
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular generation targeting desired electronic properties via deep generative models†
Qi
Yuan
 a,
Alejandro
Santana-Bonilla
a,
Martijn A.
Zwijnenburg
b and
Kim E.
Jelfs
*a
a,
Alejandro
Santana-Bonilla
a,
Martijn A.
Zwijnenburg
b and
Kim E.
Jelfs
*a
aDepartment of Chemistry, Molecular Sciences Research Hub, White City Campus, Imperial College London, Wood Lane, London, W12 0BZ, UK. E-mail: k.jelfs@imperial.ac.uk; Tel: +44 (0)207 594 3438
bDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK
First published on 9th March 2020
Abstract
As we seek to discover new functional materials, we need ways to explore the vast chemical space of precursor building blocks, not only generating large numbers of possible building blocks to investigate, but trying to find non-obvious options, that we might not suggest by chemical experience alone. Artificial intelligence techniques provide a possible avenue to generate large numbers of organic building blocks for functional materials, and can even do so from very small initial libraries of known building blocks. Specifically, we demonstrate the application of deep recurrent neural networks for the exploration of the chemical space of building blocks for a test case of donor–acceptor oligomers with specific electronic properties. The recurrent neural network learned how to produce novel donor–acceptor oligomers by trading off between selected atomic substitutions, such as halogenation or methylation, and molecular features such as the oligomer's size. The electronic and structural properties of the generated oligomers can be tuned by sampling from different subsets of the training database, which enabled us to enrich the library of donor–acceptors towards desired properties. We generated approximately 1700 new donor–acceptor oligomers with a recurrent neural network tuned to target oligomers with a HOMO–LUMO gap <2 eV and a dipole moment <2 Debye, which could have potential application in organic photovoltaics.
Introduction
The successful development of new functional molecules remains one of the most important challenges to be addressed, not only due to the vastness of the chemical space to be explored, but also given the level of specificity required in the targeted properties for each application. In many cases, serendipity has played a fundamental role in the discovery and production of new materials and molecules. Recently, there is growing interest in applying artificial intelligence (AI) to the discovery of novel functional molecules, particularly in the field of drug discovery, with the aim of both exploring larger chemical space and saving the time and cost involved in the experimental synthesis and characterisation of such molecules.1–4The use of AI in material discovery generally falls into two categories. Firstly, predictive AI models, based upon supervised machine learning, are becoming more common in the use for the computation of material properties, calculating the properties of interest at reasonable accuracy, but a fraction of the computational cost compared to widely used electronic structure calculations. Predictive AI models have been applied widely for material discovery tasks, including organic photovoltaics,5–8 bioinspired hierarchical composites9 and supercompressible polymers.10 Secondly, there are generative models, which use unsupervised machine learning such that a model learns from a dataset and can then produce data of a similar format. This can be applied in chemistry to produce novel molecules from libraries of known molecules. Generative models have been reported mainly for drug or drug-like molecules,11 with only limited application for other types of functional molecules, for example non-fullerene electron acceptors12 and thermally conductive polymers.13 Our study here focuses on developing deep generative AI models for the discovery of novel donor–acceptor oligomers with desired electronic properties with potential application as organic semiconductors.
Development of a deep generative model for material discovery can be divided into several tasks. The first task is to represent the molecules of interest in a way that is easy to be read and written by a computer. A standard representation of molecules is the simplified molecular-input line-entry system (SMILES), which encodes molecular graphs compactly as human-readable strings. The SMILES representations of molecules highly resemble that of natural language, where long range dependencies are crucial for a reasonable representation. For example, the SMILES of benzene is c1ccccc1, where the two ‘1's represent the opening and closing of the ring structure in the molecule and must both be present in the SMILES, however, the ‘1's are not neighbours to each other in the representation and a generative model needs to ‘remember’ the position of the first ‘1’ in the string and close the ring at chemical plausible positions. The generation of novel SMILES strings can thus be regarded as natural language generation and a recurrent neural network (RNN) is a typical tool for such tasks. An RNN is a class of artificial neural network that has a temporal memory, thus enabling the wcapture of long range dependencies within a message (such as a SMILES representation) and it has been widely used in many different areas, such as natural language processing14 and music generation.15 As reviewed recently by Elton et al., RNNs are also an important architecture for molecular generation, especially in drug discovery.11 For example, Bjerrum et al. reported an RNN model trained using the 1.6 million fragment-like and 1.3 million drug-like SMILES from the ZINC12 database16 of commercially-available compounds as training sets,17 and they generated 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 new SMILES structures from each of the RNN models.
000 new SMILES structures from each of the RNN models.
Another key task in the discovery of novel functional materials is to fine-tune the molecular generation models towards preferred properties. According to the chemical space project, there are at least 166.4 billion molecules that contain at most 17 heavyatoms.18 Therefore, enumerating such a vast chemical space is almost impossible, and in many cases unnecessary. One method to attempt to efficiently explore chemical space using deep generative models for molecular discovery was defined as inverse molecular design by Sanchez-Lengeling et al.,19 where existing functional molecules were used as starting point for the discovery of novel materials. The tools for optimizing towards target molecules include variational autoencoders (VAE),4 generative adversarial networks (GAN),20 reinforcement learning (RL),2 and transfer learning (TL), all of which can be incorporated with generative RNN models. TL refers to reusing the machine learning model developed for one task towards another related task with parameter fine-tuning. For the discovery of novel functional molecules from an existing molecular library of limited size, TL can be applied to fine-tune the parameters of a generative model trained from larger databases. Waller et al. developed a generative RNN model using long short-term memory (LSTM) and fine-tuned the parameters of the RNN model to generate molecules that were believed to target selected bacteria.1 Merk et al. discovered new chemical entities that are inspired by pharmacologically active natural products via fine-tuning the RNN model trained from ChEMBL database using TL.3
Recent development and design of electronic donor–acceptors has attracted significant attention due to their application in organic light emitting diodes (OLEDs),21 organic photovoltaics (OPVs),22 and non-linear optical (NLO) materials.23 The donor–acceptor oligomers need to be modified in order to obtain desired electronic properties.24,25 For example, the optical gap of donor–acceptor oligomers usually needs to be lowered towards the long wavelength region for OPVs to enhance light absorption. Discovery of novel donor–acceptor oligomers often involves chemical modification of existing donor and acceptor moieties26 and the combinatorial exploration of donor–acceptor libraries,27 which requires expertise in synthetic chemistry and careful experimental design.28,29 Such methodologies, while being robust and effective at producing a large number of possible oligomers, rely heavily on the expertise of chemists and can limit the accessible chemical space of the oligomers being generated.1 In addition, the mere combination of a ‘good’ donor and ‘good’ acceptor is known to miss potential oligomers using less ‘extreme’ donors and acceptors, due to the subtlety of the effects on electronic properties.7 The publication of open access databases for electronic materials30–32 has enabled the application of AI to the discovery of donor–acceptor oligomers, and indeed material building blocks in general, offering alternative pathways for exploring larger chemical diversity, potentially uncovering ‘wild cards’ that would not be found by other routes.
In this work, we developed deep generative models for the generation of molecular libraries of donor–acceptor oligomers with preferred electronic properties using an RNN combined with TL. TL was required as the libraries of existent donor–acceptor oligomers are not of sufficient magnitude for training a robust RNN model. We explore whether the structural and electronic properties of the donor–acceptors can be learned via TL, and whether the chemical space of the training sets can be fully explored with the TL models. As a proof of concept, we targeted the chemical space of donor–acceptor oligomers with an optical gap, as approximated by the HOMO–LUMO gap, close to 2 eV and a dipole moment smaller than 2 Debye. These oligomers can potentially form organic semiconductor crystals offering alternatives to the traditional families of molecular candidates such as oligoacenes or benzothieno[3,2-b][1]benzothiophene derivatives. A low HOMO–LUMO gap, and by extension a low optical gap, ensures the generated oligomers are promising materials for organic photovoltaics, and a low dipole moment in the ground state is desired as it has been shown that this can help with directing self-assembly towards supramolecular arrangements that promote macroscopic properties such as charge-carrier mobility.33 Our approach would be equally applicable to a focus on a different region of property space and we would note that for optoelectronic device applications such as OPVs and OLEDs, it is typical for there to be multiple material properties and device characteristics contributing to high performance, and these would need to be considered and appropriately weighted on a case-by-case basis. We discuss the challenges for further extension of the method in the discussion.
