
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA biocompatible ZnNa2-based metal–organic framework with high ibuprofen, nitric oxide and metal uptake capacity†
Meghan
Winterlich
ab,
Constantinos G.
Efthymiou
b,
Wassillios
Papawassiliou
c,
Jose P.
Carvalho
c,
Andrew J.
Pell
 c,
Julia
Mayans
d,
Albert
Escuer
e,
Michael P.
Carty
f,
Patrick
McArdle
b,
Emmanuel
Tylianakis
g,
Liam
Morrison
*h,
George
Froudakis
*i and
Constantina
Papatriantafyllopoulou
*abj
c,
Julia
Mayans
d,
Albert
Escuer
e,
Michael P.
Carty
f,
Patrick
McArdle
b,
Emmanuel
Tylianakis
g,
Liam
Morrison
*h,
George
Froudakis
*i and
Constantina
Papatriantafyllopoulou
*abj
aCÚRAM Centre for Research in Medical Devices, Ireland
bSchool of Chemistry, College of Science and Engineering, National University of Ireland Galway, H91 TK 33 Galway, Ireland. E-mail: constantina.papatriantafyllopo@nuigalway.ie; Tel: +353 91 493462
cDepartment of Materials and Environmental Chemistry, Stockholm University, Sweden
dInstituto de Ciencia Molecular (ICMol), Universidad de Valencia, Catedrático José Beltran 2, 46980 Paterna, Valencia, Spain
eDepartament de Quimica Inorgànica i Orgànica, Secció Inorgànica and Institut de Nanociencia i Nanotecnologia (IN2UB), Universitat de Barcelona, Marti i Franquès 1-11, 08028 Barcelona, Spain
fBiochemistry, School of Natural Sciences, National University of Ireland Galway, H91 TK 33 Galway, Ireland
gDepartment of Materials Science and Technology, University of Crete, Voutes Campus, GR-70013 Heraklion, Crete, Greece
hEarth and Ocean Sciences and Ryan Institute, School of Natural Sciences, National University of Ireland Galway, H91 TK 33 Galway, Ireland
iDepartment of Chemistry, University of Crete, Voutes Campus, GR-70013 Heraklion, Crete, Greece
jSSPC, Synthesis and Solid State Pharmaceutical Centre, Ireland
First published on 20th August 2020
Abstract
Metal organic frameworks (MOFs) have received significant attention in recent years in the areas of biomedical and environmental applications. Among them, mixed metal MOFs, although promising, are relatively few in number in comparison with their homometallic analogues. The employment of benzophenone-4,4′-dicarboxylic acid (bphdcH2) in mixed metal MOF chemistry provided access to a 3D MOF, [Na2Zn(bphdc)2(DMF)2]n (NUIG1). NUIG1 displays a new topology and is a rare example of a mixed metal MOF based on 1D rod secondary building units. UV-vis, HPLC, TGA, XRPD, solid state NMR and computational studies indicated that NUIG1 exhibits an exceptionally high Ibuprofen (Ibu) and nitric oxide adsorption capacity. The MCF-7 cell line was used to assess the toxicity of NUIG1 and Ibu@NUIG1, revealing that both species are non-toxic (cell viability > 70%). NUIG1 exhibits good performance in the adsorption of metal ions (CoII, NiII, CuII) from aqueous environments, as was demonstrated by UV-vis, EDX, ICP, SEM and direct and alternate current magnetic susceptibility studies. The colour and the magnetic properties of the M@NUIG1 species depend strongly on the kind and the amount of the encapsulated metal ion in the MOF pores.
