
The synthesis of interface-modulated ultrathin Ni(II) MOF/g-C3N4 heterojunctions as efficient photocatalysts for CO2 reduction†
Lina
Zhao
ab,
Zhenlong
Zhao
a,
Yuxin
Li
 *a,
Xiaoyu
Chu
ab,
Zhijun
Li
a,
Yang
Qu
a,
Linlu
Bai
*a and
Liqiang
Jing
*a
*a,
Xiaoyu
Chu
ab,
Zhijun
Li
a,
Yang
Qu
a,
Linlu
Bai
*a and
Liqiang
Jing
*a
aKey Laboratory of Functional Inorganic Materials Chemistry (Ministry of Education), School of Chemistry and Materials Science, International Joint Research Center and Lab for Catalytic Technology, Heilongjiang University, Harbin 150080, P. R. China. E-mail: liyuxin@hlju.edu.cn; lbai2@e.ntu.edu.sg; jinglq@hlju.edu.cn
bDepartment of Food & Environmental Engineering, East University of Heilongjiang, Harbin 150066, P. R. China
First published on 15th April 2020
Abstract
It is highly desirable to improve charge separation and to provide catalytic functions for the efficient photocatalytic CO2 reduction reaction (CO2RR) on g-C3N4 (CN). Here, dimension-matched ultrathin NiMOF/CN heterojunctions have been successfully constructed by the in situ growth of NiMOF nanosheets on hydroxylated and 1,4-aminobenzoic acid (AA) functionalized CN nanosheets, respectively, with ultrasonic assistance. The resultant NiMOF/CN heterojunctions exhibited excellent photocatalytic activities for the CO2RR to produce CO and CH4, especially NiMOF/CN-AA, which had photoactivity 18 times higher than that of bare CN. Based on the surface photovoltage responses, wavelength-dependent photocurrent action spectra, electrochemical impedance spectra, and CO2 electrochemical reduction data, it is clearly confirmed that the exceptional photoactivity mainly resulted from the favorable charge transport properties of ultrathin CN and coupled NiMOF, and from the greatly enhanced charge separation via excited high-level electron transfer from CN to NiMOF in the resultant intimately contacted heterojunction caused by the induction effect of AA, and also from the provided catalytic functionality of the central Ni(II) for CO2 activation. This work provides a feasible synthetic protocol to fabricate MOF-containing dimension-matched heterojunctions with good charge separation for efficient photocatalysis.
Introduction
The photocatalytic CO2 reduction reaction (CO2RR) into valuable fuels and chemicals is considered as a sustainable and promising pathway to lessen fossil fuel depletion and related environmental pollution.1–4 Among various semiconductor photocatalysts, graphitic carbon nitride (CN) possesses a moderate band gap to harvest visible light and a relatively negative conduction band (CB) with sufficient thermodynamic energy5,6 and hence becomes a suitable photocatalyst candidate.7 Nevertheless, bulk CN always exhibits unfavourable photocatalytic performance due to the low specific surface area, rapid recombination of photogenerated electron–hole pairs, along with weak adsorption and activation of reactants.8,9 Therefore, it is desirable to develop feasible strategies to fabricate highly active CN-based photocatalysts for CO2RR. Controlling the morphology to obtain ultrathin two-dimensional (2D) CN nanosheets with an enlarged surface area and shortened charge transfer distance might remedy the intrinsic drawbacks of bulk CN to a certain extent. Based on CN nanosheets the construction of heterojunctions with traditional semiconductors like TiO2 and Fe2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10,11etc. or the introduction of noble metals are strategies commonly performed to further improve the photoactivity.12–15 However, the dimension mismatching and loosely-contacted interface of the fabricated heterojunctions, still comprehensively exist, which would hinder efficient interfacial charge transfer. Accordingly, it is significant to construct novel dimension-matched g-C3N4-based heterojunctions with an intimately contacted interface and catalytic functions.
10,11etc. or the introduction of noble metals are strategies commonly performed to further improve the photoactivity.12–15 However, the dimension mismatching and loosely-contacted interface of the fabricated heterojunctions, still comprehensively exist, which would hinder efficient interfacial charge transfer. Accordingly, it is significant to construct novel dimension-matched g-C3N4-based heterojunctions with an intimately contacted interface and catalytic functions.
In recent years, metal–organic frameworks (MOFs), constructed with coordinating inorganic metal ions and organic linkers, have attracted great attention in photocatalysis as promising semiconductor materials. MOFs have characteristic merits such as uniformly scattered active sites as catalytic centres, enriched pore systems and large specific surface areas, which are all adjustable by modulating the multiscale structures.16–19 Therefore, it is feasible to incorporate suitable MOFs with CN to construct heterojunctions and meanwhile introducing uniform catalytic sites.20–22 To date, some MOF/CN heterojunctions such as UiO-66/g-C3N4,23 ZIF-8/g-C3N4,24 BIF-20@g-C3N425 and NH2-UiO-66/NHx-CN,26 have already been reported to be effective for photocatalytic CO2RR. However, the MOFs involved are mostly three-dimensional (3D), hence rendering long-range distances of photogenerated carriers and especially poor dimension-matching with CN nanosheets.27 Moreover, the post-combination of MOF with CN would lead to less contacted interfaces.28,29 Therefore, it is necessary to adopt in situ synthetic methods to construct 2D/2D MOF/g-C3N4 heterojunctions by controllably growing 2D MOFs on CN nanosheets.
Among numerous MOFs investigated, Ni2(OH)2(BDC) (BDC = benzene-1,4-dicarboxylate, CCDC no. 985792), briefly named NiMOF, is an ideal coupler choice for CN because it has a laminar morphology, Ni clusters as potential catalytic sites, and relatively good electroconductivity allowing for fast electron transfer.30–34 Then the main challenge is how to realize the in situ growth of ultrathin 2D NiMOF on CN nanosheets. Naturally, it is reasonable to think of pre-modifying the BDC ligands covalently connected with CN to induce the topological structure of NiMOF to form on the CN surface. However, the conjugated structure makes CN lack relevant binding sites for BDC.26 Thus, it is necessary to functionalize CN in advance by introducing oxygen-containing groups, like hydroxyl and carboxylic groups to avoid destroying the conjugated structure.2,35 More importantly, during the process of in situ growth of NiMOF, thickening should be prevented to obtain a 2D morphology. A lot of the synthetic routes towards 2D MOFs have been studied, like surfactant-induction or physically assisted methods. By contrast, physical methodologies like ultrasonic treatment are more facile and feasible.
Based on the above, in this work we have fabricated ultrathin dimension-matched NiMOF/CN heterojunctions with intimate interfaces by the in situ growth of NiMOF on pre-modified CN by OH and 4-aminobenzoic acid (AA) groups, respectively, with ultrasonic assistance.36–39 The as-prepared NiMOF/CN heterojunctions exhibited excellent photocatalytic activities for CO2RR to produce CO and CH4 as the main products, among which NiMOF/CN-AA notably reaches 18 times the photoactivity of that for bare CN. With the assistance of the surface photovoltage responses, wavelength-dependent photocurrent action spectra, electrochemical impedance spectra, and CO2 electrochemical reduction, the mechanism is uncovered in detail showing that the favourable charge transport of ultrathin CN and coupled NiMOF resulted from the greatly-enhanced charge separation via the excited high-level electron transfer from CN to NiMOF in the resultant intimately-contacted heterojunction due to the induction effect of AA and then to the provided catalytic functions of Ni clusters for CO2 activation. This work provides a feasible synthetic protocol to fabricate 2D/2D MOF involved heterojunctions with good charge separation for efficient photocatalysis.
