
Radical reactions promoted by trivalent tertiary phosphines
 *a
*a
Abstract
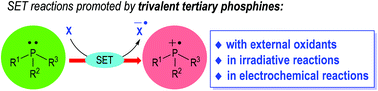
Tertiary phosphines have been extensively developed as effective organic catalysts/reagents to promote various modern organic transformations in both racemic and enantioselective fashions. However, their applications in radical generation and reactions remain relatively less explored. Phosphine-centered radical species, generated from various tertiary phosphines via phosphine–oxidant charge transfer processes, photoredox catalysis and electrochemical oxidations, can give rise to many unprecedented activation modes and reactions. Single-electron-transfer (SET) reactions associated with tertiary phosphines in particular have recently gained popularity, affording novel and promising synthetic approaches to challenging molecular structures from readily available starting materials upon simple operation. Summarized herein are the historical background and recent breakthroughs in this dynamic field of phosphine-mediated radical organic reactions.
 Please wait while we load your content...
Please wait while we load your content...

