
Cooperative photoredox and chiral hydrogen-bonding catalysis
Yanli
Yin
ab,
Xiaowei
Zhao
c,
Baokun
Qiao
*c and
Zhiyong
Jiang
 *ac
*ac
aSchool of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang, Henan 453007, P. R. China
bCollege of Bioengineering, Henan University of Technology, Zhengzhou, Henan 450001, P. R. China. E-mail: jiangzhiyong@htu.edu.cn
cHenan University, Jinming Campus, Kaifeng, Henan 475004, P. R. China
First published on 14th April 2020
Abstract
Chiral hydrogen-bonding catalysis is a classic strategy in asymmetric organocatalysis, and it has been extensively used in a variety of fundamental chemical transformations. At the same time, visible light-driven photoredox catalysis is a powerful and sustainable tool commonly used in radical chemistry. The intriguing combination of these two catalysis platforms would open a new avenue for the direct and highly efficient synthesis of enantioenriched compounds. Inspired by the conceptual breakthrough of T. Bach, in recent years, significant progress has been made in cooperative photoredox and chiral hydrogen-bonding catalysis. By developing a variety of important types of reactions, a wide range of valuable chiral compounds have been successfully synthesized. In this review, the advances in this key area are systematically described, and the examples are organized according to the distinct bond-forming patterns in the construction of the stereocentres.
 Yanli Yin | Yanli Yin received her Master Degree in Chemistry with a specialty in biochemical engineering from Huazhong University of Science and Technology in 2005. She obtained her PhD degree in 2018 from Henan University under the supervision of Professor Zhiyong Jiang. Currently, she is an associate professor at Henan University of Technology. Her research interests are centered on asymmetric photocatalysis. |
 Xiaowei Zhao | Xiaowei Zhao was born in Henan, P. R. of China in 1984. She obtained his PhD from Henan University under Professor Zhiyong Jiang in 2019. Currently, she is a faculty member at Henan University. Her research interests focus on asymmetric organocatalytic synthesis of useful chiral molecules as well as evaluations of their biological activities. |
 Baokun Qiao | Baokun Qiao was born in Henan, China, in 1987. He received his B.S. degree from Henan University in 2011. Subsequently, he joined Prof. Zhiyong Jiang at Henan University for another three years. In 2014, he pursued his Ph.D. at Tianjin University under the supervision of Professor Jun-An Ma. In 2017, he joined Henan University, where he focused his interest in asymmetric photoredox catalysis. |
 Zhiyong Jiang | Zhiyong Jiang received his B. Sc. in 1996 from Zhejiang University. He obtained his PhD from the same university under Professor Yan-Guang Wang in 2004. He first carried out 14 months of postdoctoral work at Hong Kong Baptist University. Then, he joined Professor Choon-Hong Tan at the National University of Singapore for another four years. In Aug. 2009, he began his independent research career at Henan University as a full professor. In Aug. 2018, he moved to Henan Normal University. |
Introduction
Due to the remarkable influence of chirality on processes in biology, physics and chemistry, the synthesis of optically pure compounds has always been pursued in many scientific domains, such as pharmaceuticals, functional materials and catalysis. To achieve high atom economy and produce different kinds of chiral molecules, especially those featuring new and complex structures, asymmetric catalysis has been an active field of research in the past few decades. Based on these ongoing efforts, asymmetric organocatalysis, which uses small organic molecules as catalysts, has become an important branch parallel to transition-metal catalysis and enzyme catalysis.1 Among the defined strategies (e.g., enamine catalysis, iminium catalysis, SOMO catalysis, counterion catalysis, and hydrogen-bonding (H-bonding) catalysis), H-bonding catalysis has been widely used in the synthesis of enantiomerically pure compounds.2 Chemical transformations could be promoted based on H-bonding interactions between chiral H-bonding catalysts and substrates that possess oxygen- or nitrogen-containing functional groups as H-bonding acceptors and/or protons as H-bond donors, simultaneously providing enantiocontrolling environments for the formation of diverse stereocentres. However, the rather low energy of H-bonding interactions often requires the use of highly reactive substrates, which inarguably leads to limited substrate scopes and amenable reaction types as well as tedious preparations of substrates to achieve sufficient reactivity. Furthermore, the Brønsted acid/base character of the catalyst enabling the generation of the reaction intermediates always results in non-neutral reaction systems, making the construction of labile stereogenic centres quite challenging.3 Accordingly, designing a highly reactive and mild catalytic system for chiral H-bonding catalysis would overcome this formidable bottleneck and allow new developments in this important catalysis platform.In addition to reactions of ions, radical-based transformations have been widely used in synthesis, as they enable a wide range of non-traditional bond cleavages and formations under mild reaction conditions, owing to the unique methods used to generate radicals and the high reactivity of these species.4 In particular, visible light-driven photocatalysis, which allows radical transformations to be performed in a sustainable manner, is a powerful tool in organic synthesis.5 Among the developed activation modes, photoredox catalysis, in which the catalytic cycle is triggered by an outer-sphere single-electron transfer (SET) between the excited state photosensitizer and the substrate via oxidative quenching or reductive quenching (Scheme 1), is one of the most popular due to the rapid development of both photoredox catalysts and cooperative catalytic systems based on other catalytic platforms.5,6 Notably, this catalysis platform allows the generation of a variety of H-bonding donor/acceptor-containing intermediates, such as radicals, radical ions and neutral intermediates. The use of an extrinsic chiral H-bonding catalyst could recognize these species or their reaction partners bearing H-bonding donors or acceptors through H-bonding interactions. Such a weak interaction should improve the nucleophilicity or electrophilicity of these species and alter their stability, therefore increasing the chemoselectivity of the reaction and the reactivity of the substrate and allowing the chiral catalysts to form the stereocentres with good enantiocontrol. As such, the merger of chiral H-bonding catalysis and photoredox catalysis would be a promising protocol in asymmetric synthesis.7
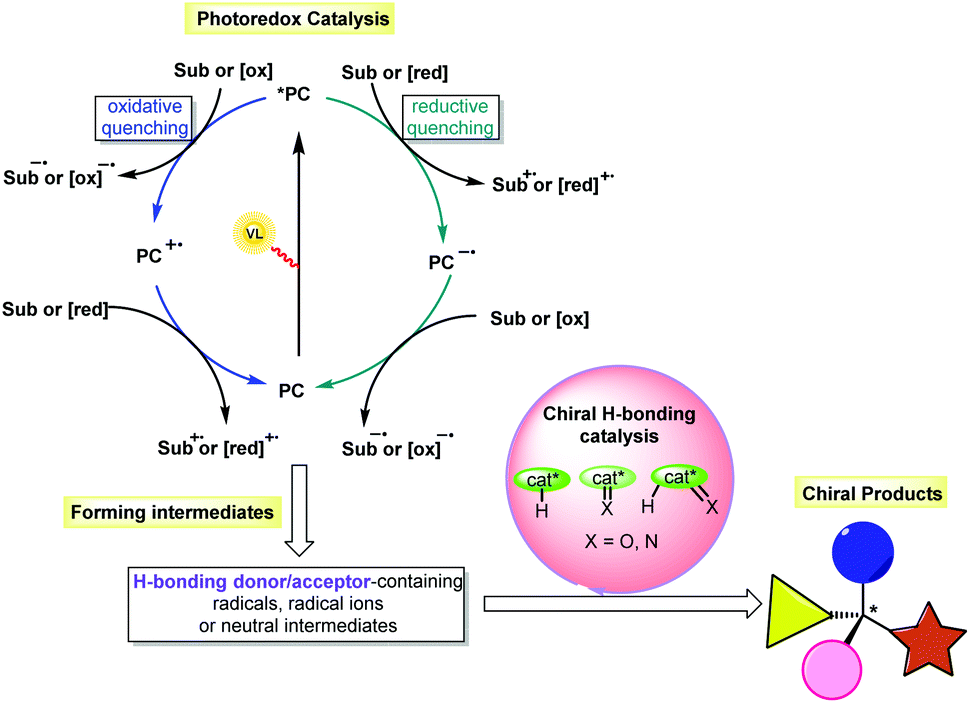 | ||
| Scheme 1 Cooperative asymmetric and H-bonding catalysis. | ||
In fact, the development of asymmetric photoredox catalysis has attracted the attention of chemists for many years. In 2005, Bach and co-workers made a breakthrough in this field; they reported an enantioselective cyclization using chiral photosensitizer 2 (Scheme 2).8 Under photoirradiation (λ > 300 nm), the ketone moiety of 2 can be excited to undergo SET with the tertiary amine of substrate 1. The generated α-amino radical species can then undergo an enantioselective Giese-type addition to the active olefin with the amide moiety of 2 acting as a bifunctional H-bonding catalyst to interact with the amide unit of 1, thus providing enantiocontrol. Although only a moderate enantiomeric excess was obtained for 3 (70% ee), the realization of enantiocontrol in this highly reactive radical-based transformation, especially when only using weak H-bonding catalysis, demonstrates the viability of asymmetric photoredox catalysis. Along with the development of SOMO catalysis, in 2008, MacMillan and co-workers reported a highly enantioselective alkylation of aldehydes by merging photoredox and chiral secondary amine catalysis.9 Both successes inspired chemists to pursue stereoselective photoredox catalysis by exploiting distinct catalytic asymmetric strategies. Cooperative photoredox and H-bonding catalysis strategies have also been developed; by establishing efficient catalytic systems that can provide robust stereocontrol for highly reactive radical transformations and overcome strong racemic background reactions, a number of valuable but challenging reactions have been achieved with high enantioselectivities.
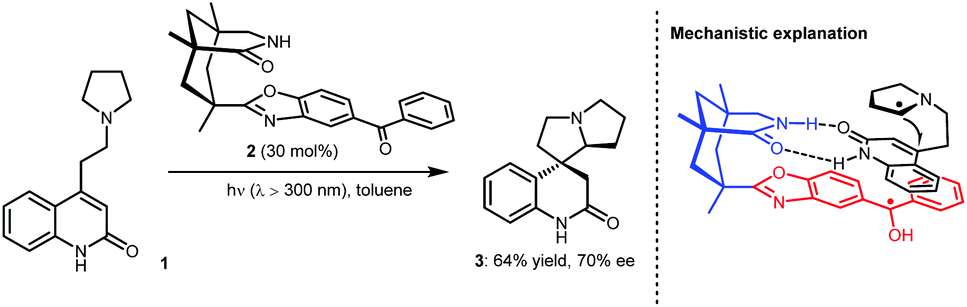 | ||
| Scheme 2 Chiral photosensitizer-catalysed asymmetric cyclization reaction. | ||
In this review, we summarize the advances in cooperative photoredox and H-bonding catalysis from the first example by Bach to March 2020. This review is organized into four parts according to the distinct bond-forming patterns in the construction of the stereocentre, namely, radical additions, radical cross couplings, C–C and C–O bond formation via ionic-type pathways and C–H bond formation via ionic-type pathways.
