
Recent advances in the total synthesis of cyclobutane-containing natural products
Jinshan
Li†
a,
Kai
Gao†
 a,
Ming
Bian
b and
Hanfeng
Ding
*ac
a,
Ming
Bian
b and
Hanfeng
Ding
*ac
aInstitute of Medicinal Natural Products, School of Advanced Study, Taizhou University, Taizhou 318000, P. R. China
bSchool of Chemistry and Environmental Engineering, Shanghai Institute of Technology, 100 Haiquan Road, Shanghai 201418, P.R. China
cDepartment of Chemistry, Zhejiang University, Hangzhou 310058, P. R. China. E-mail: hfding@zju.edu.cn
First published on 12th November 2019
Abstract
Complex natural products bearing strained cyclobutane subunits, including terpenoids, alkaloids and steroids, not only display fascinating architectures, but also show potent biological activities. Due to their unique structures as critical core skeletons in these molecules, a variety of new strategies for the construction of cyclobutane rings have greatly emerged during the last decade. In this review, we wish to summarize the recent progress in the cyclobutane-containing natural product synthesis with an emphasis on disconnection tactics employed to forge the four-membered rings, aiming to provide a complement to existing reviews.
 Jinshan Li | Jinshan Li received his Ph.D. from Tianjin University in 2018. Since then, he has been working at the Institute of Medicinal Natural Product, School of Advanced Study at Taizhou University as a lecturer. His current research interests focus on the development of new catalytic asymmetric reactions and their application in natural product synthesis. |
 Kai Gao | Kai Gao received his Ph.D. from Dalian Institute of Chemical Physics, CAS in 2012. From 2016 to 2018, he was a postdoctoral fellow working with Prof. Wei-Min Dai at Hong Kong University of Science and Technology (HKUST). Since 2018, he has been working at the Institute of Medicinal Natural Product, School of Advanced Study at Taizhou University as a lecturer. His current research interests focus on the development of hypervalent iodine-mediated reactions and their application in natural product synthesis. |
 Ming Bian | Ming Bian received his Ph.D. from Zhejiang University in 2010. After two years of postdoctoral training under the guidance of Professor Ang Li at Shanghai Institute of Organic Chemistry (SIOC), Chinese Academy of Sciences (CAS), he joined the same group as an Associate Professor. In 2014, he moved to Shanghai Institute of Technology to start his independent career. His current interests focus on radical reactions and their application in natural product synthesis. |
 Hanfeng Ding | Hanfeng Ding received his Ph.D. from Zhejiang University in 2008. From 2009 to 2011, he was a postdoctoral fellow working with Prof. David Y.-K. Chen in Prof. K. C. Nicolaou's group at CSL@Biopolis, Singapore. He began his independent career as a professor in the Department of Chemistry at Zhejiang University in September 2011. His current research interests focus on the development of novel strategies and methodologies for the total synthesis of structurally complex and biologically active natural products. |
1. Introduction
In the class of strained carbocycles, cyclobutanes have been known as intriguing structural motifs for more than one century but remained relatively less explored in parallel with their homologues.1 Due to the highly strained ring systems (ca. 26.7 kcal mol−1), construction of cyclobutane rings, especially stereoselectively, poses significant challenges in synthetic chemistry. On the other hand, cyclobutanes readily undergo a number of ring-opening reactions by virtue of their tendency to release inherent strain energies. In some cases, however, striking ring strains can be dramatically reduced by the installation of a gem-dialkyl substituent (through the Thorpe–Ingold effect),2 a carbonyl group, a heteroatom, or other functionalities (Fig. 1).3 Owing to their improved stability, these structures could exist in not only artificial molecules but also many complex natural products.4 The novel chemical structures as well as remarkable biological activities of the cyclobutane-containing natural products make them extremely attractive targets for total synthesis. Therefore, the development of new strategies for the assembly of these unique structures in a rapid and efficient manner has long been highly desirable.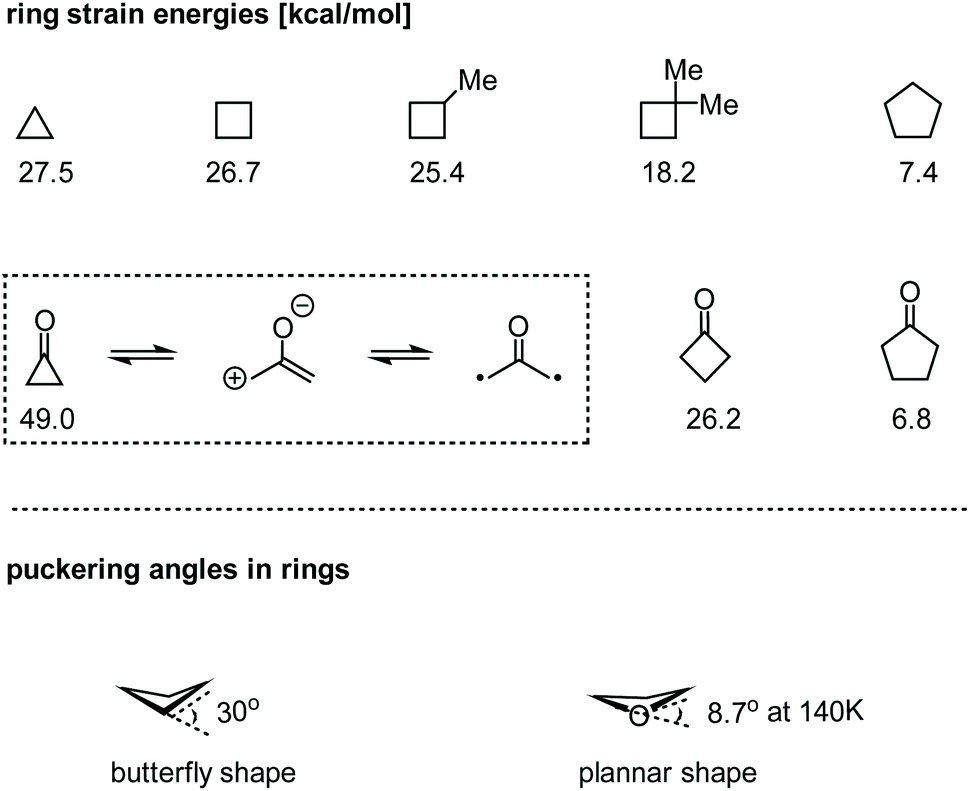 | ||
| Fig. 1 Comparison of ring strain energies and structural properties of strained carbocycles. | ||
Nowadays, grounded on the strain release, an increasing number of specialized summaries of cyclobutane chemistry have emerged, which presented the synthetic versatilities to forge acyclic and other types of cyclic skeletons. However, few systematic surveys on recent contributions to the total synthesis of natural products possessing four-membered carbocyclic scaffolds have been demonstrated.5 Therefore, we endeavour to focus on the strategic considerations in a collection of selected total syntheses that have appeared in the last decade (2008–2018, and the representative examples reported in early 2019 are also included), and divide them into four categories: intramolecular direct ring-closure, cycloaddition, ring-contraction and ring-expansion strategies. In this review, the isolation, biological activities and innovative solutions to the problems encountered during the syntheses of the cyclobutane-containing natural products are emphatically described. Due to space limitation, some other classical protocols as well as the syntheses concentrating on the reconstruction of cyclobutanes6,7 have to be neglected. Besides, the discussions of the performances on hetero four-membered ring systems such as oxetanes3a–c and azetidines3d–f are beyond the scope of this context and thus will not be covered herein.
2. Intramolecular direct ring-closure strategies
Intramolecular direct ring-closure strategies offer a facile route to the strained cyclobutane skeletons embedded in complex natural products, even though these processes are entropically disfavorable. Such approaches, which mainly include intramolecular nucleophilic cyclization, radical cyclization, cationic cyclization and transition-metal catalyzed cyclization, have become widespread and could be performed in a highly regio- or stereoselective fashion.2.1 Nucleophilic cyclization
Solanoeclepin A was isolated by Mulder in 1986 as a hatching stimulus for potato cyst nematodes Globodera rostochiensis and G. pallida, which causes significant damage to potato crops.8 Structurally, solanoeclepin A contains a heptacyclic ring system with each of the ring sizes from three to seven. In 2011, Tanino and Miyashita reported the first total synthesis of solanoeclepin A (4) featuring an intramolecular nucleophilic cyclization to assemble its highly strained tricyclo[5.2.1.01,6]decane skeleton (Scheme 1).9 Their synthesis commenced with preparation of the crucial precursor 2, which was derived from the known bicyclic acetoxy nitrile 1 in nine steps. Treatment of 2 with LDA initiated an intramolecular cyclization via the carbanion mediated ring opening of the epoxide, followed by silylation to afford cyclobutane 3 in nearly quantitative yield. The above transformation allowed the total synthesis of 4 in 52 steps and 0.18% overall yield.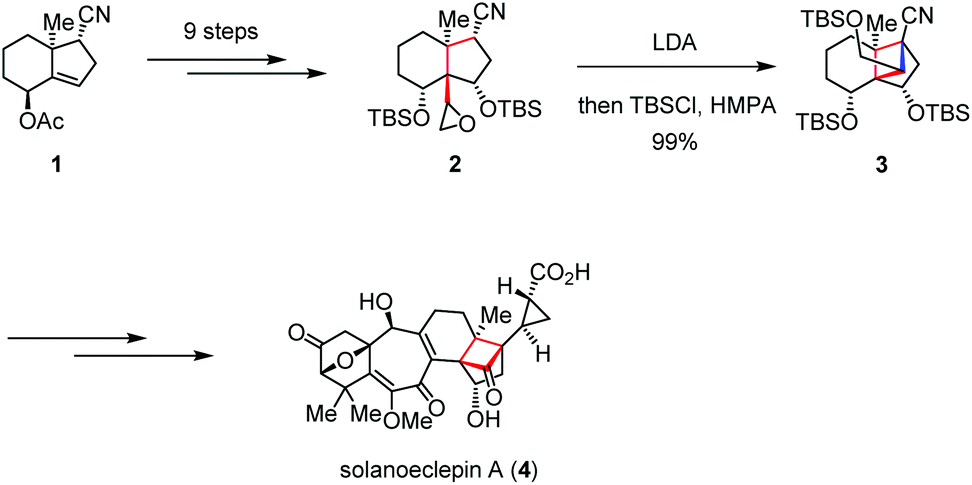 | ||
| Scheme 1 Intramolecular nucleophilic cyclization in the total synthesis of solanoeclepin A. | ||
Dendrobium species (Orchidaceaeare) have been widely used in traditional Chinese medicine for fever reduction and promotion of saliva secretion.10 (+)-Dendrowardol C, isolated from the stems of Dendrobium wardianum Warner, is a sesquiterpenoid bearing an unprecedented 4/5/5/6 tetracyclic ring skeleton decorated with nine contiguous stereogenic centers.11 Recently, Carreira and co-workers described the total synthesis of (+)-dendrowardol C (11) relying on an intramolecular aldol reaction to forge the central bicyclic scaffold and subsequent cyclization of a γ-triflyoxy ketone to construct the cyclobutane subunit (Scheme 2).12 Their synthesis began with the preparation of allylic alcohol 6 from known ester 5 in six steps. Direct epoxidation of 6 using VO(acac)2/TBHP and TMS protection proceeded in one pot to give epoxyketone 7 in 76% yield. Treatment of the latter with LDA/HMPA generated a lithium enolate intermediate and induced the following regioselective epoxide opening. Triflation of the resultant primary alcohol provided 8, which then underwent ring closure in the presence of lithium naphthalenide to deliver the corresponding cyclobutanol. After silylation, 9 was formed in 29% yield over two steps. Finally, a regio- and enantioselective Co(I)-catalyzed hydroboration of 9 followed by oxidative work-up and global deprotection provided (+)-11 in 60% yield.
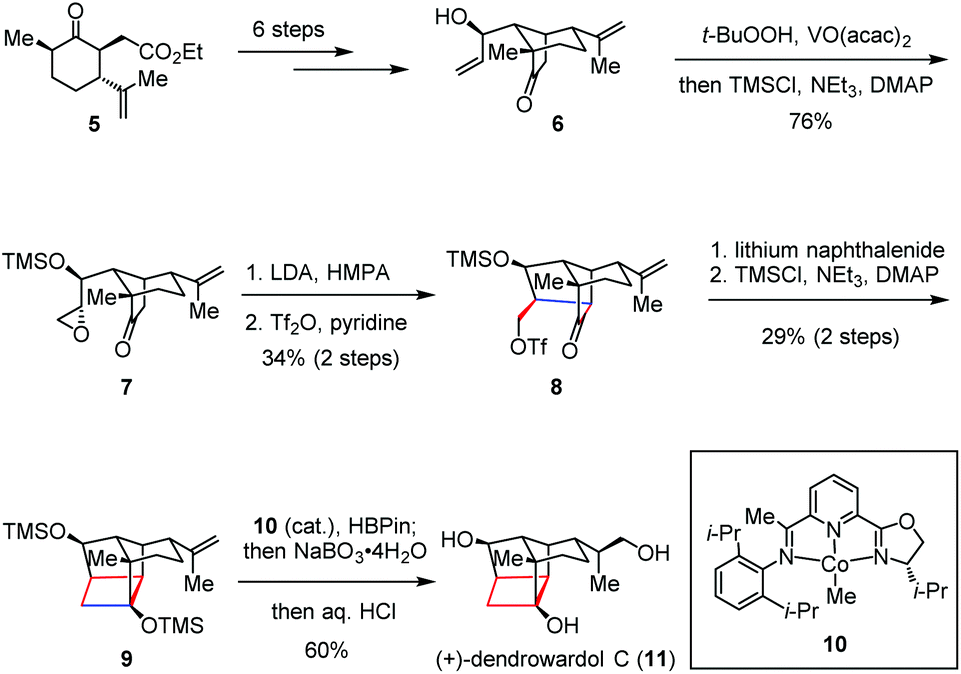 | ||
| Scheme 2 Stepwise nucleophilic cyclization in the total synthesis of (+)-dendrowardol C. | ||
2.2 Radical cyclization
Radical addition has seldom been used to construct four-membered carbon frameworks, probably because the inherent ring strain of the generated cyclobutane would easily reverse the reaction back to the corresponding acyclic form. To address this problem, electron-withdrawing groups such as carboxylic esters and sulfones can be introduced to stabilize the cyclic radical intermediate, and the gem-disubstituents could also be pre-installed on the precursor with the aim of enhancing the velocity of the intramolecular cyclization.With this concept in mind, the groups of Adachi and Nishikawa,13 and Isobe14 independently reported the construction of the tricyclo[5.2.1.01,6]decene skeleton of solanoeclepin A (4) from ent-Hajos–Parrish ketone (12). As shown in Scheme 3, upon treatment with SmI2, both enone 13 and enoate 15 underwent a 4-exo-trig ketyl-olefin radical cyclization to afford cyclobutanols 14 and 16 in 76% and 63% yield, respectively. On the other hand, Procter and co-workers also devised a similar strategy to build up the cyclobutanol skeleton and completed the total synthesis of (−)-14-O-methyl pestalotiopsin A (19).15
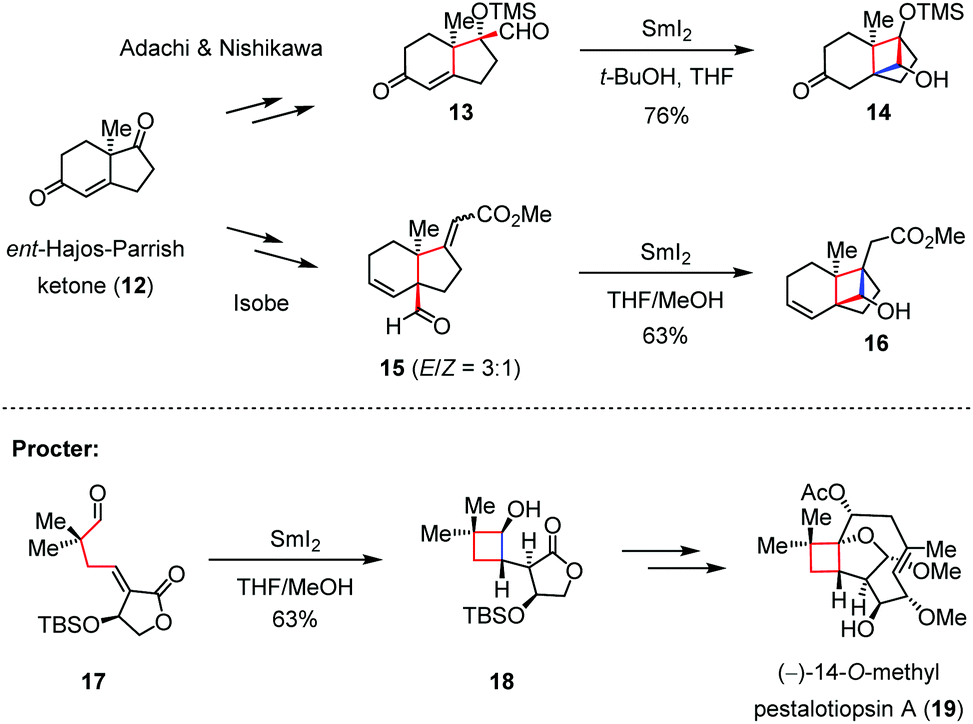 | ||
| Scheme 3 Ketyl-olefin radical cyclizations in the syntheses of solanoeclepin A and (−)-14-O-methyl pestalotiopsin A. | ||
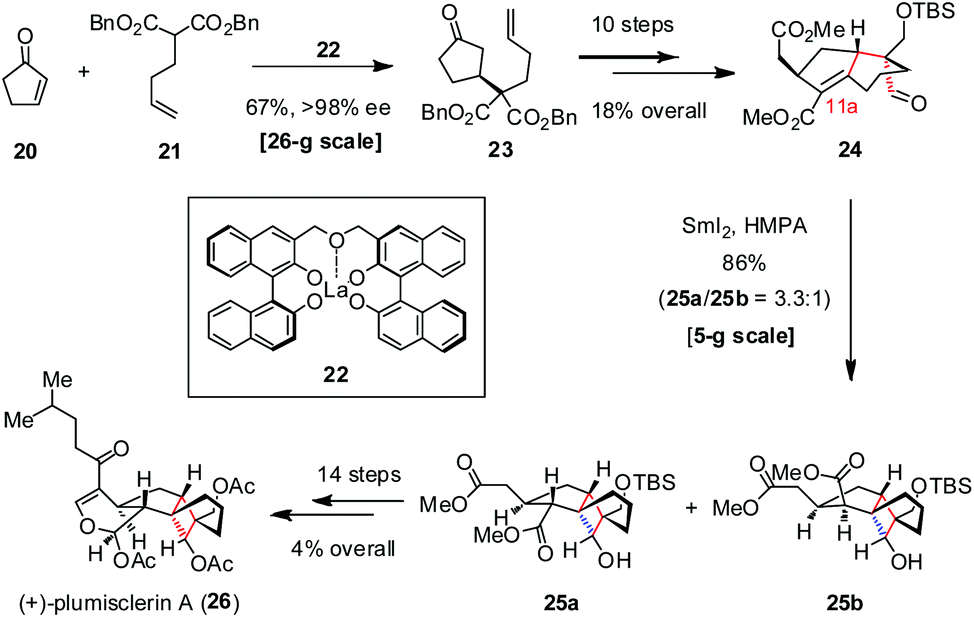
Xenicane diterpenoid (+)-plumisclerin A was isolated by Reyes and co-workers from crude extracts of soft coral Plumigorgia terminosclera collected on Mayotte Island in 2010.16 (+)-Plumisclerin A exhibits low-micromolar cytotoxicity against multiple tumor cells, such as A549, HT29 and MDA-MB-231. Featuring a compact 6/5/4/6 tetracyclic ring system, (+)-plumisclerin A is structurally more complex than other xenicane congeners. The highly congested tricyclo[4.3.1.01,5]-decane in (+)-plumisclerin A makes this molecule a challenging target for synthesis. In 2018, Yao and co-workers reported the first total synthesis of (+)-plumisclerin A (26), which proceeded enantioselectively and thus allowed the determination of its absolute configuration (Scheme 4).17 Their synthesis commenced with the asymmetric conjugate addition of malonate 21 to cyclopentenone 20 catalyzed by (S,S)-La-bis-BINOL 22 (10 mol%), providing 23 with 67% yield and >98% ee on a large scale. The following conversion of 23 to aldehyde 24 was realized in 18% overall yield over ten steps, setting the stage for the subsequent ketyl-olefin radical cyclization. In the presence of SmI2 and HMPA, the expected 1,4-addition occurred well to give 25a and 25b as a 3.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereomers at C11a in 86% combined yield on a gram scale. Further elaboration of the desired isomer 25a gave rise to (+)-26 uneventfully.
