
Trifluoroacetimidoyl halides: a potent synthetic origin
Zhengkai
Chen
 a,
Sipei
Hu
a and
Xiao-Feng
Wu
*ab
a,
Sipei
Hu
a and
Xiao-Feng
Wu
*ab
aDepartment of Chemistry, Zhejiang Sci-Tech University, Hangzhou 310018, China. E-mail: xiao-feng.wu@catalysis.de
bLeibniz-Institut für Katalyse e.V. an der Universität Rostock, Albert-Einstein-Straße 29a, 18059 Rostock, Germany
First published on 2nd December 2019
Abstract
The preparation of fluorine-containing compounds has attracted considerable attention due to the important applications of their related chemicals. Among the candidates, trifluoroacetimidoyl halides are considered as potent trifluoromethyl synthons to construct a wide variety of trifluoromethyl-containing compounds and trifluoromethyl-substituted N-heterocycles, which have found extensive applications in the fields of organic synthesis, pharmaceuticals, agrochemicals, and materials science. In the review, recent advances in the synthetic applications of trifluoroacetimidoyl halides are summarized. We specially focused on two different reaction modes upon trifluoroacetimidoyl halides, namely, coupling and annulation reactions to illustrate their synthetic applications and potentials in the construction of valuable trifluoromethyl-containing molecules. Their preparations were covered as well.
 Zhengkai Chen | Zhengkai Chen was born in 1988 in Henan, China. He obtained his B.S. degree from Zhengzhou University in 2010. He had received a Ph.D. degree at Zhejiang University under the supervision of Prof. Yuhong Zhang in 2015. Then he joined the Department of Chemistry, Zhejiang Sci-Tech University. His current research interests focus on transition metal-catalyzed carbonylative transformations and new methods for the synthesis of functionalized nitrogen-containing heterocycles. |
 Sipei Hu | Sipei Hu was born in 1995 in Zhejiang, China. He obtained his B.S. degree from Heilongjiang University of Science and Technology in 2017. He is currently studying towards a M.S. degree at Zhejiang Sci-Tech University under the supervision of Dr Zhengkai Chen and Prof. Xiao-Feng Wu. His current research interests focus on transition metal-catalyzed or metal-free synthesis of nitrogen-containing heterocycles. |
 Xiao-Feng Wu | Xiao-Feng Wu was born in China. He studied chemistry in Zhejiang Sci-Tech University (China), where he got his bachelor's degree in science (2007). In the same year, he went to Rennes 1 University (France) and earned his master's degree in 2009. Then he joined Matthias Beller's group in Leibniz-Institute for Catalysis (Germany), where he completes his PhD defense in January 2012. Subsequently, he started his independent research at LIKAT and ZSTU, where he was promoted to professor in 2013. In March 2017, Xiao-Feng defended his Habilitation successfully from Rennes 1 University (France). Xiao-Feng has authored >290 publications in international journals, meanwhile he is also the editor or author of 10 books. In 2019, Xiao-feng was invited to publish his Author Profile in Angew. Chem., Int. Ed. (Angew. Chem., Int. Ed., 2019, 58, 8624). |
1. Introduction
The properties of organic compounds with C–F bonds including electronegativity, bioavailability, metabolic stability and lipophilicity have gained significant interest.1 It is known that approximately 20–25% of the current approved drugs and 30–40% of agrochemicals contain at least one fluorine atom.2 Therefore, the incorporation of fluorine atoms or fluorinated groups into building blocks to assemble fluorine-containing compounds has emerged as a major research focus.3 Among these, considerable efforts have been devoted to construct trifluoromethylated compounds,4 which have raised widespread concerns in the fields of organic synthesis, pharmaceuticals, agrochemicals, and materials science.5The most popular pathway to access trifluoromethyl-containing molecules involves transition-metal-catalyzed or metal-free trifluoromethylation reactions with versatile trifluoromethyl reagents such as Togni's reagent,4b,6 Umemoto's reagent,7 Ruppert-Prakash reagent8 and Langlois reagent.3f,9 Besides, another mainstream approach is the reaction of diverse trifluoromethyl-containing synthons with suitable coupling substrates. It is worth mentioning that 2,2,2-trifluorodiazoethane (CF3CHN2) is recognized as the most frequently used trifluoromethyl-containing synthon.10 Trifluoroacetimidoyl halides, which seemingly have not received enough attention, are also usually applied as useful building blocks for the assembly of trifluoromethyl-substituted scaffolds in synthetic organofluorine chemistry. The present review highlights the recent advancements enabling the synthesis of diverse trifluoromethyl-containing molecules by using trifluoroacetimidoyl halides and their related compounds.
2. Preparation of trifluoroacetimidoyl halides
The traditional methods for the synthesis of trifluoroacetimidoyl halides are based on two pathways. One is the reaction of N-substituted trifluoroacetamides with phosphorus pentachloride (PCl5)11 and the other one is the thermal-induced or copper-catalyzed addition of perfluoroalkyl iodide to isocyanides.12 However, the above-mentioned methods suffer from poor yields, harsh reaction conditions and not readily available reagents. Until 1993, Uneyama and co-workers developed an efficient and streamlined one-pot approach for the synthesis of trifluoroacetimidoyl halides in high yields (Scheme 1, eqn (a)).13 Trifluoroacetimidoyl chlorides/bromides can be afforded by heating a mixture of trifluoroacetic acid and a primary amine in CCl4/CBr4 in the presence of PPh3 and NEt3. Trifluoroacetimidoyl iodides can be easily prepared by the substitution of the chloride group of trifluoroacetimidoyl chlorides by iodide ions, in which almost quantitative yields are achieved (Scheme 1, eqn (b)). In 2005, Saidi and co-workers further applied the reaction to prepare trifluoroacetimidoyl chlorides containing additional functional groups.14 By using this method, a wide range of structurally diverse trifluoroacetimidoyl halides could be readily obtained in moderate to excellent yields. In addition, other perfluoroalkaneimidoyl chlorides were also smoothly prepared by the same method using the corresponding perfluoroalkanoic acids as starting materials.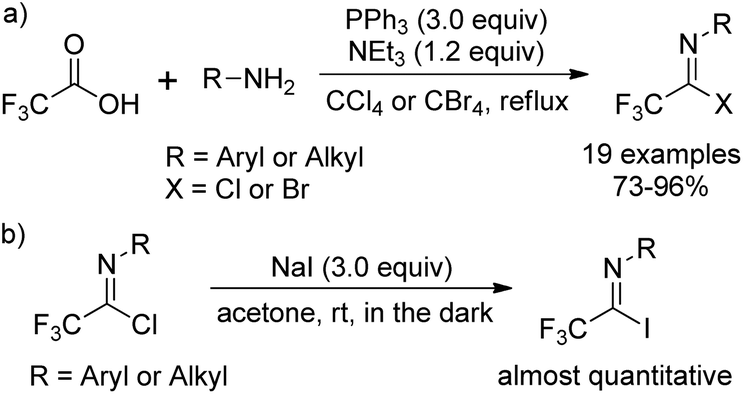 | ||
| Scheme 1 The preparation of trifluoroacetimidoyl halides. | ||
3. Coupling reaction of trifluoroacetimidoyl halides
The trifluoroacetimidoyl halides can be applied as useful synthetic blocks to participate in diverse coupling reactions with suitable coupling partners for the construction of a range of trifluoromethyl-containing compounds.3.1. C–C bond formation
As early as in 1991, Uneyama and co-workers developed a palladium-catalyzed coupling reaction of trifluoroacetimidoyl iodides with olefins and 1-alkynes to yield trifluoromethylated α,β-unsaturated imines (Scheme 2a and b).15 It was found that trifluoroacetimidoyl iodides exhibit higher reactivity and undergo palladation process smoothly to form imidoylpalladium intermediates, while the corresponding chlorides remain intact under the same conditions. The transformation tolerates diverse 1-alkenes and 1-alkynes, whether activated or non-activated ones. The resultant trifluoromethylated α,β-unsaturated imines could be easily transformed into nitrogen-containing heterocycles bearing a CF3 group.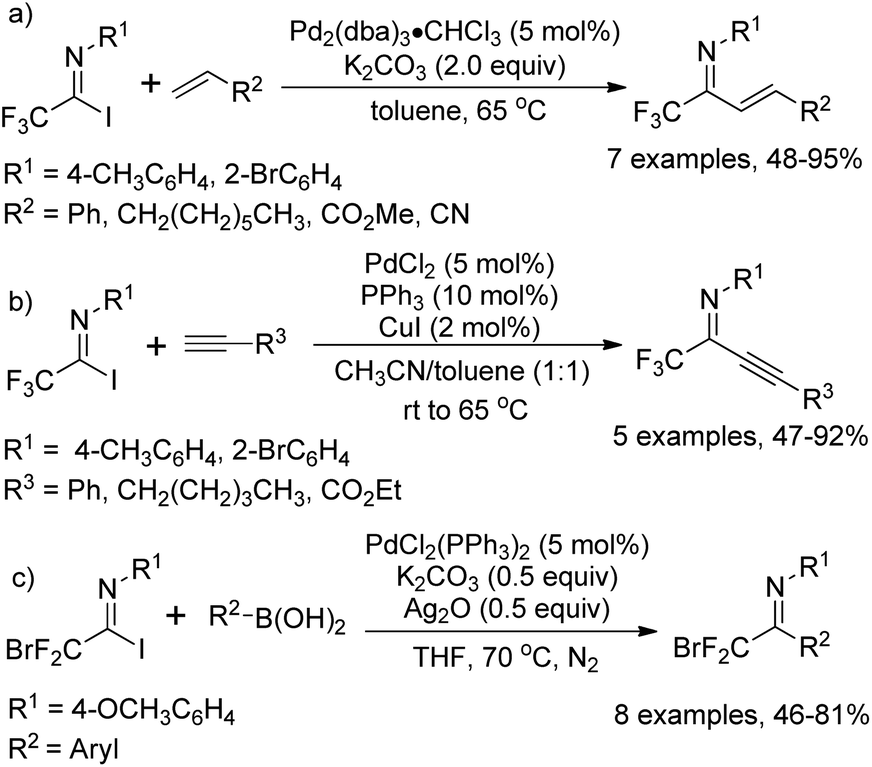 | ||
| Scheme 2 Pd-Catalyzed coupling of trifluoroacetimidoyl iodides with olefins, 1-alkynes and arylboronic acids. | ||
A palladium-catalyzed Suzuki cross-coupling reaction of bromodifluoroacetimidoyl halides with arylboronic acids to synthesize bromodifluoromethyl ketimines was realized by Wu and co-workers (Scheme 2c).16 The bromodifluoroacetimidoyl halides were prepared through the reaction of bromodifluoroacetic acid, triphenyl phosphine, arylamine and triethylamine in tetrachloromethane. In the reaction, the reactivity of bromodifluoroacetimidoyl chloride was found to be moderate even at prolonged reaction time.
In 1992, Uneyama and co-workers reported an efficient route to the formation of α-imino perfluoroalkanoates via palladium-catalyzed carboalkoxylation of fluorinated imidoyl iodides with various alcohols (Scheme 3).17 The obtained α-imino perfluoroalkanoates can be further transformed into α-amino perfluoroalkanoic acids. The mechanism of the carboalkoxylation reaction was considered to be initiated by the oxidative addition of palladium(0) to carbon–halide bonds, followed by the insertion of carbon monoxide into the palladium–carbon bond of 3-A to give an acylpalladium(II) intermediate 3-B. Then, a nucleophilic attack of the alcoholic hydroxyl group to 3-B can afford ester products, and the active palladium catalyst will be regenerated under the assistance of a base to accomplish the catalytic cycle. However, the reaction proceeded sluggishly with bulky alcohols, such as tert-butyl alcohol.
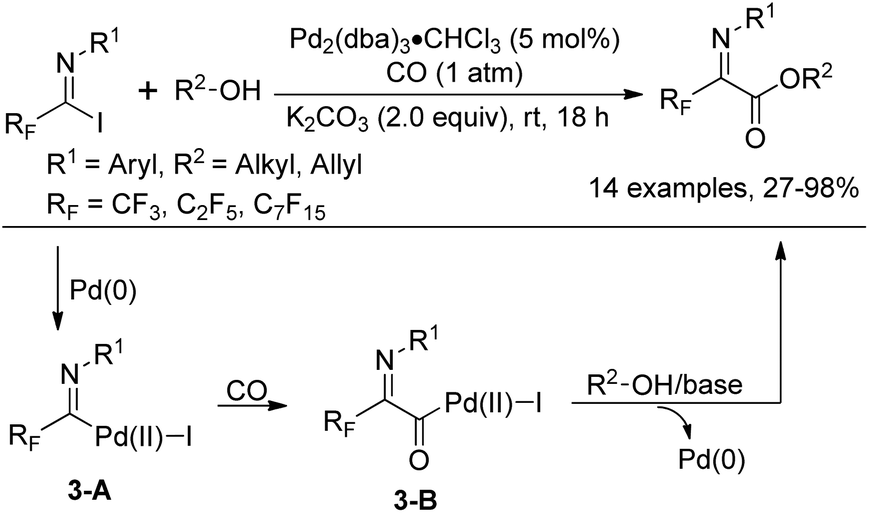 | ||
| Scheme 3 Palladium-catalyzed alkoxycarbonylation of fluorinated imidoyl iodides. | ||
In 2000, a modified protocol upon the reaction was proposed by the same group (Scheme 4).18 The efficiency of the reaction was improved by the addition of aprotic polar solvents (DMF or DMI) as an additive. It was speculated that the nucleophilicity of tert-butyl alcohol could be enhanced in the presence of DMF or DMI (1,3-dimethyl-2-imidazolidinone). However, the polar solvent might act as a weak ligand to reduce the aggregation of the intermediate palladium species and to accelerate the coordination of carbon monoxide to palladium. More importantly, a series of fluorinated α-amino acids were afforded by the reaction of fluorinated iminoester with a range of nucleophiles and the subsequent removal of an N-protecting group (PMP) and an O-protecting group (t-Bu).
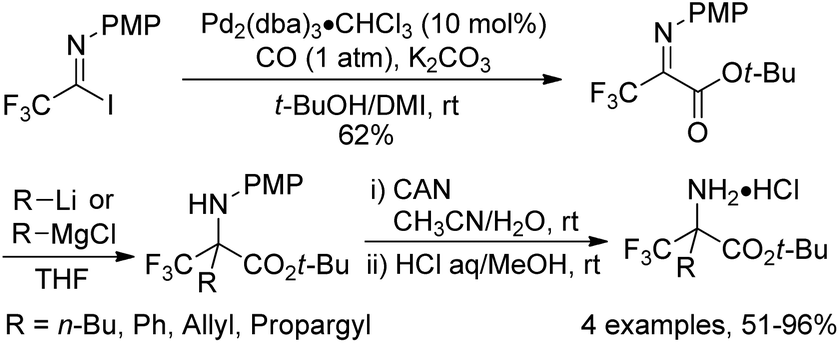 | ||
| Scheme 4 Palladium-catalyzed tert-butoxycarbonylation of trifluoroacetimidoyl iodides and the synthesis of fluorinated α-amino acids. | ||
The highly enantioenriched β-fluorinated α-amino esters could be obtained by the asymmetric hydrogenation of α-fluorinated iminoester.19 The transformation proceeded smoothly in the presence of a Pd(OCOCF3)2-BINAP complex in fluorinated alcohol solvents under hydrogen pressure with high efficiency and enantioselectivity.
Trifluoroacetimidoyl lithium, a synthetic equivalent of trifluoroacetyl carbanion, could be prepared from the iodine-lithium exchange of N-aryl trifluoroacetimidoyl iodides with n-BuLi in ether at −78 °C (Scheme 5).20 Nucleophilic reactions of the obtained imidoyl lithiums 5-A with various electrophiles took place to deliver trifluoromethyl-containing molecules 5-C. The 5-A species were very unstable and prone to isomerize to N-lithium carbene species 5-B, which underwent dimerization to afford diamine 5-D in the absence of other reactive electrophiles. Substituents on the N-aryl ring of trifluoroacetimidoyl iodides, solvent and reaction temperature greatly influenced the efficiency of nucleophilic reactions.
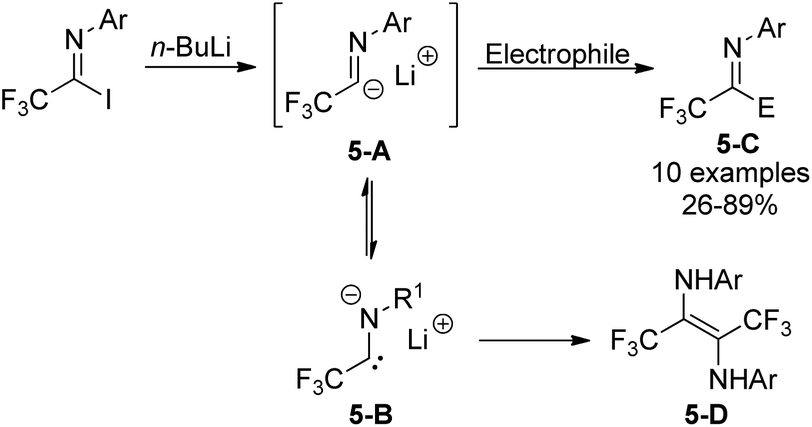 | ||
| Scheme 5 Generation and nucleophilic reactions of trifluoroacetimidoyl lithium. | ||
Uneyama and co-workers further investigated the generation and chemical properties of the trifluoroacetimidoyl lithiums (Scheme 6).21 The common imidoyl lithiums are stable only below −60 °C due to the highly ionic nature of a carbon–lithium bond. As for the benzoylation reaction with benzoyl chloride, the solvent of the reaction was complex and the lower temperature was beneficial for the reaction because the yield sharply dropped with the increase in temperature. Substituents on the aryl ring exerted a profound effect on the stability of the imidoyl lithium, where a large steric factor could promote the benzoylation reaction. Therefore, nucleophilic reactions of imidoyl lithium species bearing a 2,6-dimethylphenyl group with various electrophiles were tested to determine the scope and limitation of the transformation. Most of electrophiles could be compatible with the present reaction to give the corresponding products in moderate to good yields.
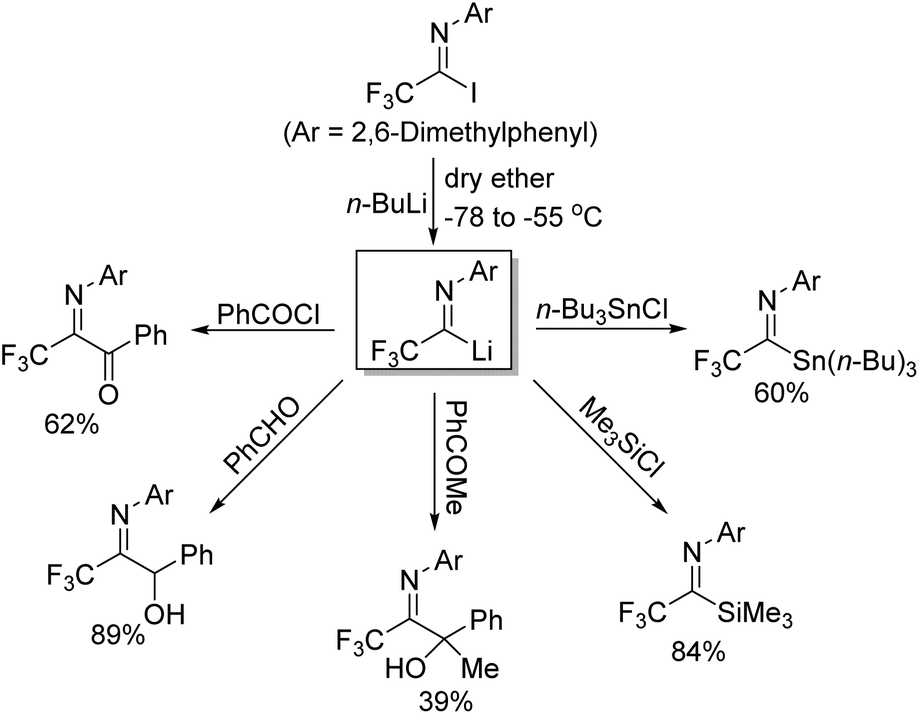 | ||
| Scheme 6 Nucleophilic reactions of trifluoroacetimidoyl lithium with diverse electrophiles. | ||
Uneyama's group also disclosed an approach to access trifluoroacetimidoyl zinc species, another trifluoroacetimidoyl metal, which were readily generated by the oxidative addition of imidoyl halides to activated zinc powder at room temperature (Scheme 7).22 In contrast to the imidoyl lithium mentioned above, the imidoyl zinc was more stable and could be handled at room temperature. The reaction was accelerated in the presence of ultrasound irradiated mixtures of zinc powders and aluminum powders in HMPA.
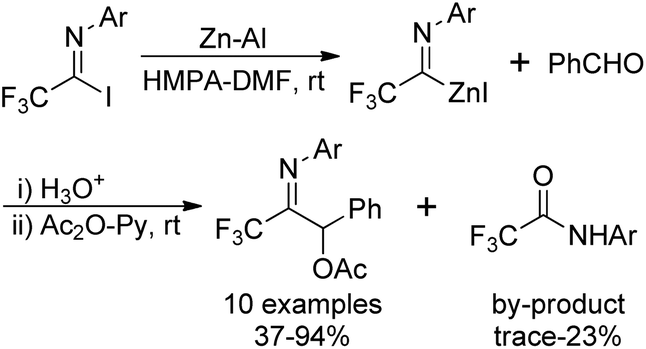 | ||
| Scheme 7 The reaction of N-aryl trifluoroacetimidoyl zinc halide with benzaldehyde. | ||
A metal-free trifluoroacetimidoyl carbanion was prepared by Uneyama and co-workers through the reaction of trifluoroacetimidoyl chlorides with trimethylsilyl cuprate and the subsequent fluoride ion-catalyzed generation of carbanions (Scheme 8).23 In the reaction, silyl cuprate was formed by the metal exchange of (trimethylsilyl)lithium with copper(I) and the replacement of lithium with tetraalkylammonium cation could stabilize the carbanion 8-B and promote the C–C bond formation at the imine carbon. With the employment of tetrabutylammonium as a counter cation, the stability of the imidoyl carbanions was enhanced and the reaction could be performed at room temperature with relatively high efficiency.
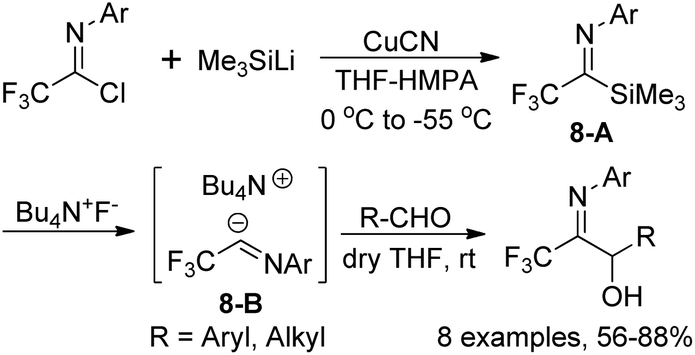 | ||
| Scheme 8 Preparation of (trifluoroacetimidoyl)trimethylsilanes and the reaction with electrophiles. | ||
In the presence of different metal species, trifluoroacetimidoyl halides are readily transformed into trifluoroimidoyl metals, which could be applied as versatile carbanion equivalents to react with most of the electrophiles. The stabilities of trifluoroimidoyl metals are quite different on the basis of the covalency between carbon and the metal, which is attributed to the electronegativity difference between carbon and the metals.24
Yuan and co-workers reported a simple and efficient one-pot reaction for the synthesis of α,β-unsaturated trifluoromethyl ketones (Scheme 9).25 In this transformation, diethoxyphosphorylmethyllithium was in situ generated from diethyl methylphosphonate and butyllithium, which reacted with N-phenyltrifluoroacetimidoyl chloride at −70 °C and quenched to afford a mixture of imino phosphonate and enamine. The treatment of the mixture with butyllithium gave a lithium salt, followed by the reaction with aldehydes though Horner–Emmons olefination and subsequent acid hydrolysis to provide the α,β-unsaturated trifluoromethyl ketone products in good yields. The protocol represents a convenient and economic route to access α,β-unsaturated trifluoromethyl ketones in contrast to Ishihara's method, which requires a four-step preparation of the imino phosphonate from pentafluoropropionic acid.26
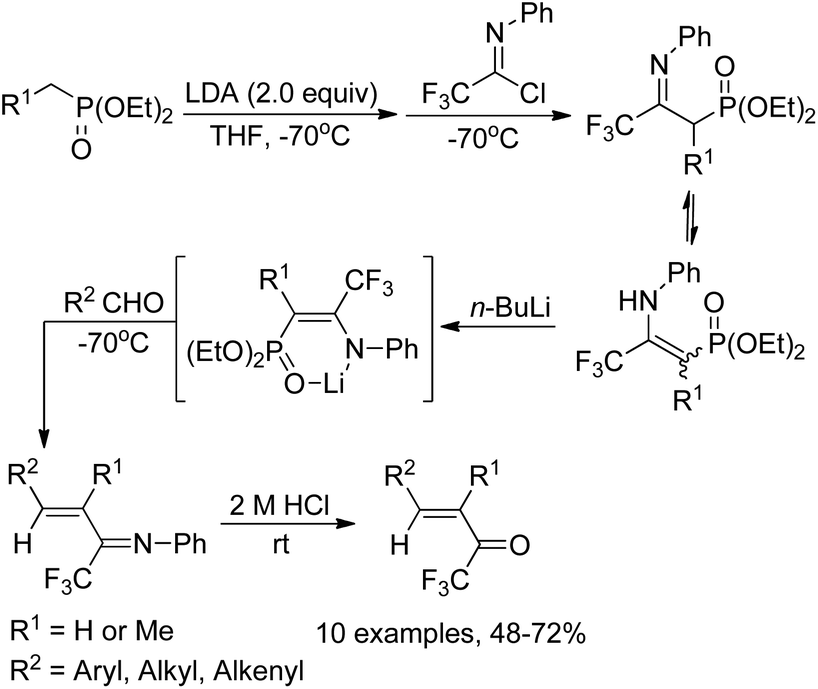 | ||
| Scheme 9 Synthesis of α,β-unsaturated trifluoromethyl ketones. | ||
The synthesis of diverse N-substituted trifluoroacetimidoyl aryl ketones by 1,3-dimethylimidazolium iodide-catalyzed aroylation of trifluoroacetimidoyl chlorides with aromatic aldehydes in the presence of sodium hydride was disclosed by Yuan and co-workers as well (Scheme 10).27 In the reaction, the aroyl anions attacked the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond of the imidoyl chloride to yield trifluoroacetimidoyl ketones with the displacement of the chlorine atom. Uneyama and co-workers also reported a pathway to produce N-aryltrifluoroacetimidoyl aryl/alkyl ketone in the early stages,21 where N-aryltrifluoroacetimidoyl lithium was generated by an iodine-lithium exchange reaction between N-aryltrifluoroacetimidoyl iodide and n-BuLi, and then trapped by the acyl chloride. The methodology suffered from low reaction temperatures (−100 °C to −78 °C) and limited substrate scope.
N bond of the imidoyl chloride to yield trifluoroacetimidoyl ketones with the displacement of the chlorine atom. Uneyama and co-workers also reported a pathway to produce N-aryltrifluoroacetimidoyl aryl/alkyl ketone in the early stages,21 where N-aryltrifluoroacetimidoyl lithium was generated by an iodine-lithium exchange reaction between N-aryltrifluoroacetimidoyl iodide and n-BuLi, and then trapped by the acyl chloride. The methodology suffered from low reaction temperatures (−100 °C to −78 °C) and limited substrate scope.
 | ||
| Scheme 10 Synthesis of N-substituted trifluoroacetimidoyl aryl ketones. | ||
The plausible reaction mechanism was depicted in Scheme 11. The interaction of NaH on the C-2 hydrogen of 1,3-dimethylimidazolium iodide formed the ylide (B−), which attacked the aldehyde to give an O-anion species 11-A. The latter could isomerize to carbanion 11-B, followed by the displacement of the chlorine of imidoyl chloride to afford the intermediate 11-C, which was treated with NaH to give a new O-anion intermediate 11-D. The release of the ylide (B−) from O-anion could deliver the final aroyl product.
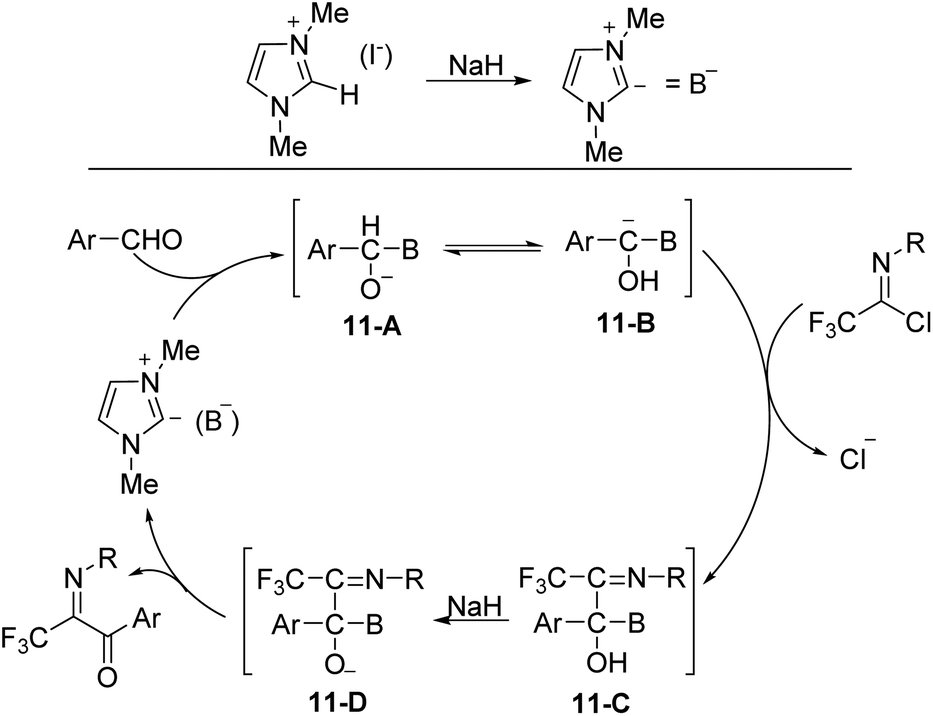 | ||
| Scheme 11 Plausible reaction mechanism. | ||
The nucleophilic substitution of N-aryl polyfluoroalkyl imidoyl iodides with the carbanion of an α-methyl ketone yields β-polyfluoroalkyl enaminones, which serve as β-dicarbonyl equivalents to synthesize fluorinated heterocyclic compounds (Scheme 12).28 Treatment of β-polyfluoroalkyl enaminones with an excess of hydrazine monohydrate could furnish 3-polyfluoroalkyl pyrazoles. 6-Polyfluoroalkyl pyrimidines were obtained in satisfactory yields by the reaction of β-polyfluoroalkyl enaminones with amidines.
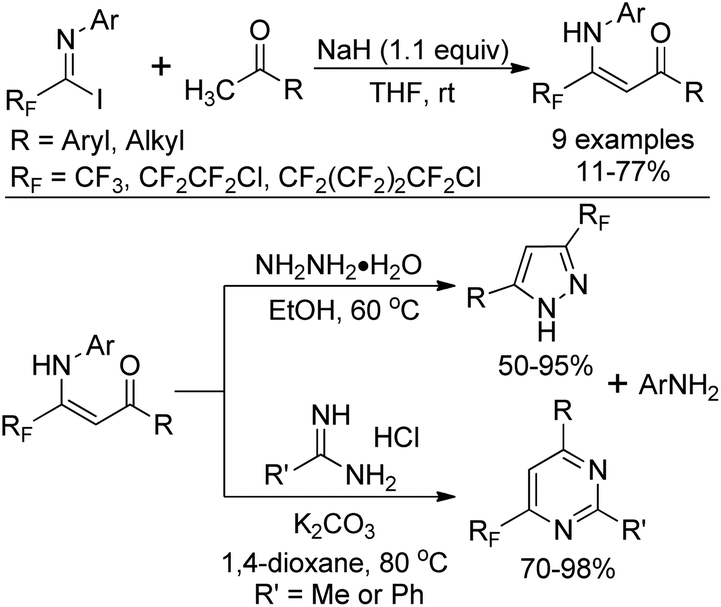 | ||
| Scheme 12 Preparation of β-polyfluoroalkyl enaminones and their application in the synthesis of fluorinated heterocycles. | ||
As early as 1989, Uneyama and co-workers have reported the coupling reaction of trifluoroacetimidoyl chlorides with various carbon nucleophiles, including Grignard reagents and active methylene compounds.29 As for Grignard reagents, the alkylation of the trifluoroacetimidoyl chlorides by the replacement of the chlorides was faster than the addition of Grignard reagents to the product imines. With respect to active methylene compounds, the obtained products were enamines rather than imines because of the relative stability of the enamine derivatives.
