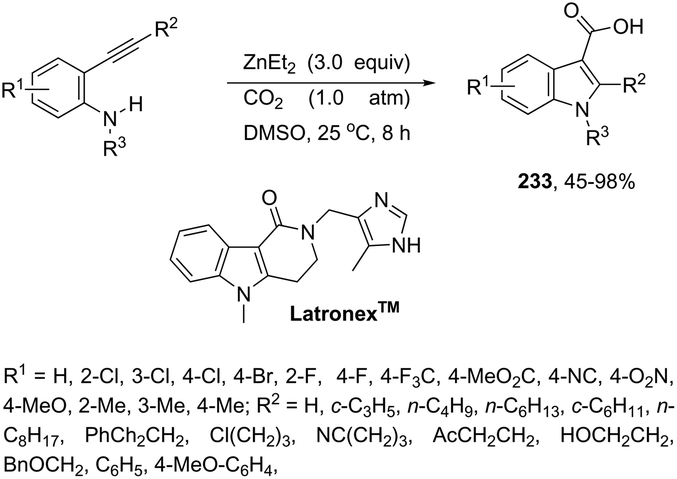
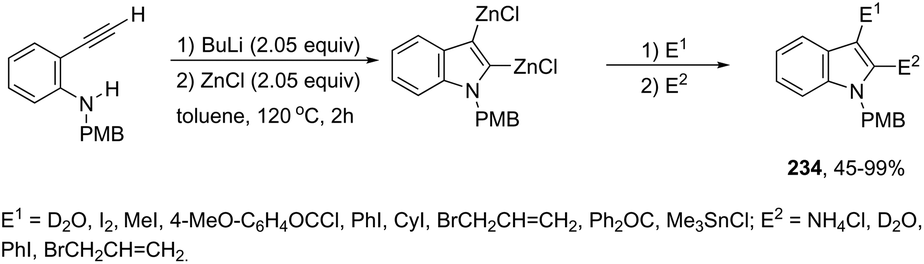
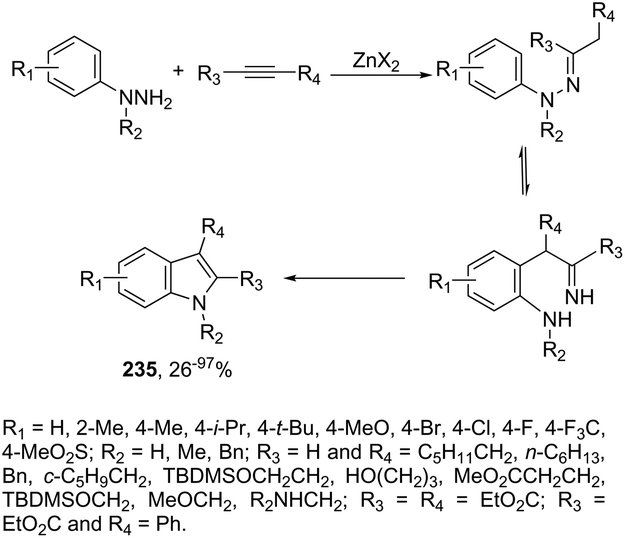
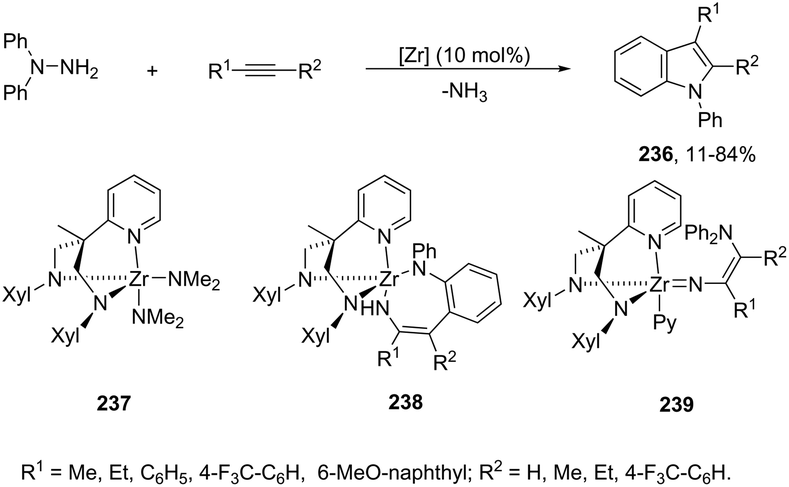
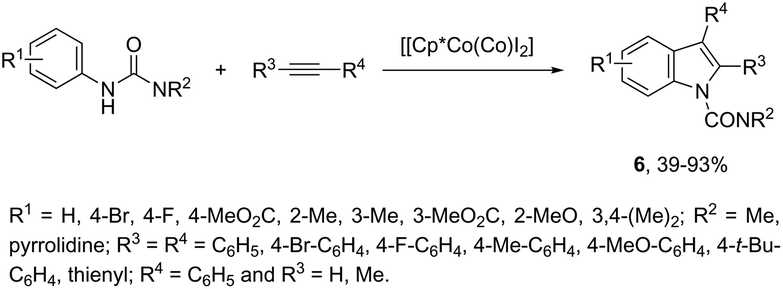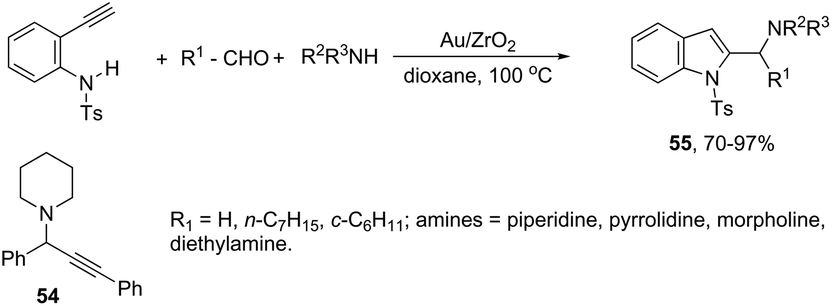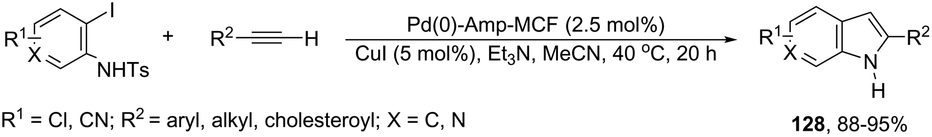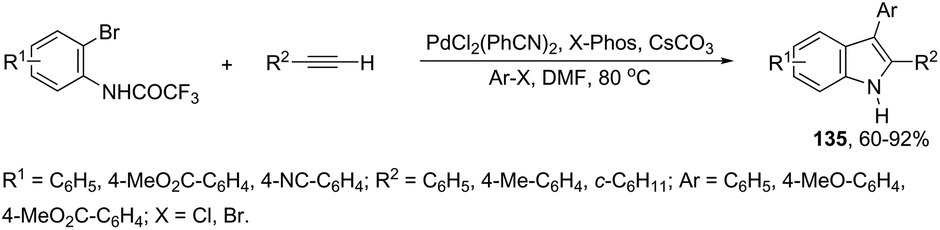DOI:
10.1039/C9QO01315F
(Review Article)
Org. Chem. Front., 2020,
7, 155-210
Recent advances in the synthesis of indoles from alkynes and nitrogen sources
Received
28th October 2019
, Accepted 22nd November 2019
First published on 22nd November 2019
Abstract
Transition metal-catalyzed cyclization reactions of unsaturated substrates have become one of the most important and useful methodologies for the preparation of heterocycles. To this end, the association between alkynes and nitrogen compounds results in versatile substrates for the synthesis of N-heterocycles, including indole derivatives. In order to demonstrate the growth in this area, this review highlights ten years of success in the synthesis of indoles using alkynes and nitrogen sources as substrates in the cyclization reactions catalyzed by metals. We believe that summarizing these methods would be very useful for the chemists who are interested in the synthesis of N-heterocycles.
 José Sebastião Santos Neto | José Neto completed his graduation in Chemistry at the Federal University of Santa Maria (UFSM) in 2010. He obtained his MSc (2012) and Ph.D. (2016) degrees at the UFSM under the supervision of Prof. Gilson Zeni. After a postdoc period at the Federal University of Pelotas under the supervision of Prof. Diego Alves, he joined Prof. Antonio Luiz Braga's group at the Federal University of Santa Catarina, where he is a postdoctoral researcher. His research interests include organochalcogen studies with environmental synthetic methodologies and pharmacological applications. |
 Gilson Zeni | Gilson Zeni received his M.S. degree from the Federal University of Santa Maria-RS-Brazil, working under the direction of Prof. A. L. Braga, and his Ph.D. under the direction of Professor J. V. Comasseto (University of São Paulo). He did his postdoctoral studies at the Iowa State University-USA with Prof. R. C. Larock. He then moved to the Federal University of Santa Maria, where he is now a full professor. The synthesis and reactivity of organochalcogen compounds and the development of new synthetic methods for application of organochalcogen substrates in cyclization reactions are among his main current research interests. |
1. Introduction
Indoles are the most important core structures in many biologically active natural products, exhibiting therapeutic properties over a wide range of targets.1–5 Indole systems exhibit pharmacological activities, and are also present in commercially important products such as agrochemicals, essential oils, cosmetics, flavoring fragrance, dyes, and photosensitizer compounds.6–8 Because they have been studied for more than one hundred years, an impressive number of well-established classical methods are now available for their synthesis.2,9–19 The methodologies for the synthesis of indoles are normally associated with the name of the discoverer, such as Fischer indole synthesis,20 Bischler indole synthesis,21 Reissert indole synthesis,22 Madelung indole synthesis,23 Nenitzescu indole synthesis,24 Sundberg indole synthesis,25 Hemetsberger indole synthesis,26 Gassman indole synthesis,27 Leimgruber–Batcho indole synthesis,28 Julia indole synthesis,29 Bartoli indole synthesis,30 Larock indole synthesis,31 and Fukuyama indole synthesis32 (Scheme 1).33 Among all the existing synthetic methods to obtain indoles, those using alkynes as substrates are particularly attractive ones because of the wide variability in alkyne reactivity, substituent tolerance, chemoselectivity and ability to react with both nucleophiles and electrophiles. The cooperative action between alkynes and nitrogen compounds as substrates in transition metal-catalyzed cyclization reactions is a useful alternative to the synthesis of indoles. We believe that summarizing these methods would be very useful for chemists. Because many advances in the synthesis of these compounds have been published in the last few years, the purpose of this review was to highlight ten years of these achievements. In addition, the number of articles found in the literature on this topic is very large; therefore, to cover as many as possible we summarized the results in equations and tables, avoiding in some cases lengthy discussions. To facilitate the presentation, the review is organized in alphabetical order of the metal used for the cyclization reaction.
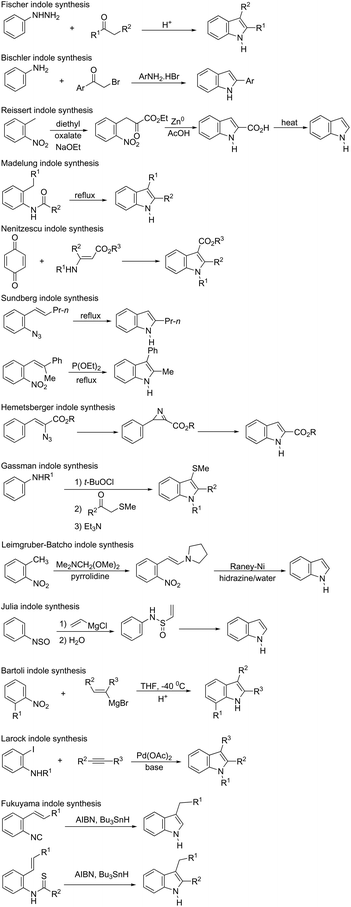 |
| | Scheme 1 | |
2. Transition-metal catalyzed synthesis of indoles
Transition metal-catalyzed activation of alkynes, followed by an intramolecular or intermolecular nucleophile addition, has become a powerful tool for the construction of the indole ring. Alkyne complexes containing bismuth, cobalt, copper, gold, indium, iridium, iron, manganese, mercury, nickel, palladium, platinum, rhenium, ruthenium, scandium, silver, titanium, tungsten, zinc, and zirconium have been employed as active catalysts for the synthesis of indole derivatives. In the next subsections, the utility of these reactions will be reviewed focusing on specific examples that appeared in the literature in the last ten years.
2.1. Bismuth-catalyzed synthesis of indoles
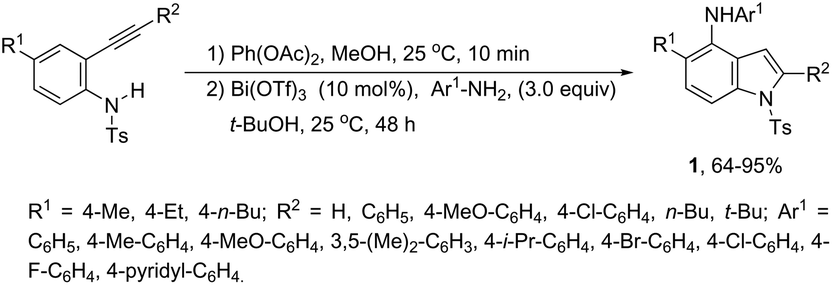
Bismuth salts have been shown to act as suitable catalysts for the cyclization reactions involving alkynes and nitrogen substrates for the preparation of indoles. Thus, the direct addition of NH from anilines across carbon–hydrogen of the 2-alkynylcyclohexadienimines was efficiently catalyzed by a Bi(OTf)3/t-BuOH system, resulting in the formation of N-arylindoles 1 (Scheme 2).34 The required starting 2-alkynylcyclohexadienimines were prepared by oxidative dearomatization reaction promoted by PhI(OAc)2.
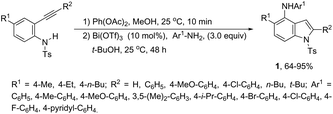 |
| | Scheme 2 | |
2.2. Cobalt-catalyzed synthesis of indoles
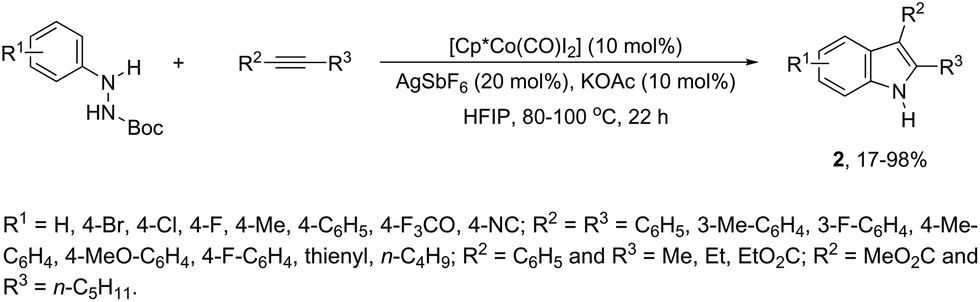
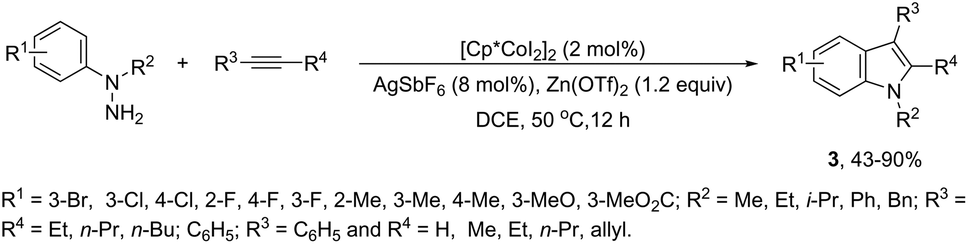
In contrast to palladium, copper, rhodium, and other transition-metal catalysts only a few reports on cobalt-catalyzed reactions of alkynes and nitrogen substrates for the preparation of indoles appeared in the literature in the last decade. In one of these reports, Boc-N-protected hydrazines were utilized as internal oxidizing directing groups for cobalt(III)-catalyzed annulation reaction with symmetrical and unsymmetrical alkynes for the synthesis of unprotected indoles 2 (Scheme 3).35 The authors developed a careful study to determine which hydrazine shows the best activity as a redox-neutral directing group, by comparing acetyl hydrazide, pivaloyl hydrazide, benzoyl hydrazide, and ethyl hydrazinecarboxylate, and Boc-phenylhydrazine led to the formation of products with best yields and selectivity. The Co(III)-catalyzed carbon–hydrogen functionalization/cyclization reactions can be also applied to 1-substituted-arylhydrazines and internal or terminal alkynes for the preparation of indoles 3 (Scheme 4).36 The results indicated that a meta-substituent in the arylhydrazine and a branched chain in the alkynes governed the reaction regioselectivity.
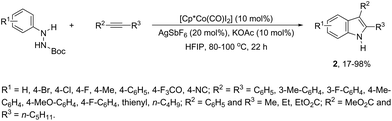 |
| | Scheme 3 | |
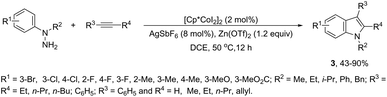 |
| | Scheme 4 | |
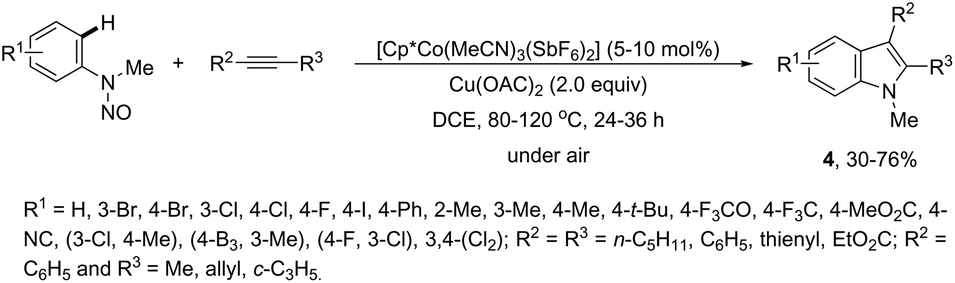
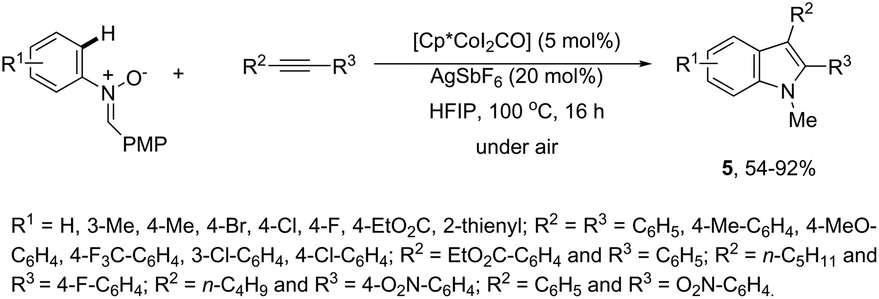
The N-nitrosoanilines have also been reported as directing groups in the cationic cobalt(III) catalyzed intermolecular cyclization of alkynes for the regioselective preparation of N-substituted indoles 4 (Scheme 5).37 A versatile cationic cobalt(III) catalyst using nitrones, as a nitrogen source, was also used in combination with internal alkynes for the preparation of 2,3-substituted indoles 5 (Scheme 6).38 The authors carried out a comprehensive mechanistic study and concluded that this redox-neutral carbon–hydrogen/nitrogen–oxygen cyclization proceeds through carbon–hydrogen activation by carboxylate assistance.
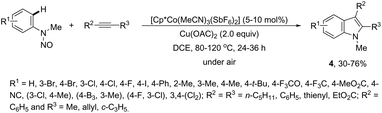 |
| | Scheme 5 | |
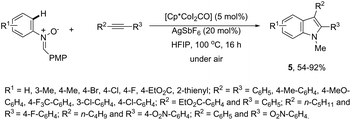 |
| | Scheme 6 | |
N-Arylureas reacted with internal alkynes in a Cp*Co(III)-catalyzed oxidative annulation to afford indoles 6 (Scheme 7).39 The authors observed that the presence of acetamides directly bonded to the nitrogen atom was crucial as directing groups. The methodology was very efficient for unsymmetrical internal alkynes giving the desired indoles with high regioselectivity.
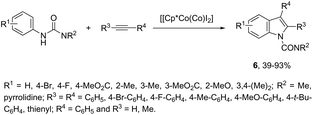 |
| | Scheme 7 | |
2.3. Copper-catalyzed synthesis of indoles
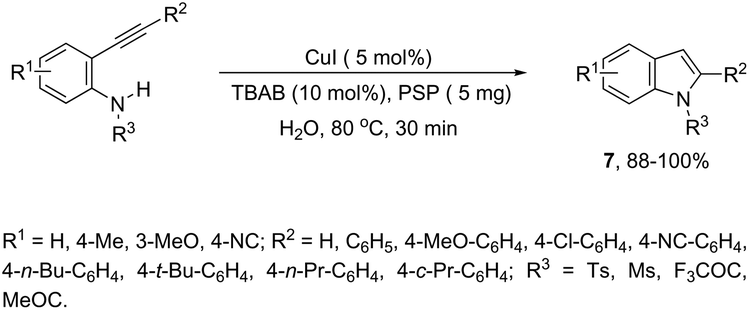
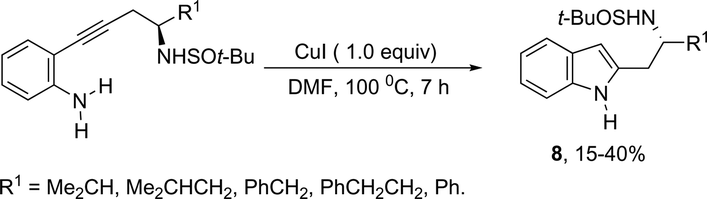
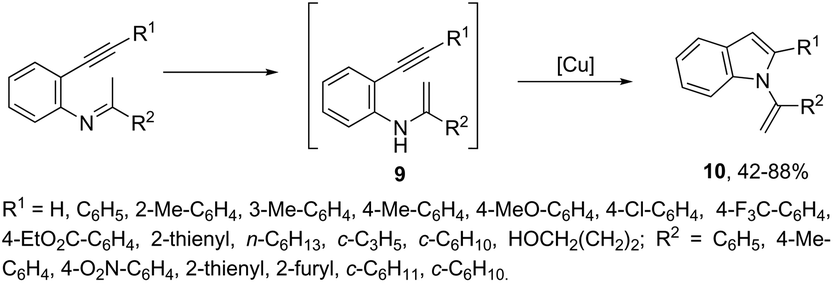
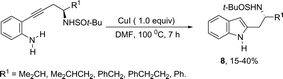
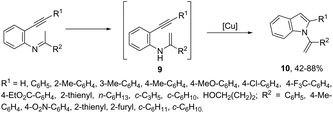
Copper salts constitute one of the most useful and powerful catalysts in organic synthesis, especially for carbon–carbon and carbon–heteroatom bond formation. This usefulness is particularly highlighted because of their lower costs, good accessibility and compatibility with a wide range of functional groups. However, at the beginning of their use, the reactions were conducted with equivalent amounts of copper salts, at high temperature, and with long reaction times. Over the past few years, great advancements have been made in copper-catalyzed reactions which include the use of a number of ligands such as aliphatic diamines, 1,10-phenanthroline, amino acids and their derivatives, and others.40–42 These important outcomes permit the use of milder reaction conditions such as catalytic amounts of copper salts, lower temperature, and short reaction times. Several N-heterocyclic systems have been obtained through copper catalyzed intramolecular and intermolecular cyclization reactions.43 A range of synthetic approaches involving alkynes in carbon–nitrogen bond formation, catalyzed by copper salts, has been applied for the generation of indoles. In one of these, a CuI-polystyrene-supported pyrrole-2-carbohydrazide catalytic system, in water, was applied for the cyclization of N-(2-ethynylphenyl)-sulfonamides for the preparation of indoles 7 (Scheme 8).44 The catalytic system was successfully reused and recycled up to seven times, without any significant loss of activity. In another approach, the treatment of ortho-alkynylanilines with copper iodide, in DMF, at 100 °C, led to the 2-(2-aminoalkyl)indoles 8 (Scheme 9).45 Although the authors observed from the TLC analyses a single spot corresponding to the products, they were obtained in low isolated yields after column chromatography, probably because of decomposition. When an imine is incorporated into the aromatic ring at the ortho-position of the alkyne, such as N-(2-alkynylphenyl)imines, the endo-cyclic products, N-vinylindoles 10, are formed (Scheme 10).46 The authors suggested that the enamine 9 is the N-nucleophilic source, which is generated via tautomerization of the imine.
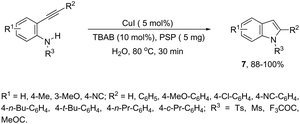 |
| | Scheme 8 | |
 |
| | Scheme 9 | |
 |
| | Scheme 10 | |
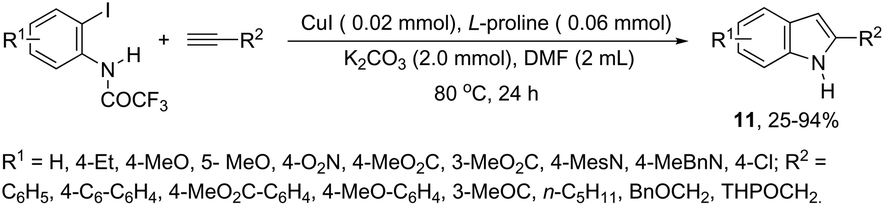
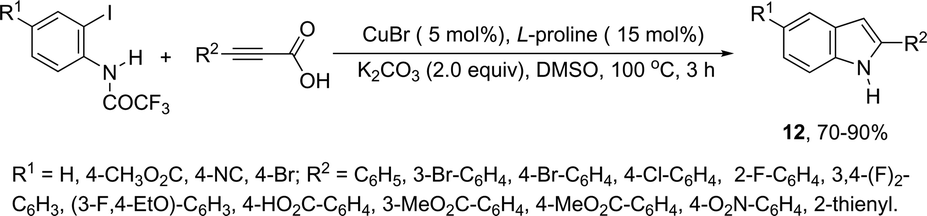
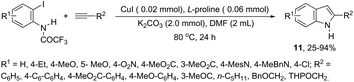
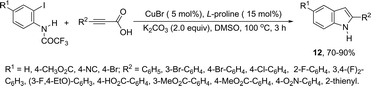
The synthesis of a number of indoles has been achieved by the copper-catalyzed domino Sonogashira coupling/cyclization reaction of N-substituted ortho-haloanilines with terminal alkynes. For example, the copper-catalyzed one-pot reaction of ortho-bromotrifluoroacetanilides with terminal alkynes, using CuI/L-proline as the catalytic system and K2CO3 as a base, in DMF, at 80 °C, gave 2-substituted indoles 11 (Scheme 11).47 In an extension of this work, ortho-arylindoles 12 were prepared by copper catalyzed domino sp–sp2 decarboxylative cross-coupling following cyclization reactions of arylpropiolic acids with ortho-iodotrifluoroacetanilides (Scheme 12).48 Subsequently, analogous to these strategies, the reaction of N-substituted ortho-iodoanilines with terminal alkynes catalyzed by CuI, using DBU as a base, in EtOH, in the absence of a ligand, gave the corresponding indoles in good yields.49 Even though the authors developed the work using copper as a catalyst, they concluded that a palladium contaminant as low as 100 ppb was the promoter of the reaction.
 |
| | Scheme 11 | |
 |
| | Scheme 12 | |
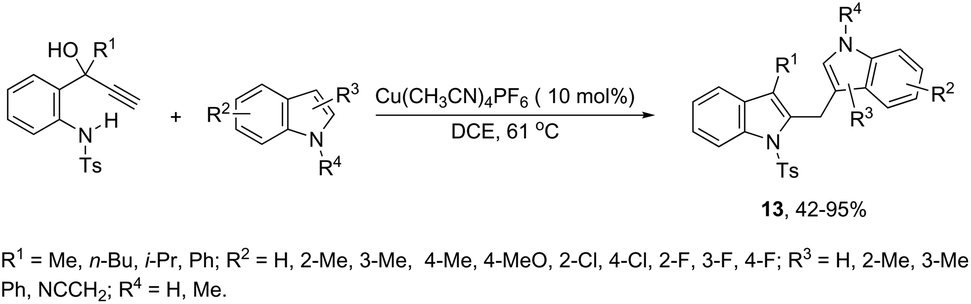
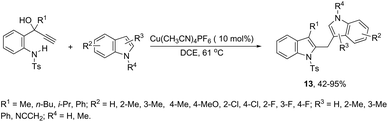
A copper-catalyzed tandem annulation/arylation has been developed for the synthesis of diindolylmethanes 13 from propargyl alcohols (Scheme 13).50 The investigation of the reaction mechanism indicated that the formation of indoles 13 involves a key allyl cation intermediate, which is attacked by an indole nucleophile to form the product. The indole nucleophiles can be replaced by other carbon or oxygen nucleophiles such as 1,3-dimethoxybenzene and alcohols.
 |
| | Scheme 13 | |
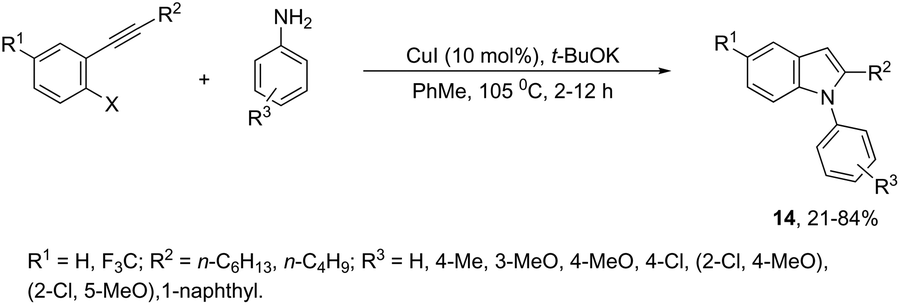
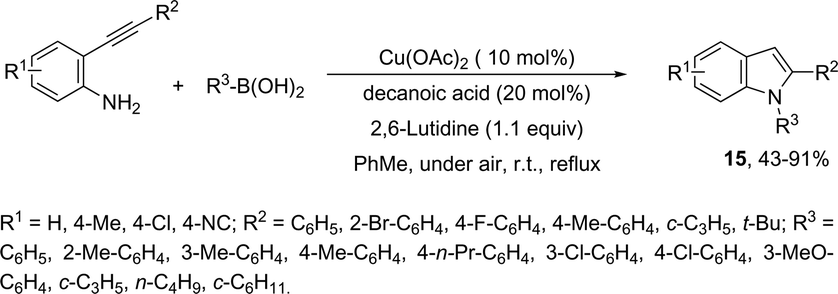
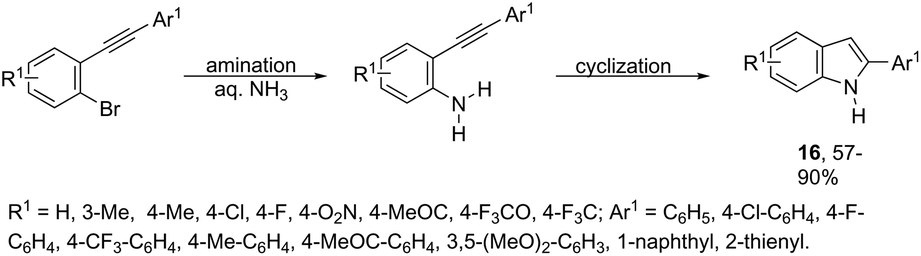
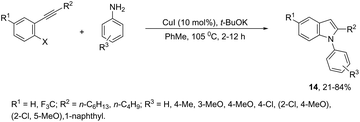
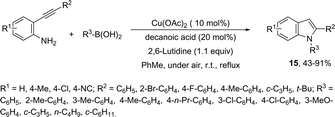
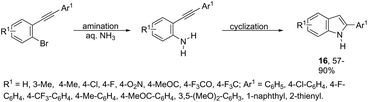
The domino N-arylation/hydroamination strategy has found many applications for the synthesis of functionalized indoles. This reaction occurs between ortho-alkynylhaloarenes and a nitrogen substrate in the presence of copper catalysts. In this case, the copper salt plays a dual role of both promoting the formation of the carbon–nitrogen bond and catalyzing the cyclization. These reactions were applied to the synthesis of several N-arylindoles 14 starting from ortho-alkynylbromoarenes (Scheme 14).51 The reaction utilized anilines as an N-nucleophilic source and copper iodide as a catalyst in a ligand-free system. Other N-nucleophiles, such as amides, required the presence of a diamine as a ligand for the preparation of the corresponding N-acylindoles. The N-arylation/hydroamination described above was extended to construct the 1,2-disubstituted indoles 15 through Cu(II)-catalyzed domino coupling/cyclization reactions of ortho-alkynylanilines and boronic acids (Scheme 15).52 This sequence was also applied to the synthesis of free N-H-2-arylindoles 16, which included copper(II)-catalyzed amination of ortho-bromoarylalkynes with aqueous ammonia (Scheme 16).53 The advancement of the methodology was due to the convenience and atom economy of using aqueous ammonia.
 |
| | Scheme 14 | |
 |
| | Scheme 15 | |
 |
| | Scheme 16 | |
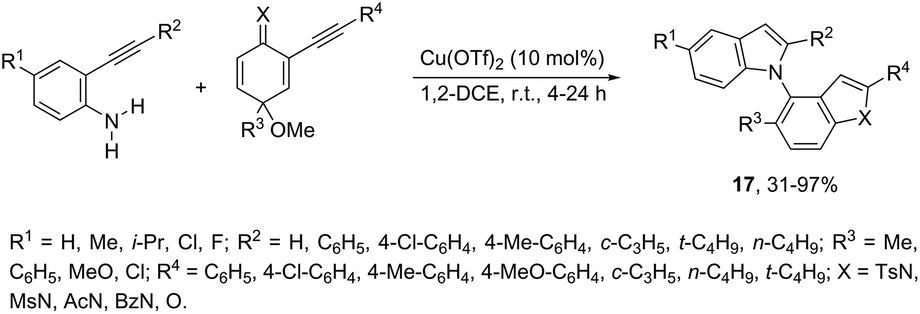
N-Alkylation reaction employing 2-alkynyl cyclohexadienimines, as the alkylating agents, has been proven to be very effective at the construction of N-heteroarylated indoles 17 (Scheme 17).54 The use of 2-alkynyl cyclohexadienones, as the alkylating agents, led to the formation of 1-(benzofuran-4-yl)-1H-indoles. The optimization of the reaction conditions revealed that higher yields of indoles were obtained using Cu(OTf)2 in 1,2-dichloroethane as the catalytic system.
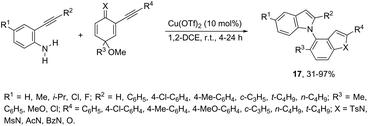 |
| | Scheme 17 | |
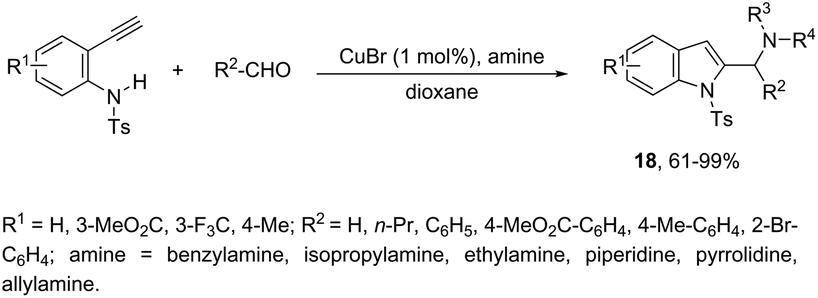
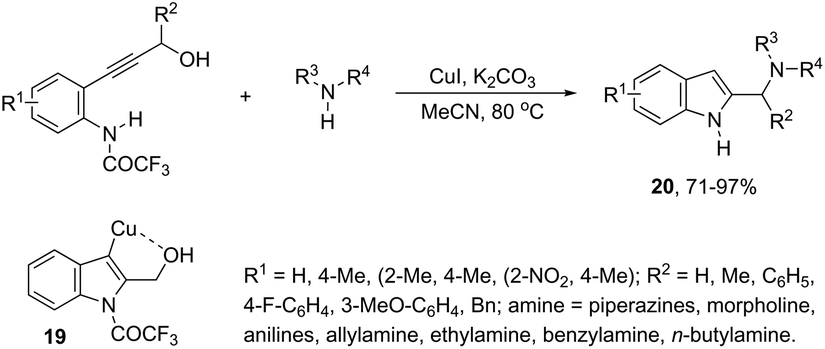
Copper-catalyzed domino three-component coupling-cyclization of ortho-alkynylanilines with secondary amines and aldehydes can also yield indoles. In the former case, cyclic or acyclic secondary amines, aldehydes and ortho-alkynylanilines allowed the copper(I)-catalyzed formation of 2-(aminomethyl)indoles 18 in high yields (Scheme 18).55,56 The reaction involved the formation of two carbon–nitrogen bonds and one carbon–carbon bond producing water as the only waste. In addition, the copper-catalyzed cyclization of 3-(ortho-trifluoroacetamidophenyl)-1-propargyl alcohols, in the presence of primary or secondary amines, gave free NH-2-(aminomethyl)indoles 20 (Scheme 19).57 The proposed key intermediate is a σ-indolyl copper 19, which can undergo substitution of the hydroxyl group for the nucleophilic amino group, assisted by the coordination of oxygen to copper (Scheme 19).
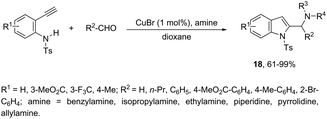 |
| | Scheme 18 | |
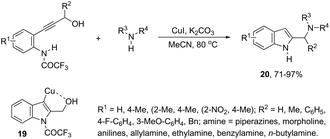 |
| | Scheme 19 | |
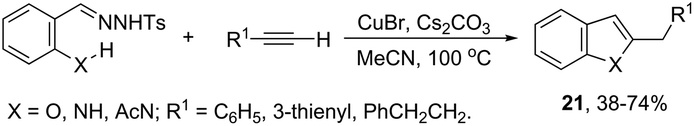
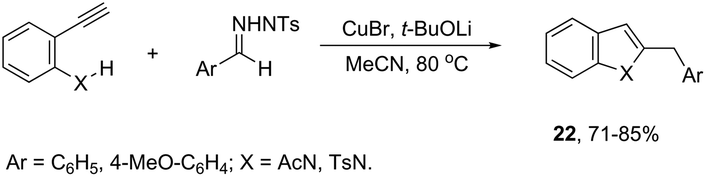
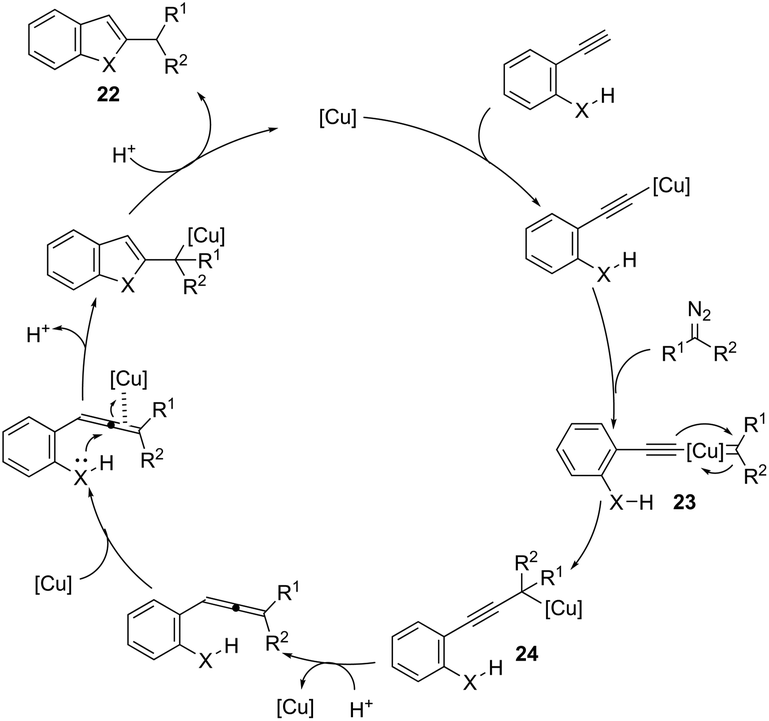
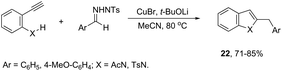
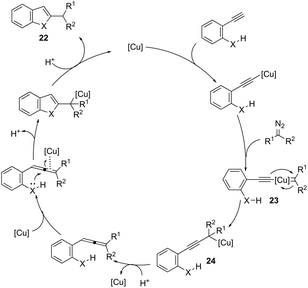
ortho-Amino-N-tosylhydrazones, which are prepared using the corresponding aldehydes, can be also applied to copper(I)-catalyzed cyclization with terminal alkynes to afford 2-substituted indoles 21 (Scheme 20).58 In additional studies, the authors used N-tosylhydrazones and ortho-aminoarylalkynes in a reaction catalyzed by CuBr to form 2-substituted indoles 22 (Scheme 21).59 The proposed mechanism consists of the in situ preparation of a diazo compound, followed by a copper acetylide attack on the diazo compound to give the copper carbine 23. The migratory insertion of the carbenic carbon into the alkynyl group affords the propargyl cuprate intermediate 24. The formation of an allene and an intramolecular cyclization promoted by the copper catalyst afford the desired indoles (Scheme 22).