Experimental
All the chemicals and reagents were of analytic grade and used without further purification. Deionized water was used throughout the reaction.Synthesis of materials
xNiMOF/CN-OH and xNiMOF/CN-AA were produced by centrifuging, washing by ethanol and water, and freeze-drying under vacuum with the assistance of a large amount of water.
Characterization of materials
The X-ray powder diffraction (XRD) patterns of the samples were conducted by using a Bruker D8 advance diffractometer with CuKα radiation. The ultraviolet-visible diffuse reflectance spectra (UV-Vis DRS) were tested on a Model Shimadzu UV2550 spectrophotometer with BaSO4 as the reference. The Fourier-transform infrared (FT-IR) spectra were recorded with a Bruker Equinox 55 spectrometer with KBr as the diluent. The thickness of the samples was analysed by atomic force microscopy (AFM) on a multimode nanoscope VIII instrument (Bruker) with mica as the base. Transmission electron microscopy (TEM) images and data documented the morphology of the samples by using a FEI Tecnai G2 S-Twin instrument with a 200 kV acceleration voltage. The surface photovoltage spectroscopy (SPS) measurements for the samples were implemented on a home-built apparatus, equipped with a lock-in amplifier (SR830, USA) synchronized with a light chopper (SR540, USA). The electron paramagnetic resonance (EPR) measurements were conducted by a Bruker EMX plus model spectrometer operating at the X-band frequency. The X-ray photoelectron spectroscopy (XPS) was carried out with Kratos-AXIS ULTRA DLD, in which Al (Mono) was the X-ray source. CO2 TPD (temperature-programmed desorption) was tested by the Chemisorption Analyzer (Tp 5080 Chemisorb) with a TCD detector.Hydroxyl radical measurements
The detection of hydroxyl radicals (˙OH) was carried out by a coumarin fluorescence method. A 50 mL coumarin solution with a concentration of 1 × 10−3 M was firstly prepared. 0.05 g catalyst was added into the coumarin solution and stirred for 30 minutes, which ensured the realization of the adsorption–desorption equilibrium. Upon irradiation for 1 h, the suspension was centrifuged and then transferred into a Pyrex glass cell. The fluorescence data was recorded by a PerkinElmer LS55 spectrofluorometer. A 420 nm light filter was utilized to cut off UV light.Photoelectrochemical and electrochemical measurements
A traditional three-electrode system was utilized to measure the photoelectrochemical (PEC) and electrochemical (EC) reduction, except that the working electrode of the prepared sample, a platinum plate (99.9%) and a saturated KCl Ag/AgCl electrode were used as the counter electrode and reference electrode, respectively. During the experiments, 0.5 M Na2SO4 solution was used as the electrolyte, and high purity nitrogen gas was bubbled through. PEC experiments were performed in a quartz cell using a 300 W xenon lamp with a cut-off filter (λ > 420 nm) as the illumination source. The PEC and EC performance of a series of catalysts was tested on an IVIUM V13806 electrochemical workstation.Photocatalytic activity evaluation for CO2 conversion
The photocatalytic device contains a cylindrical steel reactor (100 mL volume and 3.5 cm2 area), a light source (300 W xenon arc lamp), and a gas system (high pure CO2). Then, the reactor was added by a suspension solution of 0.1 g catalyst and 5 mL water. CO2 gas was passed through the water and then into the reaction setup. After reaching ambient pressure, the photocatalyst was equilibrated in the CO2/H2O system for 1 hour, followed by irradiation for 4 h. During irradiation, 250 μL of the gas produced was taken from the reaction cell at given time intervals for CO and CH4 concentration analysis using a gas chromatograph (GC-7920 with TCD, Au Light, Beijing) and for O2 concentration analysis using a gas chromatograph (GC-7900 with TCD, Perfect Light, Beijing).Results and discussion
Structural characterization
As depicted in Scheme 1, the preparation of NiMOF/CN heterojunction photocatalysts contained two steps. The functionalized CN nanosheets, CN-OH and CN-AA, were firstly obtained under nitric acid and AA treatment, respectively. Next, the NiMOF nanosheets were in situ grown on the surface of CN-OH or CN-AA after adding the BDC ligand and NiCl2·6H2O in sequence under ultrasonication. It should be pointed out that the functionalization treatments with OH or AA to CN rendered different structures and morphologies of the NiMOF grown on CN in situ.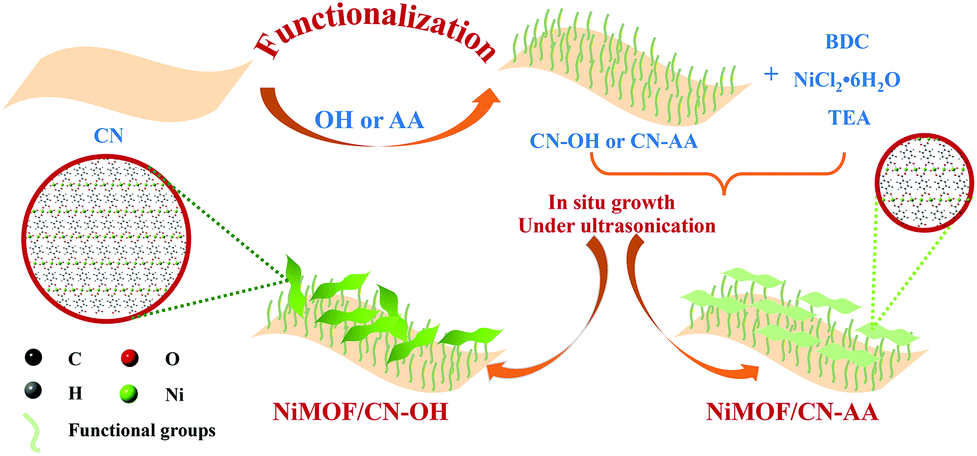 | ||
| Scheme 1 A schematic illustration of the formation of NiMOF/CN-OH and NiMOF/CN-AA heterojunctions. | ||