Methods
We present an overview of our approach for generating novel donor–acceptor oligomers with targeted electronic properties in Fig. 1. In the following sections, we will describe the origin of our datasets, how we first trained a general RNN and then used unsupervised cluster detection to uncover different clusters of molecules in a database of donor–acceptor oligomers. Next, we compared the performance of transfer learning models built from different transfer databases and finally validated the performance of the newly generated oligomers.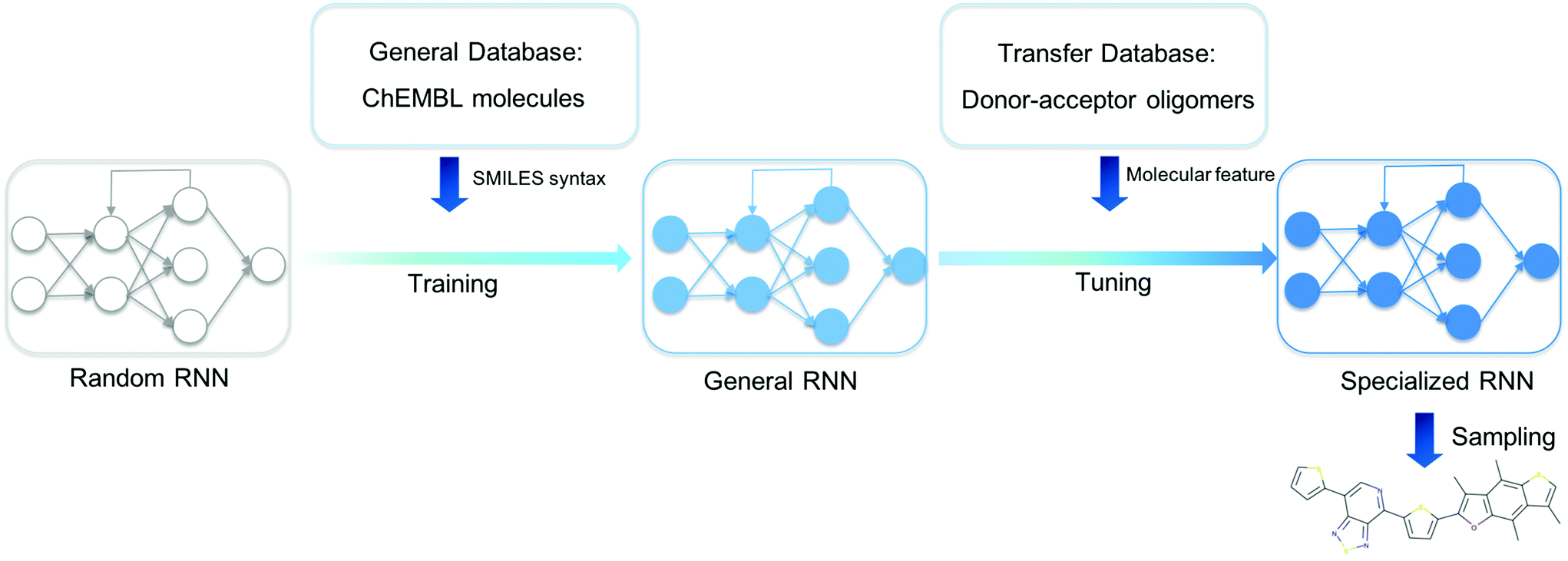 | ||
| Fig. 1 Our workflow for the discovery of novel donor–acceptor oligomers. An RNN with randomized parameters was built, and the General RNN was obtained by training the random RNN with the General Database of the ChEMBL library. The specialized RNN was developed by tuning the General RNN using the Transfer Database that only contains donor–acceptor oligomers, and then used to generated novel donor–acceptor oligomers. | ||
Datasets
The task for molecular discovery using deep generative models can be divided into two parts; (i) learning the correct representation of molecules, for instance the valid SMILES representation, and (ii) learning the required structural property relationship of the molecules in order to generate novel functional molecules. To successfully carry out the first stage of the task, it is necessary to train the model on an extensive and varied database from which the syntax of valid SMILES can be learned. For the training of such an RNN model, which will hereafter be refered to as the ‘General RNN’, we used the GuacaMol Training SMILES database34 as published by Brown et al.,35 which contains 1.2 million SMILES string from the ChEMBL database. And we refer to the training set for the General RNN hereafter as the ‘General Database’. For the TL model, the ‘Database of organic donor–acceptor molecules’ from the Computational Materials Repository31 was used and is referred to hereafter as the ‘Transfer Database’. The Transfer Database originally contained 5419 molecules, but after removing candidates that contained characters not present in the General Database and those for which density functional theory (DFT) calculations we carried out to characterise the molecular properties (see below) failed to converge, we were left with a final Transfer Database of 5024 molecules. The average length of the SMILES in the General Database is 47.6, while the average length of SMILES strings in the Transfer Database is 103.3, so it is of interest how well the General RNN will be able to be applied to the more complex molecules in the Transfer Database.The values of the Kohn–Sham HOMO–LUMO gaps, which we use to approximate the optical gaps, and dipole moments in the database of organic donor–acceptor molecules were previously calculated using the B3LYP functional.36–38 However, due to the exponentially decaying nature displayed by this exchange–correlation, instead of the correct r−1 behaviour at long distances, the excitation energies can be expected to be underestimated and the polarizability overestimated.39,40 We therefore performed additional calculations to evaluate the electronic properties of the oligomers in the Transfer Database with a long-range corrected DFT functional that does not suffer from this issue Firstly, we generated 100 conformers of each oligomer using the RDKit ETKDG method41 and evaluated the conformers’ energies using UFF.42 The lowest energy conformation was then geometry optimised by using the GFN-xTB2 method as implemented in xTB.43 The optimisation was considered complete when a threshold of 8 × 10−4 Hartrees per atomic unit length in forces and 1 × 10−6 Hartree in energies was reached, all conformations were confirmed to be true minima. We opted to optimise the geometry of the molecules using GFN-XTB2 rather than with DFT calculations due to the large number of molecules in the Transfer Database. A benchmark between geometries optimised using GFN-xTB2 and DFT (ωB97X-D3/def2-TZVP) is reported in section 5 of the ESI,† showing that GFN-xTB2 provides reasonable geometries at a much reduced computational cost. The optical and electronic features such as the HOMO–LUMO gap and polarizabilities were then computed by employing the long-range corrected ωB97X-D3 functional44 as implemented in ORCA.45 The ground and excited state properties were computed using the def2-TZVP basis set and by employing the simplified Tamm–Dancoff approach (sTDA).46–49 Excited states with an excitation energy of lower than 10.0 eV were computed, corresponding to approximately 300 singlet states and 300 triplet states. This methodology has been shown to produce reliable results in the case of co-polymers where optoelectronic properties have been computed and employed to train machine learning models.7 Further analysis of the differences can be found in the ESI.† Our computational approach, using GFN-xTB first for geometries, cut the computational cost of obtaining structures and properties by a factor of about 3–4, with each molecule on average taking about 1 day to run on a 24-core node on a university supercomputer.
We found that now only 269 of the 5024 molecules in the ‘Database of organic donor–acceptor molecules’ were promising candidates for our target of an optical gap close to 2.0 eV and dipole moment smaller than 2.0 Debye, making them possible candidates for organic semiconductors. These results highlight the importance and need for TL to explore the chemical space, with the main objective of generating many more new molecules with the target properties.
The General RNN model
The RNN architecture used in this paper is adapted from the work of Olivecrona et al.2 The molecular SMILES were encoded to numeric vectors suitable for machine learning using ‘one-hot’ tokenization. In this study, there were 78 unique symbols in the SMILES strings of the Transfer Database. Two additional tokens, ‘GO’ and ‘EOS’ were added to each string to denote the beginning and end of a SMILES sequence. As a result, a SMILES string with n symbols was represented by an (n × 80) dimensional vector. The specific RNN type used in this study was the Gated Recurrent Unit (GRU),50 where each node in the RNN was designed to learn long range dependency within the SMILES string by keeping a weighted sum of information each character in the SMILES string possesses. Three stacked GRU layers were used to process the vectors generated from the SMILES strings in this study. The model was trained using the standard Adam optimizer, which is an extension of the gradient descent algorithm designed for the training of deep neural networks.51 The generative RNN model in this work was implemented using Python 3.6 in combination with the PyTorch library.52Unsupervised cluster detection in the transfer database
In order to explore the chemical space of the Transfer Database, cluster detection within the Transfer Database was performed based upon their structural similarity. The Transfer Database was considered as a graph, where each molecule in the Transfer Database formed a node in the graph. The graph nodes were represented using the Morgan molecular fingerprints of the corresponding molecules. A Morgan fingerprint is a vector that indicates the presence of specific substructures within a molecule, and was computed here using the RDKit cheminformatics package.53 The edges in the graph were defined using the pairwise Tanimoto similarities between the fingerprints of the molecules, which quantifies how similar a pair of molecular fingerprints are. In order to reduce the complexity of the graph, a cut-off of 0.25 was applied to the molecular similarities, which means that molecules with similarity larger than 0.25 were considered ‘connected’ by an edge. The Louvain method54 was applied to the graph to detect clusters within the Transfer Database; the method detects clusters by maximising the density of edges within each cluster compared to edges connecting different clusters. The Louvain modularity cluster detection was performed using Python 3.6. After the clusters were detected, the HOMO–LUMO gaps and dipole moments of molecules in each of the clusters were analysed to examine the shared properties of the clusters and determine the most suitable clusters for TL.Transfer learning
We wanted to examine whether using subsets of the Transfer Database that contained clusters of promising candidates with the desired properties would be a more effective way of generating new candidates with promising properties than using the entire Transfer Database for TL. Therefore, we carried out the cluster detection for the entire Transfer Database (5024 molecules) and separately for the set of ‘promising’ candidates (269 molecules). As a result, three subsets of both the Transfer Database and the ‘promising’ candidates were supplied to the General RNN for parameter fine-tuning, resulting in 6 TL models. The training sets for Models 1, 2 and 3 were subsets of the whole Transfer Database, while training sets for Models 4, 5 and 6 were subsets of the 269 ‘promising’ candidates. All parameters in the General RNN were retrained during TL, with 15 epochs of training performed, which means that each of the TL models were fine-tuned by passing the corresponding training sets 15 times through the General RNN. 1024 SMILES strings were sampled from each epoch during the TL, resulting in 15360 strings being generated from each TL model.