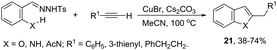 |
| | Scheme 20 | |
 |
| | Scheme 21 | |
 |
| | Scheme 22 | |
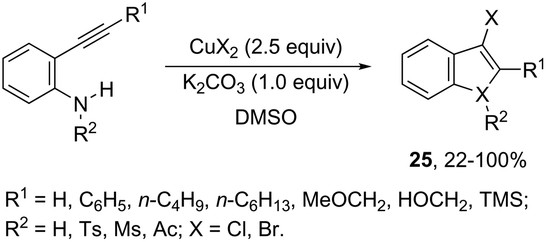
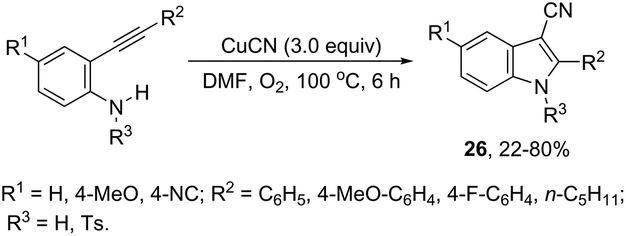
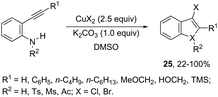
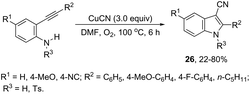
The intramolecular cyclization between ortho-alkynylanilines and copper salts is an established method for the synthesis of 3-haloindoles. It has been demonstrated that through this methodology 3-chloro- and 3-bromoindole derivatives 25 can be prepared in good yields by the reaction of ortho-alkynylanilines with cupric halide in dimethyl sulfoxide (Scheme 23).60 The authors determined that CuBr2 instead of Br2 promotes the bromocyclization reaction and that the inactive Cu(0), formed in the reaction, can be oxidized by CuX2 to produce CuX, which activates the alkynes. By using CuCN as a promoter, N-protected ortho-alkynylanilines gave 3-cyanoindoles 26via a sequential cyclization–cyanation reaction (Scheme 24).61 In the proposed mechanism, the molecular oxygen promotes the oxidation of a Cu(I)–alkyne complex intermediate to the corresponding Cu(II) or Cu(III). It has been reported that the use of camphorsulfonic acid (CSA), as an additive, was effective in increasing the yield of 3-cyanoindoles.62
 |
| | Scheme 23 | |
 |
| | Scheme 24 | |
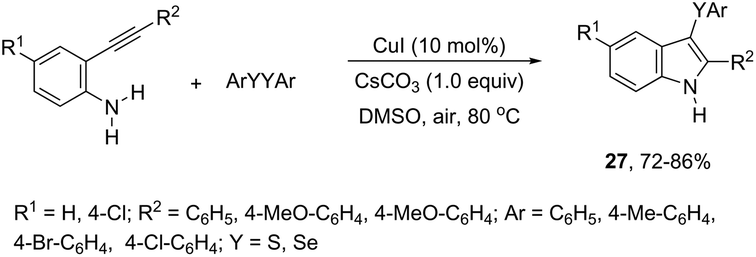
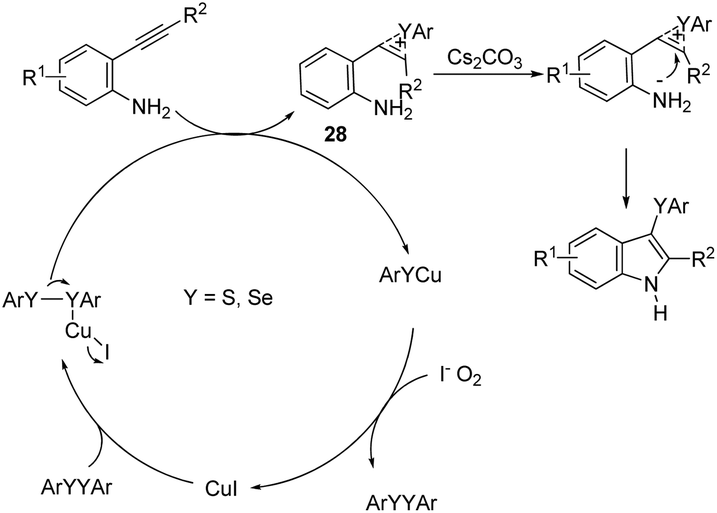
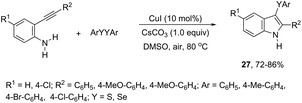
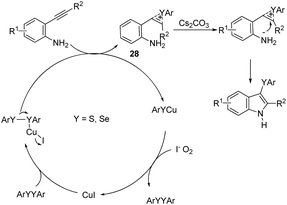
The copper catalyzed tandem cyclization of ortho-alkynylanilines with dichalcogenides provided an expedient approach for the synthesis of 3-chalcogenindoles 27 (Scheme 25).63 The results indicated that the copper catalyst promotes the chalcogen–chalcogen bond cleavage to form an electrophilic chalcogen species, which activates the carbon–carbon triple bond of the ortho-alkynylanilines via chalcogenonium cation 28. The nitrogen intramolecular attack on the cation 28 produces the corresponding 3-chalcogen-indoles (Scheme 26).
 |
| | Scheme 25 | |
 |
| | Scheme 26 | |
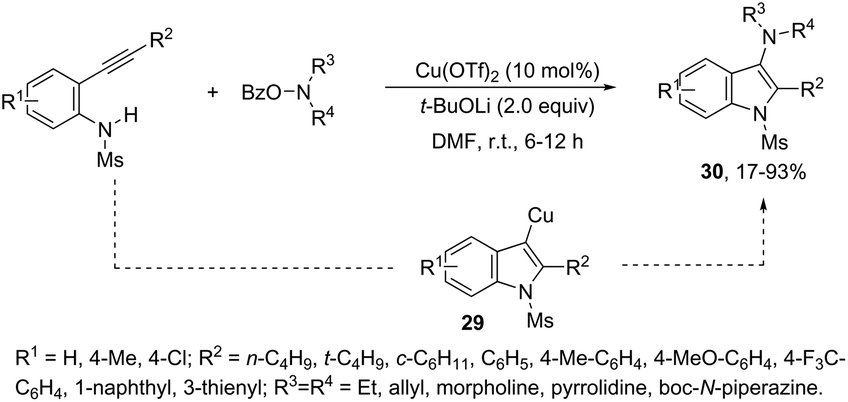
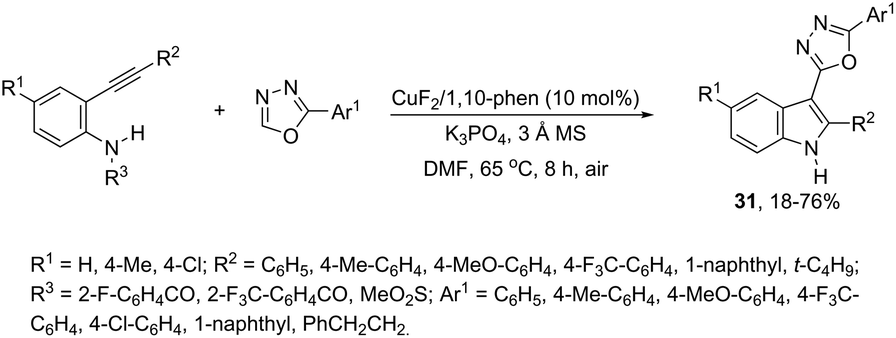
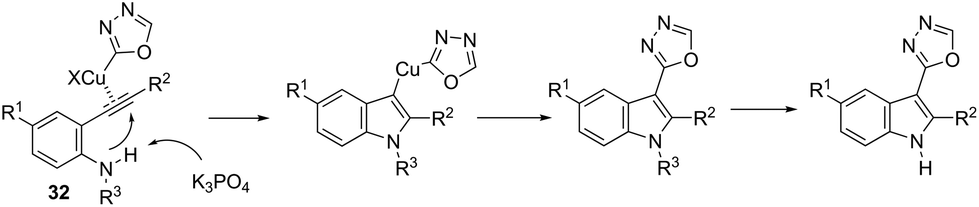
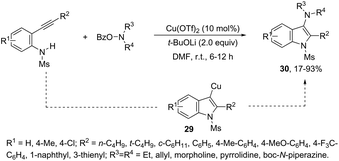
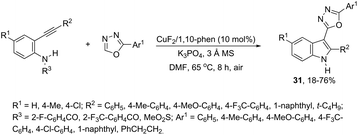
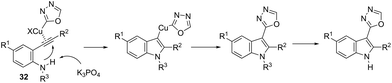
This tandem reaction was also attempted by using O-benzoyl hydroxylamines to capture the copper intermediate. Thus, the copper catalyzed cyclization of ortho-alkynylanilines produced the indolyl copper intermediate 29, which reacted with O-benzoyl hydroxylamines to give 3-aminoindoles 30 (Scheme 27).64 The investigation of the mechanism carried out by the authors indicated that a nonradical electrophilic amination of the heteroarylcopper species and a carbon–nitrogen bond-forming step were involved. Following the same reaction sequence, C3-azolylindoles 31 were synthesized from copper-promoted annulative direct coupling of ortho-alkynylanilines with 1,3,4-oxadiazoles (Scheme 28).65 The authors suggested that the carbon–carbon triple bond of anilines is activated by an oxadiazoyl copper intermediate 32, which promotes the annulative aminocupration leading to the product after reductive elimination (Scheme 29).
 |
| | Scheme 27 | |
 |
| | Scheme 28 | |
 |
| | Scheme 29 | |
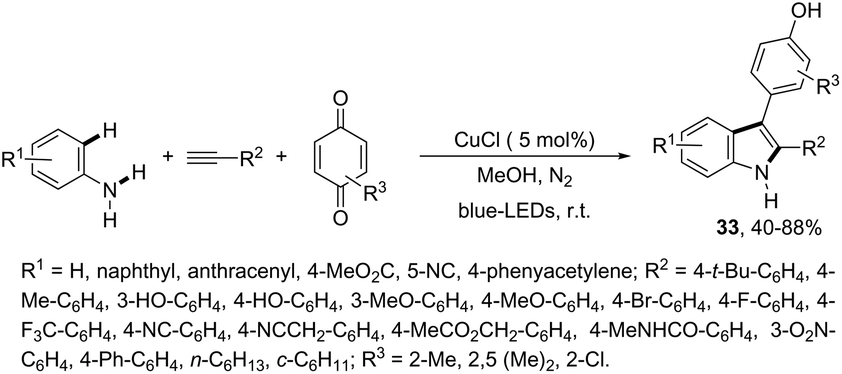
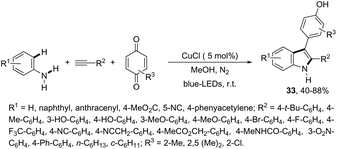
Another approach to obtain 3-substituted indole derivatives employed a visible-light-induced copper-catalyzed process for carbon–hydrogen annulation of arylamines with terminal alkynes and benzoquinone. Thus, the visible-light irradiation of aniline derivatives with alkynes in the presence of benzoquinone using a mixture of CH3CN/CH3OH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) as the solvent and CuCl (5 mol%) as the catalyst, at room temperature, gave 3-para-hydroxyphenyl substituted indoles 33 (Scheme 30).66
:1 v/v) as the solvent and CuCl (5 mol%) as the catalyst, at room temperature, gave 3-para-hydroxyphenyl substituted indoles 33 (Scheme 30).66
 |
| | Scheme 30 | |
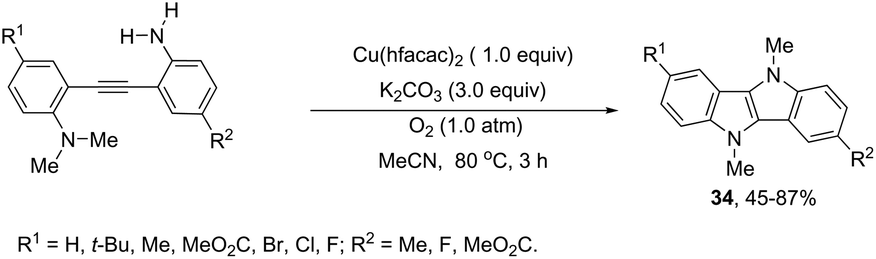
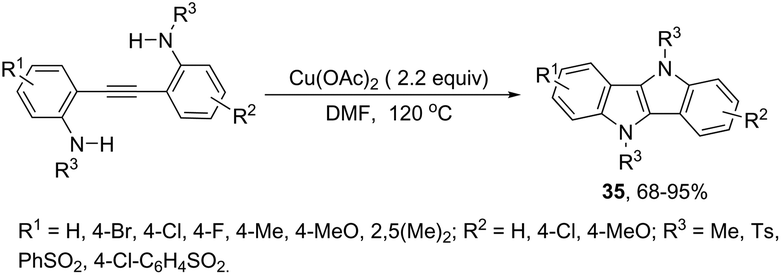
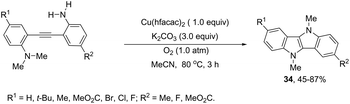
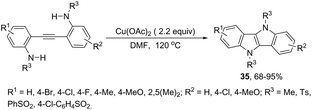
The intramolecular oxidative diamination of bis-(2-aminophenyl)acetylenes promoted by a Cu(hfacac)2/O2 oxidation system was used for the preparation of 5,10-dihydroindolo[3,2-b]indoles 34 (Scheme 31).67 The presence of both N,N-dimethylamine and primary amine groups in the bis-(2-aminophenyl) acetylenes was crucial for the success of the cyclization. The key step of the annulation process was the intermolecular N-methyl transfer from the nitrogen atom of N,N-dimethylamine to the primary amine. Subsequently, the cascade reaction annulation of diarylalkyne sulfonamides catalyzed by Cu(OAc)2, in the absence of an oxidant, led to the formation of 5,10-dihydroindolo[3,2-b]indoles 35 (Scheme 32).68 The authors stated that the process involves two sequential cyclizations with two carbon–nitrogen bond formation processes, in which Cu(OAc)2 works as a catalyst in the first step and as an oxidant in the second step.
 |
| | Scheme 31 | |
 |
| | Scheme 32 | |
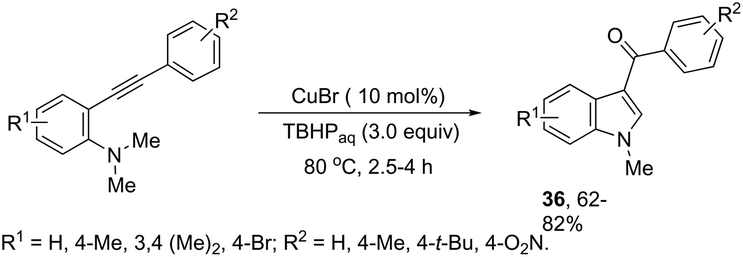
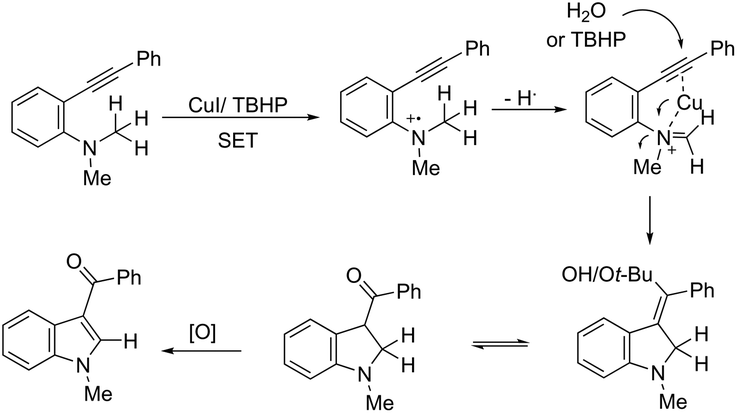
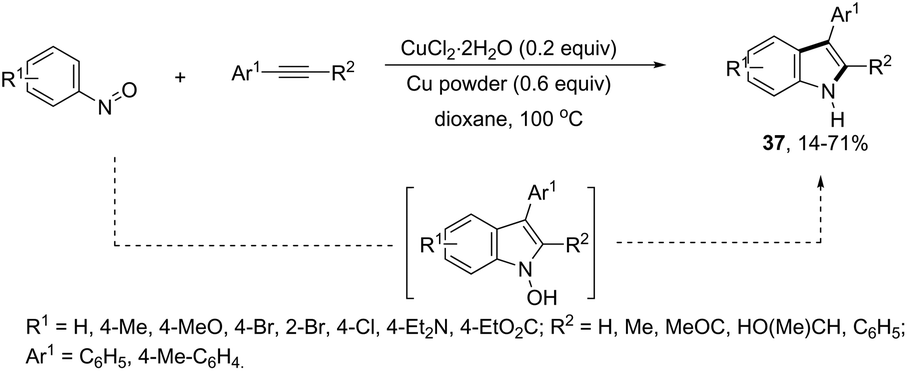
ortho-Alkynyl N,N-dimethylamines have been also utilized for the functionalization of the 3-position of the indole ring for the preparation of 3-aroylindoles 36, via a copper catalyzed tandem process (Scheme 33).69 In this cyclization, one carbon–carbon bond and one carbon–oxygen bond are formed while two sp3 carbon–hydrogen bonds are cleaved. The reaction involves an sp3 carbon–hydrogen bond activation of the methyl group bonded to nitrogen, followed by an intramolecular nucleophilic attack of water on the alkyne, which promotes the cyclization onto iminium carbon to give, after oxidation, the product (Scheme 34). The copper-catalyzed annulation of nitrosoarenes and alkynes, followed by deoxygenation, was also employed to obtain 3-substituted indoles 37 (Scheme 35).70 It was proposed that the in situ formation of the Cu(I) species, by the reaction of CuI(I) with Cu(0), is responsible for deoxygenation of the N-hydroxyindole intermediate returning the CuI(II) species to the cycle.
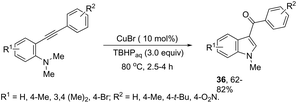 |
| | Scheme 33 | |
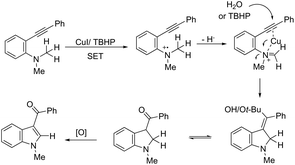 |
| | Scheme 34 | |
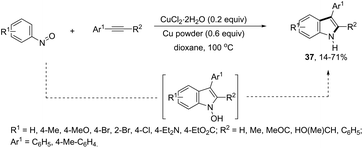 |
| | Scheme 35 | |
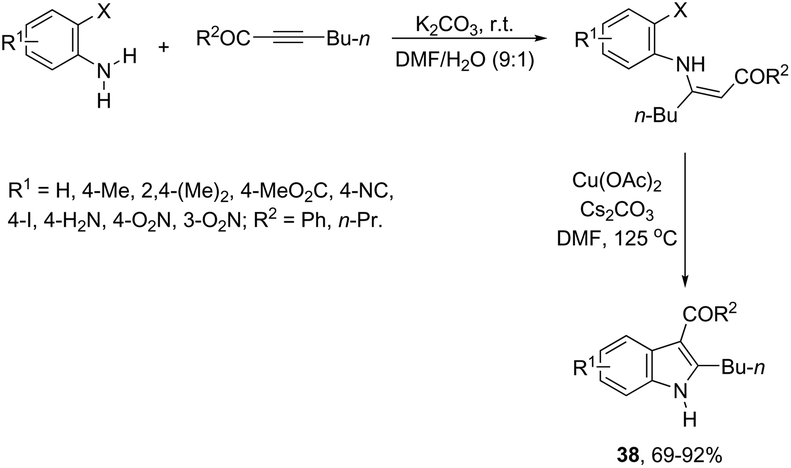
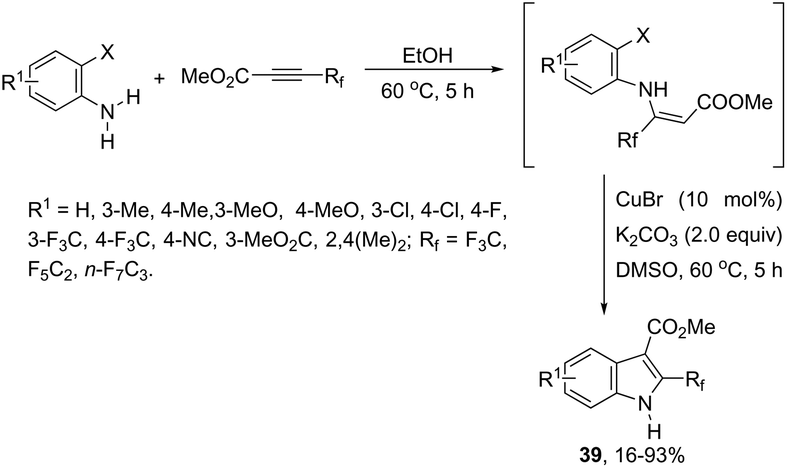
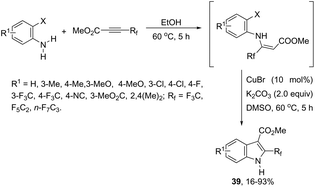
The 3-acylated indole derivatives 38 have been prepared by the conjugate addition of N-formyl-2-haloanilines to acetylenic sulfones, ketones, and esters, followed by a copper-catalyzed intramolecular C-arylation (Scheme 36).71 The C-arylation step was achieved by employing copper(II) acetate as the catalyst in the complete absence of ligands, without the need to protect from air or water. Mechanistic studies carried out by the authors indicated that the formate ion, produced by the base-catalyzed hydrolysis of DMF, reduces copper(II) to the active copper(I) species and recycles any copper(II) that is produced in the reaction, preventing the deactivation of the catalyst. A similar strategy has been also successfully employed in the synthesis of 2-(perfluoroalkyl)indoles 39 (Scheme 37).72 The reaction was carried out by the initial Michael addition of ortho-haloanilines to methyl perfluoroalkynoates giving N-(ortho-haloaryl)enamines, which afforded the corresponding indoles via a copper-catalyzed annulation reaction.
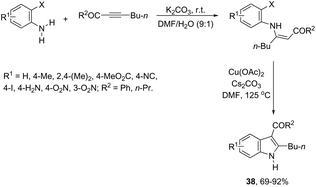 |
| | Scheme 36 | |
 |
| | Scheme 37 | |
2.4. Gold-catalyzed synthesis of indoles
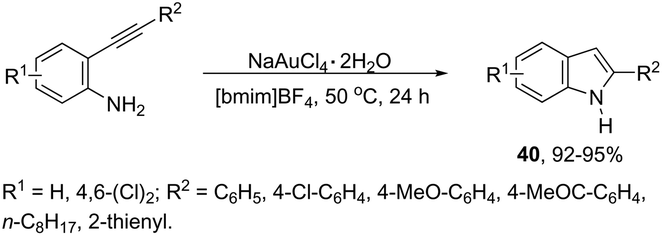
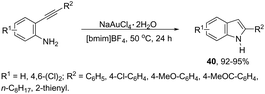
Gold-catalyzed reactions have attracted attention because of their useful applications in organic synthesis. In particular, gold salts have been extensively used as catalysts for reactions with a series of unsaturated precursors, such as alkynes, alkenes, and allenes.73 The reactions of gold(I) and gold(III) salts with alkynes produce reactive complexes, which afford products with high selectivity, in most cases in a single step. Particularly, the gold activation of carbon–carbon bonds of alkynes towards attack by nitrogen nucleophiles is one of the most important methods to prepare indoles via carbon–nitrogen bond formation. The methodologies involving the preparation of indoles via hydroamination, cycloaddition, annulation, and cycloisomerization, among other reactions with alkynes, catalyzed by gold salts, will be shown below. Thus, several types of intramolecular gold-activated alkynes were easily reduced with simultaneous carbon–nitrogen bond formation affording indoles. For example, ortho-alkynylanilines when treated with NaAuCl4·H2O, using [bmim]BF4 as the reaction medium, afforded 2-substituted indoles 40 in high yields (Scheme 38).74 The catalytic system could be recycled using n-Bu4NAuCl4. In addition, 2,3-disubstituted indoles could also be prepared by the reaction of ortho-alkynylanilines and 3-butenone in a one pot annulation–alkylation sequence or via an aza-Michael addition–annulation–alkylation process, which afforded the 1,2,3-trisubstituted indoles.
 |
| | Scheme 38 | |
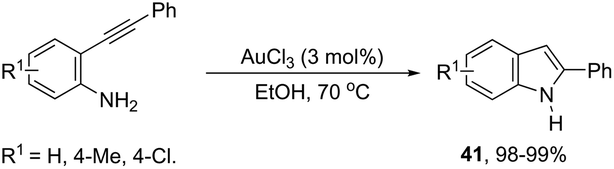
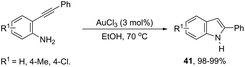
In protic solvents, AuCl3 catalyzed the cyclization of ortho-alkynylanilines to generate 2-substituted indoles 41 (Scheme 39).75 When carried out using ethanol as the solvent at 70 °C, the reaction was very fast without needing to protect the amino group. Under the same reaction conditions, 2-amine-3-alkynylpyridines led to substituted pyrrolopyridines in good yields.
 |
| | Scheme 39 | |
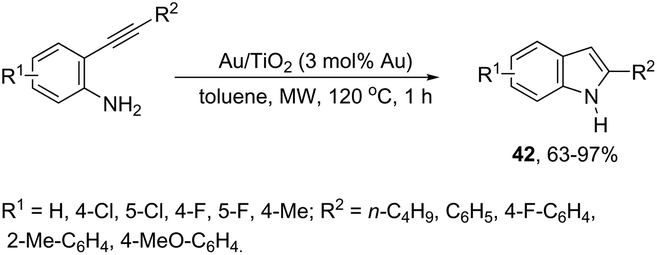
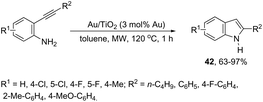
The catalytic activity of cationic gold could be hampered in the presence of basic amines. The cationic gold has much stronger affinity towards basic amines than towards the activation of carbon–carbon bonds of the alkyne substrates; thus the corresponding catalytic cycle could be inactivated. However, gold nanoparticles supported on titanium dioxide were found to be a highly efficient catalytic system for the intramolecular hydroamination of ortho-alkynylanilines to afford the corresponding indole derivatives 42 (Scheme 40).76 The reaction conditions required low catalyst loading (0.2 mol% Au) and toluene as the solvent under microwave heating. The catalytic system was reused through five straight runs without loss of catalytic activity.
 |
| | Scheme 40 | |
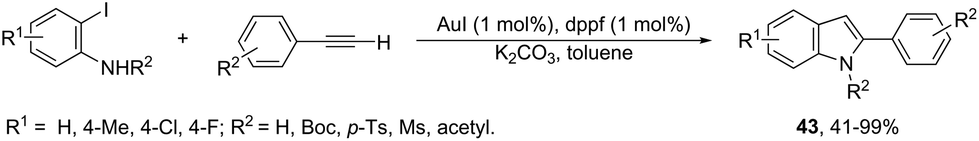
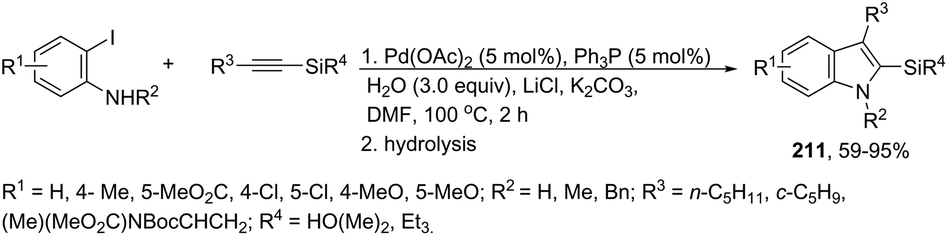
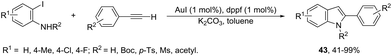
To avoid the previous preparation of ortho-alkynylanilines, one strategy is the use of a cross-coupling–cyclization reaction sequence of terminal alkynes with ortho-haloanilines. Thus, the gold(I) iodide catalyzed Sonogashira reactions of terminal alkynes with ortho-iodoanilines, in the presence of dppf as a ligand and toluene as a solvent, generated the corresponding 2-substituted indoles 43 in good to excellent yields (Scheme 41).77 The reaction conditions worked well with aromatic terminal alkynes and ortho-iodoanilines; however, moderate yields were obtained with aliphatic terminal alkynes, whereas ortho-bromoanilines did not give the desired indoles. N-Substituted ortho-iodoanilines, such as N-Boc, N-p-Ts, N-Ms and N-acetyl ortho-iodoanilines, could also react with phenylacetylene, giving the coupling–cyclization products in good yields.
 |
| | Scheme 41 | |
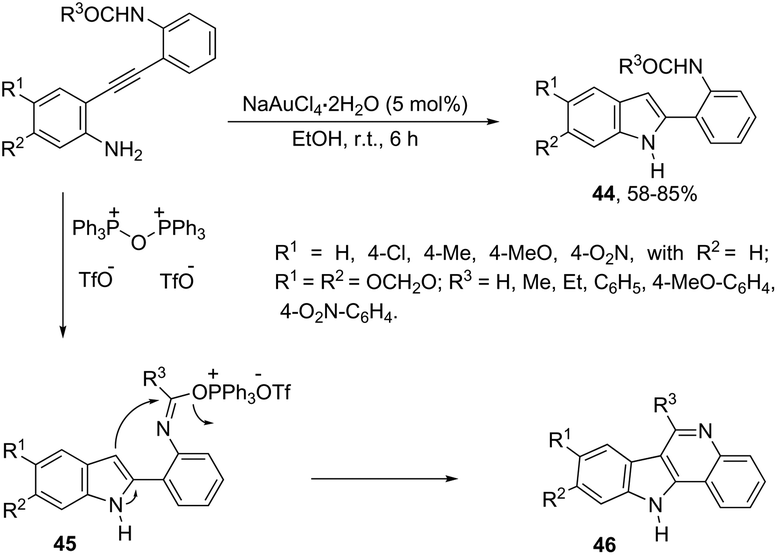
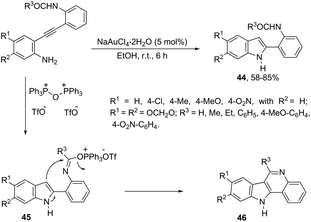
The catalyst NaAuCl4·2H2O promoted a chemoselective cyclization of aryl alkynes having two nitrogen nucleophiles in their structure with similar reactivities. Aryl alkynes having both amine and amide groups as nucleophiles, located in the ortho-position to the carbon–carbon triple bond, could undergo a competitive cyclization reaction to give 1H-indole or N-acetyl-indoles, respectively. However, the reaction of N-(2-((2-aminophenyl)ethynyl)phenyl)acetamide with NaAuCl4·2H2O, in ethanol as the solvent, at room temperature, led to the exclusive formation of N-[2-(1H-indol-2-yl)phenyl]acetamides 44 (Scheme 42).78 Alkynes containing alkylamide, formamide, benzamide, and carbamate groups were also transformed into the corresponding 1H-indole derivatives. The authors showed that treating N-[2-(1H-indol-2-yl)phenyl]acetamides 46 with bis-(triphenyl)oxodiphosphonium trifluoromethanesulfonate (Hendrickson reagent) gave 1H-indolo[3,2-c]quinolones, which were obtained via in situ preparation of amidate 45 (Scheme 42).
 |
| | Scheme 42 | |
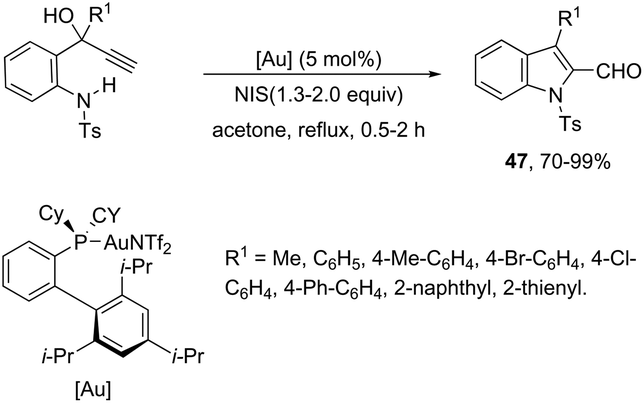
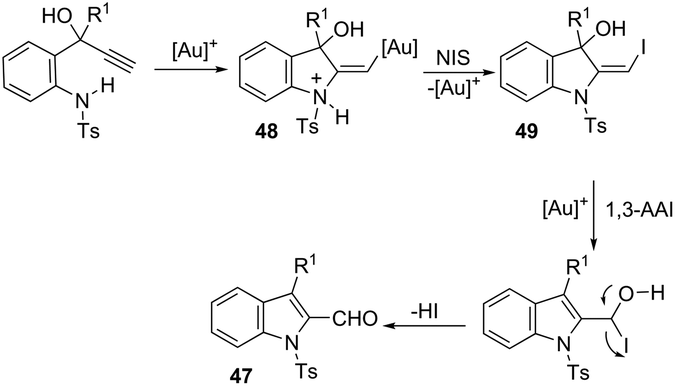
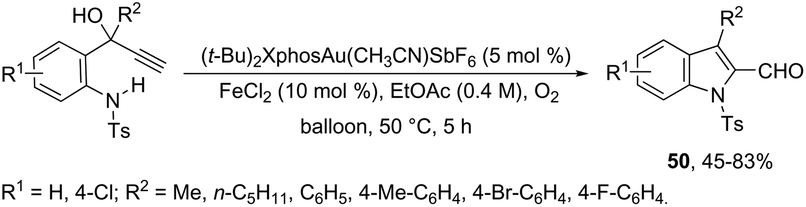
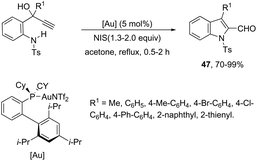
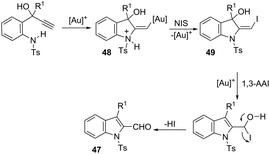
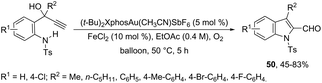
The 1H-indole-2-carbaldehydes 47 were efficiently prepared by gold(I)-catalyzed cycloisomerization of 1-(2-(tosylamino)phenyl)propynols in the presence of N-iodosuccinimide (Scheme 43).79 The reaction conditions tolerated a wide variety of substrates, even with a bulky group at the propargyl position, affording the corresponding products from 70 to 99% yields. The reaction was highly regioselective occurring via an exclusive 5-exo-dig mode. Regarding the reaction mechanism, the authors’ hypothesis is that the alkyne moiety is activated by the gold(I) catalyst, which promotes the intramolecular nucleophilic attack of aniline giving a vinyl gold intermediate 48, followed by an iododeauration promoted by NIS to give the (E)-2-(iodomethylene)indolin-3-ol adducts 49. The successive 1,3-allylic alcohol isomerization and formylation reaction give the 1H-indole-2-carbaldehydes 47 (Scheme 44). One of the major drawbacks of these cyclization reactions is the deauration step that could lead to the formation of by-products and a decrease in the yields. However, the cooperative use of gold and iron catalysts, in the presence of one atom of oxygen, was very efficient at promoting the cyclization of 1-(2-(tosylamino)phenyl)propynols to TsN-indole-2-carbaldehydes 50 in good yields (Scheme 45).80 The success of the cyclization is credited to the presence of oxygen radicals that react with the vinyl–gold intermediate forming a new reactive species, helping in the gold catalyst turnover.
 |
| | Scheme 43 | |
 |
| | Scheme 44 | |
 |
| | Scheme 45 | |
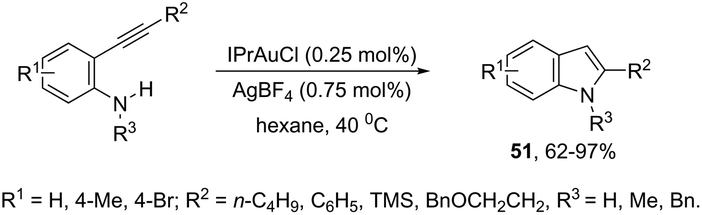
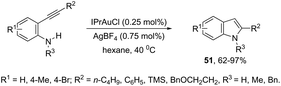
N-Heterocyclic carbenes (NHC) have been used as ligands in gold complexes for the successful synthesis of indole derivatives. Thus, the cyclization of ortho-alkynylanilines catalyzed by (IPr)AuCl afforded the 2-substituted indoles 51 (Scheme 46).81 The gold(I) salt stabilized with an NHC bulky carbene was found to be more efficient than other potential catalysts, such as NaAuCl4·2H2O, AuCl3, and AuCl. The authors also reported that the counter anions had a strong effect on the efficiency of the catalyst, whereas BF4 proved to be much more effective than Cl, TsO, and NTf2.
 |
| | Scheme 46 | |
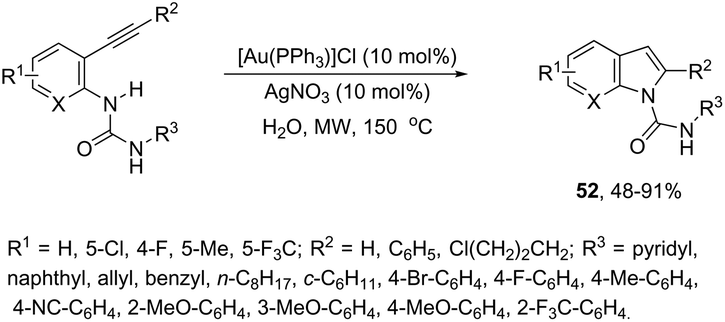
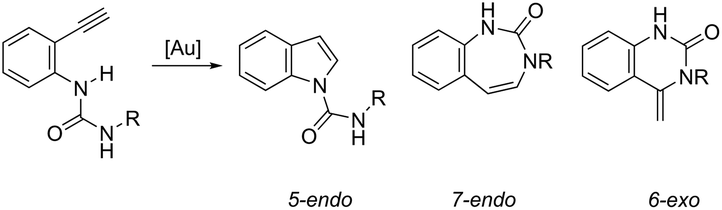
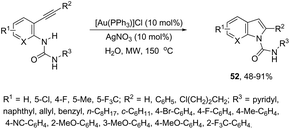
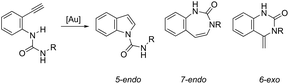
ortho-Alkynylureas were also successfully used for the synthesis of indole-1-carboxamides 52 (Scheme 47).82 The reaction of N′-substituted N-(2-alkynylphenyl)ureas, catalyzed by [Au(PPh3)]Cl/Ag2CO3, in water under microwave irradiation, gave indole-1-carboxamides 52. The authors stated that three different products could be formed via a 5-endo, 6-exo, 7-endo-cyclization mode; however, the reaction was highly selective, giving only indole derivatives, formed via 5-endo-dig cyclization (Scheme 48). The substrate scope of this reaction was studied and it indicated that the reaction conditions were tolerant to a variety of functional groups such as N′-aryl, alkyl, and heterocyclic groups on the ortho-alkynylureas.
 |
| | Scheme 47 | |
 |
| | Scheme 48 | |
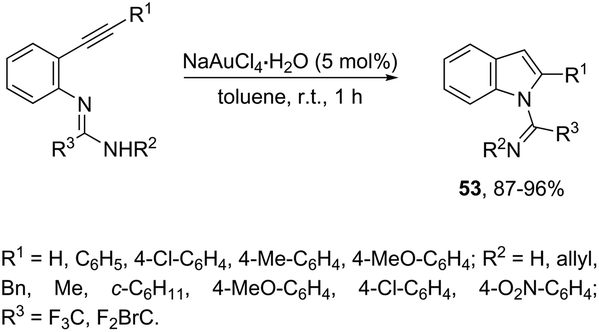
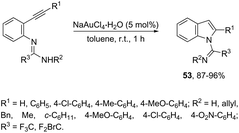
N-(ortho-Alkynyl)aryl-N′-substituted trifluoroacetamidines were also applied as the nucleophilic nitrogen source for the gold-catalyzed intramolecular hydroamination reactions. When ortho-alkynylarylamidines were treated with NaAuCl4·2H2O as the catalyst, in toluene as the solvent, at room temperature, a 5-endo-dig cyclization proceeded to give the corresponding indoles 53 (Scheme 49).83 The authors demonstrated that the same substrate, when treated with K2CO3 as a base, gave the quinazolines in a 6-exo-dig process.
 |
| | Scheme 49 | |
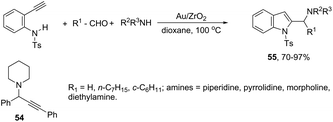
A three-component version of the gold(III)-catalyzed cyclization reaction employing aldehydes, piperidines, and alkynes was described for the preparation of indoles. The reaction carried out with phenylacetylene as the alkyne, under gold supported on nanocrystalline ZrO2, gave the propargyl amines 54 (Scheme 50). The use of terminal ortho-alkynylanilines instead of phenylacetylene, under the same reaction conditions, afforded the 2-(aminomethyl)indole derivatives 55 in high yields with no formation of propargylamines (Scheme 50).84
 |
| | Scheme 50 | |
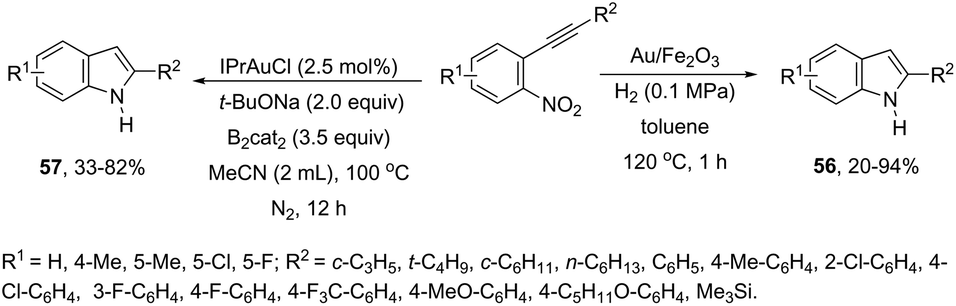
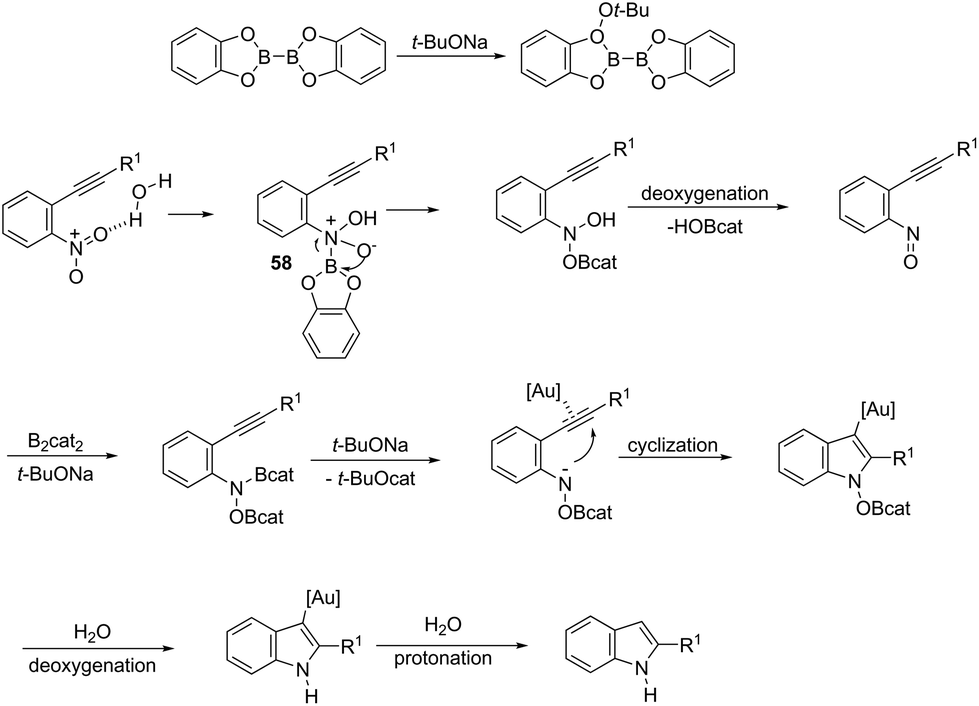
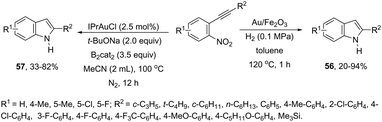
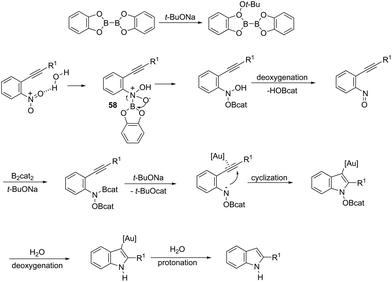
ortho-Alkynylnitroarenes can be reduced to amino derivatives, which are then cyclized under gold catalysis to yield 2-substituted indoles. The major challenge of these reactions is the reduction of the nitro group without reaching other functional groups of the starting material. The reduction of ortho-alkynylnitroarenes, using hydrogenation conditions, under gold nanoparticles supported on Fe2O3, led to the formation of 2-substituted indoles 56 (Scheme 51).85 It has also been reported that the synthesis of 2-substituted indoles 57, via a diboron/base promoted a tandem reductive cyclization of ortho-alkynylnitroarenes, under 1,3-bis-(2,6-diisopropylphenyl)imidazol-2-ylidene gold(I)chloride (IPrAuCl) catalysis conditions (Scheme 62). In this last methodology, the reductive cyclization was proposed to occur via an initial attack of a nucleophilic boronic species on the nitro group, affording the nitroso intermediate 58, via debromation/deoxygenation processes. Intermediate 58 is converted to borate-nitroso anions, which are cyclized with the help of IPrAuCl (Scheme 52).
 |
| | Scheme 51 | |
 |
| | Scheme 52 | |
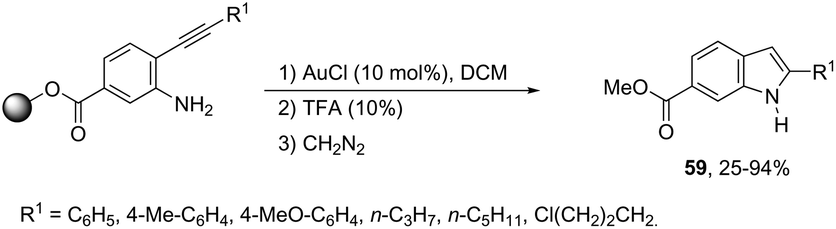
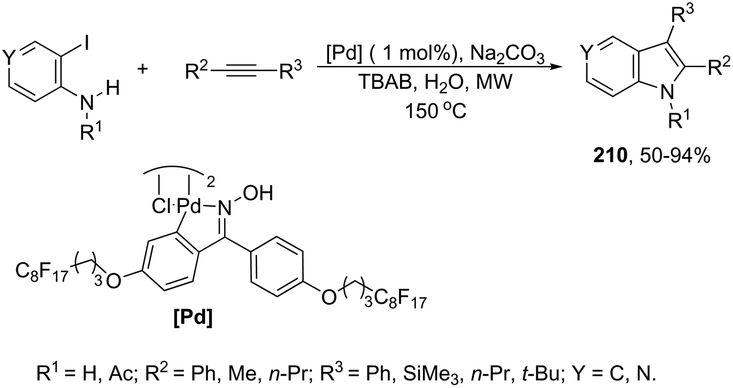
The cyclization of immobilized ortho-alkynylanilines with a gold catalyst is particularly attractive because of the advantage of easy purification. The use of ortho-alkynylanilines immobilized in Wang resin in the reaction with AuCl was the method of choice for the preparation of resin-bound indoles, which after reaction with 10% TFA in DCM followed by methylation with diazomethane gave the resin-free indole 59 (Scheme 53).86 The reaction conditions developed were applicable to a series of ortho-alkynylanilines, giving the corresponding indoles in 25–94% yields. The authors demonstrated that ortho-alkynylanilines having an aromatic substituent at the alkyne gave the indoles in better yields than those with an alkyl chain.
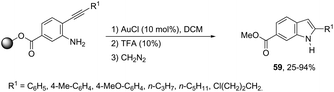 |
| | Scheme 53 | |
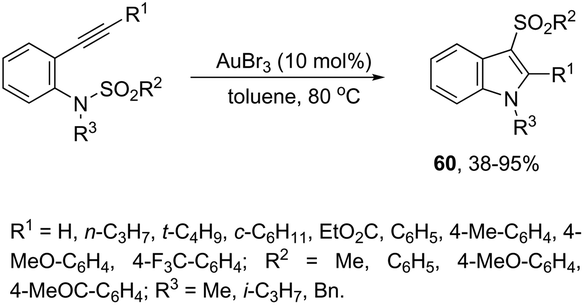
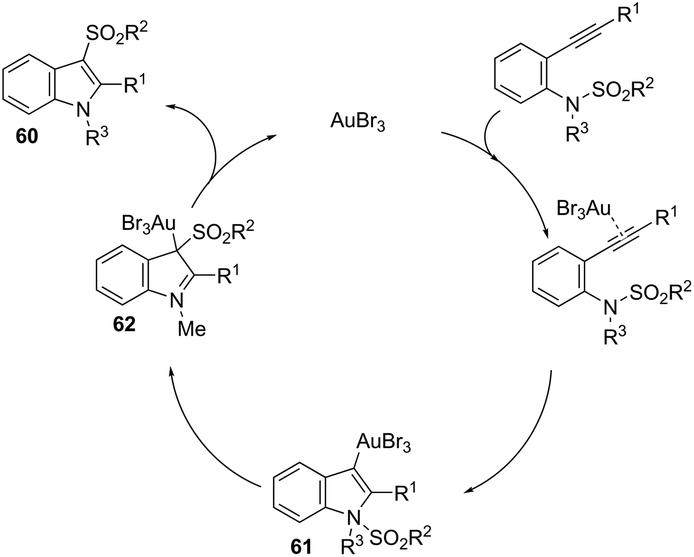
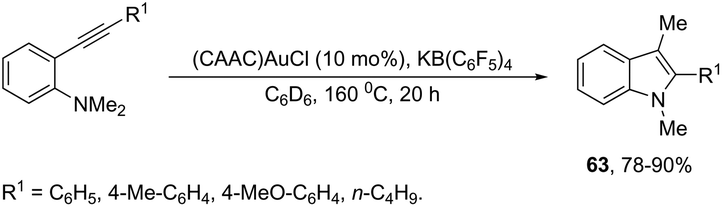
The gold-catalyzed cyclization of ortho-alkynylanilines, having a migrating group on the nitrogen atom, provides a successful route for preparation of 3-substituted indoles. This method for the synthesis of 3-sulfonylindoles 60 has been well investigated using different ortho-alkynyl-N-sulfonylanilines in the presence of a catalytic amount of AuBr3 (Scheme 54).87 The authors performed additional crossover experiments, which revealed that the migration of the sulfonyl group occurs in an intramolecular instead of intermolecular manner. Further experimental results led the authors to propose that the cyclization occurs through a nucleophilic attack of the nitrogen atom on the gold-active alkyne, leading to the cyclized intermediate 61. The sulfonyl group intramolecularly migrates to the 3-position of the indole affording the intermediate 62, which gives the 3-sulfonylindole 60, after the elimination of AuBr3 (Scheme 55). The strategies based on the methylamination reaction of 2-alkynyl-N,N′-dimethyl-benzenamines, in the presence of a 1:1 mixture of (CAAC)AuCl/KB(C6F5)4 (10 mol%), were used in the preparation of 2,3-disubstituted indoles 63 (Scheme 56).88 In this rearrangement, a methyl group bonded to nitrogen migrates to the 3-position of the indole ring. However, in the case of the N-ethyl substituted benzenamines, only the cleavage of the relatively weak carbon–nitrogen bond was observed with loss of ethylene, and the 3-substituted indoles were not formed.