The reaction of fluorinated imidoyl chlorides with lithium derivatives of enantio-pure methyl sulfoxides to produce chiral β-imino sulfoxides was described by Fustero and co-workers (Scheme 13).30 The reaction proceeded well with moderate to high efficiency under mild conditions and could be completed within 20 minutes. The configurational stability of tautomers and geometric isomers of β-imino sulfoxides 13-B were also investigated. Notably, some β-imino sulfoxides were afforded via the aza-Wittig reaction of γ-fluoro-β-keto sulfoxides with N-aryl imino triphenylphosphoranes.
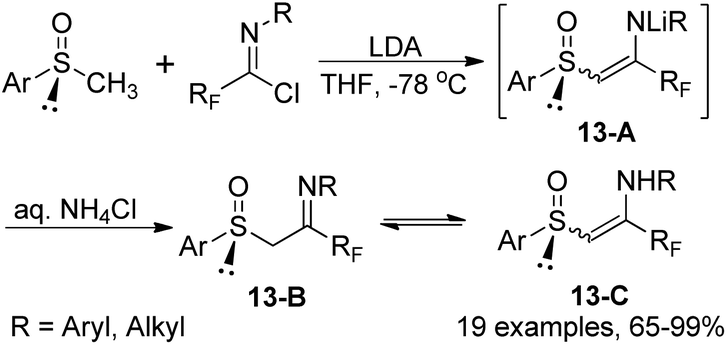 | ||
| Scheme 13 Synthesis of fluorinated chiral β-imino sulfoxides. | ||
Fustero and co-workers reported an efficient method for the assembly of fluorinated vinylogous amidines starting from imidoyl chlorides and enolizable ketimines (Scheme 14).31 In the presence of two equivalents of LDA, the fluorinated 1,3-diimine products 14-A were formed, which could be exclusively isolated as vinylogous amidine tautomers 14-B. The reaction scope was broad, as demonstrated by the compatibility of structurally various ketimines and even chiral substituents at one or both nitrogen atoms. Regioselective hydrolysis mediated by 6 N H2SO4 occurred to release the corresponding fluorinated β-enamino ketones 14-D, where only the imine group bearing the fluoroalkyl substituent (RF) could be regioselectively hydrolyzed (Scheme 14).
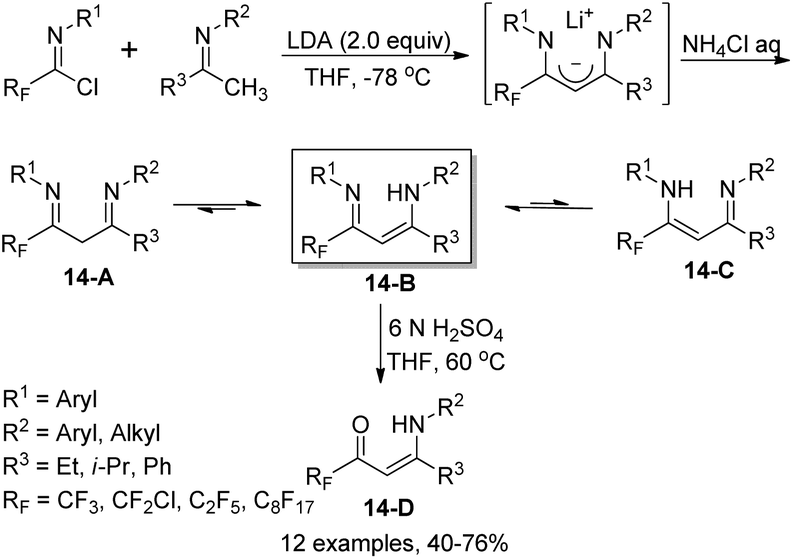 | ||
| Scheme 14 Synthesis of fluorinated vinylogous amidines and β-enamino ketones. | ||
The fluorinated β-enamino ketones could also be generated by the reaction of imidoyl chlorides and ketones with the modification of the reaction temperature. The isolated fluorinated β-enamino ketones exist as a mixture of enamino-imino tautomers 15-A and 15-B, which could be readily hydrolyzed to yield the corresponding 1,3-dicarbonyl derivatives in the enol form. However, the β-enamino ketones 14-D remain unchanged under the same hydrolysis conditions, probably due to the strong intramolecular hydrogen bond (RF) O⋯H–N (Scheme 15).
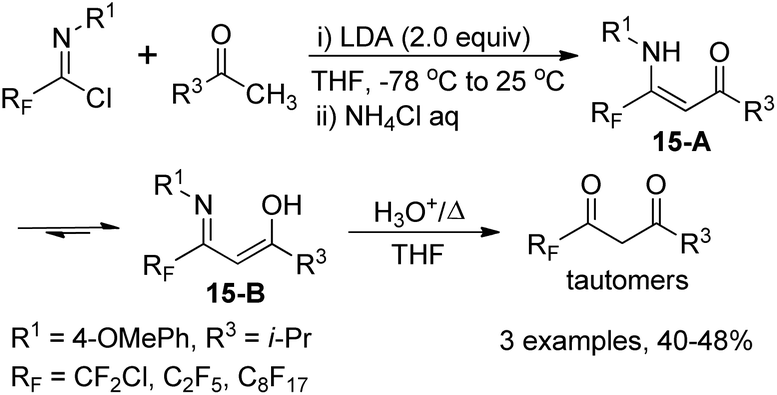 | ||
| Scheme 15 Synthesis of fluorinated β-enamino ketones and 1,3-dicarbonyl derivatives. | ||
Three years later, the same group developed a two-step route to construct racemic and/or chiral nonracemic γ-fluorinated-β-amino acid derivatives (16-D/E) through reduction of fluorinated β-enamino esters 16-C, which could be prepared from the coupling of fluorinated imidoyl chlorides and lithium enolates of alkyl esters (Scheme 16).32 In order to achieve good yield and chemo- and stereoselectivity of the fluorinated β-amino acids, optimization of the reduction conditions was performed. The optimal condition was the combination of an excess of anhydrous ZnI2 and NaBH4 in dry dichloromethane at room temperature. Mechanistic investigation implied that a metal-chelated six-membered model was involved in the reaction and the choice of (−)-8-phenylmenthol as a chiral auxiliary could enable the best diastereoselectivity to reduce the chiral β-enamino esters.
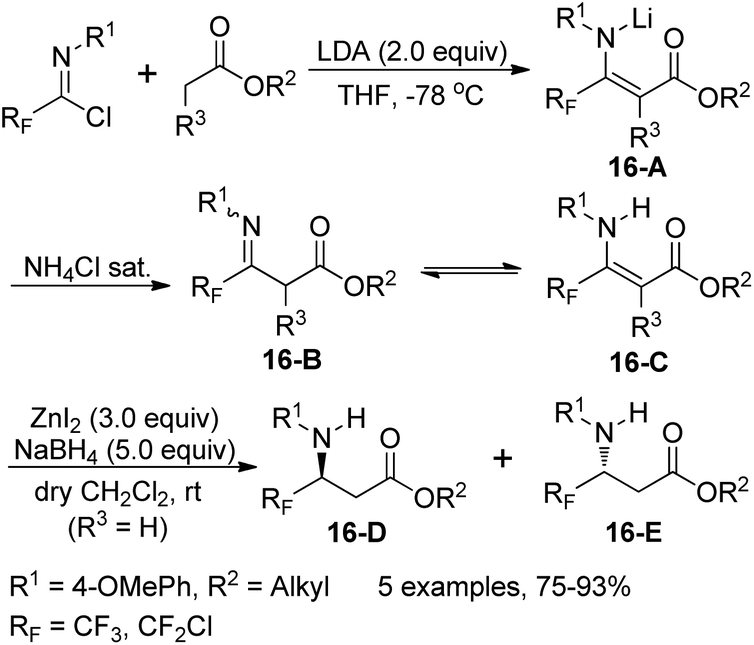 | ||
| Scheme 16 Stereoselective synthesis of fluorinated β-amino acids. | ||
Fustero and co-workers explored a simple route to the synthesis of chiral nonracemic fluorinated β-amino sulfones by the employment of imidoyl halides as fluorinated building blocks (Scheme 17).33 The reaction of fluorinated chiral imidoyl chloride with sulfonyl carbanion could afford chiral fluorinated β-imino sulfones as a tautomeric imino/enamino mixture in high yields. Then, the stereoselective reduction occurred to give the corresponding β-amino sulfones with NaBH4 as the reducing agent. The isolation of the predominant diastereoisomer through chromatographic separation and the subsequent hydrogenolysis over Pd/C produced enantiomerically pure β-amino sulfones.
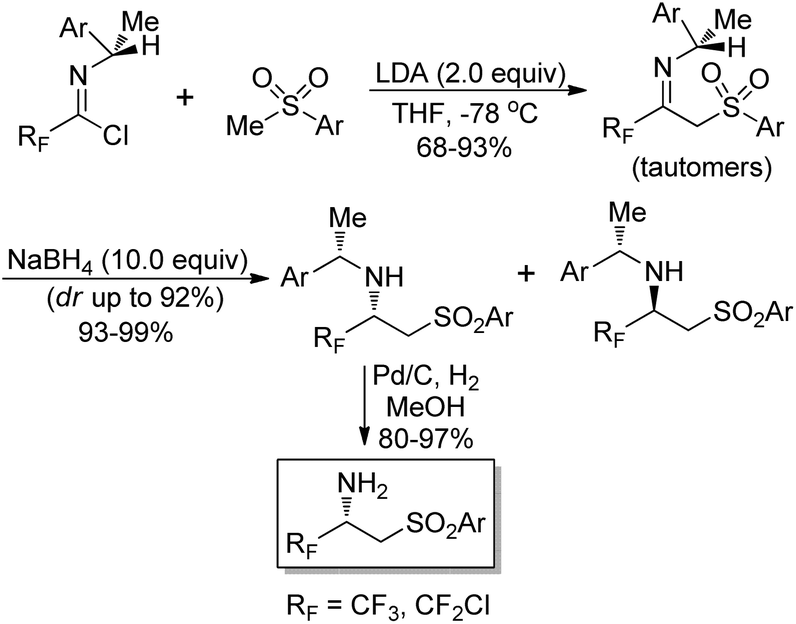 | ||
| Scheme 17 Asymmetric synthesis of fluorinated β-amino sulfones. | ||
The developed strategy was also applied to synthesize enantiomerically pure α-fluoroalkyl allylic amines, which were regarded as versatile building blocks (Scheme 18). A Julia-type methylenation-desulfonylation reaction of β-amino sulfones was involved in the reaction. In the presence of n-butyllithium and the consecutive addition of ClCH2I and methyllithium, the organolithium intermediate 18-A was formed, which underwent β-elimination process to furnish fluorinated allylic amines in good yields. It is worth mentioning that the author described a new synthesis of fluorinated allylamines through the reaction of 2-(trimethylsilyl)ethyl sulfones and sulfoxides with imines and imino esters, which utilized TBAF to enable the fragmentation of 2-(trimethylsilyl)ethyl sulfones to release the desired allylic amines.34
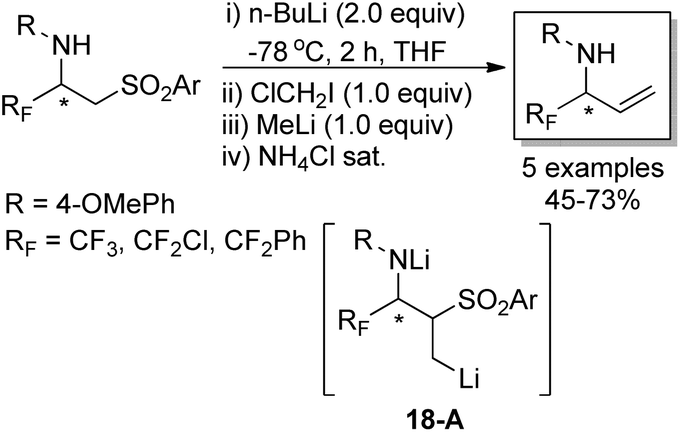 | ||
| Scheme 18 Asymmetric synthesis of allylic amines. | ||
Uneyama and co-workers presented a palladium(0)-catalyzed reductive dimerization of the fluorinated imidoyl iodides in the presence of carbon monoxide to yield fluorinated α-diimines, which were considered as a new class of catalyst ligands (Scheme 19).35 In the transformation, carbon monoxide acted as a reducing agent for regeneration of the catalytically active Pd(0) species. The α-diimines could be flexibly designed and synthesized to meet the catalytic activity through tuning of the electronic and steric properties of the N-substituent of imine moieties. Notably, a chiral diimine ligand could also be generated by the use of this method.
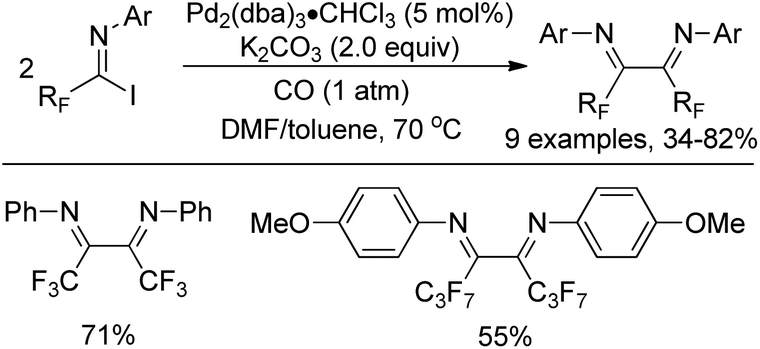 | ||
| Scheme 19 Carbon monoxide-promoted reductive homocoupling of fluorinated imidoyl iodides. | ||
The fluorinated α-diimines can also be produced from samarium iodide-mediated reductive coupling of imidoyl iodide and the subsequent oxidation by manganese dioxide (Scheme 20).36 X-ray diffraction analysis showed the imine groups in the E,E-geometry, with a substantial nonplanarity of the NC–CN moiety. Therefore, the resulting α-diimines failed to form chelate complexes of palladium and platinum.
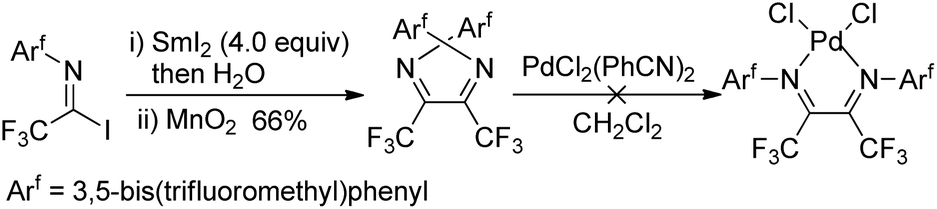 | ||
| Scheme 20 Sm-Mediated reductive coupling of trifluoroacetimidoyl iodide. | ||
In 2011, Wu and co-workers demonstrated a Cu(I)-catalyzed coupling reaction of fluorinated imidoyl halides with terminal alkynes to construct fluorinated alkynyl imines (Scheme 21).37 Compared with a similar work of synthesizing fluorinated alkynyl imines via a Pd/Cu-catalyzed coupling reaction of bromodifluoroacetimidoyl iodide with terminal alkynes previously reported by the same group,38 the reaction could be realized by a Cu(I) catalyst only. The transformation proceeds smoothly with respect to bromodifluoromethylated imidoyl halides and trifluoromethylated imidoyl halides, whereas the latter required stoichiometric amounts of Cu(I). The plausible pathway is initiated by the insertion of the generated copper alkynide 21-A to C–X bond of fluorinated imidoyl halides. The intermediate 21-B is herein formed, followed by the reductive elimination to deliver fluorinated alkynyl imines.
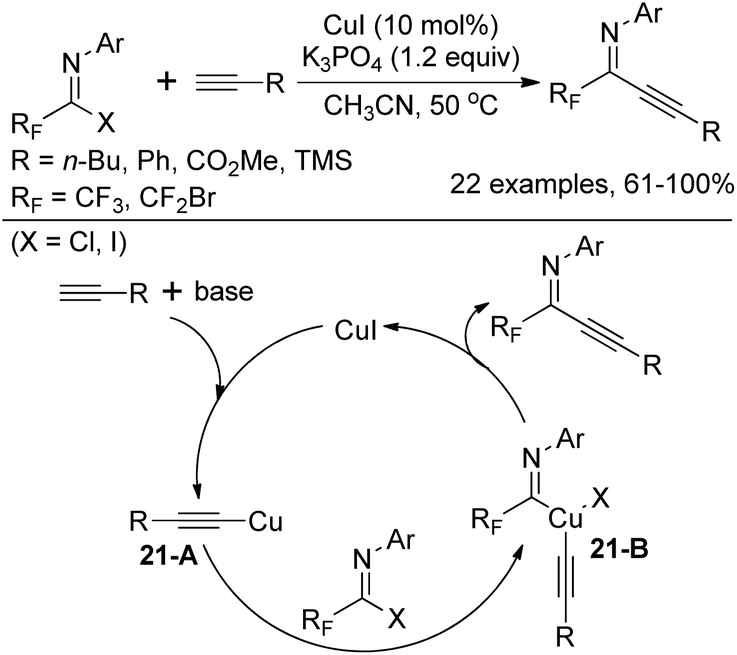 | ||
| Scheme 21 Cu(I)-Catalyzed coupling reactions of fluorinated imidoyl halides with terminal alkynes. | ||
It is worth noting that the fluorinated alkynyl imines were applied to synthesize fluorinated propargylamines via selectively asymmetric reduction. In the transformation, phosphoric acid- and ruthenium-catalyzed chemoselective biomimetic hydrogenation of the carbon–nitrogen double bond of fluorinated alkynyl ketimines with the vicinal carbon–carbon triple bond intact was achieved by Zhou and co-workers in 2016.39
The non-hydrogen-bridged 6-arylamino-3-imidoylfulvenes 22-B/C were rapidly prepared from the regioselective reaction of sodium cyclopentadienide with electrophilic trifluoroacetimidoyl chlorides (Scheme 22).40 The reaction was kinetically controlled and could be completed in two hours at room temperature. The monosubstitution of cyclopentadienide by an imidoyl chloride occurred to give imidoylcyclopentadiene, which was further deprotonated by another sodium cyclopentadienide to deliver a sodium imidoylcyclopentadienide 22-A. Electrophilic attack of trifluoroacetimidoyl chlorides at the less hindered 3-position of the 22-A furnished 1,3-bis-(imidoyl)cyclopentadienes 22-B/C with no intramolecular N–H–N hydrogen bond. The 1,3-bis(imidoyl)-pentamethylruthenocene metalloligands 22-D were generated from the deprotonation of 22-B/C by n-butyllithium and the subsequent reaction with Cp*Ru(CH3CN)3PF6.
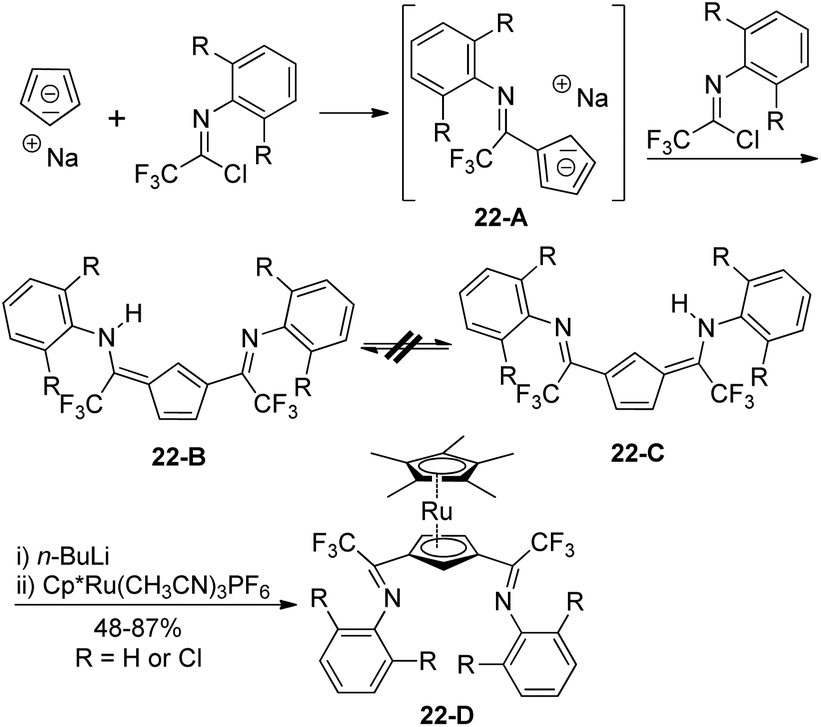 | ||
| Scheme 22 Synthesis of 6-arylamino-3-imidoylpentafulvenes. | ||
3.2. C–N bond formation
Uneyama and co-workers reported the preparation and thermolysis of N-[2,2,2-trifluoro-1-(tritylazo)ethylidene]anilines and the related azo compounds (Scheme 23).41 The reaction of imidoyl chlorides with hydrazine monohydrate could give 23-A almost quantitatively, which reacted with Ph3CCl and underwent oxidative dehydrogenation in the presence of Pb(OAc)4 to deliver tritylazo-compounds 23-C. Imidoyl radicals 23-D could be generated via thermolysis of tritylazo-compounds 23-C. The kinetics of thermal decompositions and electronic effects of substituents on the N-aryl group were investigated. The N-aryl-2,2,2-trifluoroacetimidoyl radicals, generated from the photochemical homolysis of the imidoyl iodides or the thermal homolysis of imidoyl azo-compounds, were smoothly utilized to synthesize 2-trifluoromethylated indole and quinoline derivatives by the same author.42 | ||
| Scheme 23 Preparation and thermolysis of N-[2,2,2-trifluoro-1-(tritylazo)ethylidene]anilines. | ||
Saidi and co-workers demonstrated a base promoted synthesis of N-aryltrifluoroacetimidoyl phthalimide and succinimide from the C–N bond coupling reaction of trifluoroacetimidoyl chlorides with phthalimide and succinimide (Scheme 24).43 In the reaction, the chlorine was readily replaced with nitrogen nucleophiles in the presence of NaH under mild conditions.
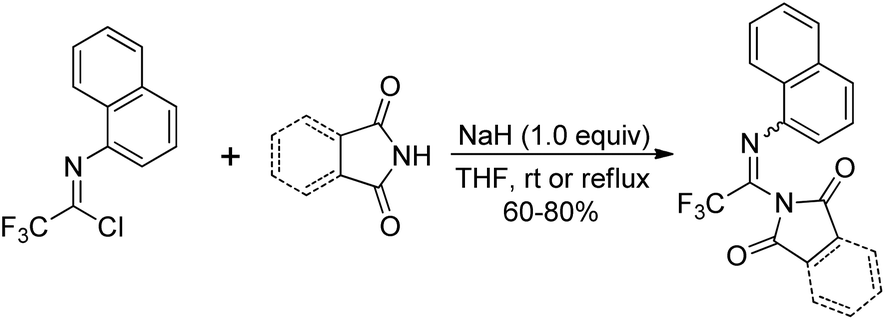 | ||
| Scheme 24 Synthesis of N-aryltrifluoroacetimidoyl phthalimide and succinimide. | ||
Wu and co-workers described a nucleophilic substitution reaction of imidoyl chlorides with different amines to synthesize α-fluoro-substituted amidines in good to excellent yields (Scheme 25).44 With regard to the active amine, an addition–elimination route was involved in the reaction. As for unreactive amines, the coupling promoted by palladium is necessary. The obtained fluorinated amidine could be used as a component to perform Hantzsch three-component synthesis in the presence of ZnCl2, and 1,4-dihydropyridine product was delivered in reasonable yields.
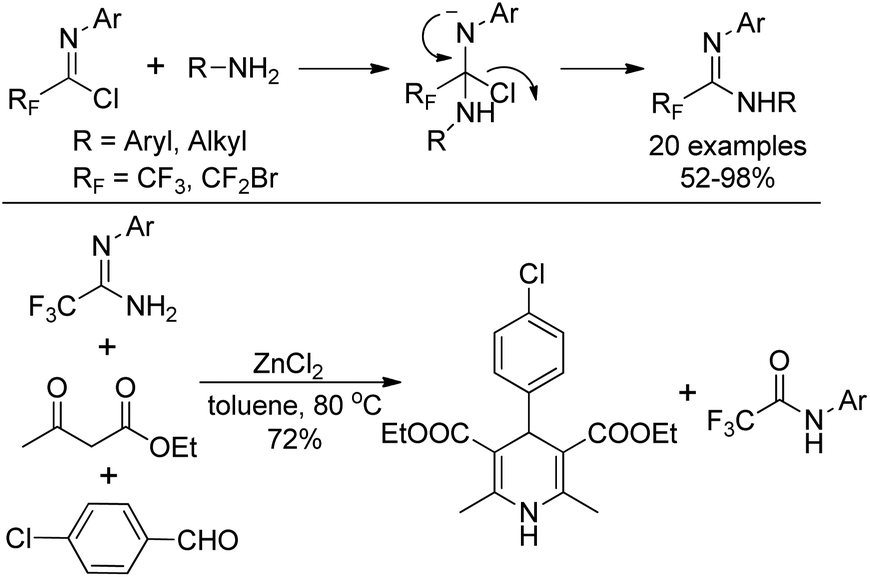 | ||
| Scheme 25 Synthesis of α-fluoro-substituted amidines from imidoyl chlorides. | ||
Darehkordi and co-workers applied N-aryltrifluoroacetimidoyl chloride as N-linkers to quinolones to construct diverse trifluoroacetimidoyl piperazinylquinolone derivatives (Scheme 26).45 The trifluoromethyl group was successfully incorporated via the coupling reaction of trifluoroacetimidoyl chlorides with norfloxacin, ciprofloxacin and enoxacin under mild conditions. The selected trifluoroacetimidoyl ciprofloxacin and norfloxacin were found to display promising antibacterial activities.
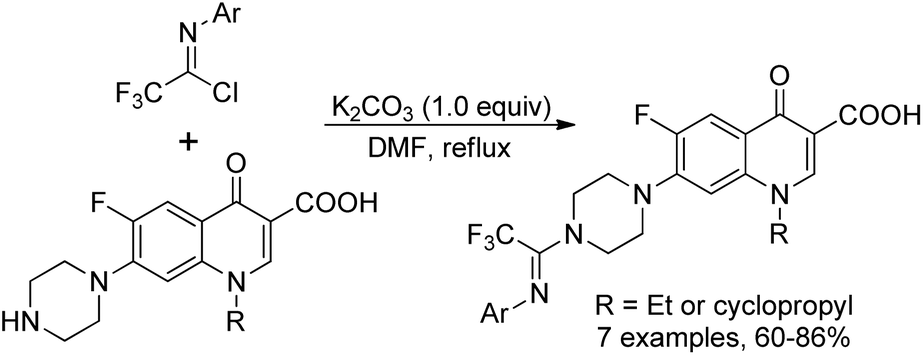 | ||
| Scheme 26 Synthesis of trifluoroacetimidoyl piperazinylquinolone derivatives. | ||
The reaction of indole molecules with different trifluoroacetimidoyl chlorides in the presence of NaH as a base to construct new trifluoromethylated indole derivatives was developed by the same group (Scheme 27).46 In the coupling reaction of common indole with trifluoroacetimidoyl chlorides, the nitrogen of pyrrole acted as a nucleophile to displace chlorine to provide the corresponding product 27-A. With regard to less reactive indole-3-carbaldehyde, the carbon atom at position 2 served as the nucleophile to yield the product 27-B. When indole-3-carboxylic acid was used, trifluoromethylated 4,9-dihydropyrano[3,4-b]indol-3(1H)-one 27-C was produced. The cyclization process was achieved by the intramolecular nucleophilic addition of oxygen of carboxylic acid into the carbon–nitrogen double bond of fluorinated imines.
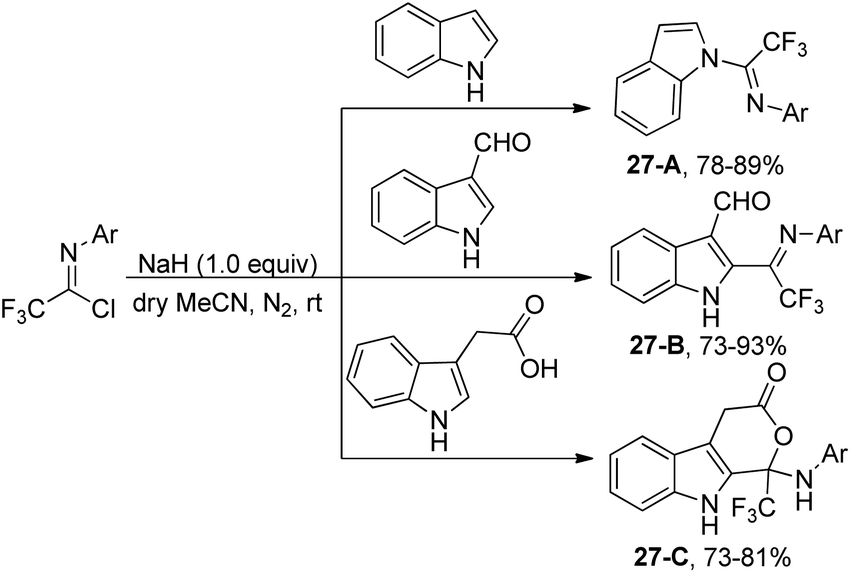 | ||
| Scheme 27 Synthesis of different indole trifluoromethyl derivatives. | ||
After three years, the same author reported a similar method for the synthesis of trifluoromethylated benzimidazole, imidazole and benzothiazole derivatives via a base-promoted coupling reaction of trifluoroacetimidoyl chlorides with the corresponding N-heterocycles (Scheme 28).47 As for the benzothiazole derivatives with unequal N and S bidentate nucleophilic sites, only an N-substituted isomer was obtained in good yields in the presence of NEt3.
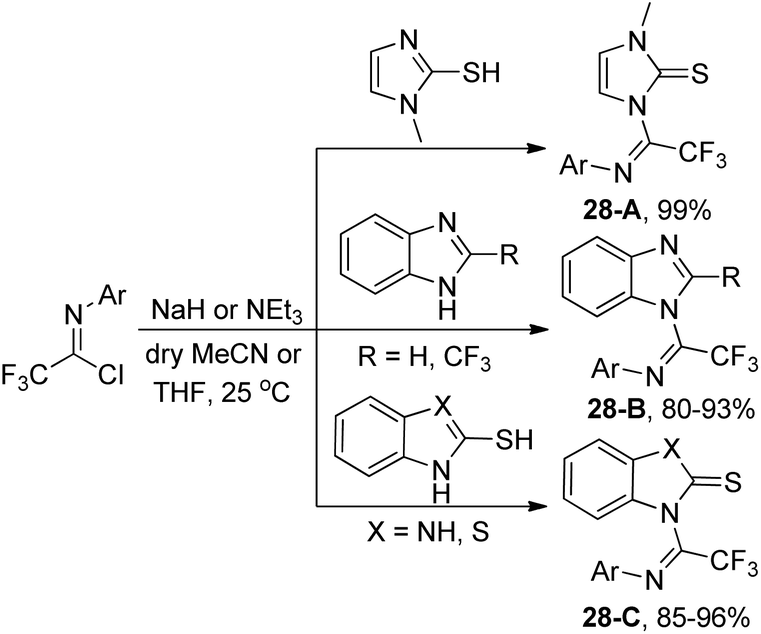 | ||
| Scheme 28 Synthesis of a new α-trifluoromethyl-substituted formamidine framework. | ||
3.3. C–O(S) bond formation
Benzyl N-phenyl-2,2,2-trifluoroacetimidate, which served as a new O-benzylation reagent, was reported by Yamada and co-workers (Scheme 29).48 The O-benzylation reagent was readily prepared by the NaH-promoted coupling reaction of benzyl alcohol with trifluoroacetimidoyl chlorides. The present trifluoroacetimidate was very stable and soluble in various solvents, which could react with sterically hindered alcohols and base-sensitive hydroxyl esters apart from common alcohols to deliver benzyl ethers in the presence of catalytic TMSOTf. The O-benzylation reagent features good stability and can be stored under air for two months without the loss of the reactivity.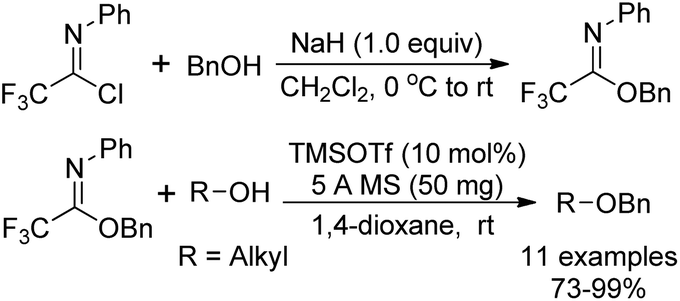 | ||
| Scheme 29 Synthesis and application of a stable reagent for O-benzylation. | ||
Barroca-Aubry and co-workers also developed a O-benzylation reagent PMB-NPTFA, which was prepared from p-methoxybenzyl alcohol and trifluoroacetimidoyl chlorides in the presence of NaH (Scheme 30).49 The PMB-NPTFA was presented in crystalline form, stable and soluble in a variety of solvents. The corresponding PMB ethers could be rapidly afforded in high yields by Bi(OTf)3 catalyzed reactions of primary or secondary alcohols with PMB-NPTFA in the presence of conventional 4 Å MS. Acid- and base-sensitive protecting groups were tolerated under the mild reaction conditions.