As shown in Fig. 1a, the XRD patterns showed that both 4NiMOF/CN-OH and 4NiMOF/CN-AA showed the characteristic peaks of CN located at 13.1° (100) and 27.3° (002), indicating that the structure of CN was preserved during functionalization treatment and in situ growth of NiMOF (Fig. 1a).40 It was also observed in 4NiMOF/CN-OH and 4NiMOF/CN-AA that tiny peaks at 8.9°, 14.1°, 15.8°, and 17.8° respectively reflected the (200), (001), (201) and (−201) planes of NiMOF (Fig. S1†).41 The weak XRD signals of NiMOF resulted from the low weight ratio. The coexistence of the characteristic peaks respectively assigned to CN and NiMOF clearly confirmed the successful combination of the two components. Noteworthily, the XRD peak at 8.9° of 4NiMOF/CN-AA was obviously weaker than that of 4NiMOF/CN-OH, which implied the thinner morphology and better dispersibility of the NiMOF nanosheets in 4NiMOF/CN-AA.42,43
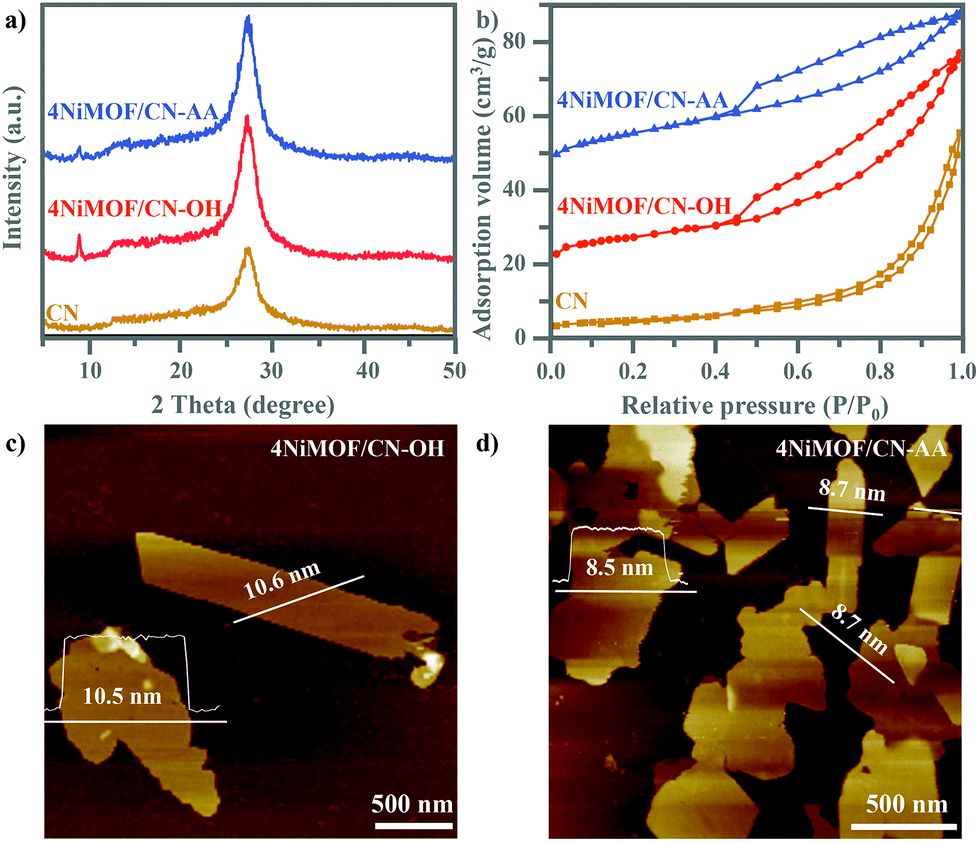 | ||
| Fig. 1 XRD patterns (a) and N2 sorption isotherm curves (b) from CN, 4NiMOF/CN-OH and 4NiMOF/CN-AA. AFM images of 4NiMOF/CN-OH (c) and 4NiMOF/CN-AA (d). | ||
Given the porous structure of NiMOF, the N2 physisorption properties were investigated, as shown in Fig. 1b. According to the classification by IUPAC, the N2 isotherm of 4NiMOF/CN-OH and 4NiMOF/CN-AA were identified as type-II curves with H4-type hysteresis loops, simultaneously possessing the micropores in the NiMOF frameworks and the mesopores originating from the slit-like pores formed by stacking of nanosheets.44 The as-measured Brunauer–Emmett–Teller (BET) surface areas of 4NiMOF/CN-OH and 4NiMOF/CN-AA were respectively 40.16 and 52.58 m2 g−1, much higher than the 35.14 m2 g−1 of CN. These results on the N2 physisorption properties, along with the above XRD patterns, indicated that the microporous NiMOF is successfully introduced to construct nanocomposites with functionalized CN. In these nanocomposites, CN possessed UV-vis absorption with edges at 460 nm (Fig. S2†), while the OH and AA functionalization and NiMOF coupling did not change the light absorption range. This was further verified by the UV-vis DRS spectrum of the single NiMOF nanosheets as fabricated by the controlled experiment where an indiscernible absorption peak appeared in the visible region (λ > 420 nm). The band gap energies (Eg) of CN and NiMOF were accordingly calculated to be 2.7 eV and 3.6 eV, respectively. Thus, under visible light CN would act as the light absorber and photogenerated charge producer, and then transfer to the NiMOF nanosheets which could play a significant role in accepting electrons to inhibit charge recombination and then execute catalytic behaviour. To better achieve this process, the NiMOF nanosheets should display good dispersibility and a thinner morphology, which is beneficial for charge transfer among interfaces and NiMOF inner structures.
AFM images depicted the 2D layer-like morphologies of the CN and functionalized CN with lateral sizes from 0.5 to 3 μm and with corresponding heights of uniformly 4.6 nm (Fig. S3†). After growing NiMOF, the height of 4NiMOF/CN-AA reached about 8.6 nm, with a thickness 4.0 nm thicker than that of CN-AA and a thickness 2.0 nm thinner than that of 4NiMOF/CN-OH. This result illustrated that 2D NiMOF nanosheets grew on the surfaces of functionalized CN nanosheets. In other words, 4NiMOF/CN-OH and 4NiMOF/CN-AA were classified to the dimension-matched heterojunctions. Moreover, given that the cell height of NiMOF is ∼1.0 nm,30 the NiMOF in the 4NiMOF/CN-OH and 4NiMOF/CN-AA was estimated as six and four layers, respectively. Integrating the layer numbers and weight ratios, it could be inferred that AA functionalization benefited the uniform dispersion of NiMOF on the surface of CN, which was consistent with the above-mentioned result on the XRD patterns. Predictably, the thinner NiMOF nanosheet would be favourable for charge separation and transfer.27
Electron microscope images revealed finer morphological features within the micro-nano scales. In the TEM images, all the prepared samples featured 2D sink-like structures, in which the edges were slightly curled (Fig. 2 and S4†). For 4NiMOF/CN-OH and 4NiMOF/CN-AA, it was hard to obviously observe the existence of bulky NiMOF, which indicated good dispersibility, without obvious aggregation, of NiMOF on the surface of CN at the sub-micrometre scale. Noteworthily, NiMOF was clearly observed in the HRTEM images of the 4NiMOF/CN-OH and 4NiMOF/CN-AA heterojunctions. Fig. 2b presented the nanoscale morphology of 4NiMOF/CN-OH, in which NiMOF nanosheets tightly adhered to the surface of CN-OH. Comparatively, 4NiMOF/CN-AA presented better dispersibility with a larger size and more uniform grayscale, both of which implied a higher dispersion of NiMOF on the surface of CN-AA (Fig. 2d). Predictably, the better dispersibility for 4NiMOF/CN-AA would be helpful for charge transfer from the interface to the whole surface and provided more accessible active sites for the dimension-matched heterojunction photocatalysts.27,45 In addition, elemental mapping analysis further proved that the 4NiMOF/CN-AA sample consisted of C, N, O and Ni elements (Fig. 2e–h). The dispersed Ni element confirmed that the NiMOF was uniformly dispersed on the CN surface. All the above information on morphology indicated the successful preparation of the dimension-matched heterojunctions by in situ growth of well-dispersed NiMOF onto the functionalized CN.
