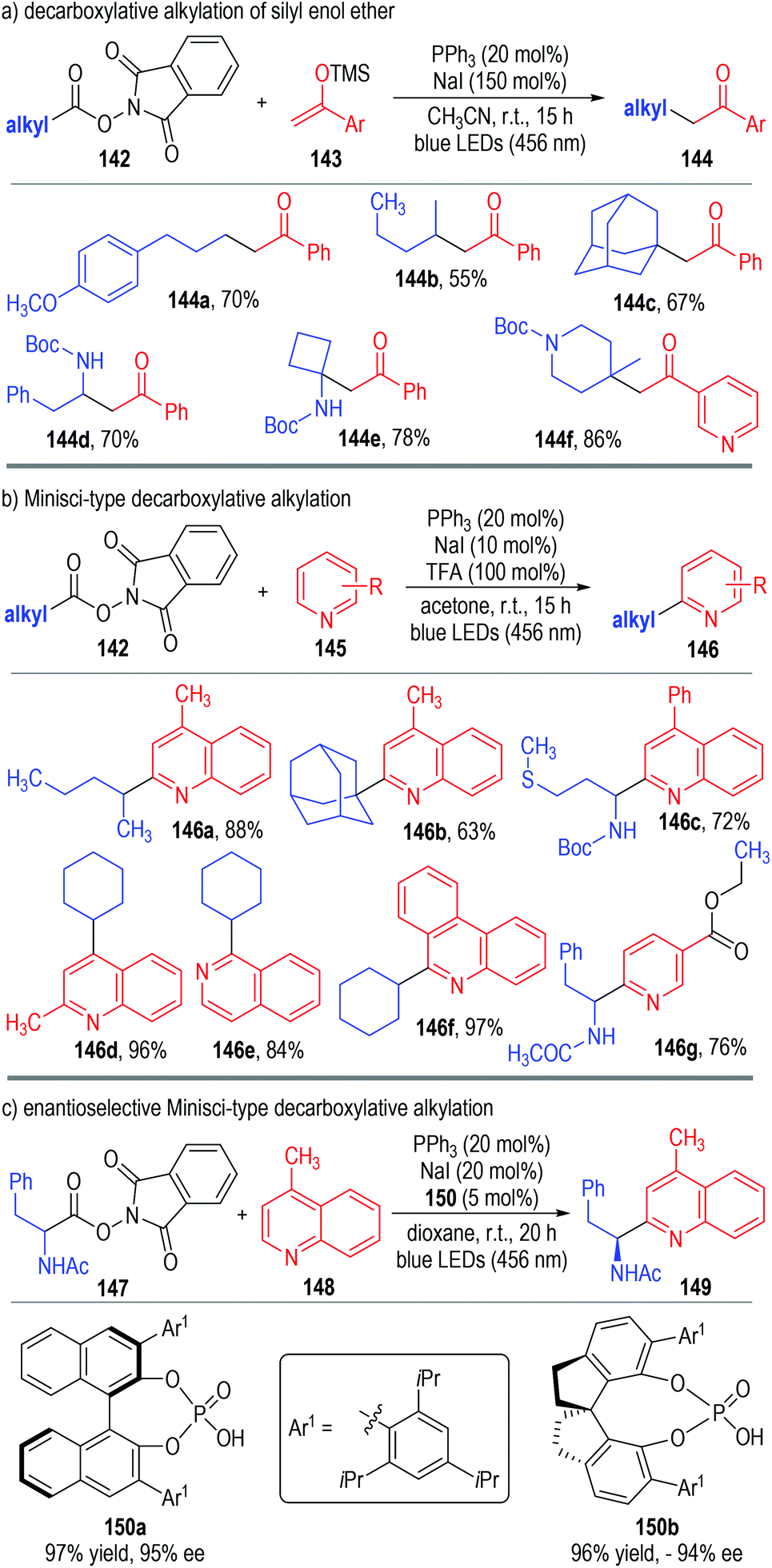 | ||
| Fig. 22 Triphenylphosphine and sodium iodide mediated photocatalytic decarboxylative alkylations. | ||
The authors have studied the reaction mechanism through both experimental and computational methods (Fig. 23). Control experiments have confirmed the essential roles of NaI, PPh3 and irradiation. The non-covalent complex 151 was formed reversibly from NaI and PPh3 and was ready to coordinate with the aliphatic redox-active ester 142 to afford the charge transfer complex 152. The complex 152 fragmented under visible light irradiation to give the phosphine-iodine radical 153 and the radical anion 154. The radical anion 154 was not stable and was readily to fragment into the alkyl radical 155 and the anion 156 with the elimination of CO2. The alkyl radical 155 reacted with the protonated quinoline 157 through a radical addition process to give the radical cation 158, which then reacted further with the phosphine-iodine radical 153 through a SET process to afford the desired product 159 with elimination of the corresponding counter acidic by-products (160 and 161).
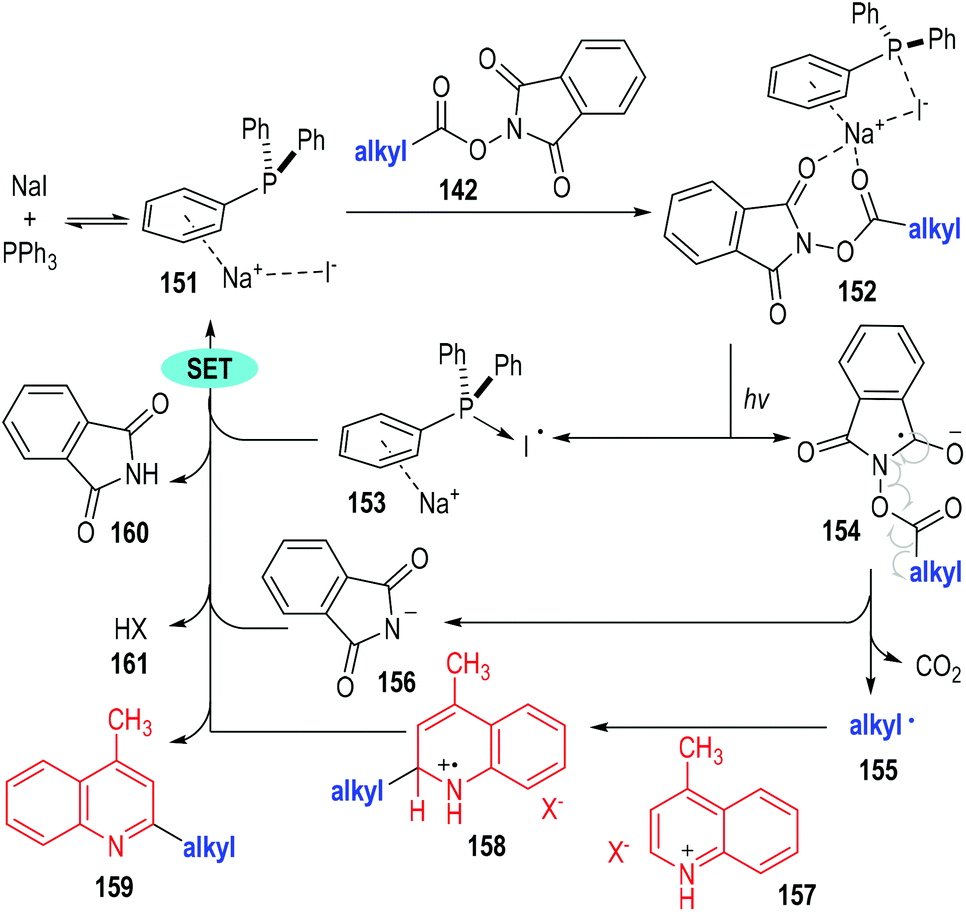 | ||
| Fig. 23 Postulated reaction mechanism for the photocatalytic decarboxylative alkylation. | ||
3.4. Photo-induced radical reactions of tertiary phosphines with transition metal complexes
Tertiary phosphines can also transfer one electron to the excited state of various transition metal complexes and give the reactive phosphinium radical cation intermediates for SET transformations. The combination of the transition metal-catalyzed photo-redox transformations with tertiary phosphine associated radical reactions can provide effective protocols for several otherwise challenging synthesis tasks.In 2018, Xie, Zhu and co-workers reported a phosphine-associated photoredox approach for the deoxygenative functionalization of carboxylic acids (Fig. 24).48 Aryl carboxylic acids 162 could be effectively deoxygenated through this protocol and react with alkenes 163 to give the aryl alkyl ketones 164 as the final products. The iridium complex salt 165 was used as the photoredox catalyst. Both the stoichiometric amount of triphenylphosphine and the irradiation with blue LED were essential for this transformation. A wide range of substrates bearing different substituents and substitution patterns could be used in this approach, with the target ketone products being afforded in moderate to good yields (Fig. 24a). Of note, this method was useful in the late-stage modification of complex structures. A variety of structurally sophisticated bioactive molecules could be effectively transformed to their corresponding ketone derivatives with moderate to good yields and stereoselectivities (e.g., 164′).