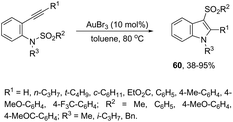 |
| | Scheme 54 | |
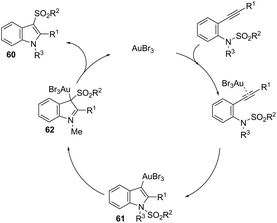 |
| | Scheme 55 | |
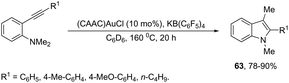 |
| | Scheme 56 | |
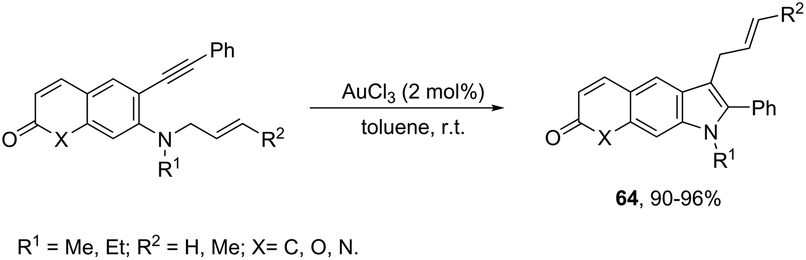
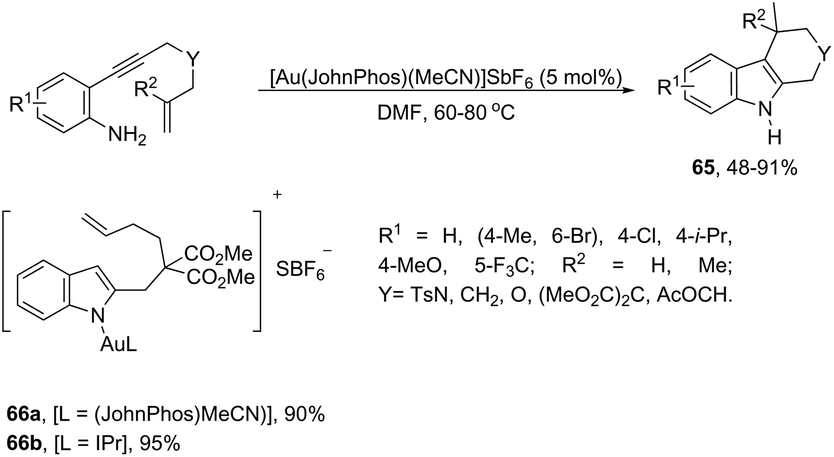
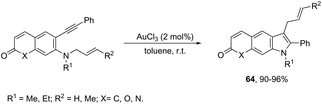
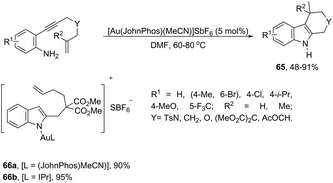
The intramolecular migration of alkenes has also been found to give 2,3-fused indole derivatives. The reaction of N-(allyl(methyl)-ortho-alkynylanilines) with a catalytic amount of AuCl3, in toluene, at room temperature, afforded the pyranoindolones 64via a cyclization/3,3 allyl migration sequence (Scheme 57).89 The 2,3-fused indole derivatives 65 were prepared in high yields by reacting ortho-alkynylanilines having an unsaturated chain bonded to the alkyne, using [Au(JohnPhos)(MeCN)]SbF6 as the catalyst (Scheme 58).90 It was determined that the indole ring formed in the first reaction step reacts with the gold catalyst to form gold stable non-catalytic intermediates 66. The intermediate 66 was isolated and the structure was proven by single crystal X-ray diffraction.
 |
| | Scheme 57 | |
 |
| | Scheme 58 | |
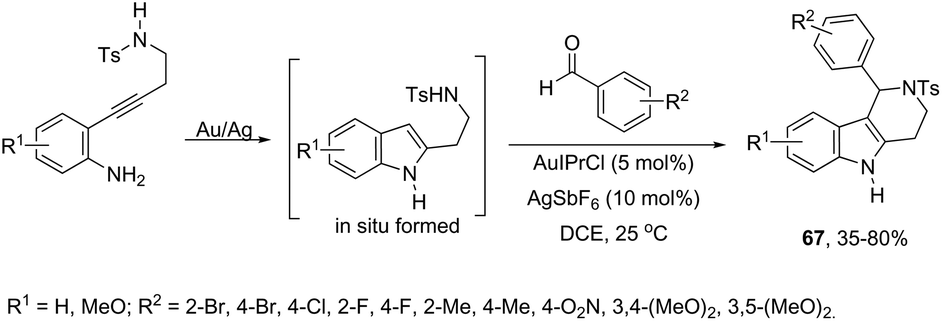
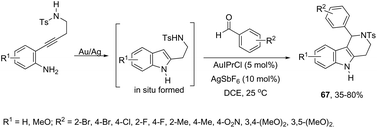
Aldehydes can be also used in combination with amines for the intramolecular functionalization of imines in the gold-catalyzed cyclization of 2-(4-aminobutynyl)anilines. The combination of catalytic amounts of AuIPrCl and AgSbF6 was found to be the most efficient catalytic system, which promoted the formation of 1-aryl-N-tosyl-2,3,4,5-tetrahydropyrido[4,3-b] indole derivatives 67 in moderate to good yields (Scheme 59).91 Further studies determined that the Au(I) species activates the alkyne via π-coordination with the carbon–carbon triple bond and Ag(I), facilitating the second step of the cyclization. Alternatively, the gold-catalyzed reaction of 2-[(2-aminophenyl)ethynyl]phenylamine derivatives with aldehydes is an efficient strategy used to prepare 1H-indolo-[3,2-c]quinolones.92
 |
| | Scheme 59 | |
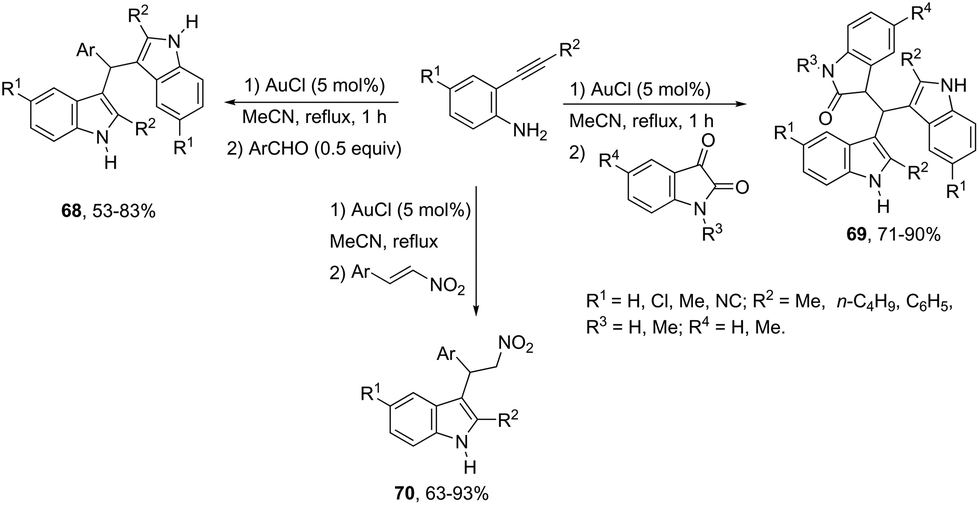
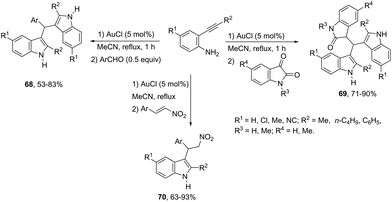
The intermolecular version for gold-catalyzed cyclization/functionalization of the 3-position of ortho-alkynylanilines was also developed for the synthesis of 3-substituted indoles. For example, starting from ortho-alkynylanilines, three different classes of 3-substituted indoles were obtained, applying the same reaction conditions, just by changing the electrophile source.93 The reaction of ortho-(phenylethynyl)aniline with aldehydes, catalyzed by 5 mol% AuCl in acetonitrile, under reflux, gave the bis-(indolyl)methanes 68. The electrophilic source exchange for isatin instead of aldehydes led to di(indolyl)indolin-2-ones 69, while the use of nitroalkenes as electrophiles gave 2-indolyl-1-nitroalkanes 70via a cycloaddition/Michael addition sequence (Scheme 60).
 |
| | Scheme 60 | |
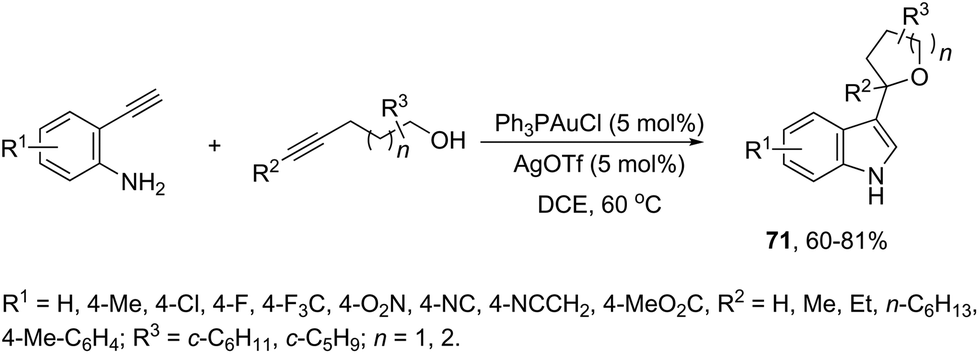
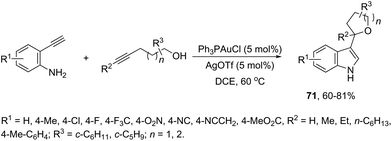
The gold(I)-catalyzed cyclization of ortho-alkynylanilines in the presence of alkynols provided an attractive route to a one-pot synthesis of 2-tetrahydrofuranyl indoles 71 (Scheme 61).94 The studies carried out to determine the optimal reaction conditions indicated that Ph3PAuCl in combination with AgOTf was the best choice to promote the cyclization.
 |
| | Scheme 61 | |
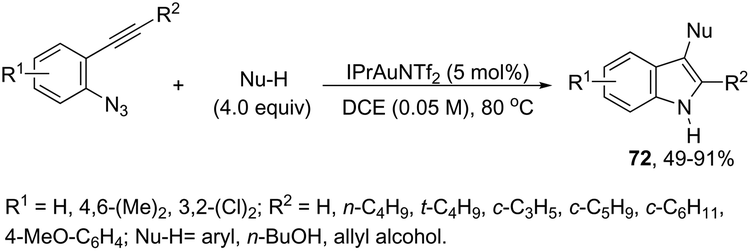
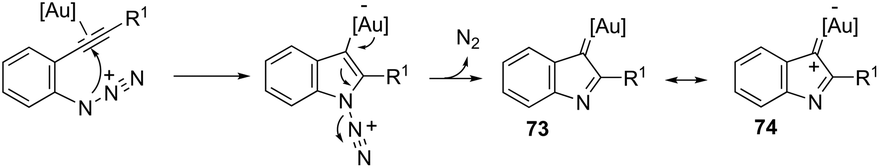
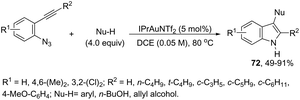
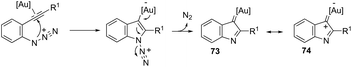
The nitrene precursors derived from an azido group, located in the ortho-azidoarylalkynes, were successfully utilized as electrophilic centers, for the general synthesis of 3-substituted indoles 72 (Scheme 62).95 When substituted ortho-azidoarylalkynes was used as a nucleophile, a mixture of regioisomers was obtained. The reaction conditions dramatically influenced the distribution of products, whereas high temperature (80 °C), using 1,2-dichloroethane as the solvent and IPrAuNTf2 as the catalyst, led to the best yield and ratio of regioisomers. The reaction is believed to proceed via gold carbene 73 and its cationic resonance form 74, which could act as an electrophilic source (Scheme 63).
 |
| | Scheme 62 | |
 |
| | Scheme 63 | |
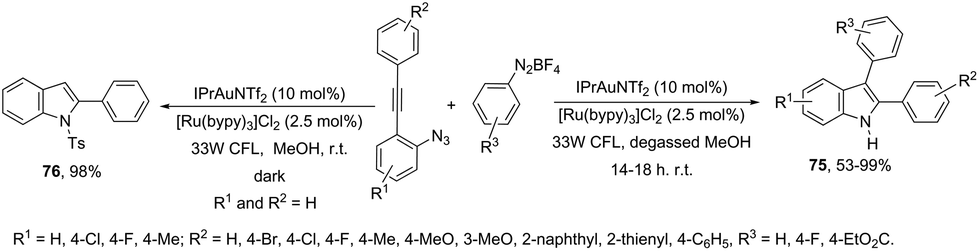
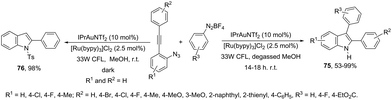
In addition to the ortho-azidoarylalkynes, the introduction of an aryl group at the 3-position of indoles was recently carried out through the reaction of N-Ts-ortho-alkynylanilines with phenyldiazonium catalyzed by dual Au/Ru catalysts under irradiation of a 33 W compact fluorescent light (CFL) bulb (Scheme 64).96 Under these experimental conditions, the 3-arylindole products 75 competed with protodeauration product 76. The chlorine anion present in the catalyst and the use of photoredox catalysis were essential for the formation of 3-arylindoles instead of 3-hydrogenated indoles. A conceptually similar approach employed heterogeneous gold nanoparticles to catalyze the cyclization of ortho-alkynylanilines in the preparation of 3,3′-bisindolyl products via domino cycloisomerization/oxidative homocoupling.97
 |
| | Scheme 64 | |
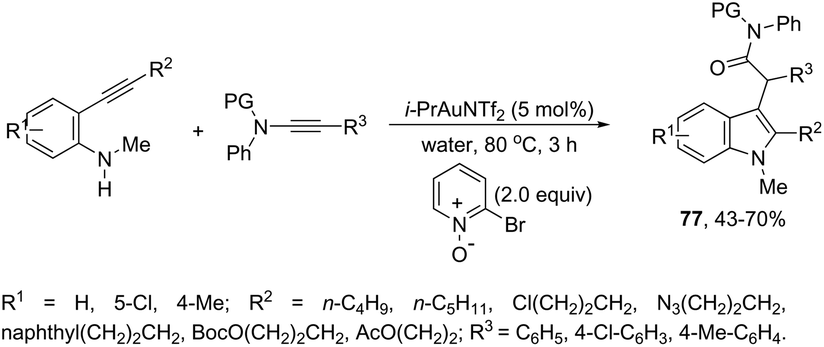
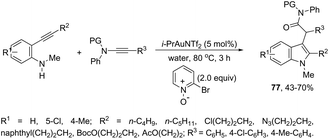
The use of gold-catalyzed tandem cycloisomerization/functionalization reaction of ortho-alkynylanilines and ynamides has also been explored to access functionalized 2-(1H-indol-3-yl)acetamides 77 (Scheme 65).98 The reaction was carried out in water and the IPrAuNTf2 catalyst promoted the cyclization of ortho-alkynylanilines and the intermolecular oxidation of ynamides.
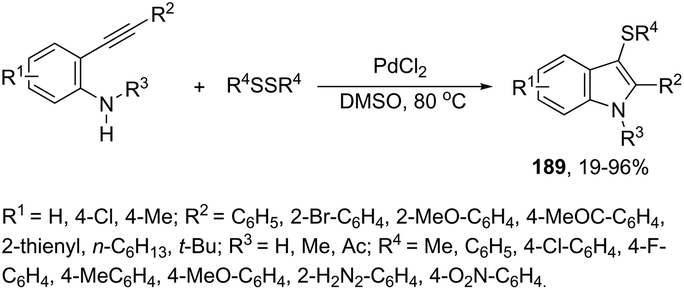
 |
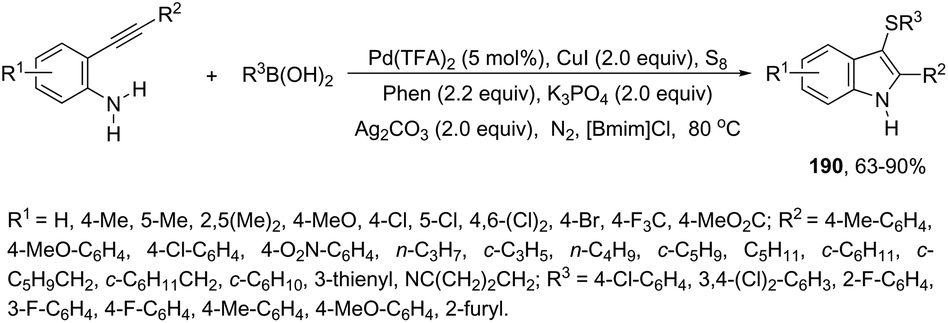
| | Scheme 65 | |
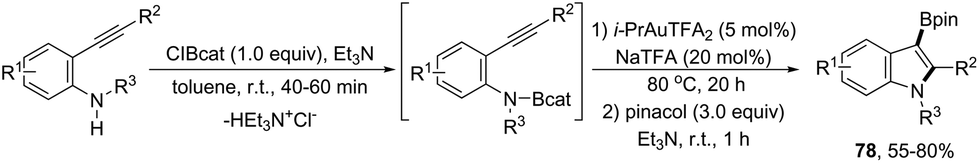
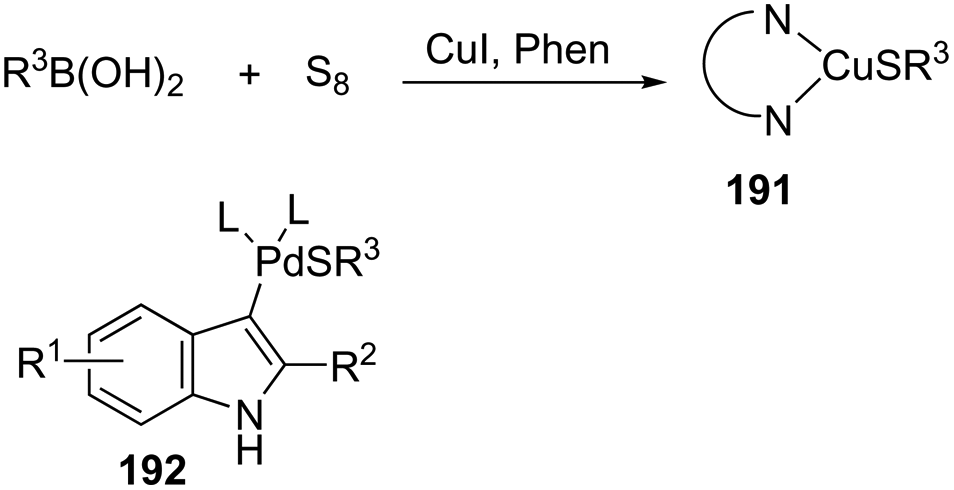
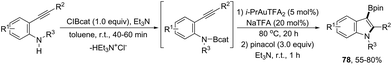
An alternative gold-catalyzed system constituted by a carbophilic gold cation and a trifluoroacetate anion allowed the formation of 3-borylated indoles 78 (Scheme 66).99 The reaction proceeds via formation of carbon–boron and carbon–nitrogen bonds in a single step from catalytic aminoboration of carbon–carbon triple bonds by boron–nitrogen σ bonds. Primary anilines led to the formation of 3-unsubstituted indoles via protodeauration promoted by N–H. Secondary anilines such as HNBn, HNTs, and HNMbs provided the product in very similar yields. The use of catecholborane, for the in situ formation of boron–nitrogen σ bonds, failed in the formation of 3-borylated indoles; however, the reaction starting from ortho-alkynylanilines and β-chlorocatecholborane gave the requisite boron–nitrogen intermediate, which afforded the products in good yields. The presence of sodium trifluoro acetate was also decisive for the success of the reaction, reactivating the catalyst, which was inhibited by the in situ formed Et3NHCl.
 |
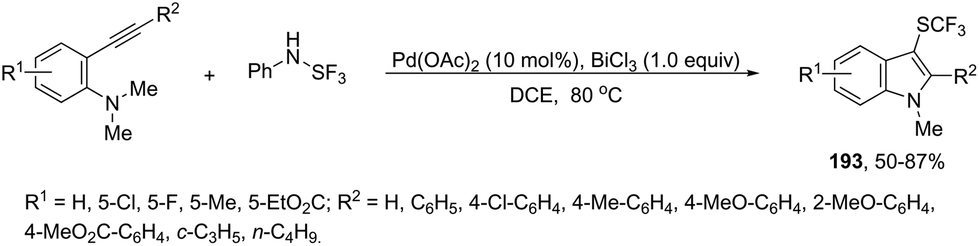
| | Scheme 66 | |
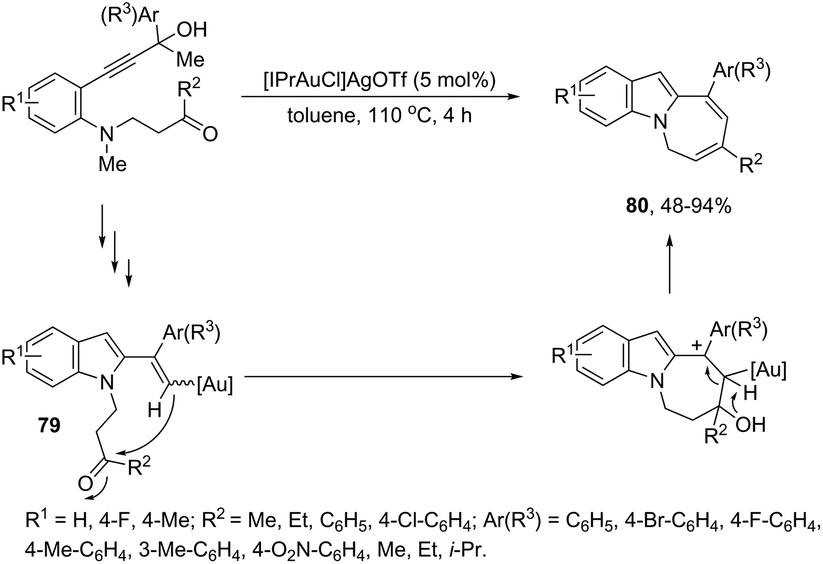
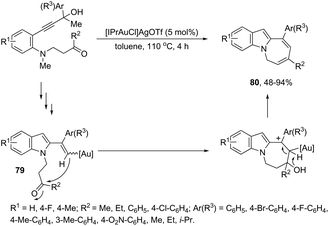
The gold-catalyzed cyclization of ortho-alkynylanilines, having a reactive group bonded to the aniline nitrogen, is a versatile process for the N1–C2 connection to give fused indoles. Under optimal reaction conditions, [Au(IPr)Cl]/AgOTf, toluene and under reflux, the azepino[1,2-a]indoles 80 were obtained from 3-(2-aminoaryl)propynols (Scheme 67).100 The possible mechanism to explain this reaction was hypothesized based on a control experiment, which indicated the initial formation of vinyl–gold intermediates 79 through a gold-triggered 5-endo-dig hydroamination/dehydration sequence. The intramolecular condensation of 79 with the ketone group would produce 80 after dehydration/1,3-proton-transfer/protodeauration sequence.
 |
| | Scheme 67 | |
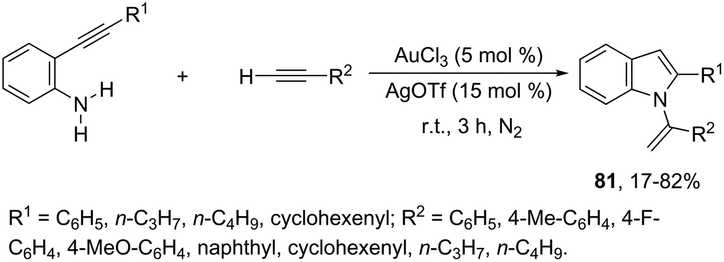
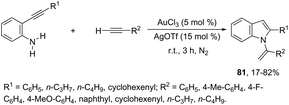
The sequential intermolecular hydroamination/cyclization reaction of anilines with alkynes was also used for the synthesis of various substituted indoles. An interesting intermolecular hydroamination sequence has been developed as an efficient route to the synthesis of N-vinylindoles 81, starting from ortho-alkynylanilines and terminal alkynes (Scheme 68).101 The process involves an efficient double hydroamination reaction catalyzed by gold(III) in the absence of a solvent. The authors determined that the first reaction step occurs via an intermolecular hydroamination, which is followed by intramolecular amination.
 |
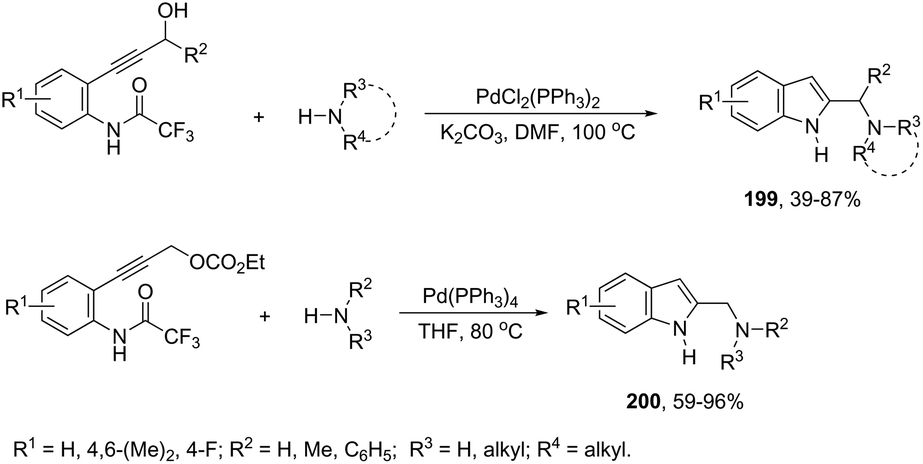
| | Scheme 68 | |
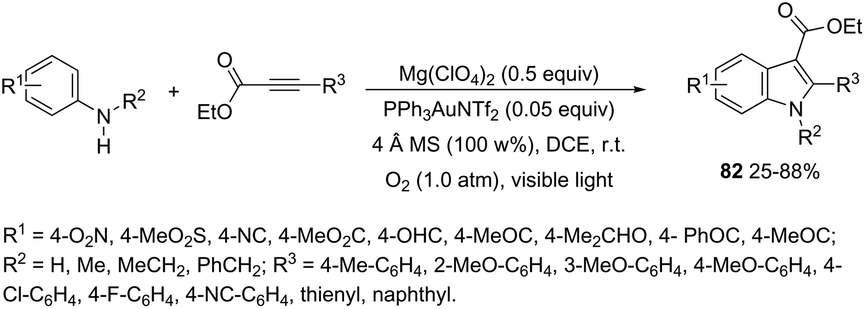
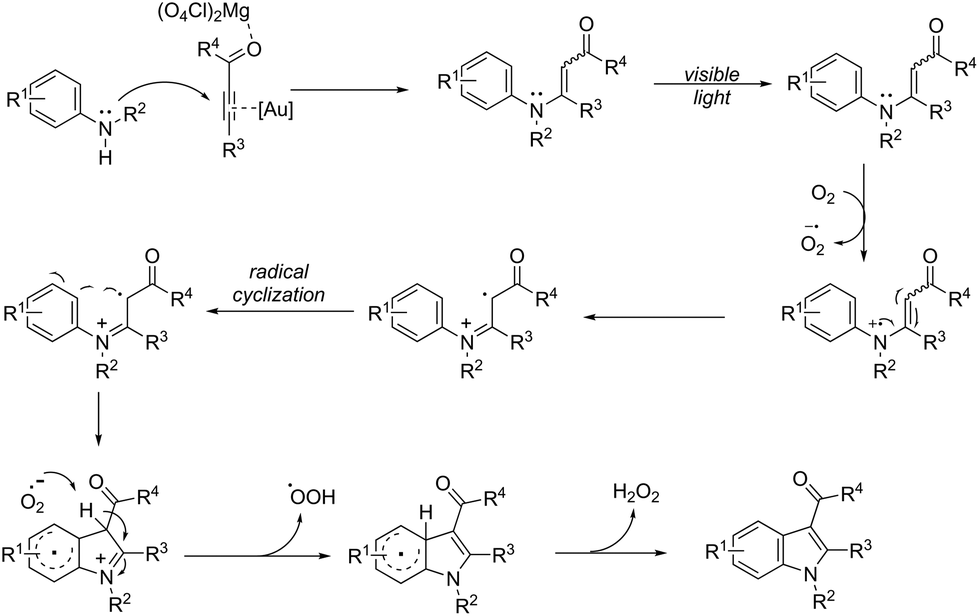
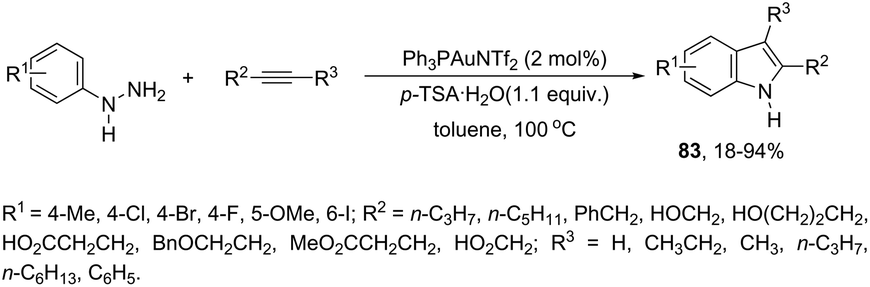
An attractive method for the generation of 2,3-functionalized indoles is the gold-catalyzed hydroamination and visible-light-enabled photoredox cross-dehydrogenative coupling of anilines and internal alkynes.102 The cyclization of anilines with arylpropiolates in the presence of 5 mol% of catalyst PPh3AuNTf2, 100 wt% of activated powdered 4 Å molecular sieves, and Mg(ClO4)2 in DCE, under a balloon oxygen atmosphere, and at room temperature, gave the indoles 82 (Scheme 69). The authors reported that the presence of the oxygen atmosphere and the use of Mg(ClO4)2 as a Lewis acid were essential to obtain good yields. The gold catalyst and a Lewis acid are fundamental for complexation with propiolate oxygen, increasing the reactivity of the triple bond towards the nitrogen nucleophilic attack (Scheme 70). A complementary method involving hydrohydrazination/indolization tandem reaction of alkynes and arylhydrazines has been developed to synthesize 2,3-disubstituted indoles 83 (Scheme 71).103 The reaction was not sensitive to moisture and required very low Ph3PAuNTf2 loading (2 mol%) in combination with p-TSA·H2O to obtain the indoles. The authors proposed two different mechanisms, which depend on the substrates; with the alkynes having OH/COOH groups in their structure, a hydroalkoxylation/hydrocarboxylation sequence forms exocyclic enol ethers/lactones that react with hydrazines to produce indoles. However, the absence of OH/COOH groups in the alkynes leads to hydration to generate ketones that react with arylhydrazines to give the desired indoles.
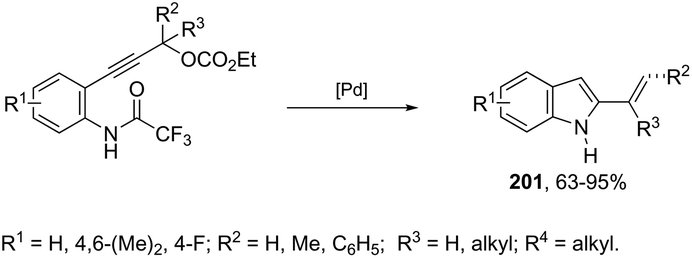
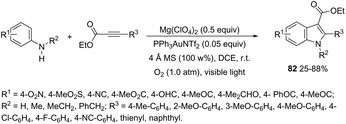 |
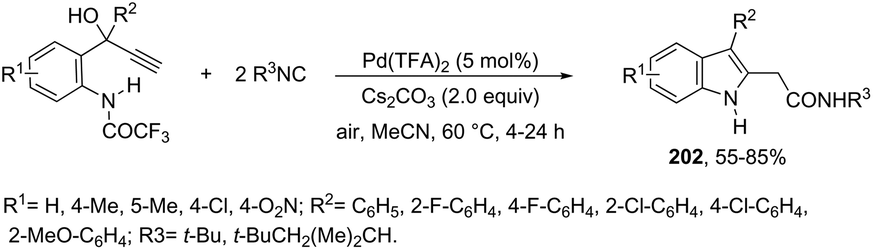
| | Scheme 69 | |
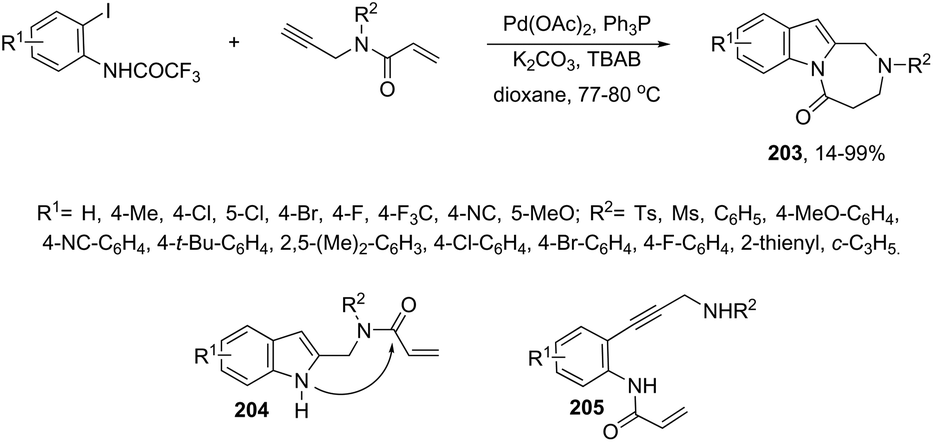
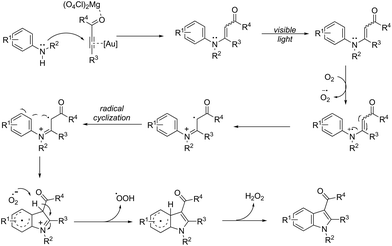 |
| | Scheme 70 | |
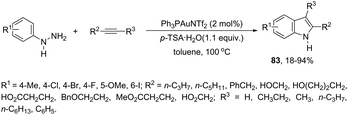 |
| | Scheme 71 | |
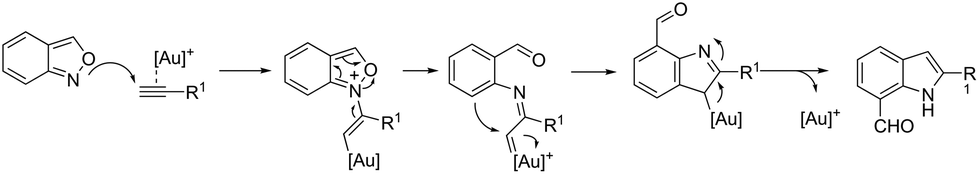
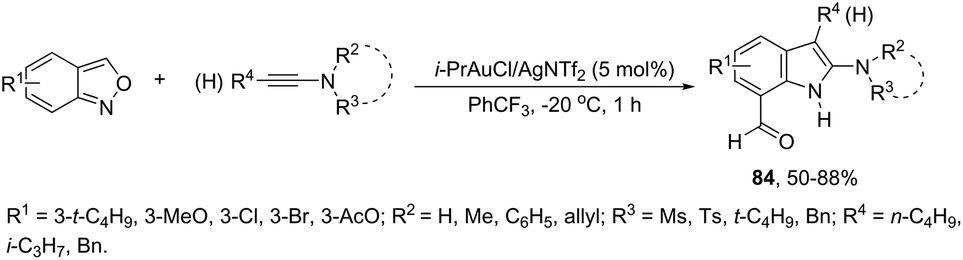
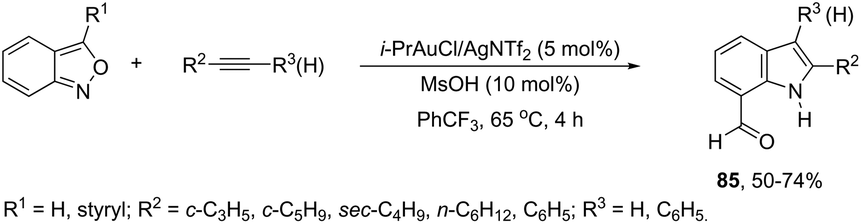
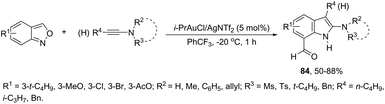
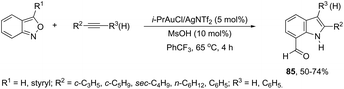
A conceptually similar approach utilized anthranil both as a source of nitrogen and for ortho-aryl carbon–hydrogen insertion in the preparation of unprotected 7-acylindoles (Scheme 72).104 The reaction of anthranil with alkynes catalyzed by gold gave an electrophilic α-imino gold carbenoid intermediate, which afforded the desired indoles via an intramolecular ortho-aryl carbon–hydrogen insertion (Scheme 73). The control experiments showed that the highest yields of 7-acylindoles 84 were obtained when IPrAuCl/AgNTf2 was used as the catalyst in PhCF3 at −20 °C. Under these reaction conditions, polarized terminal and non-terminal alkynes, ynamides and various substituted anthranils could be used as substrates in the cyclization. In the case of non-polarized alkynes, just a slight adjustment of the reaction conditions, such as raising the temperature, extending the reaction time and adding 10 mol% MsOH, to facilitate the deauration process, led to the desired indoles 85 (Scheme 74).
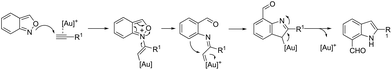 |
| | Scheme 72 | |
 |
| | Scheme 73 | |
 |
| | Scheme 74 | |
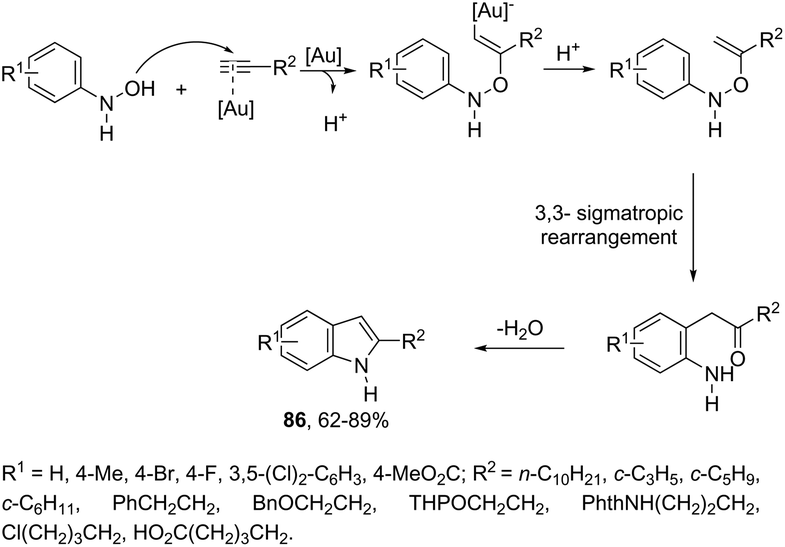
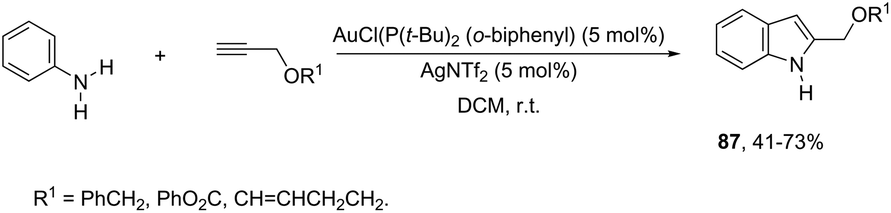
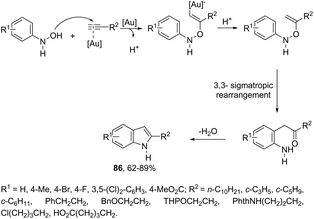
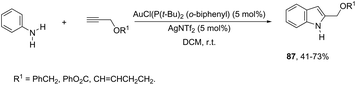
N-Arylhydroxylamines have been used as nitrogen donors in gold-catalyzed preparation of 2-alkylindoles. This reaction effectively combined the N-arylhydroxylamines with aliphatic terminal alkynes giving access to o-alkenyl-N-arylhydroxylamines, which underwent sequential 3,3-rearrangements and cyclodehydrations to afford 2-alkylindoles 86 (Scheme 75).105 The reaction was best catalyzed by (ArO)3PAuNTf2 (Ar = 2,4-di-tert-butylphenyl) in DCE. The authors reported the high regioselectivity towards 2-alkylindoles in the complete absence of 3-alkylindoles. This regioselectivity was attributed to the Markovnikov additions of hydroxamic acids to the gold-activated terminal alkynes. The reaction conditions were general for terminal alkyl alkynes with limitations to aryl terminal alkynes and internal alkynes, and the corresponding indoles were obtained in poor yields. Alternatively, AuCl(P(t-Bu)2(ortho-biphenyl) catalyzed reactions of propargyl ethers with N-hydroxyanilines yielded ethyl 2-(1H-indolyl) derivatives 87 (Scheme 76).106 The authors studied the N- versus o-attack chemoselectivity and concluded that for propargyl ethers and their benzoate derivatives the o-attack occurred selectively, whereas for 1,6-enynes the N-attack proceeded exclusively.
 |
| | Scheme 75 | |
 |
| | Scheme 76 | |
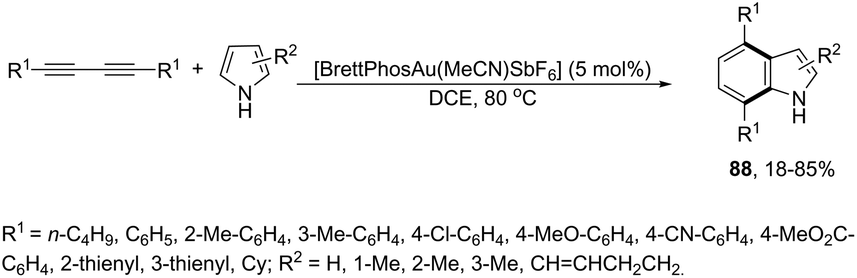
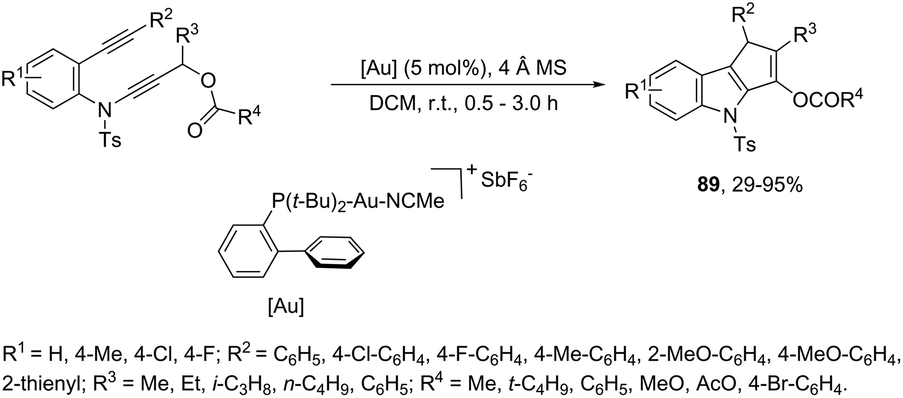
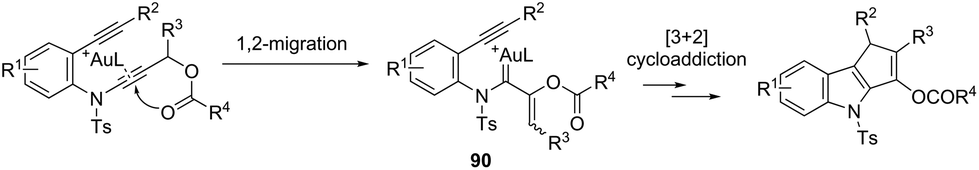
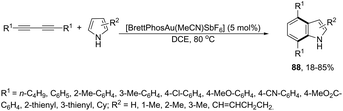
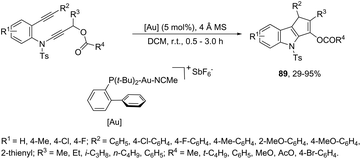
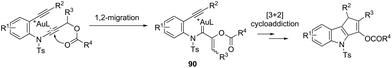
The cycloaddition reaction has been extensively applied to generation of heterocycles. For the indole synthesis a gold(I)-catalyzed intermolecular formal [4 + 2] reaction between 1,3-diynes and pyrroles has been developed. This reaction involved the intermolecular hydroarylation of 1,3-diynes with pyrroles, followed by an intramolecular hydroarylation to afford the 4,7-disubstituted indoles 88 (Scheme 77).107 A gold-catalyzed cycloisomerization/[3 + 2] cycloaddition sequence, with diynes having an ynamide propargyl ester or carbonate group, has been developed for the preparation of 3-acyloxy-1,4-dihydrocyclopenta[b]indoles 89 (Scheme 78).108 Control experiments indicated that the formation of indoles 89 proceeds through a competitive 1,2-OAc migration, followed by [3 + 2] cycloaddition of the vinyl gold-carbenoid intermediate 90 with the alkyne (Scheme 79).
 |
| | Scheme 77 | |
 |
| | Scheme 78 | |
 |
| | Scheme 79 | |
2.5. Indium-catalyzed synthesis of indoles
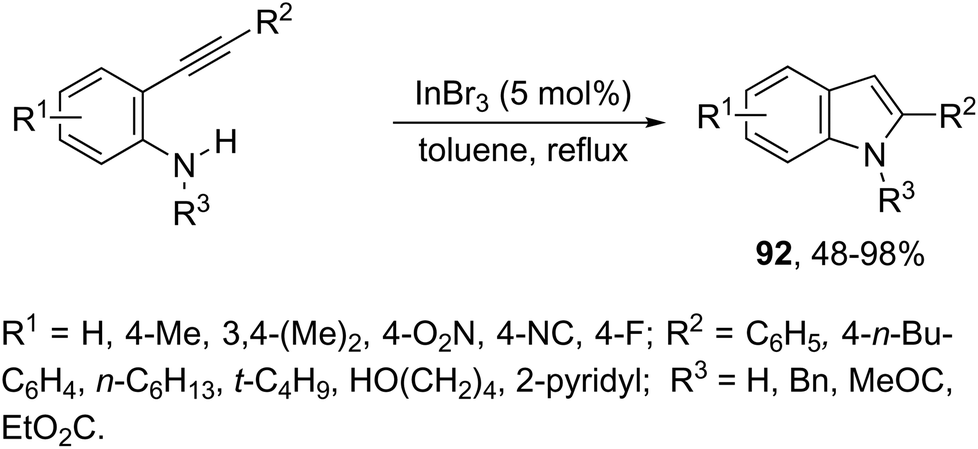
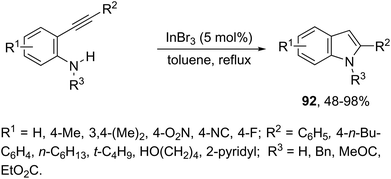
Indium-mediated cyclization reaction of alkynes with a nitrogen source was also utilized to prepare indole derivatives. When ortho-alkynylnitroarenes were reacted with indium and aqueous HI, under reflux of benzene, 2-arylindoles 91 were obtained in good yields (Scheme 80).109 The reaction carried out in the presence of indium(III) chloride, in a mixture of THF and H2O as the solvent, formed only ortho-alkynylanilines. However, when ortho-alkynylanilines having an alkyl or aryl group on the alkyne were treated with indium(III) bromide in toluene under reflux, the 2-susbtituted indoles 92 were obtained from 48 to 98% yields (Scheme 81).110 In contrast, the ortho-alkynylanilines with a trimethylsilyl group or with no substituent group on the terminal alkyne gave polysubstituted quinoline derivatives via indium-promoted intermolecular dimerization.