Enantioselective radical addition
Radical addition is a powerful strategy for rapidly constructing chemical bonds.4 In 2013, Knowles and co-workers reported an intramolecular enantioselective aza-pinacol cyclization of 4 in the presence of a dual catalyst system involving BINOL-derived chiral phosphoric acid (CPA) 5 and Ir(ppy)2(dtbpy)PF6 as a photosensitizer and using Hantzsch dihydropyridine (HEH) as a stoichiometric reductant (Scheme 3).10 The CPA was responsible for the reduction of the ketone moiety of 4via a proton-coupled electron transfer (PCET) process.11 They also verified the strong basicity of the generated ketyl intermediate, which enables CPA to closely interact with the substrate, thus providing perfect enantiocontrol for the subsequent radical addition to an imine. This work represents the first cooperative photoredox and H-bonding catalysis with high enantioselectivity (see 6). This provided a new starting point for using chiral H-bonding catalysis in radical chemistry.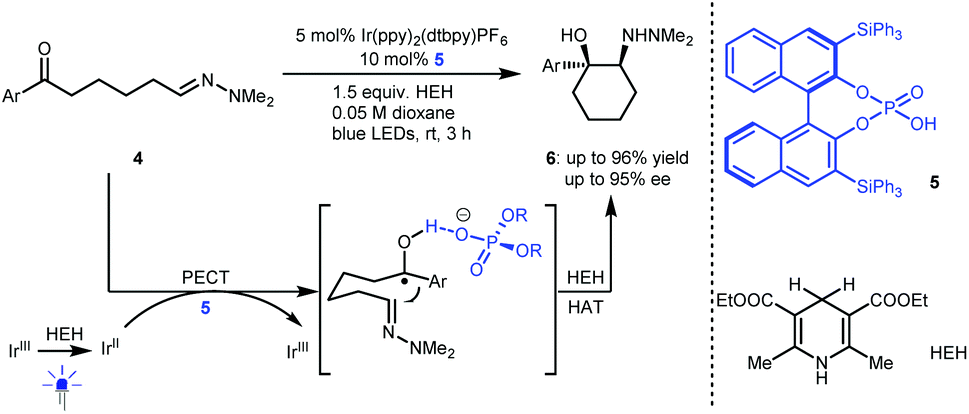 | ||
| Scheme 3 Enantioselective aza-pinacol cyclization. | ||
Imine-bearing azaarenes, such as pyridines, quinolines, isoquinolines, and imidazoles, are significant structural motifs that ubiquitous in diverse natural products, pharmaceuticals and materials.12 Given their electron-deficient properties, in recent years, exploiting these azaarenes as analogues of carbonyls to trigger asymmetric transformations of prochiral azaarene-based substrates has been widely used as a direct approach for accessing chiral azaarene derivatives.13 The fact that these azaarenes are less able than carbonyls to activate functional groups leads to limited strategies and has inspired chemists to attempt highly reactive radical pathways by using cooperative photoredox and H-bonding catalysis.
Based on the success of racemic phosphoric acid-catalysed conjugate addition of α-amino radicals generated from the single-electron oxidation of N,N-dimethyl anilines to alkenylpyridines, in 2016, the Melchiorre group attempted the enantioselective variant with alkene 7 as the representative substrate using BINOL-CPA 8 as the chiral catalyst (Scheme 4).14 Desired product 9 was obtained with 35% ee. Interestingly, both E-7 and the diastereomer Z-7 separately underwent this transformation with the same enantioselectivity, which was likely due to a Curtin–Hammett-type kinetic situation. Although the enantioselectivity was not satisfactory, only 10 mol% of the CPA yielded a product with 35% ee, which strongly supports the feasibility of asymmetric H-bonding catalysis in azaarene-involved radical reactions through the H-bonding interaction of the basic N of azaarenes with the proton of a chiral Brønsted acid.
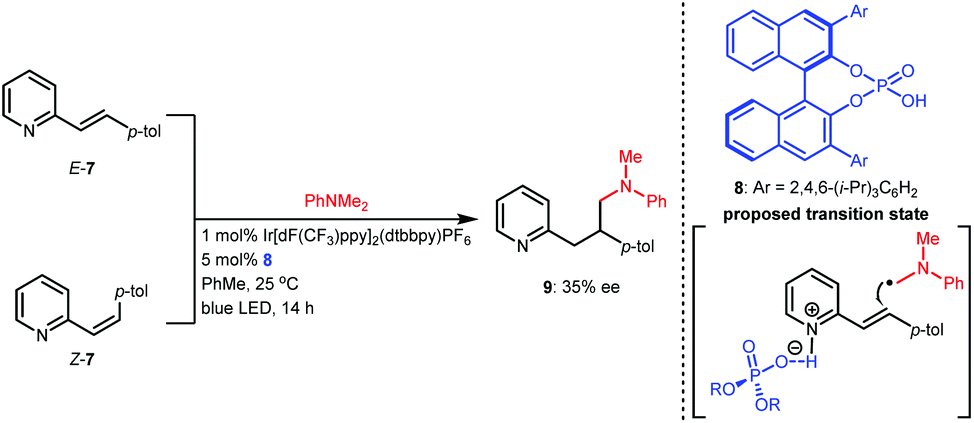 | ||
| Scheme 4 Enantioselective conjugate addition of an α-amino radical to alkenylpyridine. | ||
In 2017, Fu and co-workers described a photoredox-catalysed Minisci-type addition of α-amino acid-derived redox active esters (RAEs) 10 to azaarenes (Scheme 5a).15 In the presence of 1 mol% Ir(dF(CF3)ppy)2(dtbbpy)PF6 as a photosensitizer, RAE 10 could be reduced by an Ir(II) species. The obtained α-amino radicals would add to azaarene 12, which was activated by racemic phosphoric acid 11, leading to radical intermediate 13. After single-electron oxidation and deprotonation, a wide range of valuable azaarene-substituted secondary amines were obtained in up to 96% yield. This work revealed the possibility of realizing stereocontrolled variants.
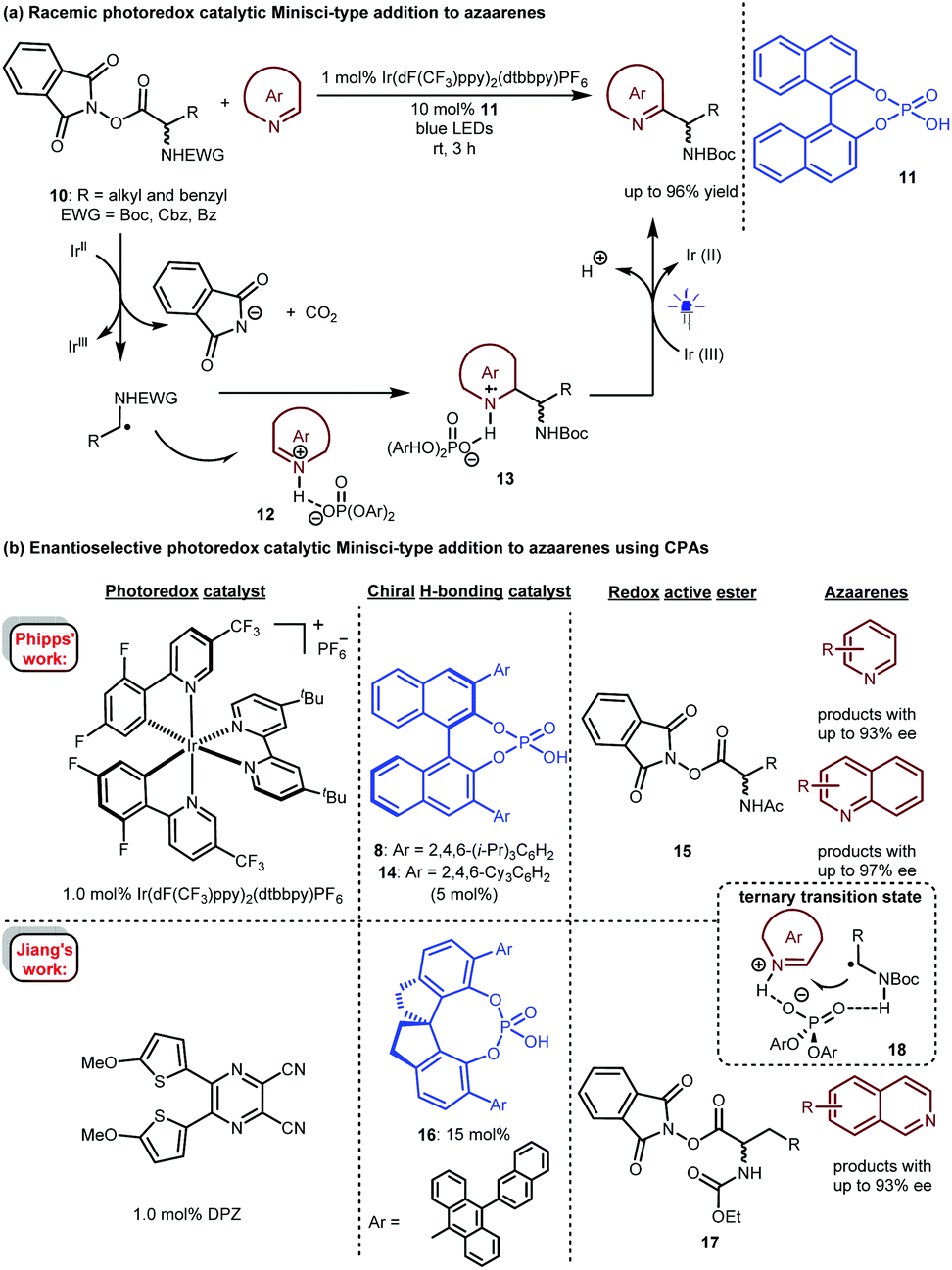 | ||
| Scheme 5 Photoredox-catalysed Minisci-type additions to azaarenes. | ||
Only one year later, Phipps and co-workers reported an enantioselective version of this important transformation (Scheme 5b).16 They used a dual catalytic system involving 1.0 mol% of Ir(dF(CF3)ppy)2(dtbbpy)PF6 and 5.0 mol% of (R)-TRIP (8) or (R)-TCYP (14), realizing the highly enantioselective addition of various RAEs 15 to a variety of pyridines and quinolines. In the same year, Jiang's group17 achieved the same reaction with their developed highly efficient visible light-absorbing organic photosensitizer, a dicyanopyrazine-derived chromophore (DPZ)18 and SPINOL-CPA 16 as the catalyst. Unlike Phipps’ work, this catalytic system was suitable for isoquinolines; with RAEs 17 as the reaction partners, products were obtainable in up to 93% ee. Both works proposed a H-bonding catalytic mechanism involving a ternary transition state (18) in which the CPA acts as a bifunctional catalyst to simultaneously interact with the proton of the radical species and the N of the azaarenes, therefore providing enantiocontrol for the new C–C bond formation. Notably, in 2019, Fu and Shang and co-workers also developed an organocatalytic system19 based on sodium iodide (20 mol%), triphenylphosphine (20 mol%) and SPINOL-CPA (5.0 mol%), and these reagents furnished azaarene-substituted chiral secondary amines with a similar substrate scope and enantioselectivity to those of Phipps.
In 2019, the Studer group elegantly extended the enantioselective Minisci-type addition into a more challenging three-component radical cascade reaction (Scheme 6).20 In the presence of Ir(dF(CF3)ppy)2(dtbbpy)PF6 and CPA 8 as a dual catalyst system, the reactions of azaarenes with enamides 19 and α-bromo carbonyl compounds 20 afforded valuable chiral heterocyclic γ-amino acid derivatives 21 in high yields and enantioselectivities. The scope of azaarenes is extremely broad, including quinolines, pyridines, and several quinolines with an additional fused benzene ring. This strategy is also compatible with the sequential addition of amidyl radicals, which are generated from oxidizing substrates 22 to the enamides and then to quinolines, leading to another kind of important chiral azaarene, derivative 23, with satisfactory results.
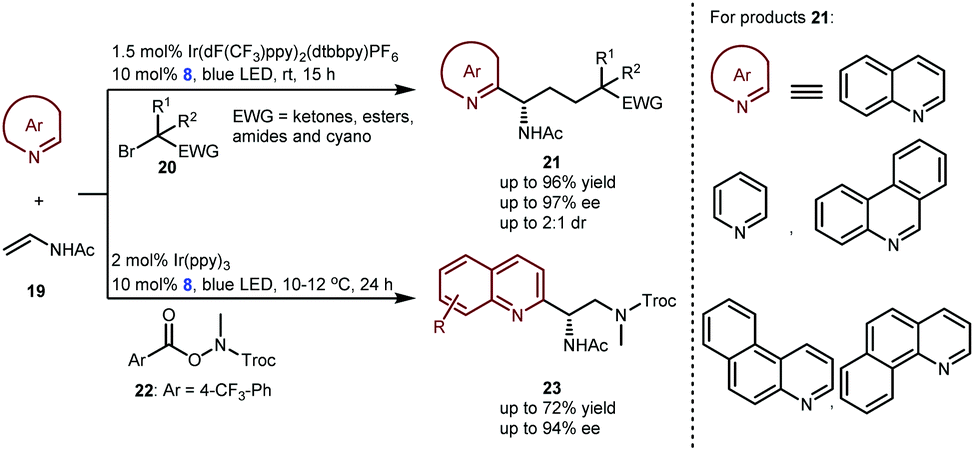 | ||
| Scheme 6 Enantioselective three-component Minisci-type addition to azaarenes. | ||
The above achievements (Schemes 5 and 6) were focused on building tertiary stereocentres α to azaarenes.16,17,19,20 As depicted by Melchiorre,14 the enantioselective formation of chiral centres at the β-position of azaarenes under the catalysis of CPA remains challenging, likely due to the greater distance between the α-amino radical and the nitrogen-coordinated chiral catalyst, which weakens the efficacy of the enantiofacial induction. Nevertheless, the Jiang group still explored the possibility of H-bonding catalysis in constructing even more distant γ-stereocentres through the conjugate addition of prochiral radicals to vinylazaarenes. Given the biological importance of the resulting chiral products, they explored the addition of aldehydes, ketones, aldimines and ketimines to vinylpyridines 24 (Scheme 7) via a reductive coupling strategy.21 As a result,22 when using DPZ with SPINOL-CPA 25 as cooperative catalysts, desired adducts 26 could be achieved in up to 90% yield with up to >99% ee. Notably, the substrate scope is very broad with respect to the both pro-nucleophiles and the pyridine electrophiles; a series of chiral secondary and tertiary alcohols/amines were embedded at the γ-position of 2-pyridines and 4-pyridines. The high enantioselectivity towards both the complex oestrone-derived ketone and 4-vinylpyridine as substrates further demonstrates the robust abilities of CPAs as bifunctional chiral H-bonding catalysts in asymmetric photoredox-catalysed reactions of prochiral azaarenes.