:1 mixture of diastereomers at C11a in 86% combined yield on a gram scale. Further elaboration of the desired isomer 25a gave rise to (+)-26 uneventfully.
 | ||
| Scheme 4 Ketyl-olefin radical cyclization in the total synthesis of (+)-plumisclerin A. | ||
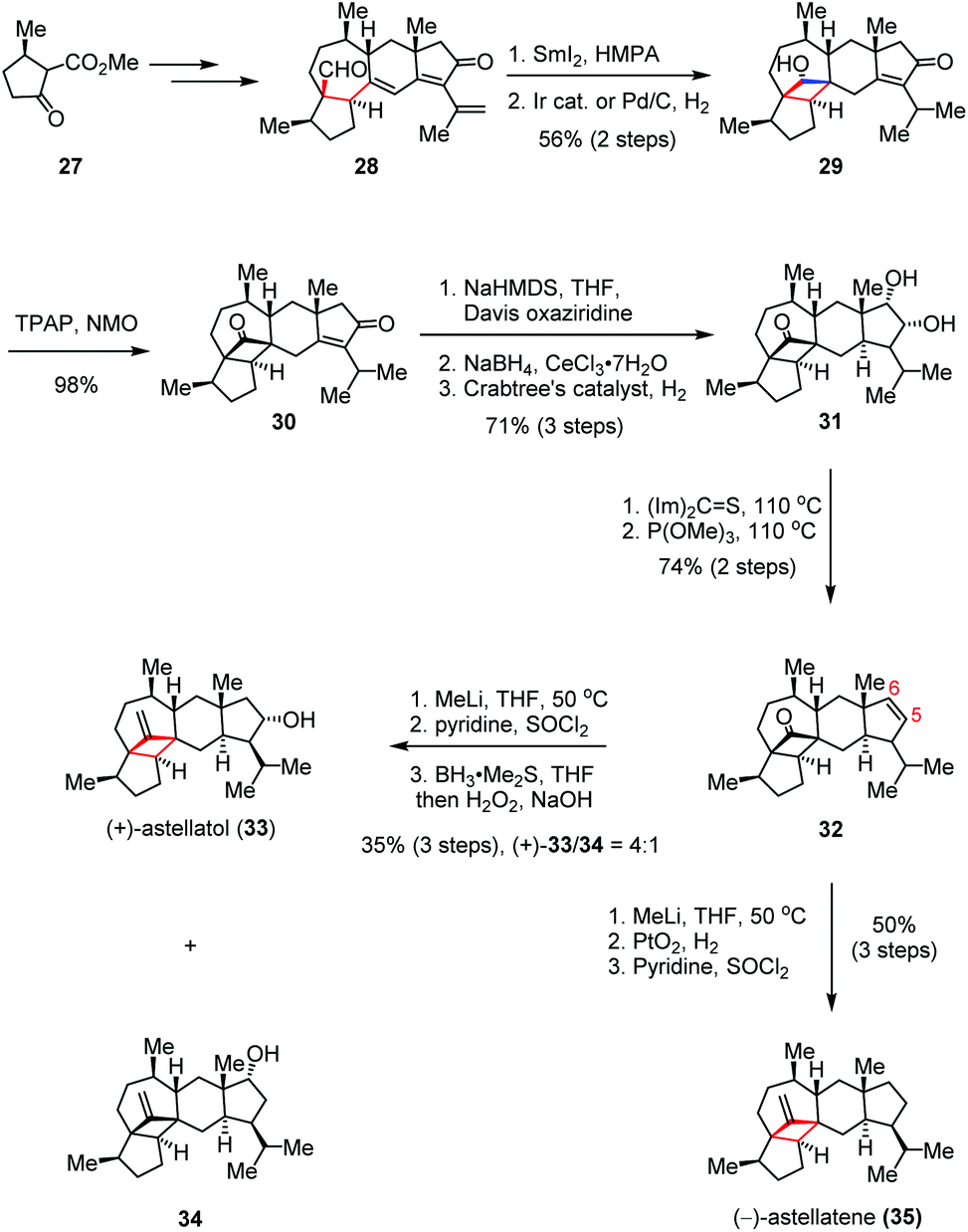
(+)-Astellatol was isolated from Aspergillus stellatus and structurally determined by Sadler and Simpson in 1989,18 while (−)-astellatene was discovered recently by the Osbourn group via the genome mining technique.19 Both of these molecules share a highly congested pentacyclic ring system, which possesses an unprecedented bicyclo[4.1.1]octane skeleton. In 2019, Xu and co-workers reported the first total syntheses of (+)-astellatol (33) and (−)-astellatene (35) featuring the SmI2-mediated intramolecular 1,6-radical additions to forge the embedding cyclobutane subunits (Scheme 5).20 The enantiomeric-enriched precursor 28 was prepared from known β-ketoester 27. In the forward sense, exposure of 28 to SmI2 and HMPA initiated the expected ketyl-olefin cyclization, followed by regioselective hydrogenation to afford cyclobutanol 29 in 56% yield over two steps. Subsequent Ley oxidation produced cyclobutanone 30, which then underwent Davis α-hydroxylation, Luche reduction and direct hydrogenation, delivering trans-hydrindane 31 in 70% yield over four steps. After being converted to 32 through a Corey–Winter elimination, an additional three-step core modification led to (+)-33 and its regioisomer 34 as a 4:1 mixture in 35% combined yield. On the other hand, further elaboration of 32 gave rise to (−)-35 in 50% yield over three steps.
 | ||
| Scheme 5 Ketyl-olefin radical cyclization in the total syntheses of (+)-astellatol and (−)-astellatene. | ||
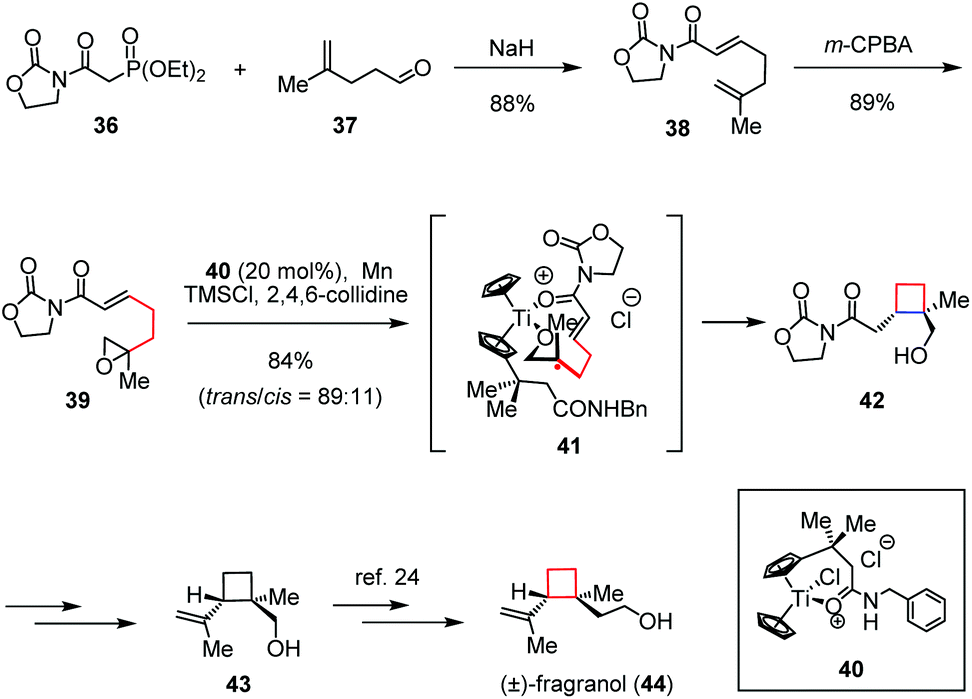
In the field of metal-mediated radical chemistry, much progress has been made toward efficient 4-exo-cyclization. Amongst, the application of titanocene(III) chloride constitutes a rapidly expanding field of research, especially for the generation of alkyl radicals from the corresponding epoxides as introduced by Nugent and RajanBabu.21 (±)-Fragranol, a monoterpene, was first isolated from Artemisia fragrans Willd. Due to its useful structure which could be considered as a potential alternative to classical pesticides, the total synthesis of this terpene is a matter of recent investigation.22 In 2009, Gansäuer and co-workers applied template catalysis to the stereoselective 4-exo radical cyclization, which led to a formal synthesis of (±)-fragranol (44).23 As depicted in Scheme 6, their synthesis began with epoxide 39, which was readily prepared through a Horner–Wadsworth–Emmons (HWE) olefination between phosphonate 36 and aldehyde 37 followed by epoxidation of the resultant alkene 38 with m-CPBA. By treatment of 39 with a catalytic amount of the cationic titanocene complex 40 in the presence of TMSCl and 2,4,6-collidine, the desired trans-cyclobutanol 42 was formed with its cis-isomer as a 89:11 mixture in 84% combined yield via the intermediacy of 41. The known cyclobutanol 43 could be obtained from 42 through late-stage functionalization, which resulted in a formal synthesis of (±)-44.24
 | ||
| Scheme 6 Ti(III)-triggered 4-exo radical cyclization in the total synthesis of (±)-fragranol. | ||
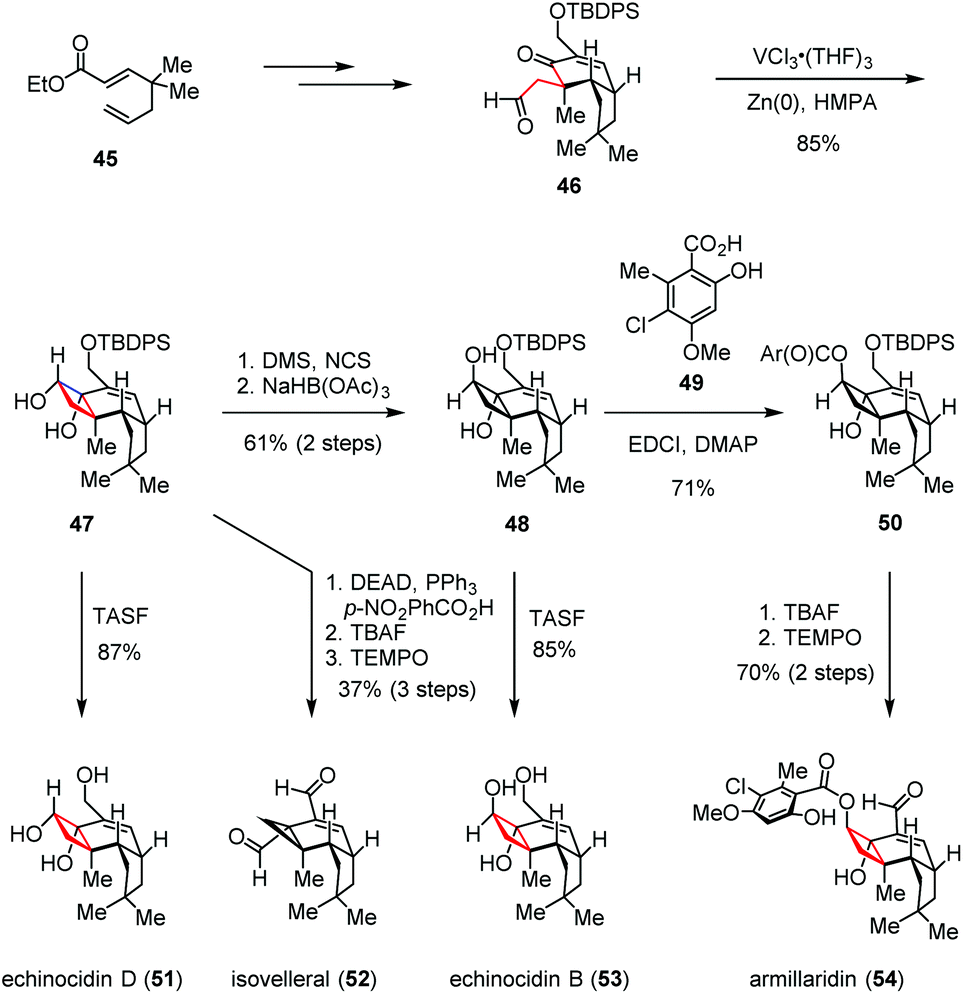
Apart from the ketyl-olefin radical cyclizations described above, the intramolecular pinacol coupling has also been exploited to assess the cyclobutane subunits. The antimicrobial protoilludane and marasmane sesquiterpenoids were isolated in the 1960s from the Armillaria mellea and Lactarius vellereus species of parasitic basidiomycetes fungi.25 Due to their densely functionalized perhydrocyclobuta[e]-indene and related carbon frameworks, great attention has been gained by these compounds. In 2017, Scheidt and co-workers reported a unified strategy to access the protoilludane, mellolide and marasmane families of natural products (Scheme 7).26 Their syntheses commenced with aldehyde 46, which could be prepared from 1,5-dienoate 45 through a series of conventional manipulations. By exposure of 46 to the in situ generated vanadium(II)/zinc(II) bimetallic complex, the expected intramolecular pinacol-type reductive coupling took place smoothly, affording the common intermediate 47 in 85% yield. Direct TASF-induced desilylation of 47 gave echinocidin D (51) in 87% yield. Under Mitsunobu conditions, a semi-pinacol ring contraction occurred as expected. After desilylation and oxidation, isovelleral (52) was obtained in 37% yield over three steps. On the other hand, 47 was conveniently transformed to trans-diol 48 through an oxidation/reduction sequence in 61% yield. Upon removal of the TBDPS group, echinocidin B (53) was formed with high efficiency. Furthermore, the esterification of 48 with orsellinic acid derivative 49 delivered orsellinate ester 50, which was then advanced to armillaridin (54) in 70% yield over two steps.
 | ||
| Scheme 7 Intramolecular pinacol coupling in the unified total syntheses of protoilludane natural products. | ||
2.3 Cationic cyclization
Although cationic cyclization has been widely applied in the construction of polycyclic skeletons, few examples were described to generate cyclobutanes.27 In 2012, Isobe and co-workers synthesized an advanced intermediate en route to solanoeclepin A (4) by using a cationic Hosomi–Sakurai cyclization as the key step (Scheme 8).28 Allylpropargyl ether 55, prepared from ent-Hajos-Parrish ketone (12), underwent TMS protection of the terminal alkyne followed by the [2,3]-Wittig rearrangement to give the trans-fused bicyclic precursor 56 as a single diastereomer in 83% yield over two steps. After in situ formation of the Nicholas-type cation 57, subsequent cyclization in the presence of TMSOTf afforded tricycle 58 bearing three quaternary stereocenters as a single diastereomer in 66% yield over three steps.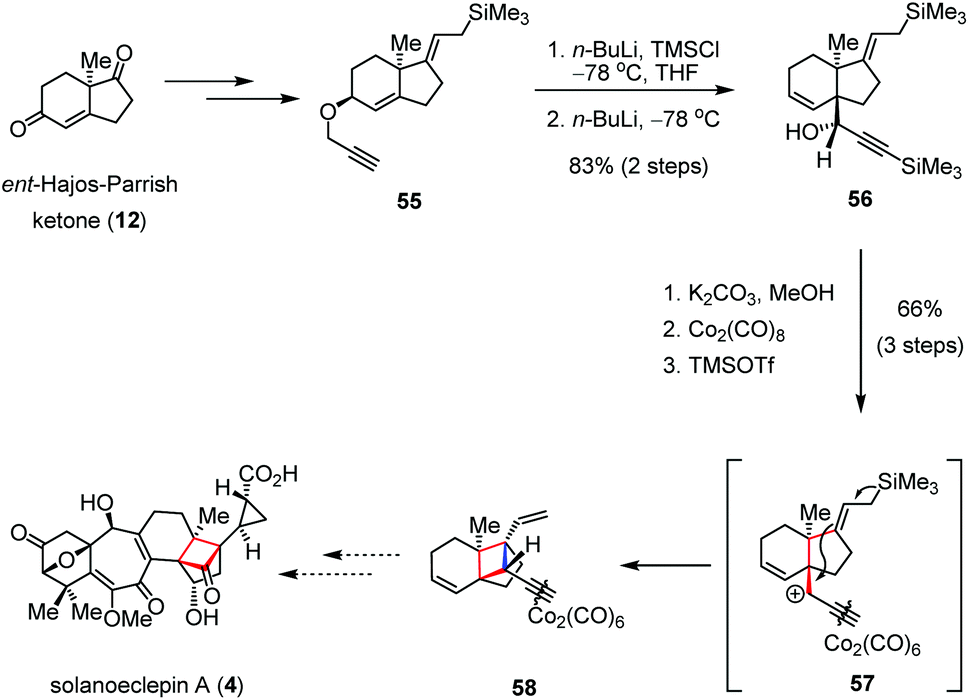 | ||
| Scheme 8 Cationic Hosomi–Sakurai type cyclization towards the total synthesis of solanoeclepin A. | ||
2.4 Transition-metal catalyzed cyclization
Transition-metal catalyzed cyclizations have been widely applied as versatile methods in C–C bond formations. Although the employment of these reactions for the generation of cyclobutane-based systems would encounter the risk of undesired ring cleavage, they still attracted considerable interest from the synthetic community due to their high efficiency in the construction of complex natural skeletons.Grandisol, a diastereomer of 44 isolated from male boll weevils and other species, is one of the active components of aggregating pheromone and thus of interest to the pesticide industry.29 The cyclobutane skeleton combined with two adjacent chiral centers makes this molecule structurally intriguing. In 2011, Suh and co-workers synthesized (±)-grandisol (65) through a Pd(0)-catalyzed intramolecular allylic alkylation in a highly regioselective manner (Scheme 9).30 The precursor 62 was prepared in 83% yield through a cross metathesis reaction between the known methyl hexanoate 59 and allyl carbonate 60 in the presence of the Grubbs II catalyst (61). Treatment of 62 with Pd(PPh3)4 in DMSO afforded the required cyclobutane 63 in 79% yield. After desulfonylation by sodium amalgam, the known intermediate 64 was formed in 78% yield, which constitutes a formal synthesis of (±)-65.31
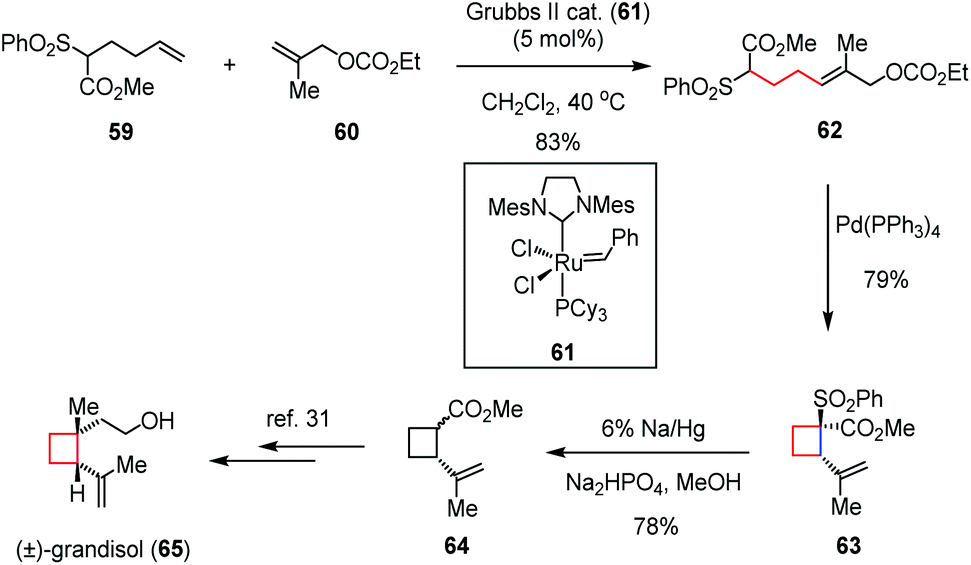 | ||
| Scheme 9 Pd-Catalyzed intramolecular allylic alkylation in the total synthesis of (±)-grandisol. | ||
A similar strategy was devised by Dong and co-workers for the construction of a more rigid spiro-fused cyclobutane skeleton en route to phainanoids,32 a class of novel triterpenoids that were isolated by Yue and co-workers from Phyllanthus hainanensis and exhibit potent immuno-suppressive activities.33 As depicted in Scheme 10, their synthesis toward phainanoid A (72) began with vinyl epoxide 67, which could be prepared from geranyl acetate (66) through several conventional transformations. A SnCl4-promoted polyene cyclization of 67 at −78 °C gave tricyclic alcohol 68 bearing a trans-decaline core in 70% yield. The latter was then converted to enol-triflate 69 in 37% yield over three steps. Subsequently, the Pd-catalyzed intramolecular ketone alkenylation occurred smoothly with the aid of QPhos (70) and t-BuOLi, affording the desired 4,5-spirocycle 71 with 83% yield and >20:1 dr. The remarkably high diastereoselectivity was presumably ascribed to the stabilizing effects of the adjacent carbonyl moiety.