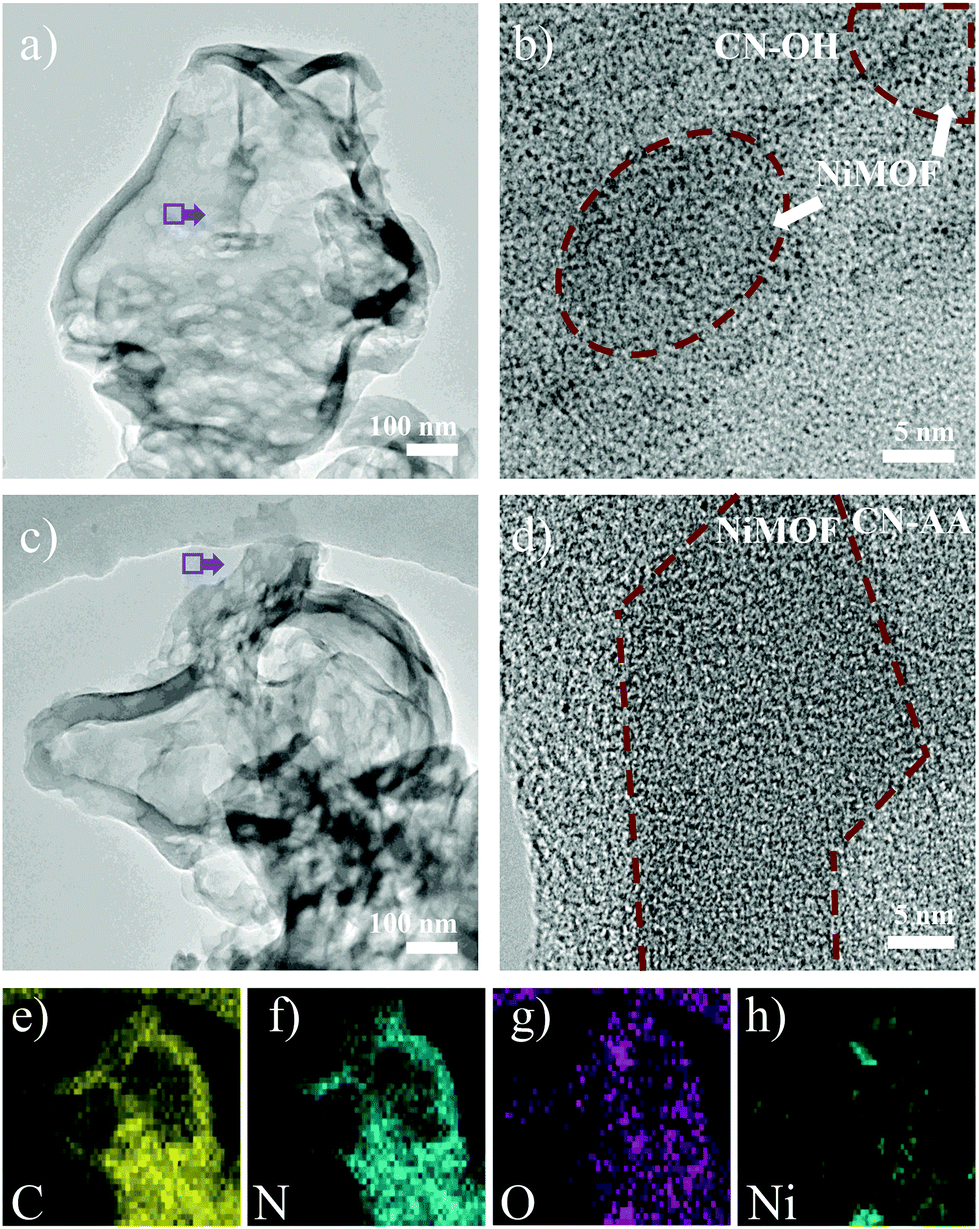 | ||
| Fig. 2 TEM (a) and HRTEM (b) images of 4NiMOF/CN-OH. TEM (c), HRTEM (d) and elemental mapping images (e–h) of 4NiMOF/CN-AA. | ||
The above information on morphology proved the successful preparation of the heterojunctions via in situ growth of well-dispersed NiMOF nanosheets onto the functionalized CN. Besides, interfacial connection is another significant consideration to influence the efficiency of charge separation and transfer in the dimension-matched heterojunctions. Therefore, the FT-IR and XPS spectra of the prepared samples were tested to finely analyse the interfacial connection between CN and NiMOF. Fig. 3a lists the FT-IR spectra of CN, CN-OH, CN-AA, 4NiMOF/CN-OH and 4NiMOF/CN-AA. All these samples exhibited similar profiles because of the low proportion of NiMOF and high coincidence of the characteristic peaks between CN and NiMOF. Still, valuable detailed information could be obtained through meticulous analysis. It could be observed in the FT-IR spectrum of CN-OH that the peak at 3040 cm−1 obviously strengthened and red-shifted after the hydroxylation, which demonstrated the successful introduction of the surface hydroxylic group on the CN.46 For CN-AA, two observations certified the success of AA functionalization. The first one is the red-shift of the peak at 1628 cm−1, which represents introduction of the carboxyl groups.35,37 Secondly, two peaks at 2874 and 2890 cm−1 were clearly observed, which were attributed to the stretching vibration of the C–H bond from the 1,4-substituted benzene of AA.47 In addition, the FT-IR spectra further revealed the connection approach between NiMOF and CN in both 4NiMOF/CN-OH and 4NiMOF/CN-AA. During the process of NiMOF growth, the BDC ligand and Ni2+ ion were introduced in sequential order, so it could be inferred that the BDC ligand is preferably connected with the functionalized CN. If not, the broad peak of the surface hydroxylic groups in the range of 3000–3200 cm−1 would obviously shift to 3600 cm−1 and be sharper under the coordination between Ni and CN-OH.48 However, a slight blue shift occurred and there was no sharpness observed due to the H-bond between the carboxylic group of the BDC ligand and the functional group on CN. This was also verified by the XPS spectra. The N 1s peaks of CN were all shifted to a lower binding energy, indicating that CN related to the NiMOF.2 The Ni 2p spectra of 4NiMOF/CN-OH and 4NiMOF/CN-AA possessed indiscernible alternation compared to that of NiMOF fabricated as reference (Fig. 3c). This demonstrated that there were few Ni ions coordinating with CN-OH or CN-AA. According to the above, it was concluded that the functionalized CN and NiMOF were chemically connected by the CN–OH(AA)–BDC–Ni mode.
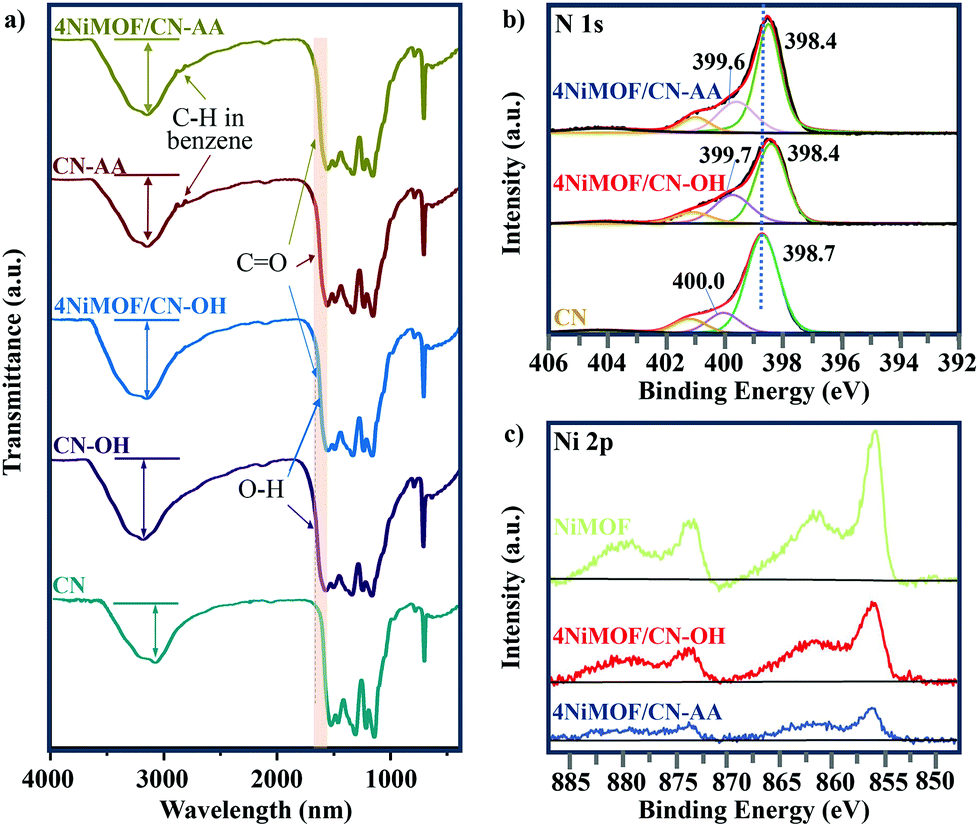 | ||
| Fig. 3 (a) FT-IR spectra of CN, CN-OH, CN-AA, 4NiMOF/CN-OH and 4NiMOF/CN-AA. (b) N 1s XPS spectra of CN, 4NiMOF/CN-OH and 4NiMOF/CN-AA. (c) Ni 2p XPS spectra of NiMOF, 4NiMOF/CN-OH and 4NiMOF/CN-AA. | ||
Based on the above results, dimension-matched NiMOF/CN-based heterojunctions with well-dispersed NiMOF and intimate CN–OH(AA)–BDC–Ni interfaces were synthesized by in situ growth of NiMOF onto the functionalized CN. The O atom introduced by functionalization provided more binding sites for the BDC ligand, which is beneficial to disperse the 2D NiMOF nanosheets. Since the AA groups with carboxylic groups have more O atoms than the OH groups, more BDC ligands could be bound on the surface of CN-AA, which renders the formation of NiMOF with better dispersibility and thinner thickness.