Evaluating the transfer learning models
We evaluated the TL models by examining the validity, uniqueness and novelty35 of the oligomer database generated by each model, as well as the ability of generating ‘promising’ molecules. The ‘validity’ measures the ability of the models to generate valid SMILE strings and was calculated by dividing the number of valid SMILES strings (as confirmed using the RDKit package) generated by each model over the total number of strings sampled by each model. The ‘uniqueness’ measures the ability of the model to generate unique SMILES that had not already been sampled and was calculated by dividing the number of unique SMILES generated over the number of valid SMILES generated by each model. The ‘novelty’ measures the models in terms of generating oligomers that were not already present in the training sets, and was calculated by dividing the number of valid and unique SMILES strings that were not already found in the corresponding training set over the total number of valid and unique SMILES generated by each model.For each TL model, about 4000 novel molecules were generated, thus the computational cost of evaluating the electronic properties of all the 24000 generated molecules using DFT was prohibitive. We estimated that this would have required approximately 24000 days on 24-core nodes, which even with access to massively parallel computer architectures, was not viable. Instead, to assess the properties of the generated molecules at acceptable computational cost, the HOMO–LUMO gaps and dipole moments were calculated using supervised gradient boosted decision tree (GBDT) models trained on the Transfer Database. The GBDT algorithm uses a weighted ensemble of decision trees to fulfill regression or classification tasks. The GBDT model was trained on the Transfer Database, with the database randomly divided into training (80%, 4019 molecules) and test (20%, 1005 molecules) sets, and the Morgan fingerprints calculated using RDKit as the molecular input feature. Morgan fingerprints with varied length (512, 1024, 2048) and radius (2, 4, 6) were used for the molecular fingerprinting. The GBDT models were optimised so as to minimise the mean squared error between the predicted values and the true values in the training set. The GBDT models were then used upon the generated unique and novel molecules from TL to predict their HOMO–LUMO gaps and dipole moments. Those molecules with a HOMO–LUMO gap less than 2 eV and dipole moment less than 2 Debye were deemed ‘promising’, as they were the target of our study. In order to validate the ‘promising’ molecules identified using the GBDT models, 90 ‘promising’ molecules were randomly sampled from the ‘promising’ generations and their HOMO–LUMO gaps and dipole moments were calculated using DFT calculations with the ωB97X-D3 functional, using the setup as described above.44
Results and discussion
Supervised machine learning model of electronic properties
Supervised GBDT models were developed to evaluate the donor–acceptor oligomers generated from the generative RNNs. It was found in this study that a GBDT model using Morgan fingerprint with length 1024 and radius 2 had the lowest error in terms of predicting the properties of the oligomers in the test set. The comparison between the ωB97X-D3-calculated electronic properties and those predicted using the GBDT models on the test set (20% of the Transfer Database, 1005 molecules) is shown in Fig. 2. The mean absolute error of the GBDT prediction for HOMO–LUMO gaps is 0.09 eV and for dipole moments is 1.31 Debye. While the GBDT model is therefore reasonably accurately predicting the HOMO–LUMO gap of the molecules, the accuracy of the prediction for the dipole moments was significantly lower. Previously, graph neural network models using molecular graphs as feature vector inputs, such as the Schnet,55 MEGNet56 were trained to predict the dipole moments of functional molecules with good accuracy. However, when we tested these models for our study, the mean absolute error of the dipole moment predictions was exceptionally large (over 10 Debye). The poor performance of the graph neural networks on the Transfer Database was probably due to the fact that such models were trained and tested against the QM9 dataset,57 which contains only molecules with no more than 9 heavy atoms. However, the number of heavy atoms in molecules in our Transfer Database ranges from 20 to 180, and application of such graph convolution networks was therefore inadequate due to the complexity of molecules in the Transfer Database. We trialled other models, such as random forest, and other fingerprints, such as Molecular ACCess System (MACCS) keys,58 but these performed less well than our selected model.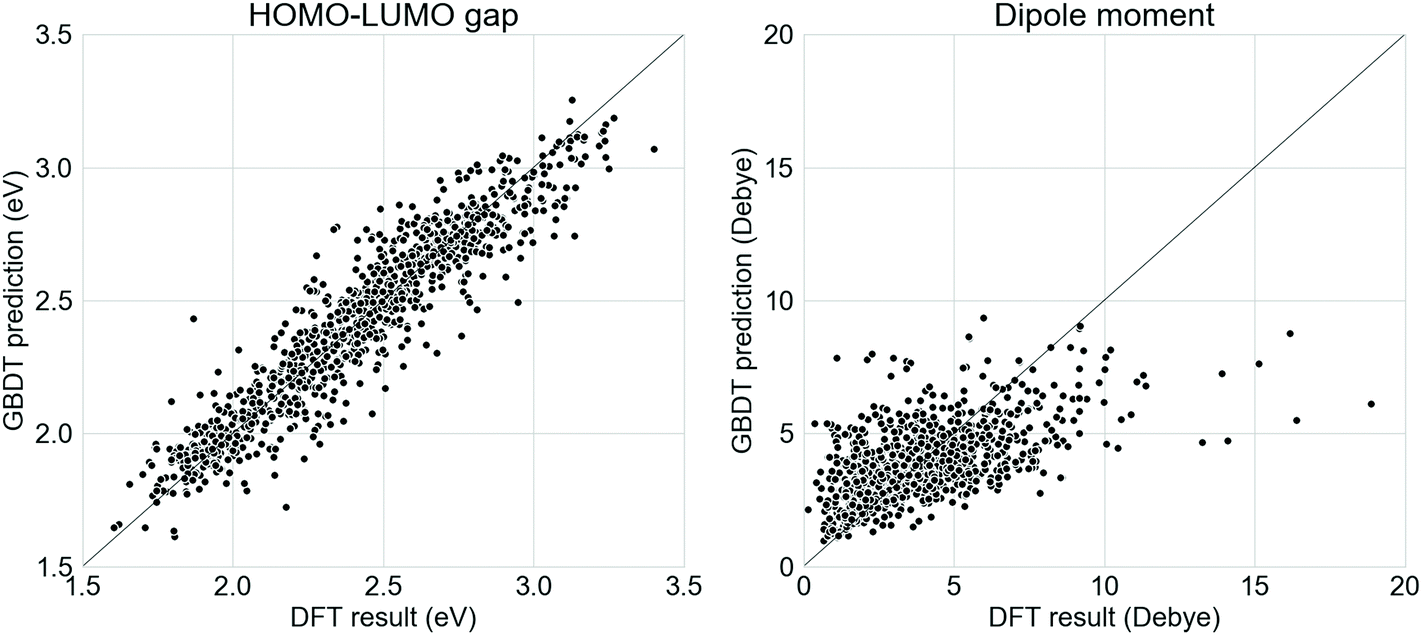 | ||
| Fig. 2 Relationship between the DFT calculated electronic properties and GBDT predictions on the test set of the Transfer Database for (left) the HOMO–LUMO gap and (right) the dipole moment. The line of y = x is shown in both plots. | ||
Here, we aim to discover as many ‘promising’ oligomers as possible for further validation, thus recall (identifying ‘promising’ oligomers) was pursued at the sacrifice of precision (labelling ‘promising’ oligomers correctly). The mean error of GBDT prediction for dipole moment was 0 Debye, with a standard deviation of 1.83 Debye. The molecules generated via TL were fed into the GBDT models to predict their electronic properties. Molecules with a GBDT-predicted dipole moment of lower than 3.66 Debye (corresponding to 0.9 standard deviation above the mean error of 0 Debye) and a HOMO–LUMO gap lower than 2.0 eV were considered as potential ‘promising’ oligomers. The choice of 3.66 Debye as cutoff for ‘promising’ oligomers implies a higher false positive rate and lower false negative rate, which met the requirement of capturing as many ‘promising’ oligomers as possible. The GBDT models for predicting the HOMO–LUMO gaps and the dipole moments, as well as the corresponding results obtained from DFT calculations can be found at github.com/qyuan7/RNN_RL_molecule/tree/master/gbdt_regressors.
Training the General RNN
The General RNN model was trained on the 1.2 million SMILES strings from the ChEMBL database. The percentage of valid SMILES strings and the logarithmic loss (log loss), which we seek to minimise here to improve the model, are shown in Fig. S2.† By the final steps, more than 90% of the SMILES strings generated were valid and the log loss for the maximum likelihood estimation fluctuated around 24, indicating that the training for the General RNN is close to convergence and it is a fairly efficient model in generating valid SMILES strings. The source code for training the General RNN, as well as the General RNN trained from this study can be found at github.com/qyuan7/RNN_RL_molecule.Training sets for TL
Different subsets of the Transfer Database representing different structural and electronic properties were used as the training sets for the TL tasks. Structurally similar oligomers in the Transfer Database tend to fall into the same cluster detected by the Louvain algorithm. Four structural clusters were identified from the Transfer Database, and representative molecules in the clusters are shown in Fig. S3.† It can be seen that molecules in clusters 1 and 3 were similar in terms of structure and size, while molecules in clusters 0 and 2 were in general larger with either longer chain length (cluster 0) or possessing complex side chains (cluster 2). The structural similarities of the oligomers can be qualitatively represented using the distances between their fingerprint vectors. In order to visualize the high-dimensional distances in two-dimensional space, the t-Distributed Stochastic Neighbour Embedding (t-SNE, a dimension reduction technique for data visualisation)59 with two dimensions was applied to the fingerprints of the molecules. The distributions of the t-SNE projections of the molecular fingerprints are shown in Fig. 3, and molecules with the target properties are shown as blue points. Almost all of the oligomers with the target properties (255 of 269) were found in cluster 3, with just 12 in cluster 2. This suggested that cluster 3 was a ‘promising’ cluster and would form a more suitable training set for TL to generate ‘promising’ oligomers with the desired electronic properties. The molecular fingerprints in cluster 2 differed greatly from those in cluster 3, it is thus possible to explore different regions of the chemical space by using cluster 2 for TL. Therefore, we used three different subsets of the Transfer Database for TL; Model 1 (containing all the molecules), Model 2 (molecules in cluster 3) and Model 3 (molecules in cluster 2).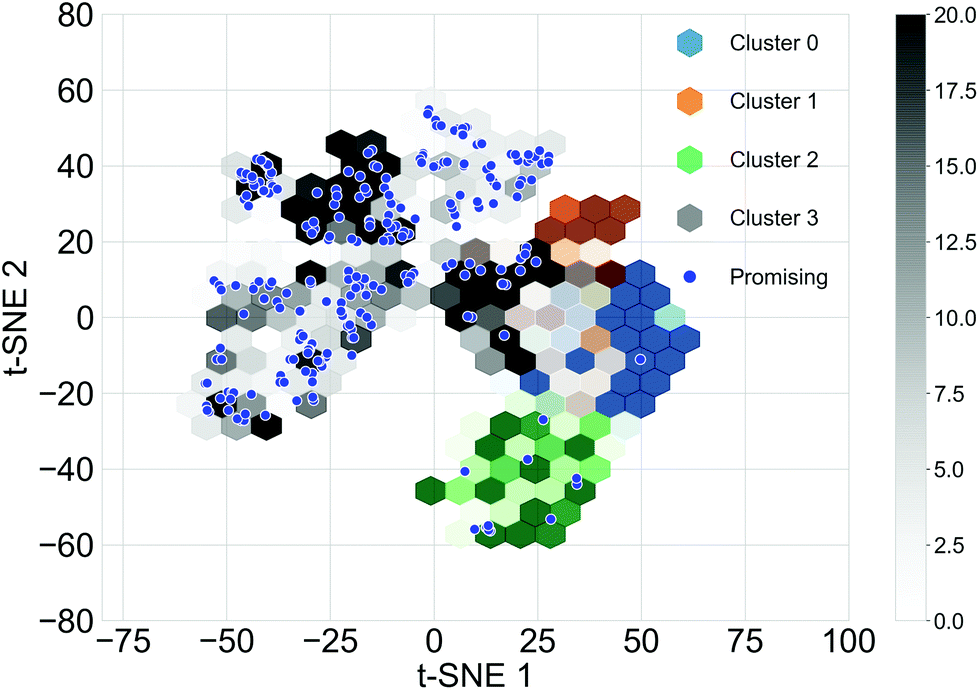 | ||
| Fig. 3 A visualisation of the different clusters found in molecules of the Transfer Database, created via a dimensionality reduction technique. Shown is a hexagonal binning plot of the t-SNE projection of the molecular fingerprints of the molecules, hexagons of different colours represents different clusters and the depth of the colours shows the density of the distribution. t-SNE 1 and t-SNE 2 correspond to the first and second dimensions of the 2-D projection of the fingerprints. The t-SNE projections of the 'promising' oligomers in the Transfer Database are plotted as blue scatter points. | ||
Cluster detection on only the oligomers with the target properties was also performed (Fig. S4†). Two major clusters with 120 and 95 molecules respectively were found and on this basis three additional training sets for TL were determined; Model 4 (all molecules with target properties), Model 5 (sub cluster 1) and Model 6 (sub cluster 2). A summary of the training sets for the six TL models are provided in Table 1, and by comparing the molecules generated using these different datasets, we can explore how to best target molecular properties with TL.