Introduction
Metal–organic frameworks (MOFs) are a family of hybrid porous materials that have attracted immense research interest in recent decades due to their appealing structural features.1,2 They often possess large surface area, high porosity, flexible structure, an amphiphilic internal microenvironment, and the possibility of introducing functional groups in the pores and frameworks in a spatially controlled way, which make these materials especially suitable for encapsulating a large variety of guest molecules. As such, MOFs can be used in a wide range of applications, e.g. gas storage and/or separation,3 magnetism, catalysis, sensing, drug delivery and imaging related applications.4,5In relation to the use of MOFs in drug delivery applications, they were introduced into this field nearly ten years ago, with one of the first reports being by Horcajada et al, who showed that the mesoporous MOFs MIL-100 and MIL-101 display remarkable ibuprofen adsorption and release performance.6 Since then, many MOFs with high drug uptake have been reported, representative examples being other members of the MIL family, Bio-MOFs, CD-MOFs, UiO-MOFs, ZIF-MOFs, Zr MOFs, etc.7–9 MOFs offer significant advantages over the currently used drug delivery systems that are based on liposomes, micelles, dendrimers, gold/iron/silica nanoparticles, carbon nanotubes, and quantum dots.10–12 MOFs often exhibit high drug upload capacity, biocompatibility, thermal stability, inexpensive and rapid scale-up synthesis, and thus can address major challenges related to the poor active pharmaceutical ingredient (API) stability and/or solubility, burst effect and toxicity.13,14 The use of MOFs in drug delivery has recently been expanded to anticancer drugs and the targeted, and controlled delivery has been achieved by a variety of different techniques, such as pH-controlled, photodynamic and magnetically triggered drug release.15–17
In a similar vein, MOFs can capture and sense toxic compounds in the environment.18–20 The adsorption process may occur via a variety of mechanisms, such as interactions between acid and base, electrostatic interactions between adsorbates and adsorbent, H-bonding and π–π stacking, and hydrophobic interactions.20c The removal of toxic metals from water is important and contemporary techniques involve chemical precipitation, ion exchange, membrane filtration and adsorption, with the latter which can employ MOFs being the most preferred as it does not require a high operating temperature. The adsorbents that are currently used either possess high porosity but are prone to aggregation, which decreases their efficiency (e.g. magnetic nanoparticles), or have high stability and efficiency, but low surface area (e.g. zeolites).21 MOFs display all the desirable features to provide an efficient solution to the removal of toxic metals from water, and as such many MOF families (MIL, UiO-66, HKUST, etc.) have now been reported for their ability to adsorb toxic metals.22 MOFs often have sensing properties, which is based on the effect of the toxic/guest metal ion to the physical properties (colour, electrochemical, luminescence, etc.) of the MOF.23,24 One recent such example is the FJI-C8 MOF,24 which exhibits a high sensitivity for Fe3+ with the detection limit being 0.0233 mM; it contains a π-conjugated aromatic ligand with free/uncoordinated N- and O- atoms, which interact strongly with the encapsulated Fe3+ ions affecting the emission spectrum of the anionic FJI-C8 MOF.
The advantages that MOFs offer in important biomedical and environmental applications, constitute an increasing need for the isolation of new such species with high stability, porosity and fine-tuning properties. One recent approach towards this direction involves the presence of a second metal ion in the MOF structure;25 this often increases the robustness of the framework, which results to high thermal and water stability. Furthermore, the synergism between the different metal ions has been proven to enhance MOF features and functionalities, including, among others, the breathing effect, catalytic activity, etc.26 It is noteworthy that although ca. 6000 new MOFs are reported per year, relatively few of them are mixed-metal, with the mixed metal MOF field being still in its infancy.25
With the above mentioned in mind, we decided to expand the family of mixed metal MOFs by using benzophenone-4,4′- dicarboxylic acid (bphdcH2, Scheme 1) as the organic linker; this ligand has been extensively used for the synthesis of homometallic MOFs, yet its employment towards new mixed metal MOFs is limited.27 In particular, the combination of bphdcH2 with other bridging ligands, including 4,4′-bipyridine (bpy), 4-diazabicyclo[2.2.2]octane (dadco), and others, in ZnII and CuII coordination chemistry has yielded new 3D MOFs with interesting properties; among them, [Cu2(bphdc)2(bpy)]n and [Zn2(bphdc)2(dadco)]n display very high H2 and CO2 adsorption.27a Herein, we report the synthesis and characterization of [Na2Zn(bphdc)2(DMF)2]n (NUIG1), which is a rare example of a mixed-metal MOF based on an 1D rod SBU. Herein, the drug uptake performance of NUIG1 have been studied and discussed in detail for Ibuprofen and nitric oxide. The metal adsorption capacity of NUIG1 was also examined using a variety of techniques, including UV-vis, EDX, ICP, SEM and magnetism.