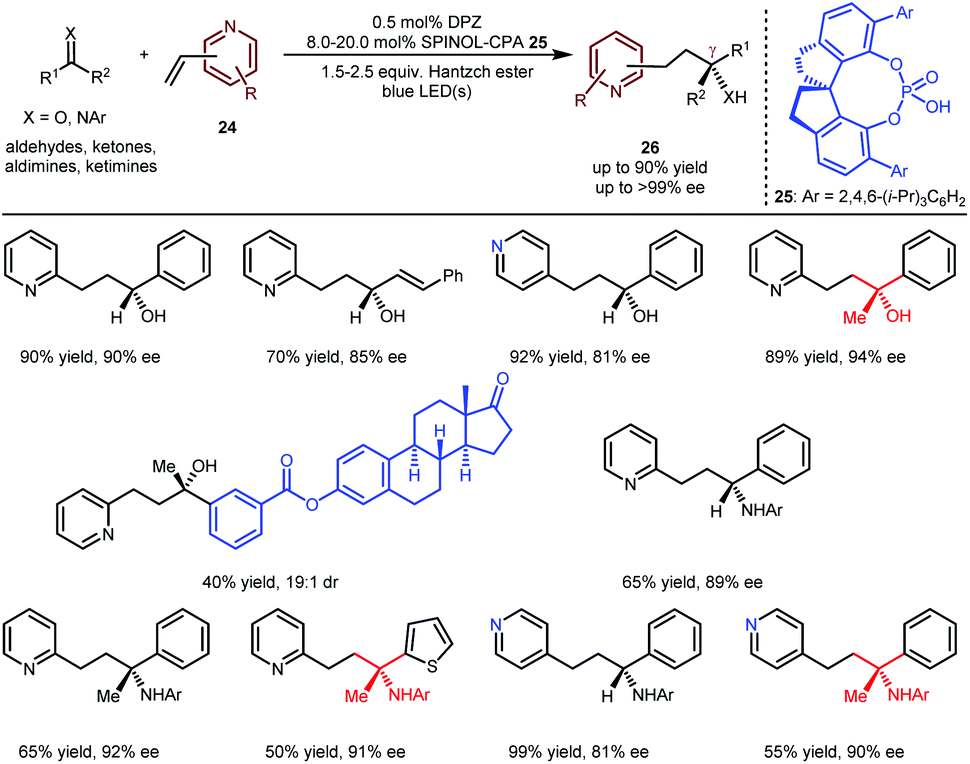 | ||
| Scheme 7 Catalytic enantioselective addition of prochiral radicals to vinylpyridines. | ||
Very recently, Knowles and co-workers reported an enantioselective intramolecular hydroamination of alkenes with sulfonamides under a dual catalyst system including [Ir(dF(CF3)ppy)2(5,5′-dCF3bpy)]PF6 (2 mol%) and a BINOL-derived phosphate (2.5 mol%).23 These transformations were proposed to occur via N-centred radicals generated by PCET activation of sulfonamide N–H bonds. The H-bonding interaction between the neutral sulfonamidyl radical and the CPA generated in the PCET event was hypothesized to be the basis for asymmetric induction in the subsequent C–N bond-forming step, providing enantioenriched pyrrolidine products with up to 96% ee.
Enantioselective radical cross coupling
Radical cross coupling reactions can generally yield new chemical bonds owing to the approximately barrierless docking of the two distinct odd-electron partners.4 In this context, this strategy can likely provide an efficient and direct synthetic approach to important molecules with high functional group tolerance. However, this synthetic advantage becomes a challenge for enantioselective variants since the high reactive radical content leads to elusive stereocontrol and, at the same time, results in a strong racemic background reaction. Furthermore, the homocoupling of radicals also makes chemoselectivity a significant challenge.24 As such, it is highly desirable to develop generic and efficient catalysis platforms for diverse enantioselective radical cross coupling reactions in both fundamental research and practical applications.In 2015, Ooi et al.25 reported a redox neutral, highly enantioselective coupling between N-arylaminomethanes 27 and N-sulfonyl aldimines 28 by harnessing the dual catalysis of P-spiro chiral arylaminophosphonium barfate (29·HBArF) and [Ir(ppy)2(Me2phen)]BArF as the photosensitizer under visible light irradiation (Scheme 8). The reaction was triggered by the reductive quenching of the photoactivated Ir(III) complex with 27, resulting in an Ir(II) intermediate and α-amino radicals. Aldimines 28 were then reduced by Ir(II) to generate radical anions, which can be captured by a chiral catalyst via counterion and H-bonding interactions (see 31). The coupling of α-amino radicals with 31 afforded valuable chiral diamine products 30 containing a tertiary stereocentre in up to 90% yield with up to 97% ee. This work represents the first enantioselective radical coupling reaction by asymmetric photoredox catalysis. Subsequently, they established a catalytic cycle initiated by the oxidative quenching of the excited photosensitizer (Ir*(ppy)3) for the same transformations.26
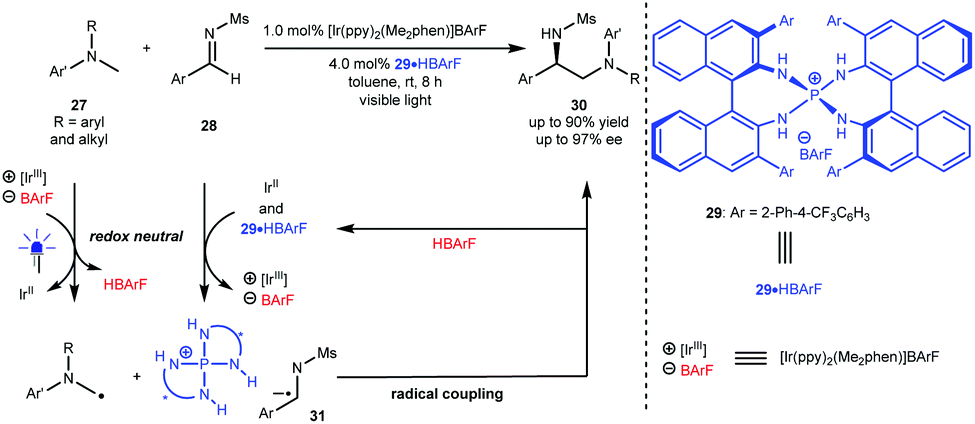 | ||
| Scheme 8 Redox neutral α-coupling of N-arylaminomethanes with aldimines. | ||
To develop a strategy for synthesizing important chiral tertiary alcohols containing a primary amine at the β-position, Jiang and co-workers developed an enantioselective radical coupling of N-aryl glycines 32 with acyclic activated ketones (i.e., 1,2-diketones 33) and cyclic activated ketones (i.e., N-Boc isatins 36) (Scheme 9).27 By exploiting a cooperative photoredox and chiral H-bonding catalytic system involving DPZ and SPINOL-CPAs 34 and 37, corresponding products 35 and 38 were synthesized in high yields and enantioselectivities. In this work, more challenging heteroquaternary stereocentres were also successfully constructed.
 | ||
| Scheme 9 Redox neutral, enantioselective radical coupling of N-aryl glycines with activated ketones. | ||
Alkyl halides are diverse, abundant and readily accessible feedstocks in organic synthesis. The nucleophilic substitution of alkyl halides is a fundamental chemical transformation for accurately delivering molecular fragments to sp3-hybridized carbon atoms utilizing the halide as the directing group.28 Although many methods, especially classic SN1 and SN2 reactions, have been established and extensively used in the synthesis of complex molecules, enantioconvergent substitutions that form a carbon stereocentre in simple alkyl halides remains challenging, thus attracting the attention of chemists.29 Given the oxidative ability of alkyl halides and a wide array of readily available and bench-stable reducible organic molecules, Jiang and co-workers developed an asymmetric photoredox catalytic strategy that offers a new and direct synthetic approach with high functional group tolerance. They explored the viability of transformations of N-aryl amino acids and α-halo carbonyl compounds by using cooperative photoredox and chiral bifunctional H-bonding catalysis (Scheme 10a).30 Two distinct radical species could be generated through this redox neutral pathway, and the H-bonding interactions of the chiral catalyst with both the α-amino radicals and the α-radicals of the carbonyl compounds would improve their nucleophilicity and electrophilicity, respectively, thus providing a stereocontrolling environment for C–C bond formation via radical coupling. As a result, by using DPZ and SPINOL-CPA (25, 37, 41 or 42), a variety of α-bromo ketones could smoothly react with diverse N-aryl amino acids (32 and 40), affording a wide range of valuable enantiomerically pure β2- and β2,2-amino ketones 43 in satisfactory yields with good to excellent enantioselectivities (Scheme 10b). Fluoro-heteroquaternary and full-carbon quaternary stereogenic centres that are otherwise difficult to prepare were successfully constructed. Notably, after the single-electron reductive debromination of 39, the generated radicals more easily undergo single-electron reduction-protonation in the absence of CPA, indicating that H-bonding catalysis is crucial for the chemoselectivity.
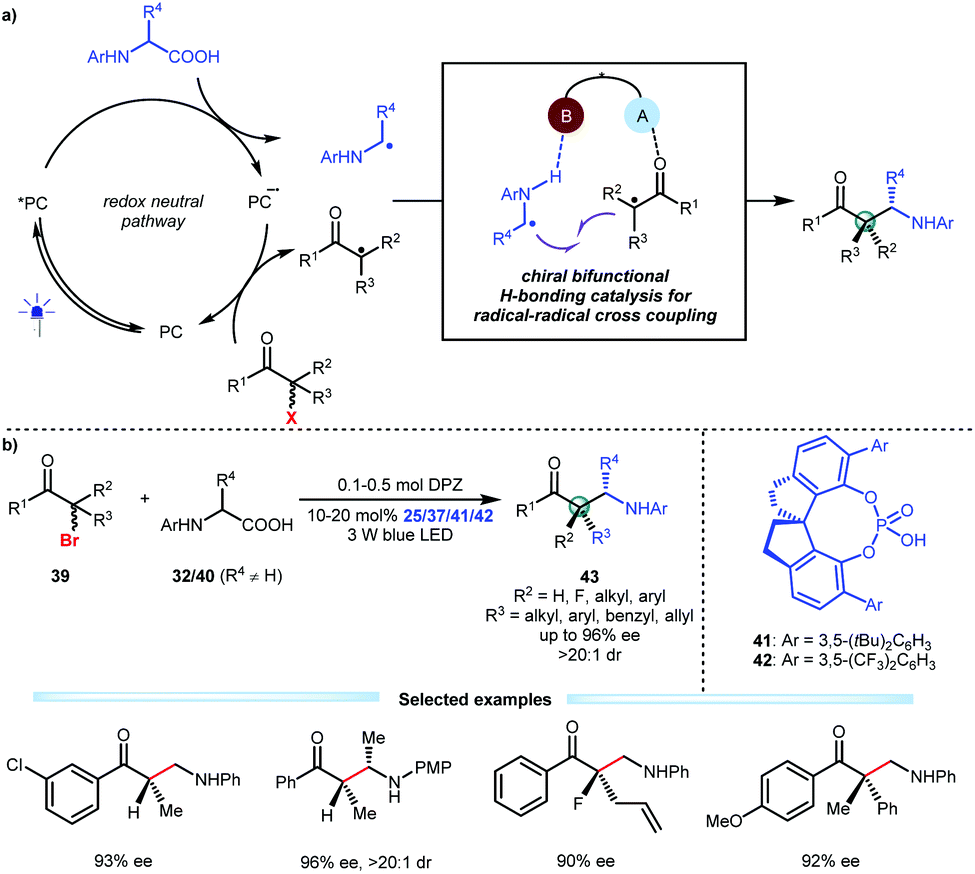 | ||
| Scheme 10 Enantioconvergent substitution of alkyl halides via catalytic asymmetric photoredox radical coupling. | ||
Upon the above success, they subsequently tested this cooperative catalysis platform in the reaction of 3-chlorooxindoles 44 with N-aryl glycines 32 given the biological importance of the resulting adducts (Scheme 11).31 In the presence of DPZ and SPINOL-CPA (45 or 46), the transformations could efficiently furnish products 47 in up to 96% yield with up to 98% ee. The substrate scope is satisfactory since 3-aryl, 3-alkyl and 3-allyl 3-chlorooxindoles were all compatible. This method enables the direct synthesis of important spiro-five- and -six-membered ring-based products, which include horsfiline and analgesic therapeutic derivatives.