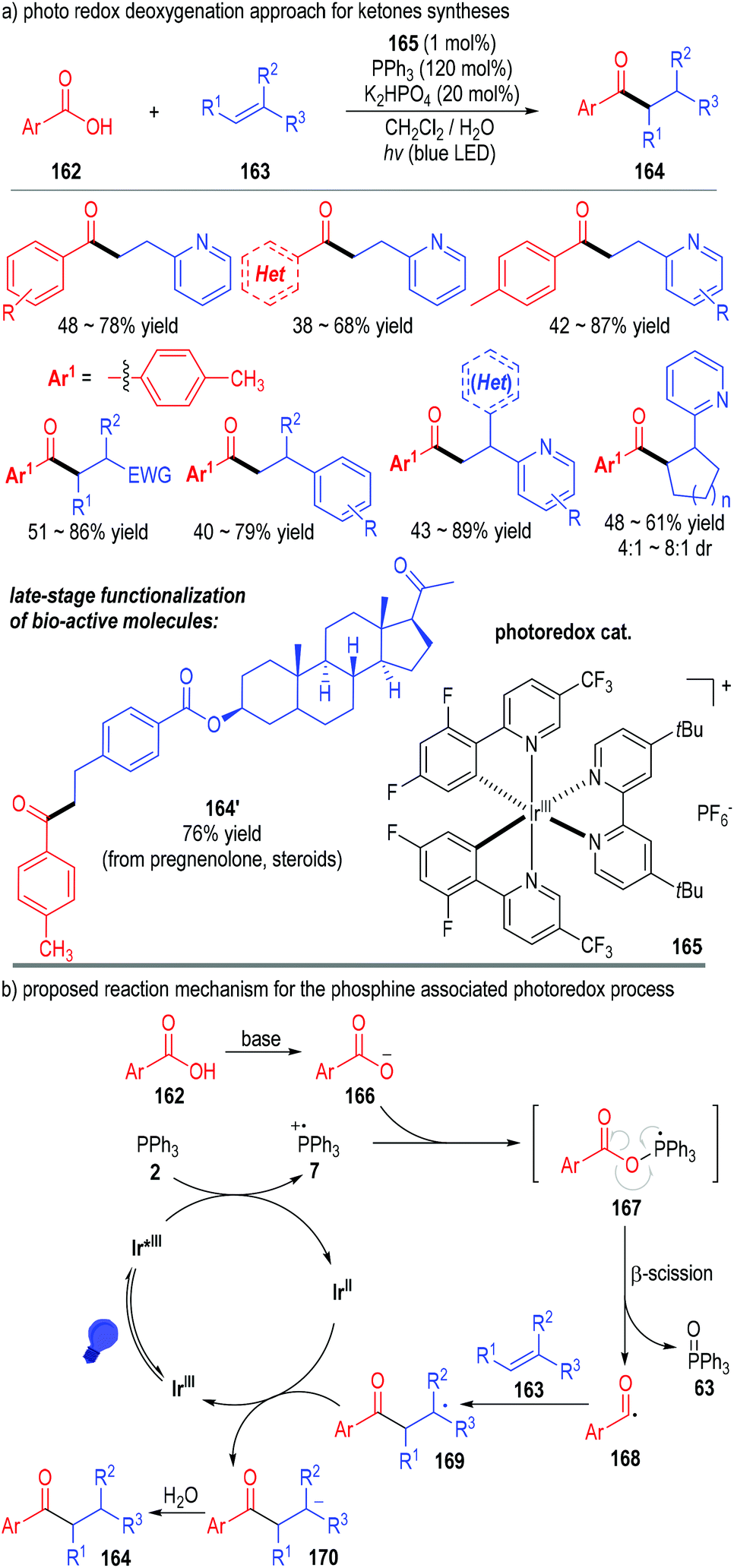 | ||
| Fig. 24 Phosphine associated photoredox catalytic deoxygenation reaction for ketone synthesis. | ||
The reaction was believed to go through a photo-redox SET process (Fig. 24b). The Ir(III) complex 165 was activated by the blue LED light to the excited state and then absorbed one electron from triphenylphosphine 2 to afford the Ir(II) complex and the phosphinium radical cation 7. The radical cation 7 reacted with the deprotonated carboxylic acid 166 through a SET process and gave the phosphoranyl radical 167. β-Scission of the phosphoranyl radical 167 led to the acyl radical 168 with elimination of the triphenylphosphine oxide 63. The acyl radical 168 could react with the alkene substrate 163 through a radical addition process to give the radical adduct 169, which absorbed one electron from the reduced Ir(II) complex to afford anion 170 with regeneration of the photo-redox catalyst Ir(III) for subsequent photocatalytic cycles. The anion 170 was protonated to give the final ketone product 164.
Almost at the same time, Doyle, Rovis and co-workers independently disclosed the generation of phosphoranyl radicals from trivalent phosphines through photoredox catalytic SET processes (Fig. 25).49 They adopted this polar/SET crossover reaction platform in the direct deoxygenation of both alcohols and carboxylic acids (Fig. 25a, 171). Of note, several structurally sophisticated natural products containing carboxylic groups could be efficiently converted to the corresponding aldehydes in good yields through this protocol (e.g., 172a).
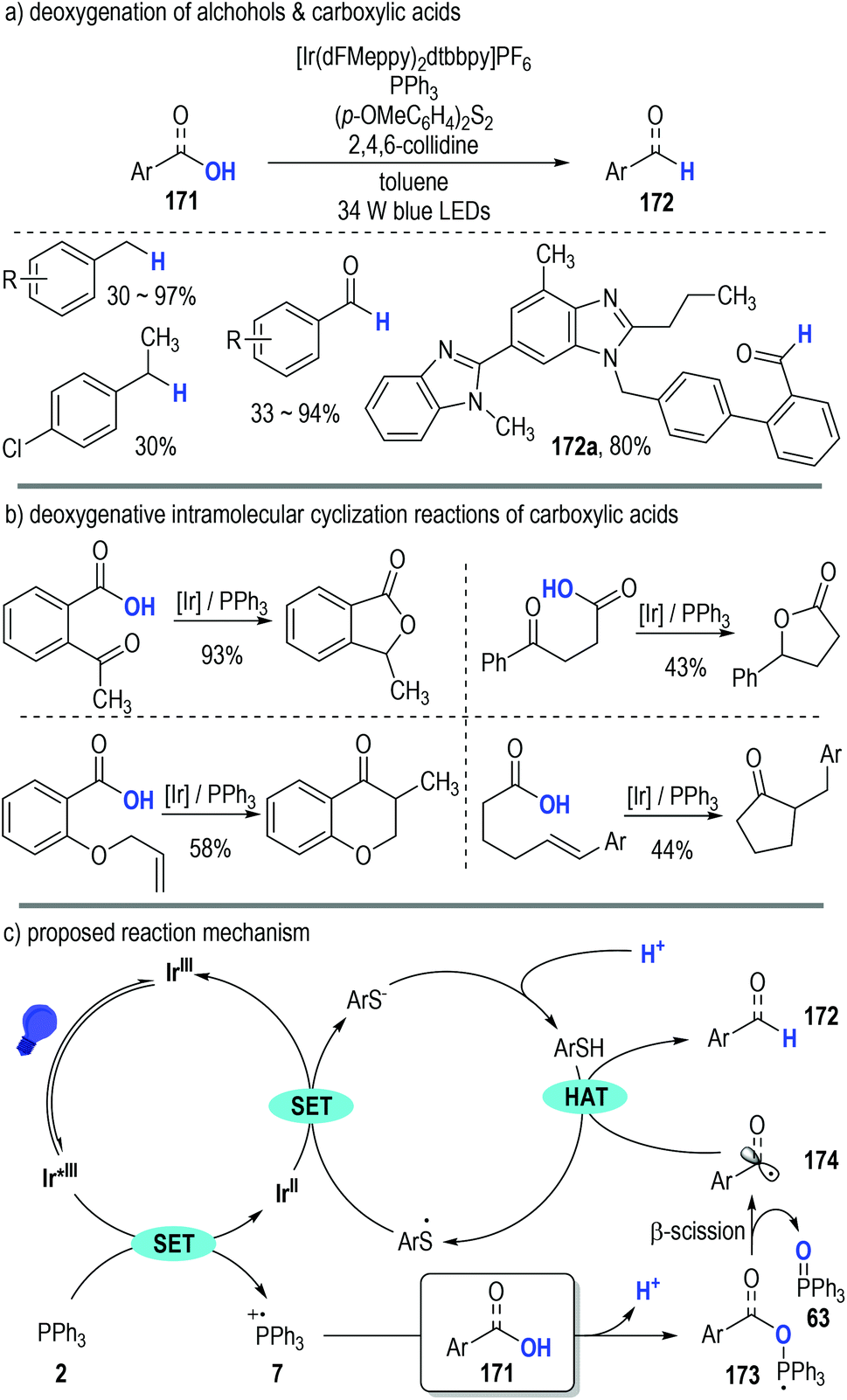 | ||
| Fig. 25 Phosphine associated photoredox catalytic deoxygenative reactions of alcohols and carboxylic acids. | ||
This phosphine-associated photoredox deoxygenation process can also be applied in the intramolecular cyclization reactions of various functionalized carboxylic acids (Fig. 25b). The acyl radicals generated during the deoxygenative process were keen to add onto the unsaturated functional groups existing in the same molecule and the corresponding cyclic carbonyl compounds were given as the final products in moderate to excellent yields.
The reaction was believed to be initiated via a reductive quenching process of the excited photoredox Ir*(III) catalyst (Fig. 25c). Triphenylphosphine 2 was effectively oxidized to the radical cation 7 by the excited Ir*(III) catalyst. A deprotonative ionic addition reaction between the substrate 171 and the radical cation 7 gave the phosphoranyl radical 173, which was readily fragmented into the carbon radical 174 and the phosphine oxide 63 through a β-scission process. An HAT took place between the radical 174 and the thiol additive (ArSH) to produce the target deoxygenative product 172. It should be noted that, the thio radicals (ArS˙) generated from the HAT process went on to oxidize the reduced Ir(II) catalyst to help complete the photoredox catalytic cycle, with the corresponding thiols (ArSH) being regenerated upon protonation for additional HAT reactions.
This phosphine-associated photo-redox catalytic process can also be used in the preparation of deuterated aldehydes (Fig. 26).50 However, due to the strong bond dissociation energy of D–O–D (118 kcal mol−1), it is very difficult for the acyl radical to directly obtain a D atom from D2O. Therefore, a thiol catalyst 175 was applied in this protocol to tune the equilibrium with D2O and the HAT rate in the catalytic process (Fig. 26a). Deuterated aromatic aldehydes were prepared in moderate to excellent yields with excellent deuterated rates of the aldehyde protons. Although aliphatic carboxylic acids also worked well in this process, the deuterated rates of the corresponding aldehyde protons were relatively lower. A variety of complex bioactive carboxylic acids were amenable in this transformation, with the structurally complex deuterated aldehydes being afforded in moderate to excellent yields with excellent aldehyde proton deuterated rates (e.g., 177a, 177b).