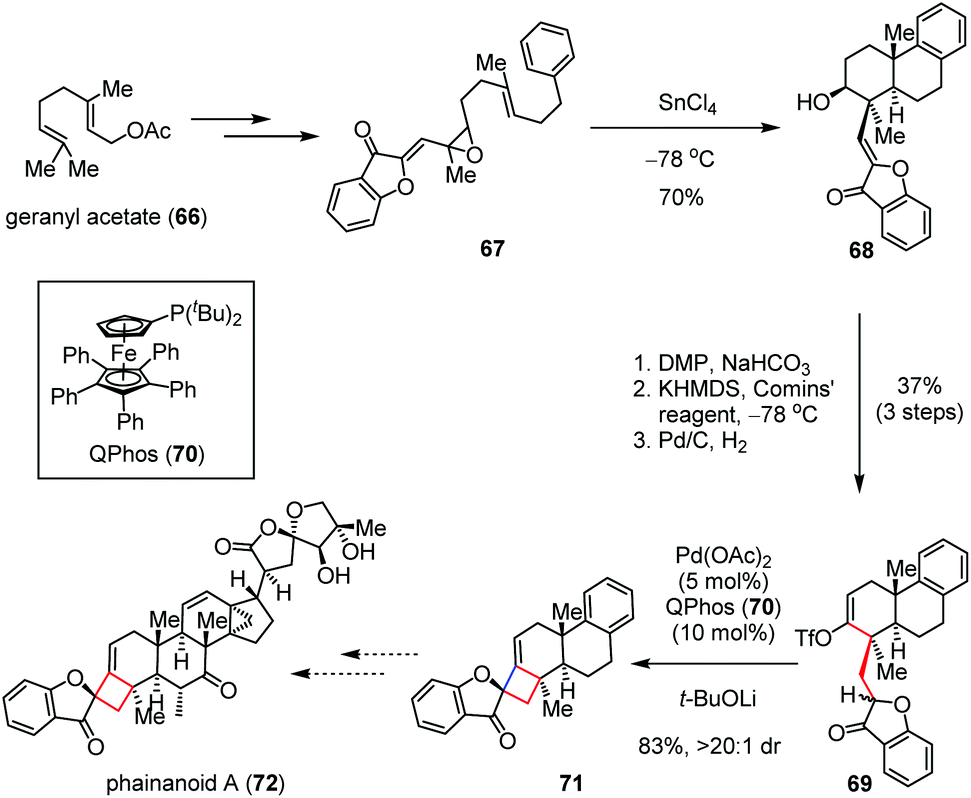 | ||
| Scheme 10 Pd-Catalyzed intramolecular coupling reaction towards the total synthesis of phainanoid A. | ||
As another representative of reactive intermediates in organometallic chemistry, the transition-metal carbenoid generated from a alkyne precursor could also be employed to construct the four-membered ring. For instance, Goess and co-workers described a second route to (±)-grandisol (65) involving a microwave-assisted enyne metathesis (Scheme 11).34 The synthesis began with the preparation of enyne 74, which could be obtained from α-methyl-γ-butyrolactone 73 in five steps. Treatment of 74 with the optimum ruthenium catalyst 75 under microwave irradiation provided vinylcyclobutene 76 in 83% yield. Subsequent regioselective hydrogenation in the presence of RANEY nickel followed by desilylation furnished (±)-65 in 63% yield over two steps.
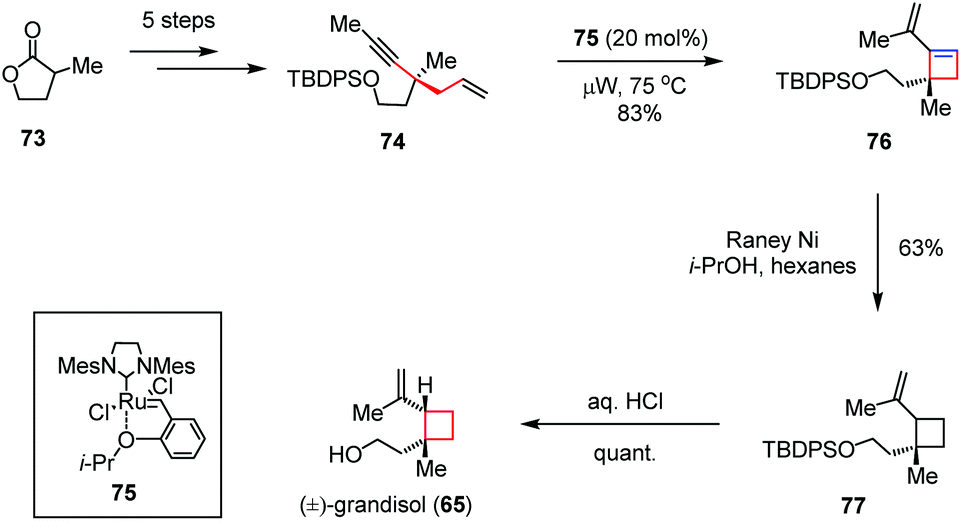 | ||
| Scheme 11 Enyne metathesis in the total synthesis of (±)-grandisol. | ||
An alternative application covered in this class of transformation is the use of gold catalysts. The sesquiterpene repraesentin F was isolated from the fruiting bodies of Lactarius repraesentaneus and shows growth regulation on plants by promoting the radicle elongation of lettuce seedlings at low concentration.35 In 2018, Echavarren and co-workers described the first total synthesis of repraesentin F (83), which allowed reassignment of the relative and absolute configuration of this molecule.36 As shown in Scheme 12, key to the success of their synthesis is a highly diastereoselective Au(I)-catalyzed cyclization cascade to assemble the tricyclic core of the natural product with the requisite syn/anti/syn ring fusion, which delivered cyclobutanes 81 and 82 as a 7.2:1 isomeric mixture in 75% combined yield from enyne 78. DFT calculations revealed that the activation energy for the C–C formation through the intermediate 80 is much lower (ΔG‡ = 10.3 kcal mol−1) than that for the activation of the keto group (ΔG‡ = 20.5 kcal mol−1), which indicated that this reaction abides by the usual activation mode as in other gold-catalyzed enyne cyclizations. It should also be noted that the use of BArF− as a counterion in the structure of the gold catalyst 79 was essential to suppress further rearrangement of cyclobutanes 81 and 82 to the corresponding cyclopropane byproduct.
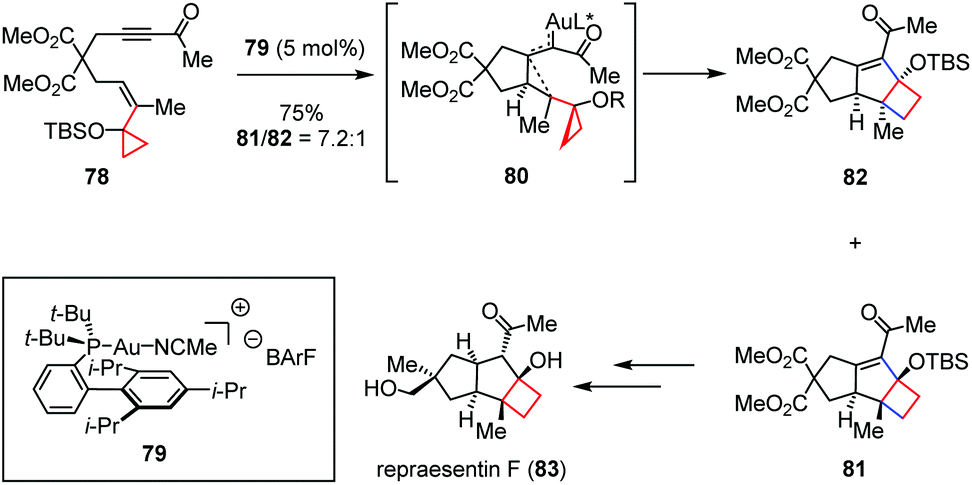 | ||
| Scheme 12 Au-Catalyzed cyclization cascade cyclization in the total synthesis of repraesentin F. | ||
3. Cycloaddition strategies
In contrast to the direct ring-closure strategies in which a single bond is formed in one step, the hallmark of the cycloadditions involves the formation of two or more bonds in a single operation.37 Such protocols have proven useful in the intramolecular context and homodimerizations, which were demonstrated by the recent applications in the assembly of four-membered carbocycles.3.1 [2 + 2] photocycloaddition
The most commonly considered and direct approaches to cyclobutanes are the [2 + 2] photocycloadditions between two alkene moieties.38 Electron-deficient olefins are favorable substrates in photo-induced cycloadditions due to their convenient excitation by widely available UV sources.Hippolachnin A was isolated from the South China Sea sponge Hippospongia lachne by Lin and co-workers in 2013.39
It shows potent antifungal activity at a minimum inhibitory concentration of 0.41 μM against three pathogenic fungi, namely Cryptococcus neoformans, Trichophyton rubrum and Microsporum gypseum. Structurally, this molecule possesses a highly substituted cyclobutane ring decorated with six contiguous stereogenic centers, four of which bear an ethyl substituent projecting in the convex orientation. The congested oxacyclobutapentalene core combined with the promising biological activities made hippolachnin A a highly desirable yet challenging synthetic target. In 2015, Carreira and co-workers accomplished the first total synthesis of (±)-hippolachnin A (90).40 As depicted in Scheme 13, the synthesis commenced with an intermolecular [2 + 2] photocycloaddition of enone 84 and hex-3-yne, followed by β-elimination to establish the key cyclobutene intermediate 85 in 54% yield over two steps. Subsequent introduction of the ethyl side chains to the bicyclic scaffold was achieved by a Cu(I) mediated 1,4-addition and a LaCl3·2LiCl promoted 1,2-Grignard addition, affording alcohol 86 with complete exo diastereoselectivity in 66% yield over two steps. After being converted to ester 88 in the presence of catalytic PPTS and (E)-methyl-3-methoxyacrylate (87) as a solvent, a BF3·2AcOH induced intramolecular ene cyclization provided tricycle 89 in 65% yield (6:1 dr). Further functionalization of the latter led to the formation of (±)-90 with good efficiency.
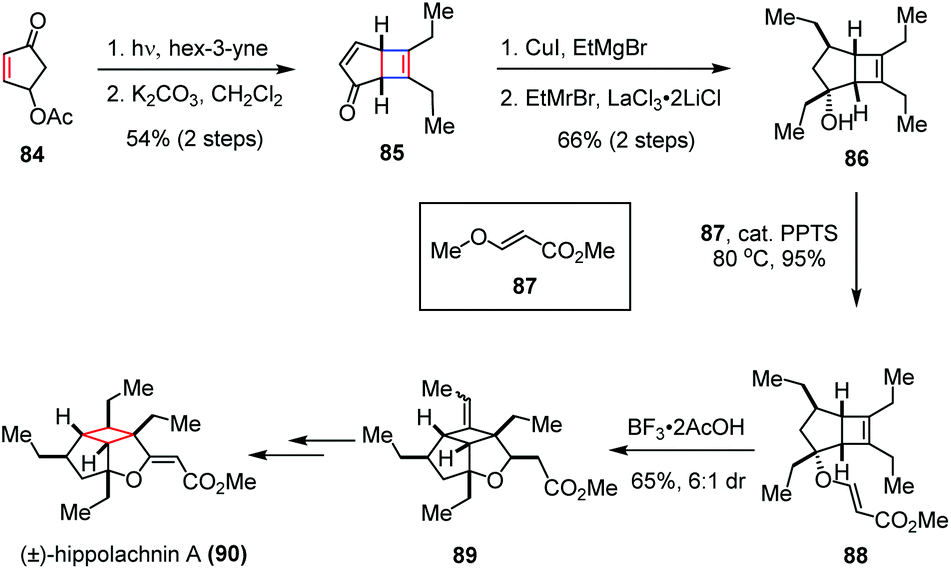 | ||
| Scheme 13 [2 + 2] photocycloaddition in the total synthesis of (±)-hippolachnin A. | ||
Recently, Tang, Enders and co-workers reported a biomimetic approach to (+)-hippolachnin A (90) involving another intramolecular [2 + 2] photocycloaddition (Scheme 14).41 In contrast to Carreira's synthetic sequence, they chose furanone 91 as the starting material and installed the required functionalities to afford the precursor 92 within six steps. By irradiation of 94 with a high pressure mercury lamp (500 W) at 45 °C for 12 h, (+)-90 was furnished in 50% yield.
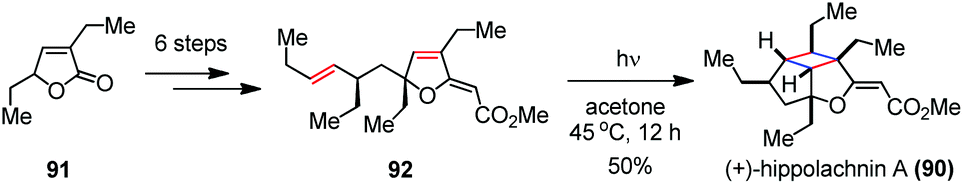 | ||
| Scheme 14 [2 + 2] photocycloaddition in the total synthesis of (+)-hippolachnin A. | ||
A similar approach was disclosed toward the synthesis of the well-known furano-cembranoid bielschowskysin (100), which was isolated from the Caribbean gorgonian octocoral Pseudopterogorgia kallos and exhibits potent antimalarial activity and selective cytotoxicity against various types of cancer cells.42 Synthesizing this diterpenoid proved to be difficult owing to its complex tricyclo[9.3.0.0]tetradecane carbon framework bearing numerous oxygen-containing functional groups and 11 stereogenic centers (Scheme 15).43 A significant advance has been made by Nicolaou and co-workers in the expedient synthesis of the highly functionalized 5/4/8/5 tetracyclic core structure of this molecule.44 The intramolecular [2 + 2] photocycloaddition of a macrocyclic precursor 95, which was prepared through a crucial oxidative coupling of β-keto ester 93 and furan 94 followed by a ring-closing metathesis, resulted in a ring contraction to form the polycyclic skeleton in one step. On the other hand, the Roche group also reported a biomimetic route to the bielschowskysin skeleton relying on a late-stage transannular [2 + 2] photocycloaddition.45 In their approach, the pivotal cycloaddition precursor (Z)-exo-enol ether 98 was prepared through a regio- and stereoselective oxidative dearomatization of the furan-containing natural product acerosolide (97). Upon UV irradiation, the advanced intermediate 99 could be obtained in 83% yield.
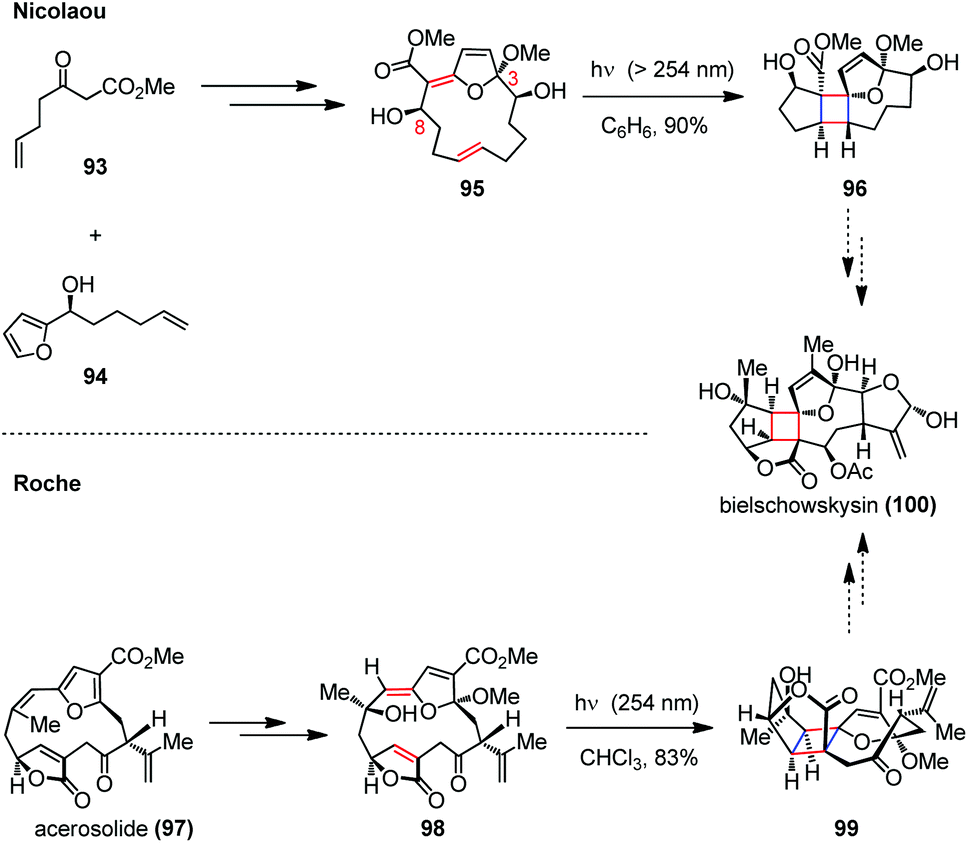 | ||
| Scheme 15 Transannular [2 + 2] photocycloadditions in the construction of the core structures of bielschowskysin. | ||
Aquatolide was first isolated from Asteriscuc aquaticus in 1989 and its structure was finally determined by single-crystal X-ray analysis in 2012.46 Structurally, aquatolide has a cyclobutane moiety embedded concurrently in a bicyclo[5.1.1]nonane and a bicyclo[2.1.1]hexane skeleton, which makes it a challenging target for synthesis. In 2019, Takao and co-workers reported a concise total synthesis of (+)-aquatolide (111) by a biomimetic transannular [2 + 2] photocycloaddition to build up its cyclobutane subunit at the late stage (Scheme 16).47 Their synthesis commenced with vinyl iodide 103, which was readily prepared from known alcohol 101 in 76% yield over four steps. The subsequent ring-opening/ring-closing/cross-metathesis (ROM/RCM/CM) reaction of 103 proceeded in a cascade manner to produce the desired aldehyde. The latter was then subjected to an intermolecular Nozaki–Hiyama–Kishi (NHK) reaction with methacrolein (104) to give macrocycle 105 as a single diastereomer in good yield. The following three-step sequence involving olefin isomerization, epoxidation and parallel [2 + 2] photocycloaddition delivered [2]-ladderane adduct 107 in 24% overall yield. Further functionalization of 107 provided the originally proposed structure of (+)-108. Furthermore, oxidation of 105 followed by 1,4-addition to mask the olefin gave 109 as a single isomer. Fortunately, the transannular crossed [2 + 2] photocycloaddition of 109 occurred successfully to construct the bicyclo[2.1.1]hexane core, affording the desired cycloadduct 110 in 37% yield. Finally, the β-elimination of MeOH from 110 proceeded smoothly and led to the completion of the total synthesis of (+)-111 in 64% yield.
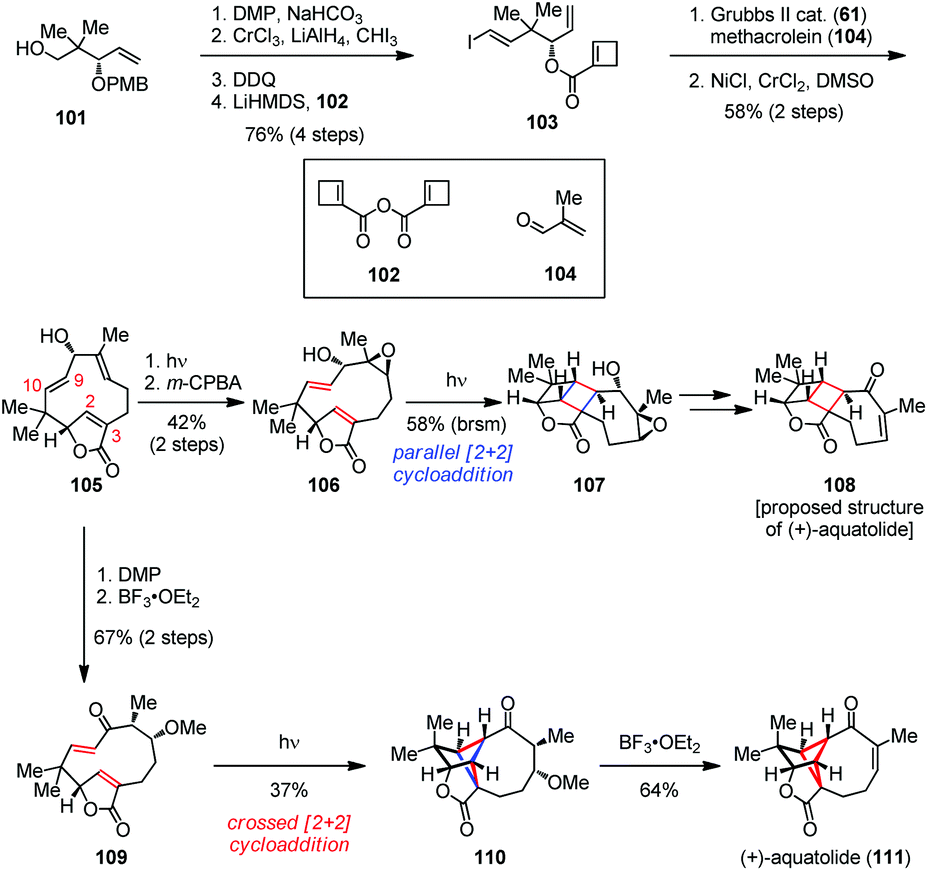 | ||
| Scheme 16 Biomimetic transannular [2 + 2] photocycloaddition in the total synthesis of (+)-aquatolide. | ||
3.2 Oxidative radical cycloaddition
For the [2 + 2] cycloaddition of two electron-rich olefins, harsh reaction conditions are normally needed. An alternative solution is to oxidize one of the substrates to generate a radical cation which is sufficiently reactive even under mild conditions. This method is typically used, for example, to dimerize the unactivated olefins.Nigramide R, bearing a tetrasubstituted cyclobutane skeleton, was isolated from the roots of Piper nigrum and exhibits inhibitory activity against cytochrome P450 2D6, as well as cytotoxicity against a mouse lymphoma cell line (L5178Y).48 In 2015, Donohoe and co-workers reported a concise synthesis of (±)-nigramide R (117) by using a hypervalent iodine-induced oxidative dimerization as the key step (Scheme 17).49 Their synthesis began with the preparation of 114, which was derived from a cross metathesis between olefins 112 and 113. The subsequent PhI(OAc)2 promoted head-to-head dimerization of olefin 114 afforded the cyclobutane 115 in 75% yield. The bis(amide) 116 was then obtained in 68% yield over two steps through oxidative cleavage to carboxylic acid and coupling with piperidine. Late-stage derivatization of 116 through additional four steps gave rise to (±)-117 in 58% overall yield.