Photogenerated charge separation and transfer
Photophysical testing with steady-state surface photovoltage spectroscopy (SS-SPS) is an eminent strategy to reflect the properties of photogenerated charge carriers and was applied to examine the as-synthesized samples. Generally, a stronger SS-SPS response reflects a more efficient charge separation. Among the NiMOF/CN-OH heterojunctions with different amounts of NiMOF, 4NiMOF/CN-OH demonstrated the strongest SS-SPS signal and thereby the most efficient charge separation (Fig. S5†). With a lower proportion of NiMOF, the quantity of charge transfer will be limited, while with a higher proportion, the quality will be negatively influenced by aggregation of NiMOF. With the same loading amount of NiMOF, 4NiMOF/CN-AA showed a further strengthened SS-SPS response in comparison to that for 4NiMOF/CN-OH, indicating an improved charge separation (Fig. 4a). This mainly resulted from more dispersed and thinner NiMOF nanosheets formed by the induction of AA groups as well as the intimate interface, both of which could significantly facilitate the photogenerated charge transfer and separation.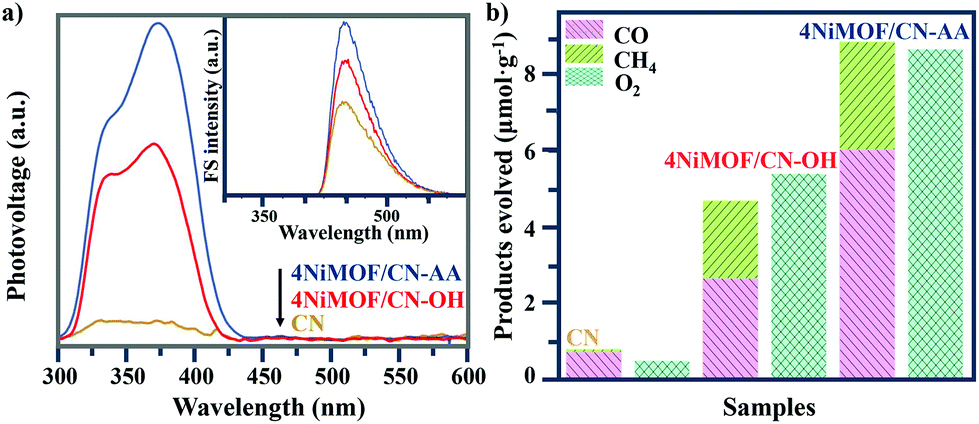 | ||
| Fig. 4 SS-SPS responses and FS spectra (inset) related to the amount of produced ˙OH (a) and the photocatalytic activities for CO2 conversion under visible-light irradiation for 4 hours (b) of CN, 4NiMOF/CN-OH and 4NiMOF/CN-AA. | ||
Photochemical analysis toward the amount of produced hydroxyl radicals (˙OH) could further verify the separation of photogenerated charge carriers in the photocatalytic process.49 A stronger fluorescent signal indicated an increased amount of ·OH, and further implied better charge separation. The coumarin fluorescence strategy is a well-accepted method to measure the amount of ·OH, where the coumarin reacts with ·OH to produce 7-hydroxyl-coumarin and emits an obvious luminescence at 430–550 nm. As shown in the Fig. 4a inset and S6,† the order of the fluorescent intensities of the NiMOF/CN-OH samples was in good agreement with the above photophysical results. In addition, the observed fluorescent intensity order was as follows: CN < 4NiMOF/CN-OH < 4NiMOF/CN-AA, which was also in good accordance with that of the SS-SPS results. Integrating the results of the photophysical and photochemical results, it could be concluded that the photogenerated charge transfer and separation was significantly improved after in situ growth of 4% NiMOF onto functionalized CN, especially for CN-AA. Additionally, the transient photocurrent–time (I–t) curves supported the above results and further presented the stability of the as-prepared photocatalysts (Fig. S7†).
Evaluation of photocatalytic activities
The photocatalytic activities for CO2RR were conducted under visible-light irradiation with a 420 nm cut-off filter. The reduction products, CH4 and CO, and the oxidation product of O2 were detected. As shown in Fig. 4b and S8,† the photoactivities on yielding CO, CH4 and O2 had low activities for CN, NiMOF and functionalized CN, but achieved a significant enhancement when the NiMOF was grown on CN-OH and CN-AA. Among the serial heterojunctions with a proper amount of NiMOF, 4NiMOF/CN-OH displayed the highest photocatalytic performance due to the improvement on charge separation and transfer (Fig. S8†). Impressively, 4NiMOF/CN-AA exhibited the best photoactivity with 11-fold and nearly 2-fold enhancements, respectively, compared with CN and 4NiMOF/CN-OH. For all the samples, the order of the photoactivities was consistent with those of the photophysical and photochemical results, indicating that the enhanced photoactivities would be tightly associated with better NiMOF dispersibility and improved charge separation.Compared with similar work reported (Table S1†),23–26 the photocatalytic activities of 4NiMOF/CN-AA in our study exhibited an excellent performance for the aqueous CO2RR among the MOF/g-C3N4-based heterojunction photocatalysts. For 4NiMOF/CN-AA, the amounts of evolved CH4 and CO linearly increased with increasing the irradiation time, and there was nearly no activity loss after five-cycle reactions (Fig. S9†). These results reflected the stable nature of the as-fabricated heterojunction photocatalysts.
