
Trifluoroacetimidoyl halides: a potent synthetic origin
 a
Xiao-Feng
Wu
*ab
a
Xiao-Feng
Wu
*ab
Abstract
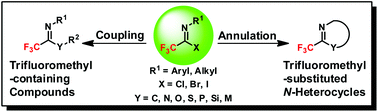
The preparation of fluorine-containing compounds has attracted considerable attention due to the important applications of their related chemicals. Among the candidates, trifluoroacetimidoyl halides are considered as potent trifluoromethyl synthons to construct a wide variety of trifluoromethyl-containing compounds and trifluoromethyl-substituted N-heterocycles, which have found extensive applications in the fields of organic synthesis, pharmaceuticals, agrochemicals, and materials science. In the review, recent advances in the synthetic applications of trifluoroacetimidoyl halides are summarized. We specially focused on two different reaction modes upon trifluoroacetimidoyl halides, namely, coupling and annulation reactions to illustrate their synthetic applications and potentials in the construction of valuable trifluoromethyl-containing molecules. Their preparations were covered as well.
 Please wait while we load your content...
Please wait while we load your content...