| Model | Training size | No. promising oligomers | Description |
|---|---|---|---|
| Model 1 | 5024 | 269 | All molecules in transfer database |
| Model 2 | 1568 | 255 | Molecules in cluster 3 of Fig. 2 |
| Model 3 | 533 | 12 | Molecules in cluster 2 of Fig. 2 |
| Model 4 | 269 | 269 | All molecules in promising lead database |
| Model 5 | 120 | 120 | Molecules in sub cluster 1 of Fig. S3† |
| Model 6 | 95 | 95 | Molecules in sub cluster 2 of Fig. S3† |
Performance of the transfer learning models
For all the TL models, 15360 strings were generated by each TL model, spread over the 15 epochs of retraining. The performance of the TL models on the six datasets in terms of the number of valid, unique, novel and ‘promising’ oligomers from the sampled SMILES strings are shown in Fig. 4 and Table S1.† The validity of molecules from all six models was enhanced as the TL progressed, but the number of unique generations for each epoch did not increase after the 10th epoch. For Models 1, 2 and 4, the number of novel SMILES dropped in the later epochs of TL. The fact that the uniqueness and novelty do not increase despite the increasing validity indicates that the TL models were trying to ‘remember’ certain valid SMILES strings in the training sets rather than learning the general rule of the SMILES format, which can be referred to as overfitting of the training set. This can be rationalised in terms of the nature of the molecules in the Transfer Database, which are considerably different from the ones observed in the ChEMBL database. Therefore, all the parameters in the General RNN had to be refined during the TL to ‘learn’ the features of the SMILES strings in the Transfer Database. Due to the limited size of the training sets for TL, a certain extent of overfitting could not be avoided. It can also be observed that Models 4, 5 and 6, which only used molecules with the target properties for the TL, were more powerful in generating ‘promising’ oligomers compared to Models 1, 2 and 3. This is an indication that the electronic properties of the training sets were ‘learned’ via TL, which will be discussed in the following section. According to the trends of performance of the models in Fig. 4, epochs beyond epoch 10 have not improved the models in generating more unique novel ‘promising’ oligomers, thus the parameters in the TL RNNs should be frozen beyond epoch 10 so that the sampling of ‘promising’ SMILES is more efficient.
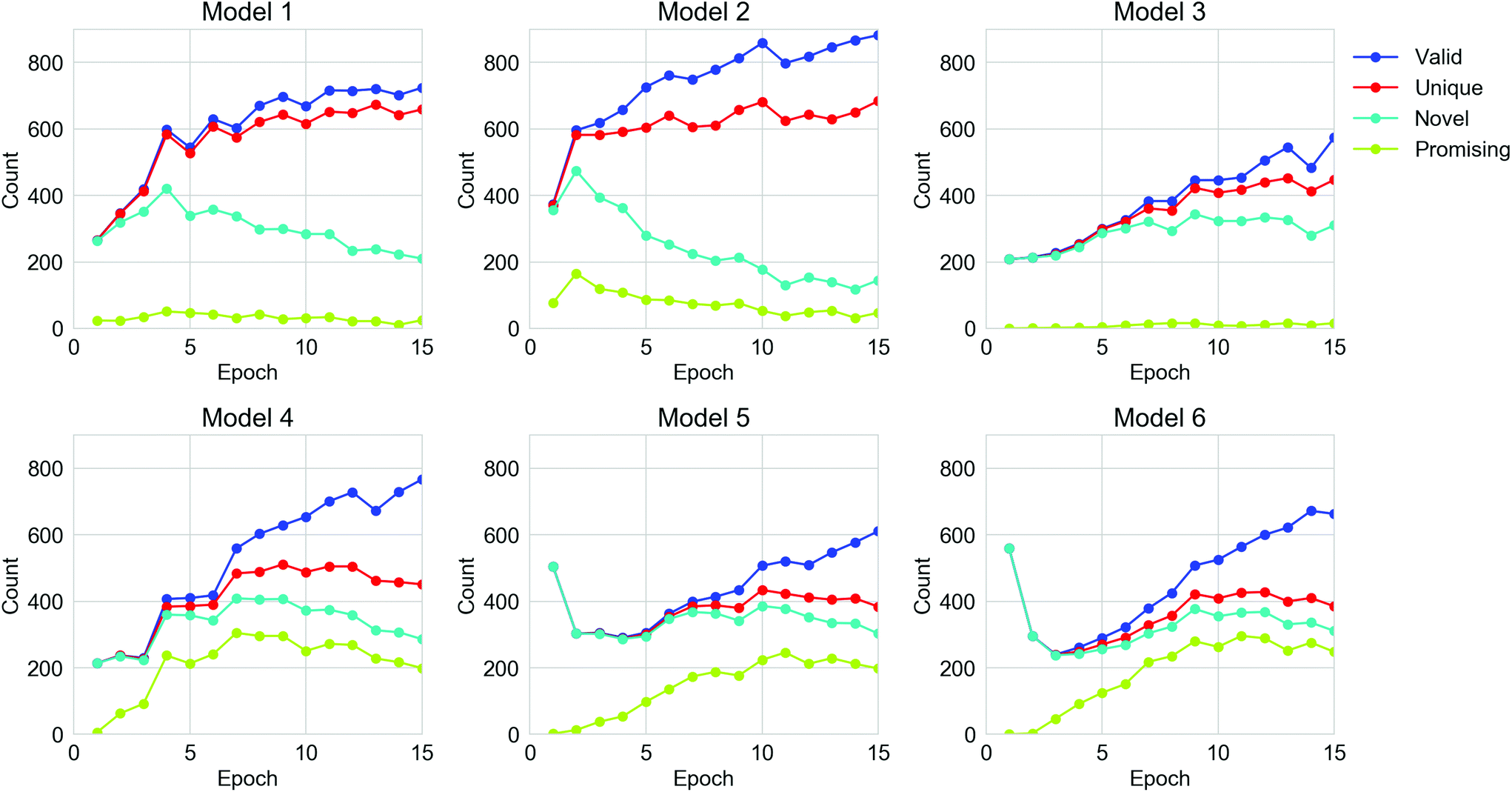 | ||
| Fig. 4 The number of valid, unique, novel and ‘promising’ oligomers generated for the six TL models along the epochs of TL. An epoch of transfer learning for a model here refers to passing the corresponding training set through the TL model once. | ||
Learning structural and electronic properties
It is interesting to assess to what extent the structural properties of the different training sets were ‘learned’ through TL. The training molecules with the highest Tanimoto similarities to the generated molecules were defined as the nearest neighbours of the generated molecules. The distribution of the neighbour similarities of all generated molecules and the ‘promising’ molecules from the six TL models (those with target properties) is shown in Fig. 5(a). The mean neighbour similarity of all the generated ‘oligomers’ and the ‘promising’ oligomers are 0.62 and 0.74, respectively. Examples of pairs of generated molecules and molecules in the Transfer Database with different levels of similarities of 0.62 and 0.74 are shown in Fig. S5,† to allow the reader to visualise what these values correspond to chemically. It can be seen that the novel oligomers with 0.62 and 0.74 neighbour similarity were generated by different levels of atom replacement on the corresponding neighbours, indicating that the molecular structural property of the training sets were ‘learned’ from the TL process, especially by the ‘promising’ oligomers. The similarities of the generated molecules and their neighbours increased as TL proceeded (Fig. S6†), and the median of the neighbour similarities at epoch 10 were larger than 0.6 for all models (Fig. 5(b)).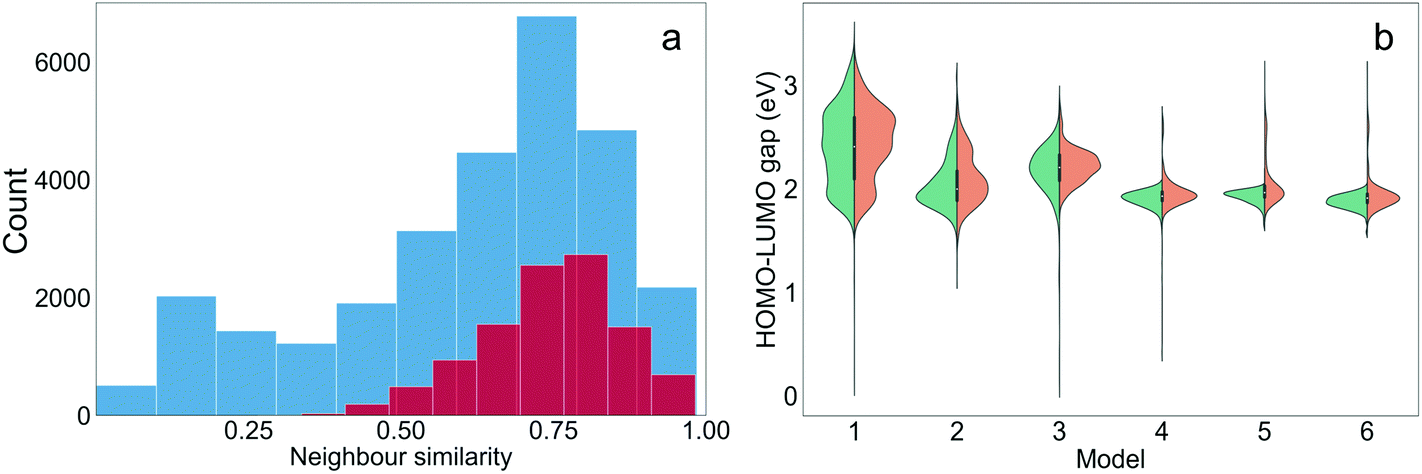 | ||
| Fig. 5 (a) Distribution of the neighbour similarities of all the generated oligomers (blue) and the ‘promising’ oligomers (maroon); (b) distribution of the HOMO–LUMO gaps of the molecules generated from the six TL models on the 10th epoch (red) compared to the corresponding training sets (green). | ||
In addition to learning the structural properties of the training sets, another task for the TL was to learn the electronic properties of the training molecules. Since the properties of the generated molecules were evaluated using the GBDT model and accurate prediction of dipole moment was not achieved in this study, only the HOMO–LUMO gaps were examined. The distributions of the HOMO–LUMO gaps of the molecules generated from the TL models at the 10th epoch are shown in Fig. 5(b) and shown for each epoch in Fig. S7.† For Models 4, 5 and 6, the HOMO–LUMO gaps of the vast majority of generated molecules are lower than 2.0 eV, and therefore the chance of obtaining ‘promising’ oligomers increased. Models 1 through 6 had varied distributions of HOMO–LUMO gaps and all of these specific distributions were ‘learned’ by the corresponding TL models. Therefore, the HOMO–LUMO gaps of the generated molecules can be tuned by sampling different subsets of the training database of interest.