 |
| | Scheme 80 | |
 |
| | Scheme 81 | |
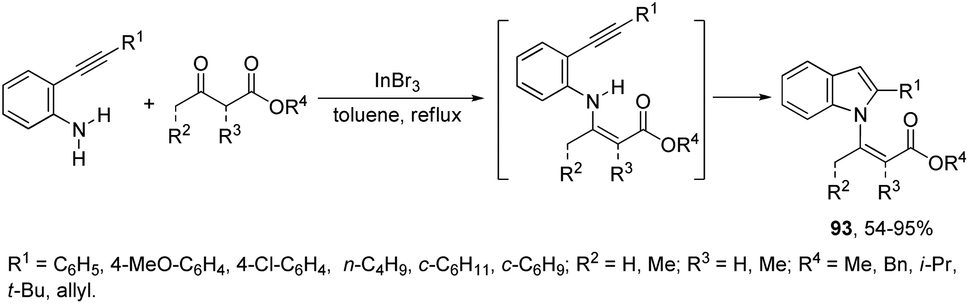
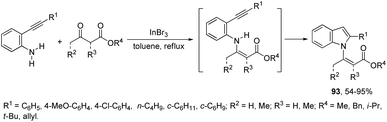
β-(N-Indolyl)-α,β-unsaturated esters 93 are readily accessible by tandem β-enamino ester formation, followed by cyclization with ortho-alkynylanilines catalyzed by indium(III) bromide (Scheme 82).111 It was observed that indium(III) bromide catalyzed the intermolecular amination with a subsequent intramolecular cyclization and promoted the formation of the β-enamino esters instead of intramolecular cyclization. The experimental results also indicated that the cyclization proceeded exclusively via a 5-endo-dig mode in place of 6-exo-dig mode.
 |
| | Scheme 82 | |
2.6. Iridium-catalyzed synthesis of indoles
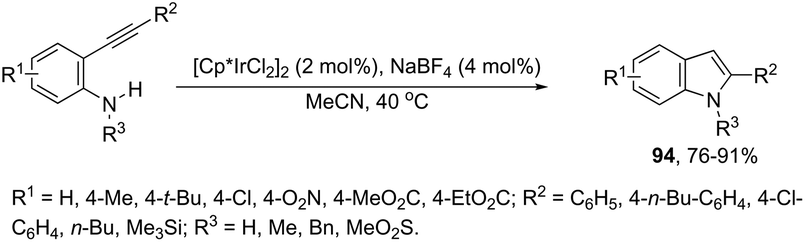
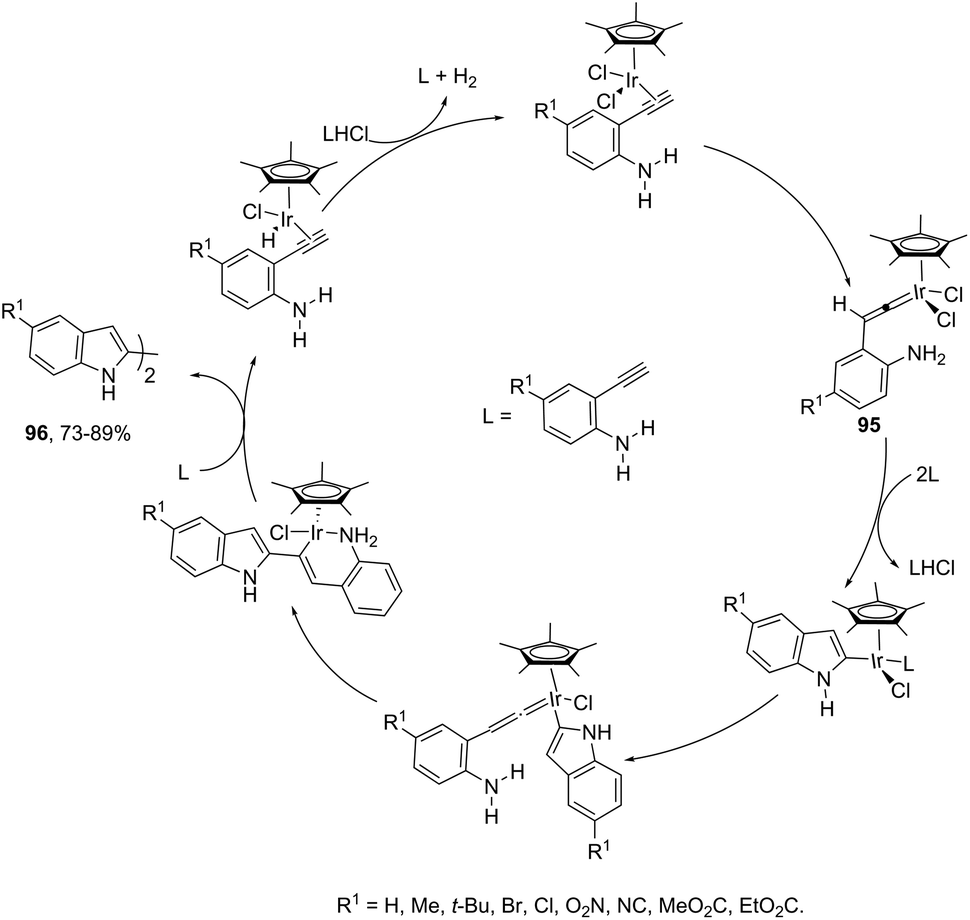
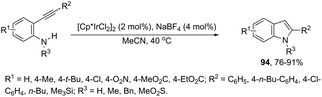
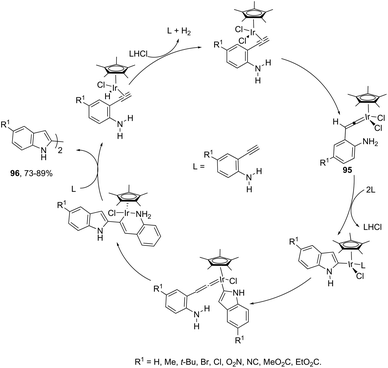
Indoles can also be synthesized from ortho-nitrogen substituted aryl alkynes under iridium-catalyzed cyclization reaction. The synthesis of 2-substituted indoles 94via [Cp*IrCl2]2-catalysed cyclization of internal ortho-alkynylanilines has been developed (Scheme 83).112 If hydrogen-substituted alkynes were used as substrates, 2,2′-biindoles 96 were obtained in good yields (Scheme 84).113 The mechanistic studies indicated that the reaction proceeds via formation of a (vinylidene)iridium intermediate 95 and an intramolecular hydroamination, followed by the insertion reaction.
 |
| | Scheme 83 | |
 |
| | Scheme 84 | |
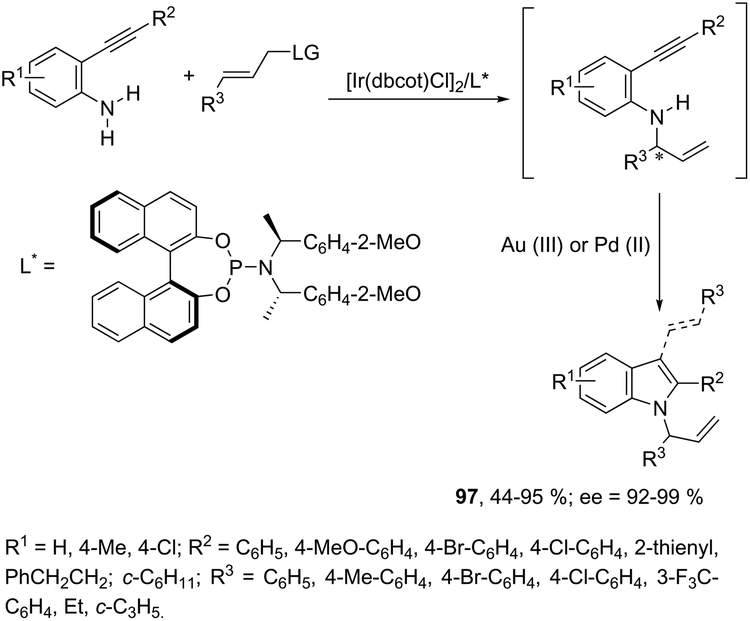
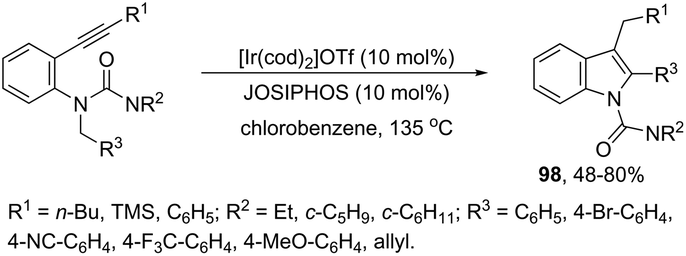
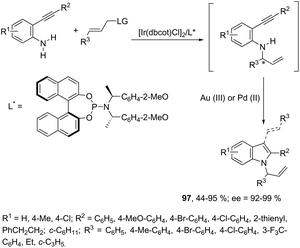
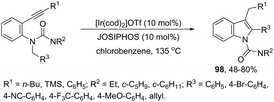
The asymmetric synthesis of functionalized N-allylindoles 97 was achieved by the iridium-catalyzed asymmetric allylic amination reaction with ortho-alkynylanilines, followed by cyclization reactions (Scheme 85).114 An interesting secondary sp3 carbon–hydrogen bond activation of N-benzylureas and an N-allyl urea, initiated by a cationic Ir(I)–JOSIPHOS catalyst, led to the preparation of 2,3-disubstituted indoles 98 (Scheme 86).115 The use of urea as a directing group, the secondary sp3 carbon–hydrogen bond cleavage and an intramolecular alkyne insertion were a key for the formation of indoles in good yields.
 |
| | Scheme 85 | |
 |
| | Scheme 86 | |
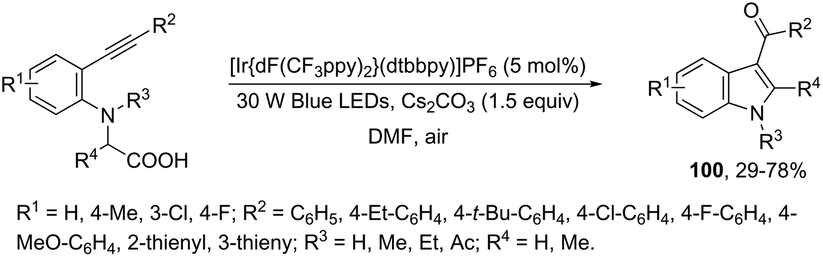
A concise route based on visible-light induced intramolecular oxidative cyclization of N,N-dibenzyl ortho-alkynylanilines, using an iridium complex as a photocatalyst, was developed for the synthesis of 3-acylindoles 99 (Scheme 87).116 The reaction proceeds via addition of an sp3 carbon–radical alpha-amino group to the carbon–carbon triple bond of the alkyne, followed by a carbon–oxygen bond formation. When ortho-alkynylanilines having a carboxylic acid as a substituent were treated with the iridium complex as the photocatalyst, the 3-acylindoles 100 were obtained in yields ranging from 29 to 78% (Scheme 88).117 The control experiments, carried out by the authors, indicated that the oxygen atom of the ketonic carbonyl group comes from the atmosphere rather than carboxylic acid.
 |
| | Scheme 87 | |
 |
| | Scheme 88 | |
2.7. Iron-catalyzed synthesis of indoles
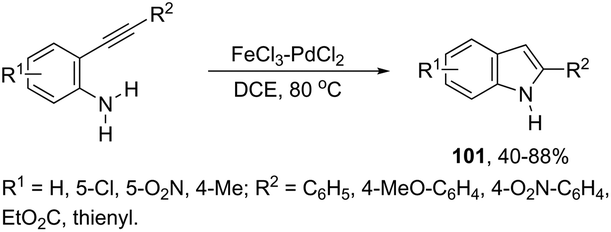
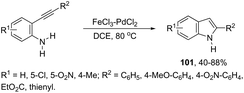
In recent years, the necessity for the development of environmentally benign protocols for the synthesis of heterocycles using green, mild, and relatively cheaper methods has grown significantly.118 Therefore, iron salts have appeared as a versatile alternative due to their low price and toxicity.119 The use of catalytic amounts of iron salts has been applied to promote the carbon–heteroatom (C–N, C–O, and C–S) bond formation.120 Catalytic and stoichiometric amounts of iron reagents also appear as a useful alternative to promote cyclization type reactions.121 Indoles have also been synthesized by several methods using catalytic or stoichiometric amounts of iron salts as discussed below. The catalytic combination between FeCl3 and PdCl2 was successfully used for the synthesis of 2-substituted indoles 101 (Scheme 89).122 In this reaction, iron(III) presented a dual activity as a cooxidant and a Lewis acid to promote the cyclization.
 |
| | Scheme 89 | |
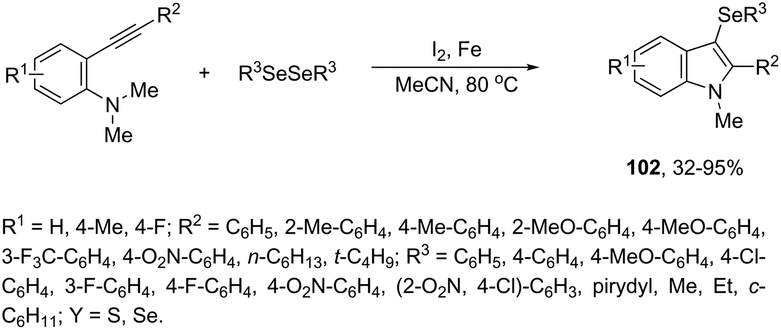
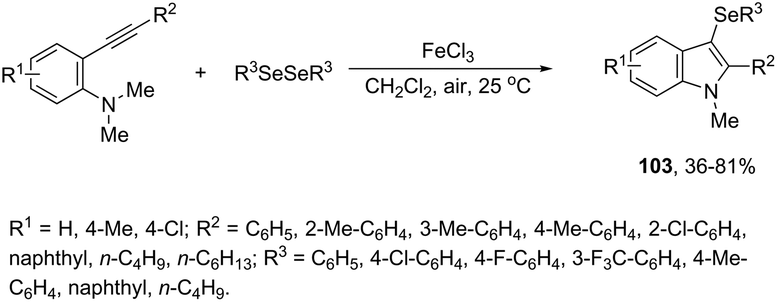
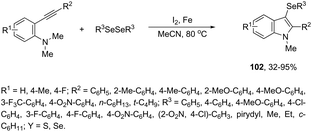
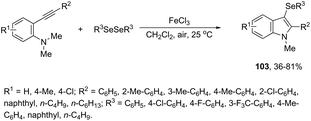
N-Methyl-3-chalcogeno-indoles 102 were prepared by iron–iodine-mediated electrophilic annulation reactions of ortho-alkynylaniline derivatives using diorganyl disulfides or diorganyl diselenides as a chalcogen source (Scheme 90).123 In this reaction, the function of iron was to form a complex with nitrogen and the alkyne enabling cyclization, whereas iodine was used to promote the in situ preparation of the chalcogen electrophilic species, responsible for the activation of the triple bond and functionalization of the 3-position of the indole. We developed an approach for the synthesis of 3-organoselenyl-(N-methyl)indoles 103via cyclization of ortho-alkynylanilines using a cooperative action between iron(III) chloride and diorganyl dichalcogenides in the complete absence of a halogen source (Scheme 91).124 The main advantage of this methodology is the incorporation of both portions of diorganyl diselenides (2 RSe) in the final product, which makes it a more useful and valuable method in terms of atom economy.
 |
| | Scheme 90 | |
 |
| | Scheme 91 | |
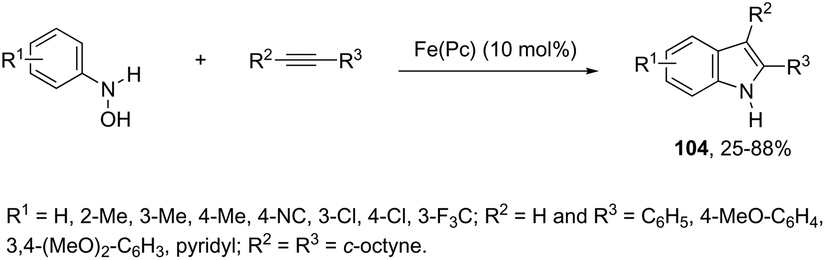
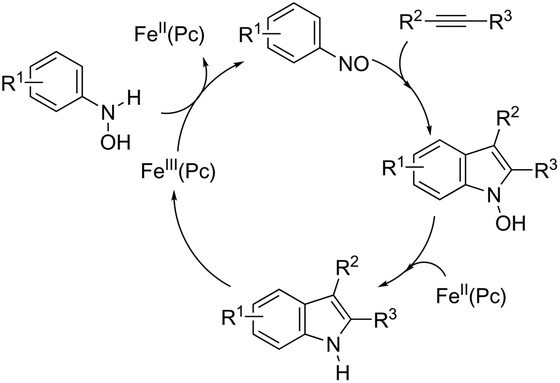
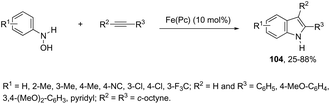
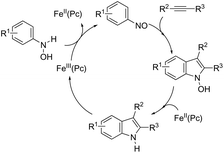
The iron-catalyzed annulation reactions of aryl hydroxylamines with terminal and internal alkynes provide another useful route to synthesize 3-arylindole derivatives 104 (Scheme 92).125 The best results were obtained with 10 mol% iron(II) phthalocyanine [Fe(Pc)] under toluene reflux. The proposed catalytic mechanism involved the initial oxidation of hydroxylamines by a Fe(III)(Pc) species to give the arylnitroso intermediate, which reacts with alkynes in a cyclocondensation reaction affording N-hydroxyindole. The catalytic cycle is completed by reducing the N-hydroxyindole to the corresponding indole with Fe(II)(Pc) (Scheme 93).
 |
| | Scheme 92 | |
 |
| | Scheme 93 | |
2.8. Mercury-catalyzed synthesis of indoles
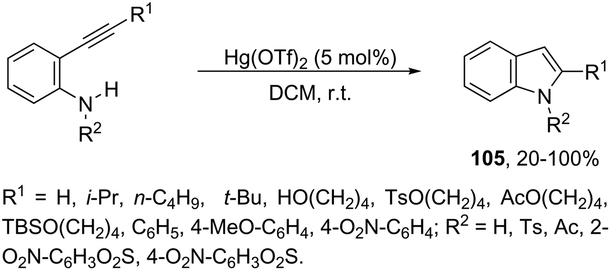
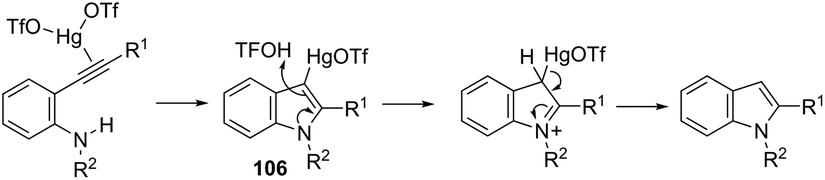
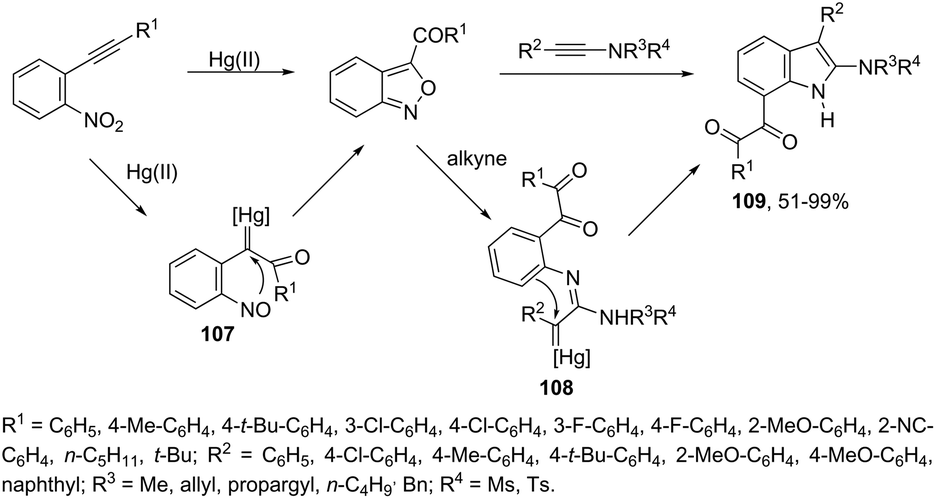
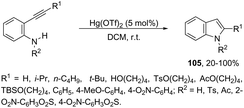
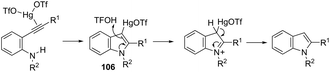
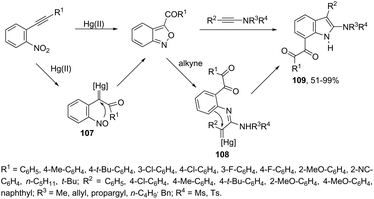
Until now, limited methods for indole synthesis via mercury-promoted cyclization reactions of alkynes with a nitrogen substrate have been developed. In 2007, it was reported that mercuric triflate-catalyzed cycloisomerization reaction of ortho-alkynylanilines afforded indole derivatives 105 (Scheme 94).126 The results support the hypothesis that an activation of the carbon–carbon triple bond by Hg(OTf)2via a π-complexation of the alkyne, followed by a nucleophilic attack of nitrogen, leads to the indol 3-mercury intermediate and TfOH. Protonation and demercuration of 106 give the indole and regenerate the catalyst Hg(OTf)2 (Scheme 95). Very recently, it has been demonstrated that Hg(OTf)2 catalyzed the cyclization of ortho-nitroalkynes to produce the corresponding indoles 109 in good yields via a regioselective 6-endo-dig process (Scheme 96).127 The structures of mercury–carbenes 107 and 108 have been proposed as the key intermediates in the reaction mechanism.
 |
| | Scheme 94 | |
 |
| | Scheme 95 | |
 |
| | Scheme 96 | |
2.9. Nickel-catalyzed synthesis of indoles
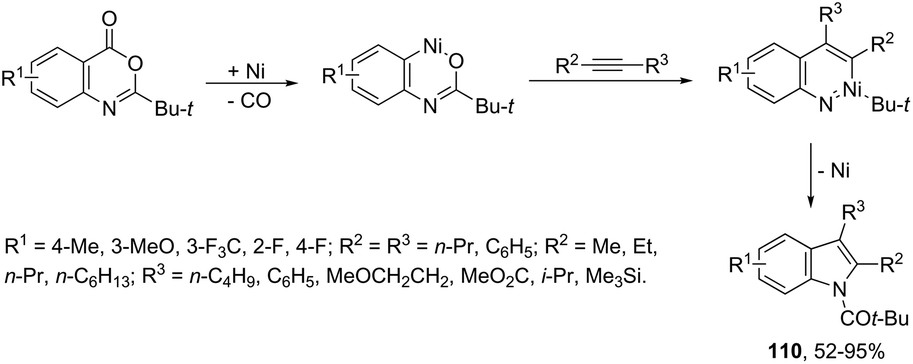
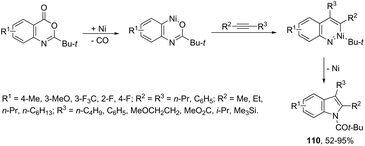
The nickel-catalyzed (6-3 + 2) cycloaddition reaction of anthranilic acid derivatives and alkynes gave indoles 110 (Scheme 97).128 The cyclization was carried out with catalytic amounts of Ni(cod)2 and xylene, under reflux. The mechanism of the reaction is believed to proceed via oxidative addition of an ester to a Ni(0) complex, decarbonylation, alkyne insertion, 1,3-acyl migration, and reductive elimination.
 |
| | Scheme 97 | |
2.10. Palladium-catalyzed synthesis of indoles
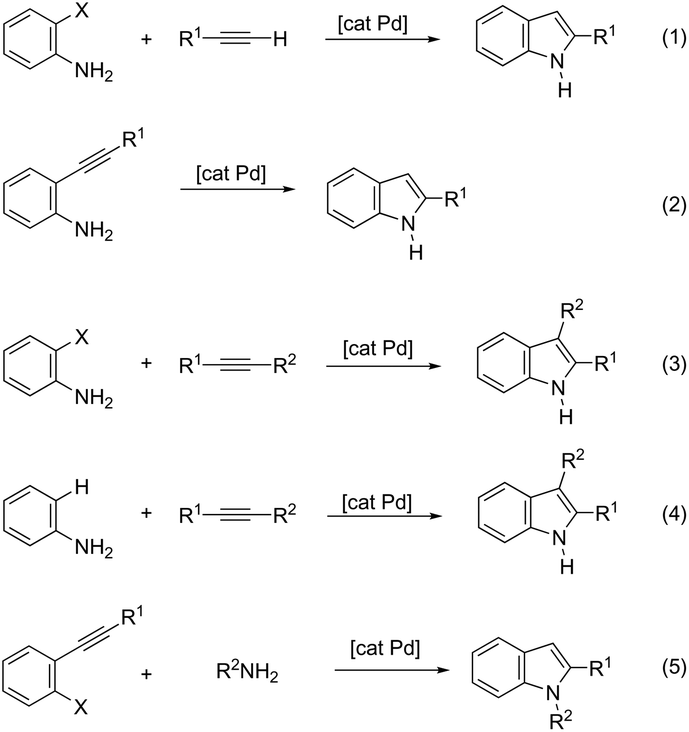
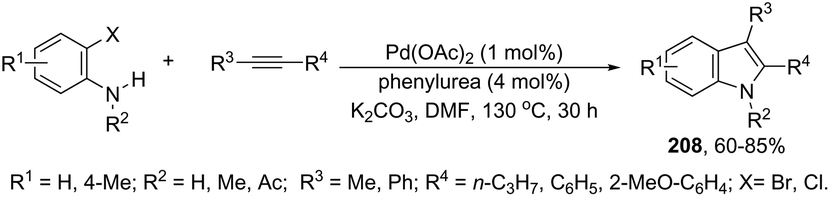
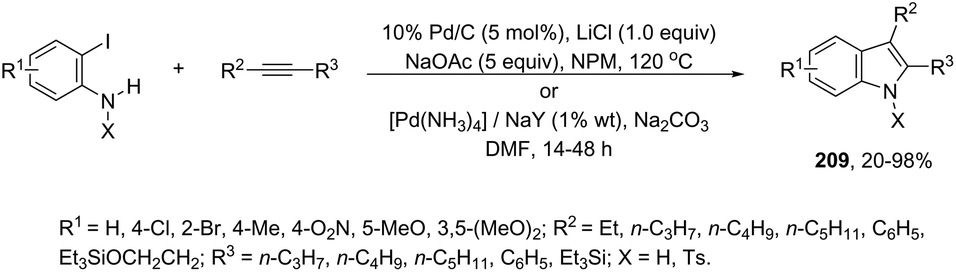
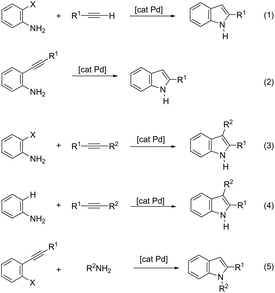
The palladium-catalyzed cyclization of alkynes is one of the most convenient methods for the preparation of indoles.129–138 Extensive efforts have been made in developing new methods for the synthesis of indoles by inter- or intramolecular palladium-catalyzed cyclization on functionalized alkynes. The principal synthetic strategies for this purpose are based on the nitrogen nucleophilic addition to the active carbon–carbon triple bond. The nitrogen atom can be a portion of the starting material or introduced in a key position of the molecule that leads to a subsequent cyclization reaction. In cases in which the nitrogen atom is a part of the molecule, the cyclization reactions may occur via: (i) palladium cross-coupling of 2-haloanilines and terminal alkynes or palladium-catalyzed cyclization of 2-alquinilanilines (Scheme 98, eqn (1) and (2)), or (ii) palladium-catalyzed annulation of 2-haloanilines and disubstituted alkynes (Scheme 98, eqn (3)), and palladium-catalyzed C–H activation of anilines, followed by inter- or intramolecular cyclization with alkynes (Scheme 98, eqn (4)). When the nitrogen atom is not present on the structure of the starting material, the palladium-catalyzed N-arylation/hydroamination sequence has been widely used to generate functionalized indoles (Scheme 98, eqn (5)). Next, the utility of these methodologies is addressed in terms of such specific examples that appear in the literature.
 |
| | Scheme 98 | |
2.10.1. Palladium-catalyzed cyclization of ortho-alkynylanilines.
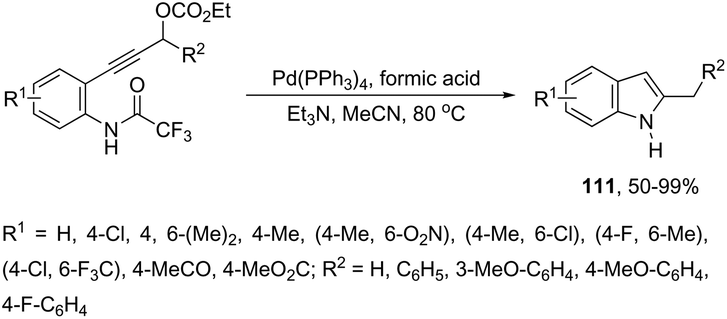
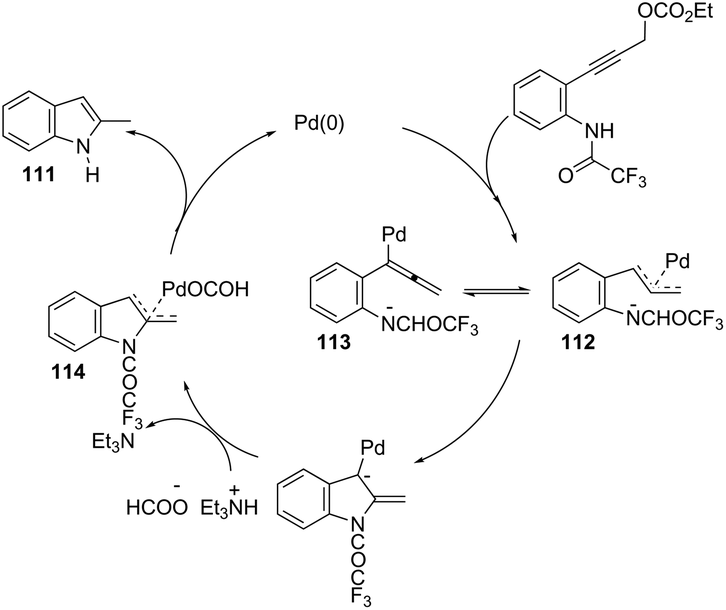
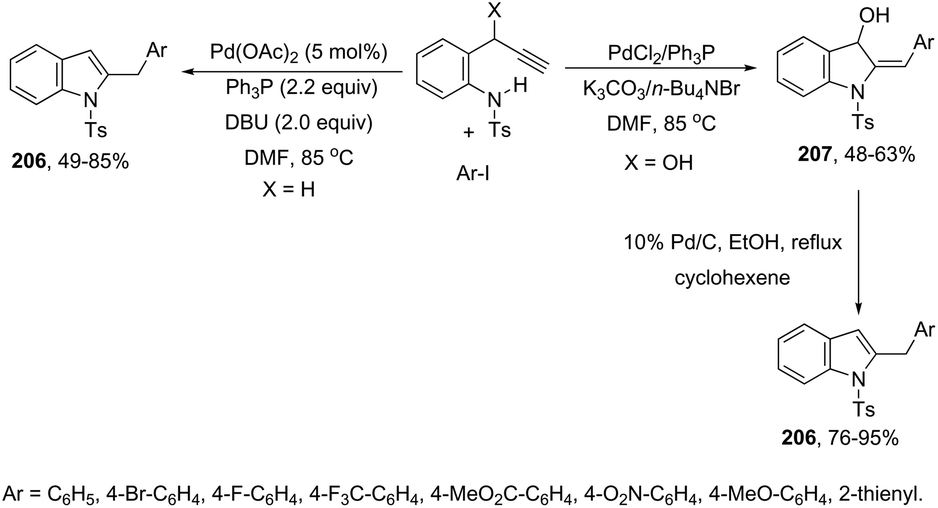
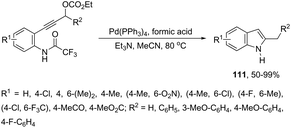
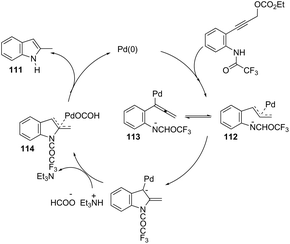
Propargyl carbonates have been used as substrates in palladium-catalyzed reductive cyclization reactions to access NH free 3-unsubstituted 2-alkylindoles 111 (Scheme 99).139 Under palladium-catalyzed conditions, the 3-(ortho-trifluoroacetamidophenyl)-1-propargyl carbonates were transformed into the π-propargyl palladium intermediate 112 and σ-allenyl palladium complex 113, which gave the carbene 114, via an intramolecular nucleophilic attack of the nitrogen atom at the central carbon of the allenyl/propargyl palladium complex. The transformation of the carbene 114 into a π-allylpalladium complex gave the alkylindoles 111via a concerted decarboxylation and hydride transfer sequence (Scheme 100).
 |
| | Scheme 99 | |
 |
| | Scheme 100 | |
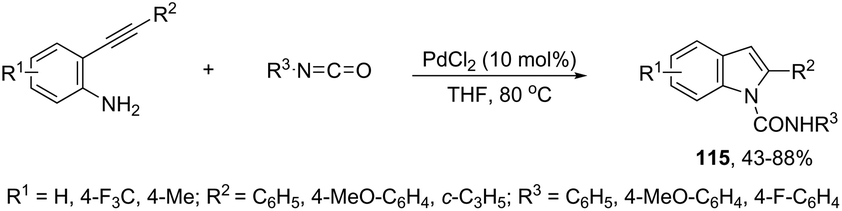
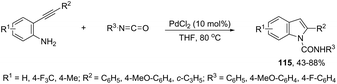
The ligand-free PdCl2 catalyst was active for the cyclization of 2-alkynyl urea affording indole-1-carboxamide derivatives 115 (Scheme 101).140 The 2-alkynyl urea was prepared in situ through reactions of 2-alkynylanilines with isocyanates. The main advantage of this protocol is the use of a simple experimental procedure (PdCl2 (10 mol%), THF and 80 °C) giving the indoles in 43–88% yields via an exclusive 5-endo-dig cyclization mode.
 |
| | Scheme 101 | |
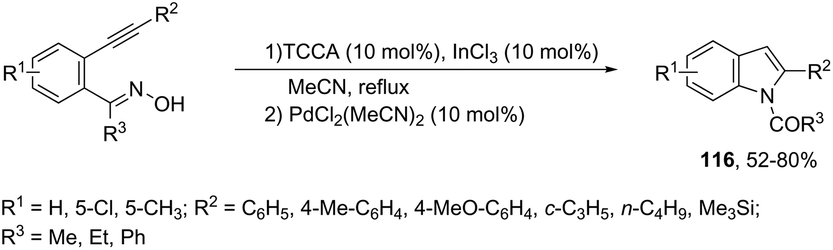
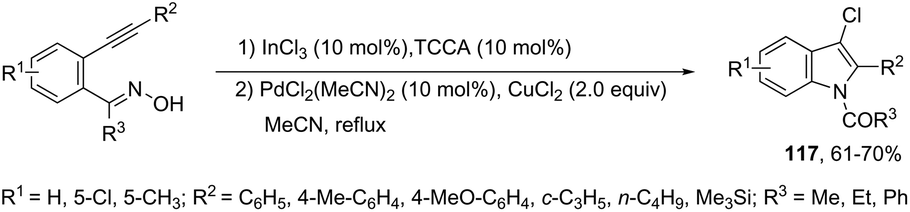
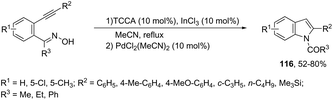
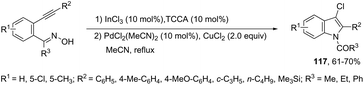
The reaction of 1-(2-alkynylphenyl)ketoximes with a palladium catalyst using cyanuric chloride and InCl3 as cocatalysts can afford the corresponding indole derivatives 116 (Scheme 102).141 The 2-alkynylanilide required as a starting material was initially formed via a Beckmann rearrangement of 1-(2-alkynylphenyl)-ketoxime. Thus, the intramolecular cyclization occurred in the presence of two catalytic systems to generate the indole derivatives. The authors also described that the simple addition of CuCl2 (2.0 equiv.) to the reaction medium led to the 3-chloroindoles 117via a multicatalytic one-pot Beckmann rearrangement/intramolecular cyclization/halogenation process (Scheme 103).
 |
| | Scheme 102 | |
 |
| | Scheme 103 | |
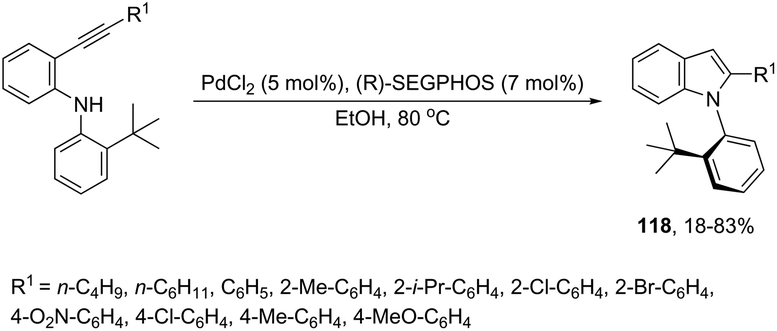
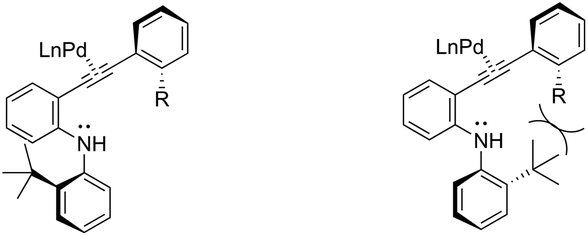
The asymmetric palladium-catalyzed reaction of 2-(t-butyl)-N-(2-ethynylphenyl)anilines has led to an enantioselective 5-exo-aminocyclization forming the nitrogen–carbon axially chiral indoles 118 in up to 83% ee (Scheme 104).142,143 The authors suggested that the enantioselectivity was strongly influenced by the bulkiness of the ortho-substituents and the electron density on the arylethynyl group due to the dynamic axial chirality generated by the twisting of the aryl substituent (Scheme 105).
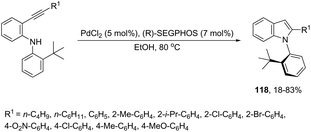 |
| | Scheme 104 | |
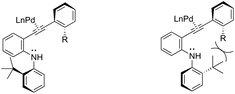 |
| | Scheme 105 | |
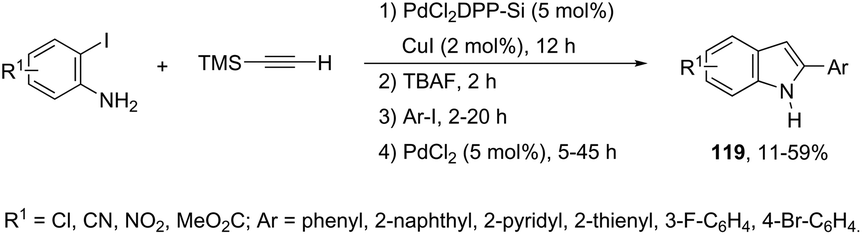
A versatile one-pot multi-step synthetic methodology, combining solid-supported heterogeneous and homogeneous palladium salts, was applied as a catalytic system in the synthesis of indoles 119 (Scheme 106).144 The sequence started with Sonogashira coupling of 2-iodoaniline with trimethylsilylacetylene giving the 2-[(trimethyl)ethynyl]aniline. The second step involved desilylation, affording the ethynylaniline. Then, Sonogashira coupling with an aromatic iodide, followed by cyclization gave the indole derivatives in yields ranging from 11 to 59% for the four-step reactions (Scheme 106).
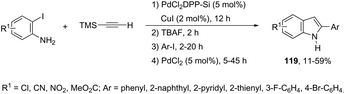 |
| | Scheme 106 | |
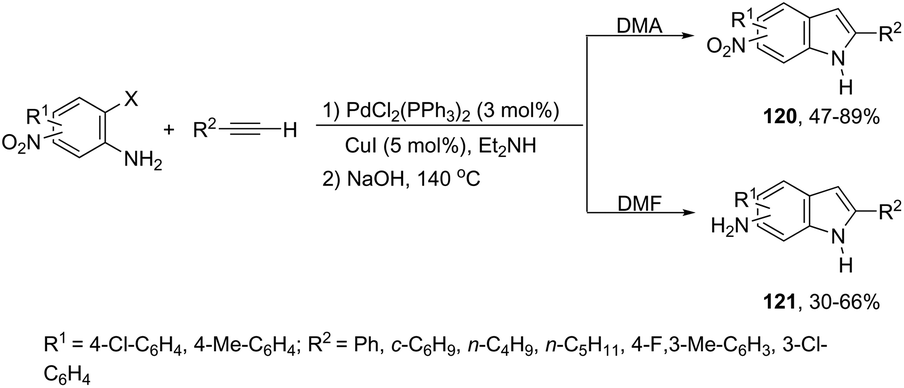
A variety of nitro- or amino-indoles have been selectively synthesized via tandem Sonogashira coupling/heteroannulation reaction of 2-halonitroanilines and terminal alkynes. The selective product formation was achieved by controlling the solvent; by choosing DMA as the solvent, nitro-indole derivatives 120 were exclusively obtained; whereas the use of DMF gave selectively the amino-indole derivatives 121 (Scheme 107).145 It was suggested that in the reaction using DMF, as the solvent, an ammonium formate derivative was in situ formed, which could act as a source of hydrogen becoming, in association with palladium salt, a reducing system.