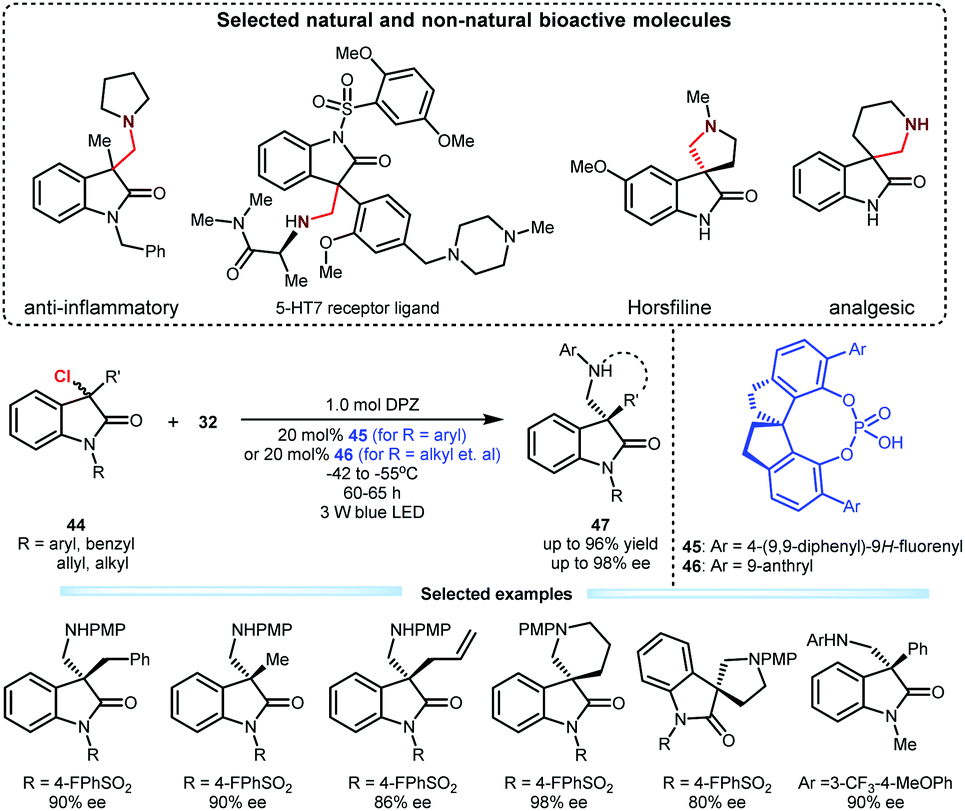 | ||
| Scheme 11 Enantioconvergent substitution of 3-chlorooxindoles. | ||
2,2-Disubstituted indolin-3-ones are privileged molecular architectures found in a number of pseudoindoxyl alkaloids. In 2014, the groups of Brasholz32a and Xiao32b independently reported an efficient synthetic method for accessing these valuable entities via a visible light-driven photoredox-catalysed aerobic oxidation and semipinacol rearrangement of 2-aryl-3-alkyl-substituted indoles (48) (Scheme 12a). Mechanistically, the two radical species obtained from 48via single-electron oxidation and from oxygen gas via single-electron reduction (superoxide anion radical) will couple to produce intermediates 49. Then, intermediates 50 were produced from the reaction between 48 and 49, and 50 could subsequently undergo a semipinacol transformation to furnish 2,2-disubstituted indolin-3-ones ((±)-51). Notably, if the radical coupling could be achieved with enantiocontrol, enantioenriched 51 would be obtained. The viability of this hypothesis was revealed by the Xiao group, although only one example (51a) with moderate enantioselectivity was achieved by using a dual catalysis system of a Ru-based photosensitizer and BINOL-CPA (52) and under a pure oxygen atmosphere (Scheme 12b).32b In 2018, the Jiang group realized the first highly enantioselective example and they used a more convenient ambient atmosphere.33 This organocatalytic system includes DPZ and SPINOL-CPA 53. Their investigation also revealed that the formed chiral 3-hydroxy intermediates 50 could affect the enantioselectivity of the radical coupling, making enantiocontrol more difficult.
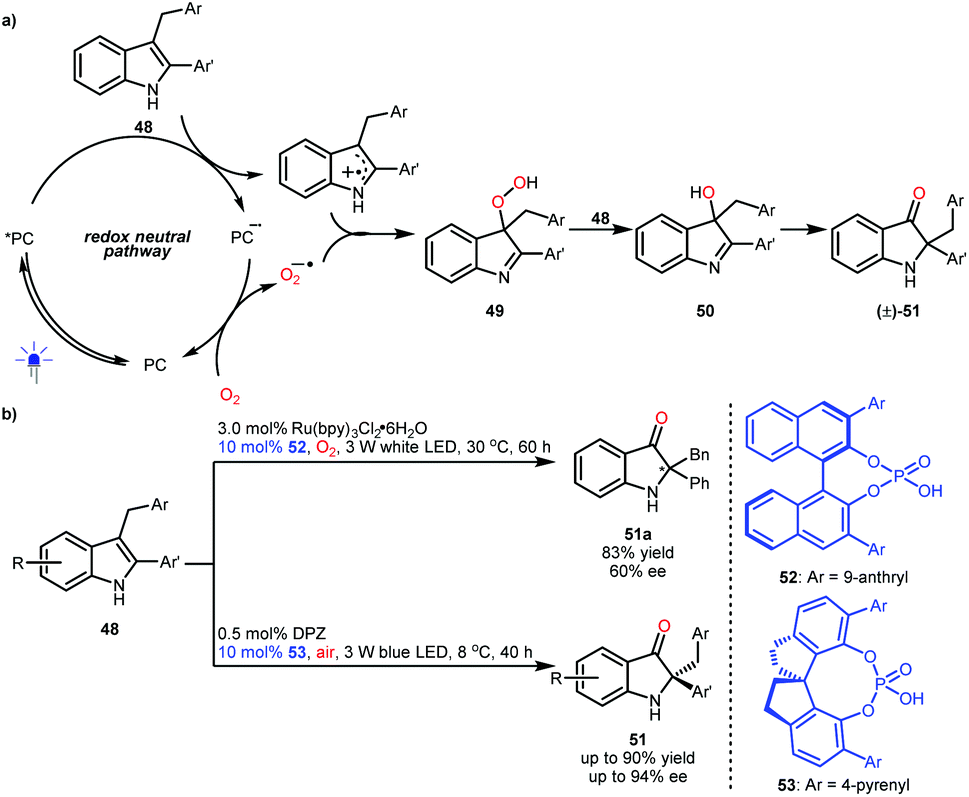 | ||
| Scheme 12 Photoredox-catalysed aerobic cascade oxidation and semipinacol rearrangement. | ||
In 2018, Knowles and co-workers used TEMPO as the radical source and TIPS-EBX as the sacrificial oxidant and proton acceptor and developed an enantioselective synthesis of pyrroloindolines from N′-Cbz-protected tryptamines (54) via a sequential single-electron oxidation, radical coupling and addition processes.34 As depicted in Scheme 13, H8-TRIP BINOL phosphate 55 acts as a Brønsted base and interacts with the radical cations generated from 54 to form radical species 57, which is crucial to constructing stereocentres via C–O bond formation (58). Obtained products 56 are valuable since they were shown to react with diverse nucleophiles, affording important derivatives, most notably in the synthesis of (−)-calycanthidine.
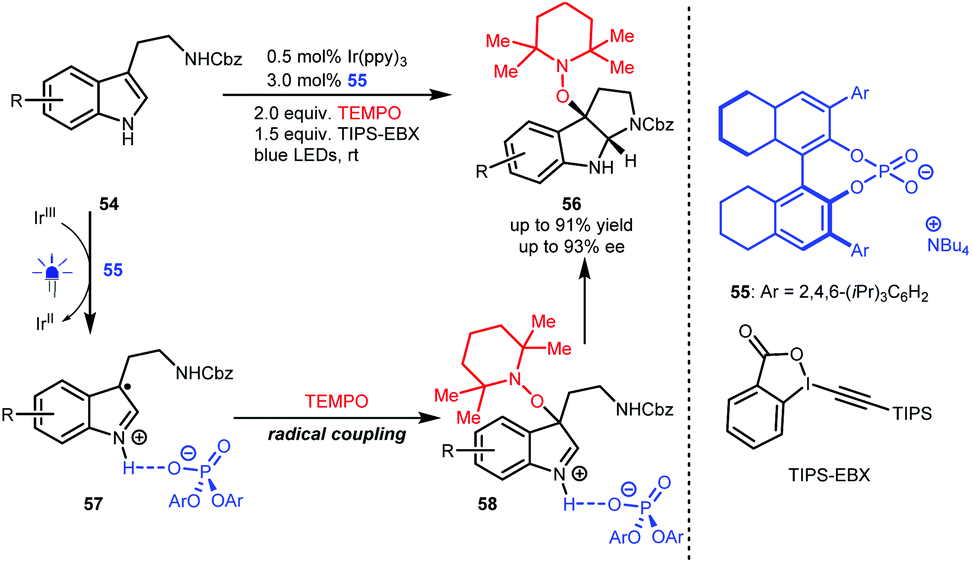 | ||
| Scheme 13 Enantioselective synthesis of pyrroloindolines. | ||
C–C and C–O bond formation via ionic-type pathways
In addition to offering highly reactive radical intermediates, photoredox catalysis can provide key electron-neutral or ionic intermediates that are difficult to prepare or are unstable. In conjunction with extrinsic asymmetric catalysis, this strategy has been widely applied in the synthesis of optically pure compounds.35 There are also several elegant examples involving the exploitation of cooperative photoredox and H-bonding catalysis.
N-Aryl α-amino acids 40 can generate α-amino radicals 61a after a single-electron oxidation (Scheme 14).26–28 In fact, these species can be further oxidized, leading to imines (e.g., 62) that are not easily purified due to their lability in acidic silica gel.36 Notably, alkyl-substituted imines are prone to tautomerization, resulting in enamines (e.g., 63). Jiang and co-workers hypothesized that N-aryl α-amino acids might undergo aerobic decarboxylative [4 + 2] cycloaddition (Povarov reaction) under photoredox catalysis, which would efficiently provide important 4-amino tetrahydroquinolines (THQs).37 After careful investigations, this synthesis was realized, and a series of 4-amino THQs and even the corresponding quinolines were obtained in satisfactory yields. Given that there were no examples of the synthesis of chiral 4-amino-2-methyl-substituted THQs, likely due to the inconvenient manipulation of acetaldehyde and the instability of acetaldehyde-derived imines, they also engaged in developing a dual catalysis enantioselective manifold for the aerobic decarboxylative Povarov reaction of N-aryl alanines 40. Finally, desired products 60 were obtained in up to 73% yield and with up to 94% ee and 5.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr by using 1.0 mol% DPZ, 10 mol% SPINOL-CPA 59 and 1.0 equiv. of tetra-n-octylammonium bromide (TOAB) as an additive and under ambient atmosphere.