Chemical space exploration
The ability for deep generative models to sample larger chemical space while obtaining oligomers with desirable properties is also important. This is a particular goal of the study here – to see whether our approach can move beyond traditional substitution strategies well established in the material chemistry community, to uncover ‘wild cards’ that might suggest alternative molecular replacements or a series of fragment alterations. The training sets of the TL models covered different regions of the chemical space, and it was expected that each of the models would cover the corresponding chemical space. In addition, covering chemical space beyond the training sets could lead to the discovery of novel donor–acceptor oligomers that had not been considered. To create a qualitative visualisation of the chemical space covered by the molecules in this study, the t-SNE projections of the fingerprints of the molecules generated from the six TL models, together with the fingerprints of the molecules in the corresponding training sets are shown in Fig. S8.† The chemical space of each training set has been thoroughly explored by the corresponding TL model, as well as unexpected regions of the chemical space of the training sets having also been covered, especially for Models 4, 5, and 6. It was thus possible to generate donor–acceptor oligomers that are not directly related to those in the Transfer Database from the TL models.Merely covering more chemical space does not ensure the discovery of novel ‘promising’ donor–acceptor oligomers; it is possible that oligomers in the newly explored regions do not exhibit the preferred electronic properties. To compare the chemical space covered by the ‘promising’ oligomers, the t-SNE projections of only the ‘promising’ training and generated oligomers are shown in Fig. 6. Representative molecules were sampled from the ‘promising’ oligomers generated from each TL model, and the structure of such molecules and their t-SNE projection values are shown in Fig. S9.† It can be seen from Fig. 6 that ‘promising’ oligomers generated from the TL explored well the corresponding chemical spaces of their respective training sets. As demonstrated in Fig. 6 and S9,† the ‘promising’ oligomers generated from Model 3 occupy a different region of chemical space compared to the other five models. In addition, oligomers generated from Models 5 and 6 occupied different sub-parts of the chemical space than Model 4. It is thus possible to explore particular regions of interest in chemical space by tuning the training sets for TL with the assistance of unsupervised cluster detection. However, the chemical spaces covered by the ‘promising’ oligomers were more conservative than that covered by all the generated molecules, as seen by higher neighbour similarity in Fig. 5a. The distribution of the neighbour similarities of generated molecules with their corresponding training sets for each of the TL models are shown in Fig. S10,† and the trend of ‘promising’ oligomers having higher neighbour similarities was found for all six models. Such results indicate that the excessive regions of chemical space explored by the TL models had limited contributions in terms of providing ‘promising’ oligomers, and the trade-off between exploring unseen regions of the chemical spaces and generating a greater percentage of more ‘promising’ oligomers is something to bear in mind.
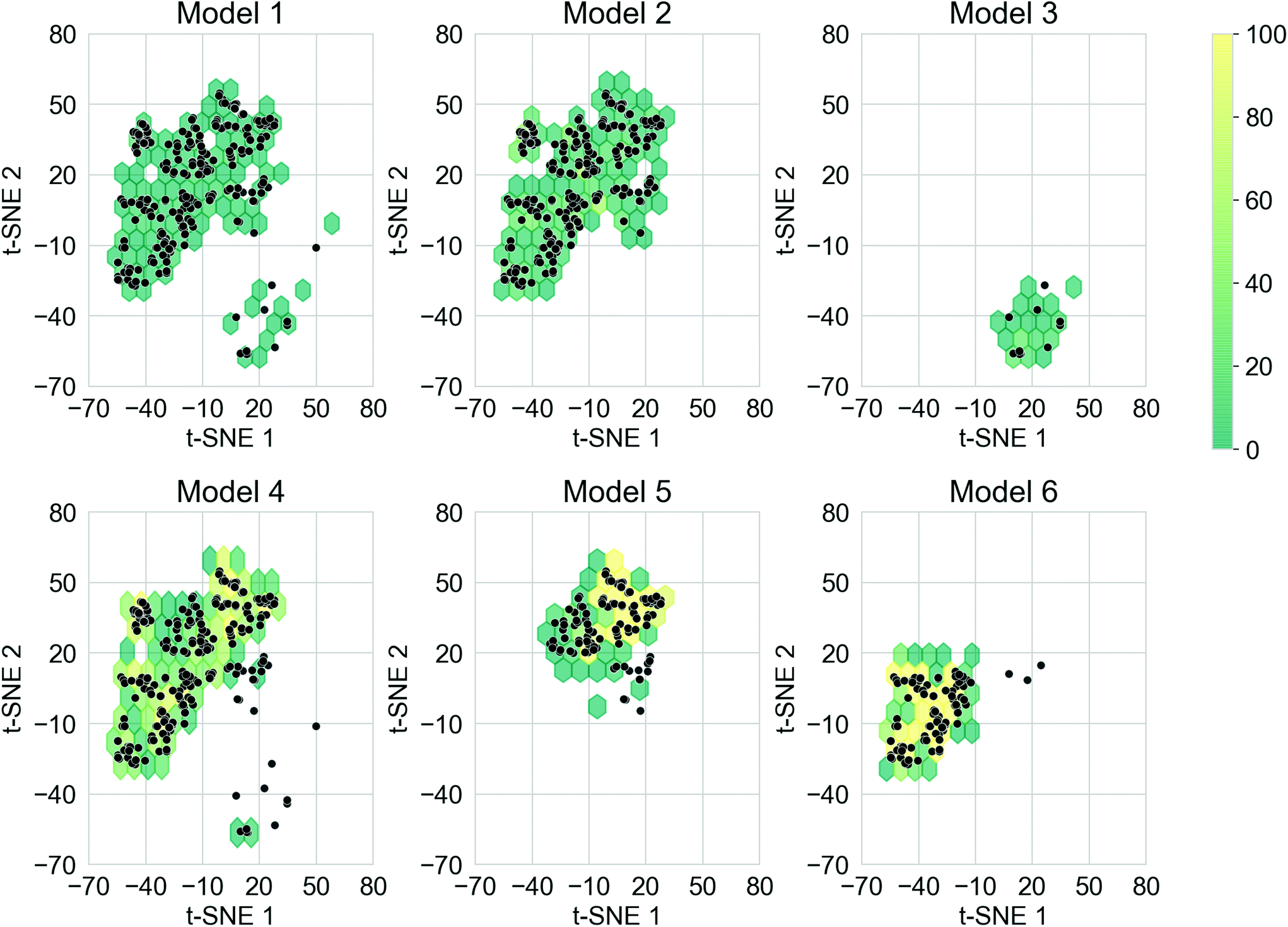 | ||
| Fig. 6 Hexagonal binning plot of the t-SNE projection of the fingerprints of the ‘promising’ molecules generated from the TL models. Colours of the hexagons represent the density of generated ‘promising’ oligomers in each hexagon according to the colour bar. The ‘promising’ molecules in the corresponding training sets are shown as black points. t-SNE 1 and t-SNE 2 correspond to the first and second dimensions of the 2-D projection of the molecular fingerprints. | ||