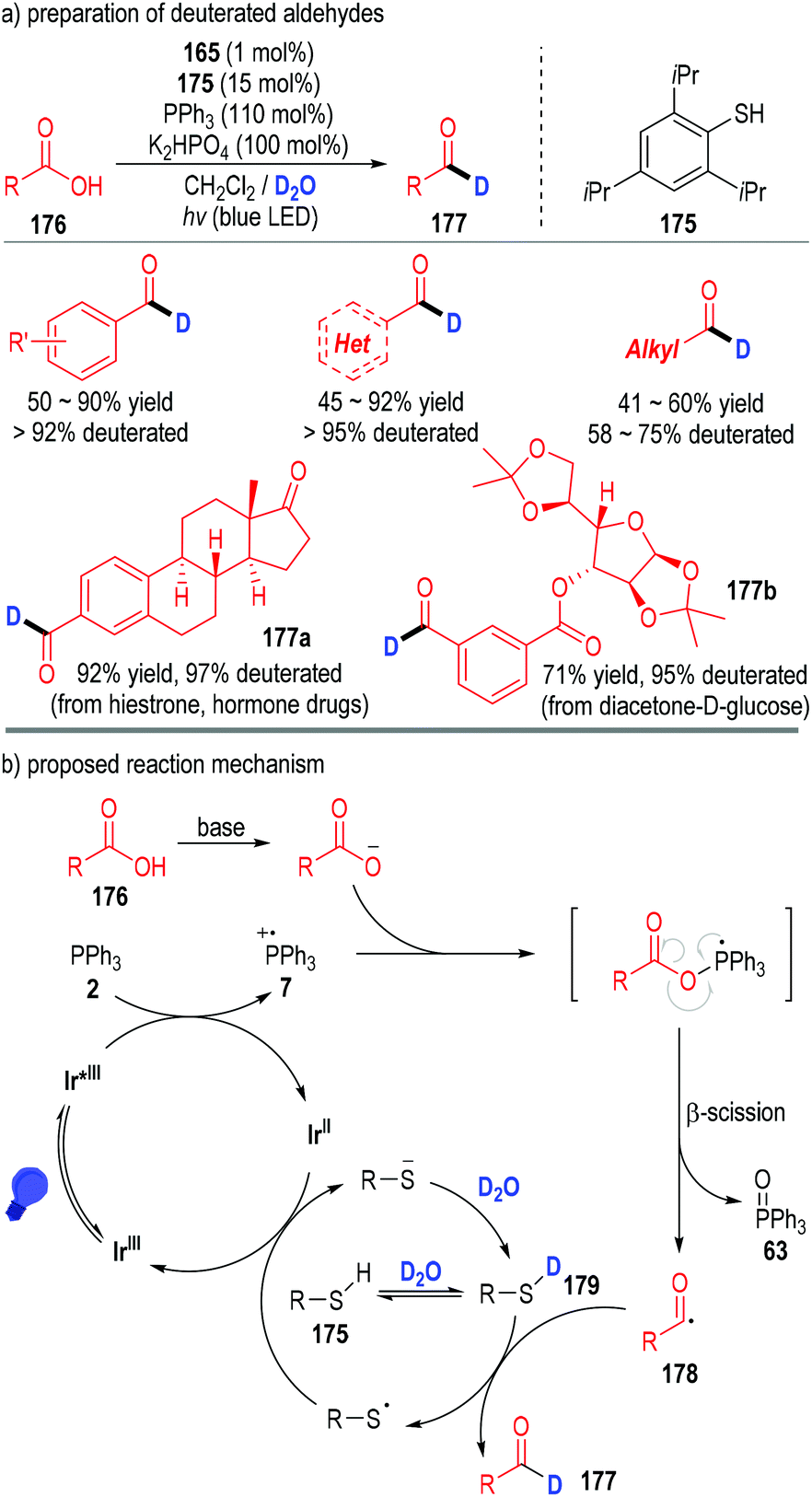 | ||
| Fig. 26 Phosphine associated photoredox catalytic deoxygenation reaction for deuterated aldehyde preparation. | ||
From the mechanistic perspective, the acyl radical 178 could be generated through similar SET processes between the carboxylic acid 176 and triphenylphosphine 2 (Fig. 26b). Then an atom transfer process took place between the radical 178 and the deuterated thiol catalyst 179 to afford the desired deuterated aldehyde product 177. The released thiol radical went on to obtain one electron from the reduced Ir(II) complex to give the thiol anion (R–S−) and the Ir(III) to complete the catalytic cycles.
Very recently, Wang and co-workers successfully synthesized a variety of γ,γ-difluoroallylic ketones through the photo-induced, triphenylphosphine-associated deoxygenative reaction of carboxylic acids (Fig. 27).51 Terminal α-trifluoromethyl alkenes 180 were used as the radical acceptors to react with the acyl radicals generated from the aromatic carboxylic acids 162. γ,γ-Difluoroallylic ketones 181 bearing various substituents were secured in moderate to excellent yields (Fig. 27a).
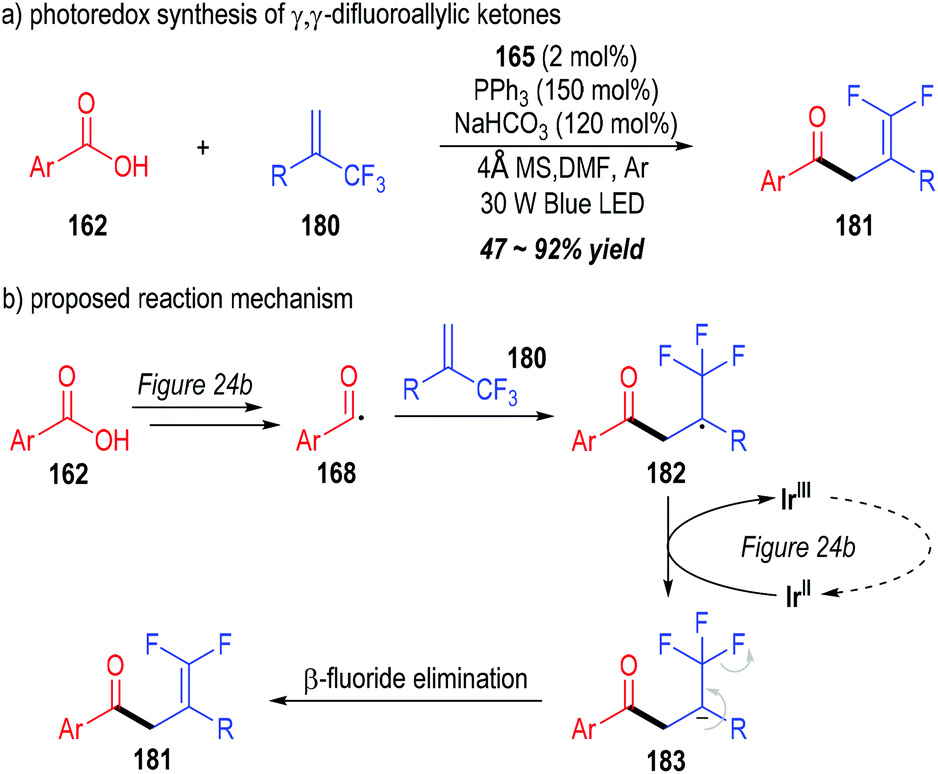 | ||
| Fig. 27 Photo-induced deoxygenation/defluorination reaction for the synthesis of γ,γ-difluoroallylic ketones. | ||
The reaction was believed to go through a similar photoredox mechanism as depicted in Fig. 24b (Fig. 27b). The aromatic acyl radical 168 was generated from the carboxylic acid 162 through a phosphinium-associated radical reducing process. The addition of the radical 168 to the terminal alkene 180 led to the formation of the radical adduct 182, which was further reduced to the carbanion 183 through a SET process with the oxidized photocatalyst Ir(II). A β-fluoride elimination process within the carbanion 183 finally gave γ,γ-difluoroallylic ketone 181 as the target product.
Besides carboxylic acids and alcohols, strained cycloketone oximes 184 are also suitable substrates for the generation of phosphoranyl radical intermediates under photoredox catalytic conditions (Fig. 28).52 The N–O bond of the oxime molecule can be directly cleaved via this protocol. Various aliphatic nitriles were afforded in moderate to excellent yields from the oxime and alkene substrates under mild photoredox catalytic conditions (Fig. 28a).
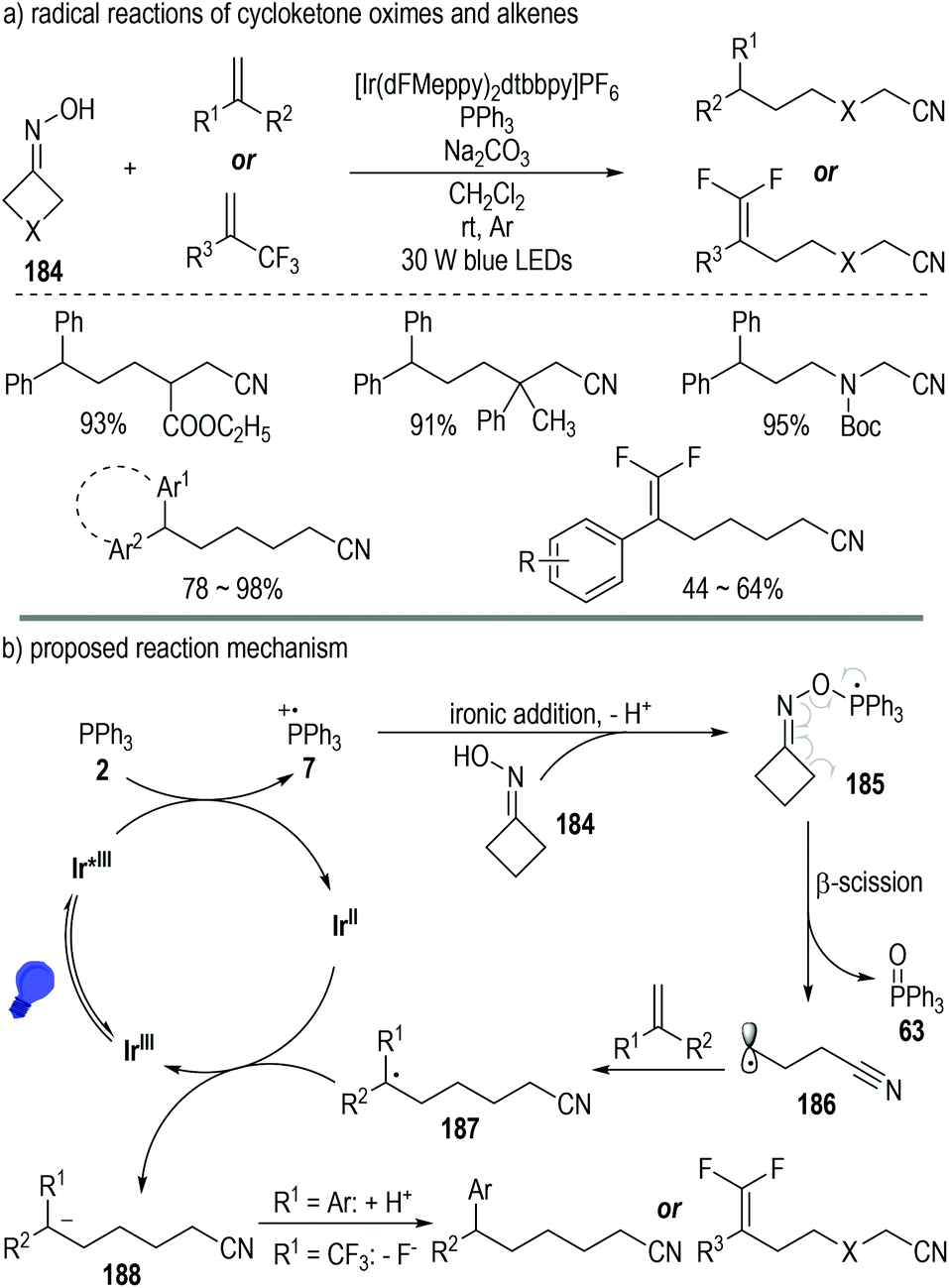 | ||
| Fig. 28 Photo-induced deoxygenation/defluorination reaction for the synthesis of γ,γ-difluoroallylic ketones. | ||
Oxime 184 could add onto the phosphinium radical 7via a deprotonative ionic addition process to give the phosphoranyl radical 185, which was prone to decompose to the ring-opening cyano radical 186 with elimination of the phosphine oxide 63 (Fig. 28b). Radical addition reactions between intermediate 186 and alkenes led to the formation of the elongated cyano radical 187, which then reacted with the reduced photoredox catalyst Ir(II) through a SET process to give the anion intermediate 188. A protonation or a fluoride elimination of intermediate 188 finally gave the desired elongated aliphatic nitrile products.