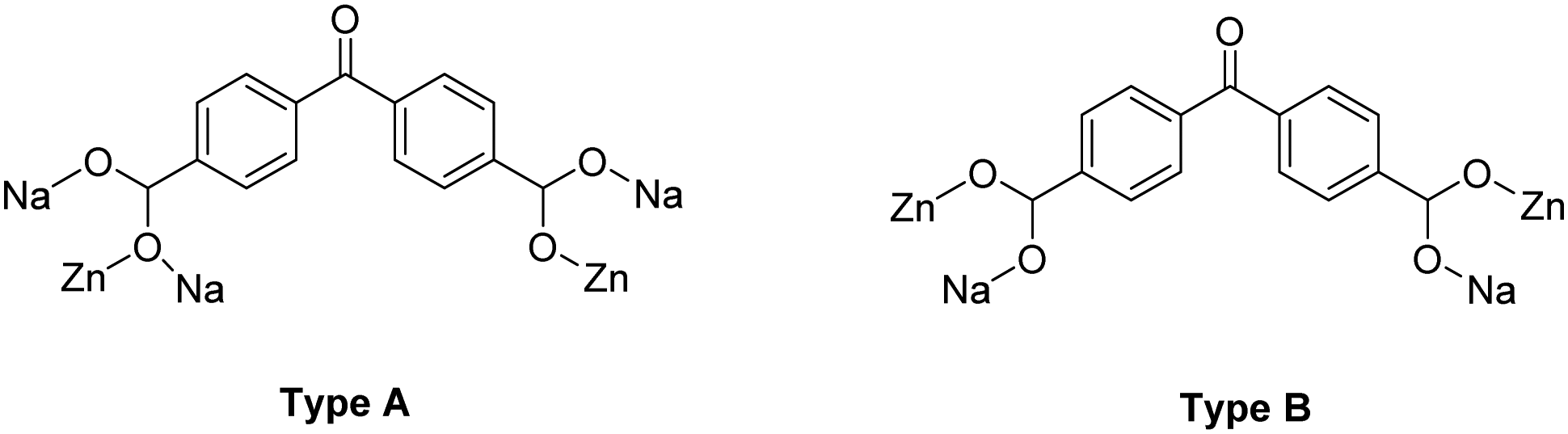 | ||
| Scheme 1 A schematic representation of benzophenone-4,4′-dicarboxylic acid (bphdcH2) and the two different coordination modes that it adopts in NUIG1: η2:η1:η1:η1:μ5 (Type A, left) and η1:η1:η1:η1:μ4 (Type B, right). | ||
Experimental
All manipulations were performed under aerobic conditions using materials as received.Synthesis of [Na2Zn(bphdc)2(DMF)2]n (NUIG1)
1 M NaOH (0.5 mL) and Zn(CH3CO2)2·2H2O (0.03 g, 0.1 mmol) were added to a solution of bphdcH2 (0.07 g, 0.3 mmol) in DMF/EtOH (10/8 mL) and left under magnetic stirring for 5 min at room temperature. Then, the vial was placed in the oven at 100 °C for 24 hours, after which time white polyhedral crystals of NUIG1 were observed. The crystals were kept in mother liquor for X-ray analysis or collected by filtration for other solid-state studies. Yield: ∼70%. Anal. calcd (found) for NUIG1: C, 54.46 (54.71); H, 3.81 (3.66); N, 3.53 (3.64)%. Selected IR data (KBr, cm−1): 3437(b), 2928(b), 1655(s), 1614(m), 1558(w), 1498(w), 1438(w), 1361(m), 1298(w), 1256(m), 1127(w), 1094(s), 1063(w), 1016(w), 933(m), 880(m), 833(s), 811(w), 786(w), 734(s), 705(w), 661(m).Physical studies
Elemental analyses (C, H, N) were performed by the in-house facilities of the National University of Ireland Galway, School of Chemistry. IR spectra (4000–400 cm−1) were recorded using a PerkinElmer 16PC FT-IR spectrometer with samples prepared as KBr pellets. Powder X-ray diffraction data (XRPD) were collected using an Inex Equinox 6000 diffractometer. Solid-state, variable-temperature and variable-field magnetic data were collected on powdered samples using an MPMS5 Quantum Design magnetometer operating at 0.03 T in the 300 to 2.0 K range for the magnetic susceptibility and at 2.0 K in the 0 to 5 T range for the magnetization measurements. Diamagnetic corrections were applied to the observed susceptibilities using Pascal's constants. TGA experiments were performed on a STA625 thermal analyser from Rheometric Scientific (Piscataway, New Jersey). The heating rate was kept constant at 10 °C min−1, and all runs were carried out between 20 and 600 °C. The measurements were made in open aluminium crucibles, nitrogen was purged in ambient mode, and calibration was performed using an indium standard.X-ray crystallography
Crystallographic data for NUIG1 were collected in an Oxford Diffraction Xcalibur CCD diffractometer using graphite-monochromatic Mo Ka radiation (λ = 0.71073 Å) at room temperature. The structures were solved using SHELXT,28 embedded in the OSCAIL software.29 The non-H atoms were treated anisotropically, whereas the hydrogen atoms were placed in calculated, ideal positions and refined as riding on their respective carbon atoms. The program SQUEEZE,30 a part of the PLATON package of crystallographic software, was used to remove contribution of highly disordered solvent molecules.Unit cell data and structure refinement details are listed in Table S1 (ESI†). CCDC 2008513.†
Drug adsorption and release experiments
The Ibu adsorption capacity of NUIG1 was investigated as described below: drug (0.23 g, 0.70 mmol) was added to a glass vial containing EtOH (10 mL) and stirred until all solid is dissolved. Solid NUIG1 (0.05 g, 0.07 mmol) was then added and the mixture was left stirring at room temperature. For the kinetic study, small volumes of aliquots were taken at designated time intervals, centrifuged, and the drug content in the supernatant solution was determined by spectroscopic (UV-vis) and chromatographic (HPLC) techniques. For the thermodynamic study, the same procedure was repeated with varying NUIG1: drug ratios; the mixture was stirred for 120 hours, filtered, and the filtrate was analysed for its drug content. The drug release properties of NUIG1 were studied in H2O and phosphate buffer solution (PBS): Ibu@NUIG1 (0.05 g) was added to a glass vial containing solvent (10 mL) and left stirring at 37 °C. Batch studies were performed at designated time intervals, and the amount of the released drug was determined by UV-vis and HPLC techniques.Metal adsorption kinetic and thermodynamic studies