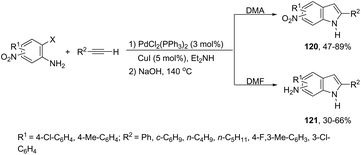 |
| | Scheme 107 | |
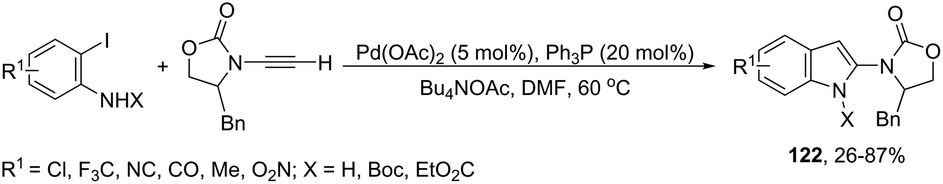
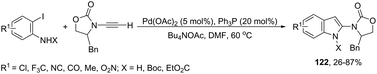
The palladium-catalyzed, one-pot, two-step reaction of ynamides with ortho-iodoanilines led to 2-aminoindoles 122 (Scheme 108).146 After the Sonogashira reaction took place, the spontaneous intramolecular hydroamination of the triple bond afforded the 2-aminoindoles from 26% to 87% yields.
 |
| | Scheme 108 | |
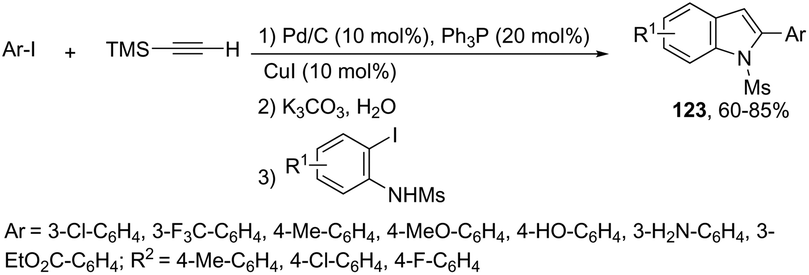
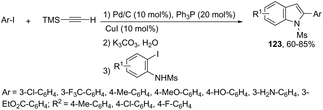
When ortho-iodoanilines are subjected to palladium-catalyzed cross-coupling with (trimethylsilyl)acetylene plus a solution of K2CO3 in aqueous MeOH, the 2-(hetero)aryl indoles 123 were isolated in good yields instead of the 2-alkynylanilines (Scheme 109).147 The power of the methodology is the use of 10% Pd/C–CuI/PPh3 to achieve the sequential (i) C–C coupling, followed by (ii) C–Si bond cleavage and subsequent (iii) C–C and (iv) C–N bond forming reactions in a single pot.
 |
| | Scheme 109 | |
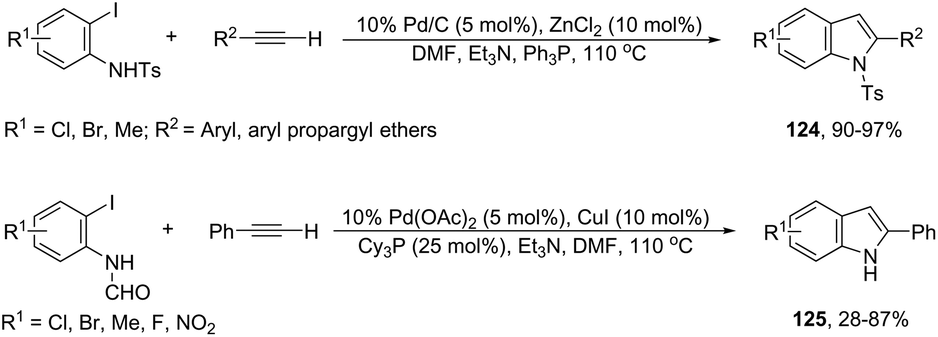
Zinc chloride can also serve as a co-catalyst in place of copper(I) iodide in the palladium-cross coupling/cyclization reaction of N-tosyl-2-iodoanilines with terminal alkynes for the synthesis of 2-substituted indoles.148 In this study, when 2-iodoanilines reacted with terminal alkynes, at 110 °C, in the presence of 10% Pd/C, moist ZnCl2, Et3N and PPh3, using DMF as the solvent, the indole derivatives 124 were obtained in high yields (Scheme 110). The authors reported that the reactions carried out under anhydrous conditions gave only the Sonogashira products in the complete absence of a cyclized product. It was suggested that the presence of HO−, liberated from moist ZnCl2, allows an in situ generation of zinc acetylide, which accelerates the cross-coupling/cyclization sequence. In order to obtain N-unsubstituted indoles, some studies were carried out utilizing N-formamide-2-iodoanilines instead of N-tosyl-2-iodoanilines. In this case, when N-(2-iodo-aryl)formamides were reacted with terminal alkynes under palladium cross-coupling/cyclization, indoles 125 were formed through an intramolecular cascade carbon–carbon/carbon–nitrogen bond formation (Scheme 110).149
 |
| | Scheme 110 | |
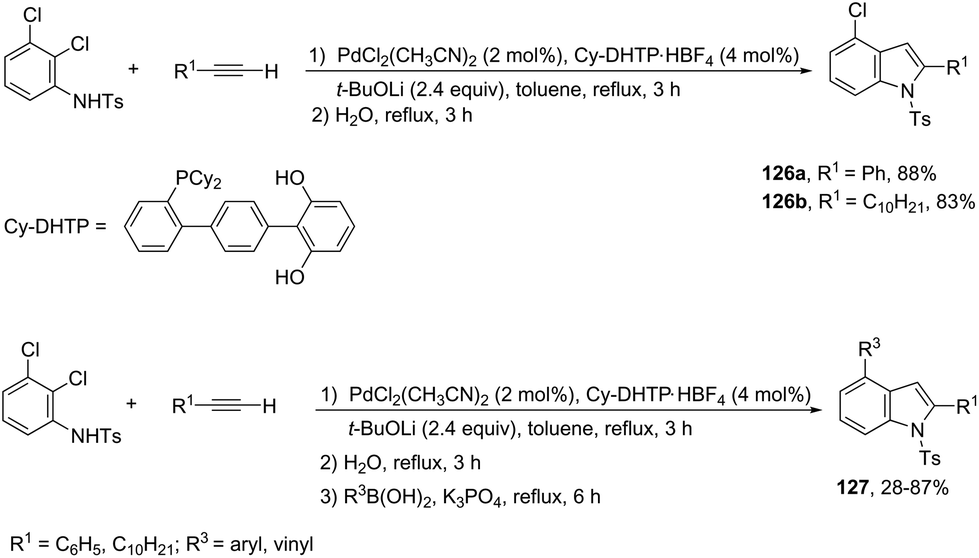
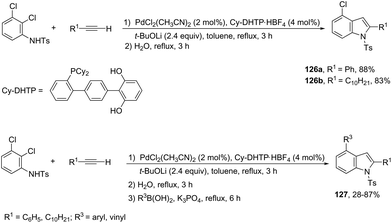
In an effort to prepare 4-chloroindoles 126, the reaction of 2,3-dichloroaniline derivatives with terminal alkynes, catalyzed by palladium and dicyclohexyl-(dihydroxyterphenyl)phosphine (Cy-DHTP), allowed the Sonogashira coupling, followed by the cyclization reaction at the ortho-position. The addition of boronic acid to the above conditions resulted in the synthesis of 2,4-disubstituted indoles 127via a one-pot Sonogashira cross-coupling/cyclization/Suzuki–Miyaura sequence (Scheme 111).150
 |
| | Scheme 111 | |
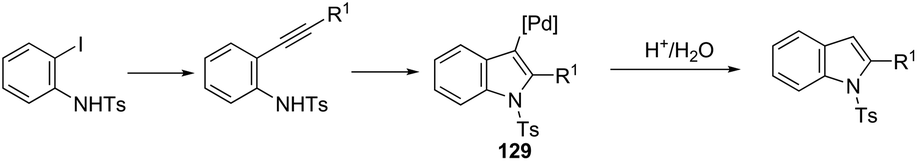
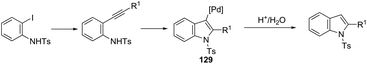
The palladium nanoparticles supported on a siliceous mesocellular foam (Pd(0)-AmP-MCF) were used as the catalytic system in the reaction of ortho-iodoanilines with terminal alkynes, which gave the indoles 128 (Scheme 112).151 This transformation was influenced by the presence of a small amount of water (1 equiv.). The authors suggested that the presence of water improved the yields of the indoles by increasing the rate of hydrolysis of the vinyl palladium intermediate 129, formed in the cyclization process (Scheme 113).
 |
| | Scheme 112 | |
 |
| | Scheme 113 | |
2.10.2. Palladium-catalyzed cascade reactions of alkynes via an aminopalladation/reductive elimination process.
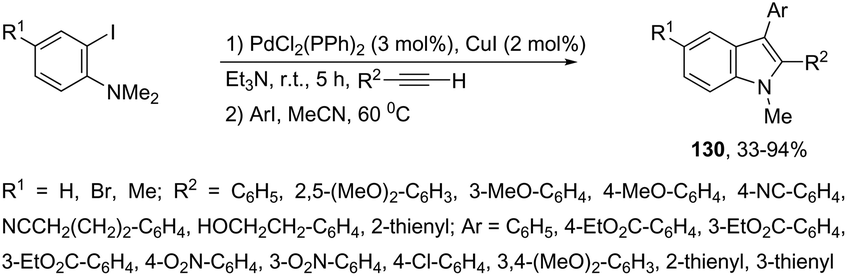
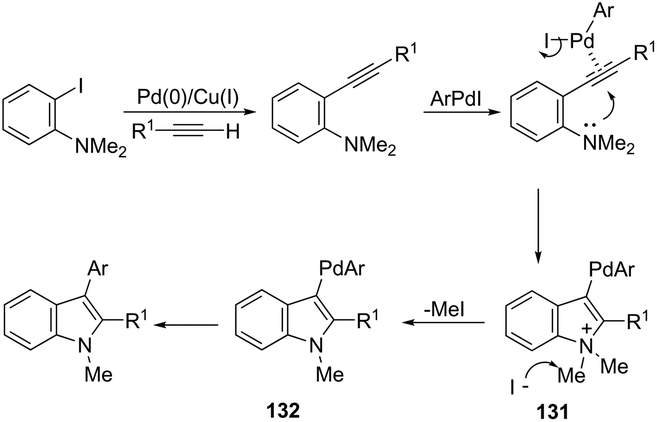
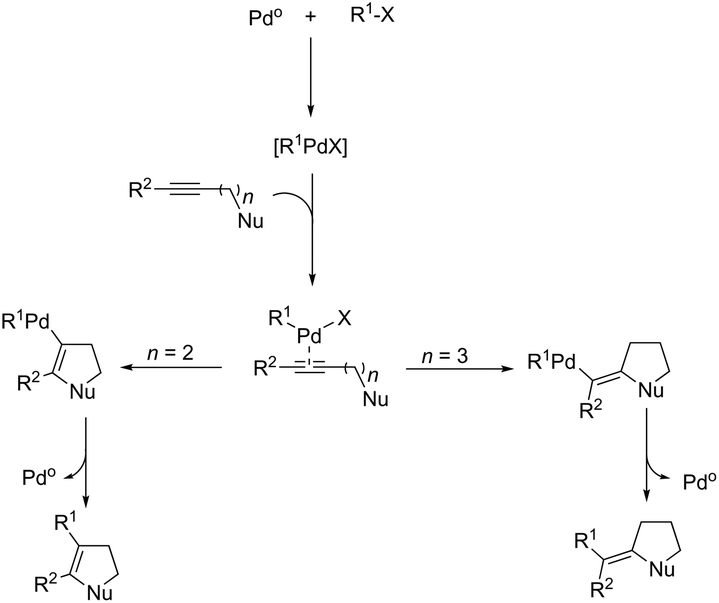
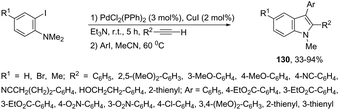
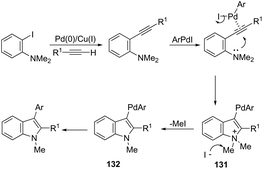
Palladium-catalyzed cascade reactions are a rapidly growing area of modern organic chemistry because their timesaving, high efficiency and atom economy. These reactions can be efficiently applied to the formation of π-alkyne–sigma-arylpalladium complexes having a proximal nitrogen nucleophile that can undergo a new carbon–nitrogen bond formation, giving consequently indoles. Extensive efforts have been also made in recent years in developing synthesis of indoles via aminopalladation/reductive elimination reactions. In 2009, Larock reported the results on the microwave-assisted, one-pot, three-component coupling reaction for the synthesis of indoles 130 (Scheme 114).152 The reaction was carried out in two steps under standard Sonogashira coupling conditions from 2-iodoanilines and terminal alkynes, followed by the addition of an aryl iodide (Scheme 114). Larock proposed a reaction mechanism for the one-pot synthesis of indoles, which is illustrated in Scheme 115. The oxidative addition of the aryl iodide to Pd(0) affords an ArPdI species, which activates the alkyne triple bond of the N,N-dialkyl-2-(1-alkynyl)aniline formed in the first step via a Sonogashira coupling. The intramolecular trans-aminopalladation gives the indolium anion 131 that loses a methyl group via SN2 displacement by the in situ generated iodide anion, leading to the indole-containing Pd(II) intermediate 132. The reductive elimination generates the indole (Scheme 115).
 |
| | Scheme 114 | |
 |
| | Scheme 115 | |
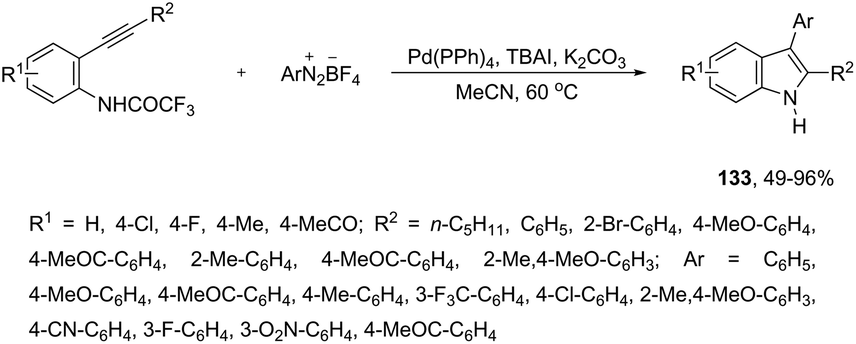
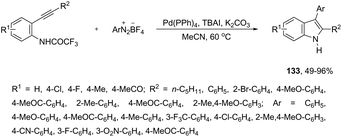
The palladium-catalyzed reaction of arenediazonium tetrafluoroborates and 2-alkynyltrifluoroacetanilides afforded free N-H-2,3-disubstituted indoles 133 (Scheme 116).153 The reaction is believed to proceed through a domino process that starts with the reaction of iododediazoniation with TBAI to the in situ formation of the aryl iodide. The reaction tolerates a variety of useful substituents, both in the starting alkyne and the arenediazonium salt, including halo substituents, and nitro, cyano, keto, ester, ether, methoxy and methyl groups.
 |
| | Scheme 116 | |
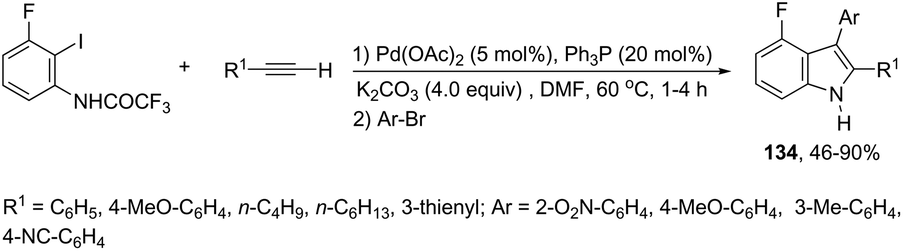
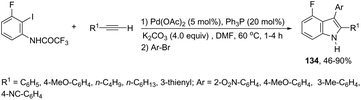
The aminopalladation/reductive elimination sequential reaction, developed by Cacchi and co-workers, has also been employed as a useful strategy for the regioselective construction of 3-aryl-4-fluoro-2-substituted-1H-indoles 134 from ortho-alkynyltrifluoroacetanilides (Scheme 117).154 The authors showed that although the use of an inorganic base, such as K2CO3 or Cs2CO3, gave good results in the formation of indoles, the cyclization worked better with Et3N as a base in a microwave-irradiated process.
 |
| | Scheme 117 | |
The scope of the aminopalladation/reductive elimination sequential reaction described by Cacchi can be extended to 2-bromoanilides instead of the corresponding iodides in the preparation of 2,3-disubstituted indoles 135 (Scheme 118).155 However, the major difficulty found in this reaction was the low reactivity of bromides, which led to the formation of a hydroamidation product without the incorporation of the aryl group at the 2-position of the indole ring. This problem was solved by the authors through a complete study of the best reaction conditions, which involved important parameters such as the base, solvent, temperature, and ligand.
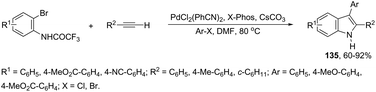 |
| | Scheme 118 | |
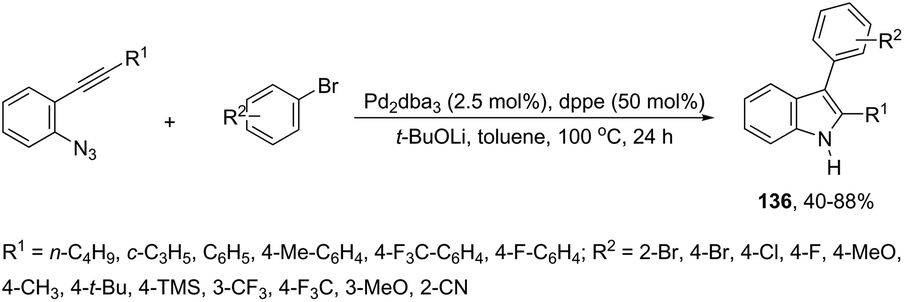
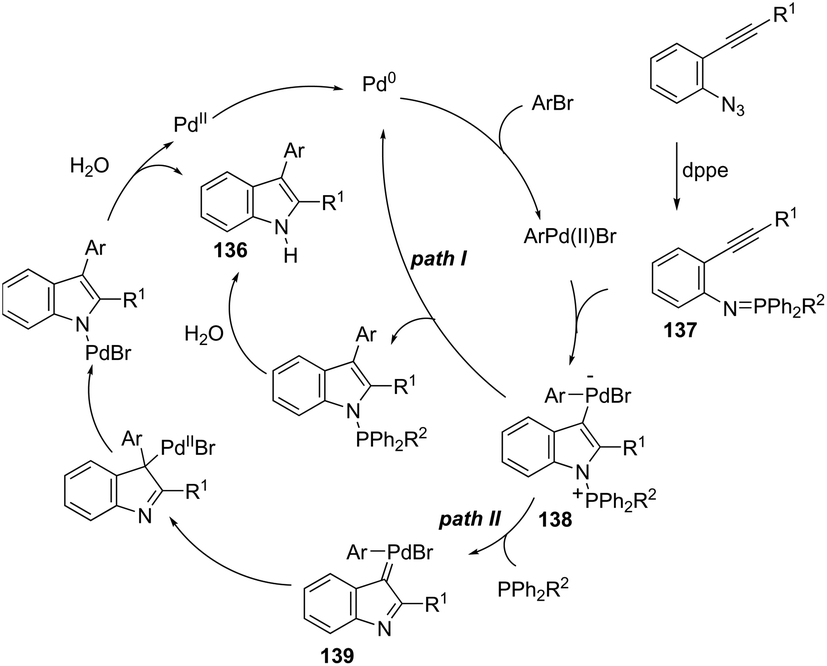
A series of 2-aryl indoles 136 has been prepared using 2-alkynyl arylazides and aryl bromides under catalysis of Pd2dba3, dppe as a ligand and t-BuOLi as a base in toluene at 100 °C (Scheme 119).156 In this cyclization, the nitrogen nucleophile is generated in situ from azides by a Staudinger reaction.157 The authors proposed two possible pathways, which are illustrated in Scheme 130. In the first one, the iminophosphorane 137 is formed in situ from 2-alkynyl arylazide by a Staudinger reaction. Then, the 5-endo-dig cyclized intermediate 138 is generated through the nitrogen nucleophilic attack on the activated triple arylpalladium(II). The reductive elimination of intermediate 138, followed by hydrolysis, gives the product. The second mechanism proposed involves the palladium(II) carbene species 139 formed from intermediate 138, which after a migratory insertion, isomerization and protonation sequence affords the indole (Scheme 120).
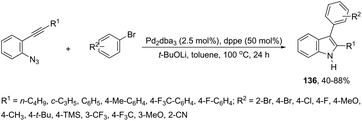 |
| | Scheme 119 | |
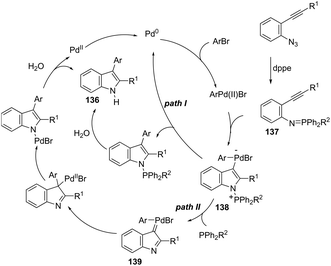 |
| | Scheme 120 | |
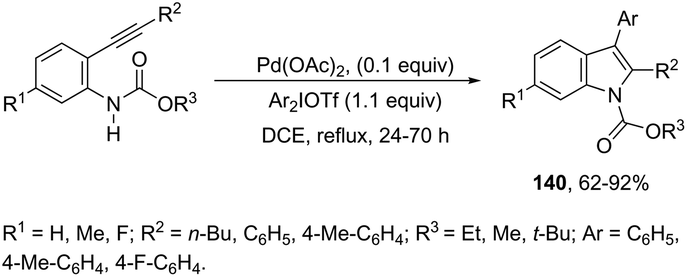
Another type of aminopalladation/reductive elimination reaction involved the regioselective palladium-catalyzed 5-endo-dig ring-closing reaction of 2-alkynylarylcarbamates with diaryliodonium salts to give 2,3-disubstituted indoles 140 (Scheme 121).158 In this reaction, diaryliodonium salts react with palladium to form a palladium(II) intermediate that acts by activating the carbon–carbon triple bond and as an arylating agent.
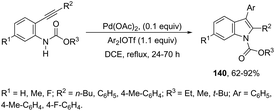 |
| | Scheme 121 | |
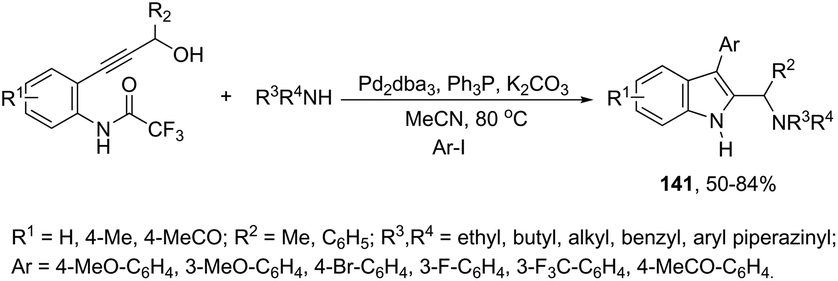
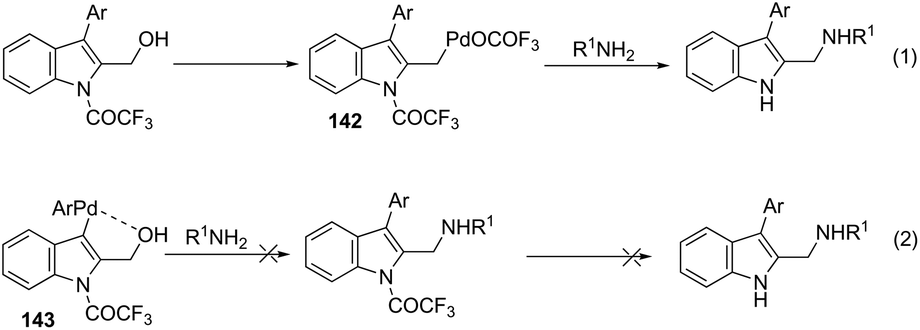
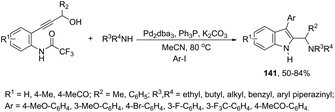
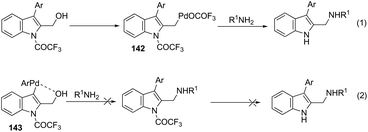
Very recently, Cacchi described the preparation of 2-(aminomethyl)-3-arylindoles 141via palladium-catalyzed reaction of 3-(ortho-trifluoroacetamidoaryl)-1-propargyl alcohols, amines, and aryl iodides through a simple procedure that allows for the formation of two carbon–nitrogen bonds and a carbon–carbon bond in a one-pot reaction (Scheme 122).159 The experimental evidence obtained by the authors supported the hypothesis that a trifluoroacetyl ester is formed in the reaction medium and that a palladium intermediate 142 is directly involved in the formation of indoles (Scheme 123, eqn (1)). The results of these experiments led the authors to discard the formation of indoles via a direct nucleophilic substitution of the amino nucleophile for the hydroxyl group assisted by the coordination of oxygen to palladium species 143 (Scheme 123, eqn (2)).
 |
| | Scheme 122 | |
 |
| | Scheme 123 | |
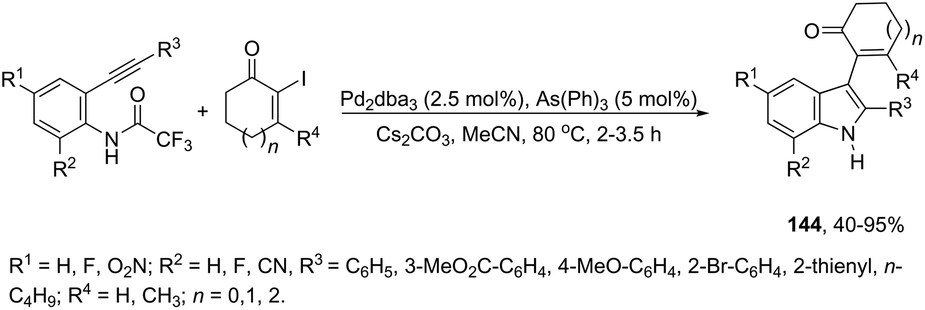
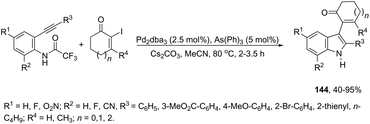
Instead of aryl halides, vinyl halides are also suitable electrophiles for the sequential aminopalladation/reductive elimination of palladium(0) to afford indoles. Alfa-iodoenones at different ring sizes were efficiently used as electrophiles in the palladium-catalyzed cyclization of 2-alkynyltrifluoroacetanilides to afford 2,3-disubstituted indoles 144 (Scheme 124).160 Various experimental conditions were tested by the authors for the cyclization, and the best results were obtained with Pd2(dba)3 as a catalyst and AsPh3 as a ligand; although, in the absence of the ligand, Pd2(dba)3 was also effective.
 |
| | Scheme 124 | |
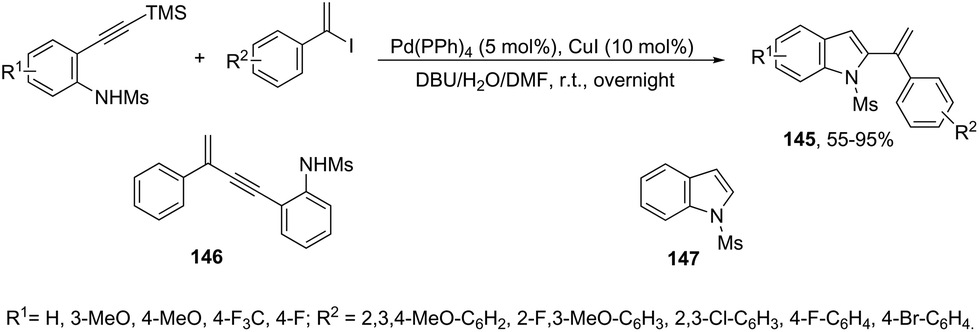
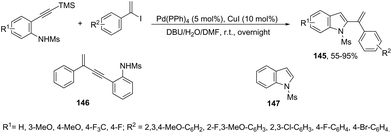
The 1-phenylvinyl iodides have also been used for the one-pot conversion of N-protected ortho-alkynylanilines to 2-(1-phenylvinyl)indoles 145 (Scheme 125).161 A mixture of unsubstituted enynes 146 and indoles 147 was obtained and the ratio between them can be greatly dependent on the substituent on the aniline nitrogen atom. Unprotected anilines gave no trace of products, N-tosylanilide and trifluoromethyl acetanilide gave a mixture of enynes 146 and indoles 147 and the best result was obtained with N-mesylanilide, which gave the desired indole 145 in 73% yield together with small amounts of unsubstituted indoles 147. Under Pd(PPh3)4 (5 mol%), CuI (10 mol%), and DBU (12 equiv.) and using a mixture of DMF and water as the solvent, the N-mesyl ortho-alkynylanilines were transformed into the corresponding 2-(1-phenylvinyl)indoles 145 from moderate to good yields.
 |
| | Scheme 125 | |
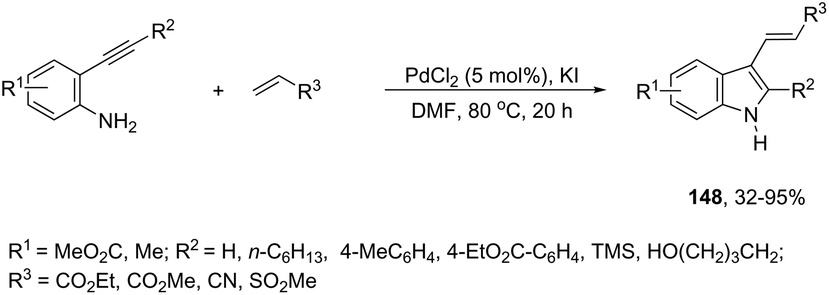
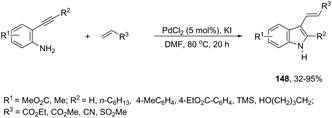
The aminopalladation reactions with the concomitant oxidative coupling have also been reported by using alkenes instead of halide derivatives. The palladium-catalyzed cascade reaction of 2-alkynylaniline and functionalized alkenes led to structurally diverse 3-alkenylindoles 148 (Scheme 126).162 The cyclization reactions follow the regioselective 5-endo-dig-mode, whereas the subsequent Heck-type coupling with alkenes occurs stereoselectively with exclusive formation of the E isomers.
 |
| | Scheme 126 | |
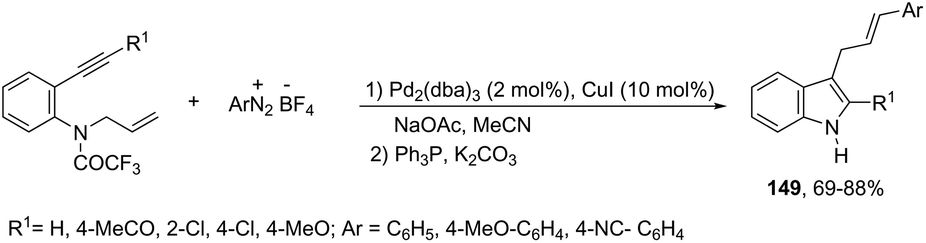
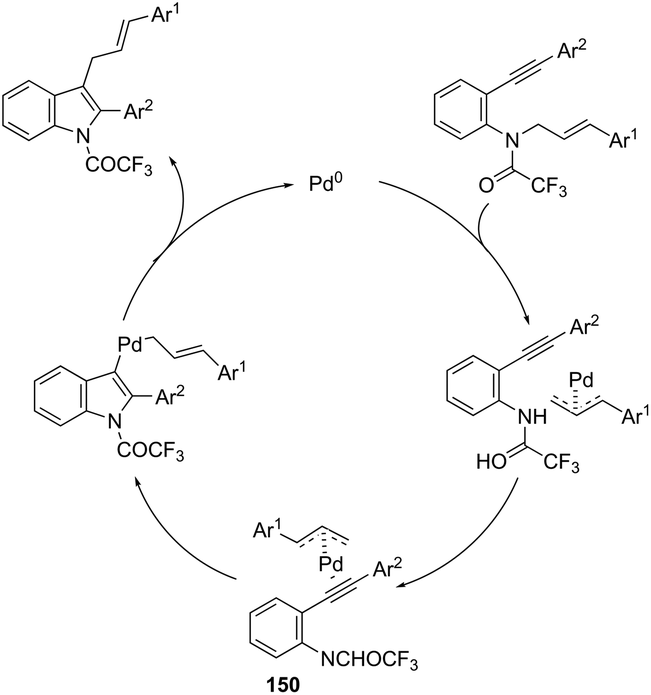
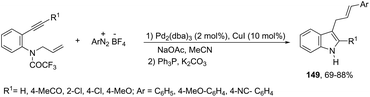
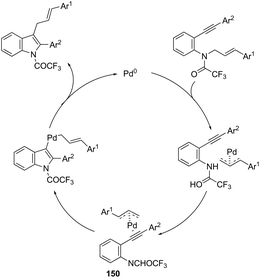
Using palladium-catalyzed reactions of arenediazonium tetrafluoroborates with N-allyl-2-alkynyl anilines, Cacchi has obtained 2-alkenyl indoles 149via an intramolecular aminocyclization/oxidative Heck-coupling sequence (Scheme 127).163 The reaction can be efficiently carried out via a simple one-pot, two-step procedure just by the addition of PPh3 and K2CO3 to the crude mixture and heating at 100 °C. Although the cyclization follows a mechanism similar to that shown in Scheme 128, it should be highlighted that the central step is the formation of π-allylpalladium species 150, which activates the carbon–carbon triple bond and introduces the functionality at the 3-position of the indoles.
 |
| | Scheme 127 | |
 |
| | Scheme 128 | |
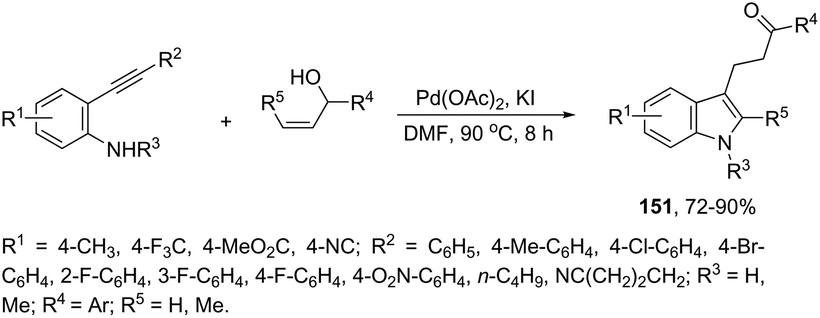
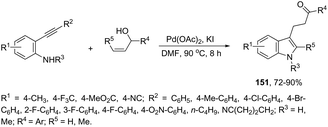
Instead of simple alkenes, allylic alcohols were efficiently used in palladium-catalyzed oxidative cyclization of 2-alkynyl anilines in the synthesis of β-indole ketones 151 (Scheme 129).164 The effectiveness of the cyclization depends on the catalyst species (Pd(OAc)2) and the presence of oxygen for the regeneration of palladium(II) active species.
 |
| | Scheme 129 | |
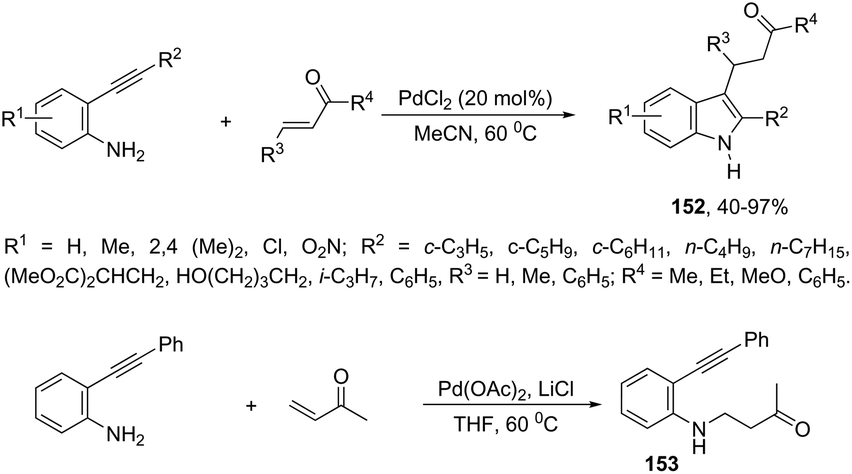
A series of 2,3-disubstituted indoles 152 have been prepared by cascade reaction of N-unprotected-2-alkynylanilines with electron-deficient alkenes using palladium as a catalyst (Scheme 130).165 The authors observed that the product distribution was affected by the nature of the palladium catalyst. The use of PdCl2 in acetonitrile at 60 °C resulted in the formation of 2,3-disubstituted indole derivatives 152, whereas the catalytic system formed by Pd(OAc)2/LiCl in THF at 60 °C produced N-alkylated-2-alkynylaniline derivatives 153.
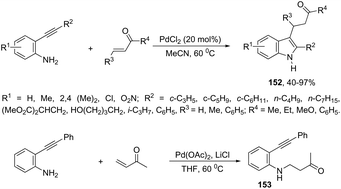 |
| | Scheme 130 | |
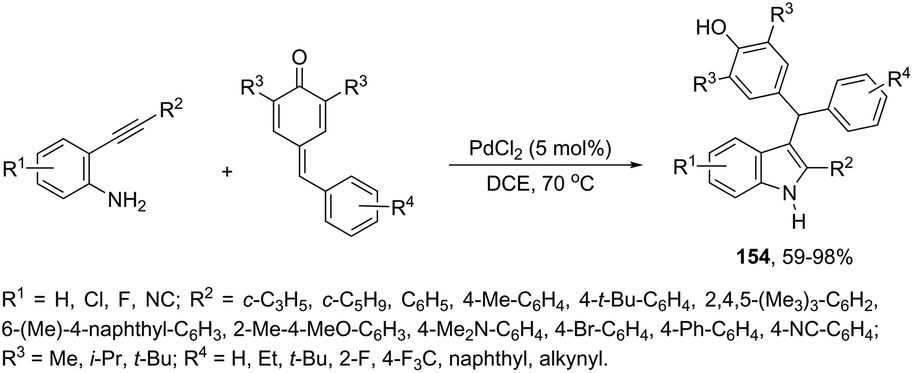
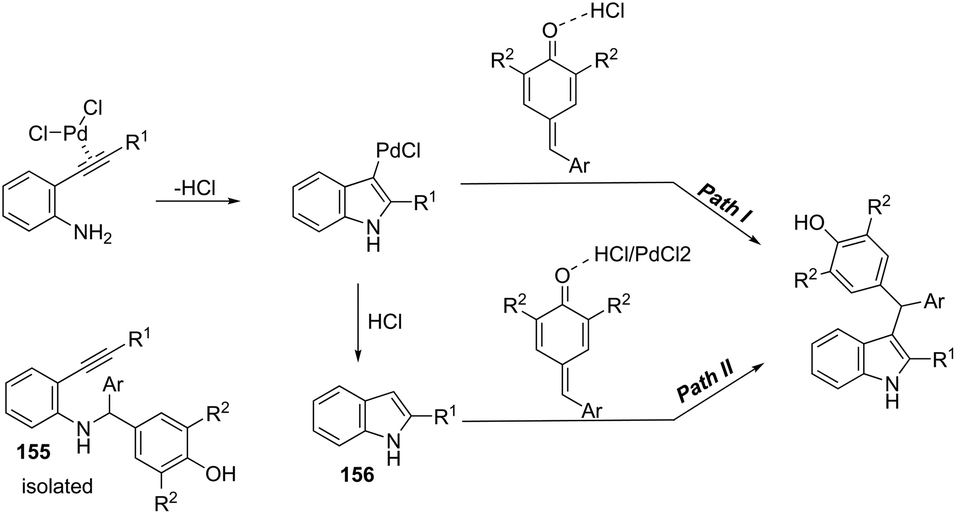
The synthesis of highly substituted unsymmetrical diarylindolylmethane derivatives 154 has been developed through palladium-catalyzed annulation of ortho-alkynylanilines, followed by 1,6-conjugate addition to para-quinones (Scheme 131).166 Various substituted ortho-alkynylanilines and para-quinones were used in the cyclization, giving the unsymmetrical diarylindolylmethanes in excellent yields. The careful monitoring of the reaction revealed that the amine addition product 155 was also formed in considerable amounts; however, it is not an intermediate of the reactions. The formation of 155 is reversible and its concentration gradually decreased with the formation of indoles 154. The authors suggested that the 2-arylindole 156 is a key intermediate and its formation is the rate-determining step of the cyclization (Scheme 132).
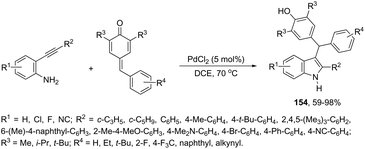 |
| | Scheme 131 | |
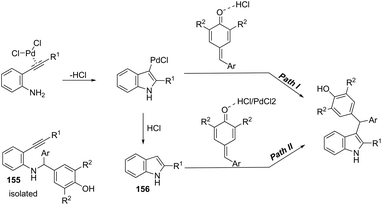 |
| | Scheme 132 | |
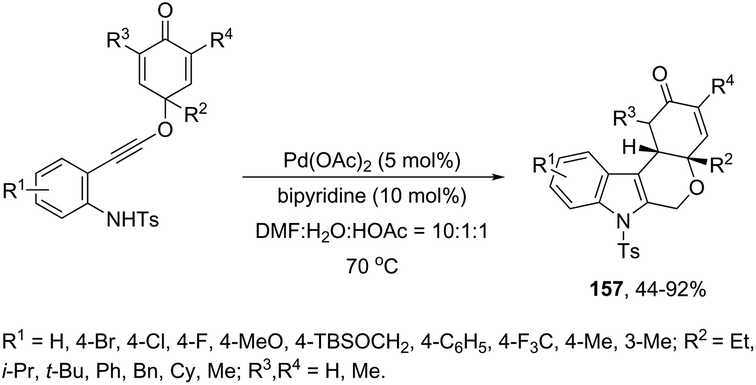
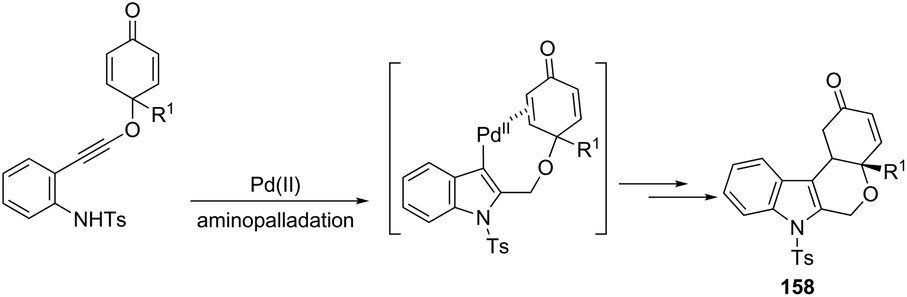
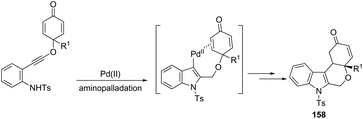
The Pd(OAc)2, bipyridine and DNF/H2O/AcOH catalytic system is active for the intramolecular cyclization of aniline-tethered alkynyl cyclohexadienones in the preparation of cyclohexenone-fused tetrahydropyrano[3,4-b]indoles 157 (Scheme 133).167 The authors suggested that the intramolecular cascade reaction would be initiated by aminopalladation of alkynes and quenched by the addition to the intramolecular cyclohexenone. The asymmetric version of this tandem cyclization was also described by using a chiral bipyridine ligand, which gave the chiral cyclohexanone fused tetrahydropyrano[3,4-b]indoles 158 in good yields with excellent enantioselectivities (93–96% ee) (Scheme 134).
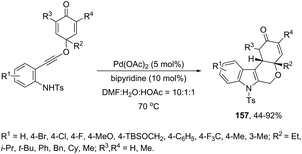 |
| | Scheme 133 | |
 |
| | Scheme 134 | |
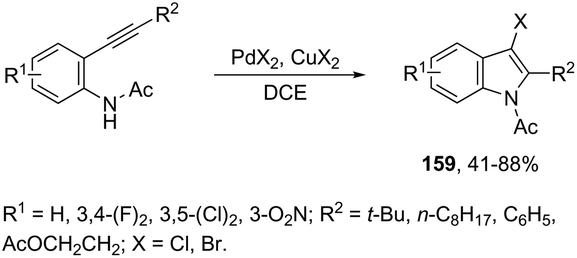
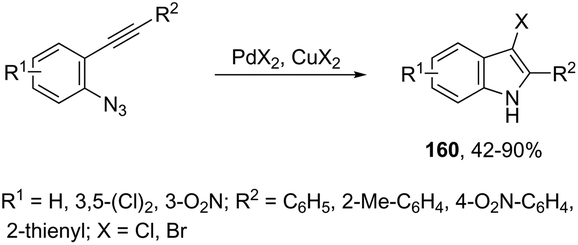
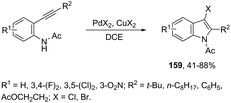
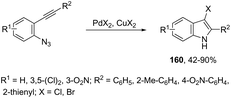
The addition of a halide source to palladium-catalyzed annulation of an ortho-alkynylaryl containing nitrogen nucleophile has been a methodology used for the synthesis of 3-halo-indoles. For example, Li and co-workers reported that in the presence of PdX2 and CuX2 (X = Cl, Br), the annulation reactions of 2-ethynylbenzeneamines gave the 2-substituted 3-haloindoles 159 (Scheme 135).168 More recently, Li and Zhang discovered that in the presence of PdX2 (X = Br, Cl) and halide sources, the 2-alkynyl aryl azides were also suitable substrates for halopalladation cyclization reactions to afford the corresponding 2-substituted 3-haloindoles 160 (Scheme 136).169
 |
| | Scheme 135 | |
 |
| | Scheme 136 | |
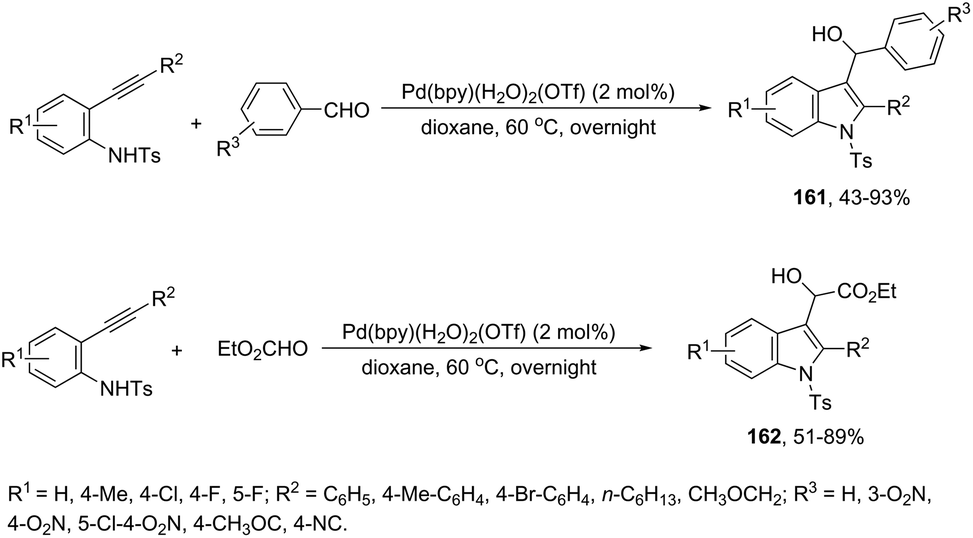
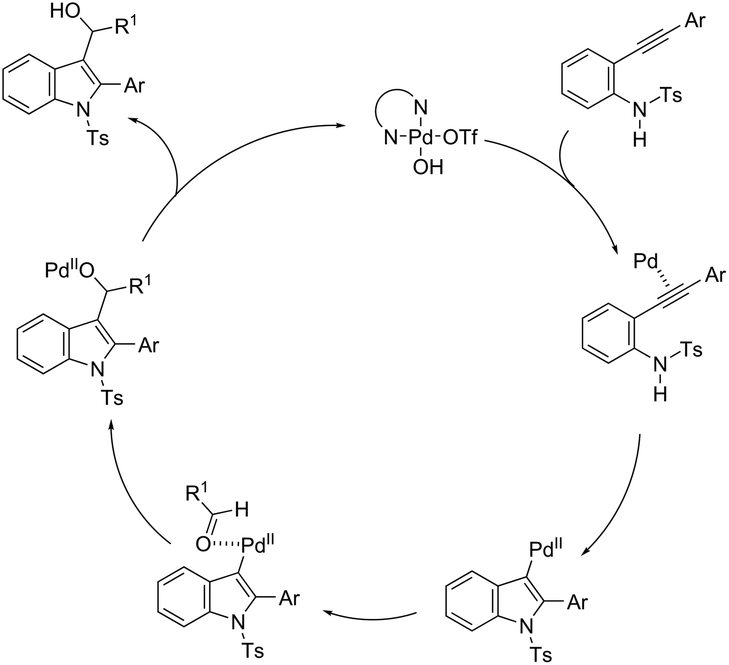
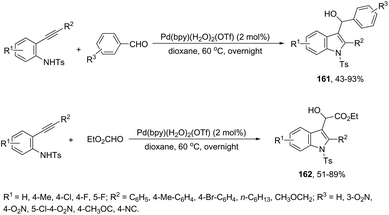
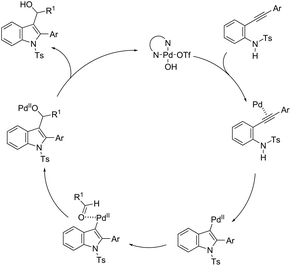
The intermolecular version of the sequence of carbon–nitrogen and carbon–carbon bond formation has been described in the course of the reaction of N-tosyl-2-arylethynylanilines with aldehydes catalyzed by the palladium(II) species in the synthesis of substituted 3-hydroxymethylindoles 161 (Scheme 137).170 The same reaction conditions were extended to ethyl glyoxylate, which afforded the alpha-hydroxyindolyl acetate 162 in good yield. According to the authors, a tandem reaction is started by an aminopalladation of 2-arylethynylanilines, followed by quenching of the carbon–palladium bond with a carbonyl group. This regenerates the active palladium(II) species without the necessity of a redox system (Scheme 138).
 |
| | Scheme 137 | |
 |
| | Scheme 138 | |
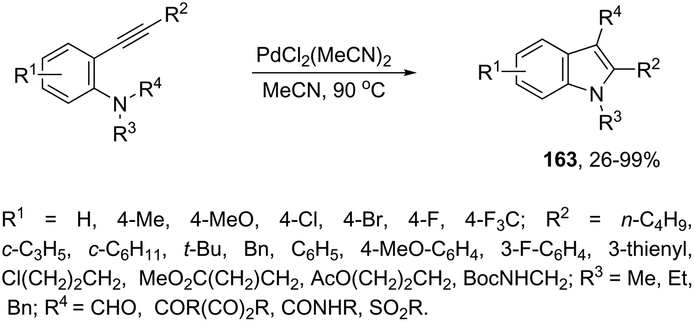
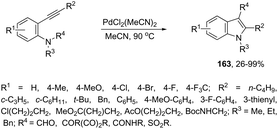
The intramolecular version of the sequence of carbon–nitrogen and carbon–carbon bond formation with the migration of the functional carbonyl group was also described. Thus, the reaction of N-acyl-2-alkynylanilines with PdCl2(CH3CN)2 in CH3CN at 90 °C gave the 3-acyl-indoles 163, resulting from the acyl migration (Scheme 139).171 The reaction conditions also promoted the migration of pyruvoyl, amide and sulfonyl groups in comparable yields.
 |
| | Scheme 139 | |
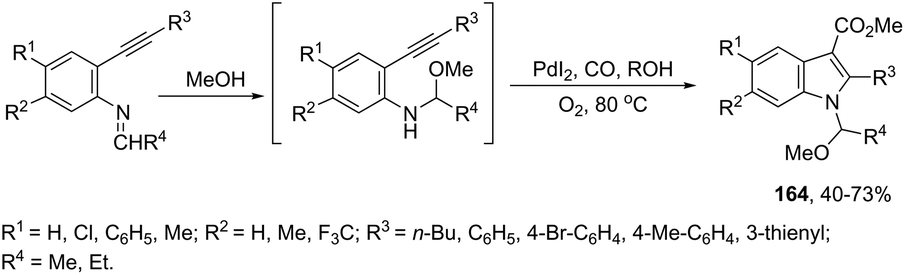
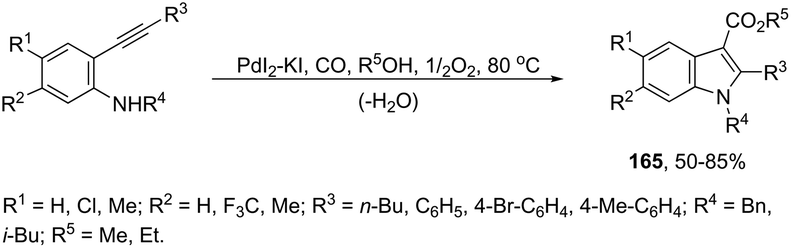
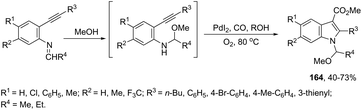
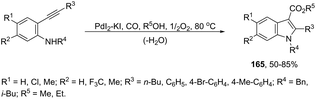
The palladium-catalyzed oxidative carbonylation of 2-alkynylaniline imines and 2-alkynylaniline derivatives in the preparation of indole-3-carboxylic acid esters has also been reported. When imine derivatives were used as the starting material, the sequential reaction with an initial nucleophilic attack on carbon of the imine group, followed by a palladium-catalyzed oxidative nitrogen-cyclization–alkoxycarbonylation, occurred to form the [1-(alkoxyarylmethyl)indole]-3-carboxylic esters 164 (Scheme 140).172 When 2-alkynylanilines were used as the starting material, in the reaction with CO, O2, and an alcohol in the presence of the PdI2/KI as the catalytic system, indole-3-carboxylic esters 165 were formed in good yields (Scheme 141).173 In addition, on changing the alcohol to trimethyl orthoformate, 2-alkynylanilines gave 1-(dimethoxymethyl) indole-3-carboxylic esters via N-(dimethoxymethyl)-2-alkynylaniline derivatives as intermediates.
 |
| | Scheme 140 | |
 |
| | Scheme 141 | |
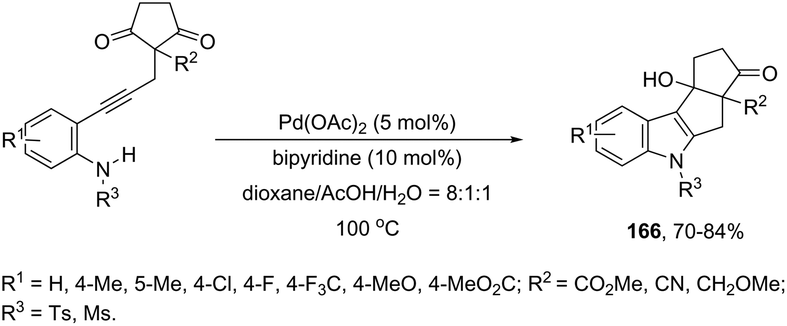
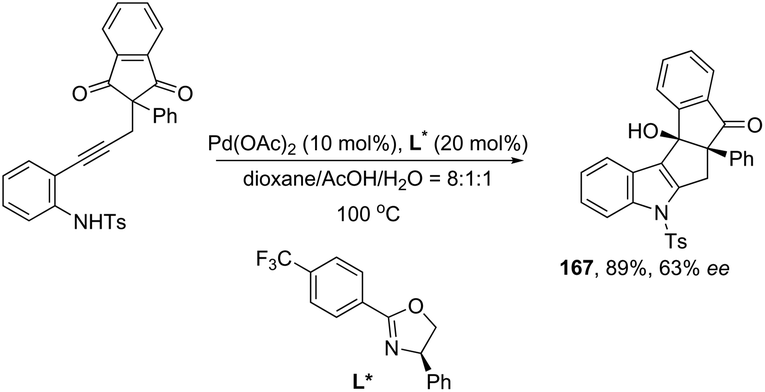
Ketones can be also used as electrophiles in the aminopalladation-initiated tandem cyclization. As reported, the Pd(OAc)2 catalyst was effective at tandem cyclization of 2-alkynylanilines having a cyclopentanone to synthesize pentaleno[2,1-b]indoles 166 (Scheme 142).174 This reaction involves the initial aminopalladation of the carbon–carbon triple bond, followed by quenching of the palladium intermediate by intramolecular addition to carbonyl groups. In the obtained tetracyclic indole framework 167, two neighboring stereocenters were formed, with excellent diastereoselectivity in a single process by using an oxazole derivative as the ligand (Scheme 143).