:1 dr by using 1.0 mol% DPZ, 10 mol% SPINOL-CPA 59 and 1.0 equiv. of tetra-n-octylammonium bromide (TOAB) as an additive and under ambient atmosphere.
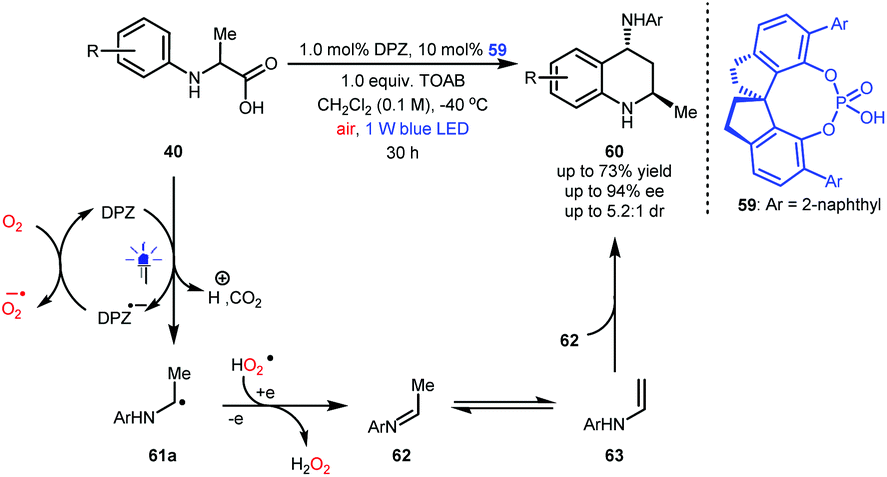 | ||
| Scheme 14 Enantioselective synthesis of chiral 4-amino-2-methyl THQs. | ||
Subsequently, Jiang et al. developed an enantioselective aerobic decarboxylative Povarov reaction of N-aryl α-amino acids 40 with methylenephthalimidines 64 in the presence of a dual catalysis system involving DPZ and SPINOL-CPA 53 (Scheme 15).38 A range of important optically pure isoindolin-1-ones 65 that feature a 3,3-spiro-tetrahydroquinoline-based stereocentre were prepared in high yields, ees and drs. This work represents the first construction of the 3,3-spiro-six-membered ring of an isoindolin-1-ones through asymmetric catalysis. Notably, the unsatisfactory results of the SPINOL-CPA 53-catalysed three-component Povarov reaction of 64 with aniline and distinct forms of formaldehyde underscore the synthetic importance of this protocol.
 | ||
| Scheme 15 Asymmetric aerobic decarboxylative Povarov reactions of N-aryl α-amino acids with methylenephthalimidines. | ||
Wang and co-workers also developed oxygen-free conditions by using oxidizing α-amino acid-derived RAEs 15 as precursors of imines 69 (Scheme 16).39 With indoles 66 as the reaction partners, an asymmetric Friedel–Crafts reaction occurred in the presence of BINOL-CPA-derived phosphate 67 as the catalyst, leading to enantioenriched 1-indolyl-1-alkylamine derivatives 68 in up to 95% yield with up to 97% ee. Ternary transition state 70 was proposed, wherein 67 acts as a Lewis acid-Brønsted base bifunctional catalyst to interact with both the N atom of the imine and the H atom of the indole.
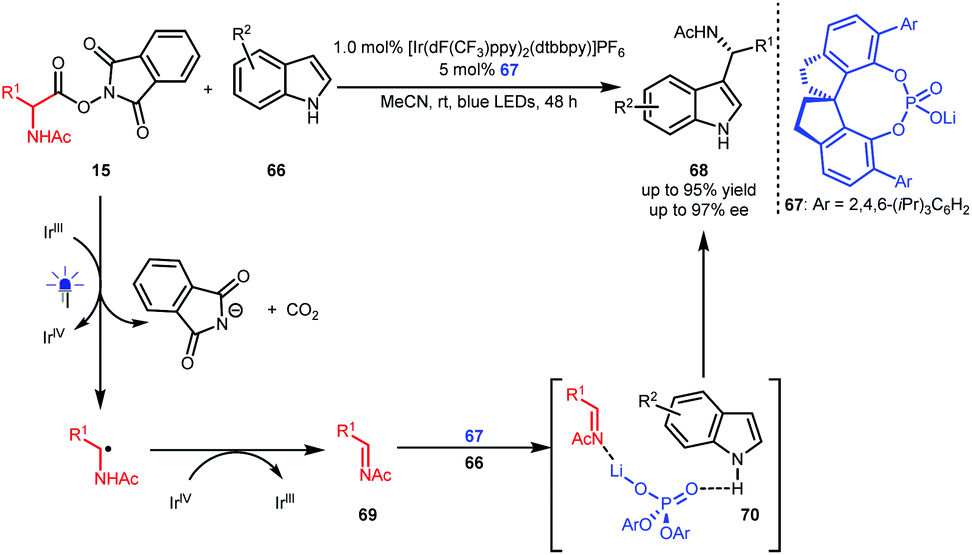 | ||
| Scheme 16 Enantioselective Friedel–Crafts reaction. | ||
As shown above (Scheme 13), Knowles reported an enantioselective synthesis of pyrroloindolines through a sequential process including a single-electron oxidation, a radical coupling and an addition.34 The formation of the stereocentre at the 3-position of the indole ring via a radical coupling occurs prior to the formation of the stereocentre at the 2-position via the aza-Mannich reaction. Very recently, You and Zhang and co-workers reported an elegant alternative method that enables the use of hydroxycarbamate 72 as the nucleophile (Scheme 17).40 In the presence of 0.2 mol% Ir(dFCF3ppy)2(bpy)PF6 as a photosensitizer and under the irradiation of blue LEDs, a variety of tryptophols and tryptamines (71) with diverse protecting groups as well as indoles containing an amide side chain first underwent a single-electron oxidation to generate a radical species, which subsequently interacts with BINOL-CPA 5 (see 74). Since the oxygen atmosphere allows the regeneration of the Ir(III) catalyst and the H-bonding interactions lower the activation energy, the enantioselective Mannich-type addition becomes the more favourable step, thus forming radical species 75 possessing a stereocentre at the 2-position of the indole ring. After the second single-electron oxidation, the generated cations would interact with the phosphate anion of 5 to offer complexes 76, which then were attacked by nucleophilic 72. As such, the formation of this C–O bond occurs via an ionic-type pathway. This umpolung method provides a general and efficient synthetic approach for accessing diverse valuable chiral pyrroloindolines and indolo[2,3-b] quinolines 73 bearing a fully substituted quaternary centre.
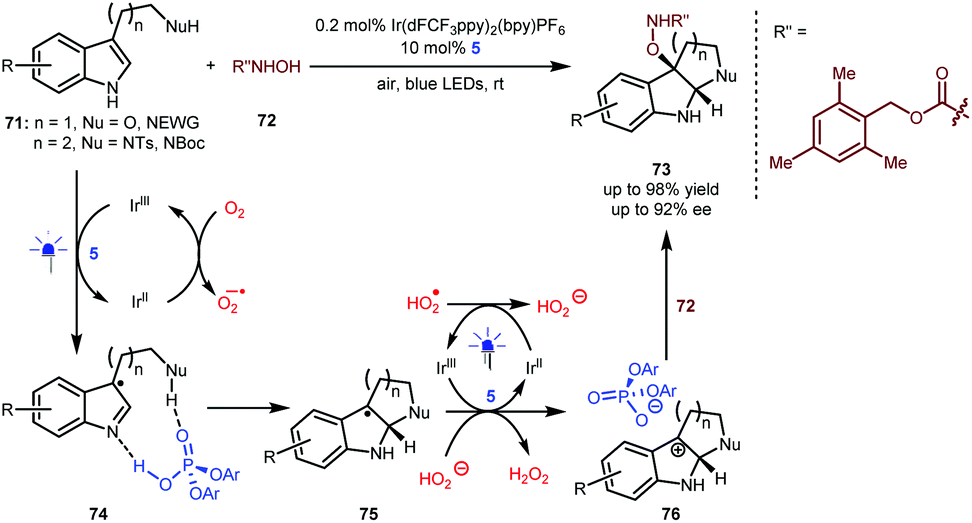 | ||
| Scheme 17 Asymmetric dearomatization of indole derivatives. | ||
C–H bond formation via ionic-type pathways
The construction of stereocentres via an ionic-type C–H bond formation process, namely, enantioselective protonation, has been widely used in the asymmetric synthesis of valuable enantioenriched compounds containing diverse tertiary carbon stereogenic centres.3 The development of novel strategies to expand the utility of this classic transformation is always attractive. In this context, the merger of radical-based photoredox catalysis and enantioselective catalytic protonation would be a significant and promising research area, as it could offer a new pathway for accessing important chiral compounds and might unlock readily accessible but poorly reactive feedstocks for direct use as substrates. However, since protons are very small and thus feature high mobility, achieving excellent enantioselectivity in enantioselective protonations is always a formidable challenge. More importantly, the rapid radical process to form the prochiral tertiary carbon anion intermediate would lead to a racemic background reaction, further increasing the difficulty of achieving high enantioselectivity. Even when using stoichiometric chiral Brønsted acid in the asymmetric visible-light-induced radical reactions of 3-alkylidene indolin-2-ones, the enantioselectivity in the generation of the corresponding stereocentres via protonation was poor.41 Nevertheless, the Jiang group focused on the development of photoredox-catalysed enantioselective protonation due to its significant advantage, that is, the ability to generate anion intermediates under controllable and near-neutral conditions via a radical process, and this would facilitate the construction of acid/base-labile tertiary stereocentres.Optically enriched α-hydroxy ketones (e.g., 78 and 82, Scheme 18) are biologically and synthetically important molecular skeletons, and the asymmetric reduction of readily available 1,2-diketones (e.g., 33a and 33b) could provide a direct approach to these valuable entities.42 However, in addition to two reductions leading to poor chemoselectivity,42b in the reaction process, the strong electron-withdrawing ability of the ketone and the high electronegativity of the oxygen make the tertiary stereocentre lead to facile racemization of α-hydroxy ketones under the non-neutral reaction conditions.42c Therefore, the catalytic asymmetric transformation of 1,2-diketones into chiral α-hydroxy ketones has always been a challenging task.