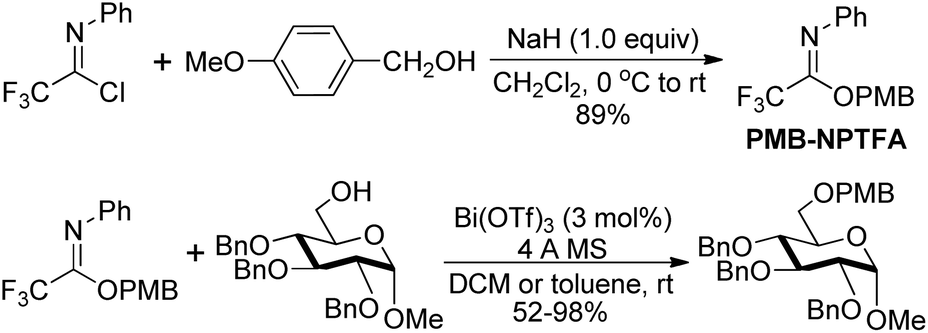 | ||
| Scheme 30 Synthesis of PMB-NPTFA and application in mild acid catalyzed etherification. | ||
Pohl and co-workers discovered O-allyl and O-benzyl N-(p-nitrophenyl)trifluoroimidates for the addition of allyl and benzyl protecting groups (Scheme 31).50 The powerful reagents 31-A/B were achieved by the exploration of the role of para-substituents on the balance between the stability and reactivity of the N-phenyl trifluoroacetimidates. With catalytic amount of TMSOTf as the promoter, the reaction of reagents 31-A/B with different alcohols to give the benzylation or allylation products in high yields compared with the less stable allyl and benzyl trichloroacetimidate reagents.
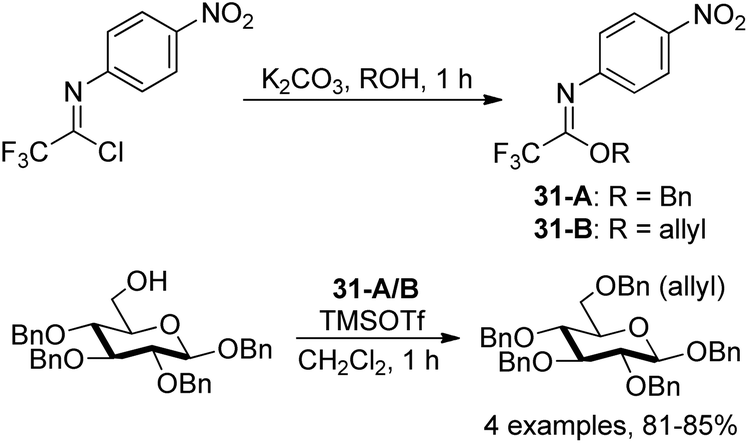 | ||
| Scheme 31 Synthesis of N-aryl trifluoroacetimidate-based benzyl and allyl protecting group reagents. | ||
It should be noted that trifluoroacetimidoyl chlorides were also successfully applied as mediators to promote the C–O bond formation in the synthesis of tetralactosaminyl O-glycothreonine51a and Lewis A (Lea) tandem repeat51b under similar conditions.
Kuniyasu, Kambe and co-workers demonstrated a Pd-catalyzed iminothiolation of alkynes by using trifluoromethylated iminosulfides to construct 4-sulfur-functionalized 1-azadienes (Scheme 32).52 The iminosulfides could be readily prepared by the base-mediated coupling of trifluoroacetimidoyl chlorides with thiols. In the iminothiolation reaction, the existence of the CF3 group was very essential for the successful transformation and the milder conditions enabled the addition of terminal alkynes compared with the internal alkynes. The reaction was initiated by the oxidative addition of iminosulfides to Pd(0) species to form Pd(II) complex 32-A, which underwent regio- and stereoselective insertion of alkynes into the Pd–S bond of 32-A to afford the intermediate 32-B. The final reductive elimination of 32-B resulted in the 1-azadiene derivatives with the release of Pd(0) complex.
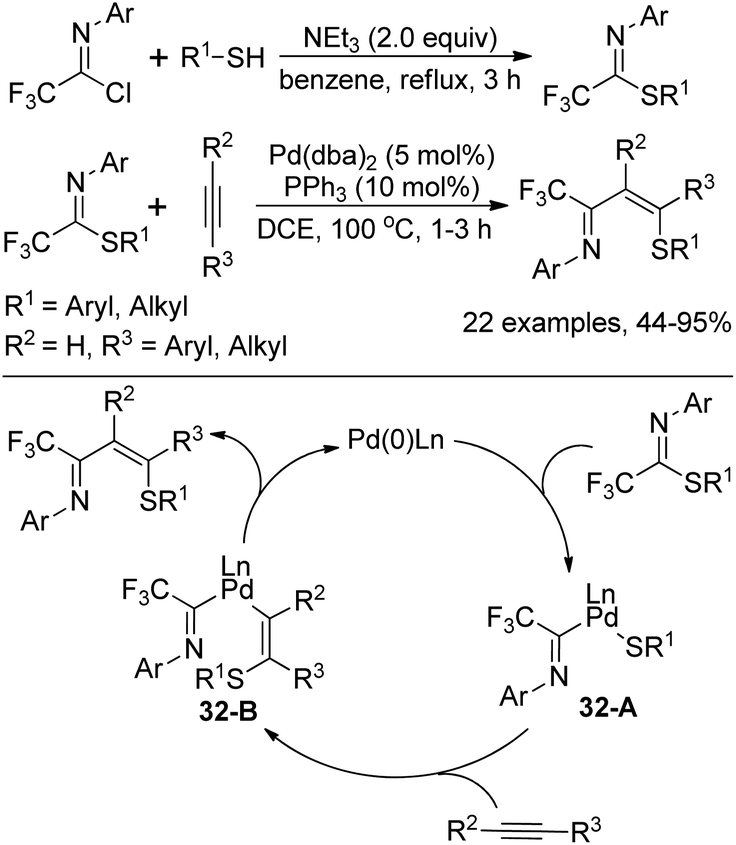 | ||
| Scheme 32 Synthesis of trifluoromethylated iminosulfides and the application in iminothiolation of alkynes. | ||
3.4. C–P bond formation
The Yuan group developed an Arbuzov-type strategy for the preparation of 1-(N-aryl/alkyl amino)-2,2,2-trifluoroethylphosphonates, which was realized by the reaction of trialkyl phosphite with trifluoroacetimidoyl chlorides followed by subsequent reduction in the presence of sodium cyanoborohydride (Scheme 33).53 Noteworthy was that low reactivity was observed by using sodium borohydride as a reducing agent, and N-substituents in the trifluoroacetimidoyl chloride exerted an obvious influence on the reaction. The experiment data implied that the transformation was initiated by the nucleophilic attack of trivalent phosphorus substrates on trifluoroacetimidoyl chlorides.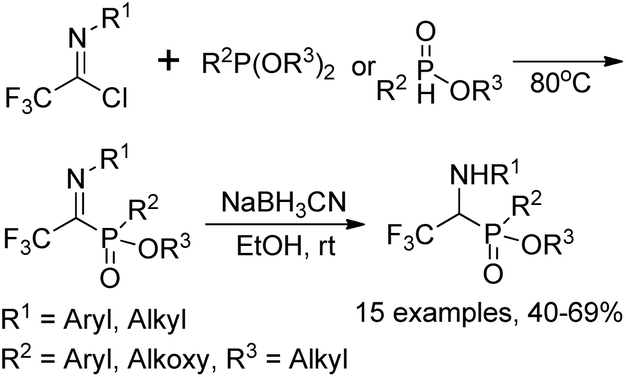 | ||
| Scheme 33 Synthesis of 1-aminophosphonic acid derivatives bearing a trifluoromethyl moiety. | ||
Onys'ko and co-workers reported a reaction of trifluoroacetimidoyl chlorides with trialkyl phosphites to give rise to N-aryltrifluoroacetimidoylphosphonates, in which existed as dynamic equilibrium mixture of Z,E isomers (Scheme 34).54 The obtained imidoylphosphonates easily reacted with O- or S-centered nucleophiles to yield the products of addition to the CN bond. The trifluoroacetimidoylphosphonates could act as dipolarophiles to undergo cycloaddition reactions with nitrile oxides, delivering the corresponding phosphorylated oxadiazolines, which included the fragment of aminophosphonic acid.
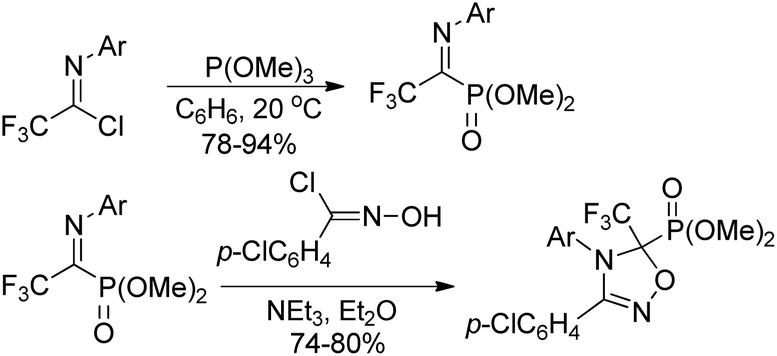 | ||
| Scheme 34 Synthesis of N-aryltrifluoroacetimidoylphosphonates. | ||
3.5. C–Si bond formation
In 2003, Uneyama, Amii and co-workers developed a Mg(0)/Me3SiCl system to promote double silylation of trifluoroacetimidoyl chlorides, providing bis-silylated difluoroenamines, which was regarded as a novel fluorinated dianion equivalent (Scheme 35).55 Initially, the Mg-mediated reductive cleavage of C–Cl bonds of the imidoyl chlorides occurred to give rise to imidoylmagnesium species 35-A, which reacted with chlorotrimethylsilane to afford imidoylsilanes 35-B. Subsequently, the two-electron reduction of the imidoylsilanes 35-B furnished the β-fluorinated organomagnesium species 35-C, followed by the β-elimination to produce bis-silylated difluoroenamines.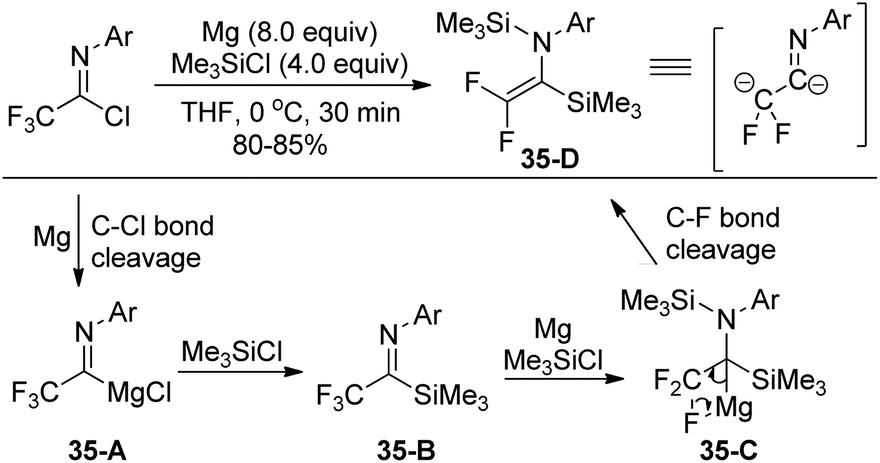 | ||
| Scheme 35 Mg-Promoted double silylation of trifluoroacetimidoyl chlorides. | ||
The resultant bis-silylated difluoroenamines could be applied as useful bifunctional synthetic blocks. The Lewis acid-catalyzed aldol-type reaction of bis(silyl)enamines with various aldehydes or aldimines in the presence of BF3·OEt2 in CH2Cl2 at 0 °C provided β-hydroxy-α,α-difluoro imidoylsilanes or β-amino-α,α-difluoro imidoylsilanes in good yields (Scheme 36). Notably, the bis(silyl)enamines could also react with two kinds of electrophiles to yield a variety of difluorinated imines.
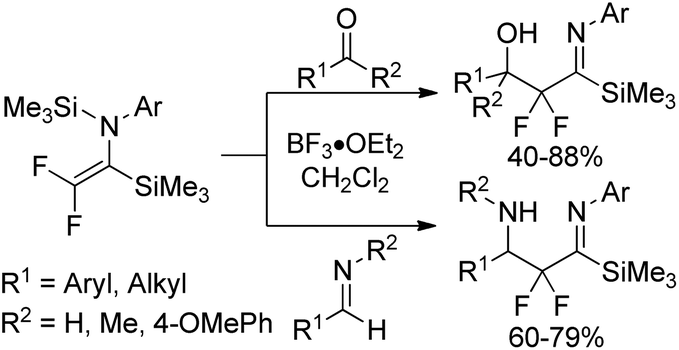 | ||
| Scheme 36 The aldol-type reactions of bis-silylated difluoroenamine with aldehydes and ketones or aldimines. | ||
A Mg-promoted selective activation of imidoyl carbon–halogen bonds to synthesize fluorinated imidoylsilanes was developed by the same group (Scheme 37).56 When excess amounts of magnesium metal and TMSCl were used, the bis(silyl)enamine 35-D was selectively formed. In order to avoid further reduction involving the C–F bond cleavage, a reduced amount of magnesium metal and a low temperature should be implemented. Moreover, the employment of a catalytic amount of ethyl bromide to activate the surface of metallic magnesium was beneficial for the reaction. Except for trifluoromethyl imidoylsilanes, difluoromethyl, perfluoroalkyl and pentafluoroethyl imidoylsilanes could be prepared in moderate to good yields, which were applied as stable and useful fluorinated acyl anion equivalents.
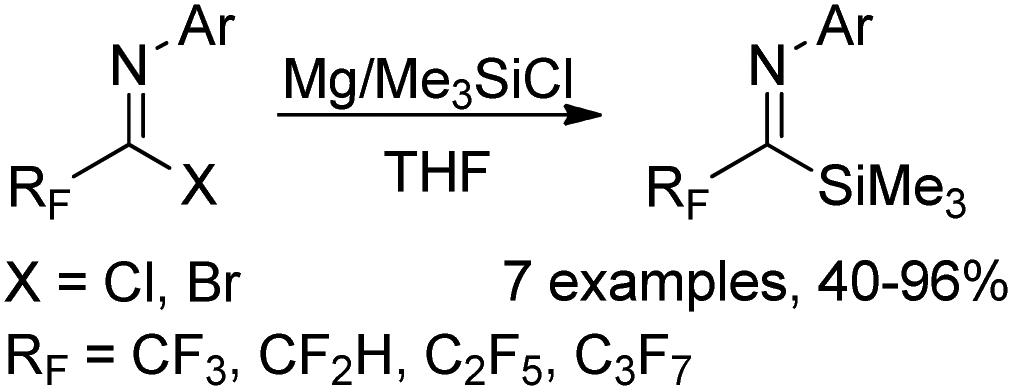 | ||
| Scheme 37 An improved synthesis of fluorinated imidoylsilanes. | ||
3.6. Formation of trifluoroacetimidoyl Pd(II) or Ti(IV) or Pt(II) complexes
The intramolecular thiopalladation of thioanisole-substituted propargyl imines to construct perfluoroalkyl-substituted benzothiophene-based palladacycles under mild reaction conditions was described by Likhar and co-workers (Scheme 38).57 The corresponding perfluoroalkyl propargyl imines could be prepared from Sonogashira coupling of perfluoroalkyl imidoyl iodides and alkynes. The structures of dimeric palladacycles were fully confirmed by 1H/13C NMR and IR. The protocol enables the formation of heterocyclic-based palladacycles in the absence of a heterocyclic ligand system.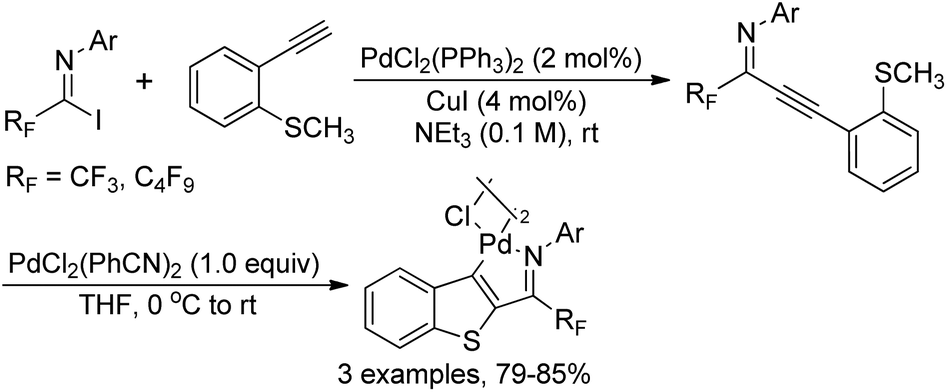 | ||
| Scheme 38 Synthesis of benzothiophene-based palladacycles. | ||
The thiopalladacycles obtained from palladium catalysts and ortho-thioanisole-substituted propargyl imines were applied to undergo Heck-type coupling of alkenes for the synthesis of 3-alkenylbenzo[b]thiophene (Scheme 39).58 The existence of imino moiety in the alkenylated benzothiophene 39-B enabled the instability of the product, so further reduction by using a combination of NaBH3CN in glacial acetic acid and methanol could give the secondary amine products 39-C. Noteworthy was that the formation and involvement of the thiopalladacycle 39-A was identified as the key step of the Heck-type coupling. The following alkene insertion and Pd–H elimination afforded the 3-alkenylbenzo[b]thiophene product.
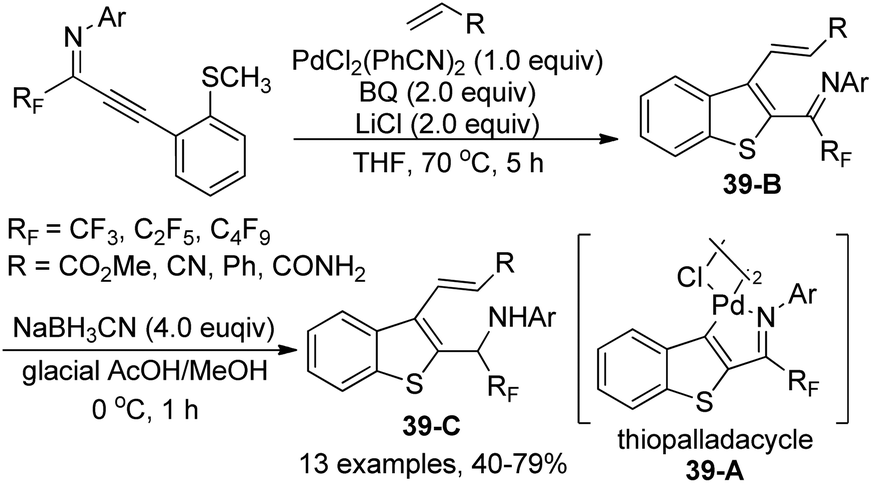 | ||
| Scheme 39 Synthesis of 3-alkenylbenzo[b]thiophenes. | ||
Diverse fluorinated imidoyl palladium complexes were synthesized and their structures were identified by Uneyama, Amii and co-workers (Scheme 40).59 Trifluoroacetimidoyl palladium(II) phosphine complexes 40-A were prepared from the oxidative addition reaction of trifluoroacetimidoyl iodide with Pd(PPh3)4 in benzene at room temperature. The obtained palladium(II) complexes 40-A could react with organotin compounds to afford trifluoromethylated imines in moderate yields. The phosphine-free trifluoroacetimidoyl palladium(II) complexes 40-B were generated from the reaction of imidoyl iodide with Pd2(dba)3·CHCl3, whereas inferior reactive imidoyl chloride did not participate in the reaction. The present palladium(II) complex 40-B could be applied as a catalyst for carboalkoxylation.
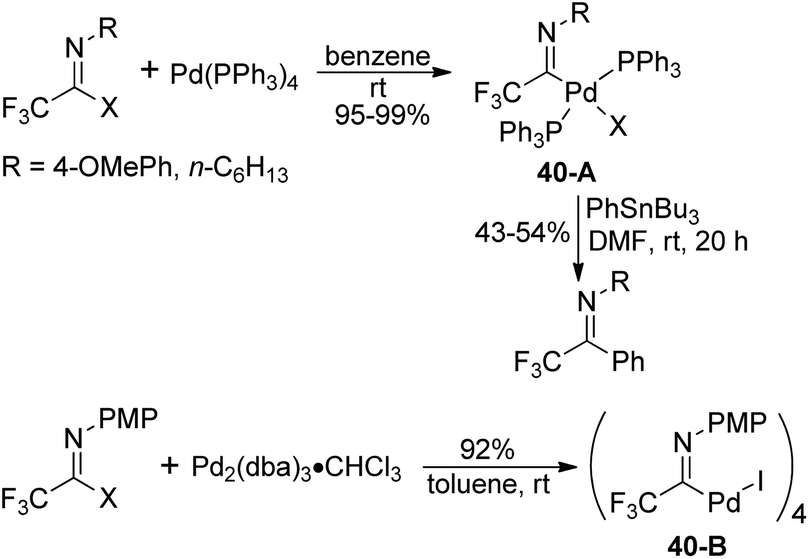 | ||
| Scheme 40 Synthesis of trifluoroacetimidoyl palladium(II) complexes. | ||
The amino(trifluoromethyl)-carbene-palladium complex 41-B was formed by the treatment of imidoyl phosphine complex 41-A with trimethyloxonium tetrafluoroborate (Meerwein's reagent) (Scheme 41). The structure of complex 41-B was verified by the control experiments, which treated 41-B with an O2 atmosphere or an excess amount of elemental sulfur (S8) under heating conditions. The production of N-methyl-N-hexyl amide or N-methyl-N-hexyl thioamide indicated that an α-(dialkylamino)-α-trifluoromethylcarbene moiety was involved in the palladium complex 41-B. In addition, the reaction of imidoyl palladium(II) complex 41-A with methyl thioglycolate could provide the N,S-acetal of fluoral 41-Cvia insertion of carbene ligands into sulfur–hydrogen σ bonds, which underwent intramolecular amide formation to release thiazolinone 41-D.
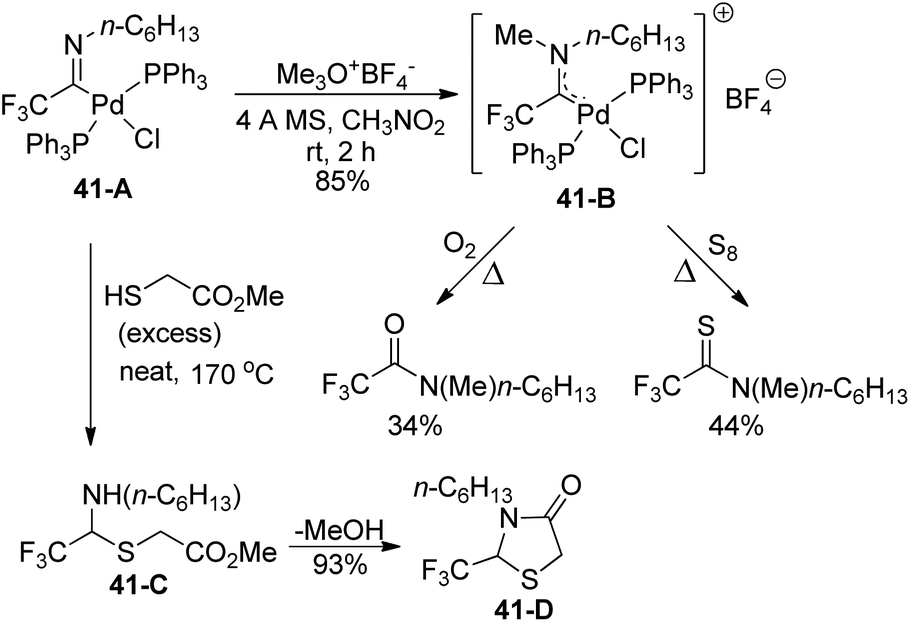 | ||
| Scheme 41 Application of trifluoroacetimidoyl palladium(II) complexes. | ||
The cyclopalladated benzoselenophenes were readily constructed from the intramolecular cyclization of selenoanisole-substituted propargyl imines in the presence of stoichiometric amounts of dichorobis(-triphenylphosphine)palladium(II) in dry THF (Scheme 42).60 The reactivity of cyclopalladated benzoselenophene complex was further explored via the insertion of different alkynes into the Pd–C bond of dimeric cyclopalladated benzoselenophenes to build benzoseleno[2,3-c]-pyridinones. The results demonstrated that the cyclopalladated heterocycles could be utilized to assemble various fused heterocycles through insertion reactions.
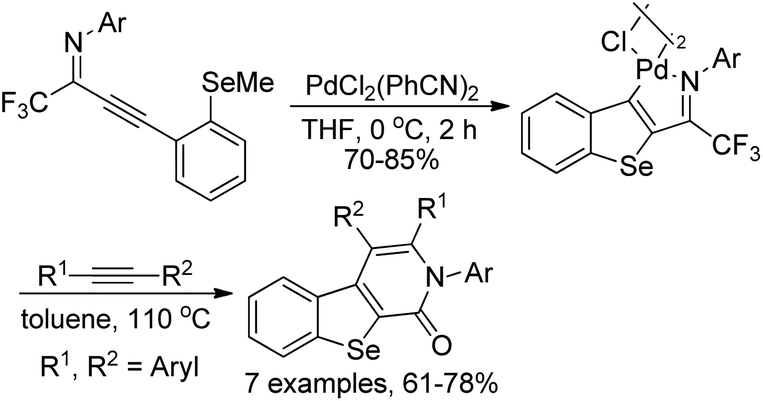 | ||
| Scheme 42 Synthesis of cyclopalladated benzoselenophene. | ||
A library of titanium complexes bearing two regioisomeric trifluoromethyl-containing enaminoketonato ligands were synthesized and characterized by Li and co-workers (Scheme 43).61 The treatment of trifluoroacetimidoyl chloride with heptanone in the presence of LDA could furnish the corresponding β-enaminoketone 43-A, which was transformed into lithium salt of β-enaminoketone 43-B to react with TiCl4 to deliver the titanium complex 43-C. Other β-enaminoketones were obtained by the Claisen condensation between methyl ketones and ethyl trifluoroacetate and the subsequent condensation of the β-diketone with aniline under acidic conditions. The present titanium complexes were active towards ethylene polymerization with modified methylaluminoxane as a cocatalyst and the substituents attached on the ligands exerted a significant influence on the catalytic activities and the behavior of ethylene polymerization.
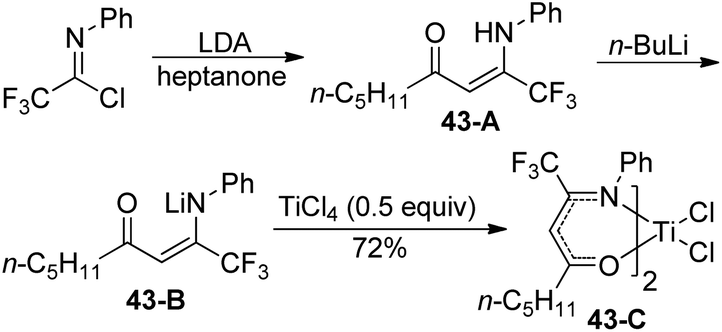 | ||
| Scheme 43 Synthesis of the titanium complex bearing trifluoromethyl-containing enaminoketonato ligands. | ||
A monomeric platinum(IV) complex, recognized as highly stable octahedral bis-ligated platinacycle, was formed by the cycloplatination of ortho thio(seleno)anisole-substituted perfluoroalkyl propargyl imines with PtCl2 (Scheme 44).62 The perfluoroalkyl propargyl imines were prepared by palladium-catalyzed Sonogashira coupling of perfluoroalkyl imidoyl iodides and alkynes. The obtained platinum(IV) complexes were fully characterized by 1H/13C NMR, IR and CHN analysis. The emission spectra of platinacycles were also tested. The reactivity of Pt(IV)-complexes was further investigated via the reaction with diphenyl acetylene or excess triphenyl phosphine, which produced benzo[4,5]-thieno-[2,3-c]-pyridine skeletons or monoligated Pt(II) complexes in quantitative yields.
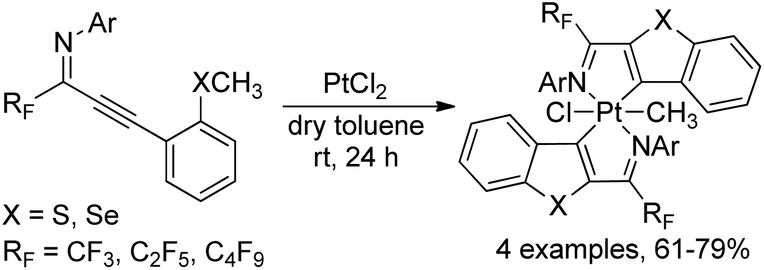 | ||
| Scheme 44 Synthesis of the titanium complex bearing trifluoromethyl-containing enaminoketonato ligands. | ||
3.7. Reduction of fluorinated imidoyl chlorides
Uneyama and co-workers reported a simple pathway for the preparation of polyfluoroalkylated aldimines through the reduction of polyfluoroalkyl imidoyl chlorides by the use of LTBA (lithium tri-tert-butoxyaluminum hydride) as a reducing agent (Scheme 45).63 Other different reducing agents, such as NaBH4, LiAlH4 or DIBAL-H, showed poor reactivity. A variety of polyfluoroalkylated imidoyl chlorides were smoothly compatible with the transformation to furnish the corresponding aldimine products, irrespective of the length of fluorinated alkyl chains.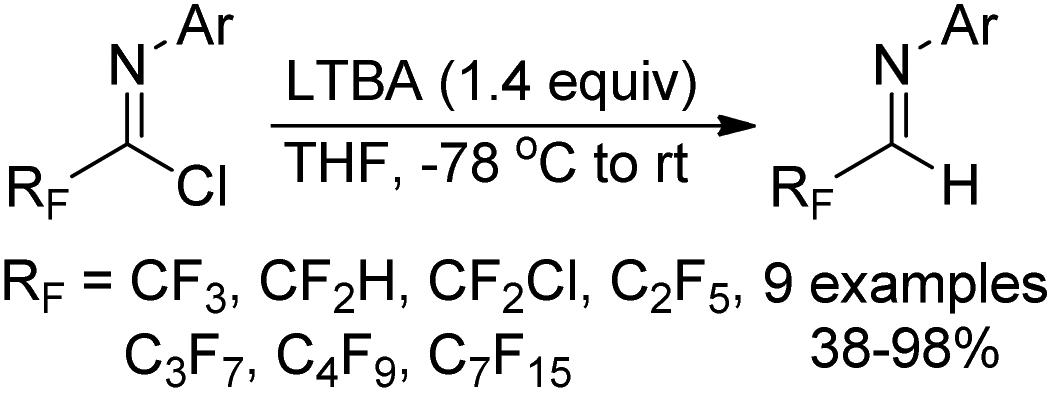 | ||
| Scheme 45 Synthesis of polyfluoroalkylated aldimines. | ||
4. Annulation reaction of trifluoroacetimidoyl halides
The trifluoroacetimidoyl halides have been known as versatile synthons to react with different substrates for the assembly of structurally diverse trifluoromethyl-substituted N-heterocycles.4.1. Synthesis of fluoroalkyl 5-membered N-heterocycles
The formal [1 + 4] cyclization reaction of N-aryl trifluoroacetimidoyl iodides with phenyl-hydrazones to access 5-trifluoromethyl pyrazoles was achieved by Huang and co-workers (Scheme 46).64 The reaction proceeded well with excess sodium hydride in anhydrous DMF at room temperature and the trifluoroacetimidoyl iodide was regarded as a CF3 carbon building block. With respect to perfluoroalkyl imidoyl iodides, the corresponding fluoroalkyl pyrazoles could not be provided under the standard conditions.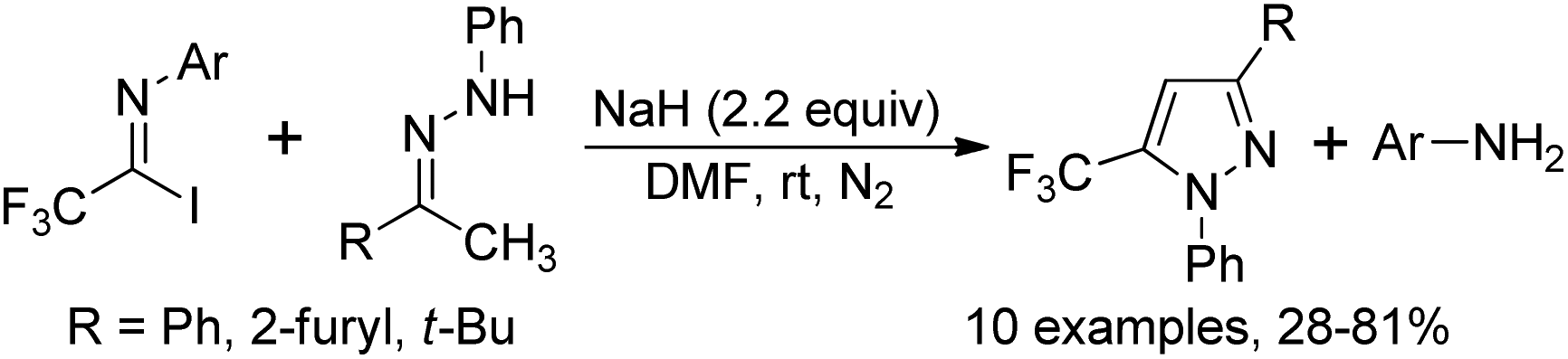 | ||
| Scheme 46 Synthesis of 5-trifluoromethyl pyrazoles. | ||
Yuan and co-workers developed a convenient method for the synthesis of 5-trifluoromethyl tetrazoles from the trifluoroacetimidoyl chlorides and NaN3 without any catalyst or additive (Scheme 47).65 The azide intermediate 47-A was formed by the nucleophilic replacement of trifluoroacetimidoyl chlorides with azide anion, and the subsequent intramolecular cyclization yielded the tetrazole products. The success of the reaction requires two factors: one is the cis configuration of the lone pair of the imino nitrogen and azido group and another is the sufficiently rich electron density on imino nitrogen.