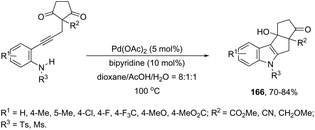 |
| | Scheme 142 | |
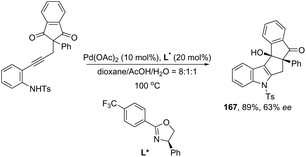 |
| | Scheme 143 | |
Results from the palladium/copper cocatalyzed oxidative cyclization of 2-alkynylanilines showed that 3-acylindoles 168 were obtained in good yields by using PdBr2 (10 mol%) and CuI (10 mol%) in the presence of t-butyl hydroperoxide (Scheme 144).175 In this cyclization reaction, t-butyl hydroperoxide was used as either the oxidant or oxygen source to introduce the carbonyl functionality. Based on the results of control experiments the authors concluded that the ketone moiety did not originate from water and molecular oxygen. These results in association with ESI/MS analysis results indicated that the reaction followed the mechanism shown in Scheme 145. The 2-alkynylaniline reacts with two molecules of t-butyl hydroperoxide under copper catalysis to give the peroxide 169, which is in equilibrium with the iminium intermediate 170. The nucleophilic attack of t-butyl hydroperoxide on the activated carbon–carbon triple bond of 170 generates the intermediate 171 that gives the 3-acylindoles after the intramolecular cyclization, oxidation, and deprotonation sequence.
 |
| | Scheme 144 | |
 |
| | Scheme 145 | |
In a recent study, 2-alkynyl arylazides and sulfonic acids were examined with regard to the synthesis of 1H-indole-3-sulfonates via palladium-catalyzed tandem reactions.176 It was found that the desired products, 1H-indole-3-sulfonates 173, were obtained in moderate to excellent yields when 2-alkynyl arylazides reacted under Pd(OAc)2 at room temperature, followed by the addition of MsOH (Scheme 146). It was proposed that the formation of the intermediate 172 is responsible for the success of the reaction. The reaction conditions could be applied to 2-alkynyl arylazides with an aryl group containing pendant electron-donating and electron-withdrawing substituents or a hetero aryl group on the alkyne carbon. 2-Alkynyl arylazides bearing the aliphatic substituted groups on the alkyne carbon were also suitable substrates, giving the desired indoles in comparable yields; however, 2-alkynyl arylazides containing a terminal alkyne did not afford the products.
 |
| | Scheme 146 | |
The application of isocyanides as electrophiles in the aminopalladation-initiated tandem cyclization was studied for the synthesis of 3-amidylindoles 176177 and 1H-indole-3-carboxamidines 177.178 In the former case, Pd(OAc)2 and silver acetate catalyzed the reaction of N,N-dimethyl-2-alkynylanilines and isocyanides, leading to 3-amidylindoles 176 in moderate to good yields (Scheme 147). When Pd(TFA)2 was used as the catalyst and silver acetate was switched to silver trifluoroacetate, N,N-dimethyl-2-alkynylanilines gave 3-cyanoindoles 177 (Scheme 147). In addition, the use of a trace amount of water under these reaction conditions led to the formation of 3-amidylindoles.179 In the mechanism proposed, the authors hypothesized that silver acetate would act as an oxidant and a reactant and that 174–176 are the key intermediates in the formation of products (Scheme 147). In the latter case,178 isocyanides, ortho-alkynyltrifluoroacetanilides and amines are involved in a three-component process for the synthesis of 2-substituted 1H-indole-3-carboxamidines 178 (Scheme 148). The reaction proceeds through intramolecular aminopalladation of alkynes activated by isocyanide-ligated palladium(II) species 179 (Scheme 149).
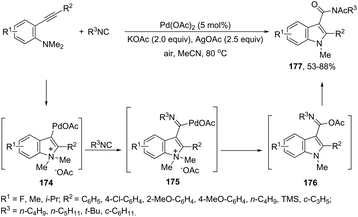 |
| | Scheme 147 | |
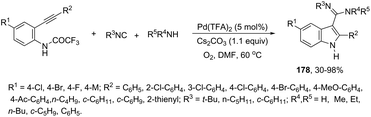 |
| | Scheme 148 | |
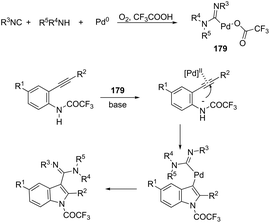 |
| | Scheme 149 | |
The same group has used diazoacetates to synthesize 3-alkylindole derivatives 180via a carbene insertion.180 In this cyclization reaction, ortho-alkynyltrifluoroacetanilides were activated by PdCl2, starting an aminopalladation triggered carbene insertion reaction via intermediates 181–184 to give aryl-substituted methyl 2-(2-phenyl-1H-indol-3-yl)acetates 180 (Scheme 150). The use of alpha-diazoacetates could result in a competition between the carbon–carbon bond formation and nitrogen–hydrogen insertion. The authors thus postulated that the migratory insertion of the carbene into the sigma-indolylpalladium intermediate was favored over the nitrogen–hydrogen insertion.
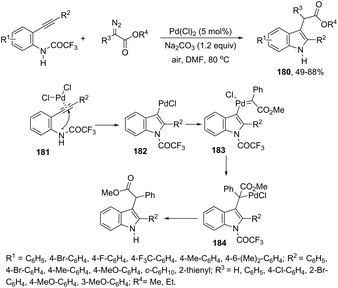 |
| | Scheme 150 | |
Terminal alkynes are useful partners for the palladium-catalyzed oxidative cyclization of ortho-alkynylanilines, have been proven to be extremely useful for the synthesis of 3-alkynylindoles via carbon–carbon bond formation. In this regard, a range of 2,3-disubstituted 3-alkynylindoles 185 were prepared by palladium(II)-catalyzed domino reaction between ortho-alkynylanilines and terminal alkynes (Scheme 151).181 The control experiments were carried out to determine the reaction mechanism, in which the authors identified as elementary steps the formation of a σ-alkynylpalladium(II) complex and aminopalladation of ortho-alkynylaniline, leading to a sigma-indolylpalladium(II) intermediate. In these experiments, they also found that the N-demethylation step occurs after the formation of the σ-alkynylpalladium intermediates (Scheme 152). Thus, the proposed mechanism for this transformation involves the aminopalladation of ortho-alkynylanilines, giving the σ-indolylpalladium intermediate. The coordination of the terminal alkyne with σ-indolylpalladium, followed by deprotonation, leads to σ-alkynylpalladium. The N-demethylation promoted by iodide affords 3-indolylpalladium intermediate and reductive elimination provides the 3-alkynylindoles 185 (Scheme 152).
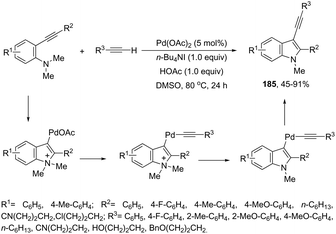 |
| | Scheme 151 | |
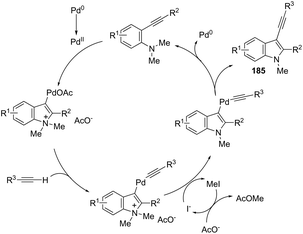 |
| | Scheme 152 | |
In a similar manner to that of 3-alkynylindoles 185, the 2,3′-bisindoles 186 were prepared by intermolecular cyclization using two ortho-alkynylanilines. The palladium-catalyzed conditions, consisting of Pd/C, n-Bu4NBr, AcOH, and DMSO at 80 °C, were very effective at promoting the cyclization of ortho-alkynylanilines and 2-ethynylaniline to 2,3′-bisindole derivatives 186 (Scheme 153).182 For the mechanism, the authors proposed two temporally separate catalytic cycles, in which the formation of 3-alkynylindole 187 occurs first, followed by a second cyclization from 187 giving the bisindoles 186 (Scheme 153).
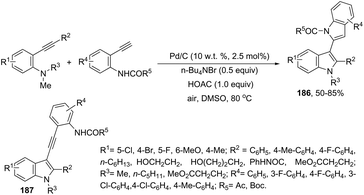 |
| | Scheme 153 | |
The palladium-catalyzed cyclization of ortho-alkynylanilines merged with cross-coupling with bis-(pinacolato)diboron was achieved upon treatment with Pd2(dba)3 and Ph3As, affording indole 3-boronic esters 188 (Scheme 154).183 The authors carried out control experiments to confirm that the borylation reaction is a process that occurs during cyclization rather than after indole formation.
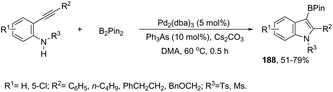 |
| | Scheme 154 | |
An efficient one-pot protocol was developed for the synthesis of the 3-sulfenylindoles 189 from diorganyl disulfides and ortho-alkynylanilines (Scheme 155).184 The results from this cyclization process, using PdCl2 in DMSO at 80 °C, indicated that a molar ratio of 2:1 between ortho-alkynylanilines and diorganyl disulfides and excellent yields were obtained. This result is significant because it indicates that the two portions (RS) of organosulfur reagents were transferred to the substrate.
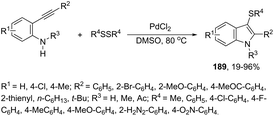 |
| | Scheme 155 | |
An important extension of this methodology was accomplished by the conversion of ortho-alkynylanilines to 3-sulfenylindoles 190via three-component cyclization reaction involving sulfur and boronic acids catalyzed by Pd(TFA)2 and CuI in ionic liquids (Scheme 156).185 The innovation of this methodology was the preparation of 3-sulfenylindoles in excellent yields by a one-pot procedure avoiding the previous preparation of diorganyl disulfides. Several control experiments indicated that the sulfur moiety is introduced via a copper thiolate 191 generated in situ from elemental sulfur, CuI and boronic acid and that palladium complex 192 is the key intermediate for the success of this cyclization (Scheme 157).
 |
| | Scheme 156 | |
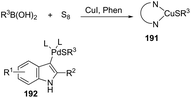 |
| | Scheme 157 | |
The exploration of the application of trifluoromethanesulfanylamide for the palladium-catalyzed cyclization/trifluoromethylthiolation of ortho-alkynylanilines was also described. The sequential cyclization and thiolation reactions between ortho-alkynylanilines with trifluoromethanesulfanylamide in the presence of Pd(OAc)2 and bismuth(III) chloride allowed a straightforward synthesis of 3-((trifluoromethyl)thio)indole derivatives 193 in excellent yields (Scheme 158).186 The presence of a quantitative amount of bismuth(III) chloride is essential for the success of this transformation because it participates in the formation of sulfur electrophilic species via an activation of trifluoromethanesulfanylamide. The transformation is believed to proceed via a palladium-catalyzed intramolecular cyclization of N,N-dimethyl-ortho-alkynylaniline giving the intermediate 194. The removal of the methyl group from intermediate 194 by chloride anions from bismuth(III) chloride with concomitant trifluoromethanesulfanyl cation formation affords the 3-((trifluoromethyl) thio)indoles 193 (Scheme 159).
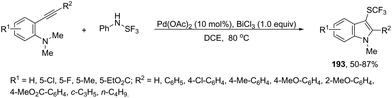 |
| | Scheme 158 | |
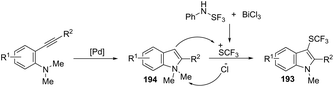 |
| | Scheme 159 | |
Another good example of a simple, but efficient, intramolecular migration of the sulfonyl group instead of allyl groups was described in the preparation of 3-sulfonyl indoles 195 (Scheme 160).187 In this reaction, the authors studied the influence of different oxidation states of palladium salts in the migratory effect. It was found that the reaction of N-allyl-N-sulfonyl-ortho-alkynylanilines with Pd(PPh3)4 led to the formation of 3-allylindoles 196, with exclusive transference of the allyl group, whereas with PdCl2(CH3CN)2 the sulfonyl group was selectively transferred, giving the 3-sulfonylindoles 195 (Scheme 160).
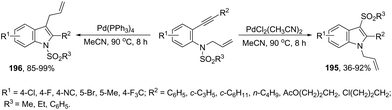 |
| | Scheme 160 | |
Moving the sequential aminopalladation reactions from functionalization of the 3-position to the 2-position of indoles, Gabriele and co-workers have described that 1-(2-aminoaryl)ynols, having a primary or secondary amino group, afforded indol-2-acetic esters 197via a carbonylation reaction promoted by the PdI2/KI catalytic system in the presence of CO, O2 and MeOH (Scheme 161).188 In addition, 1-(2-aminoaryl)ynols, having a primary amino group, gave quinoline-3-carboxylic esters, under oxidative conditions, via a 6-endo-dig cyclization.
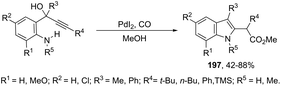 |
| | Scheme 161 | |
Cacchi and co-workers showed that the reaction of ethyl 3-(ortho-trifluoroacetamidoaryl)-1-propargyl carbonates with primary or secondary amines in the presence of Pd2(dba)3, dppf and carbon monoxide in THF at 80 °C afforded NH-indole 2-acetamides 198 (Scheme 162).189 The authors found some limitations in the methodology when aniline and benzylamine were used as substrates, whereas the indole products were formed in trace amounts.
 |
| | Scheme 162 | |
Cacchi and co-workers also demonstrated that PdCl2(PPh3)2 and Pd(PPh3)4 are effective at catalyzing the cyclization reaction of 3-(ortho-trifluoroacetamidoaryl)-1-propargyl alcohols and carbonate derivatives with amines to give 2-(aminomethyl)indoles 199 and 200, respectively (Scheme 163).190,191 This is a very useful procedure, which works well with primary and secondary amines, although primary amines give moderate yields. The author justified that primary amines could cause competitive side reactions. When the palladium-catalyzed cyclization conditions were applied to ethyl 3-(ortho-trifluoroacetamidoaryl)-1-propargyl carbonates, bearing an alkyl substituent at the propargyl position, an elimination reaction occurred to form 2-vinylic indoles 201 instead of 2-(aminomethyl)indole derivatives (Scheme 164).
 |
| | Scheme 163 | |
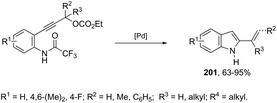 |
| | Scheme 164 | |
Results from the palladium-catalyzed cascade cyclization of (aminoaryl)propargyl alcohols and isocyanides showed that 2-indolylacetamides 202 were obtained in excellent yields, using Pd(TFA)2 as the palladium source and MeCN as the solvent, under air atmosphere at 60 °C (Scheme 165).192 Under these conditions, the intermolecular cycloaddition was successfully applied to a range of substrates, although hindered isopropyl isocyanide, phenyl isocyanide and internal alkynes were found to be unreactive, even after the prolonged reaction time and at elevated temperatures. The reaction is believed to proceed through a cascade that includes aminopalladation, isocyanide insertion, and 1,4-hydroxyl migration in which the two oxidation states of palladium promoted the activation and cyclization processes of each cycle without the use of oxidants.
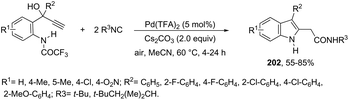 |
| | Scheme 165 | |
For intermolecular functionalization of 2-position of indoles, an interesting study has recently been developed aiming at the synthesis of tetrahydro[1,4]diazepino[1,2-a]indoles 203 from 2,2,2-trifluoro-N-(2-iodophenyl)acetamides and N-protected (prop-2-yn-1-yl)acrylamides (Scheme 166).193 It was proposed that the cascade sequence, including the palladium-catalyzed Sonogashira coupling, indole cyclization, regio- and chemoselective N-acylation, and 1,4-Michael addition, is involved in obtaining the indole derivatives. The control experiments carried out by the authors indicated that indole 204 is the key intermediate for this cyclization. They discard the participation of intermediate 205, formed via an intermolecular transamidation (Scheme 166).
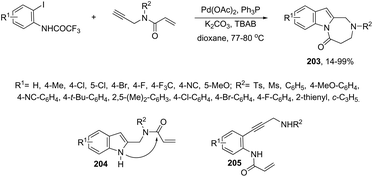 |
| | Scheme 166 | |
The palladium-catalyzed cascade cyclization can be a useful alternative to other methods to access C2 functionalized indoles. For example, the (2-propynyl)anilines and (2-aminoaryl)propynols were applied as suitable substrates in the preparation of 2-arylmethylindoles. Chowdhury and co-workers found that in the presence of a palladium catalyst (2-propynyl)anilines and (2-aminoaryl)propynols demonstrated divergent reactivity. On the one hand, the reaction of (2-propynyl)anilines with aryl halides, catalyzed by Pd(OAc)2, PPh3 and DBU, led to the direct formation of 2-arylmethylindoles 206 in good yields in a single step (Scheme 167).194 On the other hand, when (2-aminoaryl)propynols were used as substrates in the reaction with PdCl2, PPh3, K2CO3, and n-Bu4NBr in DMF, the products obtained were (arylmethylidene)indolinols 207. The hydrogenolysis employing Pd/C and cyclohexene in refluxing ethanol afforded 2-arylmethylindoles 206 in a two-step reaction (Scheme 167).195
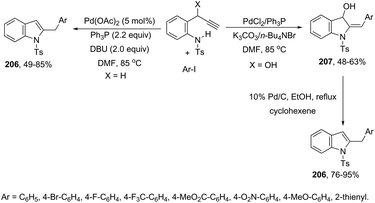 |
| | Scheme 167 | |
2.10.3. Palladium-catalyzed annulation of internal alkynes.
The palladium-catalyzed annulation of internal alkynes by aryl halides, bearing a nucleophile at an appropriate distance, is a versatile way to generate a wide variety of heterocycles. When the nucleophile is a nitrogen atom located in the ortho-position of aryl halides, the substrates deliver the indole derivatives. This reaction normally involves the addition of aryl halides to a palladium(0) complex, with cleavage of the covalent carbon–halogen bond and oxidation of palladium(0) giving a sigma-organopalladium(II) halide complex. This palladium(II) species undergoes rapid insertion of a carbon–carbon bond of the alkynes to give the palladium(II) intermediate via cis-carbopalladation. The intramolecular nitrogen nucleophile attack induces the displacement of palladium(0), most likely by prior palladacycle formation and reductive elimination giving the indole (Scheme 168). This chemistry, originally developed by Larock,31 has been an area of continuing expansion; for example a methodology using phosphine-free ligands to mediate palladium-catalyzed indolization of 2-bromoanilines with internal alkynes was developed.196 Phenylurea was found to be an optional ligand, which together with Pd(OAc)2 catalyzed the synthesis of 2,3-disubstituted indoles 208 in good yields with high regioselectivity (Scheme 169).
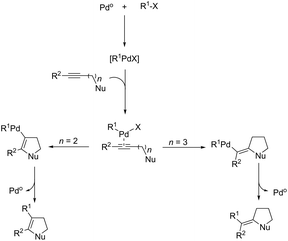 |
| | Scheme 168 | |
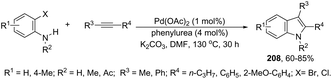 |
| | Scheme 169 | |
The use of a heterogeneous ligand and a salt-free procedure to prepare indoles was also described. In this reaction, ortho-haloanilines reacted with internal alkynes under Pd/C, Na2CO3, and DMF at 120 °C to afford the indole derivatives 209 (Scheme 170).197–199 This catalytic system could be used under aerobic conditions, and successfully re-used at least for four cycles.
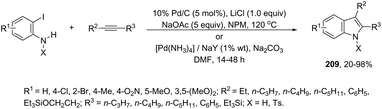 |
| | Scheme 170 | |
In an alternative method, indole derivatives 210 were efficiently prepared by palladium-catalyzed annulation of functionalized aryl iodides with internal alkynes in aqueous medium under microwave irradiation (Scheme 171).200 Both symmetrical and unsymmetrical internal alkynes bearing alkyl, aryl and silyl groups could be used as substrates for this cyclization. The annulation reactions exhibit excellent regioselectivity, where the most hindered group is located on the 2-position. The catalytic system was successfully recycled five times with a slight decrease in activity.
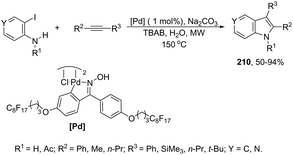 |
| | Scheme 171 | |
The alkynyldimethylsilyl t-butyl ethers reacted with ortho-iodoanilines, with excellent regioselectivity, to afford 2,3-disubstituted indoles 211 (Scheme 172).201 The regioselectivity depends mainly upon the bulkier t-butoxysilyl ether substituent on the alkynes, whereas migratory insertion could occur to minimize the steric strain. In addition, the triethylsilyl propargyl glycine derivatives were coupled with ortho-iodoanilines, catalyzed by Pd(OAc)2 and PPh3, providing useful access to N-ethyl-D-tryptophans.202
 |
| | Scheme 172 | |
Palladium-catalyzed annulation of 1-alkynylphosphine sulfides with ortho-iodoanilines gave the corresponding 2-indolylphosphine sulfides 212, which could be easily reduced to the corresponding trivalent phosphines 213 in the presence of tris-(trimethylsilyl) silane and a catalytic amount of AIBN. With the parent 1-alkynylphosphine oxides, the palladium-mediated cyclization gave, exclusively, the 2-indolylphosphine oxide derivatives, which were reduced to the trivalent phosphine 214 by treating with trichlorosilane and tributylamine (Scheme 173).203
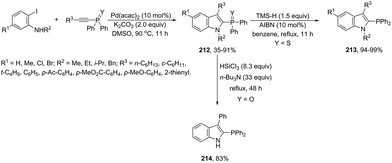 |
| | Scheme 173 | |
The reaction of ethynyloxazolidinones with ortho-iodoanilines, under Larock's heteroannulation conditions, produced optically active 2-silyl-3-indolylglycine derivatives 215, which can be induced to undergo a desilylation reaction resulting in 3-indolylglycine 216 (Scheme 174).204 Treatment of ethynyloxazolidinones and ortho-iodo-N-Ts-anilines with Pd(OAc)2, LiCl, Na2CO3, and DMF at 90 °C promoted the cyclization to give 215. In contrast, the reaction with Pd(OAc)2, PPh3, n-Bu4Cl, DIPEA, and DMF at 90 °C afforded 217.
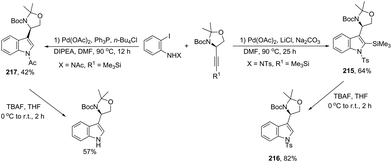 |
| | Scheme 174 | |
The generation of indoles via palladium-catalyzed three- or four-component coupling of aryl iodides, alkynes, and amines has also been described. In a one-pot protocol, the treatment of N-substituted-ortho-iodoanilines, alkynes and amines with Pd(OAc)2, cyclopentadiene–phosphine, and t-BuOLi, led to the desired indole derivatives 218 (Scheme 175).205 These conditions could be efficiently applied to a variety of ortho-iodoanilines, symmetric and unsymmetric alkynes, and cyclic and acyclic amines. The author argues that the cleavage of the C(sp3)–N bond on the intermediate 219 is the key step for the formation of desired products. After this cleavage step, the reaction could follow two pathways as illustrated in Scheme 176. The same research group also described that the indole-containing alkyl iodides 220 were obtained in the absence of amines, where palladium promoted C(sp3)–I bond formation via reductive elimination instead of syn-β-hydride elimination (Scheme 177).206 The authors observed that the use of Pd(π-allyl)Cp instead of Pd(OAc)2 facilitated the C(sp3)–I for the reductive elimination process.207
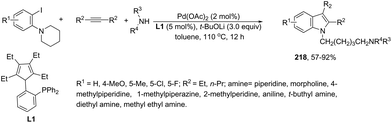 |
| | Scheme 175 | |
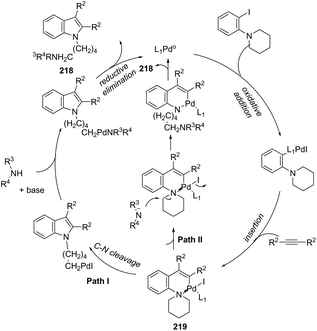 |
| | Scheme 176 | |
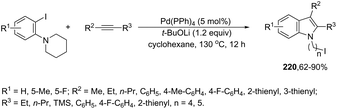 |
| | Scheme 177 | |
An important improvement in Larock's indole synthesis was accomplished by the application of propargyl bromides as alkyne sources. In this methodology, the palladium(0)-catalyzed one-pot reaction of N-Ts or N-Ms ortho-iodoanilines and propargyl bromides afforded indoles 221 (Scheme 178).208 It was assumed that the reaction involves the carbon–carbon bond coupling forming allenes and azapalladation. The substituent group of the nitrogen atom greatly influenced the reaction. The N-Ts- and N-Ms-substituted ortho-iodoanilines gave the corresponding indoles in good yields; however, N-p-Ns-substituted 2-iodoaniline (p-Ns = 4-nitrobenzenesulfonyl) afforded a trace amount of indole products.
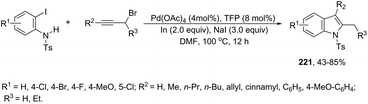 |
| | Scheme 178 | |
Expanding upon the use of alkynes in the Larock indole synthesis, very recently, the palladium(II)/N-heterocyclic naphthalimide carbene complex heteroannulation of tertiary propargyl alcohols with ortho-haloanilines, resulting in the formation of 2-alkenylindoles 222, has been reported (Scheme 179).209 A single regioisomer was obtained and this high regioselectivity was attributed to the coordination of the palladium catalyst to the propargyl hydroxyl moiety during the palladium insertion step. In addition, the authors concluded through experimental evidence that the in situ generation of HBr is the promotor of terminal double bond formation by a dehydration process. Moreover, the presence of naphthalimide units of the NHC was crucial for the success of the catalytic process.
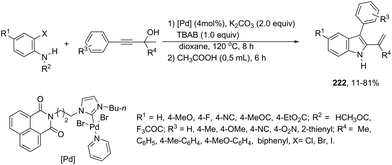 |
| | Scheme 179 | |
ortho-Iodobenzoic acid has also been studied under palladium-catalyzed annulation conditions. Under these conditions, ortho-iodobenzoic acid was converted to indole derivatives 223 by a one-pot Curtius rearrangement210/palladium-catalyzed indolization process (Scheme 180).211 In this synthetic strategy, the ortho-iodoaniline and sodium chloride, which are essential for the cyclization, are produced in situ as by-products of the Curtius rearrangement. In addition, when an acylating agent was added under the reaction conditions, indole N-carboxamide derivatives 224 were produced (Scheme 180).
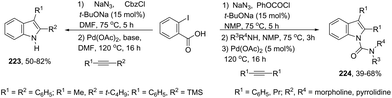 |
| | Scheme 180 | |
The intramolecular palladium-catalyzed annulation reactions have also been employed in the generation of indole systems. In this case, when N-acetyl-N-(3-phenylpropynyl)-2-iodoanilines reacted with N-tosylhydrazones in the presence of the Pd(PPh3)4 catalyst and Cs2CO3 in toluene at 80 °C, the 3-vinylindoles 225 were obtained in good yields (Scheme 181). These reaction conditions led to the desired 3-vinylindoles 225via carbon–carbon single bond and carbon–carbon double bond formation in a one-step operation. The authors suggested that the migratory insertion of palladium carbene 226 to form the intermediate 227, followed by β-hydride elimination, are the principal steps of the cyclization (Scheme 182).
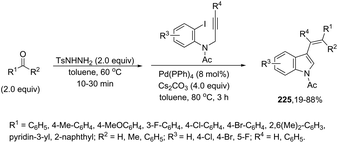 |
| | Scheme 181 | |
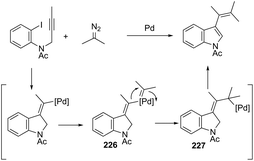 |
| | Scheme 182 | |
2.10.4. Indoles via palladium-catalyzed carbon–hydrogen activation.
Palladium-catalyzed Csp2–H bond activation has been recognized as an attractive tool in synthetic organic chemistry for carbon–hydrogen functionalization.131 The use of this concept has been considered by synthetic researchers in the preparation of indoles from alkynes. For example, palladium-catalyzed intermolecular cyclization of anilines and symmetrical diarylalkynes afforded 2,3-diarylindoles 228 or pyrrole derivatives (Scheme 183).212 Unsymmetrical diarylalkynes gave a mixture of indoles with no regioselectivity. The choice of solvents governed the product distribution, in which the reaction carried out in DMF gave the indoles, whereas the use of 1,4-dioxane as the solvent resulted in pyrrole formation.
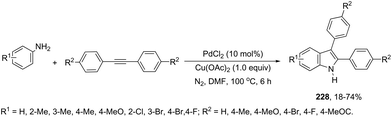 |
| | Scheme 183 | |
A high regioselectivity was achieved when unsymmetrical electron-deficient alkynes, such as methyl perfluoroalk-2-ynoates, were used in the sequential Michael-type addition and palladium(II)-catalyzed cross-dehydrogenative coupling in the preparation of 2-(perfluoroalkyl)indoles 229 (Scheme 184).213 After an electrophilic palladation and deprotonation of the nucleophilic enamine, generated by Michael-type addition of aniline and the alkyne, the palladium intermediate 230 is formed. The electrophilic aromatic palladation, followed by reductive elimination from intermediate 231, gives the desired indole (Scheme 185). The presence of O2 and an acid are crucial to regenerate the active catalyst in the cycle.
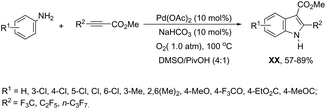 |
| | Scheme 184 | |
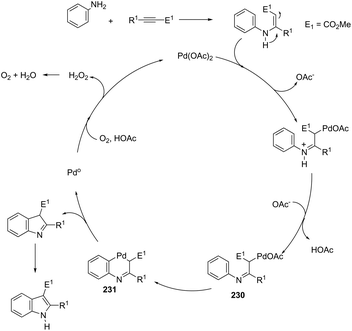 |
| | Scheme 185 | |
A similar strategy based on the direct carbon–hydrogen activation using oxygen as the oxidant was applied to regioselective reactions of anilines and internal alkynes in the preparation of indole derivatives 232 (Scheme 186).214 Because the oxidant plays an essential role in the catalytic cycle of the carbon–hydrogen activation, the authors performed a complete study to determine the activity of various oxidants, such as Cu(OAc)2, AgOAc, PhI(OAc)2, and BQ. The authors indicated dioxygen as an ideal oxidant. Under the optimal reaction conditions, the scope of the reaction was applied to anilines having electron-withdrawing or electron-donating groups and electron-deficient alkynes, and it proceeded efficiently giving the indoles in moderate to excellent yields. Additional reactions demonstrated that the carbon–hydrogen activation is not a reversible reaction and that hydroamination occurs before reductive elimination.
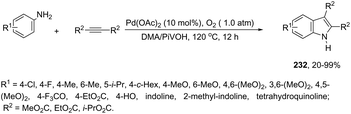 |
| | Scheme 186 | |
The use of N-acylbenzotriazoles to replace ortho-haloanilides in the preparation of indoles, via palladium-catalyzed annulation reactions, has also been reported. In the palladium-catalyzed dehydrogenative indolization sense, N-aroylbenzotriazoles reacted with disubstituted alkynes under Pd(PPh3)4, in the absence of a solvent at 130 °C to afford the corresponding polysubstituted indoles 233 in good to high yields (Scheme 187).215 Under these conditions, although asymmetric alkynes gave a mixture of regioisomers, the indoles having a bulkier substituent at the 2-position were obtained as major products. The reaction is believed to occur via an initial thermal isomerization of the benzotriazole, giving the corresponding 2-iminobenzenediazonium species 234. The oxidative addition of the diazonium moiety to palladium(0) leads to intermediate 235 or 236, which delivers the palladacycle species 236 after insertion of the alkyne. The reductive elimination of palladium(0) from 237 produces the corresponding indoles (Scheme 188).
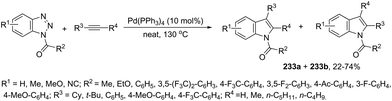 |
| | Scheme 187 | |
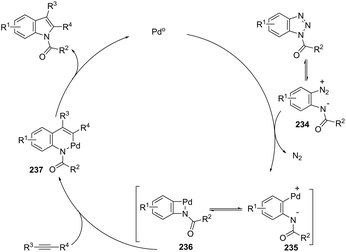 |
| | Scheme 188 | |
The use of N-acetyl anilines in the palladium carbon–hydrogen activation is an efficient method for the preparation of indoles. In this case, the acetyl group can act as a directing group and the nitrogen atom source for indole cyclization. Thus, the reaction of N-aryl amides with internal alkynes catalyzed by Pd(OAc)2 in DMA, using Cu(OTf)2 and Ag2O as oxidants, gave indoles 238 (Scheme 189).216 The author also showed that the cyclopalladated complex 239 is the key intermediate of this cyclization and its stoichiometric reaction with internal alkynes in the presence of bipyridine and DMF at 120 °C led also to the formation of indoles in moderate yields.
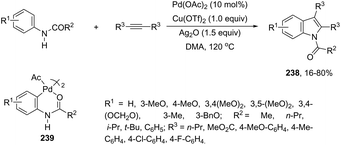 |
| | Scheme 189 | |
In order to extend the application of different N-aryl compounds as substrates for palladium-catalyzed direct functionalization of carbon–hydrogen bonds, N-aryl-2-aminopyridines were reacted with internal alkynes under catalysis by Pd(MeCH3)2Cl2, using CuCl2 as an oxidant, resulting in the direct formation of N-(2-pyridyl)indoles 240 (Scheme 190).217 When the reaction conditions were applied to internal alkynes, bearing an alkyl chain directly bonded to the carbon–carbon triple bond, a mixture of two regioisomers was obtained. On the other hand, unsymmetrically heteroaryl- and alkyl-substituted alkynes gave the corresponding indoles as a single isomer. The authors attributed the selectivity of the cyclization to the electronic and steric differences between the substituent groups. They also found that the reaction was sensitive to the steric bulk around the pyridine nitrogen and in the N-aryl ring and favored by electron-withdrawing groups in the N-aryl ring. Wu and co-workers218 have extended the scope of this reaction using the system palladium/cerium(IV) oxide and CuCl2 in the cyclization of aryl-2-aminopyridines with unsymmetrically aryl- and alkyl-substituted alkynes. N-(2-Pyridyl)indoles were obtained in excellent yields and very similar results were obtained for the selectivity, whereas the major product was the indole having the alkyl group at the 3-position.
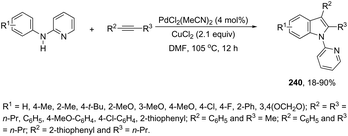 |
| | Scheme 190 | |
Alkynylimines have been also employed as substrates in the palladium-catalyzed synthesis of 2-fluoroalkyl-3-methylene-indoles 241via a domino carbopalladation/carbon–hydrogen activation procedure (Scheme 191). In order to find the suitable reaction conditions, the authors studied the influence of palladium salts, bases and solvents in this reaction. After the optimization, the best reaction conditions were established as Pd(OAc)2 and Na2CO3 in DMF at 80 °C. According to the authors, the carbon–carbon triple bond of alkynylimines could be activated by the arylpalladium(II) species formed by the oxidative addition of palladium(0) with aryl iodide. The syn carbopalladation, followed by carbon–hydrogen activation of alkynylimines, gave a six-membered palladacycle, which after reductive elimination afforded the indoles (Scheme 192).
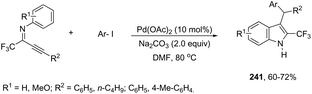 |
| | Scheme 191 | |
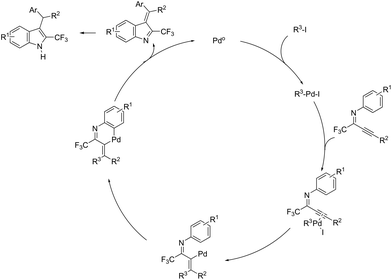 |
| | Scheme 192 | |
2.10.5. Indoles via a palladium-catalyzed N-arylation/hydroamination sequence.
The present section deals with the application of the palladium-catalyzed N-arylation hydroamination sequence-type reactions involving aryl halides, alkynes and amines in the preparation of indoles. The reaction pathway for this sequence, in general, involves a consecutive palladium-catalyzed carbon–nitrogen bond formation with a sp2-hybridized carbon, followed by the hydroamination of carbon–carbon triple bonds to promote the cyclization in a 5-endo-dig mode. As an example, the reaction of ortho-alkynyl-3-halophenyl ethers and meta-alkynyl-2-halophenyl ethers with benzylamine under Pd(OAc)2, 1,3-bis-(2,6-diisopropylphenyl) imidazolium chloride (HIPrCl) as the catalyst, in toluene under reflux, and with t-BuOK as a base, afforded the alkoxy-N-benzyl-2-substituted indoles 242 and 243via a tandem amination/cyclization reaction (Scheme 193).219 The authors observed a limitation in this protocol when an unprotected hydroxy 2-chloro-3-(phenylethynyl)phenol derivative was used as the substrate. In this case, a 7-hydroxyindole derivative could not be obtained, although, the starting material was completely consumed. This approach has been also utilized for the synthesis of an indole inhibitor of phospholipase A2 244 that is a secreted phospholipase, found at high levels in patients suffering from inflammatory diseases.
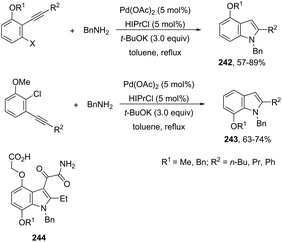 |
| | Scheme 193 | |
The palladium-catalyzed N-arylation/hydroamination sequence can be also performed with bulky alkyl amines. The palladium complex derived from an N-heterocyclic ligand was applied in the reaction of sterically hindered amines with ortho-alkynylhaloarenes in the preparation of sterically hindered N-substituted indoles 245 (Scheme 194).220,221 The authors found that the association of Pd(OAc)2 with a sterically hindered unsaturated imidazolium salt gave the best catalytic system, affording the indole derivatives in moderate to high yields. This protocol tolerated ortho-alkynylchloroarenes, 1-adamantylamine, and t-butylamine, and hindered less nucleophilic anilines.
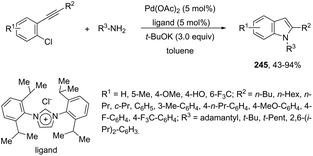 |
| | Scheme 194 | |
2-Alkynyl indoles 246 could be synthesized in a one pot procedure from ortho-bromo-(2,2-dibromovinyl)benzenes, terminal alkynes, and arylamines via a palladium-catalyzed three-component coupling process (Scheme 195).222 In this reaction, the three reagents were added at the same time in the reaction system, showing a very high chemical selectivity. Important results were obtained in the studies of the reaction mechanism, for example when the reaction was carried out in the absence of aniline, the 1,3-diyne 247 was obtained (Scheme 196). In addition, when C was reacted with aniline, under optimized reaction conditions, the corresponding 2-alkynyl indole was obtained in 65% yield. These last two experiments indicated that the reaction could follow the pathway shown in Scheme 196.
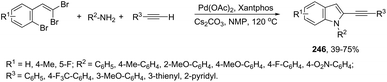 |
| | Scheme 195 | |
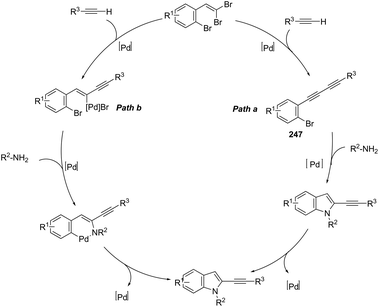 |
| | Scheme 196 | |
Ammonia has been used as the nitrogen source in the synthesis of 2-arylindoles 248 from ortho-alkynylbromoarenes through a tandem cross-coupling/alkyne amination sequence (Scheme 197).223 The authors carried out a serious study aiming to find a suitable ligand to participate together with the palladium salt as a catalyst of this reaction. They found the best results by using [Pd(cinnamyl)Cl]2 and Josiphos, delivering the indoles in 31–89% yields. They also found that heterocyclic substrates with the heteroatom ortho to the bromo or alkyne group and alkynes having silyl, alkyl, or alkenyl groups did not give the products. However, when ammonia was replaced with methylamine, the corresponding N-methyl indoles were obtained in good yields. In order to extend the above methodology, the same group found that (silanyloxyphenyl)phosphines were efficient ligands for palladium-catalyzed carbon–nitrogen cross-coupling/cyclization of ortho-alkynylhaloarenes with primary amines affording 2-substituted indole derivatives 249 (Scheme 198).224 Catalytic amounts of [Pd(cinnamyl)Cl]2 and (silanyloxyphenyl)phosphine, in the presence of t-BuOK and toluene, were able to promote the cyclization of ortho-alkynylhaloarenes bearing alkyl, aryl, and heteroaryl groups with hindered and unhindered aryl amines, including those having electron-donating and electron-withdrawing substituents, to afford indoles containing alkyl, aryl, and heteroaryl substituents at the C2 position.
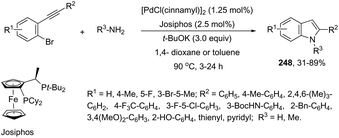 |
| | Scheme 197 | |
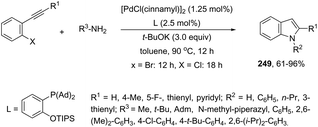 |
| | Scheme 198 | |
N,N-Disubstituted hydrazines were also used as the nitrogen source in the palladium catalyzed cross-coupling/alkyne amination reaction with ortho-alkynylhaloarenes providing a useful method for the synthesis of substituted 1-aminoindoles 250 (Scheme 199).225 For this cyclization, it was proposed that an initial palladium catalyzed coupling between the ortho-alkynylhaloarenes and hydrazines gives ortho-alkynylhydrazine, which after an alkyne amination reaction affords the N,N-disubstituted-1-aminoindole via 5-endo-dig cyclization (Scheme 200).
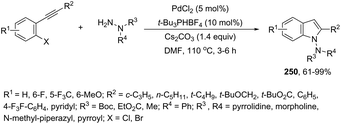 |
| | Scheme 199 | |
 |
| | Scheme 200 | |
Using t-butyl sulfonamide is a useful method for introducing nitrogen-containing moieties in the palladium catalyzed cross-coupling/alkyne amination reaction.226 In this case, the t-butyl sulfonamide, under appropriate conditions, is a suitable ammonia source. Thus, the reaction of t-butyl sulfonamides with ortho-alkynylbromoarene, in the presence of Pd(OAc)2/Xantphos/Cs2CO3 as the catalytic system in 1,4-dioxane at 110 °C, gave the 2-aryl-indoles 251 from 71 to 90% yields (Scheme 201).
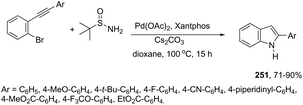 |
| | Scheme 201 | |
In the amination reaction of ortho-haloaryl alkynyl bromides, amines and amides reacted with high selectivity in Csp instead of Csp2 leading to o-haloaryl-substituted ynamides that can be useful in the construction of 2-amido indoles 252 (Scheme 202).227 Specifically, ortho-haloarylynamides reacted with p-Tol-NH2 under catalysis of Pd2(dba)3, in the presence of X-phos as a ligand, giving 2-amido-indoles in high yields. In these studies, the authors concluded that other phosphine ligands were ineffective and that aryl chlorides and bromides were better suited in this cyclization than aryl iodides.
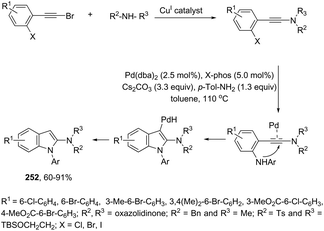 |
| | Scheme 202 | |
2.11. Platinum-catalyzed synthesis of indoles
Platinum-based catalysts have attracted considerable attention in transition metal catalysis for participating in various transformations in organic chemistry; however, their applications in the preparation of indoles have been limited. Recent reports demonstrated growing interest in this area, and platinum(II) salts have proven to be excellent catalysts for the cyclization of 2-propargyl anilines to give 3-alkoxyindoles 253 (Scheme 203).228 The reaction conditions and the substituent at the propargyl position and at the nitrogen atom had a dramatic influence on the course of the PtCl2-catalyzed cycloisomerization. For example, when the reaction was carried out at room temperature, the indole 253 was obtained exclusively, whereas on increasing the temperature to 80 °C the reaction gave the indole adduct 254.