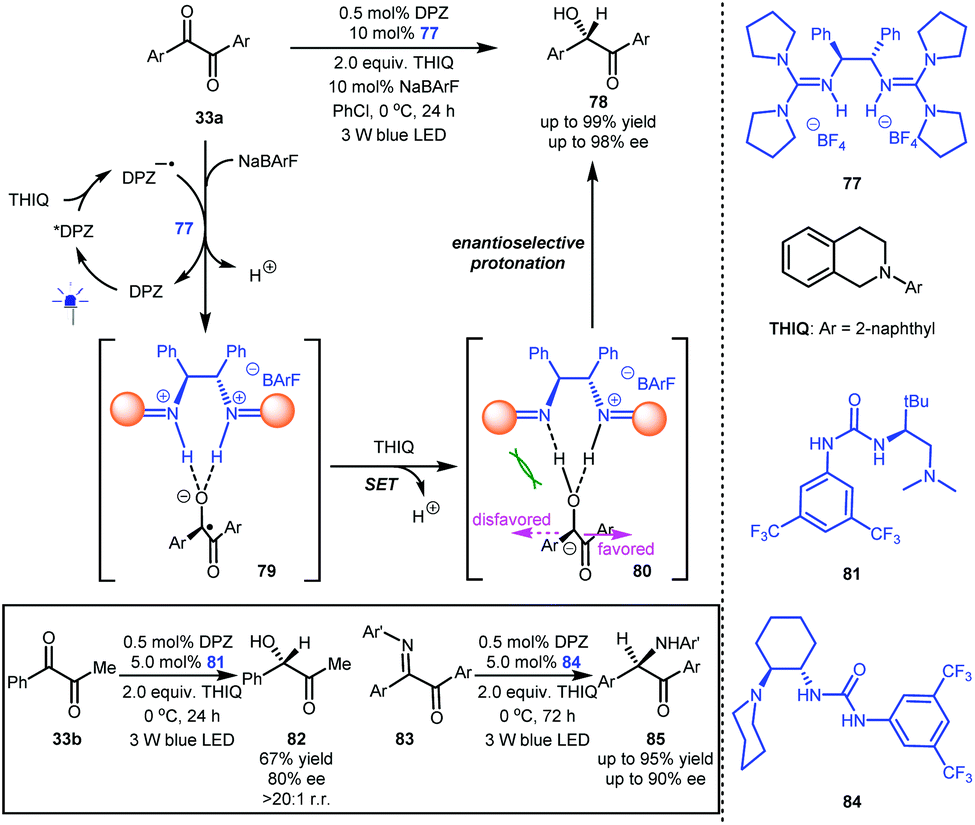 | ||
| Scheme 18 Enantioselective photoredox-catalysed reductive protonation. | ||
In 1990, Willner et al. described the reduction of benzil to benzoin with [Ru(bpy)3]Cl2 as a photoredox catalyst and Et3N as a sacrificial reductant, and protonation was suggested as the final step in the formation of the secondary alcohol.43 Inspired by this preliminary work as well as Knowles’ report10 on ketyl intermediates featuring a strong basicity, Jiang and co-workers reported the highly enantioselective reduction of 1,2-diketones (33a and 33b, Scheme 18) by establishing a dual catalytic system including DPZ and chiral guanidinium salt 77 as a Brønsted acid.44 With symmetric 1,2-diketones 33a as an example, the obtained DPZ−˙ from the reductive quenching of photoactivated DPZ by N-2-naphthyl tetrahydroisoquinoline (THIQ) could reduce 33a to generate the corresponding ketyl intermediates, which were then captured by 77 to form neutral radical species 79. The second single-electron reduction by THIQ afforded anion 80. In the stereocontrol environment provided by 78, enantioselective protonation was accomplished with good to excellent enantioselectivity (up to 98% ee). This investigation showed that the transformation occurred even without DPZ because it proceeds through an ‘electron donor-acceptor’ (EDA) mechanism, but the enantioselectivities of most of the products were lower than those obtained via the photoredox catalytic system. They next evaluated 1-phenyl-1,2-propanedione 33b as a representative unsymmetrical diketone, and tert-L-leucine-derived urea-tertiary amine4581 was determined to be the most suitable chiral catalyst; chiral alcohol 82 was obtained in 67% yield with 80% ee and excellent regioselectivity and chemoselectivity. By using Takemoto's 1,2-cyclohexanediamine-based urea-tertiary amine 84 and DPZ as the catalytic system, α-keto ketimines 83 could be efficiently reduced to α-amino ketones 85 in up to 95% yield with up to 90% ee. The fact that using three different kinds of H-bonding catalysts achieved excellent enantioselectivities suggests that similar dual catalyst systems could provide good results by carefully selecting the catalysts. Certainly, it also demonstrates chiral H-bonding catalysis in highly reactive radical-based transformations.
Their success in that reaction inspired them to exploit this dual catalysis platform for the construction of secondary α-carbon-halogen (i.e., F, Cl and Br) bonds of ketones, which are important in the pharmaceutical and synthetic fields, but there are few efficient strategies that can provide high enantioselectivity due to the lability of stereocentres. To this end, a series of α,α-dihaloketones 86 were selected as the starting substrates; in the presence of 0.5 mol% DPZ and 5.0 mol% L-tert-leucine-derived squaramide-tertiary amine 87 and using secondary amine 88 or 89 as the terminal reductant, desired products 90 containing secondary C–F, C–Cl and C–Br bonds were obtained in up to 96% yield and up to >99% ee with a broad substrate scope. Notably, the control experiment disclosed a strong competitive racemic background reaction in the reaction system (Scheme 19).46 The photoredox catalytic cycle was triggered by the single-electron transfer between photoactivated DPZ and the secondary amine (88 or 89). Subsequently, a C–Cl or C–Br bond of 86 would be reduced by DPZ−˙ due to their matched redox potentials. Because of the weaker basicity of carbonyl relative to ketyl, the two H-bonding interactions between the two N–H units of squaramide and oxygen of the carbonyl (see 91) are crucial, as they can stabilize the radical intermediates. After a secondary single-electron reduction to generate anions 92, enantioselective protonation to access products 90 was achieved with satisfactory results. Notably, their investigation revealed the considerable lability of C–Cl/Br bond-based stereocentres, since the ee value of the products deteriorated during flash column chromatography. Accordingly, a sequential strategy involving a subsequent one-pot reduction of ketones by diisobutylaluminium hydride (DIBAL-H) was established. Therefore, a series of valuable chloro/bromo-hydrins were prepared.
 | ||
| Scheme 19 Enantioselective photoredox dehalogenative protonation. | ||
Due to the biological importance of imine-bearing azaarene-substituted secondary alcohols and the lack of general and expedient catalytic methods for accessing these compounds, Jiang and co-workers also investigated the enantioselective reduction of azaarene-based ketones 93via visible light-driven photoredox asymmetric catalysis (Scheme 20). This work can be considered a logical and highly useful extension of the photoreduction of 1,2-diketones by using these azaarenes as analogues of carbonyls.47 As a result, under the established dual catalytic system involving SPINOL-CPA 94 and DPZ and using N-phenyl piperidine as the terminal reductant, various ketones 93 could efficiently undergo this tandem process involving two single-electron-transfer reductions and an enantioselective protonation. A wide range of chiral azaarene-based secondary alcohols 95 with diverse aryl and alkyl substituents and different azaarene moieties could be prepared in high yields (up to >99%) with moderate to excellent enantioselectivities (up to 97% ee). On the basis of these achievements in enantioselective reductive protonation44,47 and enantioselective reductive dehalogenative protonation,46 they subsequently developed an enantioselective reductive deuteration of azaarene-substituted ketones and an enantioselective reductive dehalogenative deuteration of racemic α-chloro-azaarenes with inexpensive D2O as the deuterium source.48 Under cooperative photoredox and H-bonding catalysis (i.e., DPZ with SPINOL-CPA), a variety of chiral α-deuterated azaarenes were obtained in satisfactory yields with good to excellent enantioselectivities (up to 99% ee) and substantial deuterium incorporation.
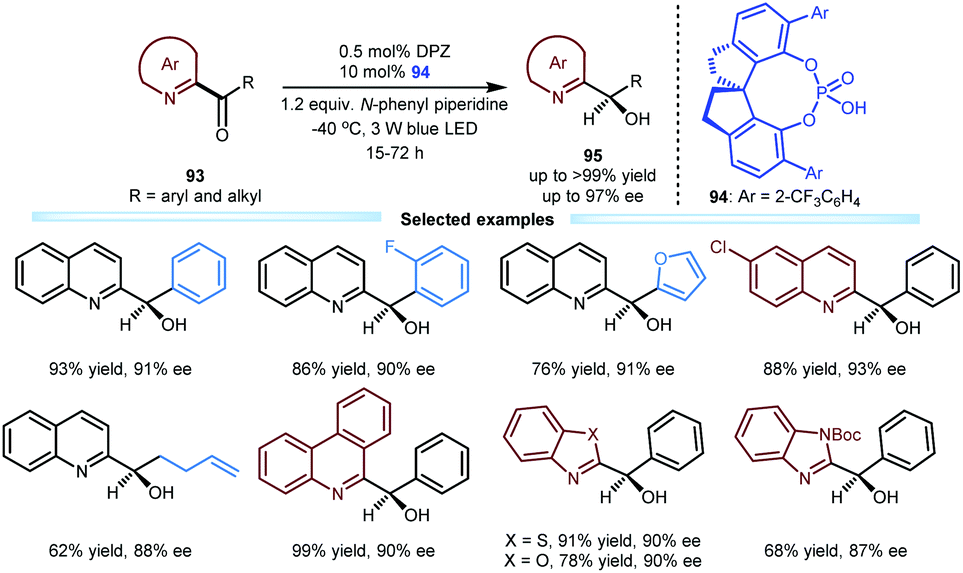 | ||
| Scheme 20 Enantioselective reduction of azaarene-based ketones. | ||
Indeed, the first example of the construction of chiral azaarene-based derivatives via enantioselective protonation was reported by the Jiang group in 2018, and they used a radical conjugate addition-enantioselective protonation of N-aryl glycine 32 to afford α-branched 2-vinylazaarenes 96 (Scheme 21).49 By using a dual catalytic system involving DPZ and SPINOL-CPA 59 or 94 mediated by visible light, the transformations afforded a variety of valuable chiral 3-(2-pyridine/quinoline)-3-substituted amines 97 in up to 97% yield with up to >99% ee. The mechanism studies revealed that α-amino radicals 61b, which are derived from 32 after a sequential process involving single-electron oxidation, deprotonation, proton transfer and decarboxylation, can smoothly attack Brønsted acid-activated olefins (98), leading to radical species 99. After the second single-electron reduction, resulting anions 100 can then undergo enantioselective protonation to generate products 97. The low reaction temperature was crucial to achieving excellent enantioselectivity since it could completely supress the racemic background reaction. Notably, the enantiomerically pure therapeutic compound pheniramine (Avil) was conveniently synthesized from the obtained product.
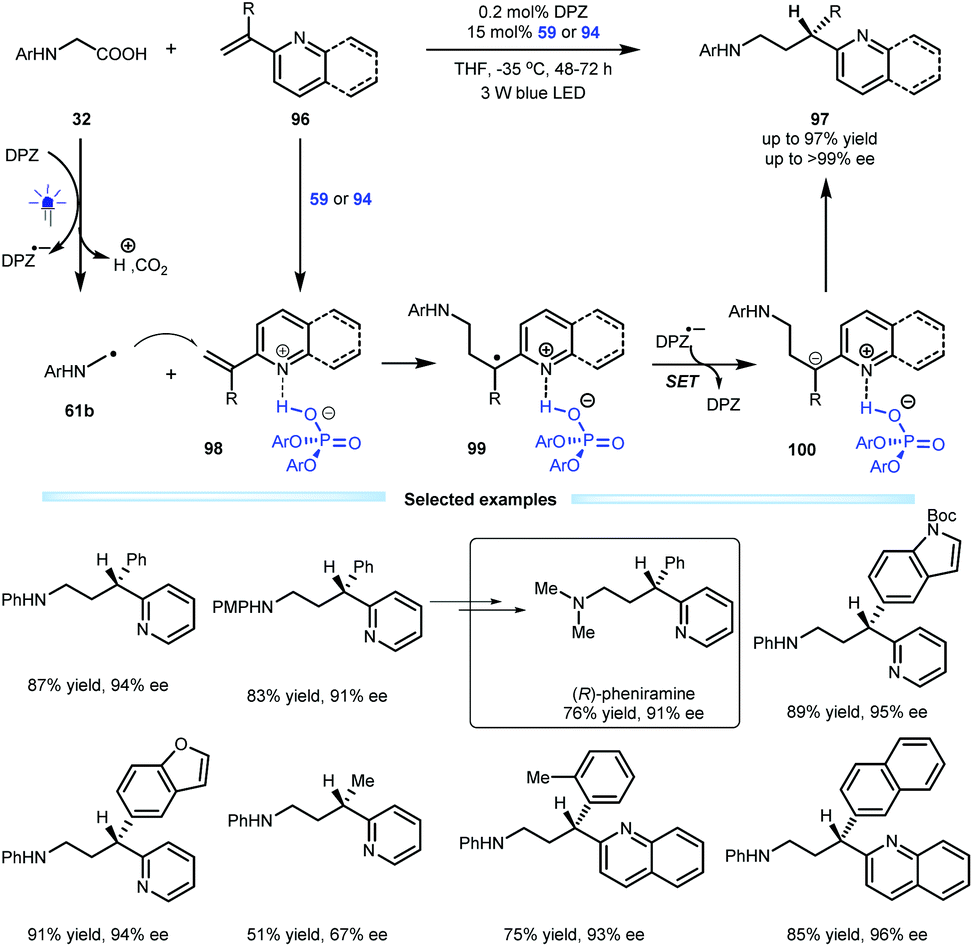 | ||
| Scheme 21 Radical conjugate addition-enantioselective protonation. | ||
Conclusions
Based on the continuous contributions of chemists in recent years, cooperative photoredox and H-bonding catalysis has become a powerful tool in asymmetric synthesis. Although radical-based transformations are highly reactive and the energy of H-bonding interactions is rather low, a number of amazing and useful asymmetric chemical transformations have been successfully developed, even though racemic background reactions are apparent in most cases. In particular, several highly challenging reactions that have been long-standing challenges in the field have been successfully conquered, such as asymmetric Minisci-type reactions, enantioconvergent substitutions of alkyl halides, and enantioselective protonations to construct labile tertiary carbon stereocentres. A large array of important enantioenriched compounds have been achieved through various bond-forming patterns, including radical addition, radical coupling and different ionic-type pathways. Among them, the successful enantioselective construction of all-carbon quaternary stereocentres via highly reactive radical couplings robustly demonstrates the powerful stereocontrol possible with chiral H-bonding catalysis. Moreover, the capacity of H-bonding catalysis to improve the poor chemoselectivity seen in photoredox catalysis, which has represented a major challenge in the past, has been disclosed. It is foreseeable that there is ample scope for the greater use of this robust catalysis platform following the development of photoredox catalysts, chiral H-bonding catalysts and new methodologies. In addition to the new reaction types and allowing the use of more kinds of readily accessible substrates, especially abundant industrial feedstocks, cooperative photoredox and H-bonding catalysis will be applied to the more challenging multicomponent reactions and the synthesis of complex chiral molecules in the future. Such discoveries should find broad application in the total synthesis of bioactive natural and non-natural products, drugs and functional materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Grants from the National Science Foundation of China (21672052 and 21925103), the PhD Research Foundation of Henan University of Technology (2019BS003) and Young Elite Scientists Sponsorship Program by CAST (2017QNRC001) are gratefully acknowledged.Notes and references
- (a) Enantioselective Organocatalysis, ed. P. Dalko, Wiley-VCH, Weinheim, Germany, 2007 Search PubMed; (b) P. Melchiorre, M. Mario, A. Carlone and G. Bartoli, Asymmetric Aminocatalysis-Gold Rush in Organic Chemistry, Angew. Chem., Int. Ed., 2008, 47, 6138 CrossRef CAS PubMed.