The metal adsorption capacity of NUIG1 was investigated using the same method to the drug adsorption: the hydrated acetate salt of a metal ion (0.10 g, 0.40 mmol for Co2+ and Ni2+; 0.05 g, 0.22 mmol for Cu2+) was added to a glass vial containing distilled H2O (10 mL, for Co2+ and Ni2+; 20 mL, for Cu2+) and stirred until all solid is dissolved. Solid NUIG1 (0.10 g, 0.14 mmol, Co2+ and Ni2+; 0.05 g, 0.07 mmol, Cu2+) was then added and the mixture was left stirring at room temperature. For the kinetic study, the sample was centrifuged at designated time intervals, and the metal content in the supernatant was determined by spectroscopic (UV-vis) techniques and by using an Elan DRC-e Inductively Coupled Plasma–Mass Spectrometry, ICP-MS, [PerkinElmer, USA] in standard mode in a class 1000 clean room (ISO 6). For the thermodynamic study, the same procedure was repeated with varying NUIG1: metal ratios; the mixture was stirred for 30 minutes, filtered, and the filtrate was analysed for its metal content.Solid state NMR
All solid-state NMR spectra were collected on a Bruker Avance III spectrometer with magnetic field strength of 9.4 T operating at frequencies of 400 MHz for 1H, 100.6 MHz for 13C, and 105.842 MHz for 23Na, respectively. For the acquisition of the data a 2.5 mm HX probe was used, employing magic-angle spinning (MAS) frequencies of up to 30 kHz.The 23Na spectra were collected at 30 kHz MAS. The one-pulse spectra were acquired using a RF pulse with a 136 kHz nutation frequency and small flip angles (0.44 μs) and a recycle delay of 16 s. Two-dimensional one-pulse double-quantum-filtered satellite transition MAS (TOP-DQF-STMAS) spectra31a,b were acquired with a sampling interval of 33.33 μs and 128 increments in the indirect dimension, with 192 scans per increment for the MOF and 96 scans for the Ibuprofen loaded MOF with a recycle delay of 4 s. Pulses with a nutation frequency of 136 kHz and lengths of 1.1 μs and 1.2 μs were used for satellite-transition coherence excitation, and central-transition reconversion respectively. The central-transition-selective pulse had a nutation frequency of 12 kHz and a length of 10.4 μs. Solid NaCl was used for RF pulse calibration and for referencing the chemical shifts at 7.21 ppm. For the one-dimensional 13C spectra, cross polarization (CP) 1H → 13C experiments were performed at 10 kHz MAS. Acquisitions employed a 1H excitation pulse of 71 kHz nutation frequency and 3.5 μs length followed by matched spin-lock fields of 81 kHz for 1H and 71 kHz for 13C, and a contact time of 1250 ms. SPINAL6431c decoupling of 71 kHz was used during acquisition. Between 8192 and 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 384 transients were collected, depending on the sample, with a 4 s recycle delay. The two-dimensional heteronuclear correlation (HETCOR) experiment31d was acquired using the same experimental conditions as CP, but with 80 increments in the indirect (1H) dimension and 256 scans per increment. Adamantane was used for rf pulse calibrations and chemical shifts referencing for both 1H and 13C.
384 transients were collected, depending on the sample, with a 4 s recycle delay. The two-dimensional heteronuclear correlation (HETCOR) experiment31d was acquired using the same experimental conditions as CP, but with 80 increments in the indirect (1H) dimension and 256 scans per increment. Adamantane was used for rf pulse calibrations and chemical shifts referencing for both 1H and 13C.