The t-SNE projections in this study are the two-dimensional projection of the 1024-bit molecular fingerprints, allowing one to visualise the distribution of the high dimensional fingerprints. The computed neighbours in the t-SNE plots could result from either structural similarity (as intended) or from a crowding effect of the low dimensional representation of high dimensional data. Examples of ‘promising’ generated molecules with high and low similarity with their nearest neighbour in the training sets are shown in Fig. 7. A ‘promising’ oligomer with high similarity to a neighbour could be generated by simple atom replacement, while a ‘promising’ oligomer with a low similarity to a neighbour less than 0.6 involves multiple alterations on the neighbour molecule, which would be difficult to suggest by using traditional experimental functionalisation strategies. We obtained about 1300 ‘promising’ oligomers with similarity to a neighbour lower than 0.6 with the TL models, which would be of interest in expanding the chemical space of the ‘promising’ oligomers. In addition, the different levels of modification to the neighbour molecules, as reflected in the generated ‘promising’ oligomers, could provide valuable insight for manual modification of the molecules in the Transfer Database.
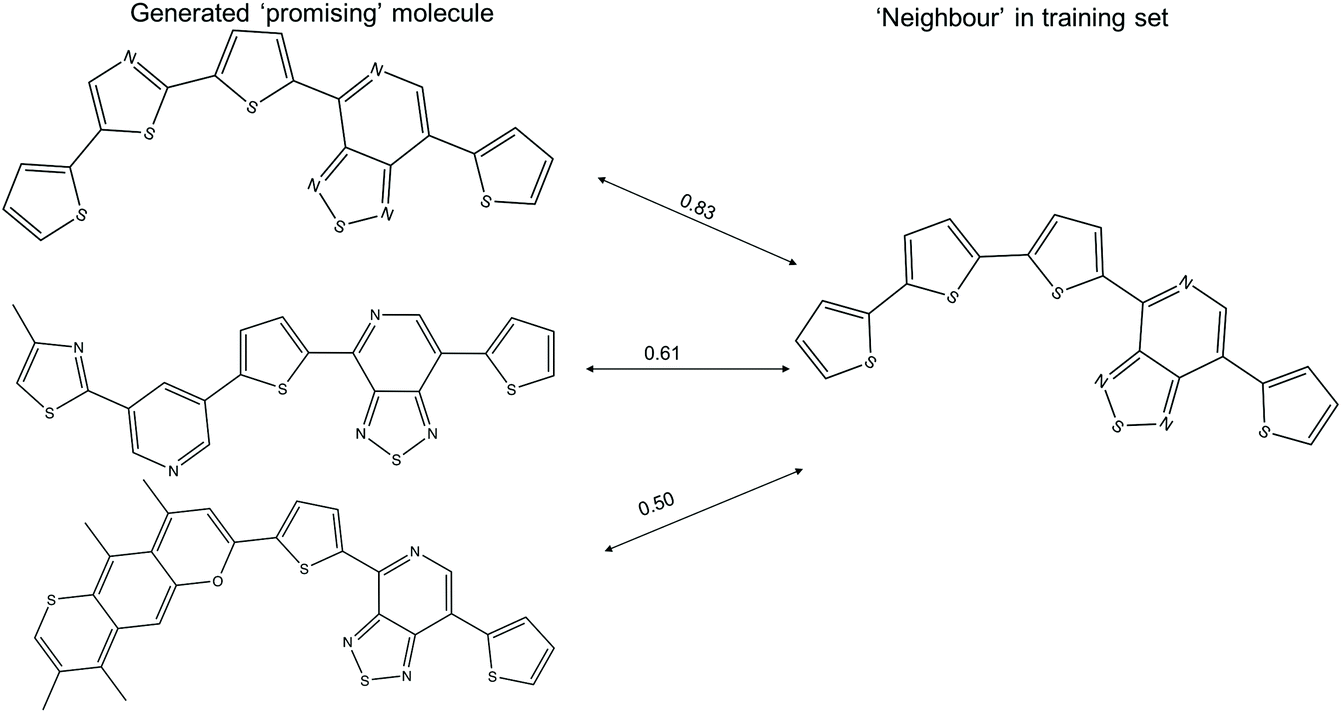 | ||
| Fig. 7 Example of three ‘promising’ oligomers generated from the TL and their nearest neighbour in the training set, as calculated using the Tanimoto similarity criterion. The Tanimoto similarities between the donor–acceptor oligomers are shown above the arrows. It should be noted that these 2-dimensional structures do not show the exact 3-dimensional conformation of the molecule used in the calculations. | ||
Validation of promising molecules
We randomly selected 15 molecules from the predicted ‘promising’ oligomers generated from each of the six TL models (90 molecules in total) for electronic property validation at the DFT level with the ωB97X-D3 functional.44 Of the 90 selected molecules, 22 have HOMO–LUMO gaps below 2.0 eV and dipole moments smaller than 2.0 Debye. The 22 structures of this subset of ‘promising’ oligomers are shown in Fig. 8. The distribution of the calculated HOMO–LUMO gaps and dipole moments of the molecules are shown in Fig. 9(a). The vast majority of this molecular subset have HOMO–LUMO gaps below 2.0 eV, while the distribution of dipole moments of the selected molecules has a larger variance, which can be ascribed to the lower accuracy of the GBDT models in dipole moment prediction. The precision of identifying ‘promising’ oligomers was thus 0.24. In total, 7224 novel unique oligomers were predicted to be ‘promising’ from the six TL models. If we assume that 0.24 of them would be truly ‘promising’, about 1700 ‘promising’ oligomers as organic semiconductors have been generated, which is 6-times larger than the original 269 ‘promising’ oligomers from a database of 5024 molecules conceptually designed for this task.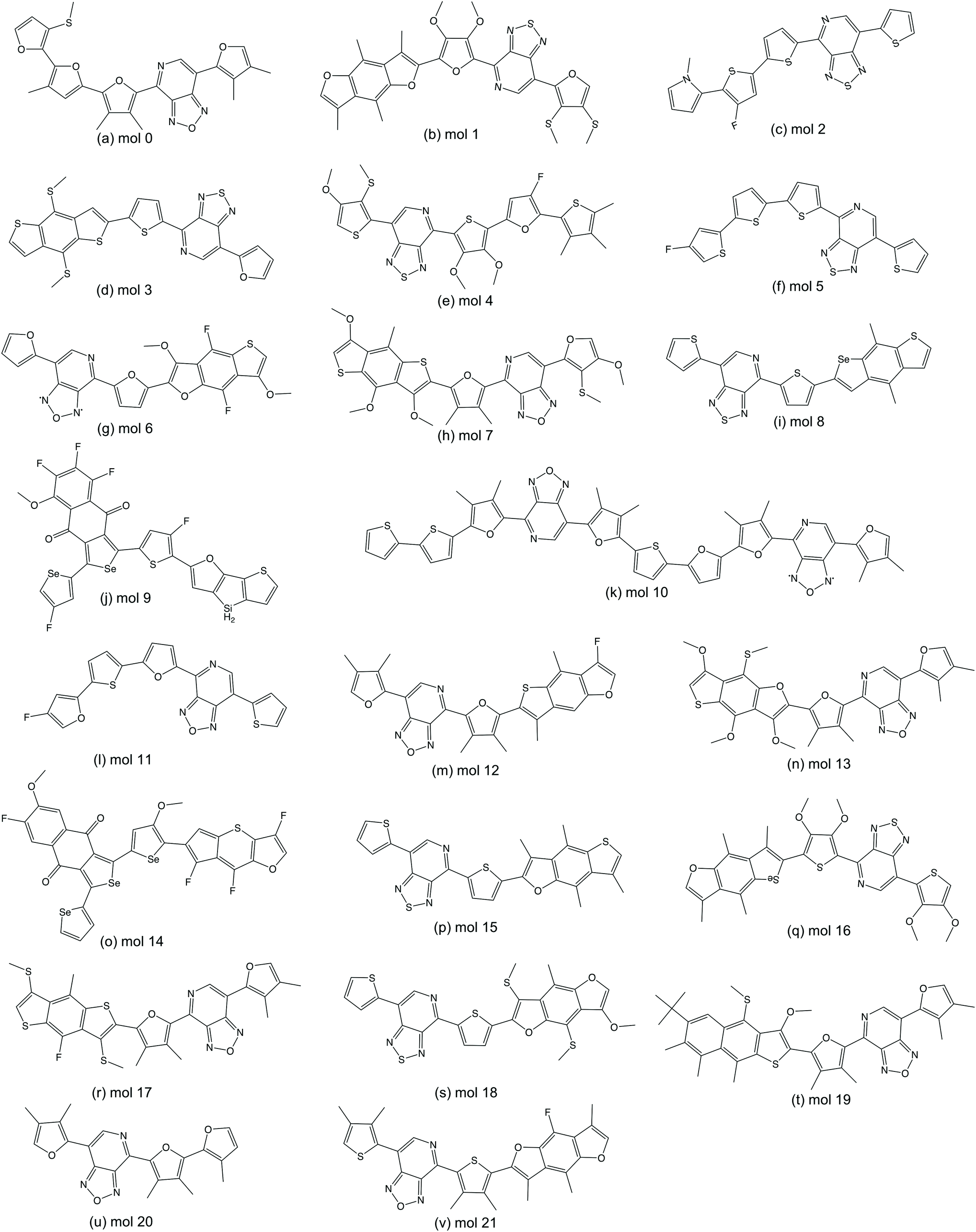 | ||
| Fig. 8 The subset of ‘promising’ oligomers validated by DFT calculations. | ||
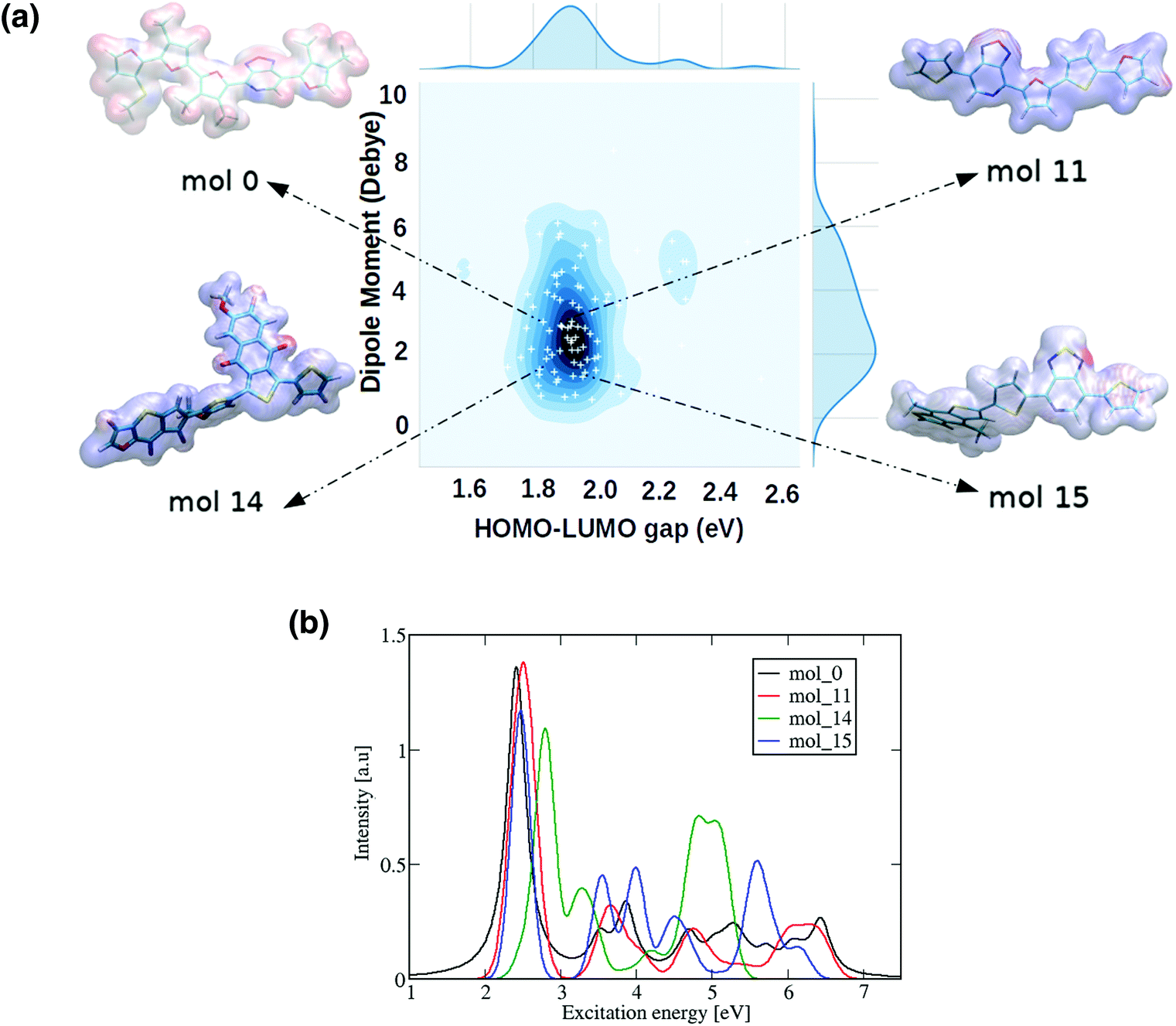 | ||
| Fig. 9 (a) Distribution of the ωB97X-D3 calculated HOMO–LUMO gaps and dipole moments of 90 randomly selected ‘promising’ oligomers generated from our trained deep generative model. Each newly produced donor–acceptor oligomer is represented as a white cross in the 2-D distribution, where darker regions represent a greater density of molecules found. The example molecules are shown with their molecular electrostatic potential, red represents more electron-rich regions and blue more electron-poor. (b) Computed UV-Vis spectra for the 4 selected oligomers. | ||
A significant issue with generative models is the generation of molecules that are not synthetically viable, for example due to the complexity or the cost of potential synthetic routes to access them. We therefore inspected the 22 ‘promising’ oligomers to compare their ease of synthetic accessibility to that of oligomers in the original Transfer Database. While the origin of all the oligomers in the Transfer Database is not clear, there are many systems in the database that have been previously synthetically reported. The SA score from Ertl and Schuffenhauer60 was calculated for each of the ‘promising’ oligomers and their corresponding neighbour oligomers, where scores can range between 1 (easy to make) and 10 (very difficult to make). All of our 22 generated molecules have scores between 3 and 5 and this reassures us that the generated molecules are not extremely difficult to make and thus plausible. The scores for each of the generated molecules is very similar to those of the Transfer database, as shown in Fig. S11,† with a mean absolute error of 0.13 between a ‘promising’ oligomer and its nearest neighbour in the Transfer Database.