- D. W. C. MacMillan, The Advent and Development of Organocatalysis, Nature, 2008, 455, 304 CrossRef CAS PubMed.
- J. T. Mohr, A. Y. Hong and B. M. Stoltz, Enantioselective Protonation, Nat. Chem., 2009, 1, 359 CrossRef CAS PubMed.
- (a) Radicals in Organic Synthesis, ed. P. and Renaud and M. P. Sibi, Wiley-VCH, Weinheim, Germany, 2001 Search PubMed; (b) H. Togo, Advanced Free Radical Reactions for Organic Synthesis, Elsevier Science, Amsterdam, 2004 Search PubMed.
- (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (b) M. H. Shaw, J. Twilton and D. W. C. MacMillan, Photoredox Catalysis in Organic Chemistry, J. Org. Chem., 2016, 81, 6898 CrossRef CAS PubMed; (c) D. Staveness, R. Bosque and C. R. J. Stephenson, Free Radical Chemistry Enabled by Visible Light-Induced Electron Transfer, Acc. Chem. Res., 2016, 49, 2295 CrossRef CAS PubMed; (d) G. Wei, C. Basheer, C.-H. Tan and Z. Jiang, Visible Light Photocatalysis in Chemoselective Functionalization of C(sp3)-H Bonds Enabled by Organic Dyes, Tetrahedron Lett., 2016, 57, 3801 CrossRef CAS; (e) N. A. Romero and D. A. Nicewicz, Organic Photoredox Catalysis, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed; (f) B. Qiao and Z. Jiang, Catalytic Photoreduction Induced by Visible Light, ChemPhotoChem, 2018, 2, 703 CrossRef CAS.
- (a) K. L. Skubi, T. R. Blum and T. P. Yoon, Dual Catalysis Strategies in Photochemical Synthesis, Chem. Rev., 2016, 116, 10035 CrossRef CAS PubMed; (b) T. P. Yoon, Photochemical Stereocontrol Using Tandem Photoredox-Chiral Lewis Acid Catalysis, Acc. Chem. Res., 2016, 49, 2307 CrossRef CAS PubMed; (c) J. Twilton, C. C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, The Merger of Transition Metal and Photocatalysis, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS; (d) Y.-Q. Zou, F. M. Hörmann and T. Bach, Iminium and Enamine Catalysis in Enantioselective Photochemical Reactions, Chem. Soc. Rev., 2018, 47, 278 RSC.
- (a) C. Wang and Z. Lu, Catalytic Enantioselective Organic Transformations via Visible Light Photocatalysis, Org. Chem. Front., 2015, 2, 179 RSC; (b) E. Meggers, Asymmetric Catalysis Activated by Visible Light, Chem. Commun., 2015, 51, 3290 RSC; (c) R. Brimioulle, D. Lenhart, M. M. Maturi and T. Bach, Enantioselective Catalysis of Photochemical Reactions, Angew. Chem., Int. Ed., 2015, 54, 3872 CrossRef CAS PubMed; (d) C. Jiang, W. Chen, W.-H. Zheng and H. Lu, Advances in Asymmetric Visible-Light Photocatalysis, 2015–2019, Org. Biomol. Chem., 2019, 17, 8673 RSC.
- A. Bauer, F. Westkämper, S. Brimme and T. Bach, Catalytic Enantioselective Reactions Driven by Photoinduced Electron Transfer, Nature, 2005, 436, 1139 CrossRef CAS PubMed.
- D. A. Nicewicz and D. W. C. MacMillan, Merging Photoredox Catalysis with Organocatalysis: The Direct Asymmetric Alkylation of Aldehydes, Science, 2008, 322, 77 CrossRef CAS PubMed.
- L. J. Rono, H. G. Yayla, D. Y. Wang, M. F. Armstrong and R. R. Knowles, Enantioselective Photoredox Catalysis Enabled by Proton-Coupled Electron Transfer: Development of an Asymmetric Aza-Pinacol Cyclization, J. Am. Chem. Soc., 2013, 135, 17735 CrossRef CAS PubMed.
- E. C. Gentry and R. R. Knowles, Synthetic Applications of Proton-Coupled Electron Transfer, Acc. Chem. Res., 2016, 49, 1546 CrossRef CAS PubMed.
- (a) A. F. Pozharskii, A. T. Soldatenkov and A. R. Katritzky, Heterocycles in Life and Society: An Introduction to Heterocyclic Chemistry, Biochemistry and Applications, Wiely, 2011 CrossRef; (b) G. Chelucci, Metal-Complexes of Optically Active Amino- and Imino-Based Pyridine Ligands in Asymmetric Catalysis, Coord. Chem. Rev., 2013, 257, 1887 CrossRef CAS; (c) E. Vitaku, D. T. Smith and J. T. Njardarson, Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals, J. Med. Chem., 2014, 57, 10257 CrossRef CAS PubMed; (d) M. C. Bagley, J. W. Dale, E. A. Merritt and X. Xiong, Thiopeptide Antibiotics, Chem. Rev., 2005, 105, 685 CrossRef CAS PubMed.
- For selected examples, see: (a) S. Yu, H. L. Sang and S. Ge, Enantioselective Copper-Catalyzed Alkylation of Quinoline N-Oxides with Vinylarenes, Angew. Chem., Int. Ed., 2017, 56, 15896 CrossRef CAS PubMed; (b) C. Xu, C. W. Muir, A. G. Leach, A. R. Kennedy and A. J. B. Watson, Catalytic Enantioselective Synthesis of α-Chiral Azaheteroaryl Ethylamines by Asymmetric Protonation, Angew. Chem., Int. Ed., 2018, 57, 11374 CrossRef CAS PubMed; (c) J. Izquierdo, A. Landa, I. Bastida, R. López, M. Oiarbide and C. Palomo, Base-Catalyzed Asymmetric α-Functionalization of 2-(Cyanomethyl)azaarene N-Oxides Leading to Quaternary Stereocenters, J. Am. Chem. Soc., 2016, 138, 3282 CrossRef CAS PubMed; (d) B. Trost and D. A. Thaisrivongs, Palladium-Catalyzed Regio-, Diastereo-, and Enantioselective Benzylic Allylation of 2-Substituted Pyridines, J. Am. Chem. Soc., 2009, 131, 12056 CrossRef CAS PubMed; (e) A. Saxena, B. Choi and H. W. Lam, Enantioselective Copper-Catalyzed Reductive Coupling of Alkenylazaarenes with Ketones, J. Am. Chem. Soc., 2012, 134, 8428 CrossRef CAS PubMed; (f) D. Best, S. Kujawa and H. W. Lam, Diastereo- and Enantioselective Pd(II)-Catalyzed Additions of 2-Alkylazaarenes to N-Boc Imines and Nitroalkenes, J. Am. Chem. Soc., 2012, 134, 18193 CrossRef CAS PubMed; (g) I. D. Roy, A. R. Burns, G. Pattison, B. Michel, A. J. Parker and H. W. Lam, A Second-Generation Ligand for the Enantioselective Rhodium-Catalyzed Addition of Arylboronic Acids to Alkenylazaarenes, Chem. Commun., 2014, 50, 2865 RSC; (h) Y.-Y. Wang, K. Kanomata, T. Korenaga and M. Terada, Enantioselective Aza Michael-Type Addition to Alkenyl Benzimidazoles Catalyzed by a Chiral Phosphoric Acid, Angew. Chem., Int. Ed., 2016, 55, 927 CrossRef CAS PubMed; (i) S. Wang, X. Li, L. Xu, J. Zhuang, J. Li, H. Li and W. Wang, Organocatalytic Enantioselective Direct Additions of Aldehydes to 4-Vinylpyridines and Electron-Deficient Vinylarenes and Their Synthetic Applications, J. Am. Chem. Soc., 2015, 137, 2303 CrossRef CAS PubMed; (j) X. Bai, G. Zeng, T. Shao and Z. Jiang, Catalytic Enantioselective γ-Selective Additions of 2-Allylazaarenes to Activated Ketones, Angew. Chem., Int. Ed., 2017, 56, 3684 CrossRef CAS PubMed.
- H. B. Hepburn and P. Melchiorre, Brønsted Acid-Catalysed Conjugate Addition of Photochemically Generated α-Amino Radicals to Alkenylpyridines, Chem. Commun., 2016, 52, 3520 RSC.
- W.-M. Cheng, R. Shang and Y. Fu, Photoredox/Brønsted Acid Co-Catalysis Enabling Decarboxylative Coupling of Amino Acid and Peptide Redox-Active Esters with N-Heteroarenes, ACS Catal., 2017, 7, 907 CrossRef CAS.
- S. J. Proctor, H. J. Davis and R. J. Phipps, Catalytic Enantioselective Minisci-Type Addition to Heteroarenes, Science, 2018, 360, 419 CrossRef PubMed.
- X. Liu, Y. Liu, G. Chai, B. Qiao, X. Zhao and Z. Jiang, Organocatalytic Enantioselective Addition of α-Aminoalkyl Radicals to Isoquinolines, Org. Lett., 2018, 20, 6298 CrossRef CAS PubMed.
- (a) Y. Zhao, C. Zhang, K. F. Chin, O. Pytela, G. Wei, H. Liu, F. Bureš and Z. Jiang, Dicyanopyrazine-Derived Push–Pull Chromophores for Highly Efficient Photoredox Catalysis, RSC Adv., 2014, 4, 30062 RSC; (b) X. Liu, X. Ye, F. Bureš, H. Liu and Z. Jiang, Controllable Chemoselectivity in Visible-Light Photoredox Catalysis: Four Diverse Aerobic Radical Cascade Reactions, Angew. Chem., Int. Ed., 2015, 54, 11443 CrossRef CAS PubMed; (c) T. Shao and Z. Jiang, Visible Light Mediated Photocatalytic Aerobic Dehydrogenation: A General and Direct Approach to Access 2,3-Dihydro-4-Pyridones and 4-Quinolones, Acta Chim. Sin., 2017, 75, 70 CrossRef CAS; (d) C. Zhang, S. Li, F. Bureš, R. Lee, X. Ye and Z. Jiang, Visible Light Photocatalytic Aerobic Oxygenation of Indoles and pH as a Chemoselective Switch, ACS Catal., 2016, 6, 6853 CrossRef CAS.
- M.-C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Photocatalytic Decarboxylative Alkylations Mediated by Triphenylphosphine and Sodium Iodide, Science, 2019, 363, 1429 CrossRef CAS PubMed.