Computational studies
In order to explore the ability of NUIG1 to adsorb NO and Ibu, Grand Canonical ensemble Monte Carlo simulations were conducted. According to these simulations, the chemical potential (μ), the volume of the system (V), and the temperature (T) are kept constant, whereas the number of particles of the system, N could fluctuate. During the simulations trial moves are attempted: particle insertion or deletion, particle translation and particle rotation with each trial move having the same probability of selection.32The chemical potential was determined for NO by using the Peng–Robinson equation of state33a with the appropriate physical constants i.e. critical pressure, Pc, critical temperature, Tc, and acentric factor. Temperature was set to 121 K. The Lennard Jones potential was used to describe the van der Waals interactions between framework atoms and guest molecules. Parameters set for the various atoms were taken from the Dreiding force field,33b whereas the Lorentz–Berthelot mixing rules were used for cross parameters. The minimum image convention was used, placing the atom under examination at a centre of a sphere having radius equal to the potential cut off distance, i.e. 12.8 Å. Interactions of the atom under examination with atoms lying outside this sphere were not taken into consideration. The potential beyond this distance was truncated and no tail corrections were used. A super cell of 2 × 2 × 2 unit cells was used for the simulations to ensure that each direction, x, y and z is at least twice the cut off distance.
Together with Lennard Jones for the van der Waals interactions, the Coulomb potential was considered for describing the electrostatic interactions between the framework and fluid atoms. Point charges for the atoms of the framework were calculated using the Gaussian 03 software package and applying the cluster method applied: An appropriate part of the framework was cut and this part was first geometrically optimized applying Density Functional Theory at the B3LYP level.34 The charges of the atoms for the optimized part were then calculated using the CHELPG method.35
N2: The TraPPE force field36a was used for describing the interactions of nitrogen molecules according to which the molecule is described as a three site model, one for each atom and one for the centre of mass. A charge of q = −0.482 was assigned to each atom with Lennard Jones parameters ε = 36 K and σ = 3.31 Å. A q = +0.964 was placed to the centre of mass without any LJ interactions.