A more detailed study of the 22 promising donor–acceptor oligomers started with a visual inspection of their computed molecular electrostatic potential (MEP), as shown in Fig. 9 and S11.† This quantity has been employed as a useful indicator to identify the electrostatic interaction between molecules, in particular non-covalent molecular interactions that play an important role in the formation of condensed phases.61 The oligomers can be classified into three different categories based on the distribution of the electron density: oligomers with a homogeneous electron density distribution, oligomers with a small accumulation and depletion of electronic charge, and oligomers with visible regions where charges have been strongly localised. Differences in the MEP distribution suggest different underlying mechanisms used by the RNN for reducing the molecular dipole moments. Some molecules achieved the dipole constraint by creating many different regions where charge is depleted or localised, creating small local dipole moments that are cancelled out when the total dipole moment of the molecule is computed. Alternatively, for some molecules a more even distribution of charges was observed. Thus, two alternative strategies have been employed to achieve small dipole moments with the RNN. In this regard, such characteristics have been directly inherited from the Transfer database, generating a set of diverse ‘promising’ molecules without the limitation of rearranging atoms within the oligomer in a combinatorial manner. The deep generative model also provides different alternatives to fulfill the imposed constraints beyond atom redistribution, such as using the size of the oligomers as a mechanism to tune the HOMO–LUMO gaps and dipole moments.
We can observe the impact of the substitutions carried out by the deep generative model on the optical properties by computing the UV-Vis spectra, as shown in Fig. 9(b) for 4 selected oligomers and in Fig. S12† for the remaining 22 oligomers. As a general trend across all oligomers, the first and most intense bright state can be found in energy regions between 2–3 eV, suggesting that they are possible candidates for applications such as photovoltaics. The constraints imposed on the RNN model's chemical space therefore had an impact on other important molecular features such as the optical properties.
Within the predicted structures, one can find known units, such as thiophene and furan, which have been previously used in the construction of donor–acceptor oligomers. The deep generative model also performed interesting substitutions such as fluorination or methylation. These strategies are commonly found in the literature and can be categorised as traditional substitutions in donor–acceptor oligomers. Similarly, completely changed small units such as selenophene (e.g.Fig. 8o) can be found within the set of suggested molecular transformations executed by the deep generative model. Analogously, one can observe frequently employed units for donor–acceptor molecules such as benzo[1,2-b:4,5-b]dithiophene (BDT)62 or pyridal[2,1,3]thiadiazole(PyT).63 However, the original composition of the molecule is not preserved in all cases, with the molecules displaying interesting atomic substitutions in the core such as BDT selenium (Fig. 8i) or oxygen substituted (Fig. 8v). Similar molecules have been experimentally synthesised and characterised as organic semiconductor with enhanced charge-carrier mobility as a consequence of such replacements.64 These previous findings provide an argument in favour of the capability of the deep generative model not only to offer an effective procedure to explore the chemical space, but also as a tool to provide new ideas and paths for novel atomic substitutions.
The TL model in this work is not limited to the generation of novel donor–acceptor oligomers as organic semiconductors; with proper tuning of the training sets for TL, electronic materials for other applications could be obtained. For example, the power conversion efficiency of organic solar cells is qualitatively related to HOMO energy of the donor, LUMO energy of the acceptor, and bandgap of the donor according to the Scharber model;65 OLED materials require low singlet–triplet gap and minimized spatial overlap of HOMO and LUMO.66,67 If such properties can be described with a supervised learning model from molecular information contained in the SMILES representation, the TL models can ‘learn’ the relevant properties with corresponding tuning of the training sets. In addition, when multiple electronic properties need to be optimized to improve device performance, the optimization can be assisted with a sampling approach such as an evolutionary algorithm (EA). In this fashion, we believe that the low precision and high recall model in discovering novel donor–acceptor oligomers can be used as a new methodology to discover and enhance selected families of new materials.
There are challenges in applying this type of approach to more complex properties beyond simple single molecule properties, particularly for applications based on combinations of properties. The first bottleneck to focus on to test expanding the approach is the generation of further training data for organic materials, such as to include excited state properties or, even more challenging, properties or behaviours beyond the single molecule level. While calculation of these properties ab initio for the requisite large number of systems is computationally demanding, it is pleasing to note recent additions to open-source databases in this area, which will open up new opportunities.32 Of course, device characteristics that are not predominantly linked to molecular features, such as preparation conditions and sample history that influence the device microstructure, are beyond the scope of what could be screened for by this approach. However, identification of promising molecules is still an important starting point.
Conclusions
In attempting to discover new molecular building blocks with promising properties for materials, a limitation can come from the ability to truly explore and optimise the vast chemical space of possibilities. Here we have focused on an approach using recurrent neural networks (RNN) combined with transfer learning (TL) to effectively discover novel molecules with targeted properties for donor–acceptor oligomers. While most previously reported deep generative models for molecule discovery have focused on drug discovery, we showed that the generative models trained from a pharmaceutical database can be transferred to relatively large and complex systems such as the donor–acceptor oligomers. Different chemical and electronic property spaces were covered using different subsets of molecules as training sets. Both structural and electronic properties can be ‘learned’ through TL, thus the RNN models suggested in this study can be used to target different property spaces to fulfill the requirement of different types of electronic materials. Many of the molecular transformations learnt mimic those used to enhance performance in donor–acceptor systems in the literature. The models developed and the ‘promising’ oligomers identified are open to future theoretical and experimental validation.An ideal generative model for molecular discovery would enable exploration of wider chemical space while retaining desired properties. However, it was found in this study that there is some degree of trade-off between exploration of chemical space and the optimisation of electronic properties. ‘Wild’ modifications to the training molecules were observed, likely modifications that would not be proposed by chemists, but such molecules did not generally exhibit satisfactory electronic properties. The generated ‘promising’ oligomers, with target properties, were more ‘conservative’ neighbours of the oligomers in the training set. The two factors need to be balanced in future molecular discovery tasks, although there is always the possibility that we are seeking a ‘needle in a hay stack’ – an extreme modification that still has the desired properties and has truly allowed us to move out of the region of chemical space that would be considered by chemists alone.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding from the European Research Council under FP7 (CoMMaD, ERC Grant No. 758370), and the Engineering Research Council and Physical Sciences Research Council (EPSRC) (EP/M017257/1, EP/P005543/1, EP/N004884/1), including the UK's HEC Materials Chemistry Consortium (EP/L000202/1) for time on the UK supercomputer, ARCHER. K. E. J. thanks the Royal Society for a University Research Fellowship. We thank Drs Andrew Tarzia, James Pegg and Liam Wilbraham for useful discussions.References
- M. H. S. Segler, T. Kogej, C. Tyrchan and M. P. Waller, Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks, ACS Cent. Sci., 2018, 4, 120–131 CrossRef CAS PubMed.
- M. Olivecrona, T. Blaschke, O. Engkvist and H. Chen, Molecular de-novo design through deep reinforcement learning, J. Cheminf., 2017, 9, 48 Search PubMed.
- D. Merk, F. Grisoni, L. Friedrich and G. Schneider, Tuning artificial intelligence on the de novo design of natural-product-inspired retinoid X receptor modulators, Commun. Chem., 2018, 1, 68 CrossRef.
- R. Gómez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernández-Lobato, B. Sánchez-Lengeling, D. Sheberla, J. Aguilera-Iparraguirre, T. D. Hirzel, R. P. Adams and A. Aspuru-Guzik, Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules, ACS Cent. Sci., 2018, 4, 268–276 CrossRef PubMed.
- S. A. Lopez, B. Sanchez-Lengeling, J. de Goes Soares and A. Aspuru-Guzik, Design Principles and Top Non-Fullerene Acceptor Candidates for Organic Photovoltaics, Joule, 2017, 1, 857–870 CrossRef CAS.
- D. Padula, J. D. Simpson and A. Troisi, Combining electronic and structural features in machine learning models to predict organic solar cells properties, Mater. Horiz., 2019, 6, 343–349 RSC.
- L. Wilbraham, R. Sprick, K. Jelfs and M. Zwijnenburg, Mapping binary copolymer property space with neural networks, Chem. Sci., 2019, 10, 4973–4984 RSC.
- P. B. Jørgensen, M. Mesta, S. Shil, J. M. García Lastra, K. W. Jacobsen, K. S. Thygesen and M. N. Schmidt, Machine learning-based screening of complex molecules for polymer solar cells, J. Chem. Phys., 2018, 148, 241735 CrossRef PubMed.
- G. X. Gu, C.-T. Chen, D. J. Richmond and M. J. Buehler, Bioinspired hierarchical composite design using machine learning: simulation, additive manufacturing, and experiment, Mater. Horiz., 2018, 5, 939–945 RSC.
- M. A. Bessa, P. Glowacki and M. Houlder, Bayesian Machine Learning in Metamaterial Design: Fragile Becomes Supercompressible, Adv. Mater., 2019, 1904845 CrossRef CAS PubMed.
- D. C. Elton, Z. Boukouvalas, M. D. Fuge and P. W. Chung, Deep learning for molecular design—a review of the state of the art, Mol. Syst. Des. Eng., 2019, 4, 828–849 RSC.
- B. Sanchez-Lengeling, C. Outeiral, G. L. Guimaraes and A. Aspuru-Guzik, Optimizing distributions over molecular space. An objective-reinforced generative adversarial network for inverse-design chemistry (ORGANIC), ChemRxiv, 2017 DOI:10.26434/chemrxiv.5309668.v3.
- S. Wu, Y. Kondo, M. Kakimoto, B. Yang, H. Yamada, I. Kuwajima, G. Lambard, K. Hongo, Y. Xu, J. Shiomi, C. Schick, J. Morikawa and R. Yoshida, Machine-learning-assisted discovery of polymers with high thermal conductivity using a molecular design algorithm, npj Comput. Mater., 2019, 5, 5 CrossRef.
- T. Young, D. Hazarika, S. Poria and E. Cambria, Recent Trends in Deep Learning Based Natural Language Processing [Review Article], IEEE Comput. Intell. Mag., 2018, 13, 55–75 Search PubMed.
- N. Boulanger-Lewandowski, Y. Bengio and P. Vincent, Modeling Temporal Dependencies in High-dimensional Sequences: Application to Polyphonic Music Generation and Transcription, Proceedings of the 29th International Coference on International Conference on Machine Learning, USA, 2012, pp. 1881–1888 Search PubMed.
- J. J. Irwin, T. Sterling, M. M. Mysinger, E. S. Bolstad and R. G. Coleman, ZINC: A Free Tool to Discover Chemistry for Biology, J. Chem. Inf. Model., 2012, 52, 1757–1768 CrossRef CAS PubMed.
- E. Jannik Bjerrum and R. Threlfall, Molecular Generation with Recurrent Neural Networks (RNNs), arXiv e-prints, 2017, https://arxiv.org/abs/1705.04612.
- J.-L. Reymond, The Chemical Space Project, Acc. Chem. Res., 2015, 48, 722–730 CrossRef CAS PubMed.
- B. Sanchez-Lengeling and A. Aspuru-Guzik, Inverse molecular design using machine learning: Generative models for matter engineering, Science, 2018, 361, 360–365 CrossRef CAS PubMed.