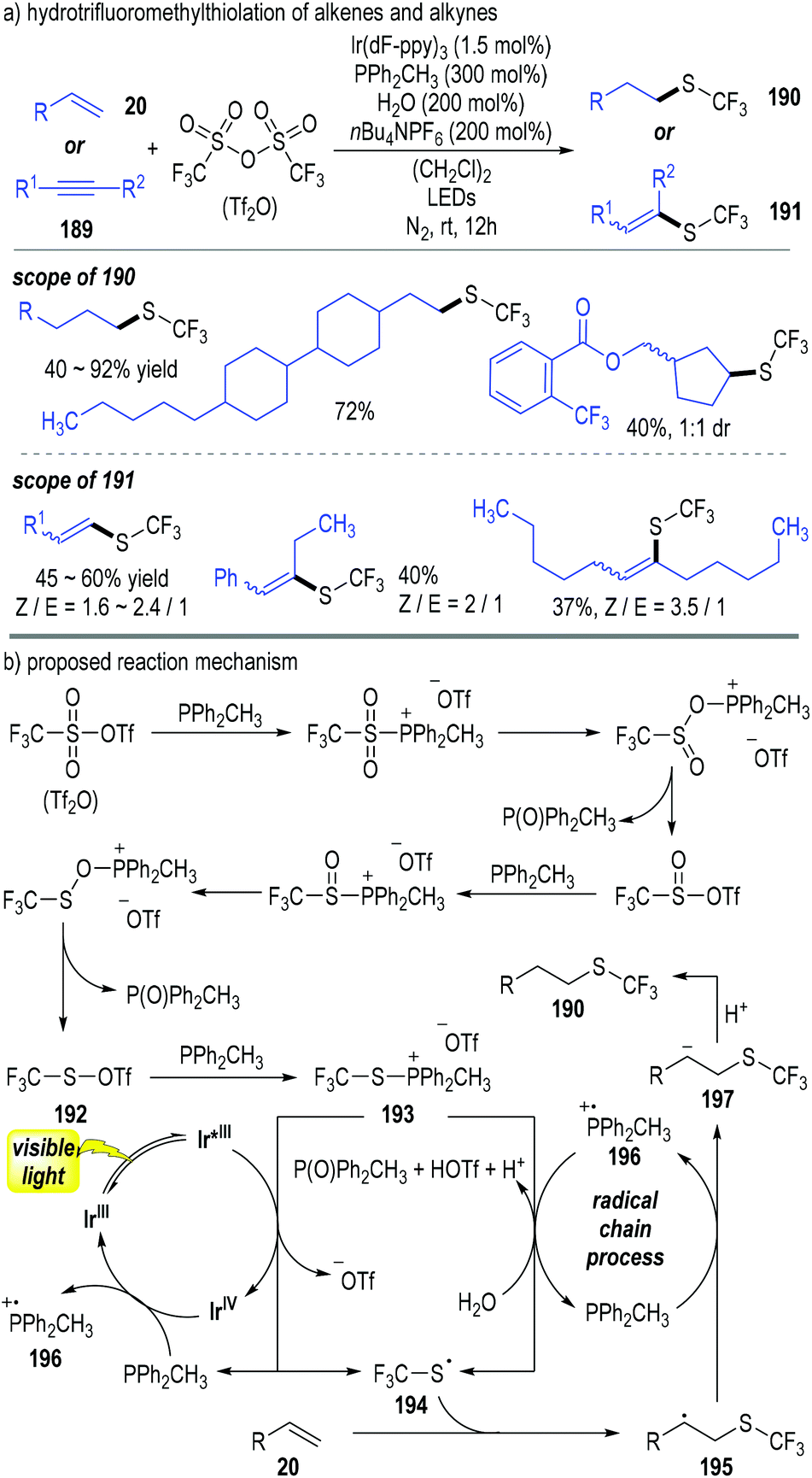
Qing and co-workers have disclosed a photoredox catalytic reaction for the hydrotrifluoromethylthiolation of alkenes 20 and alkynes 189 (Fig. 29).53 Trifluoromethanesulfonic anhydride (abbreviated as Tf2O) was used as the trifluoromethylthiolation reagent in this process; methyldiphenylphosphine (PPh2CH3) was used as both a reducing reagent for the Tf2O and a radical carrier for the radical chain reactions. A broad scope of trifluoromethylthiolated alkanes 190 and alkenes 191 were afforded through this approach in moderate to excellent yields (Fig. 29a).
 | ||
| Fig. 29 Photoredox catalytic hydrotrifluoromethylthiolation reaction. | ||
Tf2O was reduced using 2 equiv. of trivalent phosphine (PPh2CH3) through deoxygenative processes and gave the thiosulfonate 192 with elimination of 2 equiv. of phosphine oxide. The thiosulfonate 192 reacted with another trivalent phosphine molecule and generated the trifluoromethylthiophosphonium salt 193. The intermediate 193 obtained one electron from the photo-activated Ir*(III) catalyst to afford the thio radical 194 with the elimination of the phosphine (PPh2CH3). The released PPh2CH3 could help reduce the oxidized Ir(IV) to regenerate the photocatalyst Ir(III) for continuous catalytic cycles. The afforded free thio radical 194 reacted with the alkene substrate 20 and gave the radical adduct 195. Electron transfer between the radical 195 and the trivalent phosphine (PPh2CH3) made the phosphinium radical 196 and the carbanion 197. Radical cation 196 could react with the intermediate 193 in the presence of H2O and gave phosphine oxide together with trivalent phosphine (PPh2CH3) catalytically. Protonation of the carbanion 197 by the afforded acidic species achieved the trifluoromethylthiolated alkane 190 as the final product (Fig. 29b).
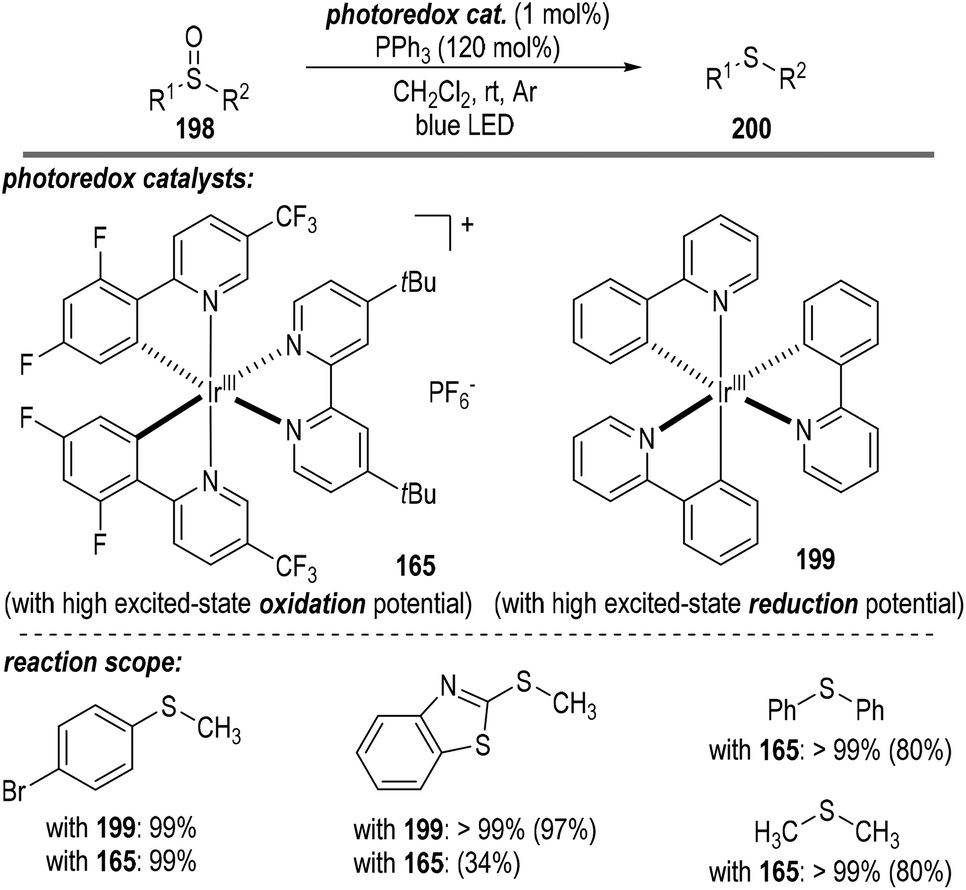
A short time later, Rossi-Ashton and co-workers reported the photoredox catalytic deoxygenation reaction of sulfoxides 198 with the assistance of triphenylphosphine (Fig. 30).54 Both the photoredox catalyst 165 possessing a high excited-state oxidation potential and the catalyst 199 with a high excited-state reduction potential were able to promote this radical process. Thioether product 200 comprising both aromatic and aliphatic substituents was afforded in good to excellent yields.