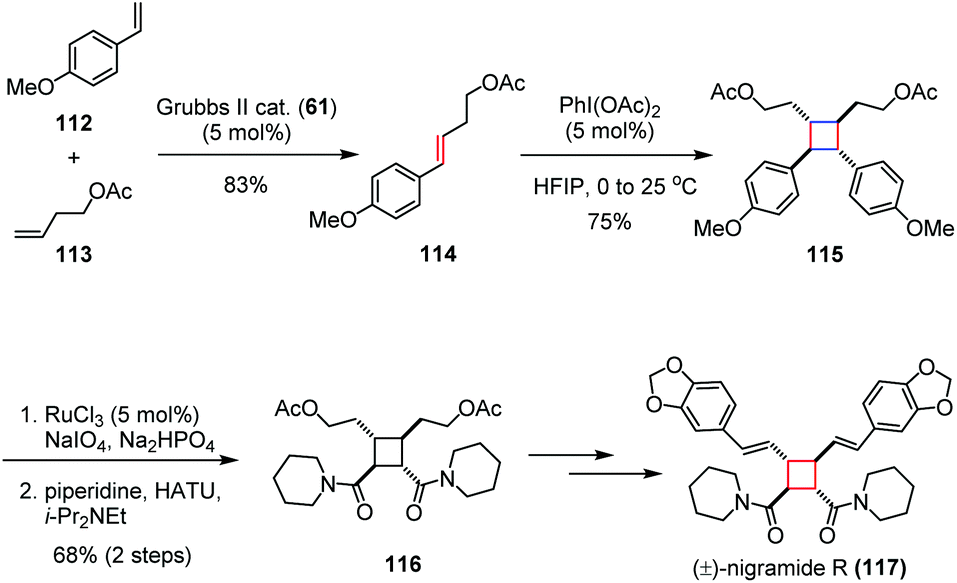 | ||
| Scheme 17 Oxidative radical dimerization in the total synthesis of (±)-nigramide R. | ||
Besides, the cyclobutane-containing natural products could also be synthesized via photo-induced single-electron transfer (SET) reactions, in which the radical cations are transiently produced by excited photosensitizers. The dimeric pyrrole-imidazole alkaloids are structurally complex molecules produced by marine sponges through reactions that are not well understood.50 These natural products are highly polar, noncrystalline, redox labile, and pH sensitive owing to their exceptionally high nitrogen content. In 2014, Chen and co-workers demonstrated the biomimetic synthesis of ent-sceptrin (124).51 As depicted in Scheme 18, the key intermediate 119 was prepared from L-glutamic acid (118) in seven steps. After Staudinger reduction, irradiation of a solution of the resultant 120 with catalytic Ir(ppy)3 (121) induced the [2 + 2] cycloaddition to give 122 and its C10-epimer as a 1.8:1 mixture. The following trans-thioketalization afforded 123 in 43% yield, which was then readily transformed to ent-124 within a few steps.
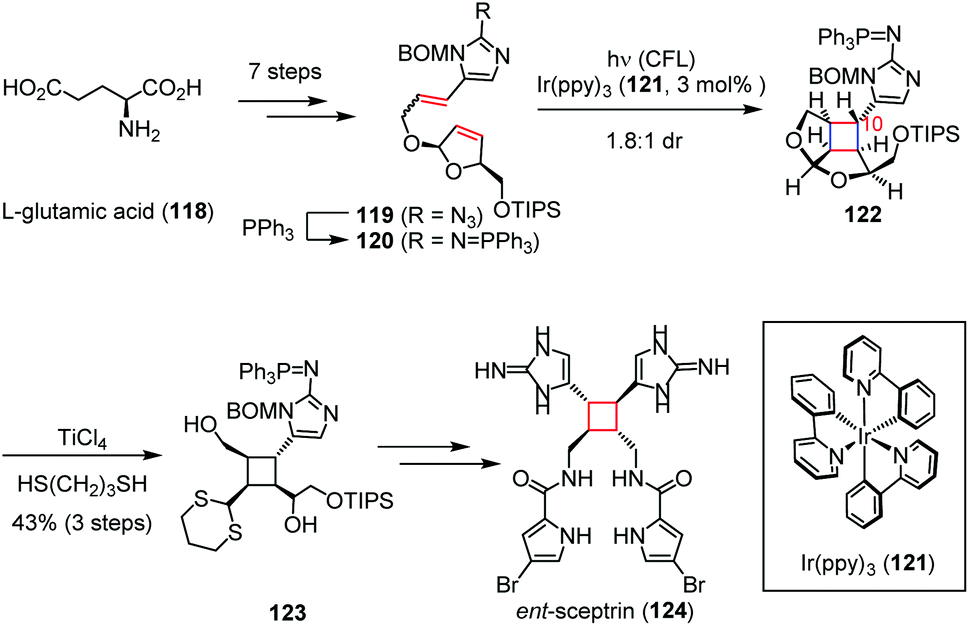 | ||
| Scheme 18 SET [2 + 2] cycloaddition in the total synthesis of ent-sceptrin. | ||
Recently, Evanno, Poupon, Smietana, Arseniyadis and co-workers developed an interesting DNA-templated cationic [2 + 2] photodimerization process.52a Its synthetic utility was further demonstrated by application to the synthesis of cycloaplysinopsin-type natural products such as dictazole B, which was isolated from the sponge Smenospongia cerebriformis.52b,c
Nyingchinoids belong to a family of polycyclic meroterpenoids that were isolated from Rhododendron nyingchiense by Hou and co-workers in 2018, with each molecule identified as a scalemic mixture by chiral HPLC.53 Very recently, George and co-workers achieved the biomimetic synthesis of (±)-nyingchinoid D (130) and its analogues by a photocatalytic aerobic [2 + 2] cycloaddition pathway (Scheme 19).54 Diene 127 was readily prepared through the known condensation of orcinol (125) and citral (126) followed by TBS protection. The pyrylium photoredox-catalyzed aerobic [2 + 2] cycloaddition of 128 according to the slightly modified procedure of Nicewicz [4-MeO-TPT (2 mol%), O2 (1 atm), LED (470 nm), DCE, 0 °C, 20 min] afforded cyclobutane 129 in 87% yield as the sole product. Desilylation of the latter completed the rapid synthesis of (±)-130 in 86% yield.
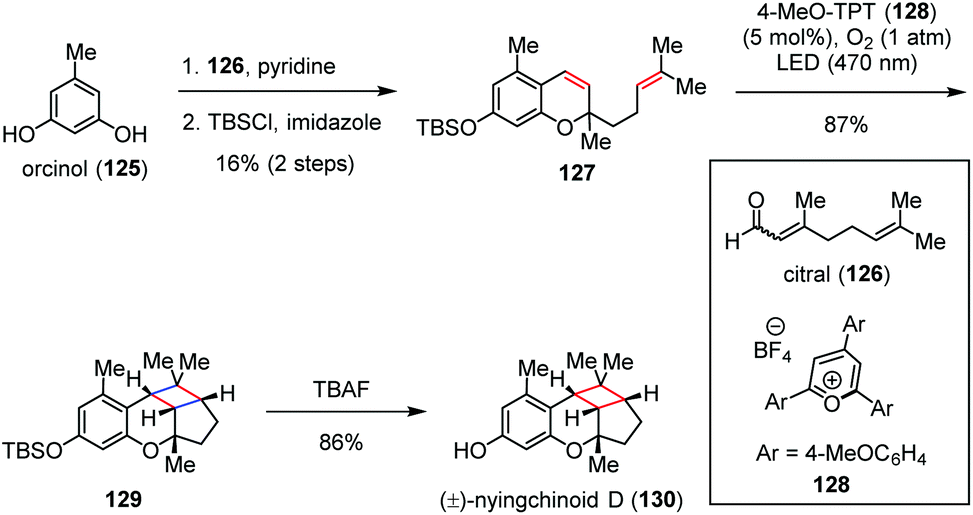 | ||
| Scheme 19 Photocatalytic aerobic [2 + 2] cycloaddition in the total synthesis of (±)-nyingchinoid D. | ||
3.3 Strain-releasing cycloaddition
The [2 + 2] cycloadditions can be driven by not only the external factors but also the inherent ring strains in the substrates. In 2016, the Wood and Brown groups independently accomplished the total synthesis of (±)-hippolachnin A (90) via a strain-releasing cyclobutanation and a late-stage C–H oxidation (Scheme 20).55 Tricycles 133 and 137 were efficiently obtained through an unusual [2π + 2σ + 2σ] cycloaddition of quadricyclane (131) with electron-deficient alkenes 132 and 136, respectively. In Brown's synthesis, 133 underwent a ring-opening metathesis with ethylene followed by reduction to give alcohol 134 in 72% yield over two steps. Subjection of the latter to Suárez conditions [I2, PhI(OAc)2, hν] afforded a tetrahydrofuran intermediate, which was oxidized by DMDO to produce the lactone 135 in 77% yield. Further attachment of the side chain furnished (±)-90.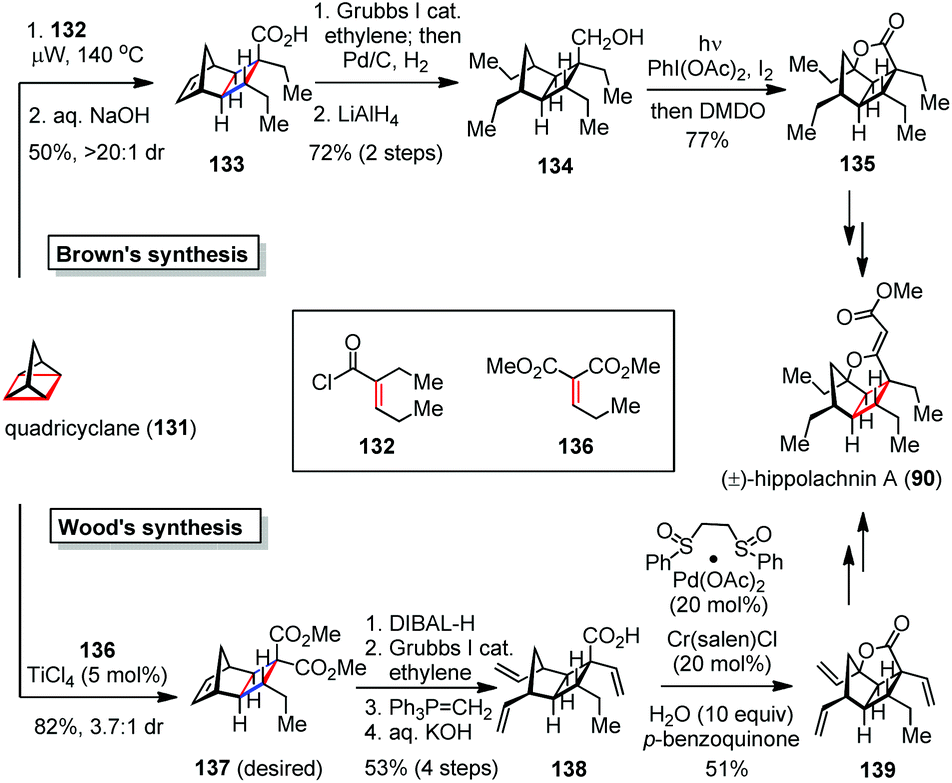 | ||
| Scheme 20 Strain-releasing cycloadditions in the total synthesis of (±)-hippolachnin A. | ||
In Wood's synthesis, 137 was converted to acid 138via a series of transformations involving regioselective reduction of ester, ring-opening metathesis, Wittig olefination and saponification. Upon exposure of 138 to modified White's conditions [Pd(OAc)2·(CH2SO2Ph)2 (20 mol%), Cr(salen)Cl (20 mol%), H2O (10 equiv.), 1,4-benzoquinone (2 equiv.)], the C–H activation/oxidation proceeded smoothly and provided lactone 139 in 51% yield. Of note, the presence of excess water was found to be crucial for the efficiency of this process. Further installation of the remaining vinylogous carbonate completed the synthesis of (±)-90 within three steps.
By providing the shortest routes to (±)-hippolachnin A, the above two syntheses demonstrated the capability of the strain-releasing cycloadditions in concise total syntheses of natural products.
3.4 Ionic stepwise [2 + 2] cycloaddition
The acid-catalyzed [2 + 2] cycloaddition reaction of silyl enol ethers with α,β-unsaturated carbonyl compounds provides a promising tool for the syntheses of donor–acceptor (D–A) cyclobutane derivatives in a different stepwise fashion. The strategy involves an intramolecular aldol reaction of δ-ketoenolate, which was formed by the Michael addition of silyl enol ethers with α,β-unsaturated carbonyl compounds in the former step.Takasu and co-workers have made great efforts on the triflic imide (Tf2NH) catalyzed [2 + 2] cycloaddition reactions of silyl enol ethers for the construction of various fused cyclobutane rings bearing a silyloxy group at the bridgehead position.56 This effective method has been successfully applied to the synthetic studies toward penitrems, a family of indole diterpene alkaloids isolated from Penicillium cyclopium and Penicillium crustosum with strong neurotoxicity.57
Most recently, the same group reported the first total synthesis of cytotoxic paesslerin A (146) through a Tf2NH-catalyzed multicomponent reaction between cyclopentenone 140, 2-siloxy-butadiene 141 and methyl acrylate 144 (Scheme 21).58 This catalytic three-component reaction consists of a [4 + 2] cycloaddition, an alkene isomerization and an ionic stepwise [2 + 2] cycloaddition. The reaction cascade enables the assembly of the desired tricyclo[6.3.0.02,5]-undecanone 145 as the major product in 34% yield. Further core functionalization led to 146 with good efficiency. The above synthesis led to the revision of the originally proposed tricyclic structure of paesslerin A (147).
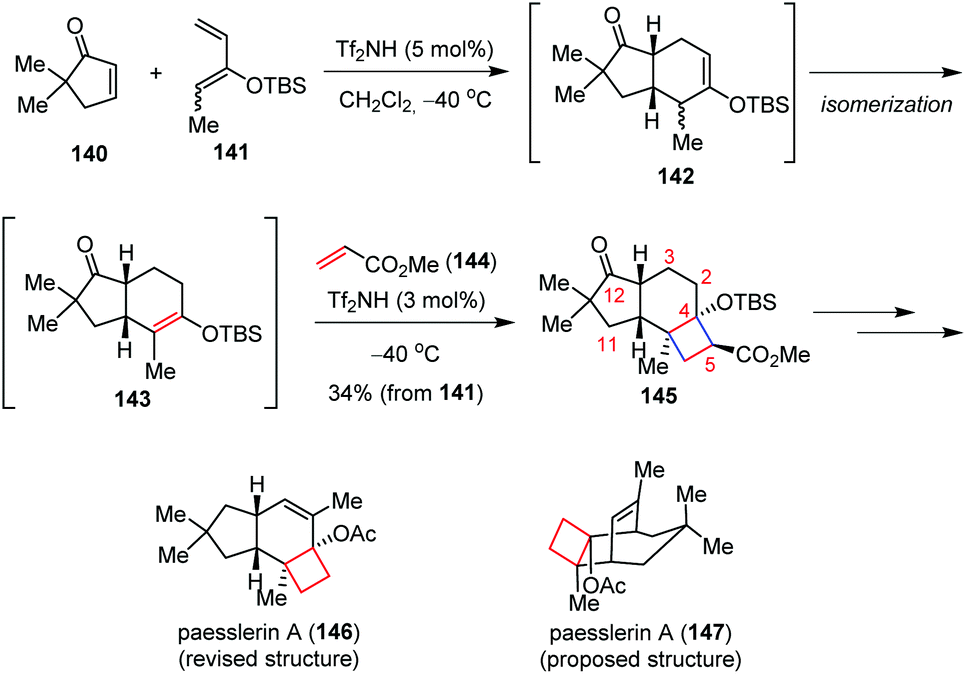 | ||
| Scheme 21 Multicomponent domino cycloaddition in the synthesis of paesslerin A. | ||
4. Ring contraction strategies
Aside from the aforementioned cycloaddition reactions, the four-membered cyclic scaffolds can also be accessed from the existing rings by modification of their sizes. In this regard, the ring contraction is one important and common strategy to address this issue. Although such reactions would inevitably intensify the ring strains, appropriate transient reactive intermediates could be devised to trigger the reactions toward the desired direction.4.1 Electrocyclization
Electrocyclization has been widely used to build up various complex polycyclic scaffolds in a highly stereocontrolled manner, in which the electrocyclic contraction of 4π- and 6π-electron systems can rapidly generate cyclobutanes.59The highly cytotoxic cyclobutane-fused piperaborenines were isolated from the stems of Piper arborescens.60 These molecules were classified into two stereochemical classes including cis–trans–cis and trans–cis–trans. Both of them were biosynthetically generated through head-to-tail dimerization of piplartine-type monomers with varying degrees of oxidation on the aryl rings. Syntheses of such cyclobutanoid amides through direct coupling of monomeric olefins are challenging due to the difficulties encountered in ensuring the desired heterodimerization to provide the final targets. Baran and co-workers found an ingenious solution by using monoarylated cyclobutane 154 as a common intermediate followed by conducting sequential C(sp3)–H functionalization (Scheme 22).61 Inspired by Maulide's creative work on the cyclobutene synthesis62a and Corey's pioneering research on pyrone photochemistry,62b the desired precursor 149 was obtained from the commercially available methyl coumalate (148) via a photochemical 4π electrocyclization. This unstable intermediate obtained was immediately hydrogenated and coupled with o-thioanisidine 151 to provide cyclobutanoid amide 152 in 61% yield. Selective C–H cross-coupling of 152 with 3,4,5-trimethoxy-iodobenzene 153 delivered the desired monoarylated cyclobutane 154 in 52% yield. Divergent epimerization of 154 in the presence of t-BuOLi and KHMDS gave C1- and C3-epimers 155 and 158, respectively. Further functionalization delivered piperarborenine B (156) and the proposed structure of piperarborenine D (159).
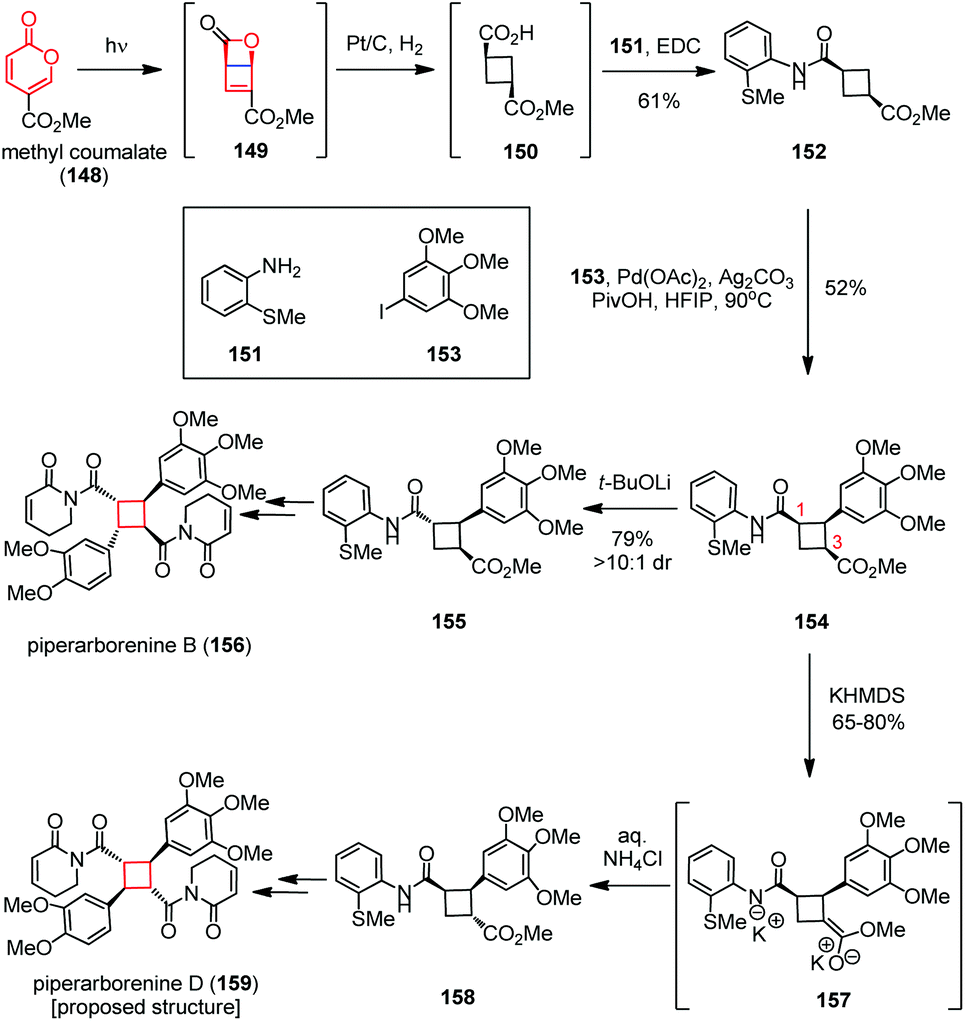 | ||
| Scheme 22 4π electrocyclization in the total syntheses of piperaborenines. | ||
The Trauner group used another 4π electrocyclization to forge the cyclobutane subunit in the total synthesis of (±)-hippolachnin A (Scheme 23).63 Based on earlier results from Dauben and colleagues,64 the researchers obtained the key bicyclo[3.2.0]heptadiene 162 in 52% yield by irradiation of the readily available tropolone ether 160 with a high pressure mercury lamp (150 W). The reaction process involves a disrotatory 4π electrocyclization, followed by excited state rearrangement. A three-step sequence including a stereoselective 1,4-addition and a Wittig olefination followed by hydrolysis gave 163 as a 10:1 mixture of Z- and E-isomers in 59% yield. Subsequent formation of enol-triflate using Comins’ reagent and Pd(0)-catalyzed CO insertion afforded eonate 164 in 80% yield over two steps. The latter underwent a [3 + 2] cycloaddition with thiocarbonyl ylide in situ generated from 165 and DMPU at elevated temperature to provide tetrahydrothiophene 166 as a single diastereomer in 68% yield. Final derivatization of 166 accomplished the synthesis of (±)-90.