- I. Goodfellow, J. Pouget-Abadie, M. Mirza, B. Xu, D. Warde-Farley, S. Ozair, A. Courville and Y. Bengio, Generative adversarial nets. Advances in neural information processing systems, 2014, pp. 2672–2680 Search PubMed.
- Y. Tao, C. Yang and J. Qin, Organic host materials for phosphorescent organic light-emitting diodes, Chem. Soc. Rev., 2011, 40, 2943–2970 RSC.
- A. Mishra and P. Bäuerle, Small molecule organic semiconductors on the move: promises for future solar energy technology, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS PubMed.
- M. J. Cho, D. H. Choi, P. A. Sullivan, A. J. Akelaitis and L. R. Dalton, Recent progress in second-order nonlinear optical polymers and dendrimers, Prog. Polym. Sci., 2008, 33, 1013–1058 CrossRef CAS.
- G. S. He, L.-S. Tan, Q. Zheng and P. N. Prasad, Multiphoton Absorbing Materials:? Molecular Designs, Characterizations, and Applications, Chem. Rev., 2008, 108, 1245–1330 CrossRef CAS PubMed.
- L. R. Dalton, P. A. Sullivan and D. H. Bale, Electric Field Poled Organic Electro-optic Materials: State of the Art and Future Prospects, Chem. Rev., 2010, 110, 25–55 CrossRef CAS.
- P. Kautny, F. Glöcklhofer, T. Kader, J.-M. Mewes, B. Stöger, J. Fröhlich, D. Lumpi and F. Plasser, Charge-transfer states in triazole linked donor–acceptor materials: strong effects of chemical modification and solvation, Phys. Chem. Chem. Phys., 2017, 19, 18055–18067 RSC.
- S. K. M. Nalluri and R. V. Ulijn, Discovery of energy transfer nanostructures using gelation-driven dynamic combinatorial libraries, Chem. Sci., 2013, 4, 3699–3705 RSC.
- Y. Li, T. Miao, P. Li and L. Wang, Photo-Driven Synthesis of C6-Polyfunctionalized Phenanthridines from Three-Component Reactions of Isocyanides, Alkynes, and Sulfinic Acids by Electron Donor–Acceptor Complex, Org. Lett., 2018, 20, 1735–1739 CrossRef CAS PubMed.
- A. Postigo, Electron Donor-Acceptor Complexes in Perfluoroalkylation Reactions, Eur. J. Org. Chem., 2018, 6391–6404 CrossRef CAS.
- J. Hachmann, R. Olivares-Amaya, S. Atahan-Evrenk, C. Amador-Bedolla, R. S. Sánchez-Carrera, A. Gold-Parker, L. Vogt, A. M. Brockway and A. Aspuru-Guzik, The Harvard clean energy project: large-scale computational screening and design of organic photovoltaics on the world community grid, J. Phys. Chem. Lett., 2011, 2, 2241–2251 CrossRef CAS.
- https://cmr.fysik.dtu.dk/ .
- B. G. Abreha, S. Agarwal, I. Foster, B. Blaiszik and S. A. Lopez, Virtual Excited State Reference for the Discovery of Electronic Materials Database: An Open-Access Resource for Ground and Excited State Properties of Organic Molecules, J. Phys. Chem. Lett., 2019, 10, 6835–6841 CrossRef CAS PubMed.
- S.-H. Chou, H.-W. Kang, S.-T. Chang, K.-Y. Wu, G. C. Bazan, C.-L. Wang, H.-L. Lin, J.-H. Chang, H.-W. Lin, Y.-C. Huang, C.-S. Tsao and K.-T. Wong, Cofacial Versus Coplanar Arrangement in Centrosymmetric Packing Dimers of Dipolar Small Molecules: Structural Effects on the Crystallization Behaviors and Optoelectronic Characteristics, ACS Appl. Mater. Interfaces, 2016, 8, 18266–18276 CrossRef CAS PubMed.
- https://figshare.com/projects/GuacaMol/56639 .
- N. Brown, M. Fiscato, M. H. Segler and A. C. Vaucher, Guacamol: benchmarking models for de novo molecular design, J. Chem. Inf. Model., 2019, 59, 1096–1108 CrossRef CAS PubMed.
- A. D. Becke, A new mixing of Hartree–Fock and local density-functional theories, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- K. Raghavachari, Perspective on “Density functional thermochemistry. III. The role of exact exchange”, Theor. Chem. Acc., 2000, 103, 361–363 Search PubMed.
- P. J. Stephens, F. Devlin, C. Chabalowski and M. J. Frisch, Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- T. Körzdörfer and J.-L. Brédas, Organic Electronic Materials: Recent Advances in the DFT Description of the Ground and Excited States Using Tuned Range-Separated Hybrid Functionals, Acc. Chem. Res., 2014, 47, 3284–3291 CrossRef PubMed.
- S. Maekawa and K. Moorthi, Polymer Optical Constants from Long-Range Corrected DFT Calculations, J. Phys. Chem. B, 2016, 120, 2507–2516 CrossRef CAS PubMed.
- S. Riniker and G. A. Landrum, Better informed distance geometry: using what we know to improve conformation generation, J. Chem. Inf. Model., 2015, 55, 2562–2574 CrossRef CAS PubMed.
- A. K. Rappé, C. J. Casewit, K. Colwell, W. A. Goddard III and W. M. Skiff, UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef.
- C. Bannwarth, S. Ehlert and S. Grimme, GFN2-xTB—An accurate and broadly parametrized self-consistent tight-binding quantum chemical method with multipole electrostatics and density-dependent dispersion contributions, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- Y.-S. Lin, G.-D. Li, S.-P. Mao and J.-D. Chai, Long-Range Corrected Hybrid Density Functionals with Improved Dispersion Corrections, J. Chem. Theory Comput., 2013, 9, 263–272 CrossRef CAS.
- F. Neese, Software update: the ORCA program system, version 4.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- S. Grimme, A simplified Tamm-Dancoff density functional approach for the electronic excitation spectra of very large molecules, J. Chem. Phys., 2013, 138, 244104 CrossRef PubMed.
- C. Bannwarth and S. Grimme, A simplified time-dependent density functional theory approach for electronic ultraviolet and circular dichroism spectra of very large molecules, Comput. Theor. Chem., 2014, 1040–1041, 45–53 CrossRef CAS , Excited states: from isolated molecules to complex environments.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- K. Cho, B. Van Merriënboer, C. Gulcehre, D. Bahdanau, F. Bougares, H. Schwenk and Y. Bengio, Learning phrase representations using RNN encoder-decoder for statistical machine translation, arXiv preprint arXiv:1406.1078, 2014.
- D. P. Kingma and J. Ba, Adam: A method for stochastic optimization, arXiv preprint arXiv:1412.6980, 2014.
- A. Paszke, S. Gross, S. Chintala, G. Chanan, E. Yang, Z. DeVito, Z. Lin, A. Desmaison, L. Antiga and A. Lerer, Automatic differentiation in PyTorch. NIPS-W, 2017 Search PubMed.
- G. Landrum, RDKit: Open-source cheminformatics, 2006 Search PubMed.
- V. D. Blondel, J.-L. Guillaume, R. Lambiotte and E. Lefebvre, Fast unfolding of communities in large networks, J. Stat. Mech.: Theory Exp., 2008, 2008, P10008 CrossRef.
- K. T. Schütt, P. Kessel, M. Gastegger, K. A. Nicoli, A. Tkatchenko and K.-R. Müller, SchNetPack: A Deep Learning Toolbox For Atomistic Systems, J. Chem. Theory Comput., 2019, 15, 448–455 CrossRef PubMed.
- C. Chen, W. Ye, Y. Zuo, C. Zheng and S. P. Ong, Graph Networks as a Universal Machine Learning Framework for Molecules and Crystals, Chem. Mater., 2019, 31, 3564–3572 CrossRef CAS.
- R. Ramakrishnan, P. O. Dral, M. Rupp and O. A. Von Lilienfeld, Quantum chemistry structures and, properties of 134 kilo molecules, Sci. Data, 2014, 1, 140022 CrossRef CAS PubMed.
- J. L. Durant, B. A. Leland, D. R. Henry and J. G. Nourse, Reoptimization of MDL keys for use in drug discovery, J. Chem. Inf. Comput. Sci., 2002, 42, 1273–1280 CrossRef CAS PubMed.
- L. v. d. Maaten and G. Hinton, Visualizing data using t-SNE, J. Mach. Learn. Res., 2008, 9, 2579–2605 Search PubMed.
- P. Ertl and A. Schuffenhauer, Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions, J. Cheminf., 2009, 1, 8 Search PubMed.
- P. Politzer, J. Martinez, J. S. Murray and M. C. Concha, An electrostatic correction for improved crystal density predictions of energetic ionic compounds, Mol. Phys., 2010, 108, 1391–1396 CrossRef CAS.
- H. Yao, L. Ye, H. Zhang, S. Li, S. Zhang and J. Hou, Molecular design of benzodithiophene-based organic photovoltaic materials, Chem. Rev., 2016, 116, 7397–7457 CrossRef CAS PubMed.
- S. Chaurasia, C.-Y. Hsu, H.-H. Chou and J. T. Lin, Synthesis, optical and electrochemical properties of pyridal[2,1,3]thiadiazole based organic dyes for dye sensitized solar cells, Org. Electron., 2014, 15, 378–390 CrossRef CAS.
- H. Takenaka, T. Ogaki, C. Wang, K. Kawabata and K. Takimiya, Selenium-Substituted β-Methylthiobenzo [1, 2-b:4, 5-b] dithiophenes: Synthesis, Packing Structure, and Transport Properties, Chem. Mater., 2019, 31, 6696–6705 CrossRef CAS.
- M. C. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger and C. J. Brabec, Design rules for donors in bulk-heterojunction solar cells—Towards 10% energy-conversion efficiency, Adv. Mater., 2006, 18, 789–794 CrossRef CAS.
- R. Gómez-Bombarelli, J. Aguilera-Iparraguirre, T. D. Hirzel, D. Duvenaud, D. Maclaurin, M. A. Blood-Forsythe, H. S. Chae, M. Einzinger, D.-G. Ha, T. Wu, G. Markopoulos, S. Jeon, H. Kang, H. Miyazaki, M. Numata, S. Kim, W. Huang, S. I. Hong, M. Baldo, R. P. Adams and A. Aspuru-Guzik, Design of efficient molecular organic light-emitting diodes by a high-throughput virtual screening and experimental approach, Nat. Mater., 2016, 15, 1120–1127 CrossRef PubMed.
- H. Bronstein, C. B. Nielsen, B. C. Schroeder and I. McCulloch, The role of chemical design in the performance of organic semiconductors, Nat. Rev. Chem., 2020, 1–12 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/C9NR10687A |
| This journal is © The Royal Society of Chemistry 2020 |