 | ||
| Fig. 30 Photo-induced deoxygenation reaction of sulfoxides. | ||
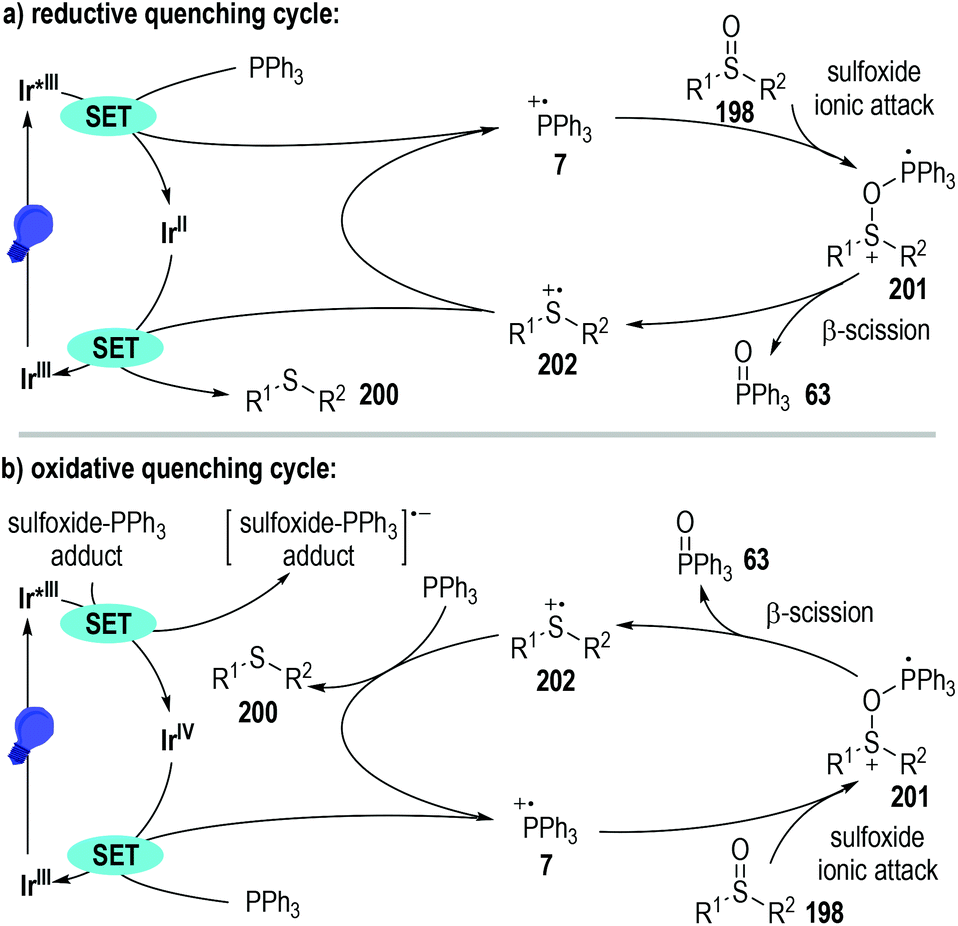
Since both the photoredox catalysts 165 and 199 bearing entirely different excited-state potentials promoted this radical reaction well, the authors proposed two distinct reaction pathways for this transformation (Fig. 31). The photoredox catalytic sulfoxide deoxygenation reaction could be initiated either by a phosphine oxidation process or by a phosphine-sulfoxide adduct reduction process. Given that the excited-state oxidation potential of the photo-redox catalyst 165 is greater than that of the PPh3, effective single-electron oxidation of the PPh3 was enabled to give the trivalent phosphine radical cation 7 through a reductive quenching process of the excited Ir*(III) catalyst (Fig. 31a). Nucleophilic attack of the sulfoxide 198 to the radical cation 7 led to the formation of a new radical cation 201, which was ready to crack into the sulfide radical cation intermediate 202 with the elimination of the phosphine oxide 63. A SET process between the reduced Ir(II) catalyst and the radical cation intermediate 202 finally afforded the target thioether product 200 with the regeneration of the photoredox Ir(III) catalyst at the ground state.
 | ||
| Fig. 31 Two reaction pathways for photo-induced sulfoxide deoxygenation. | ||
Alternatively, when the photoredox Ir(III) catalyst 199 with a highly excited-state reductive potential was used, an oxidative quenching cycle might operate in this radical transformation (Fig. 31b). In this scenario, an unclarified sulfoxide-PPh3 adduct might be initially formed, which could accept one electron from the excited Ir*(III) catalyst to give a highly reactive oxidated Ir(IV). The oxidative Ir(IV) then absorbed one electron from the PPh3 to initiate the following radical deoxygenation reaction of sulfoxides and regenerate the photoredox Ir(III) catalyst at the ground state for additional photo-induced catalytic cycles.
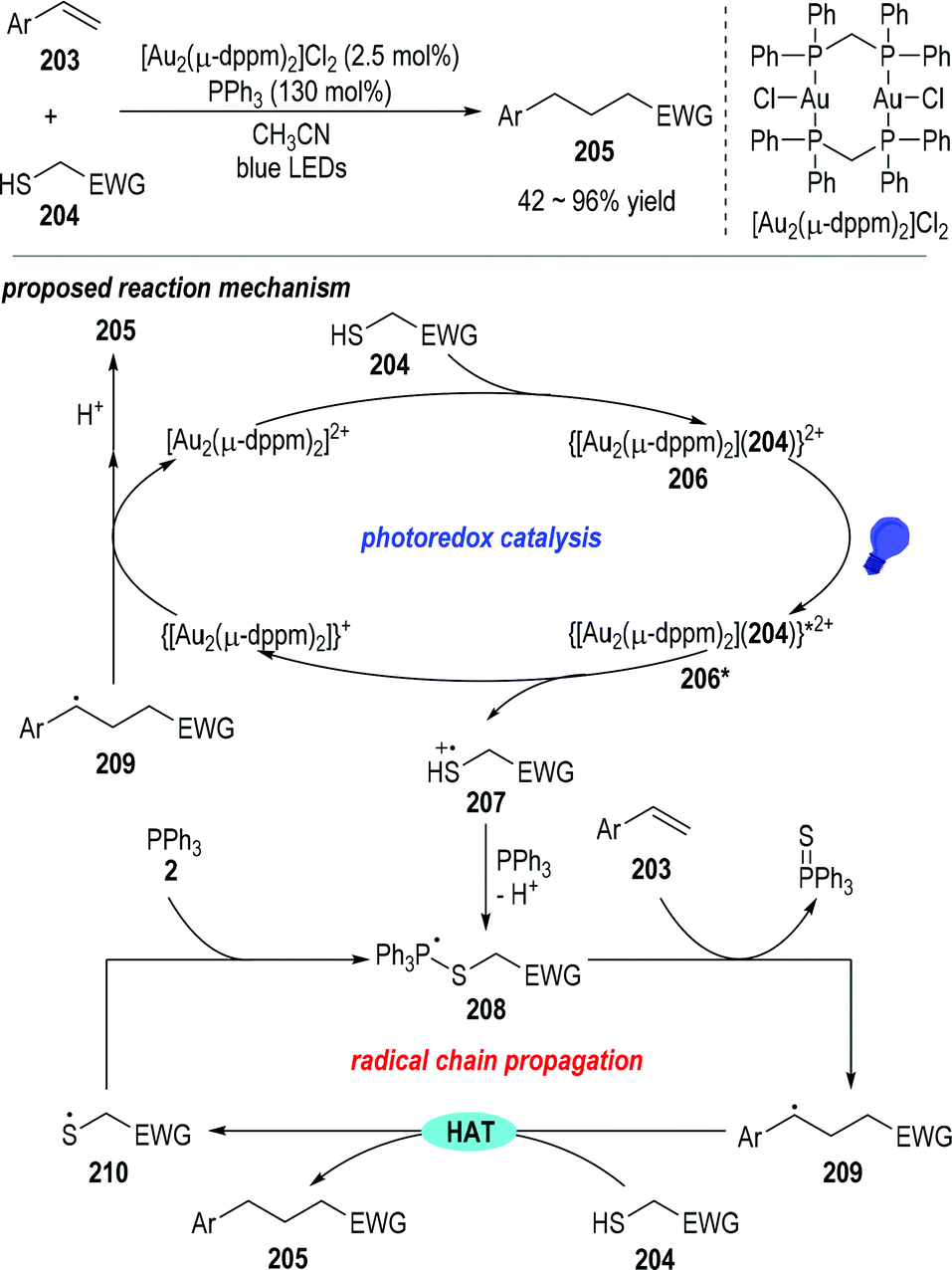
Mercaptans can be used as the alkyl radical precursor through the combination of a photoredox catalytic process and radical chain reactions.55 Hashmi and co-workers reported in 2019 that trivalent phosphines could act as the desulfurization reagent in the photocatalyzed reductive C–C coupling reaction between terminal arylalkenes 203 and the electron-deficient mercaptans 204 (Fig. 32).56 Various substituents could be installed on the aryl rings of the terminal alkenes 203, and the electron-withdrawing groups on the mercaptan 204 could be ester groups, ketone groups, cyano groups, or electron deficient aryl/alkyl groups. Functionalized alkanes 205 were obtained as the final products in moderate to excellent yields.
 | ||
| Fig. 32 Photoredox catalytic desulfurizing C–C coupling reaction. | ||
Mechanistically speaking, the reaction was postulated to go through a combination of a photoredox catalytic cycle with a chained radical propagation process. The electron-deficient mercaptan substrate 204 could first react with the gold catalyst to give a new gold complex 206, which could be activated by blue light to give the excited state 206*. The excited gold complex 206* was ready to fragment into the reduced gold species and the thio radical cation 207 through a ligand-to-metal charge transfer process. The thio radical cation 207 could be coupled with the triphenylphosphine and gave the phosphoranyl radical 208 after deprotonation. Phosphoranyl radical 208 could then react with the terminal alkene 203 through a β-scission/radical addition process and give the radical adduct 209 with the elimination of triphenylphosphine sulfuride. The radical adduct 209 could absorb a hydrogen atom from the mercaptan substrate 204 and afford the thio radical 210 and the final product 205. The thio radical 210 could couple with another triphenylphosphine and give the phosphoranyl radical 208 for other rounds of radical propagation reactions. On the other hand, the reduced gold complex could transfer one electron to the radical adduct 209, thereby regenerating the gold catalyst and releasing another equiv. of the final product 205 after an additional protonation step.
The CN double bonds of aromatic imines 211 could be alkylated by mercaptans 212 through the photoredox catalytic radical reactions (Fig. 33).57 The functionalized secondary amines 213 could be afforded as the final products in moderate to good yields (Fig. 33a). Likewise, the mercaptan 212 could be oxidized by the excited state of the photocatalyst Ir(III)* to give the sulfinium radical cation 214, which could be coupled with the triphenylphosphine to form the phosphoranyl radical 215 after deprotonation. β-Scission of the phosphoranyl radical 215 led to the alkyl radical 216 with the elimination of triphenylphosphine sulfuride. The alkyl radical 216 could be coupled with the amino radical anion 217 that was generated from the SET reaction between the imine 211 and the reduced photocatalyst Ir(II), delivering the amino anion 218 (Fig. 33b, pathway A). Alternatively, the amino anion 218 could be afforded through the SET process between the reduced photocatalyst Ir(II) and the amino radical 219 that was generated from the radical addition reaction of 216 and the imine substrate 211 (Fig. 33b, pathway B). Protonation of the amino anion 218 provided the desired functionalized amine product 213.