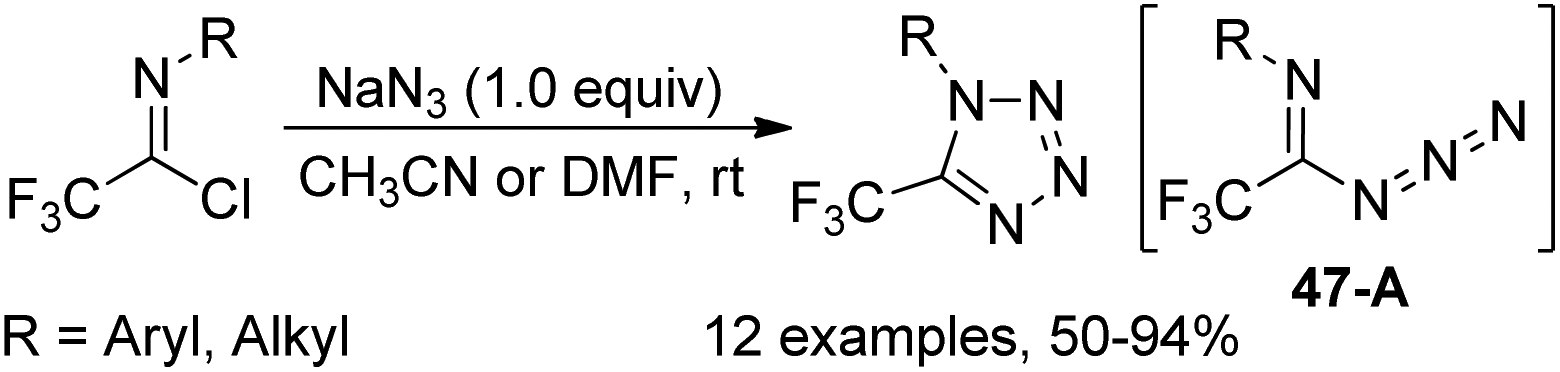 | ||
| Scheme 47 Synthesis of 5-trifluoromethyl tetrazoles. | ||
An efficient and green strategy for the preparation of bioactive 5-trifluoromethyl tetrazoles via microwave-assisted 1,3-dipolar cycloaddition reaction of trifluoroacetimidoyl chlorides and NaN3 was demonstrated by Darehkordi and co-workers (Scheme 48).66 Compared with the common reaction at room temperature, the microwave-assisted protocol provided a remarkable improvement in yields and the tetrazole products were easily purified by simply washing with n-hexane.
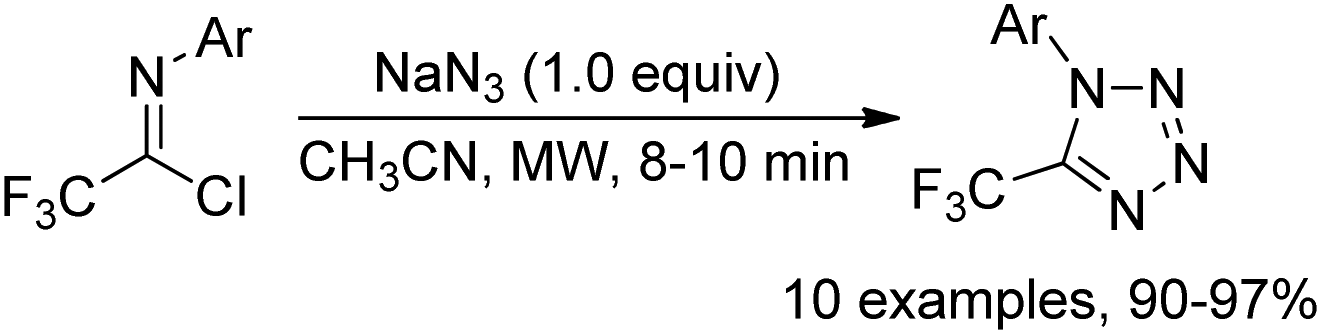 | ||
| Scheme 48 Synthesis of 5-trifluoromethyl tetrazoles. | ||
Wu and co-workers presented a gold(I)-catalyzed synthesis of 2-fluoroalkyl imidazoles from a 5-exo-dig cyclization of fluorinated propargyl amidines, which were obtained by the coupling of fluorinated imidoyl chlorides with propargyl amines (Scheme 49).67 The transformation proceeded smoothly with regard to a series of fluorinated imidoyl chlorides. The proposed mechanism is outlined in Scheme 49. The nucleophile attack of amidinonyl nitrogen to the C–C triple bonds activated by cationic Au(I) led to intermediate 49-A, which isomerized to the more stable imidazole 49-B by an H-1,3-shift. The photolysis of 49-B could give rise to the imidazole products 49-C. Noteworthy was that when 3-phenyl propargyl amine was employed as a starting reagent, a 6-endo-dig cyclization occurred to provide 1,4-dihydropyrimidine product in 73% yield.
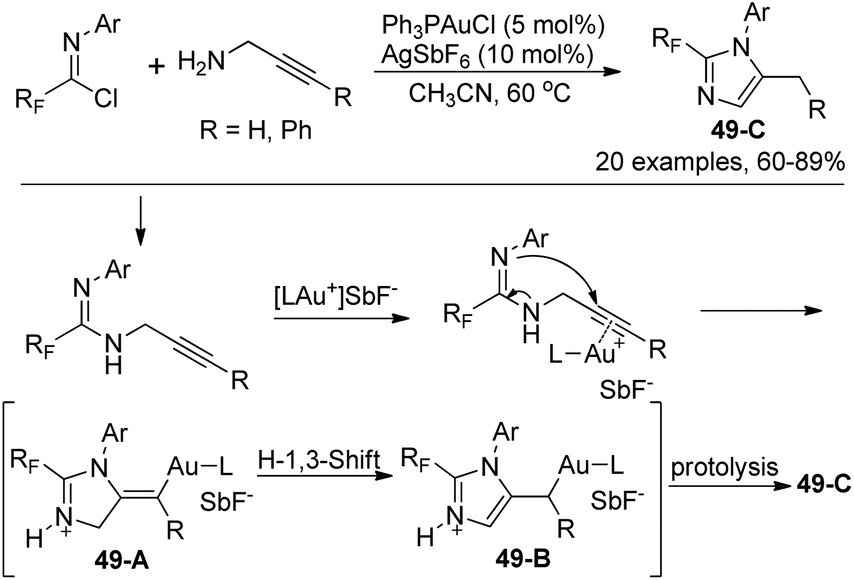 | ||
| Scheme 49 Synthesis of 2-fluoroalkyl imidazoles. | ||
When the fluorinated propargyl amidine was treated with electrophile NIS in the presence of Ph3PAuCl/AgBF4, imidazole-5-carbaldehyde was produced (Scheme 50). The transformation tolerated various functional groups and only terminal alkynes were suitable for the reaction. With 3-phenyl propargyl amine as a substrate, 5-iodo-1,4-dihydropyrimidine was delivered in moderated yields. Mechanistic study revealed that the carbonyl oxygen was derived from O2. Activated propargyl amidine reacted with NIS affords 5-iodomethyl imidazole 50-B, which underwent the dissociation of the C–I bond to give the radical intermediate 50-C and an iodine radical. Subsequently, the peroxy-intermediate 50-D was formed in the presence of O2. The final aldehyde product was provided by the removal of a hydroxyl radical from 50-D.
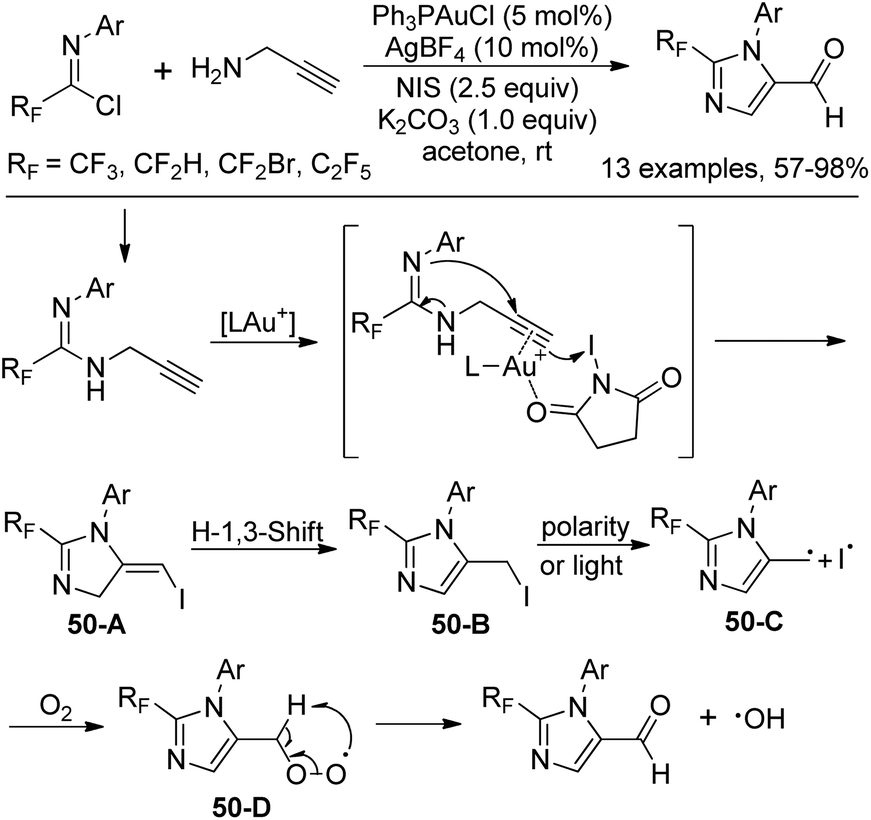 | ||
| Scheme 50 Synthesis of 2-fluoroalkyl imidazole-5-carbaldehyde. | ||
A three-component reaction of trifluoromethylimidoyl chlorides with isocyanides and dimethyl or diethyl acetylene dicarboxylate without any catalyst for the construction of trifluoromethylated pyrroles was reported by Darehkordi and co-workers (Scheme 51).68 The reaction was initiated by the nucleophilic addition to DMAD (dimethyl acetylene dicarboxylate) to generate zwitterion 51-A, which underwent nucleophilic addition to the imidoyl carbon to give the intermediate 51-B. Intramolecular cyclization of 51-B delivered 51-C, followed by the elimination of chlorine and protonation to yield the pyrrole product.
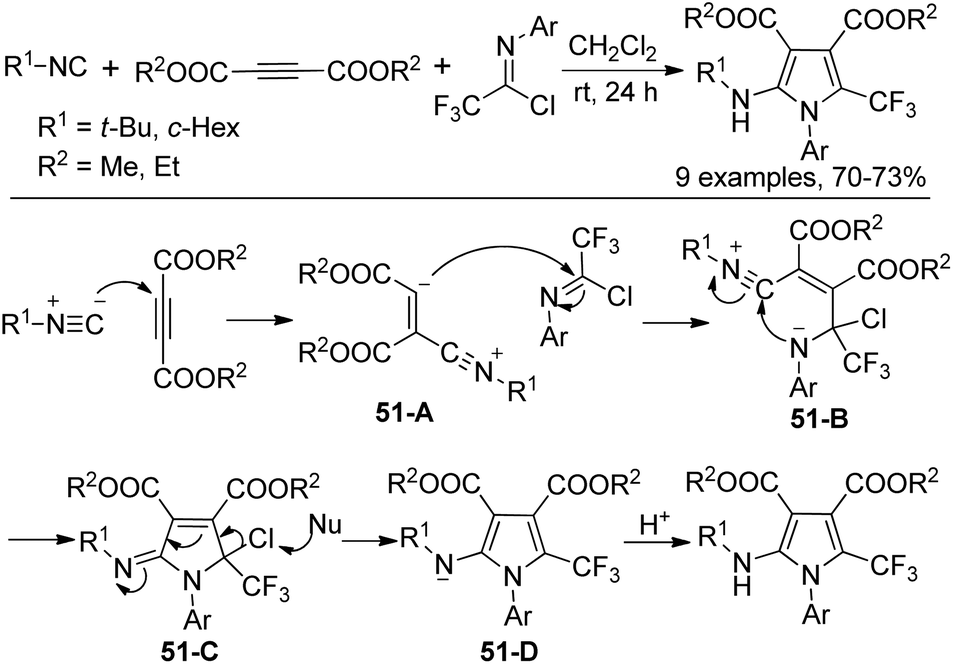 | ||
| Scheme 51 Synthesis of trifluoromethylated pyrroles. | ||
Darehkordi and co-workers disclosed a TiO2-nanoparticle-catalyzed synthesis of trifluoromethyl-4,5-dihydro-1,2,4-oxadiazoles and trifluoromethyl-1,2,4-oxadiazoles from the reaction of trifluoroacetimidoyl chlorides and amidoximes (Scheme 52).69 Changing the reaction condition regarding solvent and base, different trifluoromethyl-substituted N-heterocycle products were obtained. The reaction was considered to be initiated by the activation of imidoyl chlorides by TiO2-NPs to result in a TiO2-imidoyl complex, which was attacked by the nucleophilic oxygen atom of amidoxime. The following intramolecular attack of the –NH anion on the imino group afforded 5-(trifluoromethyl)-4,5-dihydro-1,2,4-oxadiazol-5-amine 52-A. The elimination of aryl amine from 52-A led to trifluoromethyl-1,2,4-oxadiazole 52-B.
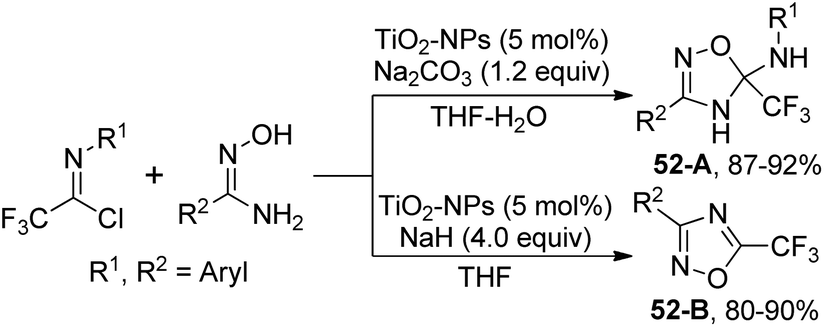 | ||
| Scheme 52 Synthesis of trifluoromethyl-4,5-dihydro-1,2,4-oxadiazoles and trifluoromethyl-1,2,4-oxadiazoles. | ||
Our group developed an efficient approach for the assembly of 5-trifluoromethyl-1,2,4-triazoles through iodine-mediated annulation of trifluoroacetimidoyl chlorides and hydrazones under mild reaction conditions (Scheme 53).70 The scope of the protocol was broad, as illustrated by more than 40 successful examples with moderate to high efficiency, including other fluorinated imidoyl chlorides. The reaction proceeded in a consecutive one-pot manner, which presumably involved base-promoted intermolecular C–N bond formation via coupling of trifluoroacetimidoyl chlorides and hydrazones to give the trifluoroacetamidine derivative 53-A. Then, the base-promoted oxidative iodination of 53-A furnished an iodo species 53-B and subsequent intramolecular nucleophilic attacks of nitrogen on the iodo-substituted carbon atom afforded the intermediate 53-C. The final base-promoted deprotonation and rearomatization of 53-C led to the desired 1,2,4-triazole product.
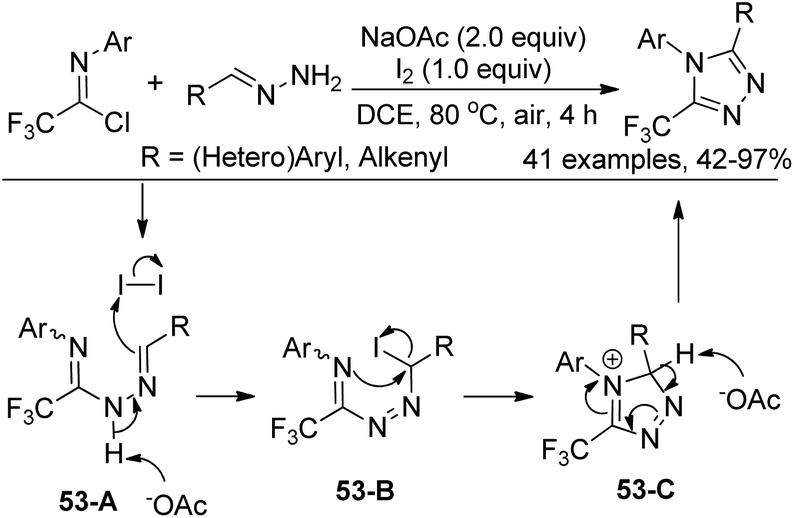 | ||
| Scheme 53 Synthesis of 5-trifluoromethyl-1,2,4-triazoles. | ||
4.2. Synthesis of 2-fluoroalkyl benzimidazoles
An approach to produce N-substituted 2-(trifluoromethyl)benzimidazoles via electrochemical oxidation of N,N’-disubstituted trifluoroethanimidamides was disclosed by Uneyama and co-workers (Scheme 54).71 The benzimidazoles 54-A could be smoothly formed under electrolysis conditions in dry acetonitrile with respect to N,N’-diaryl derivatives, whereas complex mixtures of trifluoroethanimidamides, p-benzoquinone imine derivatives 54-B and polymeric compounds were observed with regard to N-alkyl-N’-(4-methoxyphenyl) derivatives. When the reaction was conducted in wet acetonitrile, p-benzoquinone imine derivative 54-B was the major product. A two-electron-oxidation process was considered to be involved in the reaction.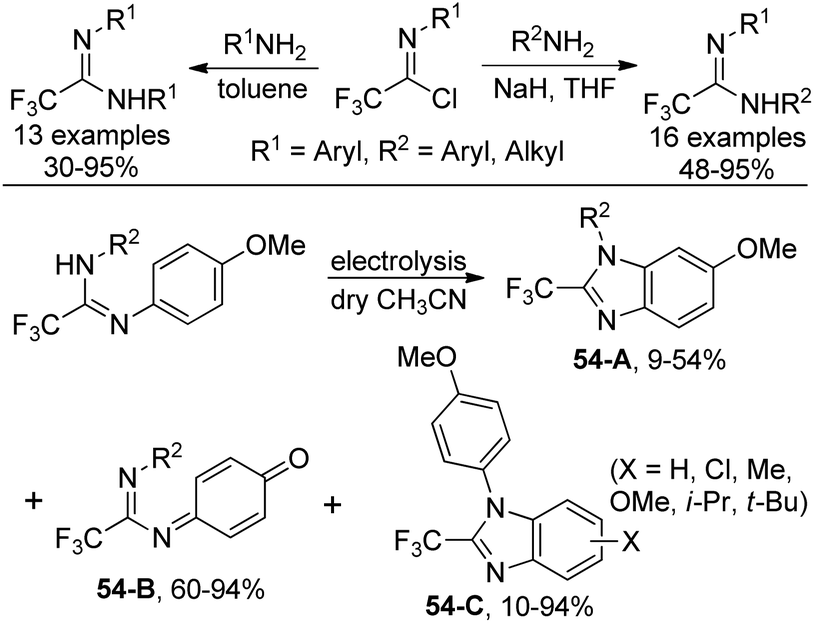 | ||
| Scheme 54 Synthesis of N-substituted 2-(trifluoromethyl)benzimidazoles. | ||
Based on the previously reported synthesis of 2-(trifluoromethyl)benzimidazoles, Uneyama and co-workers further investigated the conversion of p-benzoquinone imines into trifluoromethylated 6-hydroxybenzimidazoles (Scheme 55).72 The p-benzoquinone imines 55-A were obtained by electrochemical oxidation of 2,2,2-trifluoroethanimidamides. Lewis acid-catalyzed intramolecular cyclization of 55-A gave rise to the desired trifluoromethylated 6-hydroxybenzimidazole products, and BF3·OEt2 was the best choice. The scope of the reaction was relatively broad and different substituted p-benzoquinone imines were applicable to afford the desired products in reasonable yields.
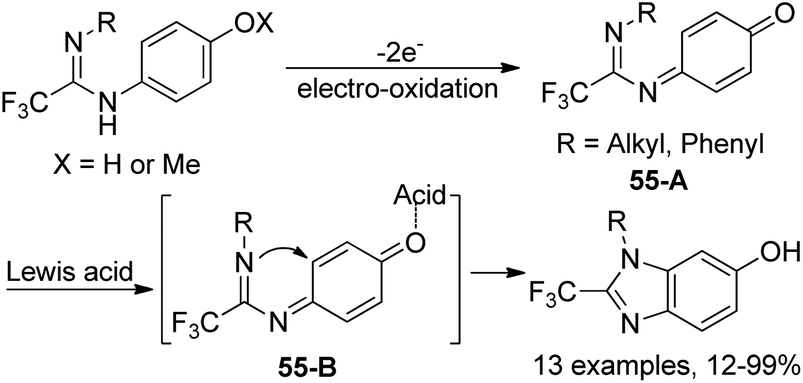 | ||
| Scheme 55 Synthesis of trifluoromethylated 6-hydroxybenzimidazoles. | ||
One year later, a more detailed investigation on the electrochemical and the oxidant-promoted reactions of perfluoroalkyl/aryl imidamides for the preparation of 2-(perfluoroalkyl/aryl)benzimidazoles was presented by the same group.73 The protocol provided a deeper understanding of the transformation of imidamides into benzimidazoles.
Wu and co-workers developed a copper(I)-catalyzed tandem reaction for the synthesis of 2-fluoroalkylbenzimidazoles starting from N-aryl trifluoroacetimidoyl (or bromodifluoroacetimidoyl) chlorides and primary amines (Scheme 56).74 In the transformation, amidines could be easily formed via the coupling of imidoyl chlorides and amines. The subsequent intramolecular C–N bond formation was enabled in the presence of copper catalyst to deliver the 2-fluoroalkylbenzimidazole products. The employment of K2CO3 or K3PO4 was essential for the success of the reaction and ligands were unnecessary for improving the yield.
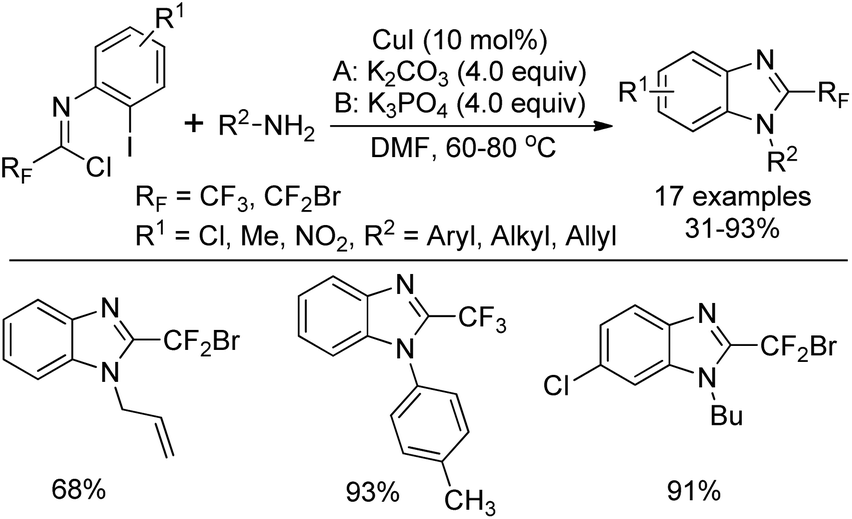 | ||
| Scheme 56 Synthesis of 2-fluoroalkylbenzimidazoles. | ||
In the meantime, Zhang and co-workers reported a similar reaction for the construction of 2-(trifluoromethyl)benzimidazoles by Cu(I)/TMEDA-catalyzed cross coupling reaction of N-(2-haloaryl)trifluoroacetimidoyl chlorides with primary amines (Scheme 57).75 Different from the Wu's reaction, the protocol required suitable ligand to promote the reaction and the Cl, Br, or I substituent at the 2-position of the phenyl group of imidoyl chlorides were all tolerated under the reaction conditions, albeit the slightly inferior reactivity of the aryl bromides and aryl chlorides.
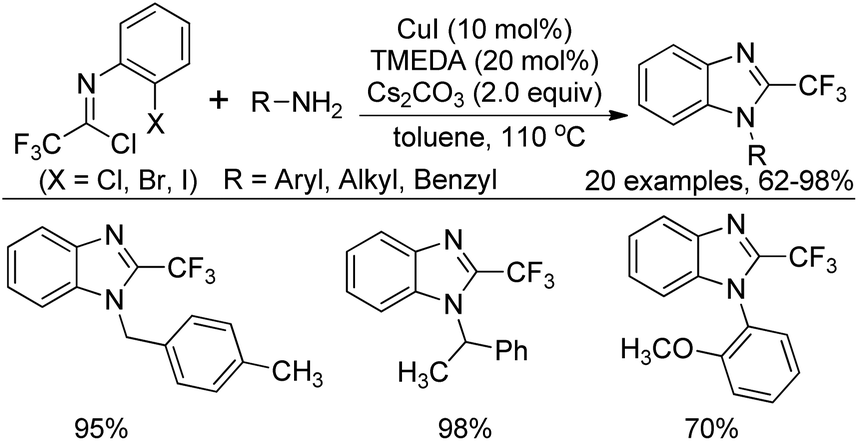 | ||
| Scheme 57 Synthesis of 2-(trifluoromethyl)benzimidazoles. | ||
A practical route to the synthesis of 2-fluoroalkylbenzimidazoles via PIDA-mediated oxidative intramolecular cyclization of the fluorinated amidines was presented by Wu and co-workers (Scheme 58).76 The fluorinated amidines were generated from the coupling of fluorinated imidoyl chlorides with gas of ammonia at room temperature. Compared with the previous reported methods, the current protocol avoided the use of ortho-aminoanilines and ortho-haloanilines as the starting materials.
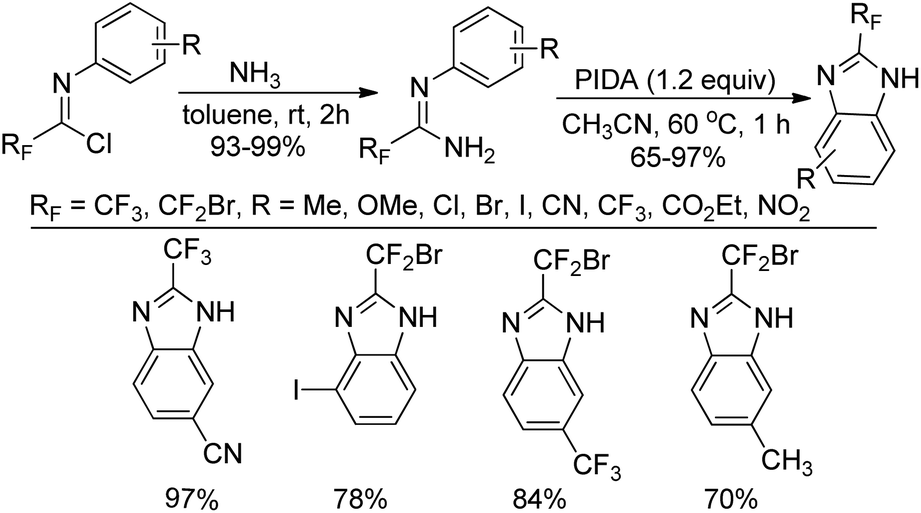 | ||
| Scheme 58 Synthesis of 2-fluoroalkylbenzimidazoles by PIDA oxidation of amidines. | ||
4.3. Synthesis of 2-fluoroalkyl benzothiazoles
A copper-catalyzed thiolation annulation reaction of 1,4-dihalides with NaHS for the synthesis of biologically active 2-trifluoromethyl benzothiazoles was disclosed by Zhang and co-workers (Scheme 59).77 In this Ullmann reaction, the Cu(I)-catalyzed formation of double carbon–sulfur bonds was realized. Less active N-(2-chlorophenyl)-trifluoroacetimidoyl chlorides were also applicable to the reaction with unsatisfactory reactivity. Notably, the 2-trifluoromethyl benzothiophenes could be afforded through the Cu(I)-catalyzed thiolation annulation of 2-chloro-1-(2-haloaryl)-3,3,3-trifluoropropylenes with Na2S.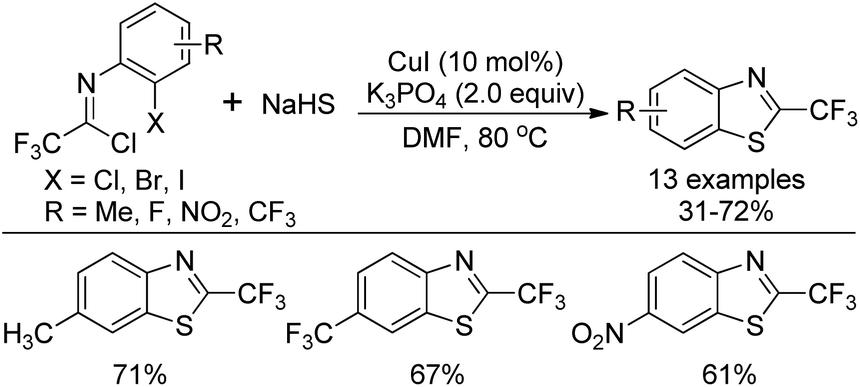 | ||
| Scheme 59 Synthesis of 2-trifluoromethyl benzothiazoles. | ||
An improved method for the synthesis of 2-trifluoromethylbenzothiazoles was described by Wu and co-workers (Scheme 60).78 The reaction proceeded with common trifluoroacetimidoyl chlorides and sodium hydrosulfide hydrate by using PdCl2 as catalyst. A thiolation/C–H bond functionalization/C–S bond formation sequence was involved in the transformation. It was envisaged that 2,2,2-trifluoro-N-phenylethanethioamide was the intermediate for the reaction, which could be in situ generated from the coupling reaction of trifluoroacetimidoyl chloride with sodium hydrosulfide hydrate.