NO: For nitric oxide a two centre model was used, both centres having LJ parameters and Coulomb interactions.36b The N–O bond length was kept fixed at 1.15 Å; for oxygen, ε = 96.986 K and σ = 2.875 Å and for nitrogen, ε = 79.538 K and σ = 3.014 Å. These parameters were able to reproduce the heat of vaporization during NVT simulations. In addition, prior to proceeding with simulations, these parameters successfully reproduced the NO bulk density at all thermodynamic states used for the GCMC simulations.
Ibuprofen: Ibuprofen molecule was modelled following the methodology reported by Bernini et al.36c The parameters for the atoms or beads of the drug were taken from the corresponding references of the TraPPe Force. The molecule was treated as flexible.
Cytotoxicity measurements using the MTT assay
The MTT assay involves the reduction of a yellow tetrazolium salt, [3-(4,5-dimethylthazol-2-yl)-2,5-diphenyltetrazolium bromide] tetrazolium, to an insoluble formazan crystal by the metabolic activity of living cells. For this analysis, MTT assay was used to investigate the cytotoxicity of NUIG1, and Ibu@NUIG1. MCF-7 cells were seeded at a density of 1000 cells per mL in a 96-well micro assay culture plate and allowed to grow over a period of 24 h at 37 °C in a 5% CO2 incubator. NUIG1 in fresh culture medium was added into each well with different concentrations from 0.01–30 μM and incubated for 72 hours. Culture medium only was employed as the control group, and wells containing culture media without cells were used as blanks. 20 μL of a solution containing 43.2 mg MTT dissolved in 8.6 mL RPMI was added to each well, and the cells were incubated for another 3 hours. The excess MTT solution was then carefully removed from each well, and the formed formazan was dissolved in 100 μL of DMSO. The optical density of each well was then measured at a wavelength of 550 nm using a microplate reader (Bio-Rad, xMark). The results from the three individual experiments were averaged. The following formula was used to calculate the viability:| Viability (%) = (mean of absorbance value of treatment group–blank)/(mean absorbance value of control–blank) × 100. |
Results and discussion
Synthesis
Various reactions with differing reagent ratios, metal sources, solvents and other reaction conditions have been systematically explored towards the isolation of new non-toxic, mixed metal MOFs. The reaction of Zn(CH3CO2)2·2H2O, bphdcH2 and NaOH in a 1:2:1.5 molar ratio in EtOH/DMF (1:1.25) at 100 °C for 24 hours provided access to colourless cubic crystals of [ZnNa2(bphdc)2(DMF)2]n (NUIG1). The stoichiometric equation of the reaction that led to the formation of NUIG1 is presented in eqn (1).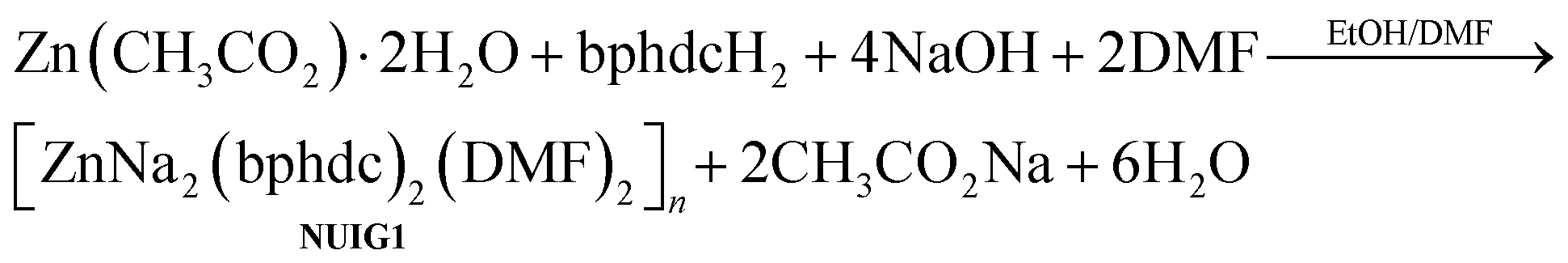 | (1) |