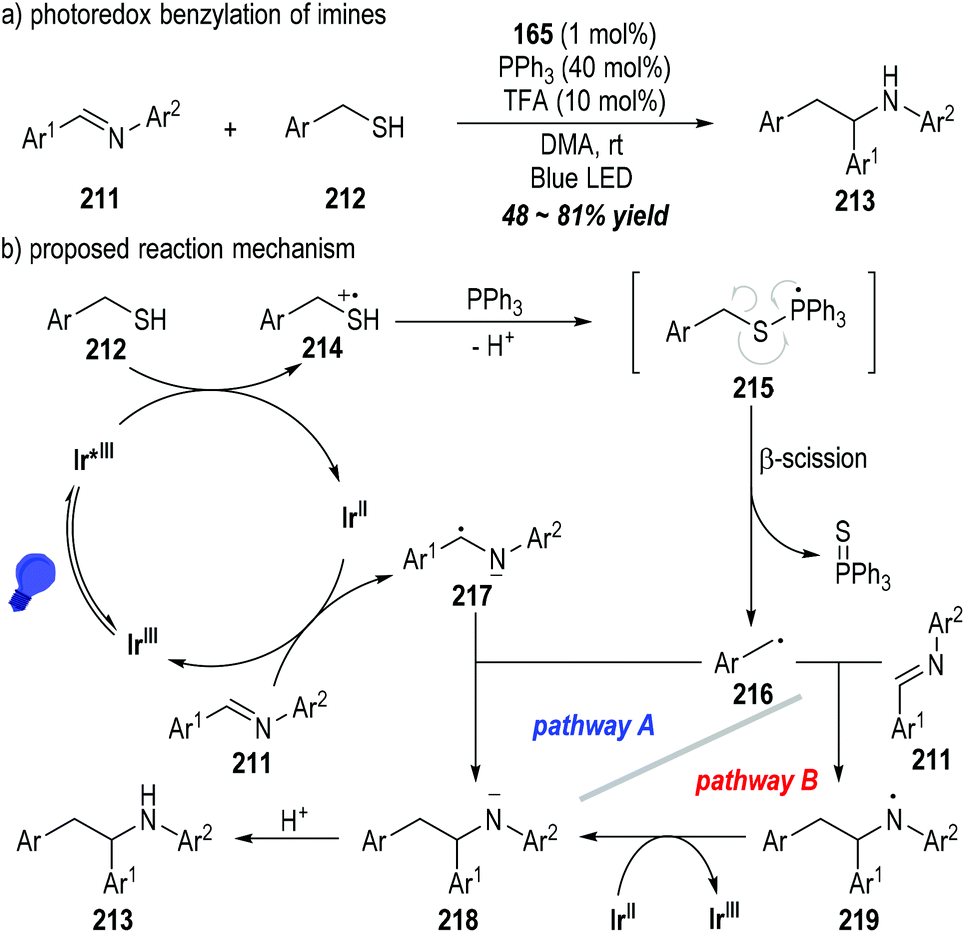 | ||
| Fig. 33 Photo-induced benzylation of imines via phosphine-associated radical processes. | ||
4. Radical reactions of tertiary phosphines in electrochemical reactions
Electrochemical oxidation can also be used for the generation of tertiary phosphine-derived radical cations 220 (Fig. 34, eqn (1)).58 The radical cations 220 are too reactive to be detected when their parent phosphines were oxidized in the cavity of an ESR spectrometer, but they are readily to dimerize in polar solvents to form a new radical cation 221 (e.g., CH3CN, eqn (2)). Clean ESR spectra of such dimeric radical cations 221 can be obtained through anodic oxidative processes. For instance, the electronic oxidative formation of the transient dimeric radical cation 222 was detected by Roberts and co-workers in 1975 via ESR analysis.58a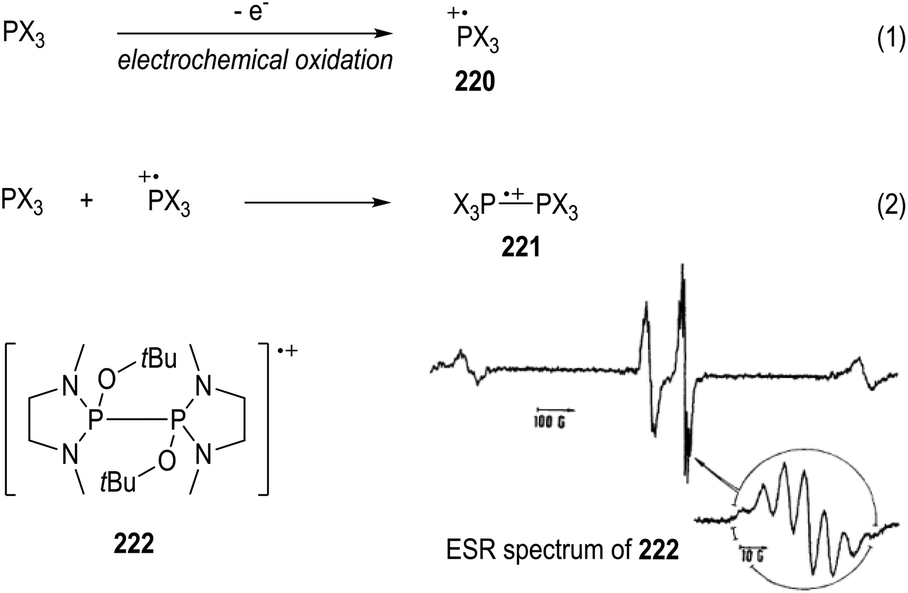 | ||
| Fig. 34 ESR studies on the electronic oxidative generation of phosphinium radical cations. | ||
It has been well established that the kinetic stabilities of a radical species rely on the steric hindrance around the radical center.59 Therefore, a variety of crowded trivalent phosphines have been synthesized and examined in the anodic oxidation reactions.60 Several of the phosphinium radical cations generated from the very bulky triarylphosphines were found persistent enough to be observed by ESR at even room temperature. An ESR signal of the phosphinium radical cation 223 generated electrochemically from the structurally crowded trimesitylphosphine could be observed at −111 °C (Fig. 35).
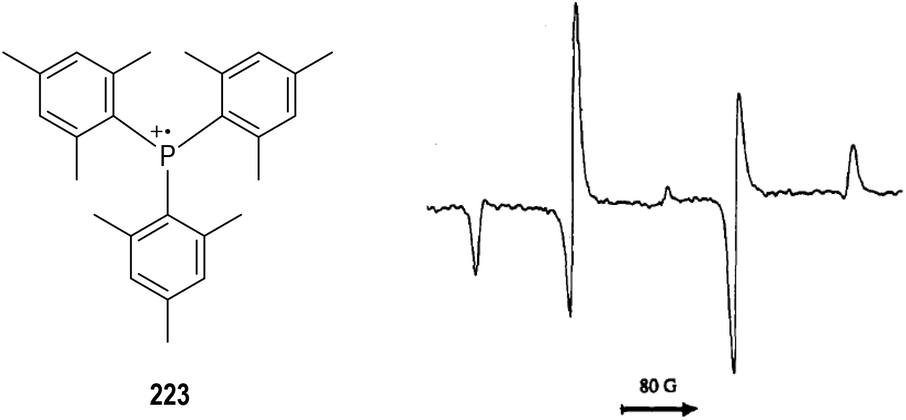 | ||
| Fig. 35 ESR spectrum of the phosphinium radical cation generated electrochemically from a bulky triarylphosphine. | ||
As expected, however, the conventional phosphinium radical cations are extremely reactive and are prone to react with various nucleophilic reagents such as H2O, arenes, and electron-enriched heteroatoms.61 For example, triphenylphosphine 2 could be oxidized electrochemically in the presence of H2O and gave the triphenylphosphine oxide 63, followed by the elimination of two protons (Fig. 36, eqn (1)). Amide 224 can react with the phosphinium radical cation 7 and give the benzonitrile 225 under electrochemical conditions. The carbodiimide 227 can be effectively afforded electrochemically through the reactions of the radical cation 7 with urea 226, in up to 92% yield (eqn (2) and (3)).
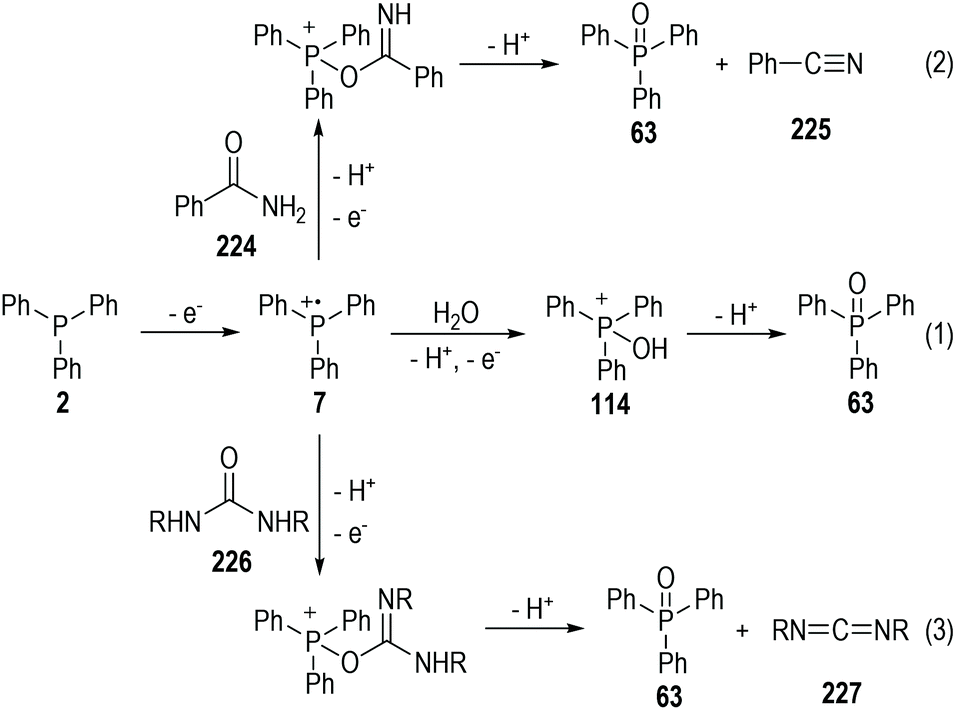 | ||
| Fig. 36 Reactions of electrochemically generated phosphinium radical cations with heteroatoms. | ||
Electron-rich cyclic alkenes 228 can react with the triphenylphosphine 2 under electrochemical conditions and produce the phosphonium salts 229 as the final products in moderate to excellent yields (Fig. 37).62 The nucleophilic addition of alkenes 228 to the electrochemically generated radical cation 7 can give a new radical cation 230, which can then be deprotonated to become the neutral radical 231. An additional one-electron anodic oxidation of the radical 231 can lead to the formation of the final product 229.