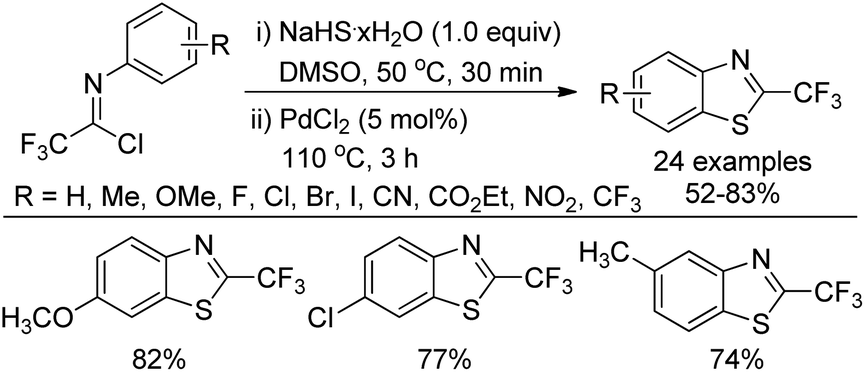 | ||
| Scheme 60 Synthesis of 2-trifluoromethyl benzothiazoles. | ||
4.4. Synthesis of 2-fluoroalkyl indoles
Uneyama and co-workers developed an electrochemical oxidation of 2-substituted 3-(N-arylamino)-4,4,4-trifluoro-2-butenoate for the production of 2-(trifluoromethyl)-indoles (Scheme 61).79 The butanoate derivatives 61-A could be readily prepared by the nucleophilic reaction of active methylene compounds with trifluoroacetimidoyl chlorides. The 61-A underwent a sequential reaction of single-electron-oxidation, deprotonation, a further single-electron-oxidation, addition of water and demethoxylation process to afford the p-benzoquinone imines 61-B. With respect to different substitutions attaching on 61-B, different reaction conditions were implemented to convert p-benzoquinone imines 61-B into 2-(trifluoromethyl)-3H-indoles.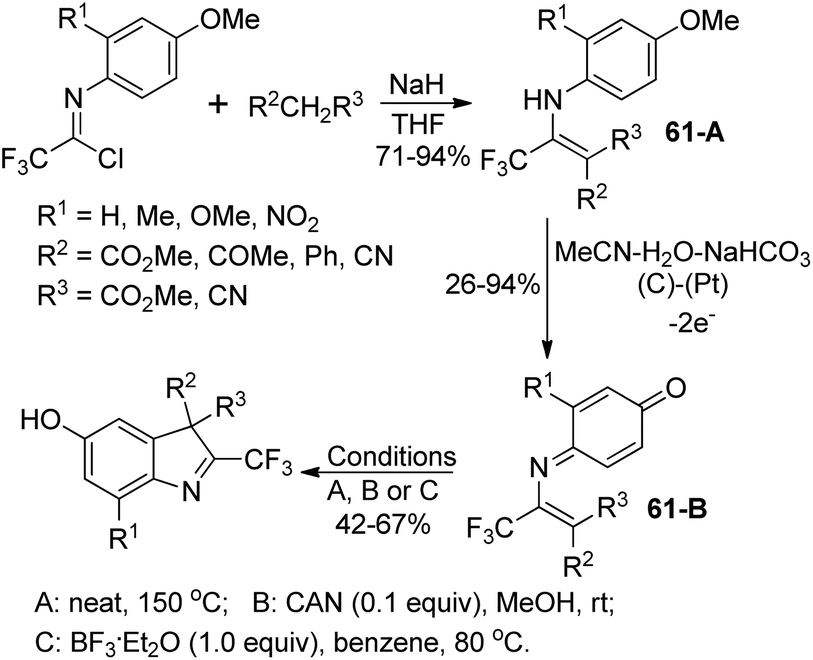 | ||
| Scheme 61 Synthesis of 2-(trifluoromethyl)-3H-indoles. | ||
Hao and co-workers established a facile strategy for the assembly of biologically important 2-fluoroalkyl-substituted indoles via the Grignard cyclization reaction of N-[2-(bromoalkyl)phenyl]imidoyl chlorides (Scheme 62).80 The α-bromination at the benzylic position of N-[2-(alkyl)phenyl]imidoyl chlorides 62-A with N-bromosuccinimide (NBS)/benzoyl peroxide (BPO) yielded 62-B. The Grignard cyclization reaction proceeded well in the presence of magnesium and was greatly influenced by the electronic effect of the substituent group on the benzene ring. The synthesized 2-fluoroalkyl indoles could undergo further derivatization to construct more complicated fluorine-containing indoles, which may constitute the precursor of the valuable biologically active molecules.
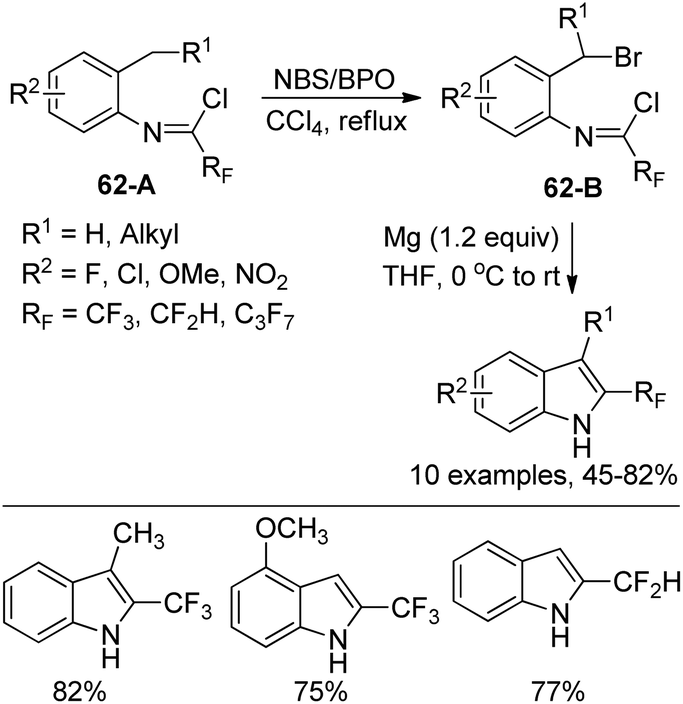 | ||
| Scheme 62 Synthesis of 2-fluoroalkyl indoles from N-[2-(bromoalkyl)phenyl]imidoyl chlorides. | ||
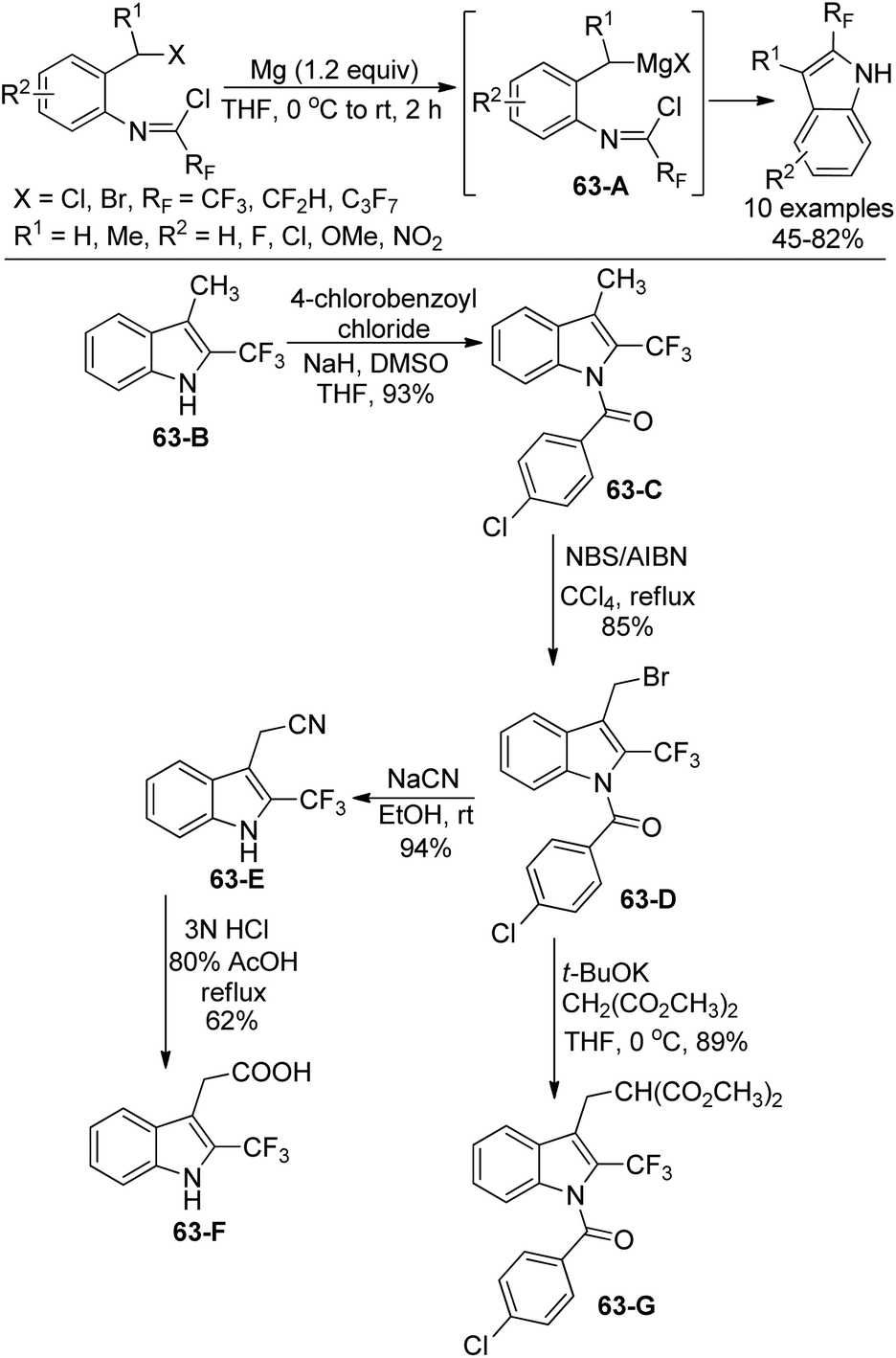
The same group further investigated the Grignard cyclization reaction to synthesize 2-fluoroalkyl-substituted indole derivatives and extended their application in the synthesis of biologically interested indoles (Scheme 63).81 The Grignard reagents were in situ generated and transformed into 2-fluoroalkyl indole products. N-[2-(Chloroalkyl)phenyl]imidoyl chlorides were also suitable for the reaction. The application of 3-methyl-2-trifluoromethyl indole was implemented. The protection of nitrogen groups in 63-B with 4-chlorobenzoyl chloride gave 63-C, which underwent bromination at 3-methyl group with NBS/AIBN to deliver 63-D. The nucleophilic substitution of 63-D with NaCN enabled the formation of N-deprotected product 63-E, followed by the subsequent hydrolysis of the cyanide group to yield 2-trifluoromethylated heteroauxin 63-F. The nucleophilic substitution of 63-D with malonate carboanion resulted in the formation of 63-G in good yields.
 | ||
| Scheme 63 Synthesis of 2-fluoroalkyl indoles and their application. | ||
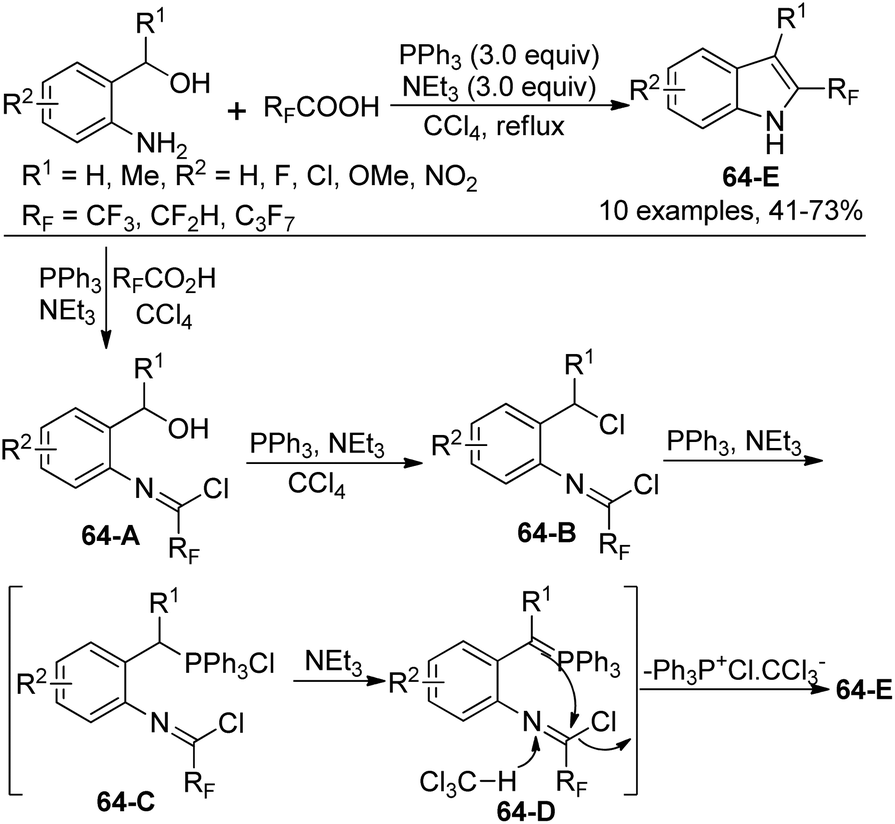
A simple approach for the formation of 2-fluoroalkyl indoles from 2-aminobenzyl alcohols with fluoroalkanoic acids in the presence of Ph3P, CCl4, and NEt3 was developed by Wang and co-workers (Scheme 64).82 It was found that N-[2-(hydroxymethyl)phenyl]-2,2,2-trifluoroacetimidoyl chloride 64-A was formed as an intermediate in the reaction. [Ph3P+Cl] CCl3−, in situ generated from the reaction of Ph3P with a large excess amount of CCl4, could enable the transformation of a hydroxyl group into a chlorine atom, thereby giving the compound 64-B. The reaction of 64-B with Ph3P formed a phosphonium salt 64-C, which was converted into active and unstable ylide 64-D. The final intramolecular cyclization of 64-D could lead to the indole product.
 | ||
| Scheme 64 Synthesis of 2-fluoroalkyl indoles and the plausible mechanism. | ||
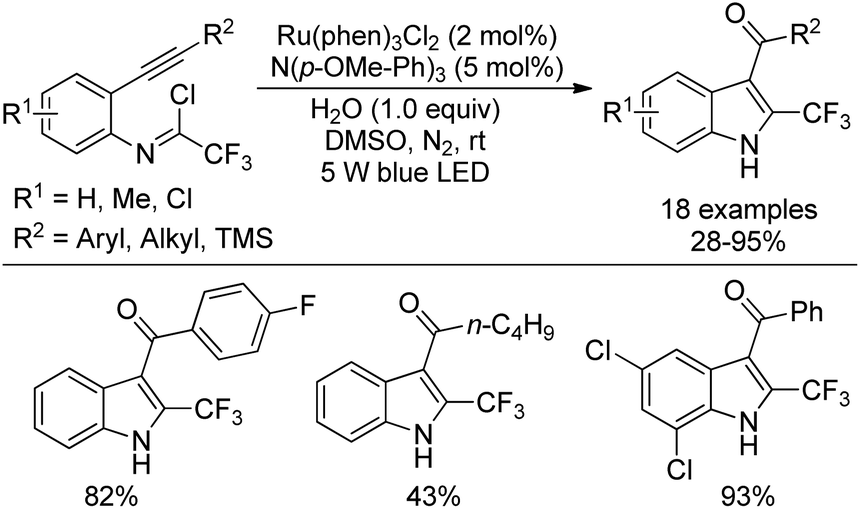
A visible-light-induced intramolecular radical cyclization of N-[2-(alkynyl)phenyl]trifluoroacetimidoyl chlorides for the assembly of 2-trifluoromethyl indoles was established by Zhou and co-workers (Scheme 65).83 In this transformation, imidoyl radical was generated through the irradiation with a 5 W blue LED in the presence of a visible-light photoredox catalyst, and a sequential C–C and C–O bond formation was involved. Triarylamine was employed as an effective additive to initiate a radical reaction. A wide range of substituents on the phenyl group attached to alkyne or nitrogen and functional groups were all smoothly tolerated, representing the good compatibility of the reaction.
 | ||
| Scheme 65 Visible-light-induced synthesis of 2-trifluoromethyl indoles. | ||
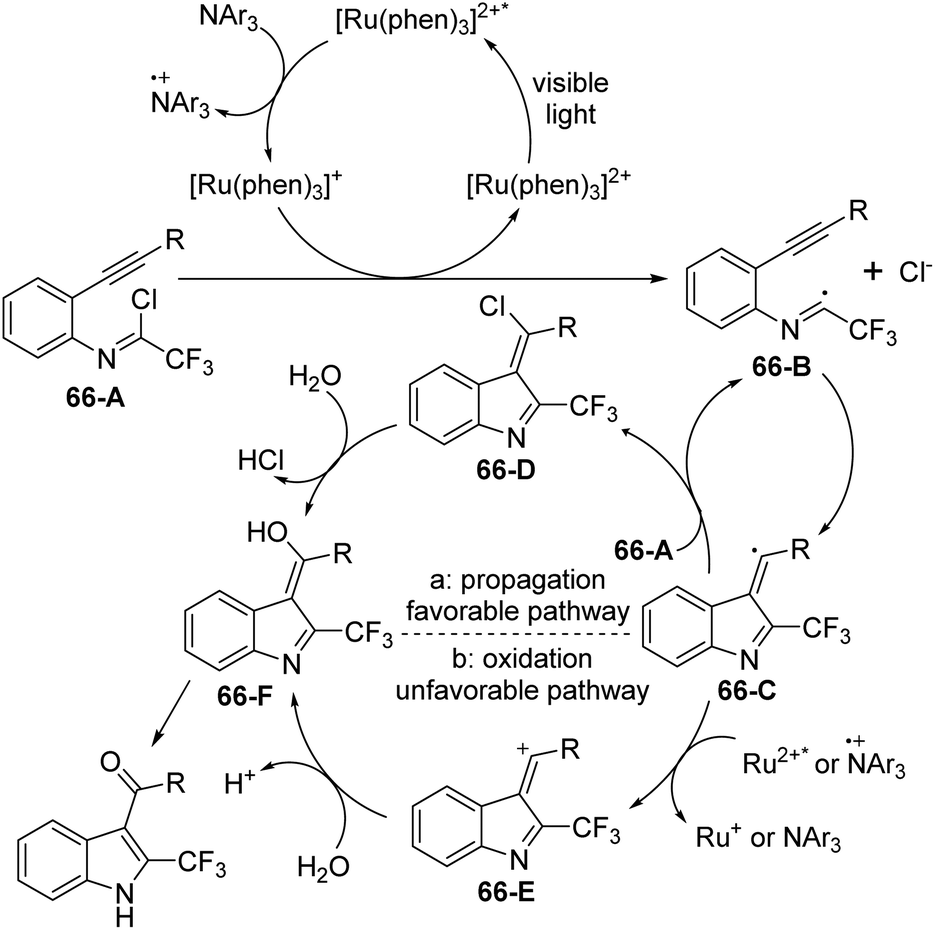
A plausible reaction mechanism was proposed as outlined in Scheme 66. At first, the excited [Ru(phen)32+]* was generated from photoexcitation of Ru(phen)32+, which accepted single-electron transfer from tris(4-anisyl)amine to yield Ru(phen)3+ and NAr3 radical cations. The imidoyl radical 66-B was formed by the reductive cleavage of the C–Cl bond of 66-A with the assistance of Ru(phen)3+. Then, intramolecular radical addition of 66-B to alkyne gave vinyl radical 66-C, which underwent the propagation process to afford vinyl chloride 66-D. The hydrolysis of 66-D resulted in the formation of 66-F and herein isomerized to 2-trifluoromethyl indole product. Another pathway involved the single-electron oxidation of 66-C by Ru(phen)32+ or NAr3 radical cation to deliver vinyl cation 66-E, followed by the hydrolysis and isomerization to give rise to the indole product. The additional mechanistic experiments indicated that the latter pathway was less likely involved in the reaction.
 | ||
| Scheme 66 Plausible reaction mechanism. | ||
A Pd(0)-catalyzed C(sp3)–H functionalization of trifluoroacetimidoyl chlorides for the synthesis of 2-trifluoromethyl indoles was reported by Cramer and co-workers (Scheme 67).84 The trifluoroacetimidoyl chlorides were derived from o-methylanilines, which attached the adjacent methyl group to be activated in the reaction. In the optimization of the conditions, the choice of the ligand and additive was important. PCy3 and S-IPr had a positive effect on the reaction with the addition of the suitable additive. The reaction pathway was proposed. Oxidative addition of the Pd(0) catalyst to the C–Cl bond of imidoyl chloride afforded the complex 67-A, which underwent ligand exchange to give the intermediate 67-B. Subsequently, palladacycle 67-C was formed by the carboxylate-assisted concerted metalation–deprotonation (CMD) process. The reductive elimination of 67-C with the intramolecular C–C bond formation and tautomerization led to the final 2-trifluoromethyl indole product.
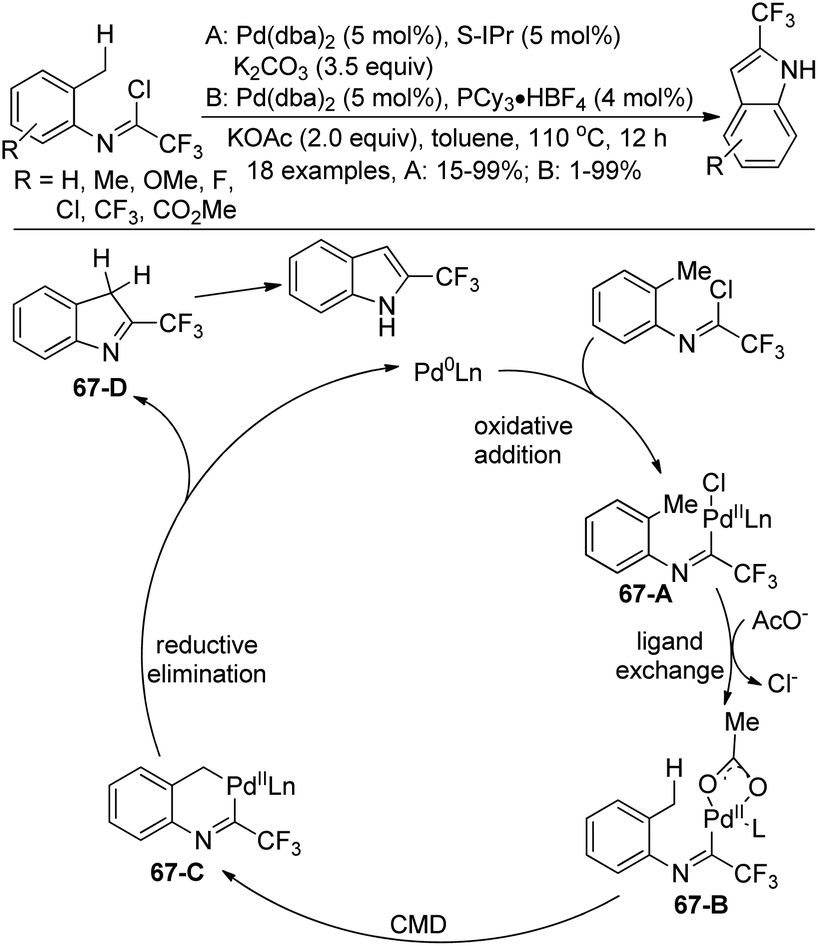 | ||
| Scheme 67 Synthesis of 2-trifluoromethyl indoles Pd(0)-catalyzed C(sp3)–H functionalization. | ||
Dolbier Jr. and co-workers disclosed a synthetic approach for the preparation of 3-arylsulfonyl-2-trifluoromethyl-indoles in two steps from methyl-arylsulfones and trifluoroacetimidoyl chlorides (Scheme 68).85 The coupling reaction of methylsulfones with trifluoroacetimidoyl chlorides in the presence of LDA led to trifluoromethylimino/enamino-β-sulfonylaryl intermediates 68A/68B as a mixture of imine and enamine tautomers. After the optimization of the copper-catalyzed cyclization reaction conditions, 3-arylsulfonyl-2-trifluoromethyl-indoles were obtained in moderate to good yields with Cu(OAc)2·xH2O as the catalyst and Cs2CO3 as the base. A Cu(I)/Cu(III) mechanism was probably involved in the transformation.
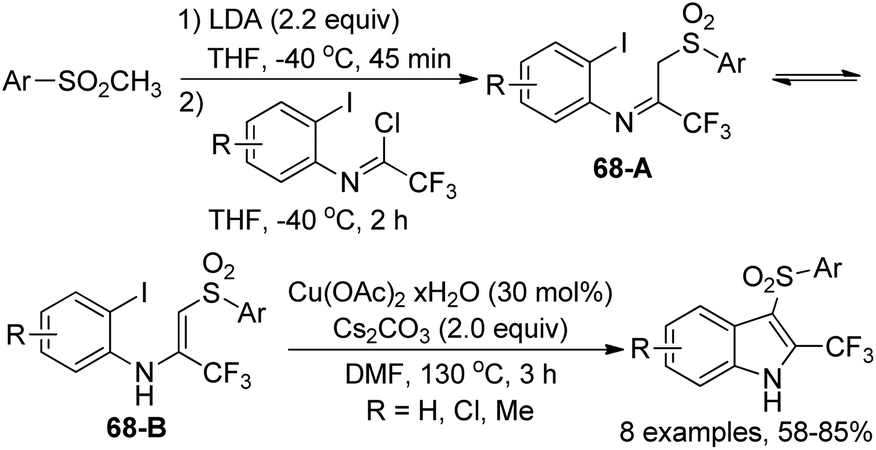 | ||
| Scheme 68 Synthesis of 3-arylsulfonyl-2-trifluoromethyl-indoles. | ||
4.5. Synthesis of 2-fluoroalkyl quinolines
A Rh(I)-catalyzed coupling-cyclization reaction of trifluoroacetimidoyl chlorides with alkynes to construct 2-trifluoromethylated quinolines was presented by Uneyama and co-workers (Scheme 69).86 The phosphine ligand TFP (P(2-furyl)3) or dppe (Ph2P(CH2)2PPh2) was found to be beneficial for the reaction. The electron effect of the reaction was obvious, which illustrated that the imidoyl chlorides possessing an electron-withdrawing group enabled a sluggish yield. With regard to unsymmetrical alkynes, a bulkier group was prone to be attached to the quinoline 4-position and an electron-withdrawing group was attached to the quinoline 3-position.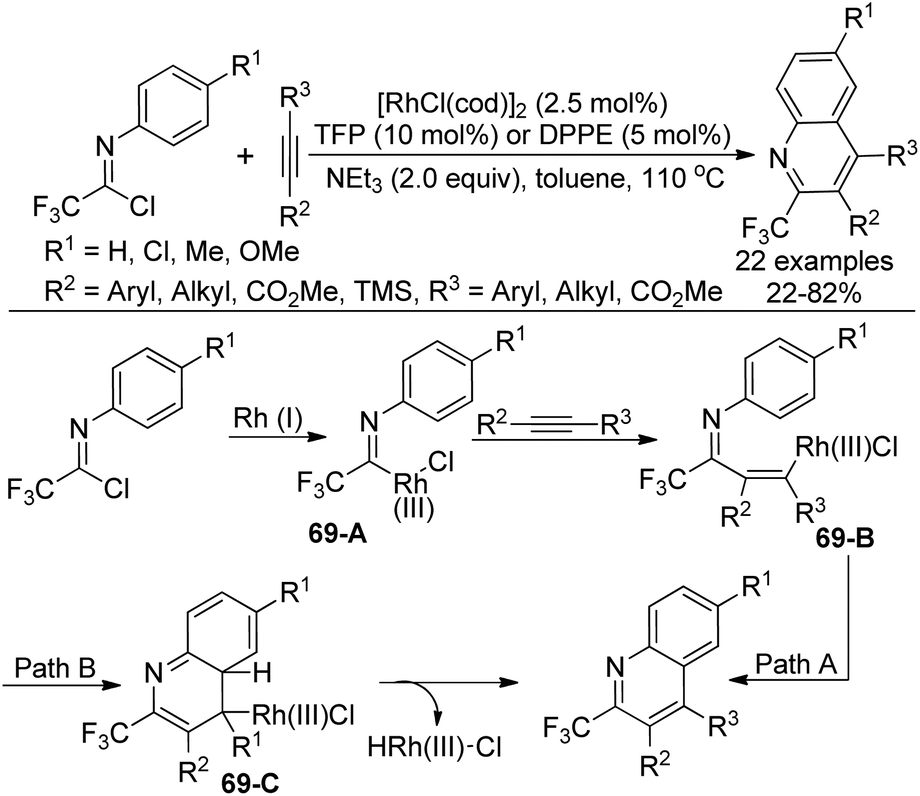 | ||
| Scheme 69 Synthesis of 2-trifluoromethylated quinolines. | ||
The possible mechanism was also depicted in Scheme 69. The oxidative addition of rhodium(I) complex to the imidoyl chloride formed trifluoroacetimidoyl rhodium(III) species 69-A. Then, the alkyne insertion into the Rh–C bond of 69-A delivered the vinylrhodium 69-B, which underwent intramolecular electrophilic substitution to release the quinoline product (Path A). Another pathway involved intramolecular thermal 6π-electrocyclization to generate intermediate 69-C, followed by the subsequent β-hydride elimination to provide the quinoline product (Path B). The experimental results supported the electrophilic substitution pathway.
Yuan and co-workers developed a palladium-catalyzed coupling reaction of trifluoroacetimidoyl iodide with dialkyl vinylphosphonates and a sequential cyclization reaction for the preparation of 2-trifluoromethyl-quinoline derivatives (Scheme 70).87 The transformation proceeded via the Heck coupling reaction to give the corresponding α,β-unsaturated imines in good yields, which were converted into quinoline derivatives with Pd/C in nitrobenzene. The protocol realized the simultaneous introduction of the trifluoromethyl group and the phosphoryl moiety into heterocycle molecules.
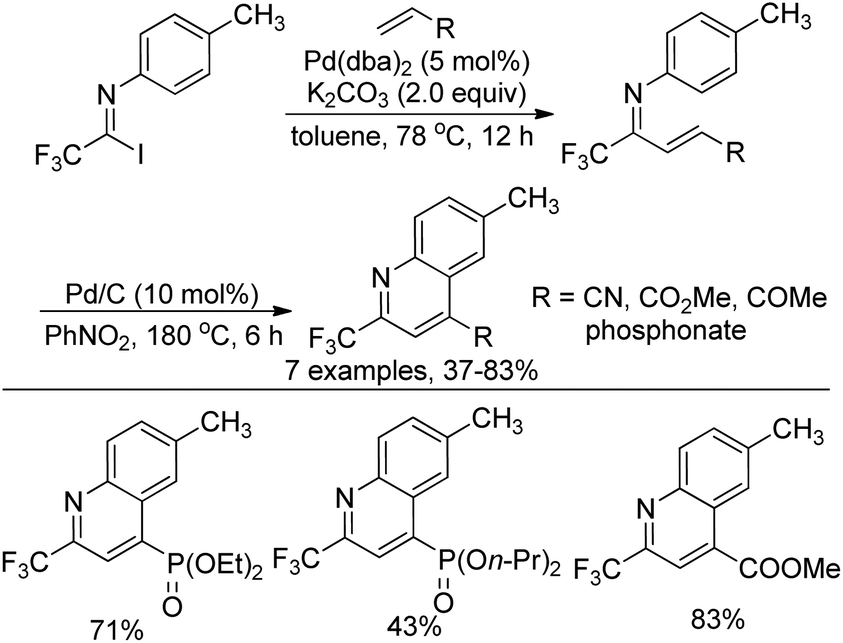 | ||
| Scheme 70 Synthesis of 2-trifluoromethyl-4-(O,O-dialkyl)-phosphoryl-quinoline. | ||
Uneyama, Katagiri and co-workers disclosed a palladium-catalyzed chloroimination of fluorinated imidoyl chlorides to a triple bond to build 4-chloro-2-perfluoroalkyl quinolines in high yields (Scheme 71).88 Noteworthy was that chloroimination of a multiple bond was recognized as a useful tool to construct nitrogen-containing heterocycles. Moreover, an additional chloride source was required to accelerate the reaction and TBAC was the best choice. Besides CF3 group, other fluorinated groups, such as C3F7, CF2H, and CF2Cl, were all tolerated in the reaction.
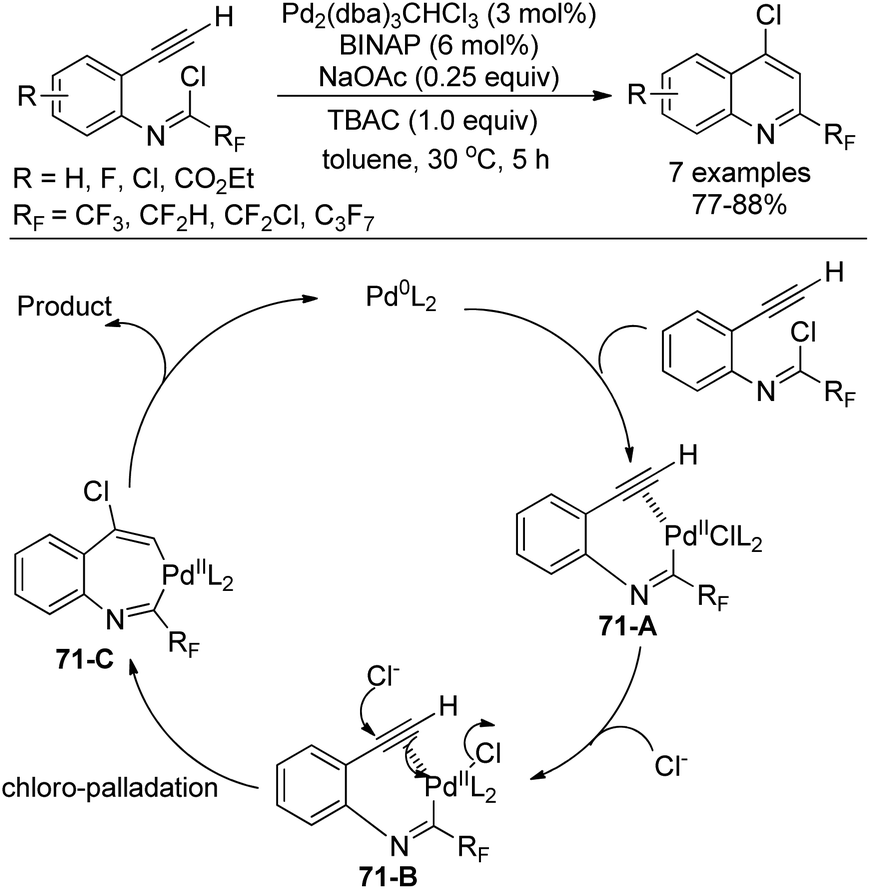 | ||
| Scheme 71 Synthesis of 4-chloro-2-perfluoroalkyl quinolines. | ||
The reaction was initiated by the similar oxidative addition of the Pd(0) species into the C–Cl bond of imidoyl chloride to give the Pd(II) complex 71-A. The triple bond was activated by the Pd(II) catalyst and attacked by an exogenous chloride to yield the seven-membered trans-chloropalladation palladacycle intermediate 71-C. Reductive elimination of 71-C could deliver the desired quinoline product.