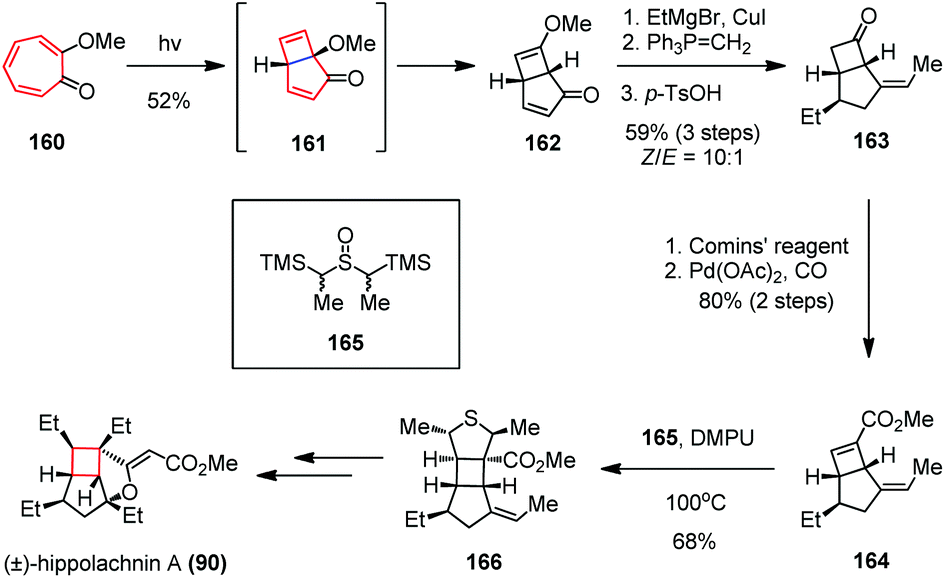 | ||
| Scheme 23 4π electrocyclization in the total synthesis of (±)-hippolachnin A. | ||
Salvileucalin C, a highly rearranged neoclerodane diterpenoid, was isolated by Takeya and co-workers from Salvia leucantha.65 The structurally closely related salvileucalin D was proposed to be a biosynthetic precursor of salvileucalin C, whose bicyclo[3,2,0]hept-6-ene core structure would be formed through bicyclization of the cycloheptadiene subunit.66 Inspired by this hypothesis, Ding and co-workers achieved the first total synthesis of salvileucalin C (174) via a late-stage photo-induced electrocyclic ring contraction (Scheme 24).67 Their synthesis began with triflate 169, which was prepared from silyloxyfuran 167 and aldehyde 168 in nine steps. Subjection of 169 to a one-pot diastereoselective Stille coupling/debromination/lactonization process delivered monobromide 172 in 69% yield via the intermediacy of 171, together with 12-epi-172 13% yield. Exposure of 172 to aqueous NaOH in the presence of Et3BnNCl led to ring-expansion and gave salvileucalin D (173) in 80% yield. Upon UV irradiation (λ = 365 nm), the expected 4π electrocyclization took place smoothly and afforded 174 in 84% yield.
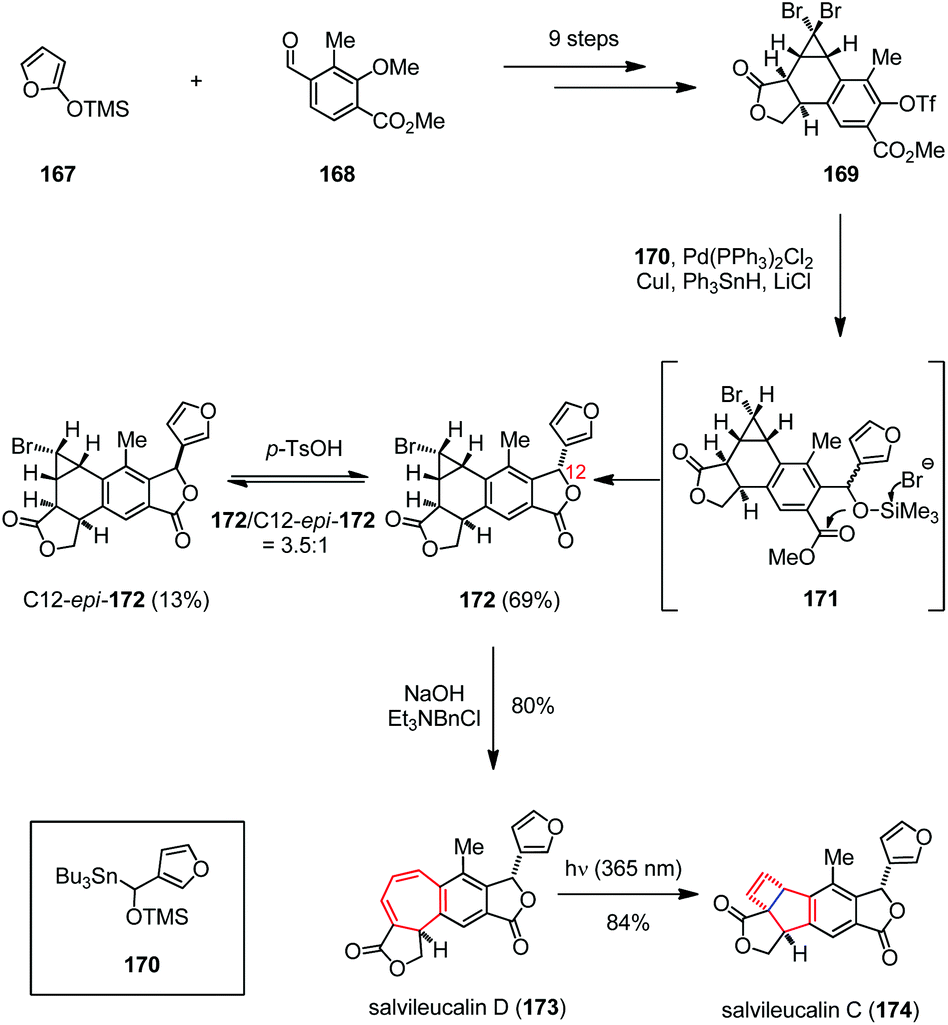 | ||
| Scheme 24 4π electrocyclization in the total synthesis of salvileucalin C. | ||
Later, Li and co-workers applied a conceptually similar strategy in their total syntheses of colchicine (178) and its derivatives (Scheme 25).68 The highly toxic tricyclic alkaloid colchicine was the first tubulin-destabilizing agent reported in the literature.69 Structurally, (−)-178 contains a single stereocenter at C-7, an aR-configured chiral axis and a highly oxidized tropolone ring. The synthesis commenced with 176, which was prepared with excellent ee from 175 by using an intramolecular oxidopyrylium-mediated [5 + 2] cycloaddition as the key step.70 Subsequently, a three-step sequence involving a diastereoselective reduction, a methylation and a regioselective Wacker oxidation afforded 177 in 53% yield. Double elimination of the oxa-bridge in 177 proceeded smoothly in the presence of TMSOTf and Me2NEt, providing (−)-178 in 81% yield with >99% ee.
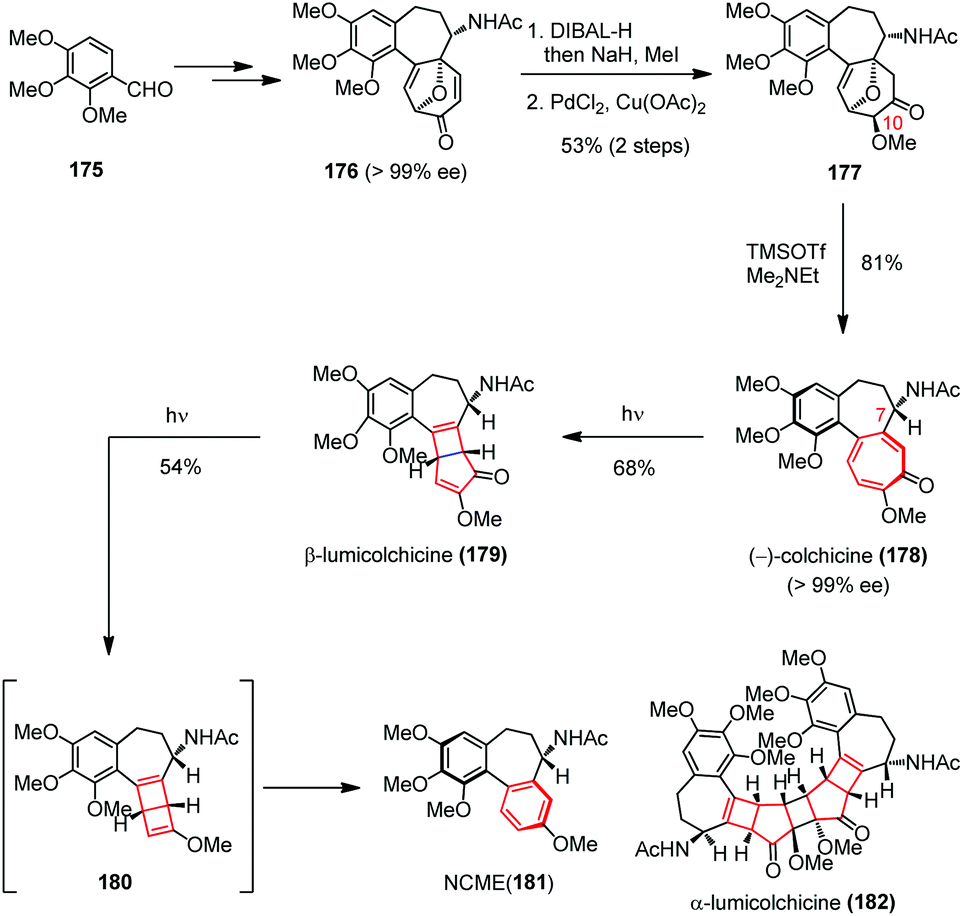 | ||
| Scheme 25 4π electrocyclization in the total synthesis of colchicine derivatives. | ||
The biomimetic conversion of (−)-178 to β-lumicolchicine (179), NCME (181) and α-lumicolchicine (182) was then investigated. Upon irradiating a solution of (−)-178 in CH3CN/acetone with a Pyrex water jacket surrounded high-pressure mercury lamp (125 W) for 25 min, the expected ring contraction occurred and afforded 179 in 68% yield. Repeating this procedure with the isolated 179 for 20 min led to a rapid decarbonylation and generated intermediate 180, which was then transformed to 181 instead of 182via a retro-4π electrocyclization.
Thermal or photo-induced electrocyclic reactions of conjugated tetraenes with 8π systems can readily provide diverse bicyclic scaffolds.71 Such operations need to be rationally incorporated into a synthesis from the beginning, given that the researchers should choose from among multiple ring-closure reactions and take into account torque and chemoselectivity in the electrocyclization steps.72 The Nicolaou group first applied an 8π–6π cascade electrocyclization in their landmark biomimetic syntheses of endiandric acids.73 The above work has inspired other groups a lot to build up related polycyclic skeletons. Kingianins were isolated from the bark of Endiandra kingiana (Lauraceae) and show promising activity against the anti-apoptotic protein Bcl-xL (Fig. 2).74 Retrosynthetically, their unique pentacyclic architecture was proposed to arise from thermal 8π–6π electrocyclization cascade and Diels–Alder dimerization.
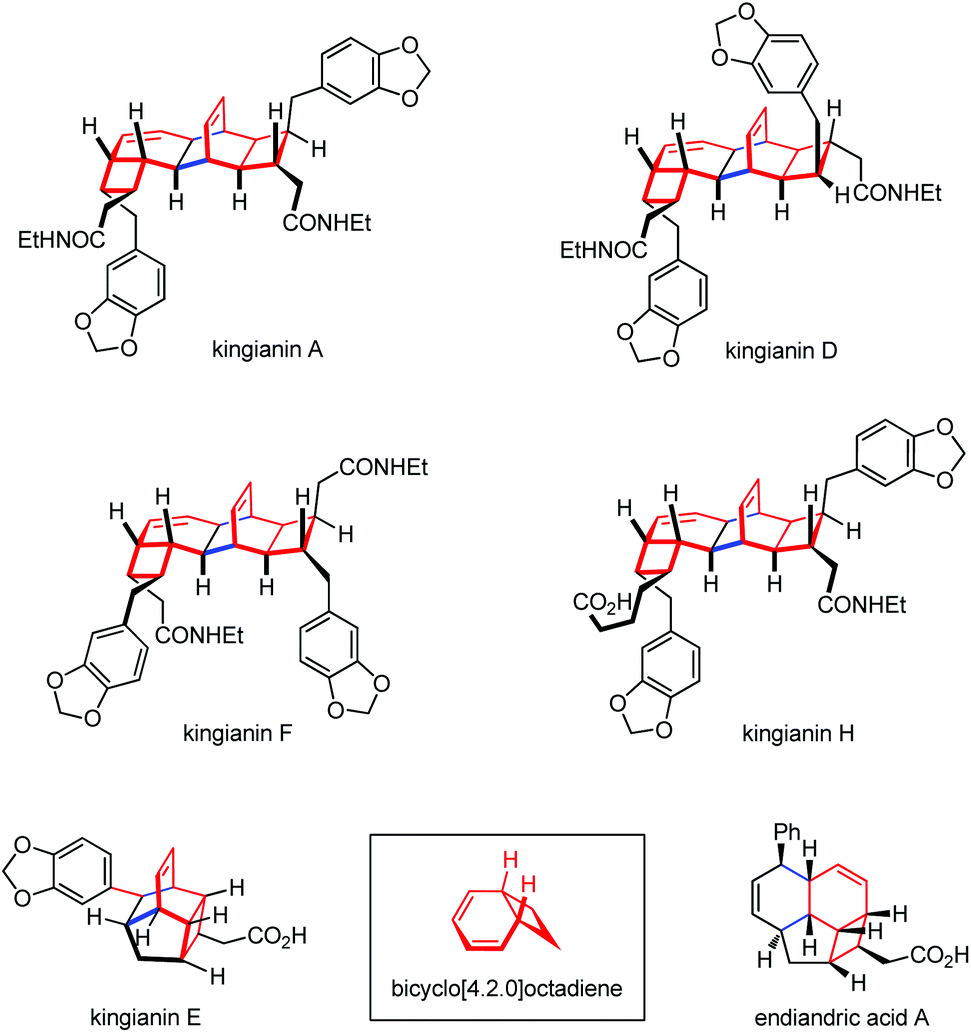 | ||
| Fig. 2 Structures of representative members of the kingianin family and endiandric acid A. | ||
In 2013, Sherburn, Lawrence and co-workers reported the total syntheses of (±)-kingianins A (190), D (191) and F (192) in a longest linear sequence (LLS) of ten steps based on a highly divergent biomimetic strategy (Scheme 26).75 The unsymmetrical tetrayne 185 was readily prepared from the alcohol 183 and 184 in six steps on a multi-gram scale. Reduction of 185 with Rieke zinc in ethanol afforded (Z,Z,Z,Z)-tetraene 186 in a completely chemoselective and highly diastereoselective manner. The latter was then immediately heated to 100 °C in toluene to bring about the expected 8π–6π electrocyclization cascade. After desilylation, alcohols 187 and 188 were obtained as a 1:1 mixture of diastereomers in 21% combined yield. Subsequent oxidation of 187 with TPAP/NMO followed by Ledwith–Weitz aminium salt (189) promoted Diels–Alder dimerization to provide a mixture of diacids, which were directly converted into the corresponding diamides and achieved the syntheses of (±)-190 and (±)-191. On the other hand, (±)-192 was also obtained through dimerization of 188 followed by double oxidation and diamide formation.
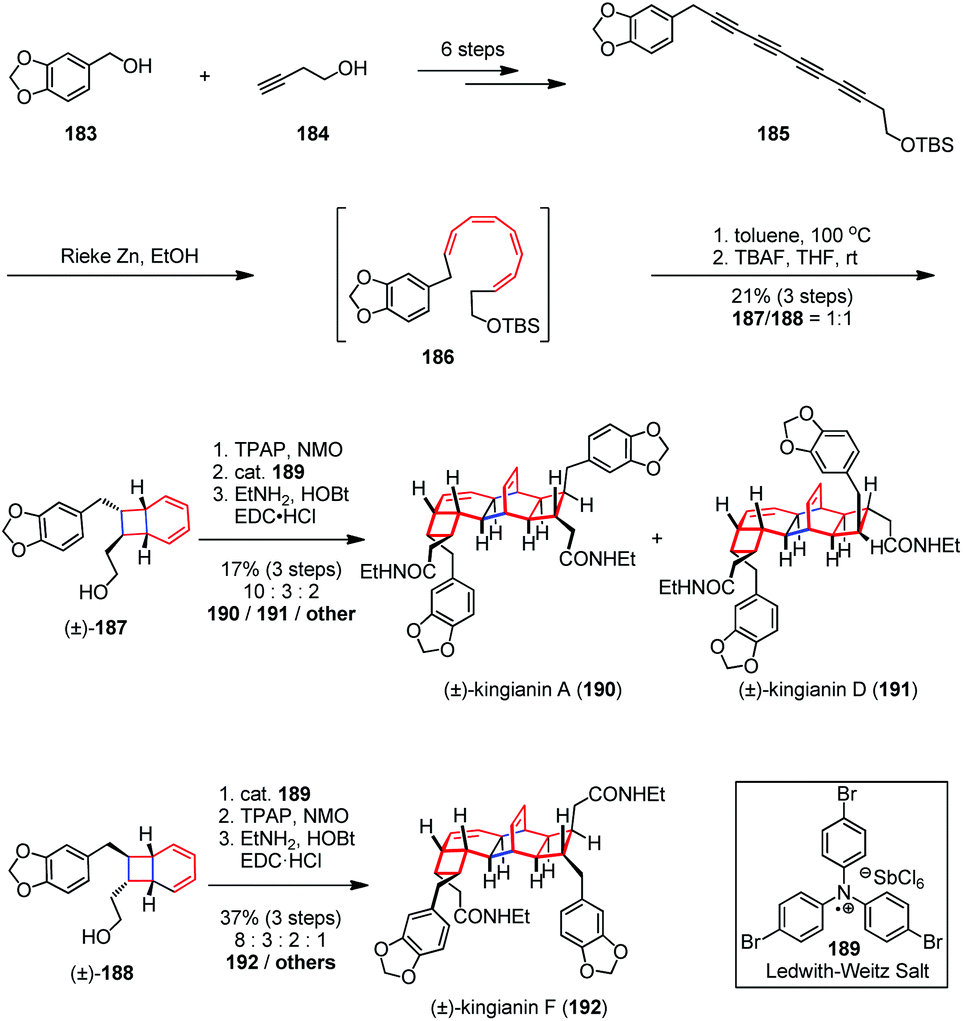 | ||
| Scheme 26 8π–6π electrocyclization cascade in the total synthesis of kingianins. | ||
4.2 Rearrangement
In addition to the concerted electrocyclic processes mentioned above, the migration of a C–C bond between two vicinal atoms has also been extensively investigated when embedding the four-membered rings into complex molecules through ring contraction. These migrations could be initiated by an electrophilic reagent or through a highly reactive neutral intermediate in order to overcome the reaction barriers arising from ring strains.Under various conditions, the α-diazo ketone would lose its nitrogen and undergo 1,2-shift to yield a ketene intermediate, a process known as the Wolff rearrangement.76 Although the mechanism of this rearrangement is still under debate,77 it can effectively generate the ring-strained systems where other approaches fail. (+)-Psiguadial B, a diformyl phloroglucinol meroterpenoid, was isolated from the leaves of Psidium guajava and found to inhibit proliferation of HepG2 human hepatoma cancer cells.78 This natural compound possesses a compact bicyclo[4.3.1]decane core fused to a trans-cyclobutane ring, which was probably derived from the biotransformation of β-caryophyllene. In 2016, Reisman and co-workers proposed a synthetic strategy toward (+)-psiguadial B (202) featuring de novo construction of the trans-fused cyclobutane via a Wolff rearrangement/asymmetric ketene addition cascade, followed by a Pd-catalyzed C(sp3)–H alkenylation reaction (Scheme 27).79 The synthesis started by irradiating a mixture of the known diazoketone 193 and 8-aminoquinoline 194 with UV light (254 nm) in the presence of 10 mol% (+)-cinchonine, providing cyclobutane 195 with 62% yield and up to 79% ee. A solution of 195 and vinyl iodide 196 (isolated as an 8:1 mixture of olefin isomers) was then subjected to Pd(OAc)2 and Ag2CO3 at 90 °C and furnished the cis-fused cyclobutane 197 in 72% yield. Reduction of 197 using Schwartz's reagent followed by treatment with KOH in methanol gave the desired trans-aldehyde 198 in 70% yield over two steps. The corresponding aldehyde was telescoped with Wittig olefination and hydrolysis to afford vinyl enone 200 in 88% yield. Subsequently, the copper-catalyzed conjugate addition under Alexakis’ conditions delivered the desired ketone 201 in 94% yield (19:1 dr), which was finally converted to (+)-202 within a few steps.