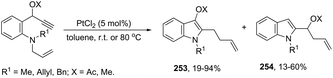 |
| | Scheme 203 | |
Indole carbamates, indole ureas and indole phosphoranes 255 were prepared by a platinum-catalyzed Hofmann-type rearrangement of ortho-alkynylbenzamides and ortho-alkynylbenzylamides. The reaction occurred via a nucleophilic addition of alcohols and amines to the isocyanate intermediates, followed by an intramolecular aminocyclization (Scheme 204).229,230
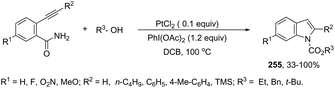 |
| | Scheme 204 | |
Another important development in the functionalized indole synthesis is the platinum-catalyzed tandem annulation/arylation reaction. Diindolylmethanes 256 were prepared using propargyl ethers and substituted indoles via a platinum-catalyzed tandem indole annulation/arylation cascade (Scheme 205).231 The electrophilic platinum carbene intermediate was proposed to be the key intermediate in this cascade reaction. This tandem reaction was also attempted using β-dicarbonyl compounds as nucleophiles in the preparation of 2-substituted indoles 257 (Scheme 206).232 The reaction generated α,β-unsaturated carbene intermediates via an intramolecular nucleophilic addition into alkynes bearing propargyl ethers, followed by vinylogous nucleophilic additions using enol as the nucleophile.
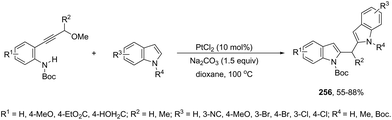 |
| | Scheme 205 | |
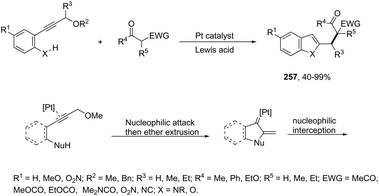 |
| | Scheme 206 | |
A sequential platinum-catalyzed cyclization/intermolecular addition was performed for the preparation of 2,3-disubstituted indoles 258 (Scheme 207). The reaction involved ortho-alkynylanilines with ethyl propiolate, dimethyl acetylenedicarboxylate and PtCl2 as the catalyst. The platinum catalyst showed a dual action by acting in catalytic π-activation of both ortho-alkynylanilines and ethyl propiolate.233 The platinum-catalyzed cyclization/intramolecular carbon–nitrogen bond formation-[1,3] shift of carbamoyl and ester groups provides another useful route to synthesize indole-3-carbamides and -carboxylates 259 (Scheme 208).234 The authors concluded, via crossover experiments, that the cyclization of ortho-alkynylphenyl ureas or ortho-alkynylphenyl carbamates, catalyzed by PtI4, occurred in an intramolecular manner instead of an intermolecular manner. In addition, the formation of 3-protonated indole was observed as a by-product in all reactions. It has also been concluded that the proton source was the methyl moiety of the migrating carbamoyl group, which would be eliminated in the [1,3] carbamoyl migration.
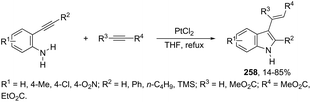 |
| | Scheme 207 | |
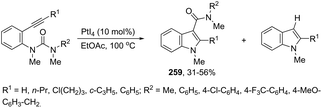 |
| | Scheme 208 | |
2.12. Rhenium-catalyzed synthesis of indoles
The cyclization of ortho-alkynylaryl piperidines catalyzed by [ReBr(CO)5] gave N-fused tricyclic indole derivatives 261 in moderate to good yields (Scheme 209).235 The catalytic process assumed by the authors involves the initial formation of metal containing ammonium ylides 260, which undergo ring expansion through a 1,2-rearrangement, followed by 1,2-alkyl migration to form N-fused tricyclic indole derivatives. In addition, the authors found that using ortho-alkynylaryl pyrrolidines the best catalytic action was achieved with [W(CO)6].
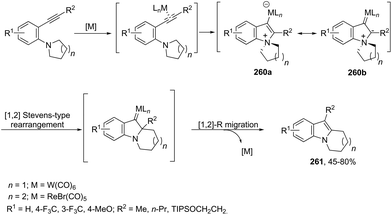 |
| | Scheme 209 | |
2.13. Rhodium-catalyzed synthesis of indoles
2.13.1. Rhodium-catalyzed cyclization of ortho-alkynylanilines.
For several decades, rhodium-catalyzed transformations have been successfully employed in the development of novel methodologies, which have found widespread use in different areas because of their versatility and diverse applications. In organic synthetic methodologies, rhodium catalysis is broadly applied in olefin functionalization, hydrogenation, carbon–hydrogen functionalization, cycloisomerization, and cycloaddition reactions.236 In particular, rhodium-catalyzed reactions with alkynes allow the synthesis of diverse carbo- and heterocycles. In this section, we will show the synthetic efforts in developing indoles based on rhodium-catalyzed reactions using alkynes and different nitrogen compounds as substrates. In an early report, rhodium-catalyzed cycloisomerization of unprotected anilines and terminal alkynes proceeded effectively to produce indoles 262 in good yields (Scheme 210).237 The reaction conditions were carefully optimized indicating that the use of Rh(cod)Cl2 (1 mol%) and PPh3 (4 mol%) in DMF at 85 °C was the optimal conditions to deliver the indoles in best yields.
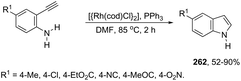 |
| | Scheme 210 | |
The intramolecular reaction of propargyl alcohol derivatives with Rh(CO)2Cl2 generated rhodium(I) carbenes 263, via a dehydrative indole annulation. Depending on the substituent on the aniline nitrogen nucleophile, either a cyclopropanation or dimerization product could be selectively obtained. The results indicated that cyclopropanation products 264 were exclusively formed with N-Ts protected aniline, whereas the N-Boc-protecting group gave the dimerization products 265 (Scheme 211).238
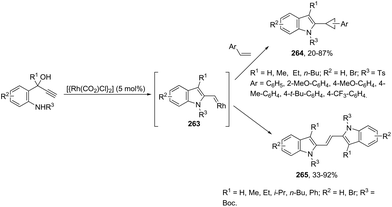 |
| | Scheme 211 | |
The methylsulfonyl-protected aniline derivatives were successfully utilized for the development of a convenient protocol for the general synthesis of siloles. The intramolecular cyclization of aniline substrates generated an organometallic intermediate 266, which after the activation of the Csp3–Si bond afforded the desired indole [3,2-b] siloles 267 (Scheme 212).239 Subsequently, 3-silylindole derivatives 268 were formed from the reaction between the 2-silylethynylanilines and cationic rhodium(I)/H8–BINAP complex via a 1,2-silicon migration. The authors observed that the use of H8–BINAP in place of BINAP led to obtaining the products in good yields (Scheme 213).240
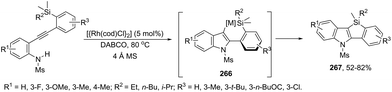 |
| | Scheme 212 | |
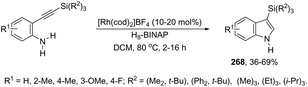 |
| | Scheme 213 | |
Several 2,3-disubstituted indoles have been obtained through an intramolecular rhodium-catalyzed tandem cyclization–addition sequence using ortho-alkynylanilines as substrates and an electrophile source. For example, the cyclization of ortho-alkynylanilines catalyzed by Rh(I)/BINAP with a subsequent reaction with electrophilic alkenes gave the indole derivatives 269 having an alkyl chain at the C3-position (Scheme 214).241 When isocyanates were used as an electrophile source in the cyclization of ortho-alkynylanilines catalyzed by [RhCl(COD)]2 (5 mmol%), K2CO3 and 2-BuOH, the indole-3-carboxamides 270 were formed in excellent yields (Scheme 215).242 The authors confirmed, via a control experiment, that the formation of a 3-rhodium indolyl intermediate occurred during the cyclization process rather than after indole formation. In the same way, the rhodium(III)-catalyzed cascade cyclization/electrophilic amidation of ortho-alkynylanilines, using N-pivaloyloxylamides as the electrophilic nitrogen source, gave 3-amidoindoles 271 (Scheme 216).243
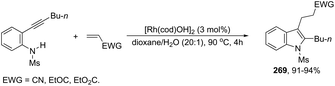 |
| | Scheme 214 | |
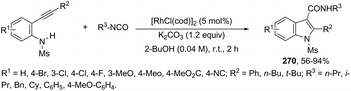 |
| | Scheme 215 | |
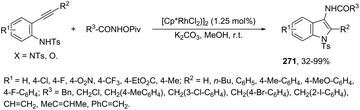 |
| | Scheme 216 | |
2.13.2. Indoles via rhodium-catalyzed carbon–hydrogen activation.
The rhodium-catalyzed annulation reaction of alkynes via carbon–hydrogen activation is a valuable method for the synthesis of heterocycles.244 The cyclization basically involves the reaction of an aromatic system with a highly electrophilic rhodium species to give a rhodacycle intermediate. Subsequently, the insertion of the rhodacycle into the carbon–carbon bond of the unsaturated substrate, followed by reductive elimination, releases the product and rhodium catalyst. In an attempt to prepare 1,2-disubstituted indoles 272, via rhodium-catalyzed annulation, simple anilines were reacted with symmetrical and unsymmetrical internal alkynes in the presence of the rhodium(III) catalyst under aerobic conditions (Scheme 217).245 The advance of this methodology is the evidence that the rhodium(I) species is oxidized to rhodium(III) species via molecular oxygen in the presence of an appropriate acid.
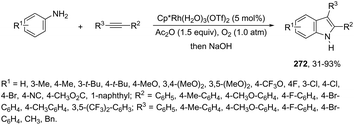 |
| | Scheme 217 | |
The acetanilide derivatives were successfully utilized for the development of a convenient protocol for the general synthesis of 1,2-disubstituted indoles 273 (Scheme 218).246–248 In the preliminary study, the reaction was carried out at high temperature (120 °C) using [Cp*RhCl2]2 as a precatalyst and a stoichiometric amount of Cu(OAc)2 as an oxidant. A broad study to improve these reaction conditions showed that the combination of [Cp*RhCl2]2, AgSbF6, and Cu(OAc)2 H2O was the best catalytic system to form the indoles in higher yields. It was also observed that molecular oxygen worked as the internal oxidant, reactivating the reduced rhodium(I), which allowed the reaction to be carried out at mild temperatures (60 °C). This permitted the expansion of the compatibility of the reaction to include a range of internal alkynes bearing useful functional groups. As an alternative to improve the yields of the less reactive alkynes, the dicationic analogue [Cp*Rh(MeCN)3][SbF6]2 was efficiently employed as a catalyst. The rhodium-catalyzed oxidative annulation of acetanilides with internal alkynes, under ambient conditions, was also carried out using an in situ generated electron-deficient dicationic rhodium(III) complex 274, derived from η5-cyclopentadienyl rhodium(III). The authors reported a high catalytic activity under ambient conditions, at room temperature, and under air, giving the desired indoles 275 in 49–99% yields (Scheme 219).249 Very recently, in an alternative strategy for the generation of indoles 276, Bolm and co-workers reported a mechanochemical rhodium(III)-catalyzed carbon–hydrogen bond functionalization, involving oxidative coupling of acetanilides and alkynes, in a planetary mill and in the absence of any solvent and heating (Scheme 220).250 The reaction conditions required [Cp*Rh(MeCN)3][SbF6]2 (5 mol%) as a catalyst, and a catalytic amount of Cu(OAc)2 in association with dioxygen as a terminal oxidant.
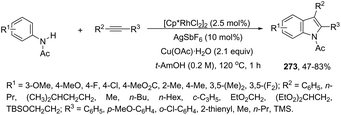 |
| | Scheme 218 | |
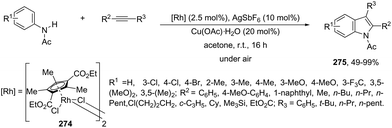 |
| | Scheme 219 | |
 |
| | Scheme 220 | |
Substituted indole derivatives 277 were synthesized from N-arylureas and internal alkynes via a rhodium-catalyzed carbon–hydrogen activation strategy, in which N-arylurea assisted the activation of the carbon–hydrogen bond and a copper salt acted as an oxidant, closing the catalytic cycle under aerobic conditions (Scheme 221).251 The optimized reaction conditions were compatible with electron-donating and electron-withdrawing functional groups in the substituted N-arylurea; however, strongly electron-withdrawing groups, such as nitro and cyano groups, did not give the corresponding indoles. The sterically hindered ortho-F and ortho-Br substituted arylureas were other limitations observed, which yielded the indoles in a trace amount. N-(2-Pyridyl)anilines were also suitable substrates for the directing group in the rhodium-catalyzed oxidative coupling with alkynes to give N-(2-pyridyl)indoles 278 (Scheme 222).252 The results indicated that groups containing the oxygen atom and the electronic effects of the aryl ring directly bonded to the nitrogen atom governed the high selectivity achieved in the cyclization. N-(2-Pyridyl)anilines can also be efficiently used in rhodium-catalyzed carboamination of unsymmetric alkynyl cycloalkanols leading to 1,2,3-trisubstituted indoles 279 (Scheme 223).253 The authors carried out a number of control experiments to elucidate the reaction mechanism, which confirmed that the formation of indoles 279 proceeds through a sequential aryl carbon–hydrogen/Csp3–Csp3 activation process, whereas the pyridyl nitrogen intermediate 280 plays an important role in chelation assistance (Scheme 224).
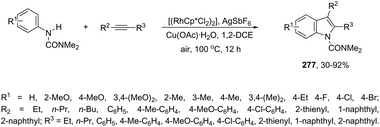 |
| | Scheme 221 | |
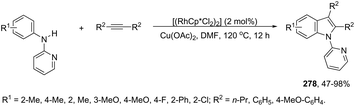 |
| | Scheme 222 | |
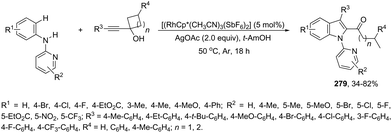 |
| | Scheme 223 | |
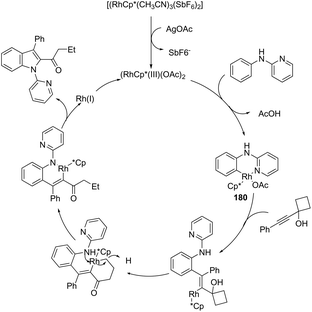 |
| | Scheme 224 | |
A regioselective synthesis of indoles 181 from primary 2-acetyl-1-arylhydrazines and internal alkynes was described to occur via a rhodium(III)-catalyzed hydrazine-directed carbon–hydrogen activation (Scheme 225).254 The advance of this methodology was the application of 2-acetyl-1-arylhydrazines as directing groups and the use of mild conditions for the nitrogen–nitrogen bond cleavage that typically requires harsh reaction conditions when it is used in redox-neutral reactions. The arylhydrazine derivatives were excellent precursors for the regioselective synthesis of indoles 182 (Scheme 226).255 The products were obtained in high yields when the reaction of arylhydrazines with internal alkynes was carried out using [RhCp*Cl2]2 (2 mol%) and AgSbF6 (8 mol%), in the presence of HOAc and MeOH as the solvent. In this reaction, the oxidant required for rhodium-catalyzed carbon–hydrogen bond activation was produced in an internal oxidation mechanism through the cleavage of the nitrogen–nitrogen bond with the formation of NH3, which was captured by AcOH. Another external-oxidant-free rhodium(III)-catalyzed carbon–hydrogen activation of aryl hydrazines with alkynes used hydrazone as a directing group to prepare indoles 183 (Scheme 227).256 The hydrazone group was formed in situ via condensation of hydrazines with isobutyraldehyde. In this methodology, the nitrogen–nitrogen bond worked as an internal oxidant without the necessity of the pre-installation and post-cleavage of the directing group. Similarly, the regioselective synthesis of indoles 184, using the hydrazone group, prepared in situ by condensation of arylhydrazine hydrochloride with diethyl ketones as a directing group and an internal oxidant, was also described (Scheme 228).257 1,3-Dinitrobenzene was also used as an external oxidant in rhodium-catalyzed oxidative annulation of hydrazines with alkynes in the preparation of 1-aminoindoles 185 (Scheme 229).258 Mechanistic studies demonstrated that 1,3-dinitrobenzene served as an oxidant during the reaction, consuming the leaving hydrogen atoms and regenerating the active rhodium catalyst.
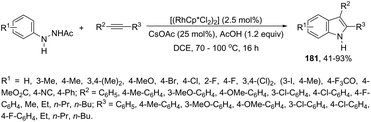 |
| | Scheme 225 | |
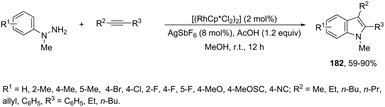 |
| | Scheme 226 | |
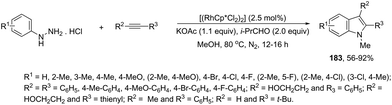 |
| | Scheme 227 | |
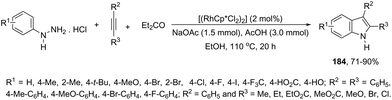 |
| | Scheme 228 | |
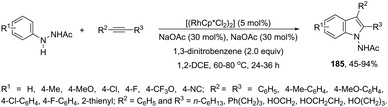 |
| | Scheme 229 | |
In another variation of the rhodium-catalyzed carbon–hydrogen bond activation, triazenes have successfully transformed in unprotected indoles 186via reaction with internal alkynes (Scheme 230).259,260 This reaction is proposed to undergo a 1,2-rhodium shift ring contraction, in which triazene behaved as an internal cleavable directing group. The studies using HRMS and theoretical calculations suggested that a 1,2-alkyl migration might be responsible for the in situ cleavage of the directing group. N-Nitrosoanilines and alkynes, in the presence of the rhodium catalyst, were employed in the synthesis of N-alkyl indoles 187, via redox-neutral carbon–hydrogen activation and annulation, using a traceless nitroso directing group (Scheme 231).261,262 The N-nitroso group serves as both a directing group for carbon–hydrogen activation and an internal oxidant for catalyst turnover. The construction of indole derivatives 188 was also successfully described using a nitrone as the oxidizing directing group (Scheme 232).263 The presence of a sterically hindered Mes group on the carbon center of the nitrones was decisive in forming indoles in high yields. In a similar reaction, nitrones were used to selectively prepare unsymmetrical 2,3-diaryl substituted indoles 189 (Scheme 233).264 The rhodium(III)-catalyzed carbon–hydrogen annulation of anilines with internal alkynes afforded the unsymmetrical 2,3-diaryl substituted indoles with poor regioselectivity. The authors used an excellent alternative to control this selectivity, in which they carried out the rhodium(III)-catalyzed annulation of nitrones with symmetrical diaryl alkynes, giving 2,3-diaryl-substituted N-unprotected indoles having two different aryl groups. Because one of the aryl substituents came from the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C-aryl group present in the nitrone and the other came from the alkyne, the methodology delivered the corresponding indoles with an exclusive regioselectivity.
C-aryl group present in the nitrone and the other came from the alkyne, the methodology delivered the corresponding indoles with an exclusive regioselectivity.
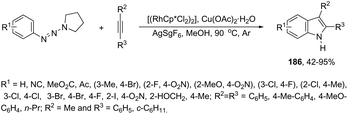 |
| | Scheme 230 | |
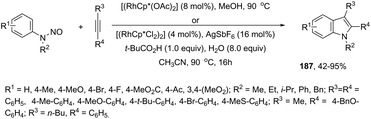 |
| | Scheme 231 | |
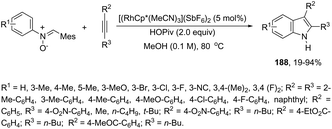 |
| | Scheme 232 | |
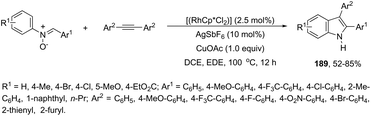 |
| | Scheme 233 | |
Another example of a rhodium-catalyzed carbon–hydrogen bond activation was the regioselective annulation of tertiary aniline N-oxides with alkynes, giving N-alkylindoles 190 (Scheme 234).265 The reaction proceeds via a sequential Csp2–H and Csp3–N activation, in which the N-oxide acts as a traceless directing group, avoiding the use of metal oxidants. A related process was employed in an approach to the synthesis of indoles bearing an N-(3-aminobutenoyl) substituent 191 through rhodium(III)-catalyzed redox-neutral carbon–hydrogen bond activation of pyrazolones and alkynes (Scheme 235).266 The key step was the formation of a bicyclic metallacycle rhodium(V) intermediate via a 1,2-rhodium migration, accompanied by the nitrogen–nitrogen bond cleavage of the pyrazolone ring, without loss of the N-terminus.
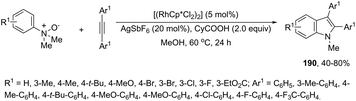 |
| | Scheme 234 | |
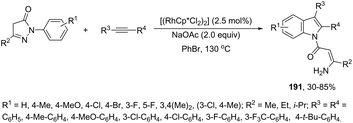 |
| | Scheme 235 | |
A concise route to substituted indoles 192 was based on a rhodium-catalyzed denitrogenative transannulation of N-sulfonyl-1,2,3-triazoles and 1-ethynylcyclohexenes (Scheme 236).267 In this reaction, the key rhodium-stabilized iminocarbene intermediate 193 was generated via ring-chain isomerization and nitrogen extrusion. The methodology based on rhodium(II)-catalyzed denitrogenative annulation of N-sulfonyl-1,2,3-triazoles was also applied to the construction of 3-indolylimines 194 using N-propargylanilines as the starting material (Scheme 237).268
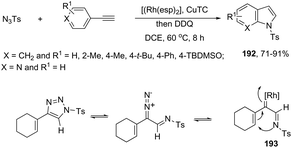 |
| | Scheme 236 | |
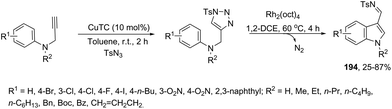 |
| | Scheme 237 | |
The amino-Claisen rearrangement of N-propargylaniline derivatives involves the [3,3]-sigmatropic shift of N-alkenyl-N-arylamine to furnish ortho-allenylanilines. An alternative version of this rearrangement has been reported in the synthesis of 2,3-substituted indole derivatives 195 using the [Rh(cod)2]OTf catalyst in hexafluoroisopropyl alcohol (HFIP) (Scheme 238).269,270 The authors confirmed by single-crystal X-ray crystallographic analysis that [Rh(CO)(Ph3P)2]OCH(CF3)2 is the active catalyst, which was generated in situ from RhH(CO)(Ph3P)3 and HFIP. In addition, they also confirmed that ortho-allenylaniline is the intermediate of the cyclization. The catalytic system was also active for the one-pot synthesis of indoles by reacting N-alkylanilines with propargyl bromide in the presence of K2CO3 in HFIP.
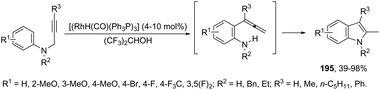 |
| | Scheme 238 | |
2.14. Ruthenium-catalyzed synthesis of indoles
Ruthenium can form a large number of different complexes with oxidation states ranging from divalent negative to octavalent positive, although the complexes with oxidation states 0, II and III are most common. This characteristic makes the salts of ruthenium versatile catalysts with extensive applications. Among them, the intramolecular carbon–hydrogen activation, hydrogenation reactions, reduction, oxidation, isomerization, carbon–carbon bond formation, and hydrogen transfer reactions have been commonly applied.271 Ruthenium complexes have been proven to be highly efficient at converting acyclic substrates in carbon- and heterocycles via metathesis and nonmetathesis reactions.272 In this section, we will cover the main methods described for the preparation of indoles using alkynes in ruthenium-catalyzed cyclization. Among them, ortho-ethynylanilines were used as substrates, in an alkyne hydration reaction catalyzed by [RuL2Cp(CH3CN)][PF6], to form indoles 197 (Scheme 239).273 The experimental evidence indicated that the ruthenium vinylidene 196 is the key intermediate for this intramolecular cyclization.
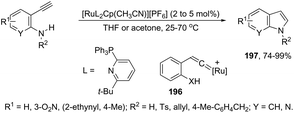 |
| | Scheme 239 | |
The ruthenium catalyst, with triphenylphosphine instead of bifunctional pyridine–phosphine as the ligand, was employed in the catalytic cyclization of ortho-alkynylanilines for the preparation of indoles 199 (Scheme 240).274 This cycloisomerization process occurred regioselectively via a 5-endo cyclization mode, where the ruthenium vinylidene 198 is the key intermediate. The catalytic activity of CpRuCl(PPh3)2 was supported by the presence of an amine/ammonium base–acid pair which can also offer higher conversions of ortho-alkynylanilines.
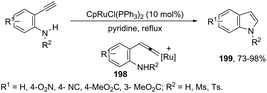 |
| | Scheme 240 | |
A very efficient method to form 2-trifluoromethyl indoles 200 involved the ruthenium-catalyzed intramolecular radical cyclization of trifluoroacetimidoyl chlorides (Scheme 241).275 This photoreaction was carried out via a reductive cleavage of the carbon(sp2)–chlorine bond of trifluoroacetimidoyl chloride by Ru(phen)3+, prepared in situ by photoexcitation and single-electron transfer of Ru(phen)32+.
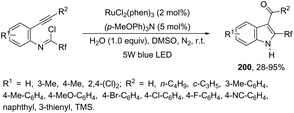 |
| | Scheme 241 | |
Recently, another example of a nonmetathesis reaction with the use of a ruthenium carbene catalyst has been described for the preparation of 2,3-disubstitued indoles 201via a cycloisomerization of N-acyl-N-vinyl-2-silylalkynylaniline derivatives (Scheme 242).276 The results obtained from the control experiments suggest that the cycloisomerization proceeds via ruthenium hydride 202, generated in situ from the Grubbs II catalyst, which activates the alkyne to give the corresponding indole (Scheme 243).
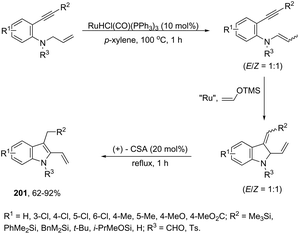 |
| | Scheme 242 | |
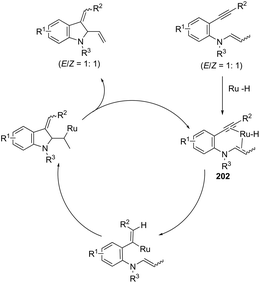 |
| | Scheme 243 | |
Intramolecular ruthenium-catalyzed annulation of nitrogen-functionalized alkynes, followed by carbon-migration has been the key to synthesize 3-substituted indoles. In one example, 1H-indole-3-carbaldehydes 203 were prepared by ruthenium-catalyzed intramolecular annulation of ortho-alkynylamides (Scheme 244).277 The reaction involved formyl translocation, which occurred via cleavage of the N–CHO bond alone, instead of a cleavage of the aldehyde C–H bond. In another example, the ruthenium-catalyzed cycloisomerization of ortho-alkynylanilides afforded 3-substituted indoles 205 (Scheme 245).278 Experimental evidence related to the reaction mechanism was addressed in detail indicating that the catalytic process proceeds via a ruthenium vinylidene intermediate 204, which is formed by 1,2-carbon migration.
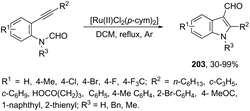 |
| | Scheme 244 | |
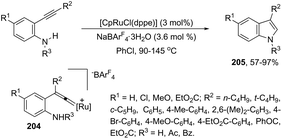 |
| | Scheme 245 | |
Ruthenium-catalyzed carbon–hydrogen activation and annulation of alkynes is an attractive method for the synthesis of indoles with concomitant introduction of two substituents at 2- and 3-positions. It has been employed in the conversion of pyrazolidinones into 2,3-substituted indoles 206 (Scheme 246).279 In this reaction, carbon–hydrogen was activated for expending a nitrogen–nitrogen bond cleavage of the pyrazolidinones, which work as both a directing group and an internal oxidant. The hydrazine group was also applied for the directed carbon–hydrogen functionalization of alkynes affording the indole derivatives. Thus, the reaction of aryl hydrazines with internal alkynes under catalysis of ruthenium(II) and Zn(OTf)2 allowed the formation of 2,3-substituted indoles 207 (Scheme 247).280 The authors determined that the best catalytic action was achieved by using Ru(p-cymene)(OTf)2 as the catalyst, which was prepared in situ by reacting RuCl2(p-cymene)2 and Zn(OTf)2 in an anion exchange reaction.
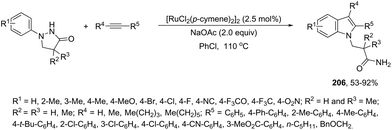 |
| | Scheme 246 | |
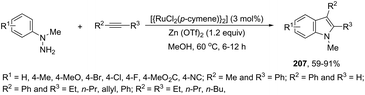 |
| | Scheme 247 | |
The use of an external oxidant in the ruthenium(II)-catalyzed carbon–hydrogen activation process has also been reported. The reaction between N-phenylacetamides and internal alkynes, under the catalysis of RuCl2(p-cymene)2, gave the best yields of the N-acyl indole derivatives 208 when Cu(OAc)2·H2O was used as an external oxidant (Scheme 248).281 Besides copper salt, the presence of the oxygen atom from the amide, to direct the cyclometalation at the ortho carbon–hydrogen activation, and the temperature were essential in the catalytic system. The 2-pyrimidyl-substituent in the anilines was also used as a directing group in oxidative annulation of alkynes. For example, the reaction of N-2-pyrimidyl-substituted anilines with alkynes, in the presence of [Ru2Cl3(p-cymene)2][PF6] as the catalyst, Cu(OAc)2·H2O as the oxidant and PEG-400/H2O as the solvent, resulted in the formation of 1-(pyrimidinyl)-1H-indole 209 (Scheme 249).282 The PEG-400/H2O catalytic system was recycled and reused six times without any loss of catalytic activity.
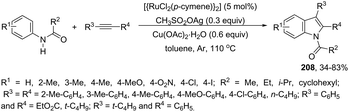 |
| | Scheme 248 | |
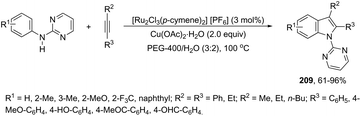 |
| | Scheme 249 | |
2.15. Scandium-catalyzed synthesis of indoles
Scandium salts have also been used as catalysts in the cyclization reaction of alkynes and nitrogen source to prepare indoles. The Friedel–Crafts reaction of 5-(arylamino)pentynones using Sc(OTf)3 as a catalyst has been described for the synthesis of 3-substituted indoles 210 (Scheme 250).283 The mechanism proposed by the authors most likely involves the coordination of the carbonyl moiety to the Sc(OTf)3 catalyst, followed by the nucleophilic attack of the aromatic ring on the carbon–carbon triple bond giving the allene intermediate, which after an isomerization, followed by a 1,3-H shift, delivers the indole derivatives (Scheme 251).
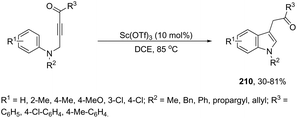 |
| | Scheme 250 | |
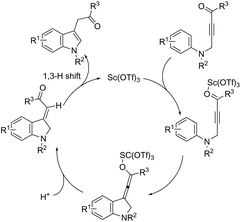 |
| | Scheme 251 | |
2.16. Silver-catalyzed synthesis of indoles
Because of their electronic configuration and mild π-acidity, silver salts are very efficient at alkyne activation.284 These special properties make silver salts highly effective at promoting cyclization reactions providing a facile access to heterocycles. An interesting application of the properties of silver was in the synthesis of indole N-carboximidamides or N-carboximidoates 211 using silver(I)-catalyzed regioselective cyclization of iminophosphoranes (Scheme 252).285 The reaction of iminophosphoranes with isocyanates gave the carbodiimides, which after reaction with secondary amines afforded the (2-alkynylphenyl)guanidine (or isourea) intermediates. The in situ addition of a catalytic amount of AgNO3, at room temperature, promoted the cyclization affording the indole N-carboximidamides or N-carboximidoates in good yields (Scheme 252). A variation of this silver-catalyzed cyclization involved the cycloisomerization of N-imidoyl-2-alkynylanilines to form indole derivatives 212 (Scheme 253).286 The authors suggested that the structure of the silver–alkyne complex 213 would be beneficial for the 5-endo-dig N-nucleophilic attack (pathway a, complex 213). In a similar way, the reaction of ortho-alkynyl isothiocyanates with secondary amines, catalyzed by silver triflate, led to the formation of N-thiourea indoles 214via a 5-endo-dig mode (Scheme 254).287
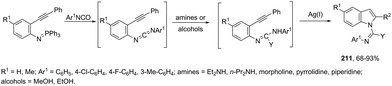 |
| | Scheme 252 | |
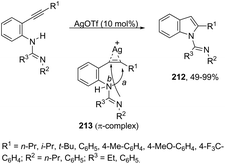 |
| | Scheme 253 | |
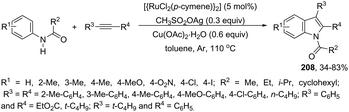 |
| | Scheme 254 | |
The 2-substituted NH-indoles 215 were synthesized by a regioselective cyclization reaction from 2-aryltriazene alkynes via a silver(I) salt mediated nitrogen–nitrogen bond cleavage (Scheme 255).288 The authors postulated that the solvolysis of the intermediate 216 by MeOH led to the corresponding indoles and AgOMe. The absence of catalytic activity of the silver salt can be attributed to the decomposition of MeOAg. A similar route was also used to describe the preparation of N-cyano-2-substituted indoles 218 using 2-aryltetrazole alkynes as the starting material in a silver(I) catalyzed intramolecular cyclization (Scheme 256).289 The advance of the methodology is the identification of a cyclopropyl argentium cationic intermediate 217, which could be responsible for the use of a catalytic amount of the silver salt.
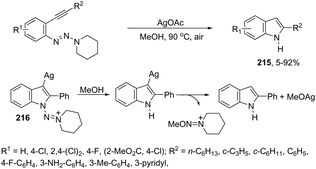 |
| | Scheme 255 | |
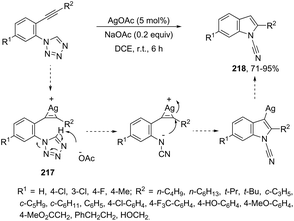 |
| | Scheme 256 | |
Silver-catalyzed cascade cyclization/3-carbon functionalization represents a versatile strategy for construction of 3-functionalized indoles because of its efficiency, atom economy and ability to deliver the product in a single reaction step. Listed below are a number of silver-catalyzed cascade cyclization examples that led to the formation of 3-substituted indoles. A silver(I)-catalyzed activation of ortho-alkynylanilines and aldehydes gave 2,2′-disubstituted bisindolylarylmethanes 219, via a domino 5-endo-dig indole annulation, addition to the carbonyl group, second indole annulation, and dehydroxylative sequence (Scheme 257). Another example applying silver-catalyzed cascade cyclization has been used in the preparation of 3-fluoroindoles 220 (Scheme 258).290 In this reaction, ortho-alkynylanilines, having an electron-withdrawing group directly bonded to the alkyne, in the presence of catalytic amounts of AgNO3 and Selectfluor, gave the corresponding 3-fluoroindoles in moderate yields, even by adding a base in the medium to prevent the formation of the 3-unsubstituted indoles.
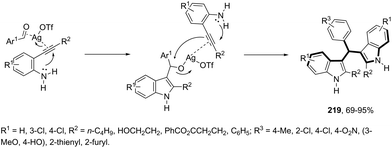 |
| | Scheme 257 | |
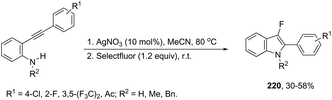 |
| | Scheme 258 | |
A recent study described the preparation of 3-phosphinoylindoles 221 through silver-mediated cycloaddition between N-Ts-ortho-alkynylanilines and H-phosphine oxides (Scheme 259).291 On the basis of experimental evidence, the authors proposed that a radical pathway is involved in this phosphinoylation–cyclization–desulfonylation sequence. Also in this topic, 2-tributylstannylfuran was used in the silver-catalyzed cascade cyclization–stannylation of ortho-alkynylanilines to introduce a stannyl substituent at the 3-carbon of the indole ring giving (3-indolyl)stannanes 222 (Scheme 260).292 The reaction mechanism studies indicated that the stannylation did not occur by the carbon–hydrogen functionalization of indole. The authors proposed that the reaction of electrophilic Bu3Sn+ with a 3-indolyl silver(I) intermediate could lead to product formation. The stannyl electrophilic species is produced via a silver-catalyzed destannylation of 2-tributylstannylfuran through transmetalation and protodemetalation.
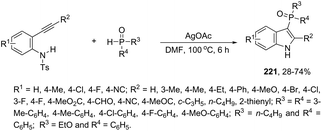 |
| | Scheme 259 | |
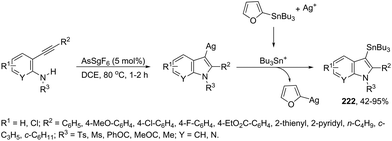 |
| | Scheme 260 | |
N-Protected ortho-alkynylanilines underwent a AgSbF6-catalyzed cascade involving the ring opening of donor–acceptor cyclopropanes to produce 2,3-disubstituted indoles 223 (Scheme 261).293 The silver catalyst presented dual action by activating the carbon–carbon bond, promoting the cyclization of the aniline and activating the cyclopropanes toward the nucleophilic attack of the 3-indolyl silver intermediate.
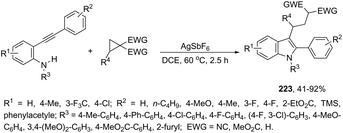 |
| | Scheme 261 | |
When iminoethers are incorporated into nitrogen of the anilines, a silver-catalyzed two-component condensation, followed by a tandem silver-induced cycloisomerization and 1,3-alkenyl shift to the silver-activated carbon, formed 2,3-disubstituted indoles 224 (Scheme 262).294 Not only can silver(I) catalyze the 5-endo-dig cyclization but also the silver carbene may be formed, which increases the electron density on the nitrogen atom driving the 1,3-alkenyl migration.
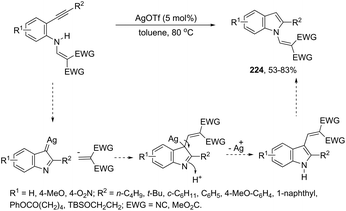 |
| | Scheme 262 | |
An oxidative dearomatization of ortho-alkynylanilines, followed by a silver(I)-catalyzed domino reaction with silyl enol ethers, provided an expedient approach to synthesize 4-acetonylindoles 225 (Scheme 263).295 In this sequence, the PhIO mediated oxidative dearomatization of ortho-alkynylanilines gave ortho-alkynylcyclohexadienimines, whereas AgOTf promoted the heterocyclization of ortho-alkynylcyclohexadienimines, which led to 4-acetonylindoles after a Mukaiyama-Michael addition with silyl enol ethers (Scheme 264).
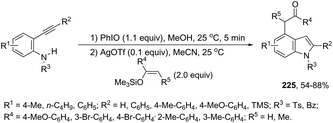 |
| | Scheme 263 | |
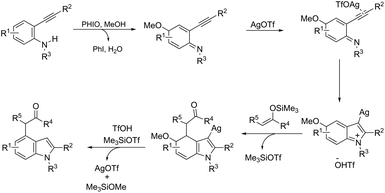 |
| | Scheme 264 | |
2.17. Titanium-catalyzed synthesis of indoles
Indoles can be directly obtained from alkynes and a nitrogen substrate in an intra- or intermolecular reaction of titanium-catalyzed cyclization. Although, titanium-catalyzed cyclization is less commonly used, the reactions show impressive reactivity and good control in the regioselectivity. A very efficient method for the synthesis of substituted 3-(tert-butyldimethylsilyloxy)indoles 227via titanium-catalyzed hydrohydrazination of a protected propargyl alcohol derivative has been developed (Scheme 265).296 The reaction gave regioselectively the key intermediate 226, via a Markovnikov addition, which afforded exclusively the 2,3-disubstituted indoles. The anti-Markovnikov hydroamination of alkynes was achieved in the reaction of a hydrazine-catalyzed bis-(amidate)-bis-(amido)titanium complex, which gave indoles 228 as products (Scheme 266).297
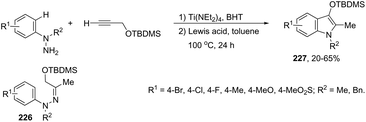 |
| | Scheme 265 | |
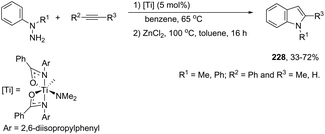 |
| | Scheme 266 | |
2.18. Zinc-catalyzed synthesis of indoles
For improving the efficiency of the transition metal-catalyzed reactions in terms of price, toxicity, availability, and production of waste, zinc chemistry is a very attractive option. Many different examples of classical processes that employ zinc have been well established, including the Reformatsky reaction, Fukuyama coupling, and Negishi coupling. In these reactions, because of the filled d-orbitals, zinc acts as a promoter rather than a catalyst. However, in the last few years many efforts have focused on increasing the effectiveness of zinc as a catalyst. As an example, the zinc-catalyzed intramolecular or intermolecular cyclization reaction between alkynes and a nitrogen moiety is efficiently used for the preparation of indoles. An interesting method using a catalytic amount of ZnBr2 as a Lewis acid to promote the cyclization of ortho-2-alkynylanilines was described for the synthesis of 2-substituted indoles 229 (Scheme 267).298 In a related study, the tandem zinc-based Sonogashira cross-coupling, followed by a 5-endo-dig cyclization of ortho-anilines with terminal alkynes, led to the formation of 1,2-disubstituted indoles 230 (Scheme 268).299 The reaction required the presence of N,N′-dimethylethylenediamine in a 1:2 ratio of zinc to form a Zn–DMEDA complex, which catalyzed the tandem process. The cascade sequence was also used to synthesize indolo[1,2-c]quinazolines. Thus, the reaction of N-2-[(2-aminophenyl)ethynyl]phenylamides with ZnBr2 gave indolo[1,2-c]quinazolines 231via a zinc bromide-promoted domino sequence involving a 5-endo-dig hydroamination and the intramolecular cyclization sequence (Scheme 269).300
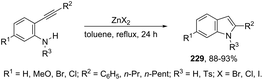 |
| | Scheme 267 | |
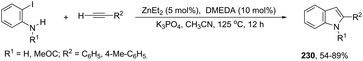 |
| | Scheme 268 | |
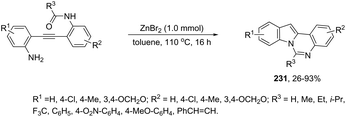 |
| | Scheme 269 | |
Another interesting cascade reaction is the zinc-catalyzed intramolecular hydroamination and cyclization-nucleophilic addition of ortho-alkynylanilines to form 2,3-disubstituted indoles. Thus, N-tosyl-protected aliphatic ortho-aminoalkynes reacted with diethylzinc in toluene, followed by the electrophile addition to give the corresponding 2,3-disubstituted indoles 232 (Scheme 270).301 When carbon dioxide was used as the electrophile, the reaction produced indole-3-carboxylic acids 233 (Scheme 271). The methodology was applied to the synthesis of Lotronex, a drug molecule used for the treatment of irritable bowel syndrome.302 More recently, in a tandem cyclization, dizinc ortho-ethynylaniline was found to act as a nucleophile in the formation of 2,3-dimetalloindole, which was functionalized with one or two electrophiles to give 2,3-disusbtituted indoles 234 (Scheme 272).303 The authors discussed the structures of zinc intermediates and concluded that the key intermediate could be a dimeric structure, where the ZnCl2 group is coordinated to the indole π-system to form a zwitterion, with the C3-carbon being tetrahedral and the C2-carbon being trigonal. The domino alkyne-amination–Fischer-indole sequence was described for the preparation of indole-2,3-dicarboxylates,304 2-methyl-3-substituted-indoles305 and 2-methyl-3-amino-indoles.306 The method was based on the reaction of phenylhydrazines with terminal or internal alkynes promoted by ZnCl2, ZnBr2, or Zn(OTf)2 affording the indole derivatives 235 (Scheme 273). In these reactions, the zinc salts participate in the intramolecular hydroamination of the arylhydrazine with terminal alkynes forming the arylhydrazone intermediate to promote the [3,3]-sigmatropic cyclization to the corresponding indoles (Scheme 273).
 |
| | Scheme 270 | |
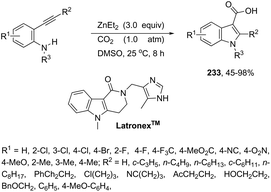 |
| | Scheme 271 | |
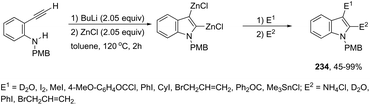 |
| | Scheme 272 | |
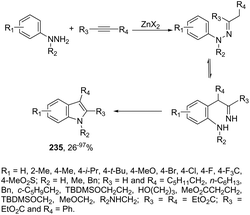 |
| | Scheme 273 | |
2.19. Zirconium-catalyzed synthesis of indoles
Reports on zirconium-catalyzed cyclization of alkynes to prepare indoles are not extensive in organic chemistry in the last ten years. A detailed study regarding the influence of the reaction mechanism on the selectivity in domino reactions of hydrazine zirconium complexes with alkynes was described in the preparation of indoles 236 (Scheme 274).307,308 The authors showed that the reaction of diarylhydrazines with terminal and internal alkynes using zirconium complex 237 at room temperature gave the corresponding indole derivatives. Although the reaction mechanism is relatively complex, the structures 238 and 239 represent the key reaction intermediates.
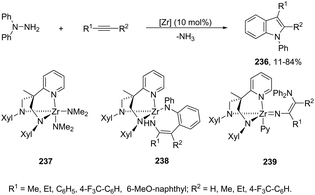 |
| | Scheme 274 | |
3. Conclusion
Transition metal-catalyzed activation of alkynes, followed by an intramolecular or intermolecular nucleophile addition, has become a powerful tool for the construction of the indole ring. Transition metal complexes containing bismuth, cobalt, copper, gold, indium, iridium, iron, manganese, mercury, nickel, palladium, platinum, rhenium, ruthenium, scandium, silver, titanium, tungsten, zinc, and zirconium have been employed as active catalysts for the activation of alkynes. The synthetic approaches to form indole derivatives developed in the last ten years using transition metal-catalyzed cyclization of alkynes with nitrogen compounds were summarized in this review. Further studies related to indole synthesis will undoubtedly lead to the discovery of new structures with biological activity and consequently the development of new medical and pharmacological applications. This will guide the new studies to overcome the challenges and keep this important class of N-heterocycles active for many years. It is our hope that this review article will assist the chemists in choosing the suitable methodology for indole preparation and stimulate the development of new synthetic methods for their synthesis.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful for financial support from the CNPq, CAPES and FAPERGS. We also want to thank the students from our research group who helped to draw the chemical structures.