Discussion of the mechanism
As described above, the photocatalytic activities of CN can be greatly improved by in situ growth of NiMOF with the induction of OH or AA to obtain a series of NiMOF/CN heterojunction photocatalysts. This has been proved as the promoted charge transfer and separation caused by the matched 2D/2D structures and efficient interfacial connection between functionalized CN and NiMOF. This is further supported by electrochemical impedance spectra of three typical samples (Fig. 5a). 4NiMOF/CN-OH and 4NiMOF/CN-AA exhibited an obviously decreased capacitive arc radius compared with CN, implying that the charge recombination was inhibited by transferring the photogenerated electrons from CN to NiMOF. Moreover, 4NiMOF/CN-AA showed the smallest arc radius, which demonstrated that the well-dispersed NiMOF on CN-AA had a better charge transfer capacity.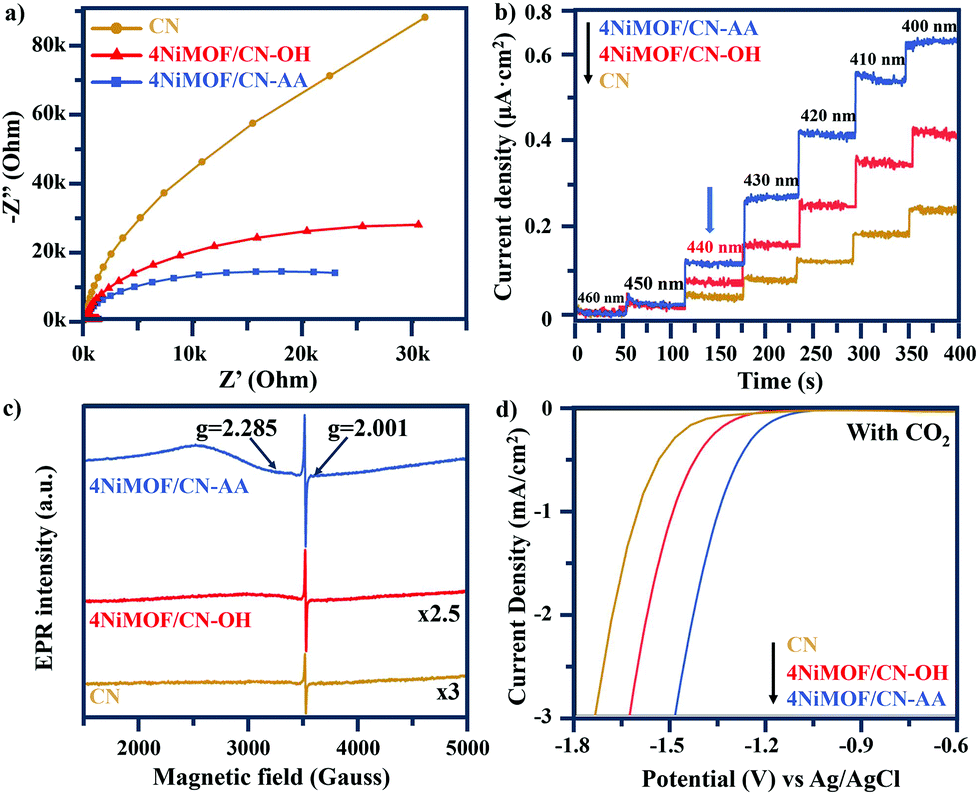 | ||
| Fig. 5 Electrochemical impedance spectra (a), normalized photocurrent action spectra under different monochromatic light irradiation (b), EPR spectra with UV-vis light irradiation at 98 K (c) and electrochemical reduction curves in the CO2-bubbled system (d) of CN, 4NiMOF/CN-OH and 4NiMOF/CN-AA. | ||
The charge separation might be attributed to the photoelectron transfer from CN to the as-grown NiMOF acting as an electron-accepting platform in the NiMOF/CN heterojunctions via the well-modulated intimate interfaces. To verify the possibility for CO2 photoreduction, Mott–Schottky spectra of CN and NiMOF were carried out at the frequency of 500, 1000 and 1500 Hz (Fig. S10†). The resulting positive slopes of the C2 – E values (vs. the applied potentials) were consistent with that of the typical n-type semiconductors, and the intersection at −1.46 eV for CN and −1.53 eV for NiMOF vs. Ag/AgCl (i.e. −1.26 eV for CN and −1.33 eV for NiMOF vs. NHE) was determined as the flat band position. Given that generally for the n-type semiconductor the bottom of the conduction band (CB) for CN or lowest unoccupied molecular orbital (LUMO) for NiMOF is approximately equal to the flat-band potential, the CB of CN and the LUMO of NiMOF were determined as −1.26 eV and −1.33 eV vs. NHE, respectively. With the bandgap energy provided by the Tauc plot (Fig. S2†), the valence band (VB) of CN was calculated as +1.49 eV, and the highest occupied molecular orbital (HOMO) of NiMOF was +2.27 eV. Given the more negative potential of CN and NiMOF in the heterojunctions than the reduction potential of CO2 reduction, it is theoretically feasible for the photocatalytic CO2 reduction to CO and CH4. Because the energy position of the LUMO of NiMOF was slightly more negative than the CB of CN, it was inferred that the electrons could transfer by means of high-level electron transfer.
In order to verify this hypothesis, monochromatic photocurrent action spectra were thus examined. As shown in Fig. 5b, the photocurrent density of 4NiMOF/CN-OH and 4NiMOF/CN-AA gradually increased as the excitation wavelength decreased from 460 to 400 nm with steps of 10 nm. Noticeably, it began to sharply increase under 440 nm excitation, which was the threshold wavelength to facilitate the transfer of visible-light excited appropriate-energy electrons from CN to NiMOF. The difference between the absorption edge at 460 nm and the threshold wavelength at 440 nm verified that the photogenerated electrons should be excited to a higher energy level of the CB of CN so as to realize the photoelectron transfer. Subsequently, a low-temperature EPR technique was employed to explore the process of photoelectron transfer from CN to NiMOF (Fig. 5c). The weak EPR signal of CN at g = 2.001 was assigned to the unpaired electrons on the conjugated CN aromatic rings.23 After growing NiMOF, this signal becomes stronger, especially for 4NiMOF/CN-AA. This indicated that the introduction of NiMOF efficiently inhibited the charge recombination and generated more free electrons. This corresponded well with the results of the fluorescent spectra. It was remarkable that the unpaired electron in Ni(I) was directly observed for 4NiMOF/CN-OH and 4NiMOF/CN-AA. The signals at g = 2.285 in the EPR spectra were attributed to the unpaired electron in the 3dx2−y2 orbital of Ni.43 However, Ni(I) complexes were extremely thermally unstable and disproportionately formed Ni(0) and Ni(II) species. The formation of Ni(I) species was due to the partially reduced central Ni(II) in NiMOF by the photoelectrons transferred from CN. In addition, 4NiMOF/CN-AA possessed a much stronger signal at g = 2.285 in comparison to 4NiMOF/CN-OH, which confirmed the better capacity of charge transfer and separation for 4NiMOF/CN-AA.
Electrochemical reduction measurements were also performed to explore the mechanism of CO2 conversion (Fig. 5d and S11†). In both of the N2 and CO2 bubbled systems, the potential values had the order 4NiMOF/CN-AA < 4NiMOF/CN-OH < CN. This result confirmed that both 4NiMOF/CN-OH and 4NiMOF/CN-AA were beneficial to improve the photocatalytic activity for the CO2 reduction. It also demonstrated that the NiMOF possessed catalytic potential. By comparison, the samples in the CO2 bubbled system had lower potential values than those in the N2 bubbled system, which illustrated that the reduction behaviour of CO2 was also much preferred compared with H2O reduction. This indicated that the coupled NiMOF could provide good catalytic functions for CO2 reduction, which was further supported by the promoted adsorption of CO2 based on the CO2 TPD spectra (Fig. S12†). Thus, it was deduced that the specific central Ni(II) species in the NiMOF/CN heterojunctions were the catalytic sites for activating CO2 and hence facilitated the reduction by the photoelectrons.