It is noteworthy that the nature of the solvent affects the crystallinity of the product; for example, reactions in DMF yielded white microcrystalline powder of NUIG1 (IR evidence). Similarly, the source of Na+ ions does not play a crucial role in the identity of the product, but it affects its crystallinity and the yield of the reaction. In particular, when Na(CH3CO2)·3H2O was used instead of NaOH, NUIG1 was produced in polycrystalline form in lower yield. The employment of significantly different molar ratios resulted in amorphous precipitates that could not be further characterized. Several bands appear in the 1655–1360 cm−1 region in the IR spectrum of NUIG1. Contributions from carboxylate νas(CO2) and νs(CO2) modes would be expected in this region, but overlap with the stretching vibrations of the aromatic ring and the carbonyl group renders assignment difficult.
Description of structure
Representations of the ZnNa2 repeating unit, the rod SBU and the 3D network of NUIG1 are shown in Fig. 1 and 2. NUIG1 crystallizes in the monoclinic space group C2/c and its structure contains three crystallographically inequivalent metal ions (Zn1, Na1 and Na2) and bphdc2− ligands that adopt two different coordination modes, η2:η1:η1:η1:μ5 (Type A, Scheme 1), and η1:η1:η1:η1:μ4 (Type B, Scheme 1). The metal ions within the ZnNa2 repeating unit are held together through two Type B and two Type A ligands with one of the latter being coordinated through its μ2 end, whereas the other through its μ3 end. Furthermore, three Type A and two Type B bphdc2− ligands are linked to Na1 and Na2 and bridge the ZnNa2 with five neighboring units. This results in the formation of a Zn/Na 1D chain, which is the secondary building unit (SBU) of NUIG1 (Fig. 1, bottom); all the Na atoms are aligned in a collinear fashion, with the Zn atoms lying in a zig-zag configuration. Each 1D chain is cross-linked to four adjacent rod SBUs through the organic bphdc2− ligands, leading to the formation of the 3D microporous network (Fig. 2).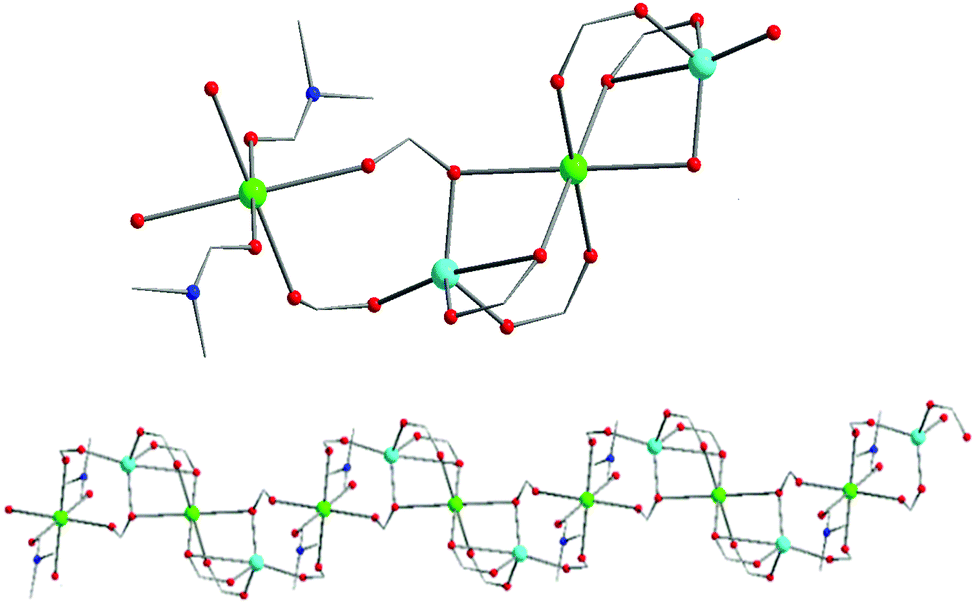 | ||
| Fig. 1 Representation of the ZnNa2 repeating unit (top) and a part of the 1D zig-zag Zn/Na SBU (bottom) in NUIG1. Color code: N (dark blue), Zn (light blue), Na (green), O (red). | ||
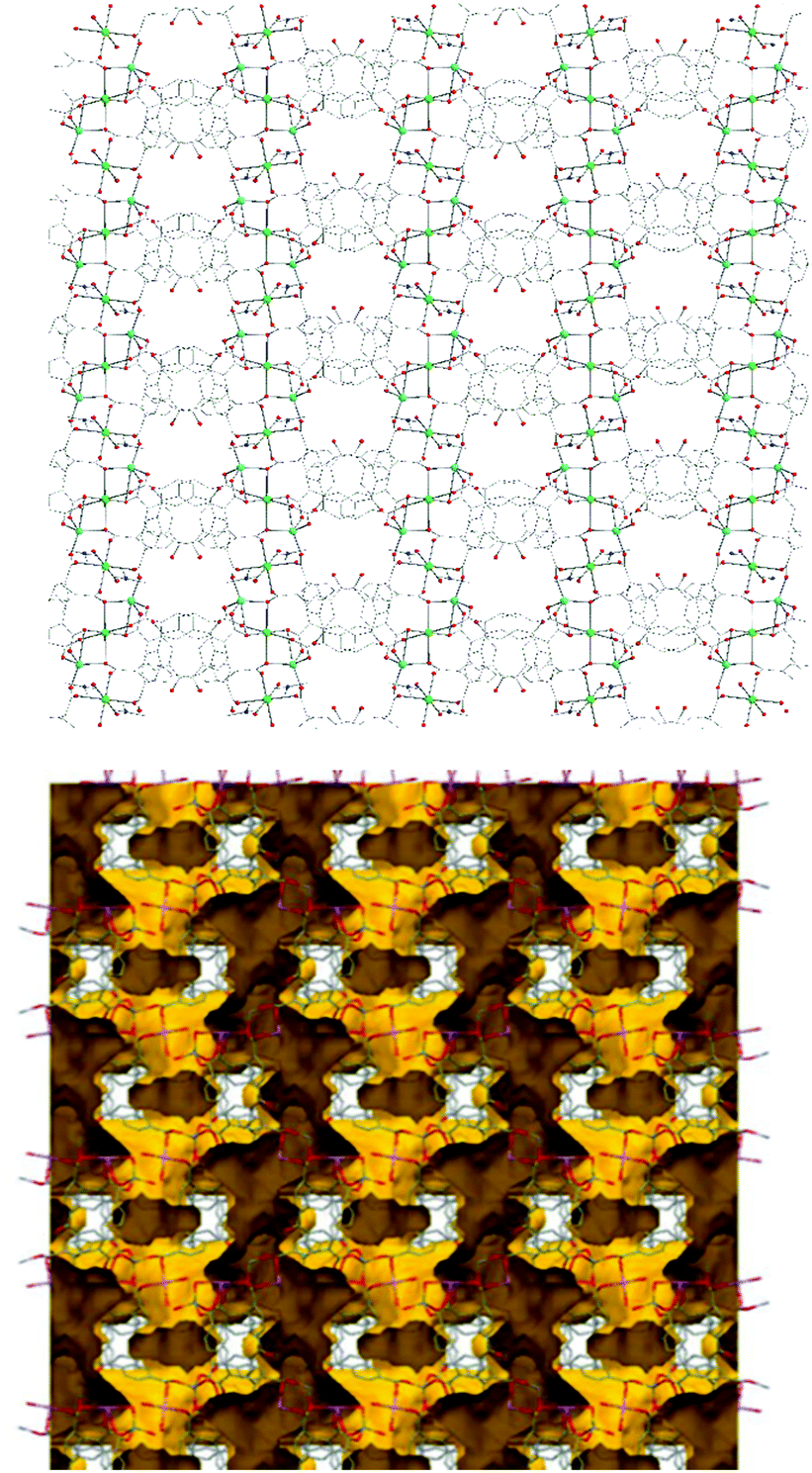 | ||
| Fig. 2 Representation of the 3d structure of NUIG1 along the a axis (top) and its pore network shown in yellow (bottom). Color code: N (dark blue), Zn (light blue), Na (green), O (red), C (grey). | ||
Zn1 adopts a tetrahedral coordination geometry being linked to four carboxylic O atoms, that come from four different bphdc2− ligands. Both Na atoms are hexacoordinate with a distorted octahedral coordination geometry. Na1 is coordinated to four oxygen atoms (equatorial plane), which come from four different organic ligands, as well as to two terminal DMF molecules in the axial positions. The coordination sphere of Na2 consists of six carboxylic O atoms from six different bphdc2− ligands.