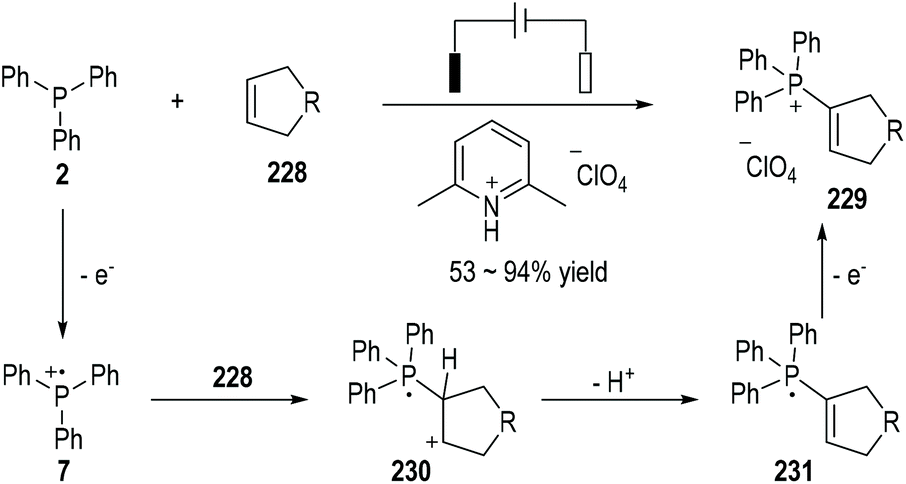 | ||
| Fig. 37 Reactions of electrochemically generated phosphinium radical cations with alkenes. | ||
5. Summary and outlook
Trivalent tertiary phosphines are one class of the most important nucleophilic reagents in organic synthesis. They could not only be used as reactive electron-pair donors, but also be used as single-electron reductants. Although the generation and detection methods of the radical species from trivalent tertiary phosphines have been established for over half a century, the applications of the SET processes associated with them in organic synthesis have been relatively scarce. However, the significance of tertiary phosphine associated radical transformations has been recognized in recent years and a number of excellent synthetic methodologies have been developed based on this strategy. In particular, the combinations of the tertiary phosphine-associated SET processes with photoredox catalysis or electrochemical synthesis have provided us with promisingly new approaches to achieve challenging molecular structures from readily available starting materials via simple operation.Nevertheless, challenges still exist in the radical transformations promoted by trivalent tertiary phosphines. One of the main shortcomings within this research field lies in the lack of chirality controls in these SET reactions. The phosphine reagents used in these radical reactions are usually oxidized to phosphinium radical cation intermediates by either the reaction substrates or the photoredox catalysts. The afforded phosphinium radical cation intermediates generally act as radical carriers or single-electron reductants that do not participate in most of the new bond forming processes. It is therefore difficult for a chiral phosphine catalyst to transfer any chiral elements into the reaction. As a result, limited success has been achieved in the phosphine-promoted enantioselective radical reactions.
The application of a chiral non-covalent co-catalyst is one of the promising protocols to solve this problem.47 H-bond donating organocatalysts and Lewis acids are both suitable candidates for the chiral co-catalysts used in the phosphine-catalyzed enantioselective radical reactions. They could not only coordinate with the electron-rich functional groups existing in the reaction substrates but also have interactions with the radical species generated from the other reactants, thus making the radical coupling/addition reactions happen in stereoselective fashions.
Alternatively, the combination of the phosphine-promoted radical reactions with other organic or transmetallic catalytic enantioselective processes may also provide us with effective ways for various asymmetric phosphine-radical-associated reactions. For example, the activation of an aldehyde substrate by a chiral amine or NHC catalyst via covalent bond formation generates chiral reactive intermediates that can react with the radical species generated through phosphine-promoted SET processes in an asymmetric fashion.
The limitation in activation modes is another drawback long-standing in the trivalent phosphine-promoted SET transformation. The ways for the generation and the very short lifetimes of the P-centered radical cation intermediates both have severely restricted their reaction modes. For example, electron-deficient iodides have frequently been used for the generation of phosphinium radicals to initiate the radical propagation chain reactions. Electron-neutral or electron-rich halides are not applicable in such reactions, which is probably due to their high bond-dissociation energies and the mismatched oxidative potentials. The design and preparation of new trivalent phosphine catalysts bearing various functional groups to change their reductive/oxidative potentials is a promising method to make breakthroughs in the scope of phosphine-promoted radical reactions. On the other hand, the newly designed functionalized trivalent phosphine molecules may extend the lifetimes of the corresponding P-centered radical species, and it would therefore be possible for less reactive substrates to have interactions with the P-radicals before they collapsed into inert species.
Although the trivalent phosphines have played significant roles in various photo-induced radical reactions, their application in electrochemistry is relatively less investigated. It has been proven that radical species could be generated under electrochemical conditions, but limited success has been achieved in the phosphine-induced electrochemical reactions. Given the rapid and remarkable development in the field of electrochemistry in the last decade, trivalent phosphines should have prospective applications within this field. For example, the generation and application of the corresponding phosphoranyl radical intermediates under electrochemical conditions would be more attractive, since the use of the costly noble metal photoredox catalysts could be avoided in the preparation of valuable functional products (e.g., the functional products prepared in section 3.4).
However, compared with the electron-pair transfer reactions catalyzed by tertiary phosphines, the phosphine-promoted radical reactions have provided us with unprecedented activation modes and achieved challenging transformations that had previously been not reliable. For instance, electron-neutral or electron-rich alkenes cannot be used in the phosphine-catalyzed electron-pair-transfer reactions, while they can be effectively functionalized through radical pathways promoted by trivalent phosphines. Functional molecules such as alcohols, carboxylic acids, and oximes cannot be activated by phosphine catalysts through electron-pair-transfer processes, but they can be efficiently transformed into various interesting products via phosphine-associated SET pathways.
Compared with the radical reactions catalyzed by amines and NHC catalysts, the trivalent phosphines bear different activation modes and could embrace a different set of reaction substrates for SET transformations. Conventionally, trivalent amines and NHC catalysts react with one of the reaction substrates through electron-pair-transfer steps to give electron-rich intermediates, which are prone to be oxidized by external oxidants through SET processes to afford reactive ionic radical species for additional transformations. Therefore, aldehydes, ketones and related carbonyl compounds took significant positions as the reaction starting materials in the radical transformations catalyzed by amines and NHC catalysts. Trivalent phosphine molecules can be independently oxidized to give the reactive radical cation intermediates at the beginning of the radical reactions, and therefore can be used in the activation of various functional molecules such as alcohols, carboxylic acids, oximes and others. The SET activation modes provided by trivalent phosphines can be developed into a widely used activation strategy with a broad scope of reaction substrates applicable for the efficient preparation of various functional molecules.
To date, the development of synthetic methods involving radical species generated from tertiary phosphines has attracted more and more attention. With the rapid development of photoredox catalysis and electrochemical synthesis, SET reactions associated with tertiary phosphines will definitely make essential contributions to the development of novel, efficient and sustainable synthetic protocols.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the National Natural Science Foundation of China (No. 21801051, 21961006) and Guizhou University (No. GZU[2017]34, KY[2017]376). We thank Dr Wai-Lun Chan from Hong Kong Baptist University for his generous help on language use and writing.Notes and references
- For selected reviews on phosphine organocatalysis, see:
(a) X. Lu, C. Zhang and Z. Xu, Reactions of Electron-Deficient Alkynes and Allenes under Phosphine Catalysis, Acc. Chem. Res., 2001, 34, 535 CrossRef CAS PubMed
; (b) J. L. Methot and W. R. Roush, Nucleophilic Phosphine Organocatalysis, Adv. Synth. Catal., 2004, 346, 1035 CrossRef CAS
- M. M. Rauhut and H. Currier, (American Cyanamid Co.), Preparation of Dialkyl 2-methyleneglutarates, U.S. Patent 307499919630122, 1963, Chem. Abstr., 1963, vol. 58, p. 11224a.
- For reviews on phosphorus radicals, see:
(a) D. Leca, L. Fensterbank, E. Lacote and M. Malacria, Recent Advances in the Use of Phosphorus-centered Radicals in Organic Chemistry, Chem. Soc. Rev., 2005, 34, 858 RSC
- For selected reviews on radical reactions catalyzed by trivalent amines, see:
(a) S. Afewerki and A. Córdova, Combinations of Aminocatalysts and Metal Catalysts: A Powerful Cooperative Approach in Selective Organic Synthesis, Chem. Rev., 2016, 116, 13512 CrossRef CAS PubMed
- For selected examples on radical reactions catalyzed by trivalent amines, see:
(a) T. D. Beeson, A. Mastracchio, J.-B. Hong, K. Ashton and D. W. C. MacMillan, Enantioselective Organocatalysis Using SOMO Activation, Science, 2007, 316, 582 CrossRef CAS PubMed
- F. Ramirez and S. Dershowitz, The Structure of Quinone-donor Adducts. I. The Action of Triphenylphosphine on p-Benzoquinone, 2,5-Dichloro-p-benzoquinone, and Chloranil, J. Am. Chem. Soc., 1956, 78, 5614 CrossRef CAS
- E. A. C. Lucken, F. Ramirez, V. P. Catto, D. Rhum and S. Dershowitz, Radical Ions in the Reactions of p-Chloranil with Triphenylphosphine and with Triethyl Phosphite. Electron Spin Resonance and Ultraviolet Absorption Spectra, Tetrahedron, 1966, 22, 637 CrossRef CAS
- P. D. Bartlett, E. F. Cox and R. E. Davies, Reactions of Elemental Sulfur. IV. Catalytic Effects in the Reaction of Sulfur with Triphenylphosphine, J. Am. Chem. Soc., 1961, 83, 103 CrossRef CAS
- V. G. Wittig, H. J. Gristau and H. Braun, Racematspaltung Unsymmetrischer Triarylphosphine, Angew. Chem., 1967, 79, 721 CrossRef
- R. L. Powell and C. D. Hall, Phosphinium Radical Cation, J. Am. Chem. Soc., 1969, 91, 5403 CrossRef CAS