Based on all the above results, a schematic process has been proposed about high-energy-level electron transfer over dimension-matched intimately contacted 4NiMOF/CN-OH and 4NiMOF/CN-AA heterojunction photocatalysts (Fig. 6a). Under visible-light irradiation, the photogenerated electrons on CN were excited to the energy level higher than the CB, and then transferred to the lowest unoccupied molecular orbital (LUMO) of NiMOF via the CN–OH/AA–BDC–Ni interface. Subsequently, the Ni(II) species could accept the high-energy-level photoelectrons from the BDC ligand and then catalyse the CO2RR. In this case, the spatially separated photogenerated holes on CN and electrons on NiMOF would possess sufficient thermodynamic energy to induce redox reactions, respectively, leading to the promoted charge separation and hence to the improved photocatalytic activities. Moreover, the photocatalytic activities for CO2 conversion could be further improved under the irradiation of UV-visible light. As shown in Fig. 6b, the photocatalytic activity on the total products of CO and CH4 was 54.5 μmol g−1 for 4NiMOF/CN-AA, an 18-fold improvement compared with that of CN. Compared with visible light irradiation, the activities under UV-vis irradiation possessed a 6-fold improvement for both 4NiMOF/CN-OH and 4NiMOF/CN-AA. This large improvement further confirmed the as-proposed mechanism on high-electron-level electron transfer in that the electrons were excited to higher energy levels under UV irradiation and were more beneficial to electron transfer.
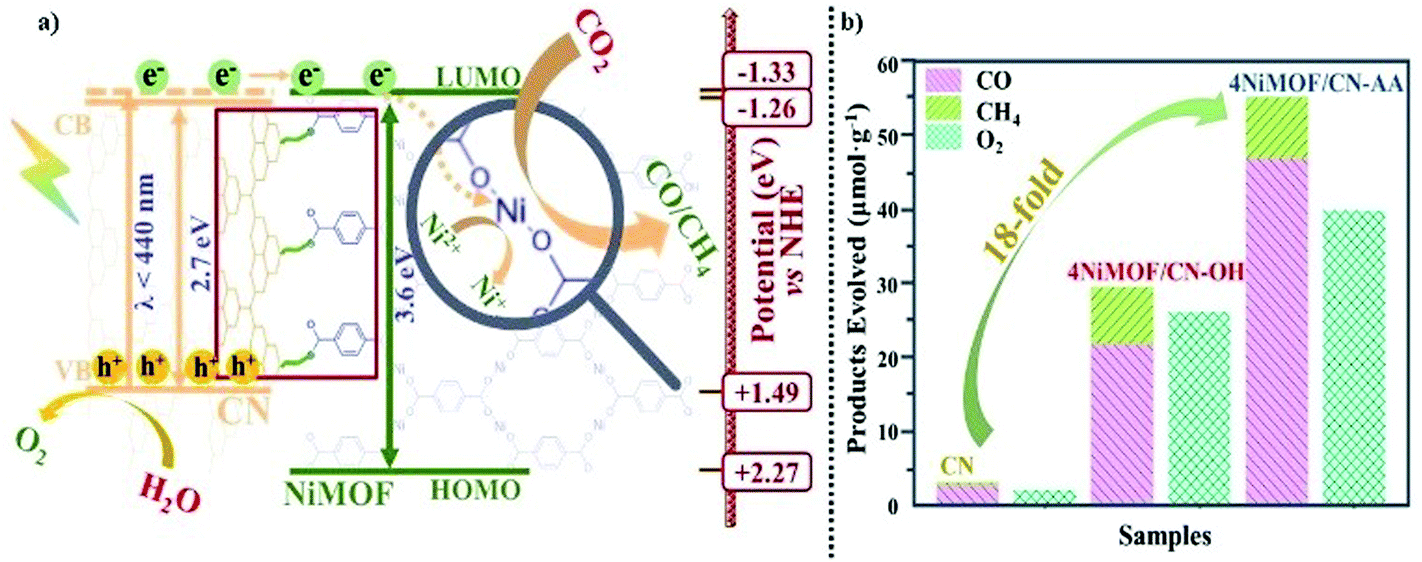 | ||
| Fig. 6 (a) A schematic diagram of the photogenerated charge transfer process and the induced photochemical reaction in the resultant NiMOF/functionalized CN nanocomposite. (b) Photocatalytic activities for CO2 conversion under UV-vis irradiation for 4 hours of CN, 4NiMOF/CN-OH and 4NiMOF/CN-AA. | ||
To clearly certify the influence on the photoactivities by dimension matching, a control experiment was carried out in which a reference sample was prepared, namely 4B-NiMOF/CN-AA, by in situ growing 4% bulky NiMOF onto CN-AA under hydrothermal conditions. Besides, 4CoMOF/CN-AA and 4ZnMOF/CN-AA were also prepared by substituting Ni2+ ions with Co2+ or Zn2+ ions during the process of in situ growth. These reference samples sharply decreased photoactivity for CO2 conversion compared with 4NiMOF/CN-AA (Fig. S13†). The SS-SPS spectra certified the weak charge separation of 4B-NiMOF/CN-AA due to the thicker morphology and bad dispersibility (Fig. S5†). The electrochemical reduction spectra revealed the decreased catalytic capacities for 4CoMOF/CN-AA and 4ZnMOF/CN-AA due to their increased potential values as seen in Fig. S14.† These results further confirmed that the excellent photocatalytic properties on CO2RR were due to the dimension-matched structures, well-modulated interfaces and introduced Ni active centres.
Conclusions
In summary, dimension-matched NiMOF/CN-OH and NiMOF/CN-AA heterojunctions with CN–OH/AA–BDC–Ni interfacial connection were successfully prepared by means of in situ growth with ultrasonic assistance. These nanocomposites exhibited excellent photocatalytic activities for the CO2RR to produce CO and CH4. It was clear that the exceptional photocatalytic activities mainly resulted from the favourable charge transport within the ultrathin CN and coupled NiMOF nanosheets and from the greatly-enhanced charge separation via excited high-level electron transfer from CN to NiMOF in the resultant intimately contacted heterojunction caused by surface oxygen-containing functionalization, and also from the provided catalytic functionality of the Ni clusters for CO2 activation. In particular, all the structural factors, such as the in situ growth approach, ultrasonic assistance, good dispersibility, well-modulated interfaces and provided active sites, are quite important for an efficient photocatalytic CO2RR. This work offers unique insight into CN-based heterojunction systems, which will be essential for the development of CO2 photocatalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for support from the National Natural Science Foundation of China (U1805255), and Scientific Research Project of East University of Heilongjiang (HDFHX180106).Notes and references
- Z. Sun, N. Talreja, H. Tao, J. Texter, M. Muhler, J. Strunk and J. Chen, Angew. Chem., Int. Ed., 2018, 57, 7610 CrossRef CAS PubMed.
- J. Bian, J. Feng, Z. Zhang, Z. Li, Y. Zhang, Y. Liu, S. Ali, Y. Qu, L. Bai, J. Xie, D. Tang, X. Li, F. Bai, J. Tang and L. Jing, Angew. Chem., Int. Ed., 2019, 58, 10873 CrossRef CAS PubMed.
- X. Li, Y. Sun, J. Xu, Y. Shao, J. Wu, X. Xu, Y. Pan, H. Ju, J. Zhu and Y. Xie, Nat. Energy, 2019, 4, 690 CrossRef CAS.
- Z. Guo, G. Chen, C. Cometto, B. Ma, H. Zhao, T. Groizard, L. Chen, H. Fan, W. Man, S. Yiu, K. Lau, T. Lar and M. Rober, Nat. Catal., 2019, 2, 830 CrossRef CAS.
- J. Fu, J. Yu, C. Jiang and B. Cheng, Adv. Energy Mater., 2018, 8, 1701503 CrossRef.
- C. Tan, X. Cao, X. Wu, Q. He, J. Yang, X. Zhang, J. Chen, W. Zhao, S. Han, G. Nam, M. Sindoro and H. Zhang, Chem. Rev., 2017, 117, 6225 CrossRef CAS PubMed.