- D. Zheng and A. Studer, Asymmetric Synthesis of Heterocyclic γ-Amino-Acid and Diamine Derivatives by Three-Component Radical Cascade Reactions, Angew. Chem., Int. Ed., 2019, 58, 15803 CrossRef CAS PubMed.
- K. N. Lee, Z. Lei and M.-Y. Ngai, β-Selective Reductive Coupling of Alkenylpyridines with Aldehydes and Imines via Synergistic Lewis Acid/Photoredox Catalysis, J. Am. Chem. Soc., 2017, 139, 5003 CrossRef CAS PubMed.
- K. Cao, S. M. Tan, R. Lee, S. Yang, H. Jia, X. Zhao, B. Qiao and Z. Jiang, Catalytic Enantioselective Addition of Prochiral Radicals to Vinylpyridines, J. Am. Chem. Soc., 2019, 141, 5437 CrossRef CAS PubMed.
- C. B. Roos, J. Demaerel, D. E. Graff and R. R. Knowles, Enantioselective Hydroamination of Alkenes with Sulfonamides Enabled by Proton-Coupled Electron Transfer, J. Am. Chem. Soc., 2020, 143, 5974 CrossRef PubMed.
- (a) H. Fischer, The Persistent Radical Effect: A Principle for Selective Radical Reactions and Living Radical Polymerizations, Chem. Rev., 2001, 101, 3581 CrossRef CAS PubMed; (b) H. Yi, G. Zhuang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Recent Advances in Radical C-H Activation/Radical Cross-Coupling, Chem. Rev., 2017, 117, 9016 CrossRef CAS PubMed; (c) J. Xie, H. Jin and A. S. K. Hashmi, The Recent Achievements of Redox-neutral Radical C–C Cross-Coupling Enabled by Visible-Light, Chem. Soc. Rev., 2017, 46, 5193 RSC.
- D. Uraguchi, N. Kinoshita, T. Kizu and T. Ooi, Synergistic Catalysis of Ionic Brønsted Acid and Photosensitizer for a Redox Neutral Asymmetric α-Coupling of N-Arylaminomethanes with Aldimines, J. Am. Chem. Soc., 2015, 137, 13768 CrossRef CAS PubMed.
- T. Kizu, D. Uraguchi and T. Ooi, Independence from the Sequence of Single-Electron Transfer of Photoredox Process in Redox-Neutral Asymmetric Bond-Forming Reaction, J. Org. Chem., 2016, 81, 6953 CrossRef CAS PubMed.
- Y. Liu, X. Liu, J. Li, X. Zhao, B. Qiao and Z. Jiang, Catalytic Enantioselective Radical Coupling of Activated Ketones with N-Aryl Glycines, Chem. Sci., 2018, 9, 8094 RSC.
- S. R. Hartshorn, Aliphatic Nucleophilic Substitution, Cambridge University Press, London, 1973 Search PubMed.
- G. C. Fu, Transition-Metal Catalysis of Nucleophilic Substitution Reactions: A Radical Alternative to SN1 and SN2 Processes, ACS Cent. Sci., 2017, 3, 692 CrossRef CAS PubMed.
- J. Li, M. Kong, B. Qiao, R. Lee, X. Zhao and Z. Jiang, Formal Enantioconvergent Substitution of Alkyl Halides via Catalytic Asymmetric Photoredox Radical Coupling, Nat. Commun., 2018, 9, 2445 CrossRef PubMed.
- G. Zeng, Y. Li, B. Qiao, X. Zhao and Z. Jiang, Photoredox Asymmetric Catalytic Enantioconvergent Substitution of 3-Chlorooxindoles, Chem. Commun., 2019, 55, 11362 RSC.
- (a) S. Lerch, L.-N. Unkel and M. Brasholz, Tandem Organocatalysis and Photocatalysis: An Anthraquinone-Catalyzed Indole-C3-Alkylation/Photooxidation/1,2-Shift Sequence, Angew. Chem., Int. Ed., 2014, 53, 6558 CrossRef CAS PubMed; (b) W. Ding, Q.-Q. Zhou, J. Xuan, T.-R. Li, L.-Q. Lu and W.-J. Xiao, Photocatalytic Aerobic Oxidation/Semipinacol Rearrangement Sequence: a Concise Route to the Core of Pseudoindoxyl Alkaloids, Tetrahedron Lett., 2014, 55, 4648 CrossRef CAS.
- L. Bu, J. Li, Y. Yin, B. Qiao, G. Chai, X. Zhao and Z. Jiang, Organocatalytic Asymmetric Cascade Aerobic Oxidation and Semipinacol Rearrangement Reaction: A Visible Light-Induced Approach to Access Chiral 2,2-Disubstituted Indolin-3-ones, Chem. - Asian J., 2018, 13, 2382 CrossRef CAS PubMed.
- E. C. Gentry, L. J. Rono, M. E. Hale, R. Matsuura and R. R. Knowles, Enantioselective Synthesis of Pyrroloindolines via Noncovalent Stabilization of Indole Radical Cations and Applications to the Synthesis of Alkaloid Natural Products, J. Am. Chem. Soc., 2018, 140, 3394 CrossRef CAS PubMed.
- For selected examples, see: (a) G. Bergonzini, C. S. Schindler, C.-J. Wallentin, E. N. Jacobsen and C. R. J. Stephenson, Photoredox Activation and Anion Binding Catalysis in the Dual Catalytic Enantioselective Synthesis of β-Amino Esters, Chem. Sci., 2014, 5, 112 RSC; (b) G. Wei, C. Zhang, F. Bureš, X. Ye, C.-H. Tan and Z. Jiang, Enantioselective Aerobic Oxidative C(sp3)-H Olefination of Amines via Cooperative Photoredox and Asymmetric Catalysis, ACS Catal., 2016, 6, 3708 CrossRef CAS.
- L. Chen, C. S. Chao, Y. Pan, S. Dong, Y. C. Teo, J. Wang and C.-H. Tan, Amphiphilic Methyleneamino Synthon through Organic Dye Catalyzed-Decarboxylative Aminoalkylation, Org. Biomol. Chem., 2013, 11, 5922 RSC.
- T. Shao, Y. Yin, R. Lee, X. Zhao, G. Chai and Z. Jiang, Sequential Photoredox Catalysis for Cascade Aerobic Decarboxylative Povarov and Oxidative Dehydrogenation Reactions of N-Aryl α-Amino Acids, Adv. Synth. Catal., 2018, 360, 1754 CrossRef CAS.
- J. Li, Z. Gu, X. Zhao, B. Qiao and Z. Jiang, Asymmetric Aerobic Decarboxylative Povarov Reactions of N-aryl α-Amino Acids with Methylenephthalimidines via Cooperative Photoredox and Chiral Brønsted Acid Catalysis, Chem. Commun., 2019, 55, 12916 RSC.
- M.-L. Shen, Y. Shen and P.-S. Wang, Merging Visible-Light Photoredox and Chiral Phosphate Catalysis for Asymmetric Friedel-Crafts Reaction with in Situ Generation of N-Acyl Imines, Org. Lett., 2019, 21, 2993 CrossRef CAS PubMed.
- Y.-Z. Cheng, Q.-R. Zhao, X. Zhang and S.-L. You, Asymmetric Dearomatization of Indole Derivatives with N-Hydroxycarbamates Enabled by Photoredox Catalysis, Angew. Chem., Int. Ed., 2019, 58, 18069 CrossRef CAS PubMed.
- D. Lenhart, A. Bauer, A. Pöthig and T. Bach, Enantioselective Visible-Light-Induced Radical-Addition Reactions to 3-Alkylidene Indolin-2-ones, Chem. – Eur. J., 2016, 22, 6519 CrossRef CAS PubMed.
- (a) Y. Ohgo, Y. Tashiro and S. Takeuchi, Asymmetric Hydrogenation Catalyzed by Bis(disubstituted glyoximato)cobalt(II)(L)-Chiral Cocatalyst System. Effect of Structural Variation of Ligands and Hydrogen Pressure, Bull. Chem. Soc. Jpn., 1987, 60, 1549 CrossRef CAS; (b) L. De Luca and A. Mezzetti, Base-Free Asymmetric Transfer Hydrogenation of 1,2-Di- and Monoketones Catalyzed by a (NH)2P2-Macrocyclic Iron(II) Hydride, Angew. Chem., Int. Ed., 2017, 56, 11949 CrossRef CAS PubMed; (c) B. Procuranti and S. J. Connon, A Reductase-Mimicking Thiourea Organocatalyst Incorporating a Covalently Bound NADH Analogue: Efficient 1,2-Diketone Reduction with in Situ Prosthetic Group Generation and Recycling, Chem. Commun., 2007, 1421 RSC.
- I. Willner, T. Tsfania and Y. Eichen, Photocatalyzed and Electrocatalyzed Reduction of Vicinal Dibromides and Activated Ketones Using Ruthenium(I) Tris(bipyridine) as Electron-Transfer Mediator, J. Org. Chem., 1990, 55, 2656 CrossRef CAS.
- L. Lin, X. Bai, X. Ye, X. Zhao, C.-H. Tan and Z. Jiang, Organocatalytic Enantioselective Protonation for Photoreduction of Activated Ketones and Ketimines Induced by Visible Light, Angew. Chem., Int. Ed., 2017, 56, 13842 CrossRef CAS PubMed.
- For a selected review, see: (a) X. Zhao, B. Zhu and Z. Jiang, Acyclic Amino Acid Based Bifunctional Chiral Tertiary Amines, Quaternary Ammoniums and Iminophosphoranes as Organocatalysts, Synlett, 2015, 26, 2216 CrossRef CAS. For selected examples, see: (b) B. Zhu, R. Lee, J. Li, X. Ye, S.-N. Hong, S. Qiu, M. L. Coote and Z. Jiang, Chemoselective Switch in the Asymmetric Organocatalysis of 5H-Oxazol-4-ones and N-Itaconimides: Addition-Protonation or [4+2] Cycloaddition, Angew. Chem., Int. Ed., 2016, 55, 1299 CrossRef CAS PubMed; (c) B. Zhu, S. Qiu, J. Li, M. L. Coote, R. Lee and Z. Jiang, Asymmetric [4+2] Annulation of 5H-Thiazol-4-ones with a Chiral Dipeptide-based Brønsted Base Catalyst, Chem. Sci., 2016, 7, 6060 RSC; (d) Y. Liu, J. Li, X. Ye, X. Zhao and Z. Jiang, Organocatalytic Asymmetric Formal Arylation of Benzofuran-2(3H)-ones with Cooperative Visible Light Photocatalysis, Chem. Commun., 2016, 52, 13955 RSC; (e) L. Jiao, X. Zhao, H. Liu, X. Ye, Y. Li and Z. Jiang, Organocatalytic Asymmetric Conjugate Addition of Diaryloxazolidin-2,4-diones to Nitroolefins: An Efficient Approach to Chiral α-aryl-α-hydroxy Carboxylic Acids, Org. Chem. Front., 2016, 3, 470 RSC; (f) S.-N. Hong, Y. Liu, R. Lee and Z. Jiang, Asymmetric Tandem Conjugate Addition-Protonation to Forge Chiral Secondary C-O Bonds for Quaternary Carbon Stereocenters at the Nonadjacent β-position, Chem. Commun., 2017, 53, 7493 RSC.
- M. Hou, L. Lin, X. Chai, X. Zhao, B. Qiao and Z. Jiang, Enantioselective Photoredox Dehalogenative Protonation, Chem. Sci., 2019, 10, 6629 RSC.
- B. Qiao, C. Li, X. Zhao, Y. Yin and Z. Jiang, Enantioselective Reduction of Azaarene-based Ketones via Visible Light-driven Photoredox Asymmetric Catalysis, Chem. Commun., 2019, 55, 7534 RSC.
- T. Shao, Y. Li, N. Ma, C. Li, G. Chai, X. Zhao, B. Qiao and Z. Jiang, Photoredox-Catalyzed Enantioselective α-Deuteration of Azaarenes with D2O, iScience, 2019, 16, 410 CrossRef CAS PubMed.
- Y. Yin, Y. Dai, H. Jia, J. Li, L. Bu, B. Qiao, X. Zhao and Z. Jiang, Conjugate Addition-Enantioselective Protonation of N-Aryl Glycines to α-Branched 2-Vinylazaarenes via Cooperative Photoredox and Asymmetric Catalysis, J. Am. Chem. Soc., 2018, 140, 6083 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2020 |