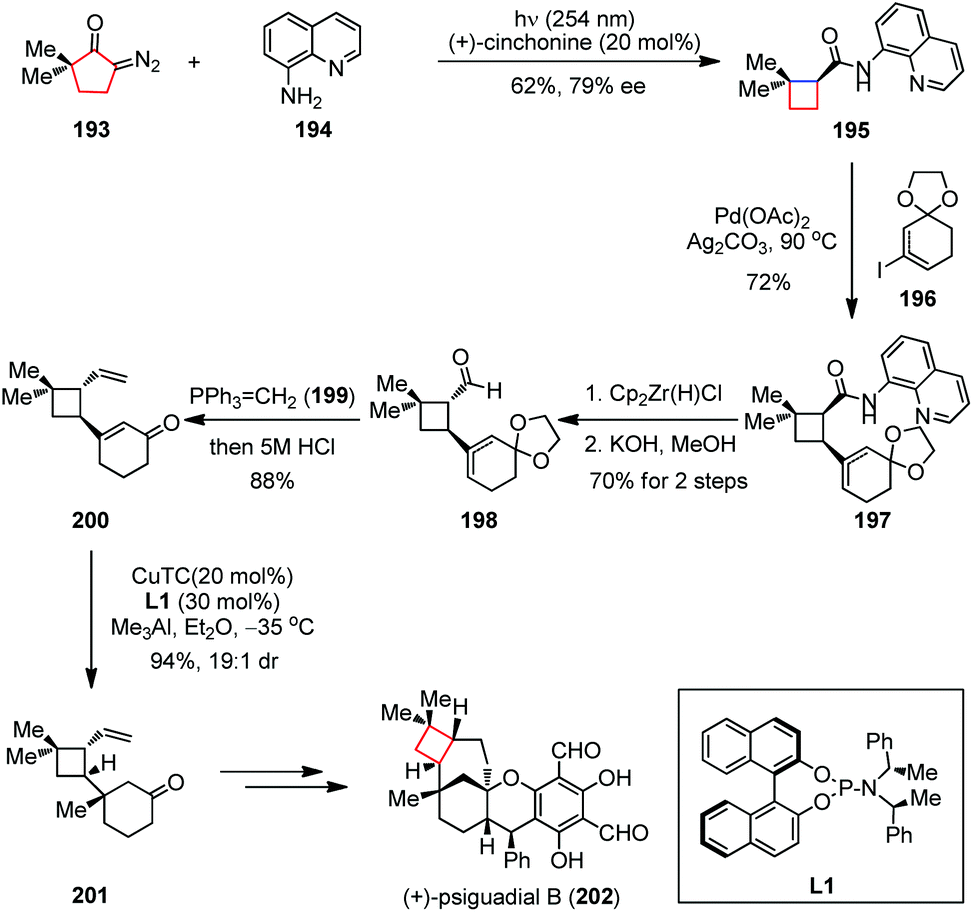 | ||
| Scheme 27 Wolff rearrangement/asymmetric ketene addition cascade in the total synthesis of (+)-psiguadial B. | ||
In 2016, Zhang and Gu reported the total synthesis of aquatolide (111) by using a Wolff ring contraction to build up the cyclobutane subunit and an intramolecular NHK coupling to forge the medium sized ring (Scheme 28).80 Their synthesis commenced with the known bromo ester 204, which was readily prepared from 2,5-norbornadiene (203) on a multigram scale.81 After being converted to 205, a two-step dihydroxylation/Swern process followed by condensation with tosyl hydrazine and detosylation delivered diazo 207 in 79% yield. Irradiation of the latter with a high-pressure mercury lamp (125 W) in the presence of NaHCO3 gave the desired cyclobutane 208 with 80% yield and a satisfactory dr value. Subsequent incorporation of functionalities led uneventfully to aldehyde 209, which underwent an intramolecular NHK cyclization followed by Swern oxidation to afford the eight-membered enone and thus completed the synthesis of 111.
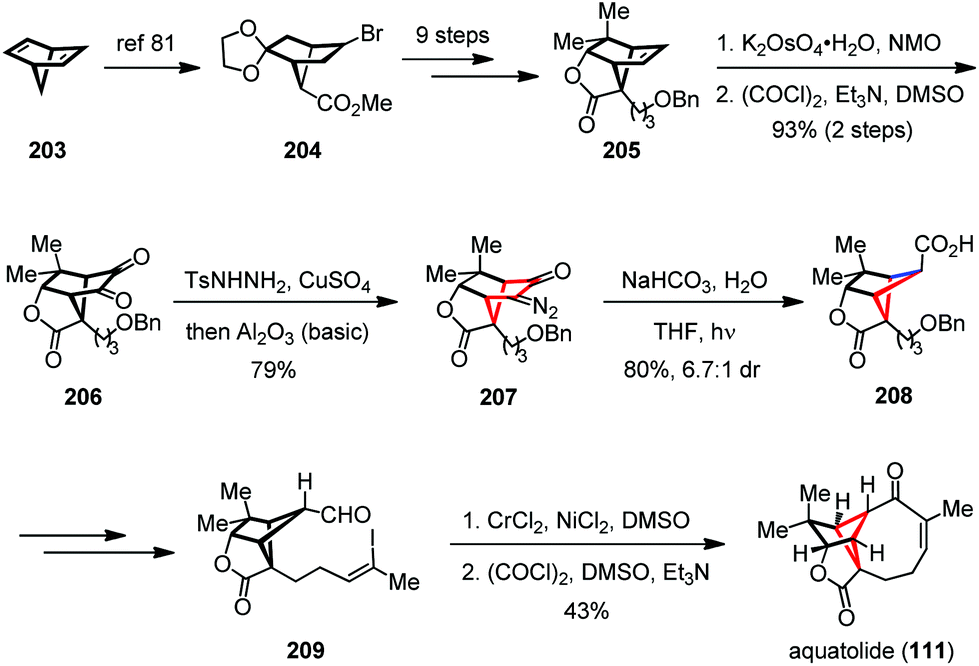 | ||
| Scheme 28 Wollf rearrangement in the total synthesis of aquatolide. | ||
Aplydactone a brominated sesquiterpenoid was isolated from the sea hare Aplysia dactylomela. It contains a novel [2]-ladderane moiety with two fused cyclobutanes connected to a cis-decalin system, which bears four quaternary centers and a secondary neopentyl bromide.82 In 2016, the Trauner group described an intriguing approach that relied on two photochemical key-steps to establish the two cyclobutane rings in the total synthesis of aplydactone (216).83 As depicted in Scheme 29, the cyclopentenone 211 was readily prepared from the known ester 210, which then upon irradiation with a high-pressure mercury lamp (150 W) gave the photochemical [2 + 2] cycloadduct 212 with decent yield and excellent diastereoselectivity. After being converted to enaminone 213, the addition of p-ABSA led to α-diazo ketone 214 in 61% yield. The expected Wolff rearrangement proceeded smoothly and generated the ladderane 215 and its C3-epimer as a 3:1 mixture in 77% combined yield. Late-stage core modification of 215 then completed the total synthesis of 216. In 2017, the Zhang group developed an alternative synthetic route to this molecule, in which the [2]-ladderane core was also installed readily by the same ring-contraction strategy at the early stage.84 Remarkably, the two fused four-membered scaffolds remained intact in continuous ring contraction courses, providing a new perspective on the inherent ring strain in cyclobutanes.
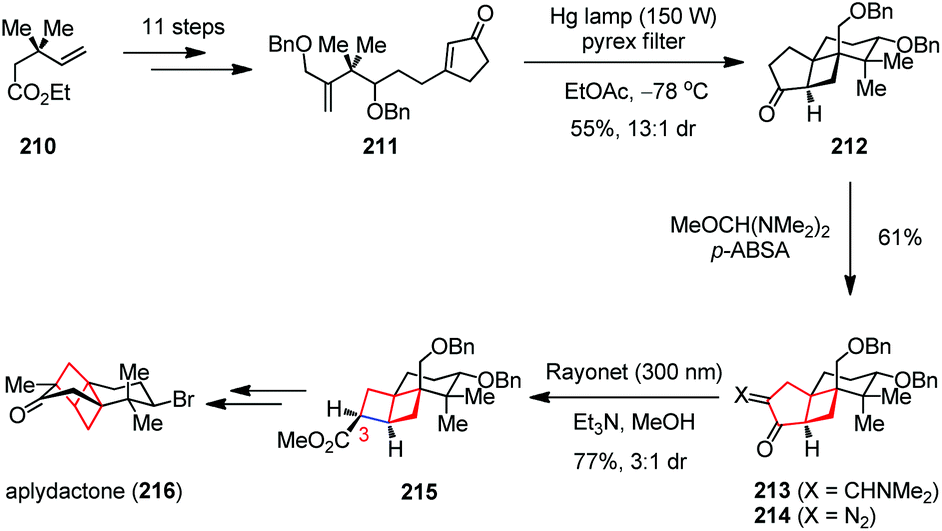 | ||
| Scheme 29 Wolff rearrangement in the total synthesis of aplydactone. | ||
Ladderane lipids are produced by anaerobic ammonium oxidizing (anammox) bacteria as a significant fraction of their membrane lipids. Ladderane phospholipid 227, isolated from anammox enrichment cultures, comprises a mixture containing [5]-ladderane tail as in acid 226 and [3]-laddrane tail as in alcohol 225.85 Attracted by the interesting ladder-like structures and potential bioactivities, Gonzalez-Martinez, Boxer, Burns and co-workers reported a highly selective total synthesis of the ladderane lipid tails 225 and 226, as well as a full phosphatidycholine to render further biophysical studies on these molecules (Scheme 30).86 Their synthesis commenced with the preparation of the key building block 219. The α-chlorocyclosulfoxide 218 was obtained from readily available diol 217 as a mixture of diastereomers in 89% yield over three steps. Treatment of 218 with excess t-BuOK effected an atypical sulfoxide Ramberg–Bäcklund ring contraction, providing olefin 219 with 49% yield. The latter was subjected to dimerization upon UV irradiation in the presence of CuOTf to afford [5]-ladderane core 220. A manganese-catalyzed C–H chlorination and elimination delivered 224. Then, an additional five-step transformation produced the [5]-ladderanoic acid 225 enantioselectively in 72% overall yield. On the other hand, the photocycloaddition between 219 and chiral dibromobenzoquinone 221 followed by selective elimination through proton abstraction from the convex face afforded vinyl bromide 223 in 73% yield over two steps. Further elaboration of the latter gave [3]-ladderanol 226 in 41% overall yield. Coupling of the two ladderane tails by a short sequence ultimately furnished ladderane phospholipid 227.
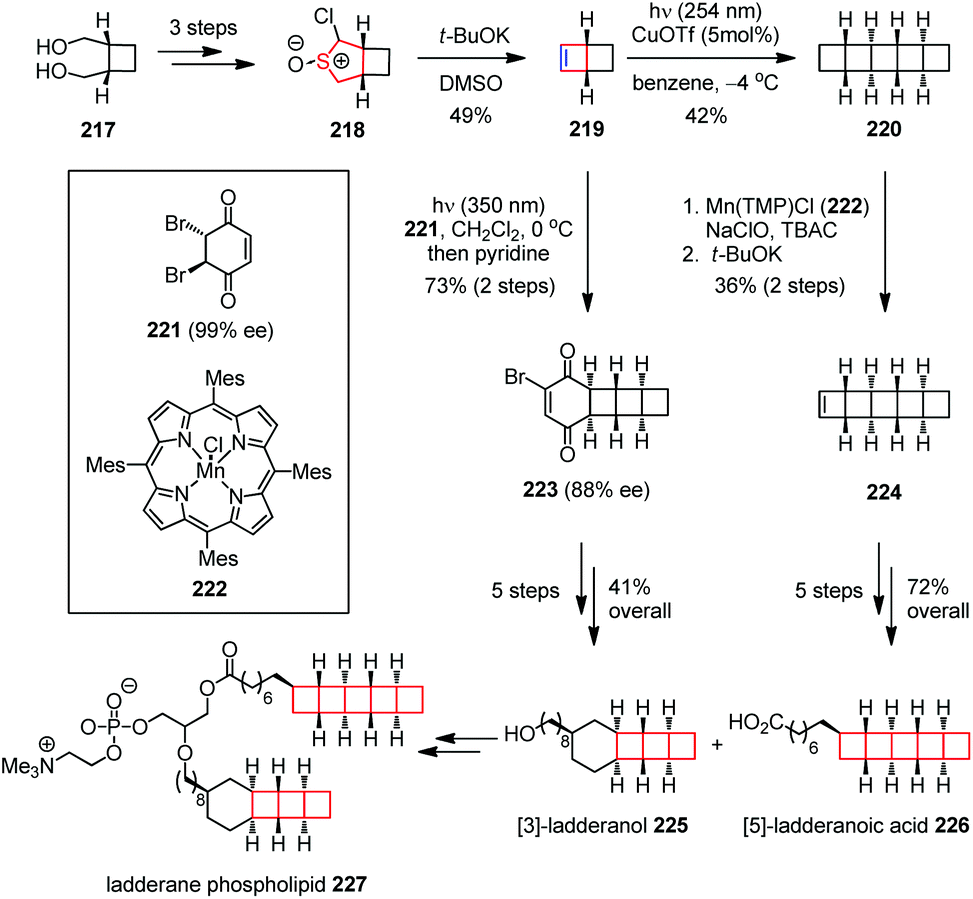 | ||
| Scheme 30 Ramberg–Bäcklund ring contraction in the total synthesis of ladderane phospholipid. | ||
Hapalindole-type natural products were discovered from soil samples in a myriad of habitats around the globe and exhibit potent anticancer and insecticidal activities.87 In 2008, Baran and co-workers accomplished the total syntheses of welwitindolinones and other hapalindole-type alkaloids (Scheme 31).88 Their synthesis towards welwitindolinone A (233) began with the multi-step conversion of carvone (228) to 12-epi-fisherindole I (229). Subsequently, a Pinacol-type oxidative ring contraction process delivered 233 in 25% yield. This transformation most likely proceeds through chlorination of 229 in the presence of t-BuOCl and Et3N to give 230, which was then transformed to 231 upon attack of water. Further elimination of chloride generated 232, followed by [1,5]-sigmatropic rearrangement to install the spirocyclobutane skeleton of 233. Given that fluorohydroxylation of indole rather than chlorohydroxylation should suppress the formation of isonitrile-derived byproducts, a milder approach was developed. Thus, by treatment of indole 229 with a solution of XeF2 in wet MeCN, the desired 233 was obtained in 44% yield via the intermediacy of 234.
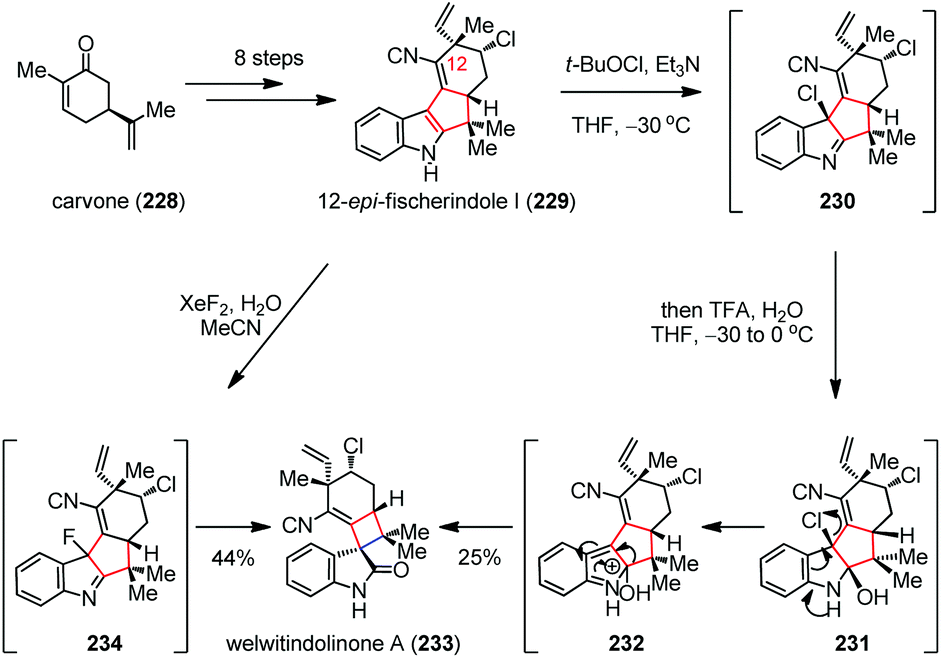 | ||
| Scheme 31 Pinacol-type oxidative ring contraction in the total synthesis of welwitindolinone A. | ||
Besides, the 1,3-acyl migration (known as the Givens rearrangement) has also been utilized for the construction of cyclobutanes in natural product synthesis.89 (+)-Armillarivin was isolated from Armillaria tabescens, a pathogenic basidiomycete that causes root disease in several commercially significant plants.90 In 2013, the group of Banwell completed the synthesis of (+)-armillarvin (241) in 20 steps from the enantiopure cis-1,2-dihydrocatechol 235.91 As shown in Scheme 32, their synthesis commenced with a high-pressure Diels–Alder cycloaddition between 235 and 236, affording 237 in 70% yield. After being converted to 238, the pivotal photochemical Givens rearrangement occurred by UV irradiation. Upon ketone reduction, cyclobutanol 239 was formed as a single isomer in 18% yield over two steps. Coupling of compound 239 with readily prepared acid 240 in the presence of DCC and DMAP provided the ester, which was then oxidized directly to (+)-241 through treatment with the in situ generated oxammonium salt via p-TsOH-induced disproportionation of 4-acetamido-TEMPO.
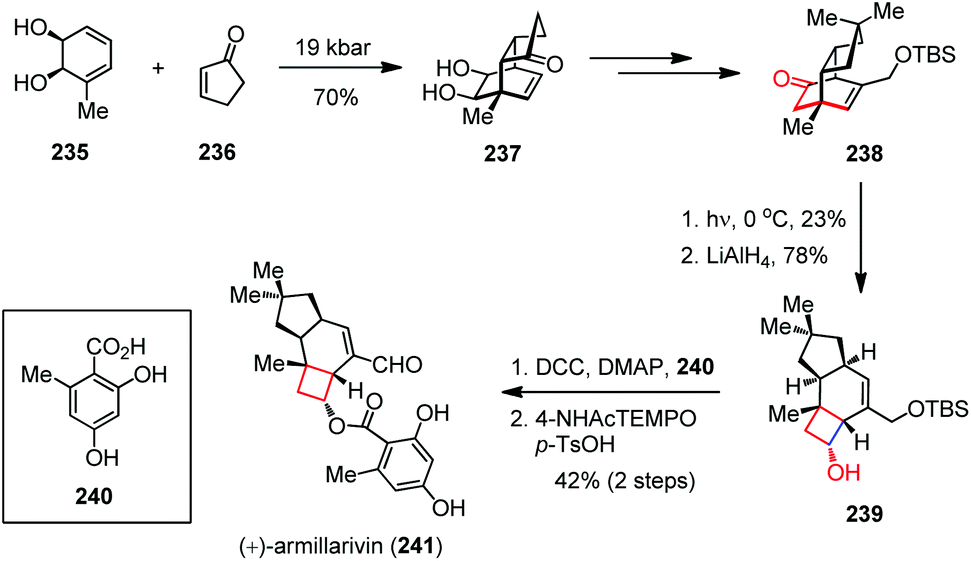 | ||
| Scheme 32 1,3-Acyl migration in the total synthesis of armillarivin. | ||
(+)-Fomannosin is the pathogen of Fomes annonsus, which was grown from still cultures of the wood-destroying Basidiomycetes fungus by Bassett and co-workers in 1967.92 It is a sesquiterpene metabolite and can cause the death of host cells prior to hyphal invasion. In 2008, Paquette and co-workers disclosed an enantiodivergent strategy for the syntheses of both naturally occurring (+)-fomannosin (249) and its (−)-antipode (Scheme 33).93 The synthesis started with the known tosylate 242, which was readily available from D-glucose. After being transformed to the advanced intermediate 243, the zirconocene-mediated ring contraction afforded a 2.4:1 diastereomeric mixture of 247 and 248 in 85% combined yield. The product distribution rests upon consideration of the ring-closure transition states. The adoption of 246 is less favored owing to the existing steric interactions between the methylene group of the developing cyclobutane and the allylic methylene group α to the zirconium, as well as the dipole–dipole interactions between the oxygen atoms resident in PMB and TBDPS ethers. Thus, 248 was formed as the less dominant product. Late-stage elaboration of 247 and 248 led to (+)- and (−)-249, respectively.