- W. Y. Huang and H. Z. Zhang, Reaction of Perfluoroalkyl Iodides with Alkenes Initiated by Organophosphine and Related Compounds, J. Fluor. Chem., 1990, 50, 133 CrossRef CAS
- M. Lumbierres, M. Moreno-Mañas and A. Vallribera, Addition of Perfluorooctyl Iodide to Alkenes. Catalysis by Triphenylphosphine, Tetrahedron, 2002, 58, 4061 CrossRef CAS
- L. Helmecke, M. Spittler, K. Baumgarten and C. Czekelius, Metal-Free Activation of C-I Bonds and Perfluoroalkylation of Alkenes with Visible Light Using Phosphine Catalysts, Org. Lett., 2019, 21, 7823 CrossRef CAS PubMed
- L. Zhao, Y. Huang, Z. Wang, E. Zhu, T. Mao, J. Jia, J. Gu, X. F. Li and C. Y. He, Organophosphine-Catalyzed Difluoroalkylation of Alkenes, Org. Lett., 2019, 21, 6705 CrossRef CAS PubMed
-
R. C. Larock, Comprehensive Organic Transformations, Wiley-VCH, New York, 2nd edn, 1999, pp. 49–52 Search PubMed
- L. Zhang and M. Koreeda, Radical Deoxygenation of Hydroxyl Groups via Phosphites, J. Am. Chem. Soc., 2004, 126, 13190 CrossRef CAS PubMed
- P. A. Lartey, H. N. Nellans, R. Faghih, A. Petersen, C. M. Edwards, L. Freiberg, S. Quigley, K. Marsh, L. L. Klein and J. J. Plattner, Synthesis of 4′′-Deoxy Motilides: Identification of a Potent and Orally Active Prokinetic Drug Candidate, J. Med. Chem., 1995, 38, 1793 CrossRef CAS PubMed
- P. A. Jordan and S. J. Miller, An Approach to the Site-Selective Deoxygenation of Hydroxy Groups Based on Catalytic Phosphoramidite Transfer, Angew. Chem., Int. Ed., 2012, 51, 2907 CrossRef CAS PubMed
- P. Yu, S. C. Zheng, N. Y. Yang, B. Tan and X. Y. Liu, Phosphine-Catalyzed Remote β-C-H Functionalization of Amines Triggered by Trifluoromethylation of Alkenes: One-Pot Synthesis of Bistrifluoromethylated Enamides and Oxazoles, Angew. Chem., Int. Ed., 2015, 54, 4041 CrossRef CAS PubMed
- S. Yasui, M. Fujii, C. Kawano, Y. Nishimura and A. Ohno, Dediazoniation of Arenediazonium Salt with Trivalent Phosphorus Compounds by One-electron Transfer Mechanism, Tetrahedron Lett., 1991, 32, 5601 CrossRef CAS
- S. Yasui, M. Fujii, C. Kawano, Y. Nishimura, K. Shioji and A. Ohno, Mechanism of Dediazoniation of Arenediazonium Salts with Triphenylphosphine and Trialkyl Phosphites. Generation of Cation Radicals from Trivalent Phosphorus Compounds and Their Reactions, J. Chem. Soc., Perkin Trans. 2, 1994, 177 RSC
- S. Yasui, K. Shioji and A. Ohno, Reaction of a Cation Radical Generated from Trivalent Phosphorus Compound through Single-electron Transfer to Arenediazonium Salt, Tetrahedron Lett., 1994, 35, 2695 CrossRef CAS
- S. Yasui, K. Shioji, M. Tsujimoto and A. Ohno, Reactivity of Cation Radicals Generated from Trivalent-phosphorus Compounds in the Reaction with Methylviologen: Kinetic Analysis, Chem. Lett., 1995, 783 CrossRef CAS
- S. Yasui, K. Shioji, M. Tsujimoto and A. Ohno, Kinetic Study on the Reaction of Tributylphosphine with Methylviologen. Reactivity of the Phosphine Radical Cation Intermediate towards Nucleophiles, J. Chem. Soc., Perkin Trans. 2, 1999, 855 RSC
-
(a) O. Mitsunobu, The Use of Diethyl Azodicarboxylate and Triphenylphosphine in Synthesis and Transformation of Natural Products, Synthesis, 1981, 1 CrossRef CAS
-
(a) D. C. Morrison, Reactions of Alkyl Phosphites with Diethyl Azodicarboxylate, J. Org. Chem., 1958, 23, 1072 CrossRef CAS
- D. Camp and I. D. Jenkins, The Mechanism of the Mitsunobu Esterification Reaction. Part I. The Involvement of Phosphoranes and Oxyphosphonium Salts, J. Org. Chem., 1989, 54, 3045 CrossRef CAS
- K. Kato and O. Mitsunobu, Oxidation of Mercaptans with Diethyl Azodicarboxylate and Trivalent Phosphorus Compounds, J. Org. Chem., 1970, 35, 4227 CrossRef
- D. Camp, G. R. Hanson and I. D. Jenkins, Formation of Radicals in the Mitsunobu Reaction, J. Org. Chem., 1995, 60, 2977 CrossRef CAS
- S. W. Lardy and V. A. Schmidt, Intermolecular Radical Mediated Anti-Markovnikov Alkene Hydroamination Using N-Hydroxyphthalimide, J. Am. Chem. Soc., 2018, 140, 12318 CrossRef CAS PubMed
-
(a) A. Horsfield, J. R. Morton and D. H. Whiffen, Electron Spin Resonance and Structure of the Ionic Radical, ˙PO3=, Mol. Phys., 1961, 4, 475 CrossRef CAS
-
(a) B. W. Fullman and M. C. R. Symons, Radiation Mechanisms. II. Electron, Spin Resonance Studies of the Mechanism of Radiation Processes in Trivalent Phosphorus Derivatives, J. Chem. Soc., Dalton Trans., 1975, 861 Search PubMed
- R. S. Davidson, The Chemistry of Exited Complexes: a Survey of Reactions, Adv. Phys. Chem., 1983, 19, 1 CAS
-
(a) D. R. Arnold, P. C. Wong, A. J. Maroulis and T. S. Cameron, Radical Ions in Photochemistry. 12. The Photoaddition of Olefins to Cyano Aromatic Compounds in Polar Solvents, Pure Appl. Chem., 1980, 52, 2609 CAS
- G. Pandey, D. Pooranchand and U. T. Bhalerao, Photoinduced Single Electron Transfer Activation of Organophosphines: Nucleophilic Trapping of Phosphine Radical Cation, Tetrahedron, 1991, 47, 1745 CrossRef CAS
-
(a) D. Rehm and A. Weller, Kinetics of Fluorescence Quenching by Electron and Hydrogen-atom Transfer, Isr. J. Chem., 1970, 8, 259 CrossRef CAS
- S. Ganapathy, K. P. Dockery, A. E. Sopchik and W. G. Bentrude, Photoinduced Single Electron Transfer Initiated Rearrangements of 2-Phenylallyl Phosphites, J. Am. Chem. Soc., 1993, 115, 8863 CrossRef CAS
- S. Yasui and A. Ohno, The Photoreduction of 10-Methylacridinium Ion by Triphenylphosphine through One-electron Transfer Mechanism, Tetrahedron Lett., 1991, 32, 1047 CrossRef CAS
- S. Yasui, K. Shioji, M. Yoshihara, T. Maeshima and A. Ohno, The Thermal- and Photo-reactions of a Diphenylphosphinite Ester with 10-Methylacridinium Iodide. Discrimination between Polar and Single Electron Transfer Processes, Tetrahedron Lett., 1992, 33, 7189 CrossRef CAS
- S. Yasui, K. Shioji, A. Ohno and M. Yoshihara, Reactivity of Phosphorus-Centered Radicals Generated during the Photoreaction of Diphenylphosphinous Acid with 10-Methylacridinium Salt, J. Org. Chem., 1995, 60, 2099 CrossRef CAS
- M. Ochiai, K. Sumi, Y. Nagao and E. Fujita, Vinyliodonium Salts: Their Stereospecific Synthesis and Reactions as the Activated Vinyl Halides, Tetrahedron Lett., 1985, 26, 2351 CrossRef CAS
- M. Ochiai, M. Kunishim, Y. Nagao, K. Fuji and E. Fujita, sp-Carbon-iodine Bond Cleavage of Alkynyl(phenyl)iodonium Salts. Novel Synthesis of (Alkylethynyl)triphenylphosphonium Salts, J. Chem. Soc., Chem. Commun., 1987, 1708 RSC
- M. C. R. Symons and R. L. Petersen, Halide Ion Capture by Radicals. Electron Spin Resonance Spectra of R3P˙-hal and R2S˙-hal σ* Radicals, J. Chem. Soc., Faraday Trans. 2, 1979, 75, 210 RSC
-
(a) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, The Merger of Transition Metal and Photocatalysis, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS
-
(a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed
- N. A. Romero and D. A. Nicewicz, Organic Photoredox Catalysis, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed
- M. C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Photocatalytic Decarboxylative Alkylations Mediated by Triphenylphosphine and Sodium Iodide, Science, 2019, 363, 1429 CrossRef CAS PubMed
- M. Zhang, J. Xie and C. Zhu, A General Deoxygenation Approach for Synthesis of Ketones from Aromatic Carboxylic Acids and Alkenes, Nat. Commun., 2018, 9, 3517 CrossRef PubMed
- E. E. Stache, A. B. Ertel, T. Rovis and A. G. Doyle, Generation of Phosphoranyl Radicals via Photoredox Catalysis Enables Voltage–Independent Activation of Strong C–O Bonds, ACS Catal., 2018, 8, 11134 CrossRef CAS PubMed
- M. Zhang, X. A. Yuan, C. Zhu and J. Xie, Deoxygenative Deuteration of Carboxylic Acids with D2O, Angew. Chem., Int. Ed., 2019, 58, 312 CrossRef CAS PubMed
- Y. Q. Guo, R. Wang, H. Song, Y. Liu and Q. Wang, Visible-Light-Induced Deoxygenation/Defluorination Protocol for Synthesis of γ,γ-Difluoroallylic Ketones, Org. Lett., 2020, 22, 709 CrossRef CAS PubMed
- P.-J. Xia, Z.-P. Ye, Y.-Z. Hu, D. Song, H.-Y. Xiang, X.-Q. Chen and H. Yang, Photocatalytic, Phosphoranyl Radical-Mediated N–O Cleavage of Strained Cycloketone Oximes, Org. Lett., 2019, 21, 2658 CrossRef CAS PubMed
- Y. Ouyang, X. H. Xu and F. L. Qing, Hydrotrifluoromethylthiolation of Unactivated Alkenes and Alkynes with Trifluoromethanesulfonic Anhydride through Deoxygenative Reduction and Photoredox Radical Processes, Angew. Chem., Int. Ed., 2019, 58, 18508 CrossRef CAS PubMed
- A. K. Clarke, A. Parkin, R. J. K. Taylor, W. P. Unsworth and J. A. Rossi-Ashton, Photocatalytic Deoxygenation of Sulfoxides Using Visible Light: Mechanistic Investigations and Synthetic Applications, ACS Catal., 2020, 10, 5814 CrossRef CAS PubMed
- F. Denes, M. Pichowicz, G. Povie and P. Renaud, Thiyl Radicals in Organic Synthesis, Chem. Rev., 2014, 114, 2587 CrossRef CAS PubMed
- L. Zhang, X. Si, Y. Yang, S. Witzel, K. Sekine, M. Rudolph, F. Rominger and A. S. K. Hashmi, Reductive C-C Coupling by Desulfurizing Gold-Catalyzed Photoreactions, ACS Catal., 2019, 9, 6118 CrossRef CAS
- J. Zhang and M. Zhang, Harnessing Thiol as a Benzyl Reagent for Photocatalytic Reductive Benzylation of Imines, Org. Lett., 2020, 22, 2112 CrossRef CAS PubMed
-
(a) W. B. Gara and B. P. Roberts, Electrochemical Generation of the Radical Cations [X3PPX3].+, J. Chem. Soc., Chem. Commun., 1975, 949 RSC
-
(a) D. Griller and K. U. Ingold, Persistent Carbon-centered Radicals, Acc. Chem. Res., 1976, 9, 13 CrossRef CAS
- M. Culcasi, Y. Berchadsky, G. Gronchi and P. Tordo, Anodic Behavior of Crowded Triarylphosphines. ESR Study of Triarylphosphoniumyl Radicals, Ar3P.+, J. Org. Chem., 1991, 56, 3537 CrossRef CAS
- H. Ohmori, K. Sakai, N. Nagai, Y. Mizuki and M. Masui, Reaction of Electrochemically Generated Triphenylphosphine Radical Cation with Amides and Ureas, Chem. Pharm. Bull., 1985, 33, 373 CrossRef CAS
- H. Ohmori, T. Takanami and M. Masui, Reaction of Triphenylphosphine Radical Cation with Cycloalkenes: Electrochemical One-step Preparation of 1-Cycloalkenyltriphenylphosphonium Salts, Tetrahedron Lett., 1985, 26, 2199 CrossRef CAS
| This journal is © the Partner Organisations 2020 |