- W. Zhang, A. Mohamed and W. Ong, Angew. Chem., Int. Ed., 2020 DOI:10.1002/ange.201914925.
- Y. Xiao, G. Tian, W. Li, Y. Xie, B. Jiang, C. Tian, D. Zhao and H. Fu, J. Am. Chem. Soc., 2019, 141, 2508 CrossRef CAS PubMed.
- S. Sun and S. Liang, Nanoscale, 2017, 9, 10544 RSC.
- X. Zhang, X. Zhang, J. Li, J. Sun, J. Bian, J. Wang, Y. Qu, R. Yan, C. Qin and L. Jing, Appl. Catal., B, 2018, 237, 50 CrossRef CAS.
- J. Wang, C. Qin, H. Wang, M. Chu, A. Zada, X. Zhang, J. Li, F. Raziq, Y. Qu and L. Jing, Appl. Catal., B, 2018, 221, 459 CrossRef CAS.
- W. Ong, L. Tan, Y. Ng, S. Yong and S. Chai, Chem. Rev., 2016, 116, 7159 CrossRef CAS PubMed.
- H. Zhang, X. Gu, P. Liu, J. Song, J. Cheng and H. Su, J. Mater. Chem. A, 2017, 5, 2288 RSC.
- L. Kong, Y. Dong, P. Jiang, G. Wang, H. Zhang and N. Zhao, J. Mater. Chem. A, 2016, 4, 9998 RSC.
- M. Chu, K. Hu, J. Wang, Y. Liu, S. Ali, C. Qin and L. Jing, Appl. Catal., B, 2019, 243, 57 CrossRef CAS.
- Q. Wang and D. Astruc, Chem. Rev., 2020, 120, 1438 CrossRef CAS PubMed.
- W. Jiang, N. Zhang, X. Zheng, C. Gao, L. Wang, Q. Zhang, Y. Zhu and Y. Xiong, J. Am. Chem. Soc., 2019, 141, 10924 CrossRef PubMed.
- S. Zhong, Y. Xi, Q. Chen, J. Chen and S. Bai, Nanoscale, 2020, 12, 5764 RSC.
- J. Duan, Y. Li, Y. Pan, N. Behera and W. Jin, Coord. Chem. Rev., 2019, 395, 25 CrossRef CAS.
- J. Wang, C. Wang and W. Lin, ACS Catal., 2012, 2, 2630 CrossRef CAS.
- Q. Yang, Q. Xu and H. Jiang, Chem. Soc. Rev., 2017, 46, 4774 RSC.
- H. Wang, X. Yuan, Y. Wu, G. Zeng, X. Chen, L. Leng and H. Li, Appl. Catal., B, 2015, 174–175, 445 CrossRef CAS.
- L. Shi, T. Wang, H. Zhang, K. Chang and J. Ye, Adv. Funct. Mater., 2015, 25, 5360 CrossRef CAS.
- S. Liu, F. Chen, S. Li, X. Peng and Y. Xiong, Appl. Catal., B, 2017, 211, 1 CrossRef CAS.
- G. Xu, H. Zhang, J. Wei, H. Zhang, X. Wu, Y. Li, C. Li, J. Zhang and J. Ye, ACS Nano, 2018, 12, 5333 CrossRef CAS PubMed.
- Y. Wang, L. Guo, Y. Zeng, H. Guo, S. Wan, M. Ou, S. Zhang and Q. Zhong, ACS Appl. Mater. Interfaces, 2019, 11, 30673 CrossRef CAS PubMed.
- M. Zhao, Y. Huang, Y. Peng, Z. Huang, Q. Ma and H. Zhang, Chem. Soc. Rev., 2018, 47, 6267 RSC.
- R. Li, W. Zhang and K. Zhou, Adv. Mater., 2018, 30, 1705512 CrossRef PubMed.
- A. Dhakshinamoorthy, A. Asiri and H. Garcia, Adv. Mater., 2019, 31, 1900617 CrossRef CAS PubMed.
- S. Zhao, Y. Wang, J. Dong, C. He, H. Yin, P. An, K. Zhao, X. Zhang, C. Gao, L. Zhang, J. Lv, J. Wang, J. Zhang, A. Khattak, N. Khan, Z. Wei, J. Zhang, S. Liu, H. Zhao and Z. Tang, Nat. Energy, 2016, 1, 16184 CrossRef CAS.
- J. Duan, S. Chen and C. Zhao, Nat. Commun., 2017, 8, 1 CrossRef PubMed.
- Y. Jiao, J. Pei, C. Yan, D. Chen, Y. Hu and G. Chen, J. Mater. Chem. A, 2016, 4, 13344 RSC.
- B. Han, X. Ou, Z. Deng, Y. Song, C. Tian, H. Deng, Y. Xu and Z. Lin, Angew. Chem., Int. Ed., 2018, 57, 16811 CrossRef CAS PubMed.
- A. Cao, L. Zhang, Y. Wang, H. Zhao, H. Deng, X. Liu, Z. Lin, X. Su and F. Yue, ACS Sustainable Chem. Eng., 2019, 7, 2492 CrossRef CAS.
- G. Zhou, M. Wu, Q. Xing, F. Li, H. Liu, X. Luo, J. Zou, J. Luo and A. Zhang, Appl. Catal., B, 2018, 220, 607 CrossRef CAS.
- H. Wang, C. Qian, J. Liu, Y. Zeng, D. Wang, W. Zhou, L. Gu, H. Wu, G. Liu and Y. Zhao, J. Am. Chem. Soc., 2020, 142, 4862 CrossRef CAS PubMed.
- M. Jahan, Q. Bao, J. Yang and K. Loh, J. Am. Chem. Soc., 2010, 132, 14487 CrossRef CAS PubMed.
- J. Xu, S. He, H. Zhang, J. Huang, H. Lin, X. Wang and J. Long, J. Mater. Chem. A, 2015, 3, 24261 RSC.
- B. Li, L. Sun, J. Bian, N. Sun, J. Sun, L. Chen, Z. Li and L. Jing, Appl. Catal., B, 2020, 270, 118849 CrossRef CAS.
- S. Yan, Z. Li and Z. Zou, Langmuir, 2009, 25, 10397 CrossRef CAS PubMed.
- A. Mesbah, P. Rabu, R. Sibille, S. Lebegue, T. Mazet, B. Malaman and M. Francois, Inorg. Chem., 2014, 53, 872 CrossRef CAS PubMed.
- Z. Liang, C. Qu, D. Xia, R. Zou and Q. Xu, Angew. Chem., Int. Ed., 2018, 57, 9604 CrossRef CAS PubMed.
- H. Yang, S. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. Chen, C. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140 CrossRef CAS.
- J. Duan, S. Chen and C. Zhao, Nat. Commun., 2017, 8, 15341 CrossRef CAS PubMed.
- H. Zhang, Adv. Mater., 2015, 27, 7372 CrossRef PubMed.
- H. Zhao, S. Wang, F. He, J. Zhang, L. Chen, P. Dong, Z. Tai, Y. Wang, H. Gao and C. Zhao, Carbon, 2019, 150, 340 CrossRef CAS.
- A. Kumar, J. Kudva, S. Kumar, U. Vishwanatha, V. Kumar and D. Naral, J. Mol. Struct., 2018, 1167, 142 CrossRef.
- S. Halder, J. Mondal, J. Ortega-Castro, A. Frontera and P. Roy, Dalton Trans., 2017, 46, 1943 RSC.
- Y. Nakabayashi and Y. Nosaka, J. Phys. Chem. C, 2013, 117, 23832 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr02551h |
| This journal is © The Royal Society of Chemistry 2020 |