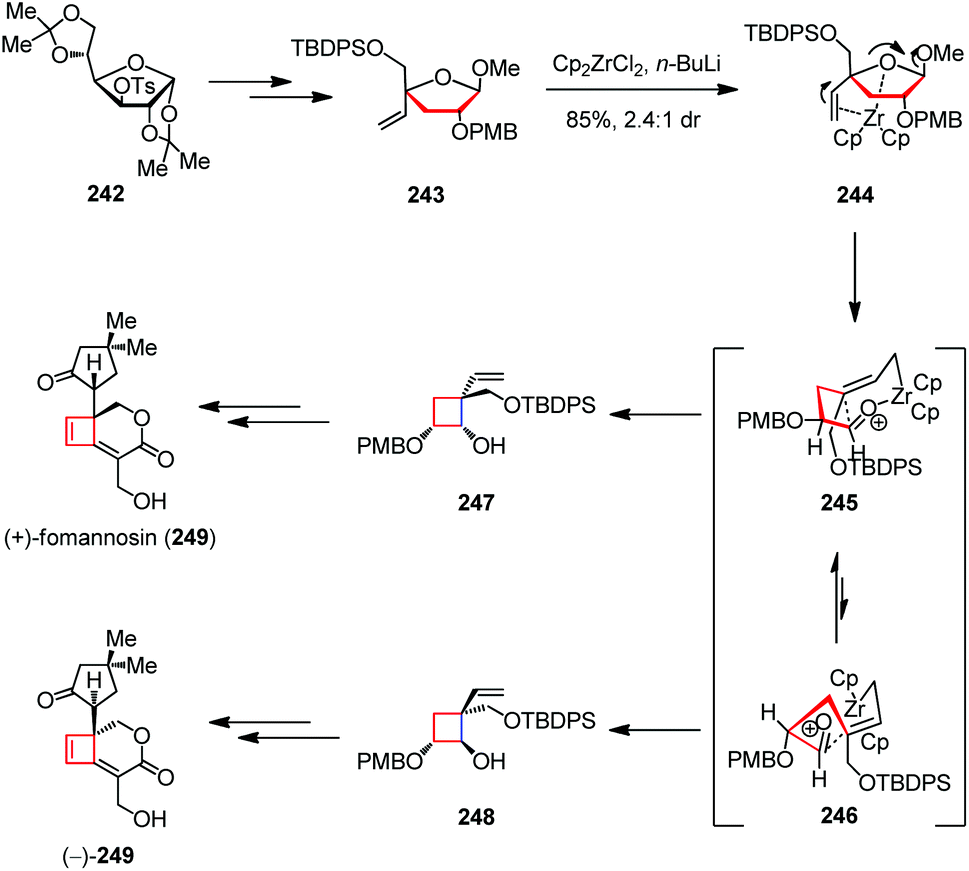 | ||
| Scheme 33 Zirconium-mediated ring contraction in the total syntheses of (+)-fomannosin and its antipode. | ||
5. Ring expansion strategies
It makes sense to examine possible routes to the cyclobutane ring systems through not only contraction of larger rings but also expansion of cyclopropanes due to their higher strain energies. Such ring expansion has rarely been explored presumably because the resultant cyclobutane intermediates are normally unstable under the strain-releasing conditions.Pipercyclobutanamides, piperchabamides, nigramides and dipiperamides are tetrasubstituted cyclobutanes that are isolated from Piper nigrum and Piper chaba.60 These natural cyclobutanes have a broad range of pharmacological activities. Among them, pipercyclobutanamide A and dipiperamide E are selective inhibitors against CYP2D6 and CYP3A4, respectively, and piperchabamide G was found to have a hepatoprotective effect and inhibit D-GalN/tumor necrosis factor-α-induced death of hepatocytes. In 2012, Tang and co-workers developed a new general strategy for the diastereo- and enantioselective introduction of four different substituents to a cyclobutane ring, and realized the total syntheses of the proposed structures of pipercyclobutanamide A (256) and piperchabamide G (258).94 As depicted in Scheme 34, the synthesis began with the preparation of ketoester 251 from the monoprotected diol 250 in six steps. Treatment of 251 with TsNHNH2 and DBU gave diazo 252 in 60% yield, which then underwent a ring expansion by subjection to catalytic AgOTf, affording cyclobutenoate 253 in 95% yield. After being converted to 254, the proposed structure of pipercyclobutanamide A (256) was obtained in 50% yield over two steps through reduction of the ester followed by olefination of the resultant aldehyde with Ando's reagent 255. On the other hand, a hydrogenation/reduction/olefination sequence afforded the proposed structure of piperchabamide G (258) in 38% yield over three steps.
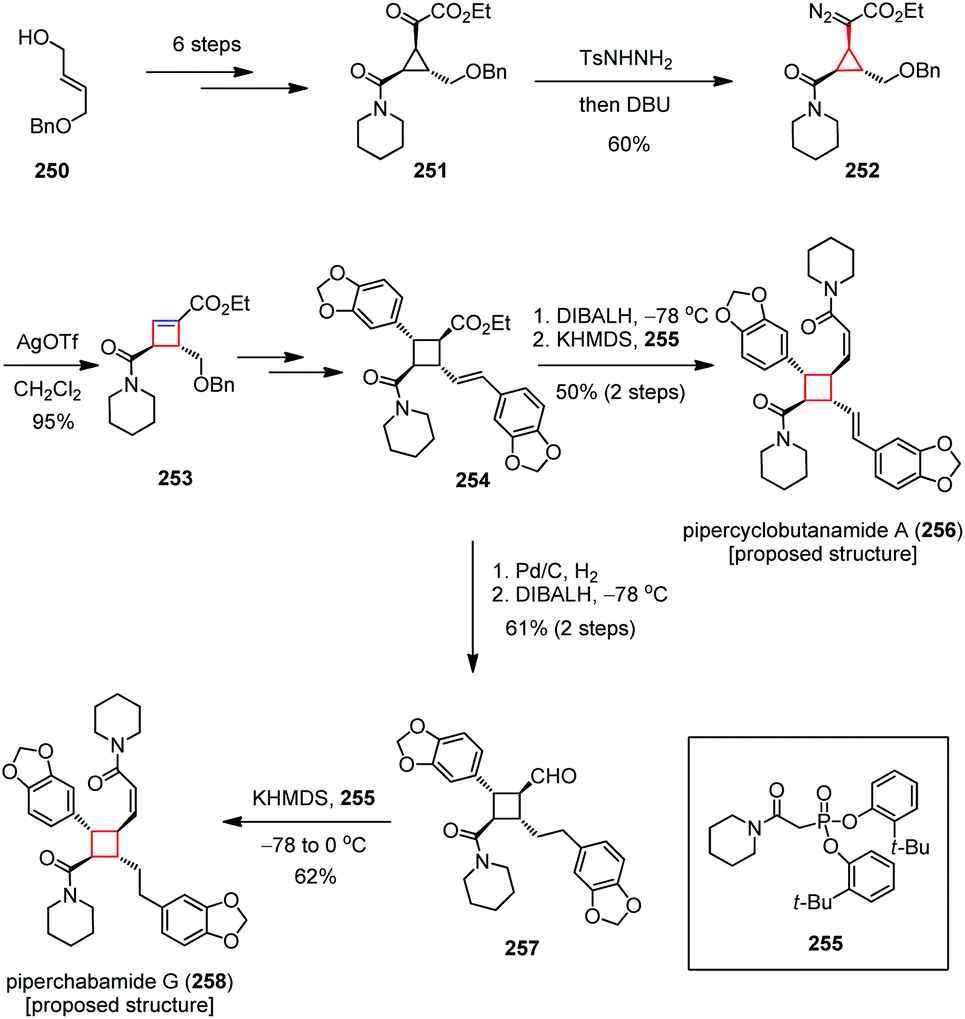 | ||
| Scheme 34 Ring expansion in the total syntheses of the proposed structures of pipercyclobutanamide A and piperchabamide G. | ||
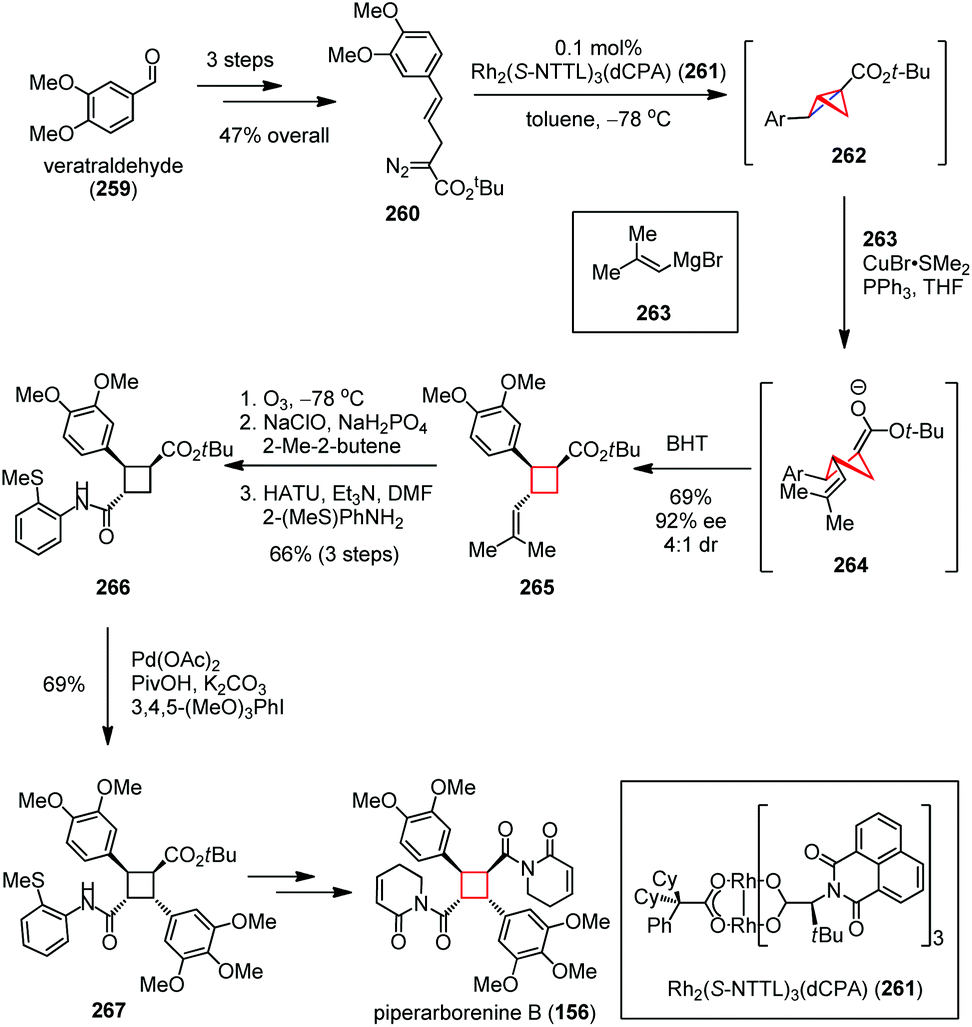
In 2016, Fox and co-workers reported an elegant total synthesis of piperarborenine B (156) featuring a bicyclobutanation/homoconjugate addition cascade to construct its trisubstituted cyclobutane core structure (Scheme 35).95 The synthesis commenced with the preparation of diazoester 260 from veratraldehyde (259) in 47% yield over three steps. Exposure of 260 to 0.1 mol% Rh2(S-NTTL)3(dCPA) (261) afforded a bicyclobutane intermediate 262, followed by copper-mediated nucleophilic homo addition of Grignard 263 and kinetic protonation using BHT to afford the enantiomerically enriched cyclobutane 265 in 69% yield (92% ee, 4:1 dr). The latter was then converted to amide 266 in 66% yield over three steps through oxidative cleavage, Pinnick oxidation and amide formation. Subsequent Pd-catalyzed C(sp3)–H functionalization delivered the densely substituted cyclobutane 267 in high yield, which could be forwarded to 156 within several conventional steps.
 | ||
| Scheme 35 Bicyclobutanation/homoconjugate addition cascade in the total syntheses of piperarborenine B. | ||
6. Conclusions
In conclusion, this review has summarized the recent advances in the total syntheses of cyclobutane-containing natural products. The rapid generation of molecular complexity in a relatively diverse manner has made the construction of cyclobutane skeletons a highly useful tool, which was highlighted with the latest progress in various strategies including direct ring closure, cycloaddition, ring contraction and ring expansion. As demonstrated in these examples, the structures of the substituted cyclobutane core are remarkably stable in a series of strain-releasing reactions, particularly in comparison with their smaller homologues. On the other hand, the biomimetic syntheses have proven to be elegant protocols for the construction of highly complex fused cyclobutane frameworks.With the deeper understanding of the cyclobutane chemistry and the development of novel synthetic methodologies, we believe that new disconnection strategies would certainly emerge and be successfully applied to the total syntheses of more structurally complex and biologically important cyclobutane-containing natural products.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the Zhejiang Natural Science Fund for Distinguished Young Scholars (LR16B020001) and the National Natural Science Foundation of China (21871230 and 21622205).References
- For a recent review on the chemistry of strained ring systems, see: (a) G. Fumagalli, S. Stanton and J. F. Bower, Chem. Rev., 2017, 117, 9404–9432 CrossRef CAS. For reviews on the chemistry of cyclopropanes, see: (b) D. J. Mack and J. T. Njardarson, ACS Catal., 2013, 3, 272–286 CrossRef CAS; (c) D. Qian and J. Zhang, Chem. Soc. Rev., 2015, 44, 677–698 RSC; (d) A. P. Thankachan, K. S. Sindhu, K. K. Krishnan and G. Anilkumar, Org. Biomol. Chem., 2015, 13, 8780–8802 RSC; (e) C. Ebner and E. M. Carreira, Chem. Rev., 2017, 117, 11651–11679 CrossRef CAS; (f) M. Bos, T. Poisson, X. Pannecoucke, A. B. Charette and P. Jubault, Chem. – Eur. J., 2017, 23, 4950–4961 CrossRef CAS; (g) W.-Q. Wu, Z.-M. Lin and H.-F. Jiang, Org. Biomol. Chem., 2018, 16, 7315–7329 RSC.
- A. L. Ringer and D. H. Magers, J. Org. Chem., 2007, 72, 2533–2537 CrossRef CAS.
- For recent reviews on the chemistry of oxetanes, see: (a) J. A. Burkhard, G. Wuitschik, M. Rogers-Evans, K. Mϋller and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 9052–9067 CrossRef CAS; (b) A. Mahal, Eur. J. Chem., 2015, 6, 357–366 CrossRef CAS; (c) J. A. Bull, R. A. Croft, O. A. Davis, R. Doran and K. Morgan, Chem. Rev., 2016, 116, 12150–12233 CrossRef CAS. For recent reviews on the chemistry of azetidines, see: (d) A. Brandi, S. Cicchi and F. M. Cordero, Chem. Rev., 2008, 108, 3988–4035 CrossRef CAS; (e) D. Antermite, L. Degennaro and R. Luisi, Org. Biomol. Chem., 2017, 15, 34–50 RSC; (f) D. Didier, A. N. Baumann and M. Eisold, Tetrahedron Lett., 2018, 59, 3975–3987 CrossRef CAS. For other related reviews, see: (g) E. M. Carreira and T. C. Fessard, Chem. Rev., 2014, 114, 8257–8322 CrossRef CAS; (h) D. Hazelard and P. Compain, Org. Biomol. Chem., 2017, 15, 3806–3827 RSC.
- (a) V. M. Dembitsky, Phytomedicine, 2014, 21, 1559–1581 CrossRef CAS; (b) Y.-Y. Fan, X.-H. Gao and J.-M. Yue, Sci. China: Chem., 2016, 59, 1126–1141 CrossRef CAS; (c) M. A. Beniddir, L. Evanno, D. Joseph, A. Skiredj and E. Poupon, Nat. Prod. Rep., 2016, 33, 820–842 RSC.
- (a) T. Seiser, T. Saget, D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2011, 50, 7740–7752 CrossRef CAS; (b) M. Luparia, D. Audisio and N. Maulide, Synlett, 2011, 735–740 CAS; (c) A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Chem. Rev., 2014, 114, 7317–7420 CrossRef CAS; (d) C. M. Rasik and K. M. Brown, Synlett, 2014, 760–765 CAS; (e) Y. Xu, M. L. Conner and K. M. Brown, Angew. Chem., Int. Ed., 2015, 54, 11918–11928 CrossRef CAS; (f) M. Wang and P. Lu, Org. Chem. Front., 2018, 5, 254–259 RSC; (g) E. N. Hancock, J. M. Wiest and M. K. Brown, Nat. Prod. Rep., 2019, 36, 1383–1393 RSC.
- For selected examples on the Favorskii rearrangements employed in the construction of cyclobutanes, see: (a) L. M. Zhang and M. Koreeda, Org. Lett., 2002, 4, 3755–3758 CrossRef CAS; (b) M. harmata and S. Wacharasindhu, Org. Lett., 2005, 7, 2563–2565 CrossRef CAS.
- For a representative review on the application of the 1,2-carbon atom migration strategies for construction of cyclobutanes, see: X.-M. Zhang, Y.-Q. Tu, F.-M. Zhang, Z.-H. Chen and S.-H. Wang, Chem. Soc. Rev., 2017, 46, 2272–2305 RSC.
- J. G. Mulder, P. Diepenhorst, P. Plieger and I. E. M. Bruggemann-Rotgans, PCT Int. Appl, WO1993002083A1, 1993 Search PubMed.
- K. Tanino, M. Takahashi, Y. Tomata, H. Tokura, T. Uehara, T. Narabu and M. Miyashita, Nat. Chem., 2011, 3, 484–488 CrossRef CAS.
- (a) C. J. Bulpitt, Y. Li, P. F. Bulpitt and J. Wang, J. R. Soc. Med., 2007, 100, 558–563 CrossRef PubMed; (b) S. Shailajan, S. Kumaria, D. Gurjar, M. Joshi, P. Paul and N. Khongthaw, J. Appl. Pharm. Sci., 2015, 5, 32–38 CrossRef CAS.
- W.-W. Fan, F.-Q. Xu, F.-W. Dong, X.-N. Li, Y. Li, Y.-Q. Liu, J. Zhou and J.-M. Hu, Nat. Prod. Bioprospect., 2013, 3, 89–92 CrossRef CAS.
- H. Wolleb and E. M. Carreira, Angew. Chem., Int. Ed., 2017, 56, 10890–10893 CrossRef CAS.
- T. Komada, M. Adachi and T. Nishikawa, Chem. Lett., 2012, 41, 287–289 CrossRef CAS.
- H. Y. Chuang and M. Isobe, Org. Lett., 2014, 16, 4166–4169 CrossRef CAS.
- T. M. Baker, D. J. Edmonds, D. Hamilton, C. J. O'Brien and D. J. Procter, Angew. Chem., Int. Ed., 2008, 47, 5631–5633 CrossRef CAS.
- M. J. Martín, R. Fernández, A. Francesch, P. Amade, S. S. de Matos-Pita, F. Reyes and C. Cuevas, Org. Lett., 2010, 12, 912–914 CrossRef.
- M. Gao, Y.-C. Wang, K.-R. Yang, W. He, X.-L. Yang and Z.-J. Yao, Angew. Chem., Int. Ed., 2018, 57, 13313–13318 CrossRef CAS.
- (a) I. H. Sadler and T. J. Simpson, J. Chem. Soc., Chem. Commun., 1989, 1602–1604 RSC; (b) I. H. Sadler and T. J. Simpson, Magn. Reson. Chem., 1992, 30, s18–s23 CrossRef CAS.
- A. C. Huang, S. A. Kautsar, Y. J. Hong, M. H. Medema, A. D. Bond, D. J. Tantillo and A. Osbourn, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6005–6014 CrossRef.
- (a) N. Zhao, S.-Q. Yin, S.-L. Xie, H. Yan, P. Ren, G. Chen, F. Chen and J. Xu, Angew. Chem., Int. Ed., 2018, 57, 3386–3390 CrossRef CAS PubMed; (b) N. Zhao, S.-L. Xie, P.-L. Tian, R.-B. Tong, C.-Q. Ning and J. Xu, Org. Chem. Front., 2019, 6, 2014–2022 RSC.