A library of highly substituted 2-perfluoroalkyl-3-iodoquinolines were prepared via electrophilic iodocyclization of perfluoroalkyl propargyl imines/amines (Scheme 72).89 In the presence of I2 and CAN, iodocyclization of perfluoroalkyl propargyl imines occurred to provide the corresponding quinoline products in high yields. As for para-substituted methoxy perfluoroalkyl propargyl imines, the azaspiro compound was obtained. In the reaction, iodine cation was generated from the activation of CAN, which attacked the carbon–carbon triple bond of the propargylic imine to result in the formation of the iodonium intermediate 72-A. Next, intramolecular cyclization of the aromatic ring with the activated triple bond led to the desired quinoline product. Perfluoroalkyl propargyl amines could be synthesized via Sonogashira coupling of perfluoroalkyl imidoyl iodides with alkynes followed by reduction with NaBH3CN. The combination of I2 and NaHCO3 or the solely stronger electrophile ICl enabled the transformation of perfluoroalkyl propargyl amines into 2-perfluoroalkyl-3-iodoquinolines.
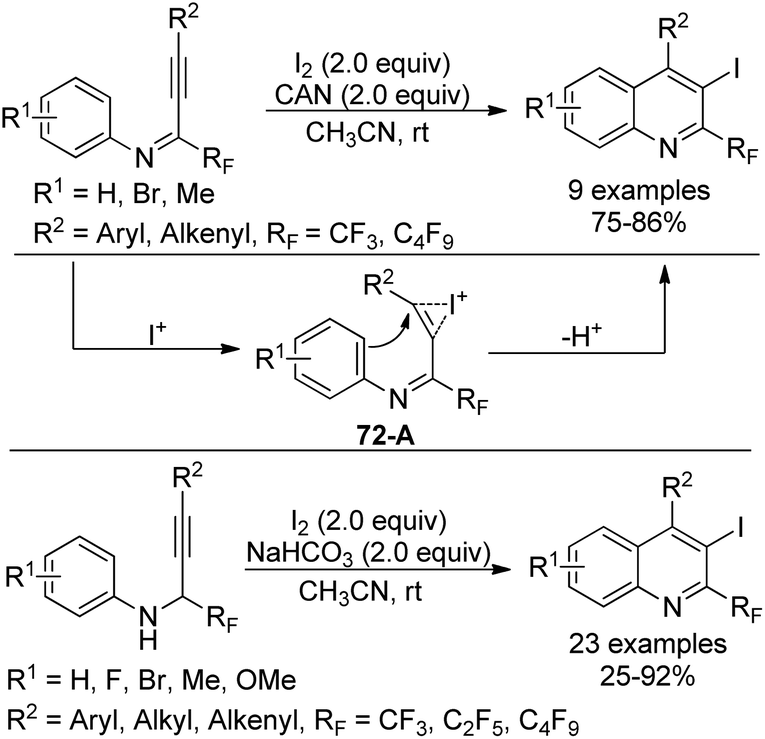 | ||
| Scheme 72 Iodocyclization of perfluoroalkyl propargyl imines/amines. | ||
Wu and co-workers demonstrated a palladium-catalyzed tandem Sonogashira–alkyne carbocyclization reaction of β-trifluoromethyl β-enaminoketones with alkynes for the construction of 2-trifluoromethylquinolines (Scheme 73).90 The β-trifluoromethyl β-enaminoketones were readily prepared from the LDA-mediated condensation of methyl ketones with trifluoroacetimidoyl chlorides. Various β-enaminoketones and terminal alkynes were applicable to the reaction, whereas aliphatic terminal alkynes failed to give the desired product under the standard conditions. Importantly, the presence of an ortho-β-acyl-enamine group probably had a strong ortho-substituent chelating effect to assist the oxidative addition of Pd(0) with aryl halide and stabilize the formed arylalkynylpalladium species, thereby promoting the Sonogashira C–C bond formation process.
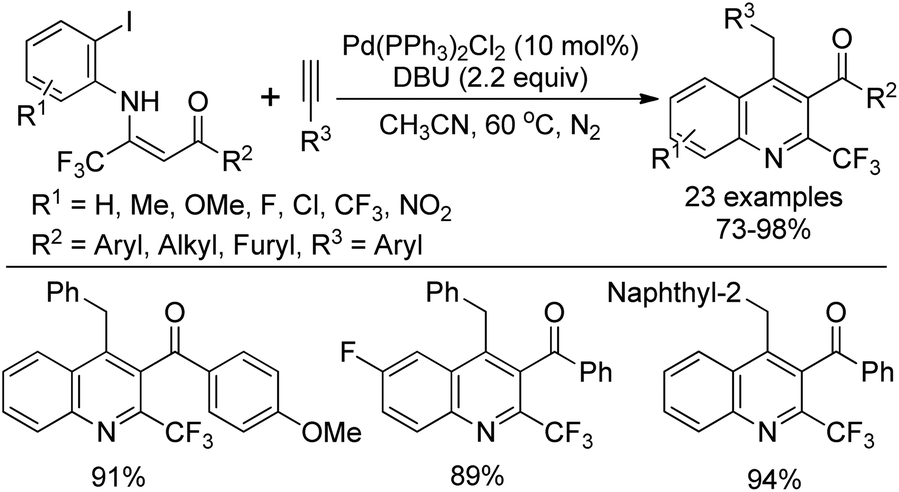 | ||
| Scheme 73 Palladium-catalyzed synthesis of 2-trifluoromethylquinolines. | ||
A Cu(I)-catalyzed coupling reaction and subsequent intramolecular cyclization from N-aryl-fluorinated imidoyl iodides and terminal alkynes for the construction of 2-fluoromethylated quinolines was achieved by Wu and co-workers (Scheme 74).91 NEt3 was considered to play a fundamental role to increase the solubility of the copper salt, whereas K3PO4 only gave the fluorinated alkynyl imine product. When phenylacetylene was used as a substrate, no quinoline product was observed, illustrating the importance of the terminal alkyne bearing α-H.
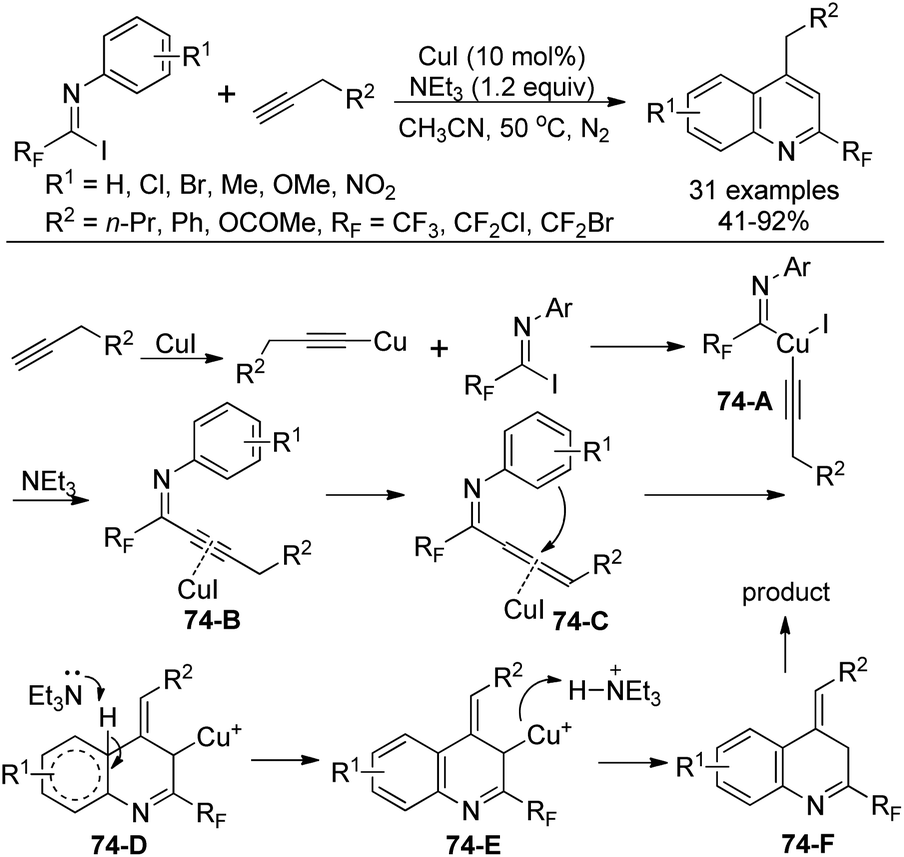 | ||
| Scheme 74 Synthesis of 2-fluoromethylated quinolines. | ||
The reaction mechanism was proposed as depicted in Scheme 74. The insertion of copper alkynide into the C–I bond of imidoyl iodides by oxidant addition generated copper complex 74-A. Then the reductive elimination of 74-A led to intermediate 74-B, which was formed in the presence of Cu(I) with the complexation effect of Et3N. A base-induced propargyl-allenyl isomerization occurred to give the allene intermediate 74-C, which undergoes a nucleophilic attack by the benzene ring to the central carbon of the allene in a Friedel–Crafts reaction manner to afford 74-D. The presence of the terminal alkyne α-H promoted the formation of an allene moiety. Finally, the protonation and isomerization of 74-D gave rise to the quinoline product 74-F.
The similar synthetic method to access 2-fluoromethylated quinoline derivatives was successfully applied to the total synthesis of all four stereoisomers of mefloquine by Leonov, Griesinger and co-workers.92 In the protocol, a domino Sonogashira-6π-electrocyclization reaction was identified as the key step.
A Cu-catalyzed tandem reaction of fluorinated terminal alkynes with sulfonyl azides for the synthesis of 2-trifluoromethylquinolines was reported by Wu and co-workers (Scheme 75).93 The azide-alkyne cycloaddition (CuAAC) reaction to construct a 1,2,3-triazole core was involved in the reaction. The electronic effect of substituents on the N-aromatic moiety of fluorinated terminal alkynes exerted an obvious influence on the reaction and the steric hinderance had a marginal effect on the reaction efficiency. The reaction pathway was proposed to be initiated by the copper-catalyzed click reaction of fluorinated terminal alkynes and sulfonyl azides to deliver the triazolo-copper intermediate 75-A, which was converted into ketenimine species 75-B with the release of N2. The subsequent cyclization and isomerization process could provide the quinoline product.
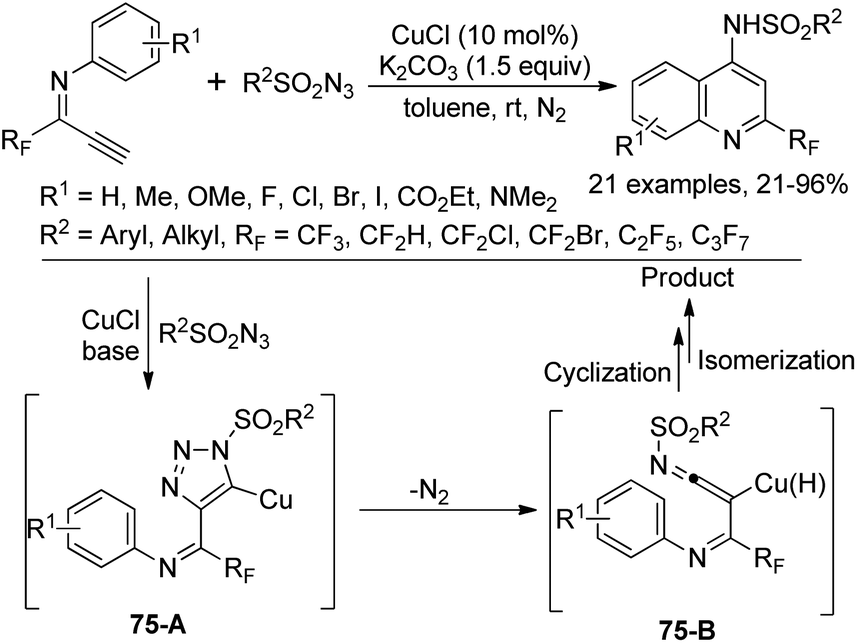 | ||
| Scheme 75 Synthesis of 2-fluoromethylated quinolines from fluorinated alkynes and sulfonyl azides. | ||
A visible-light-induced radical cyclization of trifluoroacetimidoyl chlorides and alkynes for the facile preparation of 2-trifluoromethyl quinolines was developed by Zhou and co-workers (Scheme 76).94 The generation of trifluoroacetimidoyl radicals via visible-light-irradiated activation of C(sp2)–Cl bonds was involved in the reaction, which was readily interrupted by the addition of alkynes. Besides photoredox catalyst, the addition of (nBu)3N having a stronger electron-donating ability was inevitable for the high efficiency of the reaction. The reaction exhibited a broad substrate scope and tolerated a variety of functional groups. With regard to the imidoyl chlorides with a para-substitution on the aromatic ring, two products were observed as isomers in an appropriate ratio.
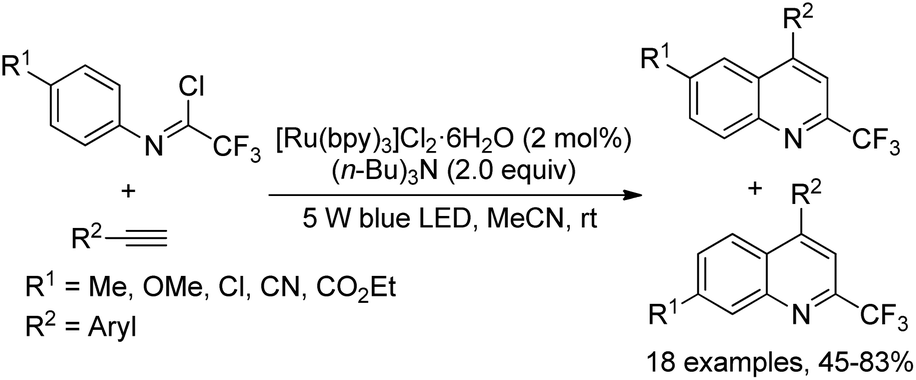 | ||
| Scheme 76 Visible-light-induced synthesis of 2-fluoromethylated quinolines. | ||
A plausible mechanism was proposed as outlined in Scheme 77. The excited [Ru(bpy)3]2+* was generated by the visible-light-induced photoexcitation of [Ru(bpy)3]2+. The formation of [Ru(bpy)3]+ and the radical cation of tributylamine was achieved through single-electron transfer from (n-Bu)3N to [Ru(bpy)3]2+*. The reductive cleavage of the C(sp2)–Cl bond by [Ru(bpy)3]+ furnished the radical intermediate 77-A, which underwent radical addition of alkynes to give vinyl radical 77-B. A homolytic aromatic substitution occurred to deliver cyclohexadienyl radical intermediate 77-C (Path A), followed by a single electron transfer from the photoexcited [Ru(bpy)3]2+* catalyst or the radical cation (n-Bu)3N to yield cation 77-D. The deprotonation of 77-D could afford the quinoline product 77-E. The isomer 77-E′ was considered to be formed through Path B, in which the reaction proceeded through ipso cyclization, C–N bond cleavage and aromatization sequence to generate cyclohexadienyl radical intermediate 77-C′.
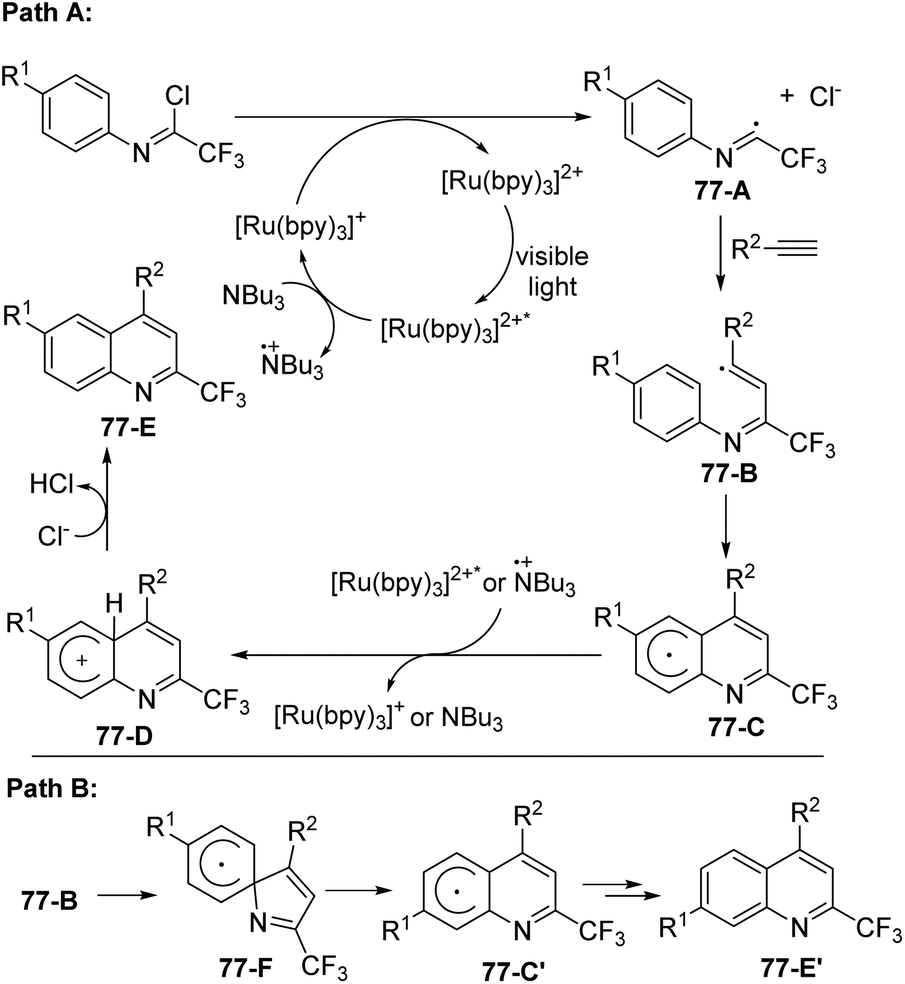 | ||
| Scheme 77 Proposed mechanistic pathways. | ||
An approach for the construction of 4-amino-2-(trifluoromethyl)quinolines by the intramolecular Friedel–Crafts reaction was presented by Darehkordi and coworkers (Scheme 78).95 At first, 2-(1-(arylamino)-2,2,2-trifluoroethylidene)malononitrile derivatives 78-A were afforded by the reaction of trifluoroacetimidoyl chlorides with malononitrile in the presence of sodium hydride at ambient temperature or under microwave irradiation. Then, an intramolecular Friedel–Crafts reaction occurred with the promotion of AlCl3 to give rise to intermediate 78-C, which underwent 1,3-H shift to yield 4-amino-2-(trifluoromethyl)quinolines. It should be noted that the protocol allowed for the preparation of a variety of 4-amino-2-(trifluoromethyl)quinolines in good yields without further purification.
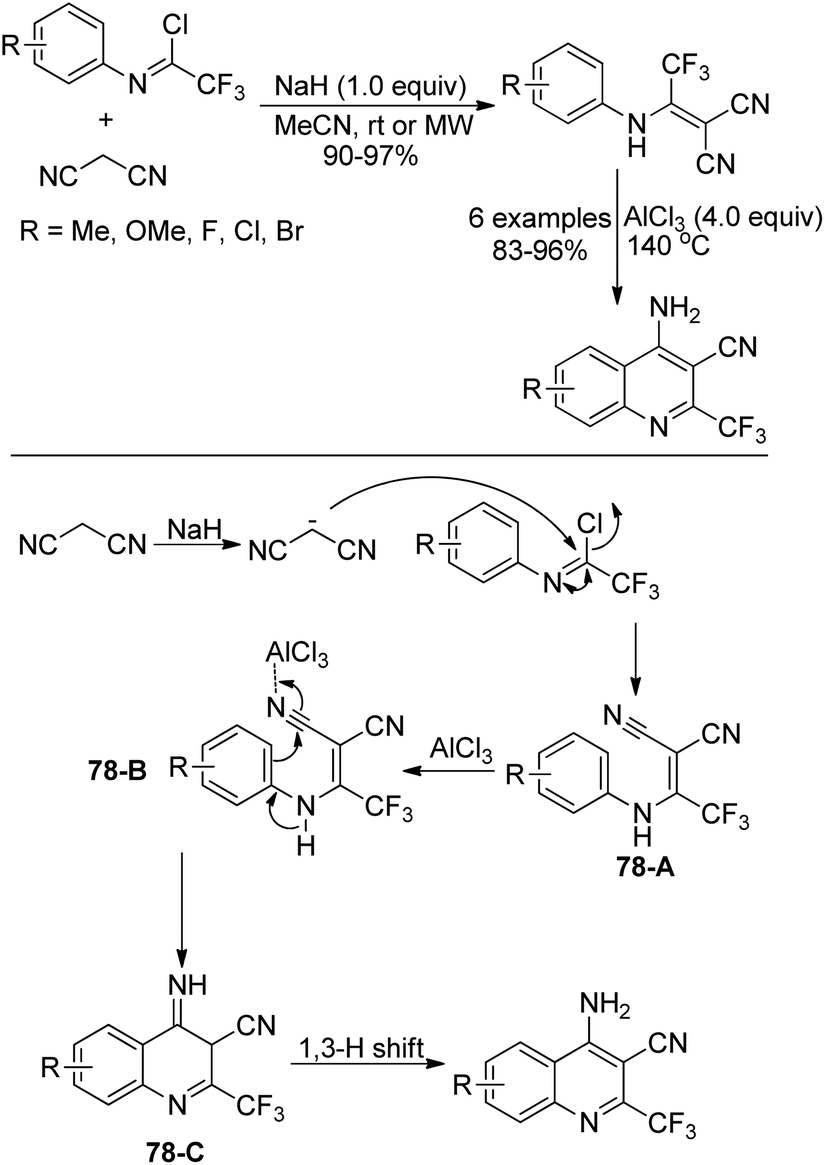 | ||
| Scheme 78 Synthesis of 4-amino-2-(trifluoromethyl)quinolines. | ||
A Pd0-catalyzed intramolecular 6-endo-trig Heck cyclization reaction of fluorinated imidoyl chlorides with a double bond for the assembly of 2-fluoroalkyl quinolines was developed by Chen, Liu and co-workers (Scheme 79).96 The scope of the reaction was broad, as verified by the good tolerance of diverse substitutions and functional groups. The transformation proceeded through a classical Heck-type reaction mechanism. Oxidative addition of the Pd0 catalyst into the C–Cl bond of the imidoyl chloride gave palladium complex 79-A, which underwent intramolecular insertion of the alkene into the C–PdII bond to furnish the intermediate 79-B. A syn-selective β-hydride elimination of the intermediate 79-B yielded the 2-fluoroalkyl quinoline product. The PdII species 79-B′ could not be formed because of the difficulty of the C–PdII–Cl reductive elimination process.
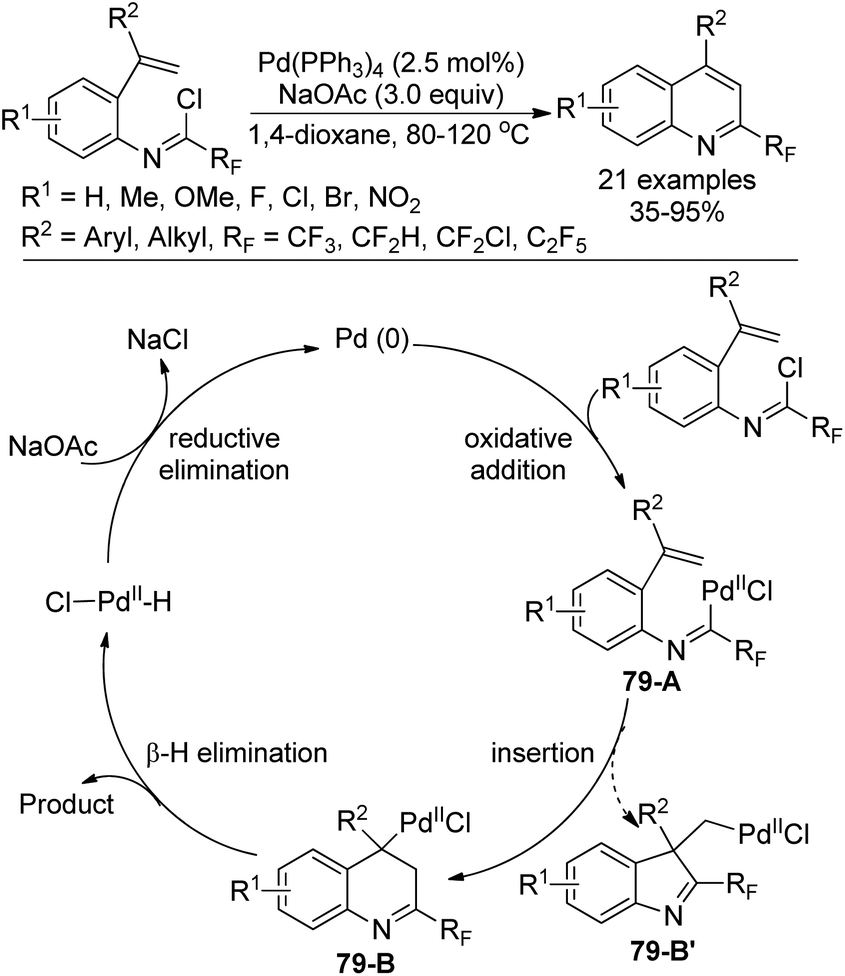 | ||
| Scheme 79 Synthesis of 2-fluoroalkyl quinolines. | ||
4.6. Synthesis of 6-fluoroalkyl-phenanthridines
Zhang and co-workers described a palladium-catalyzed tandem Suzuki/C–H arylation reaction of N-(2-bromophenyl)-trifluoroacetimidoyl chlorides with arylboronic acids for the synthesis of 6-(trifluoromethyl)phenanthridines (Scheme 80).97 A Pd(0)/bulky phosphine ligand catalytic system and a nonpolar solvent enabled the high efficiency of the transformation. The reaction was suggested to be initiated by the Suzuki reaction to provide the coupling product 80-A. The oxidative addition of 80-A to the Pd(0) catalyst yielded the Pd(II) complex 80-B, which underwent a C–H activation process to deliver palladacycle 80-C and a molecule of HBr. Finally, the reductive elimination of 80-C gave a phenanthridine product (Path a). In an alternative Pd(II)/Pd(IV) pathway (Path b), imine-directing C–H activation occurred to produce the Pd(II) complex 80-D, followed by the intramolecular oxidative addition into phenyl bromide to generate the seven-membered palladacycle 80-E. The following reductive elimination resulted in the formation of desired products and released the Pd(II) species. | ||
| Scheme 80 Synthesis of 6-(trifluoromethyl)phenanthridines. | ||
Rh-Catalyzed alkyne [2 + 2 + 2] cycloaddition reactions of diynes with various alkynes for the preparation of fluorine-containing multisubstituted phenanthridines was achieved by Wu, Gong and co-workers (Scheme 81).98 The diynes were obtained by the Sonogashira reactions between trifluoroacetimidoyl chlorides bearing an alkyne moiety and terminal alkynes. The reaction showed good functional group tolerance, and good yields were obtained with regard to most of the substrates, except for electron-deficient alkyne dimethyl acetylenedicarboxylate (DMAD). It should be noted that when a bromodifluoromethyl group was attached on the diynes, a tandem Rh-catalyzed cycloaddition/C–H difluoromethylenation process occurred to give rise to more-complicated fluorine-containing polycyclic compounds.
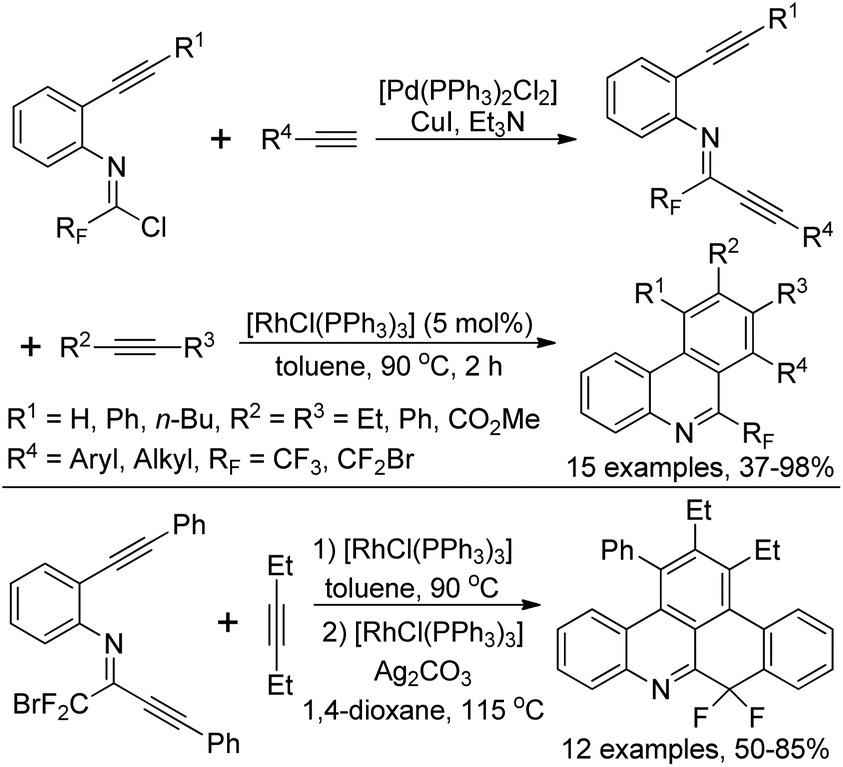 | ||
| Scheme 81 Synthesis of fluorine-containing multisubstituted phenanthridines. | ||
A visible-light-mediated intramolecular radical cyclization reaction of trifluoroacetimidoyl chlorides for the construction of 6-(trifluoromethyl)phenanthridines was presented by Fu and co-workers (Scheme 82).99 In the protocol, an imidoyl radical was in situ generated from the cleavage of C(sp2)–Cl bond induced by visible light. The plausible reaction pathway was similar to that reported in Zhou's work,94 which involved the formation of a radical intermediate and a cation intermediate via a single-electron transfer process (SET).
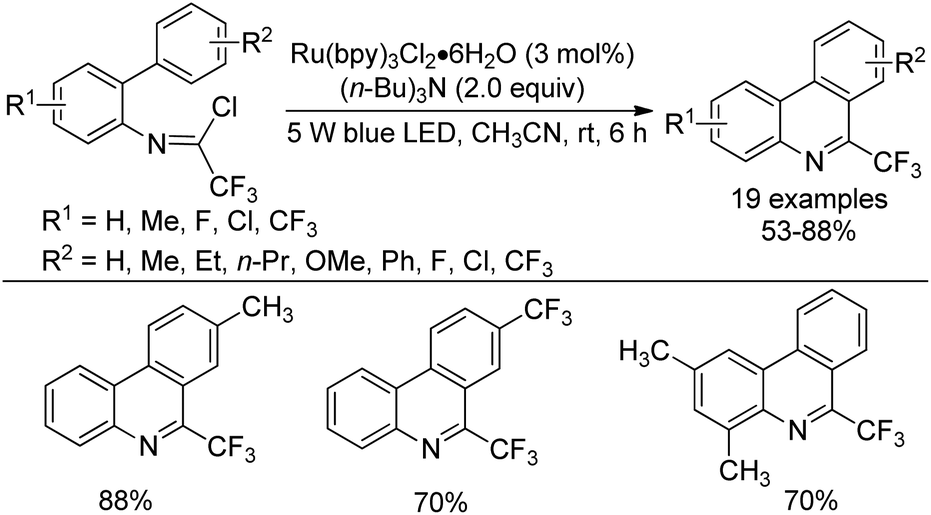 | ||
| Scheme 82 Visible-light-mediated synthesis of 6-(trifluoromethyl)phenanthridines. | ||
A palladium-catalyzed intramolecular C(sp2)–H functionalization of trifluoroacetimidoyl chlorides to access 6-trifluoromethyl-phenanthridines was achieved by the same group (Scheme 83).100 Control experiment ruled out the involvement of the radical pathway. The oxidative addition of the C–Cl bond of trifluoroacetimidoyl chloride to Pd(0) catalyst led to Pd(II) species 83-A, which occurred the intramolecular electrophilic palladation with C–H activation process to give the seven-member palladacycle 83-B. The reductive elimination of 83-B provided the phenanthridine product.
