
Cooperative photoredox and chiral hydrogen-bonding catalysis
 *ac
*ac
Abstract
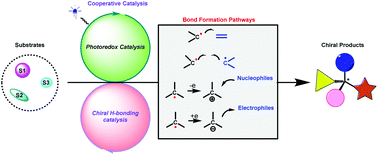
Chiral hydrogen-bonding catalysis is a classic strategy in asymmetric organocatalysis, and it has been extensively used in a variety of fundamental chemical transformations. At the same time, visible light-driven photoredox catalysis is a powerful and sustainable tool commonly used in radical chemistry. The intriguing combination of these two catalysis platforms would open a new avenue for the direct and highly efficient synthesis of enantioenriched compounds. Inspired by the conceptual breakthrough of T. Bach, in recent years, significant progress has been made in cooperative photoredox and chiral hydrogen-bonding catalysis. By developing a variety of important types of reactions, a wide range of valuable chiral compounds have been successfully synthesized. In this review, the advances in this key area are systematically described, and the examples are organized according to the distinct bond-forming patterns in the construction of the stereocentres.
- This article is part of the themed collection: FOCUS: Radical-involved chemical transformations
 Please wait while we load your content...
Please wait while we load your content...