- (a) W. A. Nugent and T. V. RajanBabu, J. Am. Chem. Soc., 1988, 110, 8561–8562 CrossRef CAS; (b) T. V. RajanBabu and W. A. Nugent, J. Am. Chem. Soc., 1989, 111, 4525–4527 CrossRef CAS; (c) T. V. RajanBabu and W. A. Nugent, J. Am. Chem. Soc., 1994, 116, 986–997 CrossRef CAS.
- (a) D. P. G. Hamon and K. L. Tuck, J. Org. Chem., 2000, 65, 7839–7846 CrossRef CAS; (b) I. Petschen, M. P. Bosch and A. Guerrero, Tetrahedron: Asymmetry, 2000, 11, 1691–1695 CrossRef CAS; (c) A. M. Bernard, A. Frongia, F. Secci, G. Delogu, J. Ollivier, P. P. Piras and J. Salaün, Tetrahedron, 2003, 59, 9433–9440 CrossRef CAS.
- A. Gansäuer, A. Greb, I. Huth, D. Worgull and K. Knebel, Tetrahedron, 2009, 65, 10791–10796 CrossRef.
- H. J. Knölker, G. Baum, O. Schmitt and G. Wanzl, Chem. Commun., 1999, 1737–1738 RSC.
- (a) J. Yang, Y. Chen, X. Feng, D. Yu and X. Liang, Planta Med., 1984, 50, 288–290 CrossRef CAS; (b) M. Bohnert, S. Miethbauer, H.-M. Dahse, J. Ziemen, M. Nett and D. Hoffmeister, Bioorg. Med. Chem. Lett., 2011, 21, 2003–2006 CrossRef CAS.
- M. T. Hovey, D. T. Cohen, D. M. Walden, P. H.-Y. Cheong and K. A. Scheidt, Angew. Chem., Int. Ed., 2017, 56, 9864–9867 CrossRef CAS.
- For selected reviews, see: (a) R. A. Yoder and J. N. Johnston, Chem. Rev., 2005, 105, 4730–4756 CrossRef CAS; (b) C. N. Ungarean, E. H. Southgate and D. Sarlah, Org. Biomol. Chem., 2016, 14, 5454–5467 RSC; (c) A. G. M. Barrett, T. Ma and T. Mies, Synthesis, 2019, 67–82 CAS.
- K. W. Tsao, C. Y. Cheng and M. Isobe, Org. Lett., 2012, 14, 5274–5277 CrossRef CAS.
- (a) J. A. Katzenellenbogen, Science, 1976, 194, 139–148 CrossRef CAS; (b) G. Rosini and E. Marotta, Tetrahedron, 1985, 41, 4633–4638 CrossRef CAS; (c) G. Rosini, E. Marotta, A. Raimondi and P. Righi, Tetrahedron: Asymmetry, 1991, 2, 123–138 CrossRef CAS.
- Y. T. Han, N.-J. Kim, J.-W. Jung, H. Yun, S. Lee and Y.-G. Suh, Arch. Pharmacal Res., 2011, 34, 1437–1442 CrossRef CAS.
- K. Mori and K. Fukumatsu, Liebigs Ann. Chem., 1992, 489–493 CrossRef CAS.
- J.-X. Xie, J.-C. Wang and G.-B. Dong, Org. Lett., 2017, 19, 3017–3020 CrossRef CAS.
- Y.-Y. Fan, H. Zhang, Y. Zhou, H.-B. Liu, W. Tang, B. Zhou, J.-P. Zuo and J.-M. Yue, J. Am. Chem. Soc., 2015, 137, 138–141 CrossRef CAS.
- T. J. A. Graham, E. E. Gray, J. M. Burgess and B. C. Goess, J. Org. Chem., 2010, 75, 226–228 CrossRef CAS.
- (a) M. Hirota, Y. Shimizu, T. Kamo, H. Makabe and H. Shibata, Biosci., Biotechnol., Biochem., 2003, 67, 1597–1600 CrossRef CAS; (b) M. Kashiwabara, T. Kamo, H. Makabe, H. Shibata and M. Hirota, Biosci., Biotechnol., Biochem., 2006, 70, 1502–1505 CrossRef CAS.
- S. Ferrer and A. M. Echavarren, Org. Lett., 2018, 20, 5784–5788 CrossRef CAS.
- (a) P. Lu and T. Bach, Angew. Chem., Int. Ed., 2012, 51, 1261–1264 CrossRef CAS; (b) S. Poplata, A. Tröster, Y.-Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748–9815 CrossRef CAS.
- V. Ramamurthy and J. Sivaguru, Chem. Rev., 2016, 116, 9914–9993 CrossRef CAS.
- (a) S.-J. Piao, H.-W. Lin, H. Lu, X.-F. Liu and W.-S. Chen, CNA 102603689, Shanghai Changzheng Hospital, P. R. China, 2012 Search PubMed; (b) S.-J. Piao, Y.-L. Song, W.-H. Jiao, F. Yang, X.-F. Liu, W.-S. Chen, B.-N. Han and H.-W. Lin, Org. Lett., 2013, 15, 3526–3529 CrossRef CAS.
- S. A. Ruider, T. Sandmeier and E. M. Carreira, Angew. Chem., Int. Ed., 2015, 54, 2378–2382 CrossRef CAS.
- Q.-G. Li, K. Zhao, A. Peuronen, K. Rissanen, D. Enders and Y.-F. Tang, J. Am. Chem. Soc., 2018, 140, 1937–1944 CrossRef CAS.
- J. Marrero, A. D. Rodríguez, P. Baran, R. G. Raptis, J. A. Sánchez, E. Ortega-Barria and T. L. Capson, Org. Lett., 2004, 6, 1661–1664 CrossRef CAS.
- For synthetic studies, see: (a) B. Doroh and G. A. Sulikowski, Org. Lett., 2006, 8, 903–906 CrossRef CAS PubMed; (b) R. Maio, S. G. Gramani and M. J. Lear, Tetrahedron Lett., 2009, 50, 1731–1733 CrossRef; (c) A. Jana, S. Mondal, M. F. Hossain and S. Ghosh, Tetrahedron Lett., 2012, 53, 6830–6833 CrossRef CAS; (d) S. D. Townsend and G. A. Sulikowski, Org. Lett., 2013, 15, 5096–5098 CrossRef CAS PubMed; (e) A. Jana, S. Mondal and S. Ghosh, Org. Biomol. Chem., 2015, 13, 1846–1859 RSC.
- K. C. Nicolaou, V. A. Adsool and C. R. H. Hale, Angew. Chem., Int. Ed., 2011, 50, 5149–5152 CrossRef CAS.
- P. Scesa, M. Wangpaichitr, N. Savaraj, L. M. West and S. P. Roche, Angew. Chem., Int. Ed., 2018, 57, 1316–1321 CrossRef CAS.
- (a) A. San Feliciano, M. Medarde, J. M. Miguel del Corral, A. Aramburu, M. Gordaliza and A. F. Barrero, Tetrahedron Lett., 1989, 30, 2851–2854 CrossRef CAS; (b) M. W. Lodewyk, C. Soldi, P. B. Jones, M. M. Olmstead, J. Rita, J. T. Shaw and D. J. Tantillo, J. Am. Chem. Soc., 2012, 134, 18550–18553 CrossRef CAS.
- K. Takao, H. Kai, A. Yamada, Y. Fukushima, D. Komatsu, A. Ogura and K. Yoshida, Angew. Chem., Int. Ed., 2019, 58, 9851–9855 CrossRef CAS.
- K. Wei, W. Li, K. Koike, Y. Chen and T. Nikaido, J. Org. Chem., 2005, 70, 1164–1176 CrossRef CAS.
- I. Colomer, R. C. Barcelos and T. J. Donohoe, Angew. Chem., Int. Ed., 2016, 55, 4748–4752 CrossRef CAS.
- A. Al-Mourabit, M. A. Zancanella, S. Tilvi and D. Romo, Nat. Prod. Rep., 2011, 28, 1229–1260 RSC.
- Z.-Q. Ma, X.-L. Wang, X. Wang, R. A. Rodriguez, C. E. Moore, S.-H. Gao, X.-H. Tan, Y.-Y. Ma, A. L. Rheingold, P. S. Baran and C. Chen, Science, 2014, 346, 219–224 CrossRef CAS.
- (a) N. Duchemin, A. Skiredj, J. Mansot, K. Leblanc, J.-J. Vasseur, M. A. Beniddir, L. Evanno, E. Poupon, M. Smietana and S. Arseniyadis, Angew. Chem., Int. Ed., 2018, 57, 11786–11791 CrossRef CAS; (b) E. M. Boyd and J. J. Sperry, Chem. N. Z., 2010, 74, 109–112 CAS; (c) J. Dai, J. I. Jimenez, M. Kelly and P. G. Williams, J. Org. Chem., 2010, 75, 2399–2402 CrossRef CAS.
- G.-H. Huang, Z. Hu, C. Lei, P.-P. Wang, J. Yang, J.-Y. Li, J. Li and A.-J. Hou, J. Nat. Prod., 2018, 81, 1810–1818 CrossRef CAS.
- J. D. Hart, L. Burchill, A. J. Day, C. G. Newton, C. J. Sumby, D. M. Huang and J. H. George, Angew. Chem., Int. Ed., 2019, 58, 2791–2794 CrossRef CAS.
- M. E. McCallum, C. M. Rasik, J. L. Wood and M. K. Brown, J. Am. Chem. Soc., 2016, 138, 2437–2442 CrossRef CAS.
- K. Takasu, Isr. J. Chem., 2016, 56, 488–498 CrossRef CAS.
- Y. Yoshii, T. Otsu, N. Hosokawa, K. Takasu, K. Okano and H. Tokuyama, Chem. Commun., 2015, 51, 1070–1073 RSC.
- Y. Mogi, K. Inanaga, H. Tokuyama, M. Ihara, Y. Yamaoka, K. Yamada and K. Takasu, Org. Lett., 2019, 21, 3954–3958 CrossRef CAS.
- For reviews on electrocyclic reactions, see: (a) W. R. Dolbier Jr., H. Koroniak, K. N. Houk and C. Sheu, Acc. Chem. Res., 1996, 29, 471–477 CrossRef; (b) C. M. Beaudry, J. P. Malerich and D. Trauner, Chem. Rev., 2005, 105, 4757–4778 CrossRef CAS; (c) S. Thompson, A. G. Coyne, P. C. Knipe and M. D. Smith, Chem. Soc. Rev., 2011, 40, 4217–4231 RSC; (d) N. S. Sheikh, Org. Biomol. Chem., 2015, 13, 10774–10796 RSC; (e) M. Bian, L. Li and H. Ding, Synthesis, 2017, 4383–4413 CrossRef CAS.
- (a) F. P. Lee, T. C. Chen, J. J. Chen, I. L. Tsai and I. S. Chen, Helv. Chim. Acta, 2004, 87, 463–468 CrossRef CAS; (b) I.-L. Tsai, F.-P. Lee, C.-C. Wu, C.-Y. Duh, T. Ishikawa, J.-J. Chen, Y.-C. Chen, H. Seki and I.-S. Chen, Planta Med., 2005, 71, 535–542 CrossRef CAS.
- (a) W. R. Gutekunst and P. S. Baran, J. Am. Chem. Soc., 2011, 133, 19076–19079 CrossRef CAS; (b) W. R. Gutekunst and P. S. Baran, J. Org. Chem., 2014, 79, 2430–2452 CrossRef CAS.
- (a) F. Frébault, M. Luparia, M. T. Oliveira, R. Goddard and N. Maulide, Angew. Chem., Int. Ed., 2010, 49, 5672–5676 CrossRef; (b) E. J. Corey and J. Streith, J. Am. Chem. Soc., 1964, 86, 950–951 CrossRef CAS.
- N. Winter and D. Trauner, J. Am. Chem. Soc., 2017, 139, 11706–11709 CrossRef CAS.
- W. G. Dauben, K. Koch, S. L. Smith and O. L. Chapman, J. Am. Chem. Soc., 1963, 85, 2616–2621 CrossRef CAS.
- Y. Aoyagi, A. Yamazaki, R. Kato, F. Tobe, H. Fukaya, T. Nishikawa, A. Nakahashi, N. Miura, K. Monde and K. Takeya, Tetrahedron Lett., 2011, 52, 1851–1853 CrossRef CAS.
- R. A. Hill and A. Sutherland, Nat. Prod. Rep., 2011, 28, 1031–1034 RSC.
- C. Fu, Y. Zhang, J. Xuan, C. Zhu, B. Wang and H. Ding, Org. Lett., 2014, 16, 3376–3379 CrossRef CAS.
- X. Liu, Y.-J. Hu, B. Chen, L. Min, X.-S. Peng, J. Zhao, S.-P. Li, H. N. C. Wong and C.-C. Li, Org. Lett., 2017, 19, 4612–4615 CrossRef CAS.
- (a) O. Boye and A. Brossi, in The Alkaloids, ed. A. Brossi and G. A. Cordell, Academic Press, San Diego, 1992, vol. 41, pp. 125–176 Search PubMed; (b) C. Le Hello, in The Alkaloids, ed. G. A. Cordell, Academic Press, San Diego, 2000, vol. 53, pp. 287–352 Search PubMed; (c) R. B. G. Ravelli, B. Gigant, P. A. Curmi, I. Jourdain, S. Lachkar, A. Sobel and M. Knossow, Nature, 2004, 428, 198–202 CrossRef CAS; (d) B. Bhattacharyya, D. Panda, S. Gupta and M. Banerjee, Med. Res. Rev., 2008, 28, 155–183 CrossRef CAS.
- (a) X. Liu, J.-Y. Liu, J. Zhao, S. Li and C.-C. Li, Org. Lett., 2017, 19, 2742–2745 CrossRef CAS; (b) J.-Y. Liu, j. L. Wu, J.-H. Fan, X. Yan, G.-J. Mei and C.-C. Li, J. Am. Chem. Soc., 2018, 140, 5365–5369 CrossRef CAS.
- (a) R. Huisgen, A. Dahmen and H. Huber, J. Am. Chem. Soc., 1967, 89, 7130–7131 CrossRef CAS; (b) E. N. Marvell, J. Seubert, G. Vogt, G. Zimmer, G. Moy and J. R. Siegmann, Tetrahedron, 1978, 34, 1307–1322 CrossRef; (c) J. E. Moses, J. E. Baldwin, R. M. Adlington, A. R. Cowleyb and R. Marqueza, Tetrahedron Lett., 2003, 44, 6625–6627 CrossRef CAS; (d) A. K. Milker and D. Trauner, Angew. Chem., Int. Ed., 2005, 44, 4602–4606 CrossRef.
- (a) G. Pohnert and W. Boland, Tetrahedron, 1994, 50, 10235–10244 CrossRef CAS; (b) G. Pohnert and W. Boland, Nat. Prod. Rep., 2002, 19, 108–122 RSC.
- (a) K. C. Nicolaou, N. A. Petasis, R. E. Zipkin and J. Uenishi, J. Am. Chem. Soc., 1982, 104, 5555–5557 CrossRef CAS; (b) K. C. Nicolaou, N. A. Petasis, J. Uenishi and R. E. Zipkin, J. Am. Chem. Soc., 1982, 104, 5557–5558 CrossRef CAS; (c) K. C. Nicolaou, R. E. Zipkin and N. A. Petasis, J. Am. Chem. Soc., 1982, 104, 5558–5560 CrossRef CAS; (d) K. C. Nicolaou, R. E. Zipkin and N. A. Petasis, J. Am. Chem. Soc., 1982, 104, 5560–5562 CrossRef CAS.
- (a) A. Leverrier, K. Awang, F. Guéritte and M. Litaudon, Phytochemistry, 2011, 72, 1443–1452 CrossRef CAS; (b) A. Leverrier, M. E. Tran huu Dau, P. Retailleau, K. Awang, F. Guéritte and M. Litaudon, Org. Lett., 2012, 12, 3638–3641 CrossRef.
- S. L. Drew, A. L. Lawrence and M. S. Sherburn, Angew. Chem., Int. Ed., 2013, 52, 4221–4224 CrossRef CAS.
- W. Kirmse, Eur. J. Org. Chem., 2002, 2193–2256 CrossRef CAS.
- G. B. Gill, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Flemming, Pergamon Press, 1991, vol. 3, pp. 887–912 Search PubMed.
- M. Shao, Y. Wang, Z. Liu, D.-M. Zhang, H.-H. Cao, R.-W. Jiang, C.-L. Fan, X.-Q. Zhang, H.-R. Chen, X.-S. Yao and W.-C. Ye, Org. Lett., 2010, 12, 5040–5043 CrossRef CAS PubMed.
- L. M. Chapman, J. C. Beck, L. Wu and S. E. Reisman, J. Am. Chem. Soc., 2016, 138, 9803–9806 CrossRef CAS.
- B. Wang, Y. Xie, Q. Yang, G. Zhang and Z. Gu, Org. Lett., 2016, 18, 5388–5391 CrossRef CAS.
- (a) R. Peel and J. K. Sutherland, J. Chem. Soc., Chem. Commun., 1974, 151–153 RSC; (b) P. A. Grieco, C. S. Pogonowski, S. D. Burke, M. Nishizawa, M. Miyashita, Y. Masaki, C.-L. J. Wang and G. Majetich, J. Am. Chem. Soc., 1977, 99, 4111–4118 CrossRef CAS.
- S. N. Fedorov, O. S. Radchenko, L. K. Shubina, A. I. Kalinovsky, A. V. Gerasimenko, D. Y. Popov and V. A. Stonik, J. Am. Chem. Soc., 2001, 123, 504–505 CrossRef CAS.
- R. Meier and D. Trauner, Angew. Chem., Int. Ed., 2016, 55, 11251–11255 CrossRef CAS.
- C.-G. Liu, R.-Z. Chen, Y. Shen, Z.-H. Liang, Y.-H. Hua and Y.-D. Zhang, Angew. Chem., Int. Ed., 2017, 56, 8187–8190 CrossRef CAS.
- J. S. S. Damsté, M. Strous, W. I. C. Rijpstra, E. C. Hopmans, J. A. J. Geevebasen, A. C. T. van Duin, L. A. van Niftrik and M. S. M. Jetten, Nature, 2002, 419, 708–712 CrossRef PubMed.
- (a) J. A. M. Mercer, C. M. Cohen, S. R. Shuken, A. M. Wager, M. W. Smith, F. R. Moss III, M. D. Smith, R. Vahala, A. Gonzalez-Martinez, S. G. Boxer and N. Z. Burns, J. Am. Chem. Soc., 2016, 138, 15845–15848 CrossRef CAS PubMed; (b) F. R. Moss III, S. R. Shuken, J. A. M. mercer, C. M. Cohen, T. M. Weiss, S. G. Boxer and N. Z. Burns, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9098–9103 CrossRef.
- (a) R. E. Moore, C. Cheuk and G. M. L. Patterson, J. Am. Chem. Soc., 1984, 106, 6456–6457 CrossRef CAS; (b) J. I. Jimenez, U. Huber, R. E. Moore and G. M. L. Patterson, J. Nat. Prod., 1999, 62, 569–572 CrossRef CAS; (c) A. Raveh and S. Carmeli, J. Nat. Prod., 2007, 70, 196–201 CrossRef CAS.
- (a) P. S. Baran and J. M. Richter, J. Am. Chem. Soc., 2005, 127, 15394–15396 CrossRef CAS; (b) P. S. Baran, T. J. Maimone and J. M. Richter, Nature, 2007, 446, 404–408 CrossRef CAS; (c) J. M. Richter, Y. Ishihara, T. Masuda, B. W. Whitefield, T. Llamas, A. Pohjakallio and P. S. Baran, J. Am. Chem. Soc., 2008, 130, 17938–17954 CrossRef CAS.
- For a recent review on the Givens rearrangement and related photochemical processes, see: M. G. Banwell and D. J.-Y. D. Bon, in Molecular Rearrangements in Organic Synthesis, ed. C. M. Rojas, Wiley, Hoboken, NJ, 2015, ch. 9, pp. 261–288 Search PubMed.
- (a) J. S. Yang, Y. L. Su, Y. L. Wang, X. Z. Feng, D. Q. Yu and X. T. Liang, Acta Pharmacol. Sin., 1991, 26, 117–122 CAS; (b) P. Cremin, D. M. X. Donnelly, J.-L. Wolfender and K. Hostettmann, J. Chromatogr., A, 1995, 710, 273–285 CrossRef CAS; (c) D. M. X. Donnelly, T. Konishi, O. Dunne and P. Cremin, Phytochemistry, 1997, 44, 1473–1478 CrossRef CAS.
- B. D. Schwartz, E. Matoušová, R. White, M. G. Banwell and A. C. Willis, Org. Lett., 2013, 15, 1934–1937 CrossRef CAS.
- C. Bassett, R. T. Sherwood, J. A. Kepler and P. B. Hamilton, Phytopathology, 1967, 57, 1046–1052 CAS.
- L. A. Paquette, X. Peng, J. Yang and H.-J. Kang, J. Org. Chem., 2008, 73, 4548–4558 CrossRef CAS.
- R.-H. Liu, M. Zhang, T. P. Wyche, G. N. Winston-McPherson, T. S. Bugni and W.-P. Tang, Angew. Chem., Int. Ed., 2012, 51, 7503–7506 CrossRef CAS PubMed.
- R. A. Pansih, S. R. Chintala and J. M. Fox, Angew. Chem., Int. Ed., 2016, 55, 4983–4987 CrossRef PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2020 |