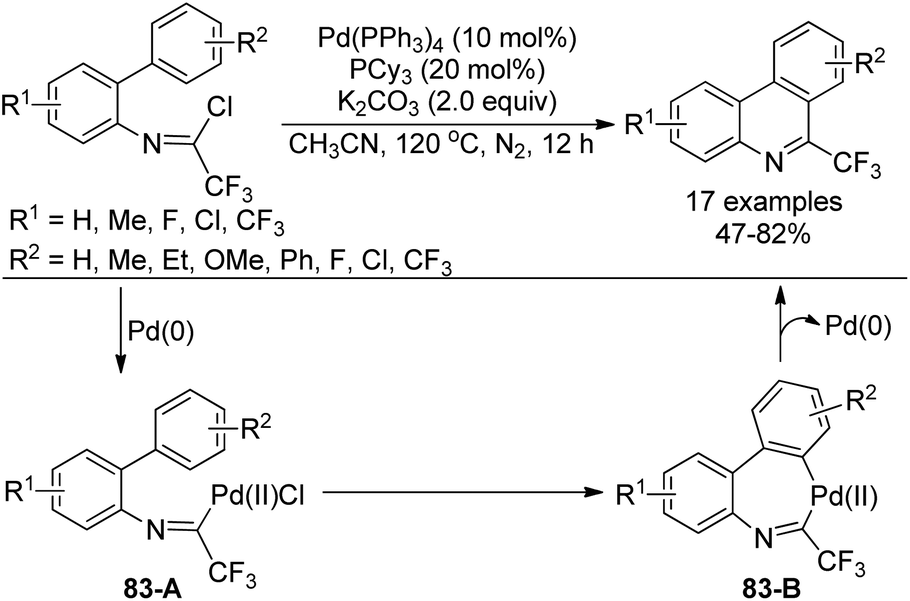 | ||
| Scheme 83 Palladium-catalyzed synthesis of 6-(trifluoromethyl)phenanthridines. | ||
Chen, Zhu and co-workers developed a palladium-catalyzed, Catellani-type dehydrogenation annulation reaction from fluorinated imidoyl chlorides and aryl iodines for the synthesis of biologically relevant 6-fluoroalkyl phenanthridines (Scheme 84).101 The norbornene (NBE) was utilized to mediate the reaction. The transformation showed broad substrate scope with respect to fluorinated imidoyl chlorides and aryl iodines. Various CF2X (X = H, Cl, Br, CF3) groups could be incorporated into phenanthridines under the optimal conditions.
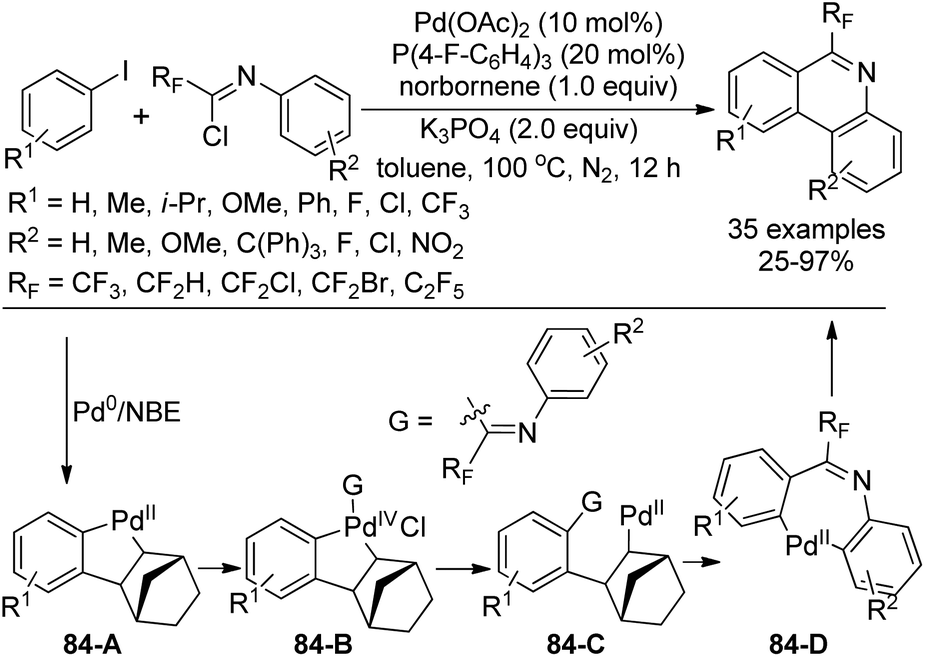 | ||
| Scheme 84 Norbornene-mediated synthesis of 6-fluoroalkyl-phenanthridines. | ||
Several control experiments were performed to shed light on the reaction mechanism and the possible reaction pathway was depicted in Scheme 84. The oxidative addition of Pd(0) with aryl iodine and the subsequent insertion of norbornene and C–H activation afforded the Pd(II) complex 84-A. Another oxidative addition of imidoyl chloride with 84-A could yield the Pd(IV) intermediate, which underwent the reductive elimination to realize the C–C bond formation, with the release of Pd(II) species 84-C. Then, another intramolecular C–H activation process occurred with the removal of norbornene to give rise to Pd(II) intermediate 84-D, followed by the reductive elimination to produce the desired phenanthridines.
The same group also reported an approach for the preparation of 6-fluoroalkyl-phenanthridines via a palladium-catalyzed tandem reaction of fluorinated imidoyl chlorides and 2-bromophenylboronic acid (Scheme 85).102 Good compatibility was obtained for the reaction, but non-fluorinated imidoyl chlorides and the styrene derivatives were not suitable for the reaction, highlighting the key role of the CF2X group and the imine group for the success of the transformation. The possible mechanism was similar to the reactions previously mentioned. The palladium-catalyzed Suzuki reaction was initiated, followed by the intramolecular C–H activation to furnish the phenanthridine product.
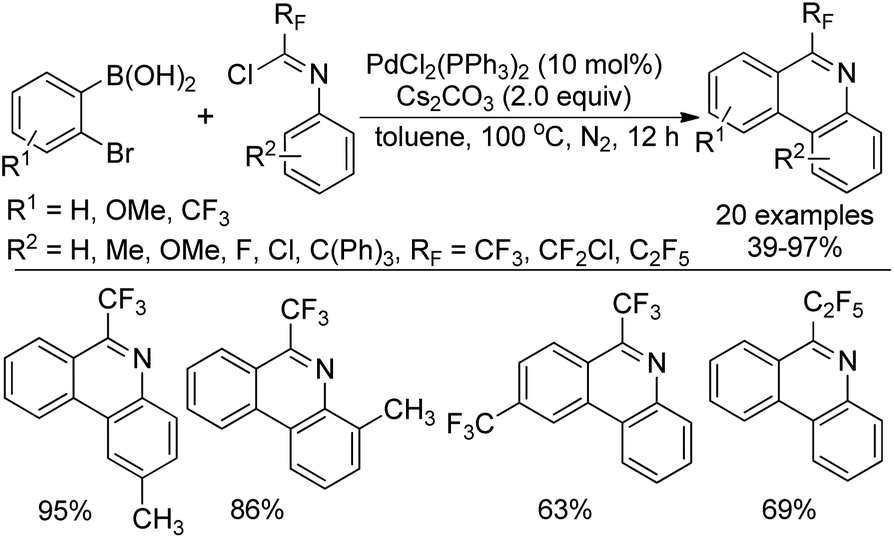 | ||
| Scheme 85 Synthesis of 6-fluoroalkyl-phenanthridines. | ||
4.7. Synthesis of other trifluoromethyl N-heterocycles
A copper(I)-catalyzed tandem reaction of N-(o-haloaryl)alkynylimines and sodium azide was developed for the preparation of biologically active 4-(trifluoromethyl)-[1,2,3]triazolo[1,5-a]quinoxalines (Scheme 86).103 The reaction proceeded well only in the presence of catalytic Cu(I) and L-proline ligand in DMSO at room temperature. Good functional group tolerance of the reaction allowed for the variation of the phenyl ring or alkyne moiety of N-(o-haloaryl)alkynylimines. A copper-catalyzed cascade [3 + 2] cycloaddition/intramolecular C–N coupling reaction sequence was involved in the reaction, providing various fluorine-containing 1,2,3-triazolo tricyclic compounds.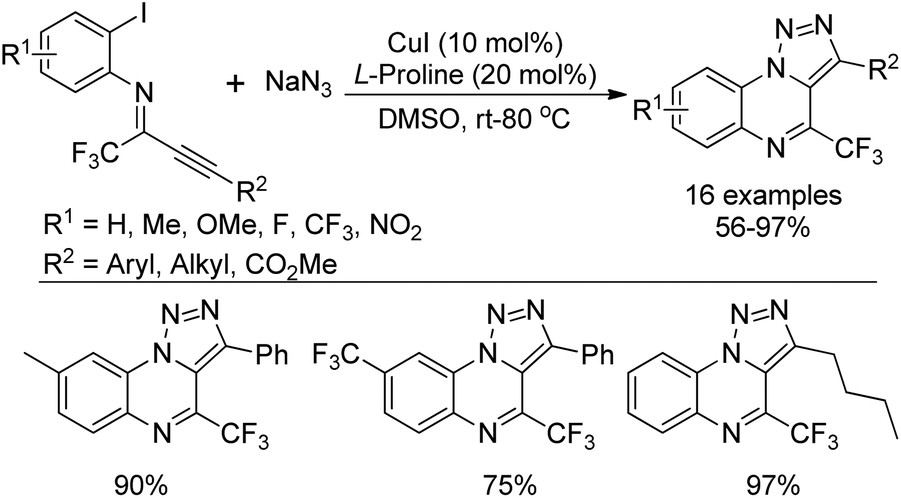 | ||
| Scheme 86 Synthesis of 4-(trifluoromethyl)-[1,2,3]triazolo[1,5-a]quinoxalines. | ||
A two-step reaction for the construction of 6-trifluoromethylindolo[1,2-c]quinazolines and other related trifluoromethylated heterocycles by the use of trifluoroacetimidoyl chlorides and indoles as starting materials was presented by Wu and co-workers (Scheme 87).104 In the protocol, 1-(N-arylimino)indole derivatives were formed by the reaction of trifluoroacetimidoyl chlorides with indoles via the addition/elimination process, which underwent palladium-catalyzed intramolecular cyclization to afford 6-trifluoromethylindolo[1,2-c]quinazolines. The scope of the reaction could also be extended to pyrrole, pyrazole, N-substituted indoles and benzene to assemble the corresponding trifluoromethylated heterocycles.
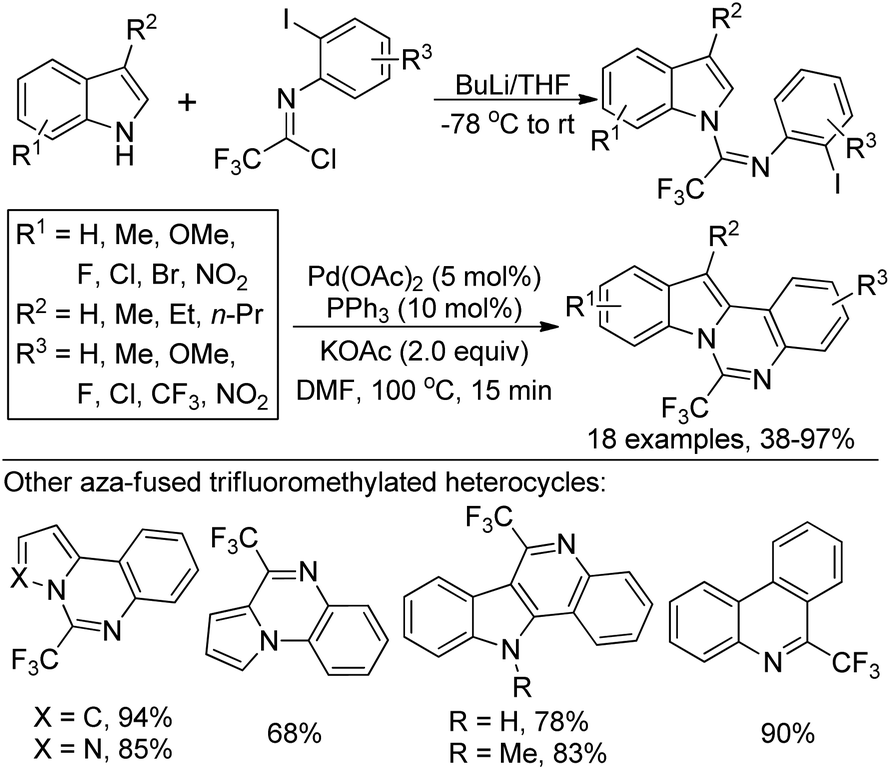 | ||
| Scheme 87 Synthesis of 6-trifluoromethylindolo[1,2-c]quinazolines and related heterocycles. | ||
The intramolecular hydroamination of fluorinated N-(ortho-alkynylaryl)amidines under different conditions for the synthesis of fluorinated N-heterocycles was explored by Wu and co-workers (Scheme 88).105 The indole derivative 88-A was smoothly produced with NaAuCl4·2H2O as catalyst through a 5-endo-dig cyclization. As for the amidine with an ethynyl substituent, various Lewis acid catalysts, including ZnCl2, BF3·OEt2, InCl3·3H2O, CuSO4 and Cu(OTf)2, could promote the reaction to deliver quinazoline 88-B as a major product with high efficiency, whereas the indole product 88-C was observed in the presence of Cu(OAc)2. Under base-mediated conditions, a 6-exo-dig pathway occurred to furnish quinazoline products 88-D, which underwent the ozonolysis process to yield quinazolone 88-E. The electrophilic cyclization of the amidines proceeded well under the I2/NaHCO3 system, offering 3-iodo-substituted indoles 88-F in good yields.
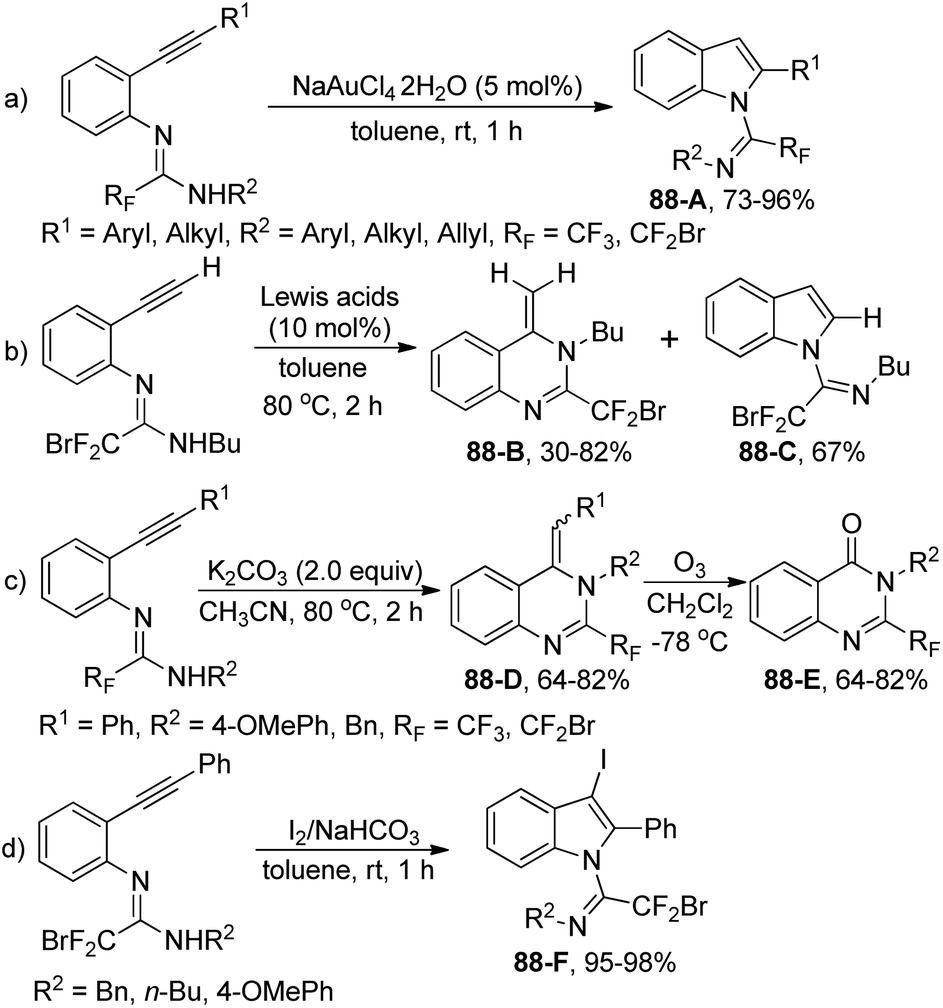 | ||
| Scheme 88 Intramolecular hydroamination of fluorinated N-(ortho-alkynylaryl)amidines. | ||
A series of structurally different C(3)-substituted benzo[e][1,2,4]triazine derivatives were synthesized by Kaszynski and co-workers (Scheme 89).106 Among these compounds, 3-(trifluoromethyl)-benzo[e][1,2,4]triazines were constructed by the Ruppert reaction of 3-iodobenzo[e][1,2,4]triazine with TMSCF3. An alternative synthetic pathway was the oxidative cyclization of amidrazone, which was obtained from the reaction of trifluoroacetimidoyl chlorides with 1,2-di(propan-2-ylidene)hydrazine and hydrazine hydrate. A similar approach had been reported by Uneyama and co-workers for the synthesis of diverse trifluoromethylated N-heterocycles.107
 | ||
| Scheme 89 Synthesis of 3-(trifluoromethyl)-benzo[e][1,2,4]triazine. | ||
Pace and co-workers realized a conceptually novel and divergent lithium halocarbenoid-mediated mono- or bis-homologation reaction to enable the formation of quaternary trifluoromethyl aziridines (Scheme 90).108 Trifluoroacetimidoyl chlorides were applied as electrophilic imine surrogates to receive the addition of one or two homologating agents. By simply modulating the stoichiometric amounts of lithium carbenoids (LiCH2Cl), the protocol could render the divergent preparation of α-halo-α-trifluoromethyl aziridines and α-halomethyl-α-trifluoromethyl aziridines. Due to the strong electrophilicity of trifluoroacetimidoyl chlorides, several functional groups sensitive to organolithium reagents could be well tolerated in the transformation.
 | ||
| Scheme 90 Synthesis of quaternary chloro- and halomethyl-trifluoromethyl aziridines. | ||
The reaction was recognized to be initiated by the addition of the first equivalent of LiCH2Cl to trifluoroacetimidoyl chlorides to afford the tetrahedral intermediate 90-A. An intramolecular nucleophilic displacement of 90-A generated the mono-homologated chloroaziridine 90-B. The chloroaziridine could be spontaneously transformed into a highly electrophilic azirinium ion, 90-C, in the presence of an excess amount of the homologating agent. The second homologation occurred to yield the chloromethylaziridine product 90-D.
A phosphine-catalyzed divergent [4 + 3] annulation reaction of fluorinated imidoyl chlorides with MBH carbonates for the construction of perfluoroalkylated benzazepines was disclosed by Huang and co-workers (Scheme 91).109 In the protocol, the fluorinated imidoyl chlorides were first applied as an electrophilic four-atom building block to participate in the phosphine-catalyzed [4 + 3] cycloaddition reactions, providing various seven-membered N-containing heterocycles in moderate to good yields. By the employment of PPh3 or P(p-OCH3Ph)3 as the catalyst, different perfluoroalkylated benzazepines were produced with good efficiency. The reaction began with the formation of phosphorus ylides and the subsequent nucleophilic attack on imidoyl chlorides. Then, carbanion intermediates were generated upon the delocalization process in the presence of bases, which were captured by the electrophile to afford perfluoroalkylated benzazepine products via a dearomatization/rearomatization process.
 | ||
| Scheme 91 Synthesis of perfluoroalkylated benzazepines. | ||
5 Conclusions
In conclusion, abundant and fruitful progresses in the applications of fluorinated imidoyl halides in the past few decades have been summarized and discussed. This compound provides a facile and rapid pathway to directly build valuable fluorine-containing molecules. Nevertheless, there is still ample room to explore more reaction patterns and potential applications in this area. In consideration of the unique characters of fluorinated imidoyl halides, they have been applied as versatile electrophilic synthons to participate in a wide range of reactions, offering the possibility of constructing more and more structurally diverse fluorine-containing scaffolds. Apart from the regular involvement in coupling and annulation reactions, the exploitation of more in-depth applications of fluorinated imidoyl halides to construct structurally complicated fluorine-containing molecules and natural products is highly desirable in the future.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge financial support from the National Natural Science Foundation of China (21772177 and 21602202), the Natural Science Foundation of Zhejiang Province (LY19B020016) and the Fundamental Research Funds of Zhejiang Sci-Tech University (2019Q065).References
- (a) T. Hiyama, K. Kanie, T. Kusumoto, Y. Morizawa and M. Shimizu, Organofluorine Compounds: Chemistry and Application, Springer, Berlin, 2000 CrossRef; (b) P. Kirsch, Modern Fluoroorganic Chemistry, Synthesis, Reactivity, Applications, Wiley-VCH, Weinheim, 2004 CrossRef; (c) K. Müller, C. Faeh and F. Diederich, Fluorine in Pharmaceuticals: Looking Beyond Intuition, Science, 2007, 317, 1881 CrossRef PubMed; (d) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Fluorine in medicinal chemistry, Chem. Soc. Rev., 2008, 37, 320 RSC; (e) L. Hunter, The C–F bond as a conformational tool in organic and biological chemistry, Beilstein J. Org. Chem., 2010, 6, 38 Search PubMed; (f) J. Nie, H.-C. Guo, D. Cahard and J.-A. Ma, Asymmetric Construction of Stereogenic Carbon Centers Featuring a Trifluoromethyl Group from Prochiral Trifluoromethylated Substrates, Chem. Rev., 2011, 111, 455 CrossRef CAS PubMed.
- (a) J. Wangó, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011), Chem. Rev., 2014, 114, 2432 CrossRef PubMed; (b) S. Large, N. Roques and B. R. Langlois, Nucleophilic Trifluoromethylation of Carbonyl Compounds and Disulfides with Trifluoromethane and Silicon-Containing Bases, J. Org. Chem., 2000, 65, 8848 CrossRef CAS PubMed.
- (a) T. Furuya, A. S. Kamlet and T. Ritter, Catalysis for fluorination and trifluoromethylation, Nature, 2011, 473, 470 CrossRef CAS PubMed; (b) O. A. Tomashenko and V. V. Grushin, Aromatic Trifluoromethylation with Metal Complexes, Chem. Rev., 2011, 111, 4475 CrossRef CAS PubMed; (c) X.-F. Wu, H. Neumann and M. Beller, Recent Developments on the Trifluoromethylation of (Hetero)Arenes, Chem. – Asian J., 2012, 7, 1744 CrossRef CAS PubMed; (d) Y. Ye and M. S. Sanford, Investigations into Transition-Metal-Catalyzed Arene Trifluoromethylation Reactions, Synlett, 2012, 2005 CAS; (e) A. Studer, A “Renaissance” in Radical Trifluoromethylation, Angew. Chem., Int. Ed., 2012, 51, 8950 CrossRef CAS PubMed; (f) H. Liu, Z. Gu and X. Jiang, Direct Trifluoromethylation of the C-H Bond, Adv. Synth. Catal., 2013, 355, 617 CrossRef CAS.
- (a) E. Merino and C. Nevado, Addition of CF3 across unsaturated moieties: a powerful functionalization tool, Chem. Soc. Rev., 2014, 43, 6598 RSC; (b) J. Charpentier, N. Früh and A. Togni, Electrophilic Trifluoromethylation by Use of Hypervalent Iodine Reagents, Chem. Rev., 2015, 115, 650 CrossRef CAS PubMed; (c) X. Liu, C. Xu, M. Wang and Q. Liu, Trifluoromethyltrimethylsilane: Nucleophilic Trifluoromethylation and Beyond, Chem. Rev., 2015, 115, 683 CrossRef CAS PubMed; (d) C. Alonso, E. M. de Marigorta, G. Rubiales and F. Palacios, Carbon Trifluoromethylation Reactions of Hydrocarbon Derivatives and Heteroarenes, Chem. Rev., 2015, 115, 1847 CrossRef CAS PubMed; (e) F. Meyer, Trifluoromethyl nitrogen heterocycles: synthetic aspects and potential biological targets, Chem. Commun., 2016, 52, 3077 RSC.
- (a) I. Tirotta, V. Dichiarante, C. Pigliacelli, G. Cavallo, G. Terraneo, F. B. Bombelli, P. Metrangolo and G. Resnati, 19F Magnetic Resonance Imaging (MRI): From Design of Materials to Clinical Applications, Chem. Rev., 2015, 115, 1106 CrossRef CAS PubMed; (b) D. O'Hagan and H. Deng, Enzymatic Fluorination and Biotechnological Developments of the Fluorinase, Chem. Rev., 2015, 115, 634 CrossRef PubMed; (c) M. Cametti, B. Crousse, P. Metrangolo, R. Milani and G. Resnati, The fluorous effect in biomolecular applications, Chem. Soc. Rev., 2012, 41, 31 RSC; (d) R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Organic fluorine compounds: a great opportunity for enhanced materials properties, Chem. Soc. Rev., 2011, 40, 3496 RSC; (e) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, Applications of Fluorine in Medicinal Chemistry, J. Med. Chem., 2015, 58, 8315 CrossRef CAS PubMed.
- (a) S. Kawamura, H. Egami and M. Sodeoka, Aminotrifluoromethylation of Olefins via Cyclic Amine Formation: Mechanistic Study and Application to Synthesis of Trifluoromethylated Pyrrolidines, J. Am. Chem. Soc., 2015, 137, 4865 CrossRef CAS PubMed; (b) J.-S. Lin, X.-Y. Dong, T.-T. Li, N.-C. Jiang, B. Tan and X.-Y. Liu, A Dual-Catalytic Strategy To Direct Asymmetric Radical Aminotrifluoromethylation of Alkenes, J. Am. Chem. Soc., 2016, 138, 9357 CrossRef CAS PubMed; (c) F. Wang, D. Wang, X. Wan, L. Wu, P. Chen and G. Liu, Enantioselective Copper-Catalyzed Intermolecular Cyanotrifluoromethylation of Alkenes via Radical Process, J. Am. Chem. Soc., 2016, 138, 15547 CrossRef CAS PubMed; (d) J.-H. Ye, L. Song, W.-J. Zhou, T. Ju, Z.-B. Yin, S.-S. Yan, Z. Zhang, J. Li and D.-G. Yu, Selective Oxytrifluoromethylation of Allylamines with CO2, Angew. Chem., Int. Ed., 2016, 55, 10022 CrossRef CAS PubMed; (e) S. Zhou, T. Song, H. Chen, Z. Liu, H. Shen and C. Li, Catalytic Radical Trifluoromethylalkynylation of Unactivated Alkenes, Org. Lett., 2017, 19, 698 CrossRef CAS PubMed; (f) Y.-F. Cheng, X.-Y. Dong, Q.-S. Gu, Z.-L. Yu and X.-Y. Liu, Achiral Pyridine Ligand-Enabled Enantioselective Radical Oxytrifluoromethylation of Alkenes with Alcohols, Angew. Chem., Int. Ed., 2017, 56, 8883 CrossRef CAS PubMed; (g) L. Wu, F. Wang, X. Wan, D. Wang, P. Chen and G. Liu, Asymmetric Cu-Catalyzed Intermolecular Trifluoromethylarylation of Styrenes: Enantioselective Arylation of Benzylic Radicals, J. Am. Chem. Soc., 2017, 139, 2904 CrossRef CAS PubMed; (h) F. Wang, D. Wang, Y. Zhou, L. Liang, R. Lu, P. Chen, Z. Lin and G. Liu, Divergent Synthesis of CF3-Substituted Allenyl Nitriles by Ligand-Controlled Radical 1,2- and 1,4-Addition to 1,3-Enynes, Angew. Chem., Int. Ed., 2018, 57, 7140 CrossRef CAS PubMed.
- (a) B. Sahoo, J.-L. Li and F. Glorius, Visible-Light Photoredox-Catalyzed Semipinacol-Type Rearrangement: Trifluoromethylation/Ring Expansion by a Radical-Polar Mechanism, Angew. Chem., Int. Ed., 2015, 54, 11577 CrossRef CAS PubMed; (b) X. Sun and S. Yu, Visible-light-promoted iminyl radical formation from vinyl azides: synthesis of 6-(fluoro)alkylated phenanthridines, Chem. Commun., 2016, 52, 10898 RSC; (c) M. L. Spell, K. Deveaux, C. G. Bresnahan, B. L. Bernard, W. Sheffield, R. Kumar and J. R. Ragains, A Visible-Light-Promoted O-Glycosylation with a Thioglycoside Donor, Angew. Chem., Int. Ed., 2016, 55, 6515 CrossRef CAS PubMed; (d) Y. Xu, Z. Wu, J. Jiang, Z. Ke and C. Zhu, Merging Distal Alkynyl Migration and Photoredox Catalysis for Radical Trifluoromethylative Alkynylation of Unactivated Olefins, Angew. Chem., Int. Ed., 2017, 56, 4545 CrossRef CAS PubMed; (e) S. Verhoog, C. W. Kee, Y. Wang, T. Khotavivattana, T. C. Wilson, V. Kersemans, S. Smart, M. Tredwell, B. G. Davis and V. Gouverneur, 18F-Trifluoromethylation of Unmodified Peptides with 5-18F-(Trifluoromethyl)dibenzothiophenium Trifluoromethanesulfonate, J. Am. Chem. Soc., 2018, 140, 1572 CrossRef CAS PubMed.
- (a) M. Shang, S.-Z. Sun, H.-L. Wang, B. N. Laforteza, H.-X. Dai and J.-Q. Yu, Exceedingly Fast Copper(II)-Promoted ortho C-H Trifluoromethylation of Arenes using TMSCF3, Angew. Chem., Int. Ed., 2014, 53, 10439 CrossRef CAS PubMed; (b) J.-B. Liu, C. Chen, L. Chu, Z.-H. Chen, X.-H. Xu and F.-L. Qing, Silver-Mediated Oxidative Trifluoromethylation of Phenols: Direct Synthesis of Aryl Trifluoromethyl Ethers, Angew. Chem., Int. Ed., 2015, 54, 11839 CrossRef CAS PubMed; (c) J. A. Pike and J. W. Walton, Nucleophilic trifluoromethylation of electron-deficient arenes, Chem. Commun., 2017, 53, 9858 RSC; (d) L. Li, C. Ni, Q. Xie, M. Hu, F. Wang and J. Hu, TMSCF3 as a Convenient Source of CF2=CF2 for Pentafluoroethylation, (Aryloxy)tetrafluoroethylation, and Tetrafluoroethylation, Angew. Chem., Int. Ed., 2017, 56, 9971 CrossRef CAS PubMed; (e) X. Gao, Y.-L. Xiao, X. Wan and X. Zhang, Copper-Catalyzed Highly Stereoselective Trifluoromethylation and Difluoroalkylation of Secondary Propargyl Sulfonates, Angew. Chem., Int. Ed., 2018, 57, 3187 CrossRef CAS PubMed.
- (a) C. Zhang, Application of Langlois’ Reagent in Trifluoromethylation Reactions, Adv. Synth. Catal., 2014, 356, 2895 CrossRef CAS; (b) Y. Zhao and F. Liu, Recent advance in radical fluoroalkylation with sulfinate salts, Tetrahedron Lett., 2018, 59, 180 CrossRef CAS; (c) H.-X. Song, Q.-Y. Han, C.-L. Zhao and C.-P. Zhang, Fluoroalkylation reactions in aqueous media: a review, Green Chem., 2018, 20, 1662 RSC.