References
- T. P. Singh and O. M. Singh, Mini-Rev. Med. Chem., 2018, 18, 9–25 CrossRef CAS PubMed.
- S. Suzen, Curr. Org. Chem., 2017, 21, 2068–2076 CAS.
- K. Walton and J. P. Berry, Mar. Drugs, 2016, 14, 28 CrossRef PubMed.
- Y. M. Ma, X. A. Liang, Y. Kong and B. Jia, J. Agric. Food Chem., 2016, 64, 6659–6671 CrossRef CAS PubMed.
- S. M. Li, Nat. Prod. Rep., 2010, 27, 57–78 RSC.
- Z. R. Owczarczyk, W. A. Braunecker, A. Garcia, R. Larsen, A. M. Nardes, N. Kopidakis, D. S. Ginley and D. C. Olson, Macromolecules, 2013, 46, 1350–1360 CrossRef CAS.
- G. M. Nie, Z. M. Bai, W. Y. Yu and L. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2385–2392 CrossRef CAS.
- M. Manickam, P. Iqbal, M. Belloni, S. Kumar and J. A. Preece, Isr. J. Chem., 2012, 52, 917–934 CrossRef CAS.
- D. V. Vorobyeva and S. N. Osipov, Synthesis, 2018, 50, 227–240 CrossRef CAS.
- M. Petrini, Chem. – Eur. J., 2017, 23, 16115–16151 CrossRef CAS PubMed.
- I. Szatmari, J. Sas and F. Fulop, Curr. Org. Chem., 2016, 20, 2038–2054 CrossRef CAS.
- E. Stempel and T. Gaich, Acc. Chem. Res., 2016, 49, 2390–2402 CrossRef CAS PubMed.
- W. W. Zi, Z. W. Zuo and D. W. Ma, Acc. Chem. Res., 2015, 48, 702–711 CrossRef CAS PubMed.
- A. H. Sandtorv, Adv. Synth. Catal., 2015, 357, 2403–2435 CrossRef CAS.
- R. Dalpozzo, Chem. Soc. Rev., 2015, 44, 742–778 RSC.
- H. Wu, Y. P. He and F. Shi, Synthesis, 2015, 47, 1990–2016 CrossRef CAS.
- N. Thirupathi, Y. K. Kumar, R. Kant and M. S. Reddy, Adv. Synth. Catal., 2014, 356, 1823–1834 CrossRef CAS.
- S. Lancianesi, A. Palmieri and M. Petrini, Chem. Rev., 2014, 114, 7108–7149 CrossRef CAS PubMed.
- G. Bartoli, R. Dalpozzo and M. Nardi, Chem. Soc. Rev., 2014, 43, 4728–4750 RSC.
- E. Fischer and F. Jourdan, Eur. J. Inorg. Chem., 1883, 16, 2241–2245 Search PubMed.
- A. Bischler, Eur. J. Inorg. Chem., 1892, 25, 2860–2879 Search PubMed.
- A. Reissert, Eur. J. Inorg. Chem., 1897, 30, 1030–1053 CAS.
- W. Madelung, Eur. J. Inorg. Chem., 1912, 45, 1128–1134 Search PubMed.
- C. Nenitzescu, Bull. Soc. Chim. Romania, 1929, 11, 37–43 Search PubMed.
- R. J. Sundberg and T. Yamazaki, J. Org. Chem., 1967, 32, 290–294 CrossRef CAS.
- H. Hemetsberger and D. Knittel, Monath. Chem., 1972, 103, 194–204 CrossRef CAS.
- P. G. Gassman, T. Van Bergen and G. Gruetzmacher, J. Am. Chem. Soc., 1973, 95, 6508–6509 CrossRef CAS.
- A. D. Batcho and W. Leimgruber, Org. Synth., 1985, 63, 214–220 CrossRef CAS.
- J.-B. Baudin and S. A. Julia, Tetrahedron Lett., 1986, 27, 837–840 CrossRef CAS.
- G. Bartoli, G. Palmieri, M. Bosco and R. Dalpozzo, Tetrahedron Lett., 1989, 30, 2129–2132 CrossRef CAS.
- R. C. Larock and E. K. Yum, J. Am. Chem. Soc., 1991, 113, 6689–6690 CrossRef CAS.
- T. Fukuyama, X. Chen and G. Peng, J. Am. Chem. Soc., 1994, 116, 3127–3128 CrossRef CAS.
- D. F. Taber and P. K. Tirunahari, Tetrahedron, 2011, 67, 7195 CrossRef CAS PubMed.
- L. Zhang, Z. Li and R. Fan, Org. Lett., 2012, 14, 6076–6079 CrossRef CAS PubMed.
- A. Lerchen, S. Vásquez-Céspedes and F. Glorius, Angew. Chem., Int. Ed., 2016, 55, 3208–3211 CrossRef CAS.
- S. Zhou, J. Wang, L. Wang, K. Chen, C. Song and J. Zhu, Org. Lett., 2016, 18, 3806–3809 CrossRef CAS PubMed.
- Y. Liang and N. Jiao, Angew. Chem., 2016, 128, 4103–4107 CrossRef.
- H. Wang, M. Moselage, M. A. J. González and L. Ackermann, ACS Catal., 2016, 6, 2705–2709 CrossRef CAS.
- Z.-Z. Zhang, B. Liu, J.-W. Xu, S.-Y. Yan and B.-F. Shi, Org. Lett., 2016, 18, 1776–1779 CrossRef CAS PubMed.
- S. Bhunia, G. G. Pawar, S. V. Kumar, Y. Jiang and D. Ma, Angew. Chem., Int. Ed., 2017, 56, 16136–16179 CrossRef CAS PubMed.
- A. Klapars, J. C. Antilla, X. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2001, 123, 7727–7729 CrossRef CAS PubMed.
- D. S. Surry and S. L. Buchwald, Chem. Sci., 2010, 1, 13–31 RSC.
- N. T. Patil and Y. Yamamoto, Chem. Rev., 2008, 108, 3395–3442 CrossRef CAS PubMed.
- S. Song, M. Huang, W. Li, X. Zhu and Y. Wan, Tetrahedron, 2015, 71, 451–456 CrossRef CAS.
- M. J. García-Muñoz, F. Foubelo and M. Yus, J. Org. Chem., 2016, 81, 10214–10226 CrossRef PubMed.
- Z.-Q. Wang, X. Zhang, L.-T. Yu, W.-T. Mao, C.-Z. Chen and K. Xu, Org. Biomol. Chem., 2015, 13, 6931–6934 RSC.
- F. Liu and D. Ma, J. Org. Chem., 2007, 72, 4844–4850 CrossRef CAS PubMed.
- T. Ponpandian and S. Muthusubramanian, Tetrahedron Lett., 2012, 53, 4248–4252 CrossRef CAS.
- R. Wang, S. Mo, Y. Lu and Z. Shen, Adv. Synth. Catal., 2011, 353, 713–718 CrossRef CAS.
- H. Li, X. Li, H.-Y. Wang, G. N. Winston-McPherson, H.-M. J. Geng, I. A. Guzei and W. Tang, Chem. Commun., 2014, 50, 12293–12296 RSC.
- L. Ackermann, S. Barfuesser and H. K. Potukuchi, Adv. Synth. Catal., 2009, 351, 1064–1072 CrossRef CAS.
- J. Gao, Y. Shao, J. Zhu, J. Zhu, H. Mao, X. Wang and X. Lv, J. Org. Chem., 2014, 79, 9000–9008 CrossRef CAS PubMed.
- H. Wang, Y. Li, L. Jiang, R. Zhang, K. Jin, D. Zhao and C. Duan, Org. Biomol. Chem., 2011, 9, 4983–4986 RSC.
- M. Yang, J. Tang and R. Fan, Chem. Commun., 2012, 48, 11775–11777 RSC.
- H. Ohno, Y. Ohta, S. Oishi and N. Fujii, Angew. Chem., Int. Ed., 2007, 46, 2295–2298 CrossRef CAS PubMed.
- Y. Ohta, H. Chiba, S. Oishi, N. Fujii and H. Ohno, J. Org. Chem., 2009, 74, 7052–7058 CrossRef CAS PubMed.
- S. Cacchi, G. Fabrizi, A. Iazzetti, C. Molinaro, R. Verdiglione and A. Goggiamani, Adv. Synth. Catal., 2015, 357, 1053–1059 CrossRef CAS.
- L. Zhou, Y. Shi, Q. Xiao, Y. Liu, F. Ye, Y. Zhang and J. Wang, Org. Lett., 2011, 13, 968–971 CrossRef CAS PubMed.
- T. Xiao, X. Dong and L. Zhou, Org. Biomol. Chem., 2013, 11, 1490–1497 RSC.
- Z. Shen and X. Lu, Adv. Synth. Catal., 2009, 351, 3107–3112 CrossRef CAS.
- N. K. Swamy, A. Yazici and S. G. Pyne, J. Org. Chem., 2010, 75, 3412–3419 CrossRef CAS PubMed.
- M. Yamashita, T. Noro and A. Iida, Tetrahedron Lett., 2013, 54, 6848–6851 CrossRef CAS.
- Z. Li, L. Hong, R. Liu, J. Shen and X. Zhou, Tetrahedron Lett., 2011, 52, 1343–1347 CrossRef CAS.
- N. Matsuda, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2011, 77, 617–625 CrossRef PubMed.
- Y. Oda, N. Matsuyama, K. Hirano, T. Satoh and M. Miura, Synthesis, 2012, 1515–1520 CAS.
- A. Sagadevan, A. Ragupathi and K. C. Hwang, Angew. Chem., Int. Ed., 2015, 54, 13896–13901 CrossRef CAS PubMed.
- H. E. Ho, K. Oniwa, Y. Yamamoto and T. Jin, Org. Lett., 2016, 18, 2487–2490 CrossRef CAS PubMed.
- J. Yu, D. Zhang-Negrerie and Y. Du, Org. Lett., 2016, 18, 3322–3325 CrossRef CAS PubMed.
- A. Gogoi, S. Guin, S. K. Rout and B. K. Patel, Org. Lett., 2013, 15, 1802–1805 CrossRef CAS PubMed.
- S. Murru, A. A. Gallo and R. S. Srivastava, Eur. J. Org. Chem., 2011, 2035–2038 CrossRef CAS.
- D. Gao and T. G. Back, Chem. – Eur. J., 2012, 18, 14828–14840 CrossRef CAS PubMed.
- L. Cao, D. Shen, J. Wei, J. Chen, H. Deng, M. Shao, J. Shi, H. Zhang and W. Cao, Eur. J. Org. Chem., 2014, 2014, 2460–2467 CrossRef CAS.
- B. Alcaide and P. Almendros, Acc. Chem. Res., 2014, 47, 939–952 CrossRef CAS PubMed.
- I. Ambrogio, A. Arcadi, S. Cacchi, G. Fabrizi and F. Marinelli, Synlett, 2007, 1775–1779 CAS.
- K. Majumdar, S. Samanta and B. Chattopadhyay, Tetrahedron Lett., 2008, 49, 7213–7216 CrossRef CAS.
- S. Liang, L. Hammond, B. Xu and G. B. Hammond, Adv. Synth. Catal., 2016, 358, 3313–3318 CrossRef CAS.
- P. Li, L. Wang, M. Wang and F. You, Eur. J. Org. Chem., 2008, 5946–5951 CrossRef CAS.
- M. Xu, Q. Hou, S. Wang, H. Wang and Z.-J. Yao, Synthesis, 2011, 626–634 CAS.
- P. Kothandaraman, S. R. Mothe, S. S. M. Toh and P. W. H. Chan, J. Org. Chem., 2011, 76, 7633–7640 CrossRef CAS PubMed.
- H. Peng, N. G. Akhmedov, Y.-F. Liang, N. Jiao and X. Shi, J. Am. Chem. Soc., 2015, 137, 8912–8915 CrossRef CAS PubMed.
- M. Michalska and K. Grela, Synlett, 2016, 599–603 CAS.
- D. Ye, J. Wang, X. Zhang, Y. Zhou, X. Ding, E. Feng, H. Sun, G. Liu, H. Jiang and H. Liu, Green Chem., 2009, 11, 1201–1208 RSC.
- J. Zhu, H. Xie, Z. Chen, S. Li and Y. Wu, Org. Biomol. Chem., 2012, 10, 516–523 RSC.
- X. Zhang and A. Corma, Angew. Chem., 2008, 120, 4430–4433 CrossRef.
- Y. Yamane, X. Liu, A. Hamasaki, T. Ishida, M. Haruta, T. Yokoyama and M. Tokunaga, Org. Lett., 2009, 11, 5162–5165 CrossRef CAS PubMed.
- A. La-Venia, S. A. Testero, M. P. Mischne and E. G. Mata, Org. Biomol. Chem., 2012, 10, 2514–2517 RSC.
- I. Nakamura, U. Yamagishi, D. Song, S. Konta and Y. Yamamoto, Angew. Chem., 2007, 119, 2334–2337 CrossRef.
- X. Zeng, R. Kinjo, B. Donnadieu and G. Bertrand, Angew. Chem., 2010, 122, 954–957 CrossRef.
- K. Majumdar, S. Hazra and B. Roy, Tetrahedron Lett., 2011, 52, 6697–6701 CrossRef CAS.
- A. Gimeno, A. Rodríguez-Gimeno, A. B. Cuenca, C. R. de Arellano, M. Medio-Simón and G. Asensio, Chem. Commun., 2015, 51, 12384–12387 RSC.
- B. Subba Reddy, M. Swain, S. M. Reddy, J. Yadav and B. Sridhar, J. Org. Chem., 2012, 77, 11355–11361 CrossRef CAS PubMed.
- G. Abbiati, A. Arcadi, M. Chiarini, F. Marinelli, E. Pietropaolo and E. Rossi, Org. Biomol. Chem., 2012, 10, 7801–7808 RSC.
- C. Praveen, K. Karthikeyan and P. Perumal, Tetrahedron, 2009, 65, 9244–9255 CrossRef CAS.
- N. T. Patil, V. Singh, A. Konala and A. K. Mutyala, Tetrahedron Lett., 2010, 51, 1493–1496 CrossRef CAS.
- B. Lu, Y. Luo, L. Liu, L. Ye, Y. Wang and L. Zhang, Angew. Chem., 2011, 123, 8508–8512 CrossRef.
- C. Qu, S. Zhang, H. Du and C. Zhu, Chem. Commun., 2016, 52, 14400–14403 RSC.
- J. E. Perea-Buceta, T. Wirtanen, O. V. Laukkanen, M. K. Mäkelä, M. Nieger, M. Melchionna, N. Huittinen, J. A. Lopez-Sanchez and J. Helaja, Angew. Chem., Int. Ed., 2013, 52, 11835–11839 CrossRef CAS PubMed.
- C.-H. Shen, L. Li, W. Zhang, S. Liu, C. Shu, Y.-E. Xie, Y.-F. Yu and L.-W. Ye, J. Org. Chem., 2014, 79, 9313–9318 CrossRef CAS PubMed.
- E. Chong and S. A. Blum, J. Am. Chem. Soc., 2015, 137, 10144–10147 CrossRef CAS PubMed.
- G. Cera, S. Piscitelli, M. Chiarucci, G. Fabrizi, A. Goggiamani, R. S. Ramón, S. P. Nolan and M. Bandini, Angew. Chem., Int. Ed., 2012, 51, 9891–9895 CrossRef CAS PubMed.
- Y. Zhang, J. P. Donahue and C.-J. Li, Org. Lett., 2007, 9, 627–630 CrossRef CAS PubMed.
- S. Cai, K. Yang and D. Z. Wang, Org. Lett., 2014, 16, 2606–2609 CrossRef CAS PubMed.
- N. T. Patil and A. Konala, Eur. J. Org. Chem., 2010, 6831–6839 CrossRef CAS.
- H. Jin, L. Huang, J. Xie, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 794–797 CrossRef CAS PubMed.
- Y. Wang, L. Ye and L. Zhang, Chem. Commun., 2011, 47, 7815–7817 RSC.
- D. B. Huple, B. D. Mokar and R. S. Liu, Angew. Chem., Int. Ed., 2015, 54, 14924–14928 CrossRef CAS PubMed.
- Y. Matsuda, S. Naoe, S. Oishi, N. Fujii and H. Ohno, Chem. – Eur. J., 2015, 21, 1463–1467 CrossRef CAS PubMed.
- J. Liu, M. Chen, L. Zhang and Y. Liu, Chem. – Eur. J., 2015, 21, 1009–1013 CrossRef CAS PubMed.
- J. S. Kim, J. H. Han, J. J. Lee, Y. M. Jun, B. M. Lee and B. H. Kim, Tetrahedron Lett., 2008, 49, 3733–3738 CrossRef CAS.
- N. Sakai, K. Annaka, A. Fujita, A. Sato and T. Konakahara, J. Org. Chem., 2008, 73, 4160–4165 CrossRef CAS PubMed.
- K. Murai, S. Hayashi, N. Takaichi, Y. Kita and H. Fujioka, J. Org. Chem., 2008, 74, 1418–1421 CrossRef PubMed.
- E. Kumaran and W. K. Leong, Tetrahedron Lett., 2014, 55, 5495–5498 CrossRef CAS.
- E. Kumaran, W. Y. Fan and W. K. Leong, Org. Lett., 2014, 16, 1342–1345 CrossRef CAS PubMed.
- K. Y. Ye, L. X. Dai and S. L. You, Chem. – Eur. J., 2014, 20, 3040–3044 CrossRef CAS PubMed.
- T. Shibata, H. Hirashima, M. Kasagawa, K. Tsuchikama and K. Endo, Synlett, 2011, 2171–2176 CrossRef CAS.
- P. Zhang, T. Xiao, S. Xiong, X. Dong and L. Zhou, Org. Lett., 2014, 16, 3264–3267 CrossRef CAS PubMed.
- F. Gao, J.-T. Wang, L.-L. Liu, N. Ma, C. Yang, Y. Gao and W. Xia, Chem. Commun., 2017, 53, 8533–8536 RSC.
- C. Bolm, J. Legros, J. Le Paih and L. Zani, Chem. Rev., 2004, 104, 6217–6254 CrossRef CAS PubMed.
- I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed.
- S. L. Buchwald and C. Bolm, Angew. Chem., 2009, 121, 5694–5695 CrossRef.
- K. C. Majumdar, N. De, T. Ghosh and B. Roy, Tetrahedron, 2014, 33, 4827–4868 CrossRef.
- V. Terrasson, J. Michaux, A. Gaucher, J. Wehbe, S. Marque, D. Prim and J. M. Campagne, Eur. J. Org. Chem., 2007, 5332–5335 CrossRef.
- H. A. Du, R. Y. Tang, C. L. Deng, Y. Liu, J. H. Li and X. G. Zhang, Adv. Synth. Catal., 2011, 353, 2739–2748 CrossRef CAS.
- A. Speranca, B. Godoi, P. H. Menezes and G. Zeni, Synlett, 2013, 1125–1132 CAS.
- A. A. Lamar and K. M. Nicholas, Tetrahedron, 2009, 65, 3829–3833 CrossRef CAS.
- T. Kurisaki, T. Naniwa, H. Yamamoto, H. Imagawa and M. Nishizawa, Tetrahedron Lett., 2007, 48, 1871–1874 CrossRef CAS.
- M. Zheng, K. Chen and S. Zhu, Synthesis, 2017, 4173–4182 CAS.
- N. Maizuru, T. Inami, T. Kurahashi and S. Matsubara, Org. Lett., 2011, 13, 1206–1209 CrossRef CAS PubMed.
- S. A. Patil, R. Patil and D. D. Miller, Curr. Med. Chem., 2009, 16, 2531–2565 CrossRef CAS PubMed.
- K. Krüger, A. Tillack, M. Beller, M. Zeng, Q. Kang, Q.-L. He and S.-L. You, Adv. Synth. Catal., 2008, 350, 2153–2167 CrossRef.
- D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef CAS PubMed.
-
J. J. Li and G. W. Gribble, Palladium in heterocyclic chemistry: a guide for the synthetic chemist, Elsevier, 2006 Search PubMed.
- G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875–2911 CrossRef CAS PubMed.
- S. Patil and J. K. Buolamwini, Curr. Org. Synth., 2006, 3, 477–498 CrossRef CAS.
- S. Cacchi and G. Fabrizi, Chem. Rev., 2005, 105, 2873–2920 CrossRef CAS PubMed.
- R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS PubMed.
- D. Wang and S. Gao, Org. Chem. Front., 2014, 1, 556–566 RSC.
- R. Chinchilla and C. Nájera, Chem. Soc. Rev., 2011, 40, 5084–5121 RSC.
- I. Ambrogio, S. Cacchi and G. Fabrizi, Tetrahedron Lett., 2007, 48, 7721–7725 CrossRef CAS.
- S. Ye, Q. Ding, Z. Wang, H. Zhou and J. Wu, Org. Biomol. Chem., 2008, 6, 4406–4412 RSC.
- G. Qiu, Q. Ding, H. Ren, Y. Peng and J. Wu, Org. Lett., 2010, 12, 3975–3977 CrossRef CAS PubMed.
- Y. Morimoto, S. Shimizu, A. Mokuya, N. Ototake, A. Saito and O. Kitagawa, Tetrahedron, 2016, 72, 5221–5229 CrossRef CAS.
- N. Ototake, Y. Morimoto, A. Mokuya, H. Fukaya, Y. Shida and O. Kitagawa, Chem. – Eur. J., 2010, 16, 6752–6755 CrossRef CAS PubMed.
- H. Sakai, K. Tsutsumi, T. Morimoto and K. Kakiuchi, Adv. Synth. Catal., 2008, 350, 2498–2502 CrossRef CAS.
- R. Sanz, V. Guilarte and A. Pérez, Tetrahedron Lett., 2009, 50, 4423–4426 CrossRef CAS.
- K. Dooleweerdt, T. Ruhland and T. Skrydstrup, Org. Lett., 2008, 11, 221–224 CrossRef PubMed.
- R. M. Rao, U. R. CH, N. Mulakayala, M. Alvala, M. Arunasree, R. R. Poondra, J. Iqbal and M. Pal, Org. Biomol. Chem., 2011, 9, 3808–3816 RSC.
- A. Ahmed, M. Ghosh, P. Sarkar and J. K. Ray, Tetrahedron Lett., 2013, 54, 6691–6694 CrossRef CAS.
- A. Ahmed, M. Ghosh, S. Dhara and J. K. Ray, Synlett, 2014, 2455–2458 CAS.
- M. Yamaguchi and K. Manabe, Org. Lett., 2014, 16, 2386–2389 CrossRef CAS PubMed.
- A. Bruneau, K. Gustafson, N. Yuan, C.-W. Tai, I. Persson, X. Zou and J.-E. Bäckvall, Chem. – Eur. J., 2017, 23, 12886–12891 CrossRef CAS PubMed.
- Y. Chen, N. A. Markina and R. C. Larock, Tetrahedron, 2009, 65, 8908–8915 CrossRef CAS PubMed.
- S. Cacchi, G. Fabrizi, A. Goggiamani, A. Perboni, A. Sferrazza and P. Stabile, Org. Lett., 2010, 12, 3279–3281 CrossRef CAS PubMed.
- V. Guilarte, M. P. Castroviejo, P. García-García, M. A. Fernández-Rodríguez and R. Sanz, J. Org. Chem., 2011, 76, 3416–3437 CrossRef CAS PubMed.
- B. Z. Lu, H.-X. Wei, Y. Zhang, W. Zhao, M. Dufour, G. Li, V. Farina and C. H. Senanayake, J. Org. Chem., 2013, 78, 4558–4562 CrossRef CAS PubMed.
- Q. Zhou, Z. Zhang, Y. Zhou, S. Li, Y. Zhang and J. Wang, J. Org. Chem., 2016, 82, 48–56 CrossRef PubMed.
- H. Staudinger and J. Meyer, Helv. Chim. Acta, 1919, 2, 635–646 CrossRef CAS.
- H. Minami, T. Kanayama, R. Tanaka, N. Okamoto, T. Sueda and R. Yanada, Eur. J. Org. Chem., 2016, 5990–6000 CrossRef CAS.
- S. Cacchi, G. Fabrizi, A. Goggiamani, A. Iazzetti and R. Verdiglione, Synthesis, 2017, 4163–4172 CAS.
- A. Arcadi, R. Cianci, G. Ferrara and F. Marinelli, Tetrahedron, 2010, 66, 2378–2383 CrossRef CAS.
- B. Tréguier, E. Rasolofonjatovo, A. Hamze, O. Provot, J. Wdzieczak-Bakala, J. Dubois, J. D. Brion and M. Alami, Eur. J. Org. Chem., 2011, 4868–4876 Search PubMed.
- R. Álvarez, C. Martinez, Y. Madich, J. G. Denis, J. M. Aurrecoechea and A. R. de Lera, Chem. – Eur. J., 2010, 16, 12746–12753 CrossRef PubMed.
- S. Cacchi, G. Fabrizi, A. Goggiamani and A. Sferrazza, Org. Biomol. Chem., 2011, 9, 1727–1730 RSC.
- Q. Wang, L. Huang, X. Wu and H. Jiang, Org. Lett., 2013, 15, 5940–5943 CrossRef CAS PubMed.
- D. Janreddy, V. Kavala, C.-W. Kuo, T.-S. Kuo, C.-H. He and C.-F. Yao, Tetrahedron, 2013, 69, 3323–3330 CrossRef CAS.
- V. Reddy and R. Vijaya Anand, Org. Lett., 2015, 17, 3390–3393 CrossRef CAS PubMed.
- J. Chen, X. Han and X. Lu, Angew. Chem., Int. Ed., 2017, 56, 14698–14701 CrossRef CAS PubMed.
- S. Tang, Y.-X. Xie, J.-H. Li and N.-X. Wang, Synthesis, 2007, 1841–1847 CAS.
- H.-P. Zhang, S.-C. Yu, Y. Liang, P. Peng, B.-X. Tang and J.-H. Li, Synlett, 2011, 982–988 CAS.
- X. Han and X. Lu, Org. Lett., 2010, 12, 3336–3339 CrossRef CAS PubMed.
- F. Zhao, D. Zhang, Y. Nian, L. Zhang, W. Yang and H. Liu, Org. Lett., 2014, 16, 5124–5127 CrossRef CAS PubMed.
- B. Gabriele, L. Veltri, G. Salerno, R. Mancuso and M. Costa, Adv. Synth. Catal., 2010, 352, 3355–3363 CrossRef CAS.
- B. Gabriele, L. Veltri, R. Mancuso, G. Salerno and M. Costa, Eur. J. Org. Chem., 2012, 2549–2559 CrossRef CAS.
- J. Chen, X. Han and X. Lu, J. Org. Chem., 2017, 82, 1977–1985 CrossRef CAS PubMed.
- X.-F. Xia, L.-L. Zhang, X.-R. Song, Y.-N. Niu, X.-Y. Liu and Y.-M. Liang, Chem. Commun., 2013, 49, 1410–1412 RSC.
- X. Zhang, P. Li, C. Lyu, W. Yong, J. Li, X. Zhu and W. Rao, Org. Biomol. Chem., 2017, 15, 6080–6083 RSC.
- G. Qiu, C. Chen, L. Yao and J. Wu, Adv. Synth. Catal., 2013, 355, 1579–1584 CrossRef CAS.
- Z. Hu, J. Wang, D. Liang and Q. Zhu, Adv. Synth. Catal., 2013, 355, 3290–3294 CrossRef CAS.
- G. Qiu, X. Qiu, J. Liu and J. Wu, Adv. Synth. Catal., 2013, 355, 2441–2446 CrossRef CAS.
- Z. Hu, S. Luo and Q. Zhu, Adv. Synth. Catal., 2015, 357, 1060–1064 CrossRef CAS.
- B. Yao, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2012, 51, 12311–12315 CrossRef CAS PubMed.
- B. Yao, Q. Wang and J. Zhu, Chem. – Eur. J., 2015, 21, 7413–7416 CrossRef CAS PubMed.
- J. Huang, S. J. Macdonald and J. P. Harrity, Chem. Commun., 2010, 46, 8770–8772 RSC.
- Y. J. Guo, R. Y. Tang, J. H. Li, P. Zhong and X. G. Zhang, Adv. Synth. Catal., 2009, 351, 2615–2618 CrossRef CAS.
- J. Li, C. Li, S. Yang, Y. An, W. Wu and H. Jiang, J. Org. Chem., 2016, 81, 2875–2887 CrossRef CAS PubMed.
- J. Sheng, S. Li and J. Wu, Chem. Commun., 2014, 50, 578–580 RSC.
- C. Wu, F. Zhao, Y. Du, L. Zhao, L. Chen, J. Wang and H. Liu, RSC Adv., 2016, 6, 70682–70690 RSC.
- B. Gabriele, R. Mancuso, G. Salerno, E. Lupinacci, G. Ruffolo and M. Costa, J. Org. Chem., 2008, 73, 4971–4977 CrossRef CAS PubMed.
- S. Cacchi, G. Fabrizi and E. Filisti, Synlett, 2009, 1817–1821 CrossRef CAS.
- S. Cacchi, G. Fabrizi, A. Goggiamani, C. Molinaro and R. Verdiglione, J. Org. Chem., 2013, 79, 401–407 CrossRef PubMed.
- I. Ambrogio, S. Cacchi, G. Fabrizi and A. Prastaro, Tetrahedron, 2009, 65, 8916–8929 CrossRef CAS.
- N. Thirupathi, M. Hari Babu, V. Dwivedi, R. Kant and M. Sridhar Reddy, Org. Lett., 2014, 16, 2908–2911 CrossRef CAS PubMed.
- Y. Liu, Y. Huang, H. Song, Y. Liu and Q. Wang, Chem. – Eur. J., 2015, 21, 5337–5340 CrossRef CAS PubMed.
- B. Das, P. Kundu and C. Chowdhury, Org. Biomol. Chem., 2014, 12, 741–748 RSC.
- C. Chowdhury, B. Das, S. Mukherjee and B. Achari, J. Org. Chem., 2012, 77, 5108–5119 CrossRef CAS PubMed.
- X. Cui, J. Li, Y. Fu, L. Liu and Q.-X. Guo, Tetrahedron Lett., 2008, 49, 3458–3462 CrossRef CAS.
- N. Batail, A. Bendjeriou, T. Lomberget, R. Barret, V. Dufaud and L. Djakovitch, Adv. Synth. Catal., 2009, 351, 2055–2062 CrossRef CAS.
- N. Batail, V. Dufaud and L. Djakovitch, Tetrahedron Lett., 2011, 52, 1916–1918 CrossRef CAS.
- Y. Monguchi, S. Mori, S. Aoyagi, A. Tsutsui, T. Maegawa and H. Sajiki, Org. Biomol. Chem., 2010, 8, 3338–3342 RSC.
- W. J. Ang, C.-H. Tai, L.-C. Lo and Y. Lam, RSC Adv., 2014, 4, 4921–4929 RSC.
- S. E. Denmark and J. D. Baird, Tetrahedron, 2009, 65, 3120–3129 CrossRef CAS PubMed.
- P. Danner, M. Morkunas and M. E. Maier, Org. Lett., 2013, 15, 2474–2477 CrossRef CAS PubMed.
- A. Kondoh, H. Yorimitsu and K. Oshima, Org. Lett., 2010, 12, 1476–1479 CrossRef CAS PubMed.
- K. Goswami, I. Duttagupta and S. Sinha, J. Org. Chem., 2012, 77, 7081–7085 CrossRef CAS PubMed.
- W. Hao, W. Geng, W. X. Zhang and Z. Xi, Chem. – Eur. J., 2014, 20, 2605–2612 CrossRef CAS PubMed.
- W. Hao, J. Wei, W. Geng, W. X. Zhang and Z. Xi, Angew. Chem., Int. Ed., 2014, 53, 14533–14537 CrossRef CAS PubMed.
- W. Hao, H. Wang, P. J. Walsh and Z. Xi, Org. Chem. Front., 2015, 2, 1080–1084 RSC.
- C. Zhu and S. Ma, Org. Lett., 2013, 15, 2782–2785 CrossRef CAS PubMed.
- P. K. R. Panyam and T. Gandhi, Adv. Synth. Catal., 2017, 359, 1144–1151 CrossRef CAS.
- T. Curtius, Eur. J. Inorg. Chem., 1890, 23, 3023–3033 Search PubMed.
- O. Leogane and H. Lebel, Angew. Chem., Int. Ed., 2008, 47, 350–352 CrossRef CAS PubMed.
- X. Chen, X. Li, N. Wang, J. Jin, P. Lu and Y. Wang, Eur. J. Org. Chem., 2012, 4380–4386 CrossRef.
- D. Shen, J. Han, J. Chen, H. Deng, M. Shao, H. Zhang and W. Cao, Org. Lett., 2015, 17, 3283–3285 CrossRef CAS PubMed.
- Z. Shi, C. Zhang, S. Li, D. Pan, S. Ding, Y. Cui and N. Jiao, Angew. Chem., Int. Ed., 2009, 48, 4572–4576 CrossRef CAS PubMed.
- I. Nakamura, T. Nemoto, N. Shiraiwa and M. Terada, Org. Lett., 2009, 11, 1055–1058 CrossRef CAS PubMed.
- F. Zhou, X. Han and X. Lu, Tetrahedron Lett., 2011, 52, 4681–4685 CrossRef CAS.
- J. Chen, Q. Pang, Y. Sun and X. Li, J. Org. Chem., 2011, 76, 3523–3526 CrossRef CAS PubMed.
- J. Chen, L. He, K. Natte, H. Neumann, M. Beller and X. F. Wu, Adv. Synth. Catal., 2014, 356, 2955–2959 CrossRef CAS.
- R. Sanz, M. P. Castroviejo, V. Guilarte, A. Pérez and F. J. Fañanás, J. Org. Chem., 2007, 72, 5113–5118 CrossRef CAS PubMed.
- L. Ackermann, R. Sandmann, M. Schinkel and M. V. Kondrashov, Tetrahedron, 2009, 65, 8930–8939 CrossRef CAS.
- L. Ackermann, R. Sandmann and M. V. Kondrashov, Synlett, 2009, 1219–1222 CrossRef CAS.
- Y. Liang, T. Meng, H.-J. Zhang and Z. Xi, Synlett, 2011, 911–914 CAS.
- P. G. Alsabeh, R. J. Lundgren, L. E. Longobardi and M. Stradiotto, Chem. Commun., 2011, 47, 6936–6938 RSC.
- C. B. Lavery, R. McDonald and M. Stradiotto, Chem. Commun., 2012, 48, 7277–7279 RSC.
- N. Halland, M. Nazare, J. Alonso, O. R'kyek and A. Lindenschmidt, Chem. Commun., 2011, 47, 1042–1044 RSC.
- A. Prakash, M. Dibakar, K. Selvakumar, K. Ruckmani and M. Sivakumar, Tetrahedron Lett., 2011, 52, 5625–5628 CrossRef CAS.
- P.-Y. Yao, Y. Zhang, R. P. Hsung and K. Zhao, Org. Lett., 2008, 10, 4275–4278 CrossRef CAS PubMed.
- K. Cariou, B. Ronan, S. Mignani, L. Fensterbank and M. Malacria, Angew. Chem., Int. Ed., 2007, 46, 1881–1884 CrossRef CAS PubMed.
- N. Okamoto, Y. Miwa, H. Minami, K. Takeda and R. Yanada, Angew. Chem., 2009, 121, 9873–9876 CrossRef.
- N. Okamoto, K. Takeda and R. Yanada, J. Org. Chem., 2010, 75, 7615–7625 CrossRef CAS PubMed.
- D. Shu, G. N. Winston-McPherson, W. Song and W. Tang, Org. Lett., 2013, 15, 4162–4165 CrossRef CAS PubMed.
- P. A. Allegretti, K. Huynh, T. J. Ozumerzifon and E. M. Ferreira, Org. Lett., 2015, 18, 64–67 CrossRef PubMed.
- X. Li, J.-Y. Wang, W. Yu and L.-M. Wu, Tetrahedron, 2009, 65, 1140–1146 CrossRef CAS.
- I. Nakamura, Y. Sato, S. Konta and M. Terada, Tetrahedron Lett., 2009, 50, 2075–2077 CrossRef CAS.
- J. Takaya, S. Udagawa, H. Kusama and N. Iwasawa, Angew. Chem., Int. Ed., 2008, 47, 4906 CrossRef CAS PubMed.
- D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2009, 110, 624–655 CrossRef PubMed.
- B. Trost and A. McClory, Angew. Chem., Int. Ed., 2007, 46, 2074 CrossRef CAS PubMed.
- X. Li, H. Li, W. Song, P. S. Tseng, L. Liu, I. A. Guzei and W. Tang, Angew. Chem., Int. Ed., 2015, 54, 12905–12908 CrossRef CAS PubMed.
- Q. W. Zhang, K. An and W. He, Angew. Chem., Int. Ed., 2014, 53, 5667–5671 CrossRef CAS PubMed.
- H. Kanno, K. Nakamura, K. Noguchi, Y. Shibata and K. Tanaka, Org. Lett., 2016, 18, 1654–1657 CrossRef CAS PubMed.
- N. Isono and M. Lautens, Org. Lett., 2009, 11, 1329–1331 CrossRef CAS PubMed.
- A. Mizukami, Y. Ise, T. Kimachi and K. Inamoto, Org. Lett., 2016, 18, 748–751 CrossRef CAS PubMed.
- Z. Hu, X. Tong and G. Liu, Org. Lett., 2016, 18, 2058–2061 CrossRef CAS PubMed.
- G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651–3678 RSC.
- G. Zhang, H. Yu, G. Qin and H. Huang, Chem. Commun., 2014, 50, 4331–4334 RSC.
- D. R. Stuart, M. Bertrand-Laperle, K. M. Burgess and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 16474–16475 CrossRef CAS PubMed.
- D. R. Stuart, P. Alsabeh, M. Kuhn and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 18326–18339 CrossRef CAS PubMed.
- M. P. Huestis, L. Chan, D. R. Stuart and K. Fagnou, Angew. Chem., 2011, 123, 1374–1377 CrossRef.
- Y. Hoshino, Y. Shibata and K. Tanaka, Adv. Synth. Catal., 2014, 356, 1577–1585 CrossRef CAS.
- G. N. Hermann, C. L. Jung and C. Bolm, Green Chem., 2017, 19, 2520–2523 RSC.
- S. Kathiravan and I. A. Nicholls, Chem. Commun., 2014, 50, 14964–14967 RSC.
- J. Chen, G. Song, C.-L. Pan and X. Li, Org. Lett., 2010, 12, 5426–5429 CrossRef CAS PubMed.
- X. Hu, X. Chen, Y. Zhu, Y. Deng, H. Zeng, H. Jiang and W. Zeng, Org. Lett., 2017, 19, 3474–3477 CrossRef CAS PubMed.
- D. Zhao, Z. Shi and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 12426–12429 CrossRef CAS PubMed.
- S. Zhou, J. Wang, F. Zhang, C. Song and J. Zhu, Org. Lett., 2016, 18, 2427–2430 CrossRef CAS PubMed.
- L. Zheng and R. Hua, Chem. – Eur. J., 2014, 20, 2352–2356 CrossRef CAS PubMed.
- K. Muralirajan and C. H. Cheng, Adv. Synth. Catal., 2014, 356, 1571–1576 CrossRef CAS.
- D. Y. Li, H. J. Chen and P. N. Liu, Org. Lett., 2014, 16, 6176–6179 CrossRef CAS PubMed.
- C. Wang, H. Sun, Y. Fang and Y. Huang, Angew. Chem., Int. Ed., 2013, 52, 5795 CrossRef CAS PubMed.
- H. Sun, C. Wang, Y.-F. Yang, P. Chen, Y.-D. Wu, X. Zhang and Y. Huang, J. Org. Chem., 2014, 79, 11863–11872 CrossRef CAS PubMed.
- C. Wang and Y. Huang, Org. Lett., 2013, 15, 5294–5297 CrossRef CAS PubMed.
- B. Liu, C. Song, C. Sun, S. Zhou and J. Zhu, J. Am. Chem. Soc., 2013, 135, 16625–16631 CrossRef CAS PubMed.
- Z. Zhou, G. Liu, Y. Chen and X. Lu, Adv. Synth. Catal., 2015, 357, 2944–2950 CrossRef CAS.
- H. Yan, H. Wang, X. Li, X. Xin, C. Wang and B. Wan, Angew. Chem., Int. Ed., 2015, 54, 10613–10617 CrossRef CAS PubMed.
- X. Huang, W. Liang, Y. Shi and J. You, Chem. Commun., 2016, 52, 6253–6256 RSC.
- Z. Fan, S. Song, W. Li, K. Geng, Y. Xu, Z.-H. Miao and A. Zhang, Org. Lett., 2014, 17, 310–313 CrossRef PubMed.
- J. S. Alford, J. E. Spangler and H. M. Davies, J. Am. Chem. Soc., 2013, 135, 11712–11715 CrossRef CAS PubMed.
- B. Rajagopal, C.-H. Chou, C.-C. Chung and P.-C. Lin, Org. Lett., 2014, 16, 3752–3755 CrossRef CAS PubMed.
- A. Saito, A. Kanno and Y. Hanzawa, Angew. Chem., 2007, 119, 4005–4007 CrossRef.
- A. Saito, S. Oda, H. Fukaya and Y. Hanzawa, J. Org. Chem., 2009, 74, 1517–1524 CrossRef CAS PubMed.
- B. M. Trost, Acc. Chem. Res., 2002, 35, 695–705 CrossRef CAS PubMed.
- B. Schmidt, Angew. Chem., Int. Ed., 2003, 42, 4996–4999 CrossRef CAS PubMed.
- R. N. Nair, P. J. Lee, A. L. Rheingold and D. B. Grotjahn, Chem. – Eur. J., 2010, 16, 7992–7995 CrossRef CAS PubMed.
- A. Varela-Fernández, J. A. Varela and C. Saa, Adv. Synth. Catal., 2011, 353, 1933–1937 CrossRef.
- X. Dong, Y. Hu, T. Xiao and L. Zhou, RSC Adv., 2015, 5, 39625–39629 RSC.
- K. Takamoto, S. Ohno, N. Hyogo, H. Fujioka and M. Arisawa, J. Org. Chem., 2017, 82, 8733–8742 CrossRef CAS PubMed.
- C.-Y. Wu, M. Hu, Y. Liu, R.-J. Song, Y. Lei, B.-X. Tang, R.-J. Li and J.-H. Li, Chem. Commun., 2012, 48, 3197–3199 RSC.
- T. Watanabe, Y. Mutoh and S. Saito, J. Am. Chem. Soc., 2017, 139, 7749–7752 CrossRef CAS PubMed.
- Z. Zhang, H. Jiang and Y. Huang, Org. Lett., 2014, 16, 5976–5979 CrossRef CAS PubMed.
- S. Zhou, J. Wang, P. Chen, K. Chen and J. Zhu, Chem. – Eur. J., 2016, 22, 14508–14512 CrossRef CAS PubMed.
- H. Lin, S.-S. Li and L. Dong, Org. Biomol. Chem., 2015, 13, 11228–11234 RSC.
- Y. Liao, T. Wei, T. Yan and M. Cai, Tetrahedron, 2017, 73, 1238–1246 CrossRef CAS.
- F. Yang, K.-G. Ji, S. Ali and Y.-M. Liang, J. Org. Chem., 2011, 76, 8329–8335 CrossRef CAS PubMed.
- G. Fang and X. Bi, Chem. Soc. Rev., 2015, 44, 8124–8173 RSC.
- N.-Y. Huang, M.-G. Liu and M.-W. Ding, J. Org. Chem., 2009, 74, 6874–6877 CrossRef CAS PubMed.
- T. Otani, X. Jiang, K. Cho, R. Araki, N. Kutsumura and T. Saito, Adv. Synth. Catal., 2015, 357, 1483–1492 CrossRef CAS.
- M. Kaname and H. Sashida, Tetrahedron Lett., 2012, 53, 748–751 CrossRef CAS.
- Y. Fang, C. Wang, S. Su, H. Yu and Y. Huang, Org. Biomol. Chem., 2014, 12, 1061–1071 RSC.
- S. Panaka, R. Trivedi, T. Sony, S. Prabhakar and L. R. Chowhan, Org. Chem. Front., 2017, 4, 1574–1579 RSC.
- L. Yang, Y. Ma, F. Song and J. You, Chem. Commun., 2014, 50, 3024–3026 RSC.
- Y. Gao, G. Lu, P. Zhang, L. Zhang, G. Tang and Y. Zhao, Org. Lett., 2016, 18, 1242–1245 CrossRef CAS PubMed.
- J. Liu, X. Xie and Y. Liu, Chem. Commun., 2013, 49, 11794–11796 RSC.
- R. Karmakar, A. Suneja and V. K. Singh, Org. Lett., 2016, 18, 2636–2639 CrossRef CAS PubMed.
- C. H. Oh, S. Karmakar, H. Park, Y. Ahn and J. W. Kim, J. Am. Chem. Soc., 2010, 132, 1792–1793 CrossRef CAS PubMed.
- X. Feng, H. Wang, B. Yang and R. Fan, Org. Lett., 2014, 16, 3600–3603 CrossRef CAS PubMed.
- N. Schwarz, K. Alex, I. A. Sayyed, V. Khedkar, A. Tillack and M. Beller, Synlett, 2007, 1091–1095 CAS.
- J. C.-H. Yim, J. A. Bexrud, R. O. Ayinla, D. C. Leitch and L. L. Schafer, J. Org. Chem., 2014, 79, 2015–2028 CrossRef CAS PubMed.
- K. Okuma, J.-I. Seto, K.-I. Sakaguchi, S. Ozaki, N. Nagahora and K. Shioji, Tetrahedron Lett., 2009, 50, 2943–2945 CrossRef CAS.
- A. P. Thankachan, K. S. Sindhu, S. M. Ujwaldev and G. Anilkumar, Tetrahedron Lett., 2017, 58, 536–540 CrossRef CAS.
- M. Xu, K. Xu, S. Wang and Z.-J. Yao, Tetrahedron Lett., 2013, 54, 4675–4678 CrossRef CAS.
- Z. Chai, W.-Y. Ma and G. Zhao, Synthesis, 2008, 4036–4040 Search PubMed.
- B. Miao, S. Li, G. Li and S. Ma, Org. Lett., 2016, 18, 2556–2559 CrossRef CAS PubMed.
- L. Ilies, M. Isomura, S.-I. Yamauchi, T. Nakamura and E. Nakamura, J. Am. Chem. Soc., 2016, 139, 23–26 CrossRef PubMed.
- I. A. Sayyed, K. Alex, A. Tillack, N. Schwarz, D. Michalik and M. Beller, Eur. J. Org. Chem., 2007, 4525–4528 CrossRef CAS.
- K. Alex, A. Tillack, N. Schwarz and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 2304–2307 CrossRef CAS PubMed.
- A. Pews-Davtyan and M. Beller, Org. Biomol. Chem., 2011, 9, 6331–6334 RSC.
- T. Gehrmann, J. Lloret Fillol, S. A. Scholl, H. Wadepohl and L. H. Gade, Angew. Chem., Int. Ed., 2011, 50, 5757–5761 CrossRef CAS PubMed.
- T. Gehrmann, S. A. Scholl, J. L. Fillol, H. Wadepohl and L. H. Gade, Chem. – Eur. J., 2012, 18, 3925–3941 CrossRef CAS PubMed.
|
| This journal is © the Partner Organisations 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 and
Gilson
Zeni
and
Gilson
Zeni