- (a) O. O. Grygorenko, O. S. Artamonov, I. V. Komarov and P. K. Mykhailiuk, Trifluoromethyl-substituted cyclopropanes, Tetrahedron, 2011, 67, 803 CrossRef CAS; (b) L. Mertens and R. M. Koenigs, Fluorinated diazoalkanes-a versatile class of reagents for the synthesis of fluorinated compounds, Org. Biomol. Chem., 2016, 14, 10547 RSC; (c) G. A. Molander and D. Ryu, Diastereoselective Synthesis of Vicinally Bis(trifluoromethylated) Alkylboron Compounds through Successive Insertions of 2,2,2-Trifluorodiazoethane, Angew. Chem., Int. Ed., 2014, 53, 14181 CrossRef CAS PubMed; (d) H. Luo, G. Wu, Y. Zhang and J. Wang, Silver(I)-Catalyzed N-Trifluoroethylation of Anilines and O-Trifluoroethylation of Amides with 2,2,2-Trifluorodiazoethane, Angew. Chem., Int. Ed., 2015, 54, 14503 CrossRef CAS PubMed; (e) Z. Chen, Y. Zheng and J.-A. Ma, Use of a Traceless Activating and Directing Group for the Construction of Trifluoromethylpyrazoles: One-Pot Transformation of Nitroolefins and Trifluorodiazoethane, Angew. Chem., Int. Ed., 2017, 56, 4569 CrossRef CAS PubMed; (f) S. Qin, Y. Zheng, F.-G. Zhang and J.-A. Ma, One-Pot Cascade Transformations of Zinc Trifluorodiazoethylide and α,β-Unsaturated Enones: Access to Trifluoromethylated Polycyclic Pyrazolines, Org. Lett., 2017, 19, 3406 CrossRef CAS PubMed; (g) L. Sun, J. Nie, Y. Zheng and J.-A. Ma, [3 + 2] Cycloaddition of arynes with CF3CHN2: Access to 3-trifluoromethyl-1H-indazoles, J. Fluorine Chem., 2015, 174, 88 CrossRef CAS; (h) Z. Chai, J. P. Bouillon and D. Cahard, Chiral Brønsted acid-catalyzed diastereo- and enantioselective synthesis of CF3-substituted aziridines, Chem. Commun., 2012, 48, 9471 RSC; (i) S. Wang, L.-J. Yang, J.-L. Zeng, Y. Zheng and J.-A. Ma, Silver-catalyzed [3 + 2] cycloaddition of isocyanides with diazo compounds: new regioselective access to 1,4-disubstituted-1,2,3-triazoles, Org. Chem. Front., 2015, 2, 1468 RSC; (j) Z. Chen, S.-Q. Fan, Y. Zheng and J.-A. Ma, Silver-catalyzed regioselective [3 + 2] cycloaddition of arene-diazonium salts with 2,2,2-trifluorodiazoethane (CF3CHN2): a facile access to 2-aryl-5-trifluoromethyltetrazoles, Chem. Commun., 2015, 51, 16545 RSC; (k) Z. Chen, N. Ren, X. Ma, J. Nei, F.-G. Zhang and J.-A. Ma, Silver-Catalyzed [3 + 3] Dipolar Cycloaddition of Trifluorodiazoethane and Glycine Imines: Access to Highly Functionalized Trifluoromethyl-Substituted Triazines and Pyridines, ACS Catal., 2019, 9, 4600 CrossRef CAS; (l) A. Kumar, S. Ahamad, R. Kant and K. Mohanan, Silver-Catalyzed Three-Component Route to Trifluoromethylated 1,2,3-Triazolines Using Aldehydes, Amines, and Trifluorodiazoethane, Org. Lett., 2019, 21, 2962 CrossRef CAS PubMed.
- W. P. Norris and H. B. Jonassen, The Reaction of Phosphorus Pentachloride with N-Methyltrifluoroacetamide. N-Methyltrifluoroacetimidoyl Chloride and 1,3-Dimethyl-2,2,2,4,4,4-hexachloro-1,3,2,4-diazadiphosphetidine, J. Org. Chem., 1962, 27, 1449 CrossRef CAS.
- M. Tordeux and C. Wakselman, Synthesis and chemical transformations of perfluoroalkylimidoyl iodides, Tetrahedron, 1981, 37, 315 CrossRef CAS.
- K. Tamura, H. Mizukami, K. Maeda, H. Watanabe and K. Uneyama, One-pot synthesis of trifluoroacetimidoyl halides, J. Org. Chem., 1993, 58, 32 CrossRef CAS.
- A. Darehkordi, H. Khabazzadeh and K. Saidi, Facile synthesis of 2,2,2-trifluoroacetimidoyl chloride derivatives, J. Fluorine Chem., 2005, 126, 1140 CrossRef CAS.
- K. Uneyama and H. Watanabe, Palladium-catalyzed coupling reactions of trifluoroacetimidoyl iodides with olefins and 1-alkynes, Tetrahedron Lett., 1991, 32, 1459 CrossRef CAS.
- Y.-M. Wu, Y. Li and J. Deng, One-pot synthesis of bromodifluoroacetimidoyl halides and its Suzuki coupling reactions with aryl boronic acids, J. Fluorine Chem., 2005, 126, 791 CrossRef CAS.
- H. Watanabe, Y. Hashizume and K. Uneyama, Homologation of trifluoroacetimidoyl iodides by palladium-catalyzed carbonylation. An approach to α-amino perfluoroalkanoic acids, Tetrahedron Lett., 1992, 33, 4333 CrossRef CAS.
- H. Amii, Y. Kishikawa, K. Kageyama and K. Uneyama, Palladium-Catalyzed tert-Butoxycarbonylation of Trifluoroacetimidoyl Iodides, J. Org. Chem., 2000, 65, 3404 CrossRef CAS PubMed.
- H. Abe, H. Amii and K. Uneyama, Pd-Catalyzed Asymmetric Hydrogenation of α-Fluorinated Iminoesters in Fluorinated Alcohol: A New and Catalytic Enantioselective Synthesis of Fluoro α-Amino Acid Derivatives, Org. Lett., 2001, 3, 313 CrossRef CAS PubMed.
- H. Watanabe, F. Yamashita and K. Uneyama, Trifluoroacetimidoyl lithium; generation and reaction with electrophiles, Tetrahedron Lett., 1993, 34, 1941 CrossRef CAS.
- H. Watanabe, F. Yan, T. Sakai and K. Uneyama, (Trifluoroacetimidoyl)lithiums and Their Reaction with Electrophiles, J. Org. Chem., 1994, 59, 758 CrossRef CAS.
- K. Tamura, F. Yan, T. Sakai and K. Uneyama, Generation and Reaction of (N-Aryltrifluoroacetimidoyl)zinc Halide, Bull. Chem. Soc. Jpn., 1994, 67, 300 CrossRef CAS.
- K. Uneyama, C. Noritake and K. Sadamune, Generation and Reaction of Metal Free Trifluoroacetimidoyl Carbanion, J. Org. Chem., 1996, 61, 6055 CrossRef CAS.
- (a) K. Uneyama, H. Amii, T. Katagiri, T. Kobayashi and T. Hosokawa, A rich chemistry of fluorinated imidoyl halides, J. Fluorine Chem., 2005, 126, 165 CrossRef CAS; (b) K. Uneyama, T. Katagiri and H. Amii, α-Trifluoromethylated Carbanion Synthons, Acc. Chem. Res., 2008, 41, 817 CrossRef CAS PubMed.
- W. S. Huang and C. Y. Yuan, A new and convenient one-pot synthesis of α,β-unsaturated trifluoromethyl ketones, J. Chem. Soc., Perkin Trans. 1, 1995, 741 RSC.
- T. Ishihara, T. Maeyawa and T. Ando, Novel synthesis of F-1-alkene-1-phosphonate derivatives from F-alkanoyl chlorides and their efficient use for preparing fluoro-ga,β-enones, Tetrahedron Lett., 1983, 24, 4229 CrossRef CAS.
- W. Huang, C. Yuan and Z. Wang, A new and simple approach to N-substituted trifluoroacetimidoyl aryl ketones, J. Fluorine Chem., 1995, 74, 247 CrossRef CAS.
- H.-B. Yu and W.-Y. Huang, A convenient synthesis of 3-polyfluoroalkyl pyrazoles and 6-polyfluoroalkyl pyrimidines from β-polyfluoroalkyl enaminones, J. Fluorine Chem., 1997, 84, 65 CrossRef CAS.
- K. Uneyama, O. Morimoto and F. Yamashita, Trifluoroacetimidoyl chlorides as a new trifluoromethyl building block for fluorinated nitrogen heterocycles, Tetrahedron Lett., 1989, 30, 4821 CrossRef CAS.
- S. Fustero, A. Navarro, B. Pina, A. Asensio, P. Bravo, M. Crucianelli, A. Volonterio and M. Zanda, Two Practical and Efficient Approaches to Fluorinated and Nonfluorinated Chiral β-Imino Sulfoxides, J. Org. Chem., 1998, 63, 6210 CrossRef CAS PubMed.
- S. Fustero, M. G. de la Torre, B. Pina and A. S. Fuentes, New Strategies for the Synthesis of Fluorinated Vinylogous Amidines and β-Enamino Ketones, J. Org. Chem., 1999, 64, 5551 CrossRef CAS PubMed.
- S. Fustero, B. Pina, E. Salavert, A. Navarro, M. C. R. de Arellano and A. S. Fuentes, New Strategy for the Stereoselective Synthesis of Fluorinated β-Amino Acids, J. Org. Chem., 2002, 67, 4667 CrossRef CAS PubMed.
- S. Fustero, J. G. Soler, A. Bartolomé and M. S. Roselló, Novel Approach for Asymmetric Synthesis of Fluorinated β-Amino Sulfones and Allylic Amines, Org. Lett., 2003, 5, 2707 CrossRef CAS PubMed.
- S. Fustero, S. Flores, A. C. Cuñat, D. Jiménez, C. del Pozo, J. Bueno and J. F. Sanz-Cervera, Synthesis of fluorinated allylic amines: Reaction of 2-(trimethylsilyl)ethyl sulfones and sulfoxides with fluorinated imines, J. Fluorine Chem., 2007, 128, 1248 CrossRef CAS.
- H. Amii, M. Kohda, M. Seo and K. Uneyama, A novel route to the fluorinated diimines: carbon monoxide-promoted reductive homocoupling of fluorinated imidoyl iodides in the presence of a palladium catalyst, Chem. Commun., 2003, 1752 RSC.
- J. P. Sadighi, L. M. Henling, J. A. Labinger and J. E. Bercaw, Synthesis of a perfluoroalkyl-substituted α-diimine by Sm-mediated reductive coupling, Tetrahedron Lett., 2003, 44, 8073 CrossRef CAS.
- S. Li, J. Zhu, H. Xie, Z. Chen and Y.-M. Wu, Cu(I)-catalyzed coupling reactions of fluorinated imidoyl halides with terminal alkynes: Convenient synthesis of fluorinated alkynyl imines, J. Fluorine Chem., 2011, 132, 196 CrossRef CAS.
- Y. Wu, M. Zhang and Y.-Q. Li, Facile synthesis of fluorinated α-imino alkynes by the coupling reaction of imidoyl iodides with terminal alkynes, J. Fluorine Chem., 2006, 127, 218 CrossRef CAS.
- M.-W. Chen, B. Wu, Z.-P. Chen, L. Shi and Y.-G. Zhou, Synthesis of Chiral Fluorinated Propargylamines via Chemoselective Biomimetic Hydrogenation, Org. Lett., 2016, 18, 4650 CrossRef CAS PubMed.
- B. Enk, D. Eisenstecken, H. Kopacka, K. Wurst, T. Müller, F. Pevny, R. F. Winter and B. Bildstein, Doubly N-Functionalized Pentafulvenes and Redox-Responsive [N,N]- and [N,C,N]-Pincer Bis(imidoyl)pentamethylruthenocene Metalloligands, Organometallics, 2010, 29, 3169 CrossRef CAS.
- K. Kadota, Y. Dan-oh and K. Uneyama, Thermolysis of N-[2,2,2-Trifluoro-1-(tritylazo)ethylidene]anilines and Related Azo-Compounds, J. Org. Chem., 1996, 61, 2364 CrossRef CAS.
- Y. Dan-oh, H. Matta, J. Uemura, H. Watanabe and K. Uneyama, Determination of Arsenic(III) and Arsenic(V) by Hydride Generation- Atomic Absorption Spectrometry Following a Rapid Coprecipitation Technique with Hafnium(IV) Hydroxide, Bull. Chem. Soc. Jpn., 1995, 65, 1497 CrossRef.
- K. Saidi and A. Darehkordi, A convenient synthesis of N-aryltrifluoroacetimidoyl phthalimide and succinimide, J. Fluorine Chem., 2000, 105, 49 CrossRef CAS.
- Y.-M. Wu, M. Zhang and Y.-Q. Li, Facile synthesis of α-fluoro substituted amidines from imidoyl chlorides and some of its application, J. Fluorine Chem., 2006, 127, 1168 CrossRef CAS.
- A. Darehkordi, M. Javanmiri, S. Ghazi and S. Assar, Synthesis of N-aryl-2,2,2-trifluoroacetimidoyl piperazinylquinolone derivatives and their antibacterial evaluations, J. Fluorine Chem., 2011, 132, 263 CrossRef CAS.
- A. Darehkordi, F. Rahmani and V. Hashemi, Synthesis of new trifluoromethylated indole derivatives, Tetrahedron Lett., 2013, 54, 4689 CrossRef CAS.
- A. Darehkordi and F. Rahmani, Synthesis of new α-trifluoromethyl substituted formamidines framework by using N-nucleophiles and N,S bidentate nucleophiles, J. Fluorine Chem., 2016, 190, 41 CrossRef CAS.
- Y. Okada, M. Ohtsu, M. Bando and H. Yamada, Benzyl N-Phenyl-2,2,2-trifluoroacetimidate: A New and Stable Reagent for O-Benzylation, Chem. Lett., 2007, 36, 992 CrossRef CAS.
- N. Barroca-Aubry, M. Benchekroun, F. Gomes and D. Bonnaffé, p-Methoxybenzyl-N-phenyl-2,2,2-trifluoroacetimidate: a versatile reagent for mild acid catalyzed etherification, Tetrahedron Lett., 2013, 54, 5118 CrossRef CAS.
- S. B. Tsabedze, D. E. K. Kabotso and N. L. B. Pohl, The development of N-aryl trifluoroacetimidate-based benzyl and allyl protecting group reagents, Tetrahedron Lett., 2013, 54, 6983 CrossRef CAS.
- (a) A. Ueki, Y. Takano, A. Kobayashi, Y. Nakahara, H. Hojo and Y. Nakahara, Solid-phase synthesis of glycopeptide carrying a tetra-N-acetyllactosamine-containing core 2 decasaccharide, Tetrahedron, 2010, 66, 1742 CrossRef CAS; (b) D. Kobayashi, A. Ueki, T. Yamaji, K. Nagao, A. Imamura, H. Ando, M. Kiso and H. Ishida, Efficient Synthesis of the Lewis A Tandem Repeat, Molecules, 2016, 21, 614 CrossRef PubMed.
- Y. Minami, H. Kuniyasu, A. Sanagawa and N. Kambe, Pd-Catalyzed Regioselective Iminothiolation of Alkynes: A Remarkable Effect of the CF3 Group of Iminosulfides, Org. Lett., 2010, 12, 3744 CrossRef CAS PubMed.
- (a) W. Huang, Y. Zhang and C. Yuan, Studies on organophosphorus compounds 93 reaction of trivalent phosphorus reagents with N-substituted trifluorocetmidoyl chlorides. A novel synthesis of 1-(N-substituted amino)-2,2,2-trifluoroethylphosphonates, Phosphorus, Sulfur Silicon Relat. Elem., 1995, 107, 21 CrossRef CAS; (b) J. Xiao, X. Zhang and C. Yuan, A Facile Asymmetric Synthesis of 1-Amino-2,2,2-Trifluoroethanephosphonic Acid, Heteroat. Chem., 2000, 11, 536 CrossRef CAS; (c) J. Xiao and C. Yuan, A Practical and Effective Asymmetric Synthesis of 2-Amino-3,3,3-Trifluoropropanephosphonic Acid, Heteroat. Chem., 2000, 11, 541 CrossRef CAS.
- Y. Y. Khomutnik, P. P. Onys'ko, Y. V. Rassukanaya, V. V. Pirozhenko and A. D. Sinitsa, N-aryltrifluoroacetimidoylphosphonates, Russ. J. Gen. Chem., 2013, 83, 445 CrossRef CAS.
- T. Kobayashi, T. Nakagawa, H. Amii and K. Uneyama, Mg-Promoted Double Silylation of Trifluoroacetimidoyl Chlorides. A New Entry to the Fluorinated Dianion Equivalents, Org. Lett., 2003, 5, 4297 CrossRef CAS PubMed.
- C. Akamatsu, A. Yamauchi, T. Kobayashi, Y. Ozeki, J. Takagi, H. Amii and K. Uneyama, An Improved Synthesis of Fluorinated Imidoylsilanes, Synthesis, 2006, 1836 CAS.
- P. R. Likhar, S. M. Salian, S. Roy, M. L. Kantam, B. Sridhar, K. V. Mohan and B. Jagadeesh, Intramolecular Thiopalladation of Thioanisole-Substituted Propargyl Imines: Synthesis of Benzothiophene-Based Palladacycles, Organometallics, 2009, 28, 3966 CrossRef CAS.
- M. V. Karkhelikar, S. S. Racharlawar, S. M. Salian, B. Sridhar and P. R. Likhar, Heck-type coupling of intramolecularly-generated thiopalladacycles with alkenes: One pot syntheses of 3-alkenylbenzo[b]thiophenes, J. Organomet. Chem., 2012, 706–707, 128 CrossRef CAS.
- H. Amii, K. Kageyama, Y. Kishikawa, T. Hosokawa, R. Morika, T. Katagiri and K. Uneyama, Preparation, Structure, and Reactions of Trifluoroacetimidoyl Palladium(II) Complexes, Organometallics, 2012, 31, 1281 CrossRef CAS.
- S. S. Racharlawar, D. Shankar, M. V. Karkhelikar, B. Sridhar and P. R. Likhar, Intramolecular heterocyclization and cyclopalladation of selenoanisole substituted propargyl imines: Synthesis and reactivity of Pd-C bond towards alkynes, J. Organomet. Chem., 2014, 757, 14 CrossRef CAS.
- W.-P. Ye, H.-L. Mu, X.-C. Shi, Y.-X. Cheng and Y.-S. Li, Synthesis and characterization of the titanium complexes bearing two regioisomeric trifluoromethyl-containing enaminoketonato ligands and their behavior in ethylene polymerization, Dalton Trans., 2009, 9452 RSC.
- D. Shankar, K. Jaipal, B. Sridhar, R. Nagarjuna Chary, S. Prabhakar, L. Giribabu and P. R. Likhar, Intramolecular cyclization assisted oxidative addition: synthesis of octahedral cycloplatinated methyl complexes, RSC Adv., 2015, 5, 20295 RSC.
- J. Takagi, R. Takihana, A. Kuwano and K. Uneyama, Facile Preparation of Polyfluoroalkylated Aldimines from Polyfluoroalkanoic Acids, Synthesis, 2007, 1624 CAS.
- H.-B. Yu and W.-Y. Huang, Regioselective Synthesis of 5-Trifluoromethyl Pyrazoles by the [1 + 4] Cyclization of Phenylhydrazones with N-Aryl Trifluoroacetimidoyl Iodides, Synlett, 1997, 679 CrossRef.
- J. Xiao, X. Zhang, D. Wang and C. Yuan, Synthesis of trifluoromethyltetrazoles via building block strategy, J. Fluorine Chem., 1999, 99, 83 CrossRef CAS.
- F. Rahmani, A. Darehkordi and B. Notash, Microwave-assisted synthesis of 1-aryl-5-(trifluoromethyl)-1H-tetrazoles, J. Fluorine Chem., 2014, 166, 84 CrossRef CAS.
- S. Li, Z. Li, Y. Yuan, D. Peng, Y. Li, L. Zhang and Y.-M. Wu, Au(I)-Catalyzed Intramolecular Hydroamination of the Fluorinated N′-Aryl-N-Propargyl Amidines: Mild Conditions for the Synthesis of 2-Fluoroalkyl Imidazole Derivatives, Org. Lett., 2012, 14, 1130 CrossRef CAS PubMed.
- F. Rahmani and A. Darehkordi, Synthesis of Trifluoromethylated Pyrroles via a One-Pot Three-Component Reaction, Synlett, 2017, 28, 1224 CrossRef CAS.
- A. Darehkordi, M. Ramezani and F. Rahmani, TiO2-Nanoparticles Catalyzed Synthesis of New Trifluoromethyl-4,5-dihydro-1,2,4-oxadiazoles and Trifluoromethyl-1,2,4-oxadiazoles, J. Heterocycl. Chem., 2018, 55, 1702 CrossRef CAS.
- S. Hu, Z. Yang, Z. Chen and X.-F. Wu, Metal-Free Synthesis of 5-Trifluoromethyl-1,2,4-Triazoles from Iodine-Mediated Annulation of Trifluoroacetimidoyl Chlorides and Hydrazones, Adv. Synth. Catal., 2019, 361, 4949 CrossRef CAS.
- K. Uneyama and M. Kobayashi, Electrochemical Oxidation of N,N’-Disubstituted Trifluoroethanimidamides. An Approach to N-Substituted 2-(Trifluoromethyl)benzimidazoles, J. Org. Chem., 1994, 59, 3003 CrossRef CAS.
- K. Uneyama and M. Kobayashi, Intramolecular Cyclization of N1-(4-Oxo-2,5-cyclohexadien-1-ylidene)-N2-substituted-2,2,2-trifluoroethanimidamides (p-Benzoquinone Imine Derivatives): Syntheses of Trifluoromethylated 6-Hydroxybenzimidazoles and Spiro Dienone Diazacarbocycles, J. Org. Chem., 1995, 60, 6402 CrossRef.
- M. Kobayashi and K. Uneyama, Synthesis of 2-(Perfluoroalkyl)- and 2-(Perfluoroaryl)benzimidazoles by Oxidative Intramolecular Cyclization of Perfluoroalkyl and Aryl Imidamides, J. Org. Chem., 1996, 61, 3902 CrossRef CAS PubMed.
- J. Zhu, H. Xie, Z. Chen, S. Li and Y.-M. Wu, Synthesis of 2-fluoroalkylbenzimidazoles via copper(I)-catalyzed tandem reactions, Chem. Commun., 2009, 2338 RSC.
- M.-W. Chen, X.-G. Zhang, P. Zhong and M.-L. Hu, Copper-Catalyzed Tandem C-N Bond Formation Reaction: Selective Synthesis of 2-(Trifluoromethyl)benzimidazoles, Synthesis, 2009, 1431 CAS.
- J. Zhu, Z. Chen, H. Xie, S. Li and Y.-M. Wu, An efficient method to access 2-fluoroalkylbenzimidazoles by PIDA oxidation of amidines, J. Fluorine Chem., 2012, 133, 134 CrossRef CAS.
- C.-L. Li, X.-G. Zhang, R.-Y. Tang, P. Zhong and J.-H. Li, Copper-Catalyzed Thiolation Annulations of 1,4-Dihalides with Sulfides Leading to 2-Trifluoromethyl Benzothiophenes and Benzothiazoles, J. Org. Chem., 2010, 75, 7037 CrossRef CAS PubMed.
- J. Zhu, Z. Chen, H. Xie, S. Li and Y.-M. Wu, A General and Straightforward Method for the Synthesis of 2-Trifluoromethylbenzothiazoles, Org. Lett., 2010, 12, 2434 CrossRef CAS PubMed.
- M. Kobayashi, K. Sadamune, H. Mizukami and K. Uneyama, Electrochemical Oxidation of 2-Substituted 3-N-(Arylamino)-4,4,4-trifluoro-2-butenoate. An Access to 3-Substituted 2-(Trifluoromethyl)-3H-indoles, J. Org. Chem., 1994, 59, 1909 CrossRef CAS.
- F. Ge, Z. Wang, W. Wan and J. Hao, Grignard Cyclization Reaction of Fluorinated N-Arylimidoyl Chlorides: A Novel and Facile Access to 2-Fluoroalkyl Indoles, Synlett, 2007, 447 CAS.
- Z. Wang, F. Ge, W. Wan, H. Jiang and J. Hao, Fluorinated N-[2-(haloalkyl)phenyl]imidoyl chloride, a key intermediate for the synthesis of 2-fluoroalkyl substituted indole derivatives via Grignard cyclization process, J. Fluorine Chem., 2007, 128, 1143 CrossRef CAS.
- Z. Wang and Q. Ma, A New and Efficient One-Pot Synthesis of 2-Fluoroalkyl Substituted Indoles, J. Heterocycl. Chem., 2015, 52, 1893 CrossRef CAS.
- X. Dong, Y. Hu, T. Xiao and L. Zhou, Synthesis of 2-trifluoromethyl indoles via visible-light induced intramolecular radical cyclization, RSC Adv., 2015, 5, 39625 RSC.
- J. Pedroni and N. Cramer, 2-(Trifluoromethyl)indoles via Pd(0)-Catalyzed C(sp3)-H Functionalization of Trifluoroacetimidoyl Chlorides, Org. Lett., 2016, 18, 1932 CrossRef CAS PubMed.
- S. E. Lopez, R. Gallagher, R. J. Gilliland, I. Ghiviriga and W. R. Dolbier Jr., Synthesis of 3-phenylsulfonyl-2-trifluoromethyl-1H-indoles: A copper catalyzed cyclization approach, J. Fluorine Chem., 2017, 193, 118 CrossRef CAS.
- H. Amii, Y. Kishikawa and K. Uneyama, Rh(I)-Catalyzed Coupling Cyclization of N-Aryl Trifluoroacetimidoyl Chlorides with Alkynes: One-Pot Synthesis of Fluorinated Quinolines, Org. Lett., 2001, 3, 1109 CrossRef CAS PubMed.
- D.-H. Gong, J.-F. Li and C.-Y. Yuan, A new and facile synthesis of 6-methyl-2-trifluoromethyl-4-(O,O-dialkyl)phosphoryl-quinolone, Chin. J. Chem., 2001, 19, 1263 CrossRef CAS.
- A. Isobe, J. Takagi, T. Katagiri and K. Uneyama, Palladium-Catalyzed Chloroimination of Imidoyl Chlorides to a Triple Bond: An Intramolecular Reaction Leading to 4-Chloroquinolines, Org. Lett., 2008, 10, 2657 CrossRef CAS PubMed.
- P. R. Likhar, M. S. Subhas, S. Roy, M. L. Kantam, B. Sridhar, R. K. Seth and S. Biswas, Synthesis of highly substituted 2-perfluoroalkyl quinolines by electrophilic iodocyclization of perfluoroalkyl propargyl imines/amines, Org. Biomol. Chem., 2009, 7, 85 RSC.
- Z. Chen, J. Zhu, H. Xie, S. Li, Y.-M. Wu and Y. Gong, Palladium catalyzed synthesis of 2-trifluoromethylquinolines through a domino Sonogashira-alkyne carbocyclization process, Chem. Commun., 2010, 46, 2145 RSC.
- S. Li, Y. Yuan, J. Zhu, H. Xie, Z. Chen and Y.-M. Wu, Efficient Synthesis of 2-Fluoromethylated Quinolines via Copper-Catalyzed Alkynylation and Cyclization of Fluorinated Imidoyl Iodides, Adv. Synth. Catal., 2010, 352, 1582 CrossRef CAS.
- N. Schützenmeister, M. Müller, U. M. Reinscheid, C. Griesinger and A. Leonov, Trapped in Misbelief for Almost 40 Years: Selective Synthesis of the Four Stereoisomers of Mefloquine, Chem. – Eur. J., 2013, 19, 17584 CrossRef PubMed.
- Y. Li, L. Zhang, L. Zhang, Y.-M. Wu and Y. Gong, Cu-catalyzed tandem reactions of fluorinated alkynes with sulfonyl azides en route to 2-trifluoromethylquinolines, Org. Biomol. Chem., 2013, 11, 7267 RSC.
- X. Dong, Y. Xu, J. Liu, Y. Hu, T. Xiao and L. Zhou, Visible-Light-Induced Radical Cyclization of Trifluoroacetimidoyl Chlorides with Alkynes: Catalytic Synthesis of 2-Trifluoromethyl Quinolines, Chem. – Eur. J., 2013, 19, 16928 CrossRef CAS PubMed.
- F. Rahmani and A. Darehkordi, Preparation of Trifluoromethylated (Arylaminomethylene)malononitriles Suitable for Synthesis of 4-Amino-2-(trifluoromethyl) quinoline Derivatives by Intramolecular Friedel-Crafts Reaction, Synlett, 2018, 50, 2124 CAS.
- F. Han, W. Yang, A. Zhao, R. Zheng, C. Ji, X. Liu, G. Liu and C. Chen, Highly Efficient Construction of 2-Fluoroalkyl Quinolines through a Palladium-Catalyzed 6-endo-trig Heck Cyclization Reaction, Asian J. Org. Chem., 2018, 7, 1124 CrossRef CAS.
- W.-Y. Wang, X. Feng, B.-L. Hu, C.-L. Deng and X.-G. Zhang, Synthesis of 6-(Trifluoromethyl)phenanthridines via Palladium-Catalyzed Tandem Suzuki/C-H Arylation Reactions, J. Org. Chem., 2013, 78, 6025 CrossRef CAS PubMed.
- Y. Li, J. Zhu, L. Zhang, Y.-M. Wu and Y. Gong, Synthesis of Fluorine-Containing Multisubstituted Phenanthridines by Rhodium-Catalyzed Alkyne [2 + 2 + 2] Cycloaddition and Tandem sp2 C-H Difluoromethylenation, Chem. – Eur. J., 2013, 19, 8294 CrossRef CAS PubMed.
- W. Fu, M. Zhu, F. Xu, Y. Fu, C. Xu and D. Zou, An approach to 6-trifluoromethyl-phenanthridines through visible-light-mediated intramolecular radical cyclization of trifluoroacetimidoyl chlorides, RSC Adv., 2014, 4, 17226 RSC.
- M. Zhu, W. Fu, G. Zou, C. Xu and Z. Wang, Palladium-catalyzed intramolecular C-H bond functionalization of trifluoroacetimidoyl chloride derivatives: Synthesis of 6-trifluoromethyl-phenanthridines, J. Fluorine Chem., 2014, 163, 23 CrossRef CAS.
- Z. Wang, T. Li, J. Zhao, X. Shi, D. Jiao, H. Zheng, C. Chen and B. Zhu, Expeditious Synthesis of 6-Fluoroalkyl-Phenanthridines via Palladium-Catalyzed Norbornene-Mediated Dehydrogenative Annulation, Org. Lett., 2018, 20, 6640 CrossRef CAS PubMed.
- Y. Bao, Z. Wang, C. Chen, B. Zhu, Y. Wang, J. Zhao, J. Gong, M. Han and C. Liu, Palladium-catalyzed tandem cyclization of fluorinated imidoyl chlorides with 2-bromophenylboronic acid: Synthesis of 6-fluoroalkyl-phenanthridines, Tetrahedron, 2019, 75, 1450 CrossRef CAS.
- Z. Chen, J. Zhu, H. Xie, S. Li, Y.-M. Wu and Y. Gong, Copper(I)-Catalyzed Synthesis of Novel 4-(Trifluoromethyl)-[1,2,3]triazolo[1,5-a]quinoxalines via Cascade Reactions of N-(o-Haloaryl)alkynylimine with Sodium Azide, Adv. Synth. Catal., 2010, 352, 1296 CrossRef CAS.
- J. Zhu, H. Xie, Z. Chen, S. Li and Y.-M. Wu, Synthesis of 6-trifluoromethylindolo[1,2-c]quinazolines and related heterocycles using N-(2-iodophenyl)trifluoroacetimidoyl chlorides as starting material via C-H bond functionalization, Chem. Commun., 2011, 47, 1512 RSC.
- J. Zhu, H. Xie, Z. Chen, S. Li and Y.-M. Wu, A detailed study of the intramolecular hydroamination of N-(ortho-alkynyl)aryl-N′-substituted trifluoroacetamidines and bromodifluoroacetamidines, Org. Biomol. Chem., 2012, 10, 516 RSC.
- A. Bodzioch, D. Pomiklo, M. Celeda, A. Pietrazk and P. Kaszynski, 3-Substituted Benzo[e][1,2,4]triazines: Synthesis and Electronic Effects of the C(3) Substituent, J. Org. Chem., 2019, 84, 6377 CrossRef CAS PubMed.
- K. Uneyama and K. Sugimoto, N-Substituted 2,2,2-trifluoroethanimidic acid 1-methylethylidene hydrazides as synthetic blocks for trifluoromethylated nitrogen heterocycles: syntheses and oxidative cyclizations, J. Org. Chem., 1992, 57, 6014 CrossRef CAS.
- L. Ielo, S. Touqeer, A. Roller, T. Langer, W. Holzer and V. Pace, Telescoped, Divergent, Chemoselective C1 and C1-C1 Homologation of Imine Surrogates: Access to Quaternary Chloro- and Halomethyl-Trifluoromethyl Aziridines, Angew. Chem., 2019, 58, 2479 CrossRef CAS PubMed.
- J. Chen, Z. Yin and Y. Huang, Phosphine-Catalyzed Divergent [4 + 3] Domino Annulations of CF3-Containing Imines with MBH Carbonates: Construction of Perfluoroalkylated Benzazepines, Org. Lett., 2019, 21, 7060 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2020 |