Employing a standard representation of the crystal structure of NUIG1 and taking into account the Na atoms, both types of ligands and all metal ions can be considered as connection points;37 this gives rise to the formation of a novel three dimensional 4,4,4,5,6-c network, with point symbol {42·83·10}{42·84}2{44·62}2{44·63·83}2{48·66·8} (Fig. S1, ESI†); the latter has never been reported in the past, thus NUIG1 exhibits a network with a unique architecture. Alternatively, the determination of the NUIG1 topology can be performed without taking into account the Na atoms as they are weakly bonded to the organic ligands. In this case, the structure consists of polycatenated 2-periodic four-coordinated networks of undulated shape with an sql topology. The latter is common with ca. 100000 such structures in different representations (complete, standard or cluster) been currently known. The solvent-accessible volume of NUIG1, calculated by PLATON (excluding all solvents from the pores), is 4001.9 Å3 and corresponds to the 46.5% of the unit-cell volume (8610.8 Å3). A representation of the voids using the structure visualization program MERCURY reveals a complex 3D pore network, where the large compartmentalized cavities communicate through relatively wide channels with a diameter ≥4 Å.
NUIG1 is a new MOF with an aesthetically pleasing crystal structure and interesting structural features. It is a new addition to the growing family of mixed-metal MOFs, being the first Zn/Na MOF that contains the ligand bphdc2−. It is a rare example of a mixed metal MOF based on a rod SBU, being the first such Zn/Na species.38 It is worth to mention that the vast majority of the rod-based MOFs are homometallic and exhibit special properties, such as breathing and forbidden catenation; their SBU can be either a simple rode, i.e. equally spaced points on a straight line, or it can be more complicated, e.g. displaying a zig-zag topology or being based on edge sharing tetrahedra, etc.38 Furthermore, NUIG1 displays a novel three dimensional 4,4,4,5,6-c network, whose topology has not appeared in the past in a MOF.
Drug adsorption and release studies
The inherent porosity in NUIG1, and the existence of open channels in its structure, prompted us to assess its potential for drug delivery applications. Ibuprofen (Ibu) was chosen as a proof of concept model drug due to the availability of literature reports that would allow the evaluation of NUIG1 as drug carrier. The MOF crystals were activated prior to the drug encapsulation in order to reduce the amount of solvent present in the pores, and thus facilitate the drug adsorption; this was performed by stirring the MOF in DMF for several hours and then exchanging this solvent with more volatile acetone, which is easily removed at 80 °C. The adsorption of Ibu by NUIG1 was initially confirmed by IR spectroscopy with the appearance of peaks at 2800 and 2900 cm−1, which correspond to the ν(C–H) vibrations of the methyl groups from the Ibu. There is an increased intensity at the broad peak at 3100–3500 cm−1 due to the additional aromatic rings coming from the Ibu (ν(Ar–H) at ∼3100 cm−1) and the increased amount of hydrogen bonding interactions. Furthermore, there is a typical increase in the intensity of the peaks at 1500, 1430 and 1040 cm−1, characteristic of the COO− groups.The impact of the Ibu: MOF molar ratio on the NUIG1 drug loading performance was studied; it was found that the Ibu uptake (mg Ibu per g NUIG1) increases as this ratio increases from 1:1 to 1:6, where it reaches its maximum capacity (Fig. 3). The latter was determined by UV-vis and HPLC (Fig. S2, ESI†), which revealed that the maximum Ibu upload is 70% by weight, corresponding to 2.8 g Ibu per g NUIG1.
 | ||
| Fig. 3 Equilibrium data for the Ibu adsorption by NUIG1 (contact time: 144 hours). The solid line is a guide to the eye. | ||
NUIG1 possesses an exceptionally high Ibu uptake capacity, which is higher than previously reported for Ibu or any other drug.6 This unusual behaviour is attributed to an enhanced breathing effect in NUIG1, i.e. enlargement of its pore diameter upon the encapsulation of a guest molecule.39,40 In order to further investigate the breathing property of NUIG1, XRPD studies were carried out; Fig. 4 shows the comparison of the XRPD pattern of a physical mixture of NUIG1 and Ibu with that of Ibu@NUIG1, as well as that of the activated NUIG1. Ibu@NUIG1 shows a similar pattern to NUIG1 with additional characteristic peaks attributed to Ibu. The peaks that correspond to NUIG1 have been shifted in the PXRD pattern of Ibu@NUIG1, indicating a change in the cell volume and/or symmetry of the MOF after the drug encapsulation.7a The latter is a strong indication of the breathing property of NUIG1, which is further supported by the slow kinetics of Ibu encapsulation (Fig. S3, ESI†); in fact only the 15% wt of the drug is adsorbed in the first 24 hours. There is a gradual increase in the adsorption in the first 40 hours, after which a sharp increase is observed in the next 24 hours, reaching the amount of 2600 mg Ibu per g NUIG1.
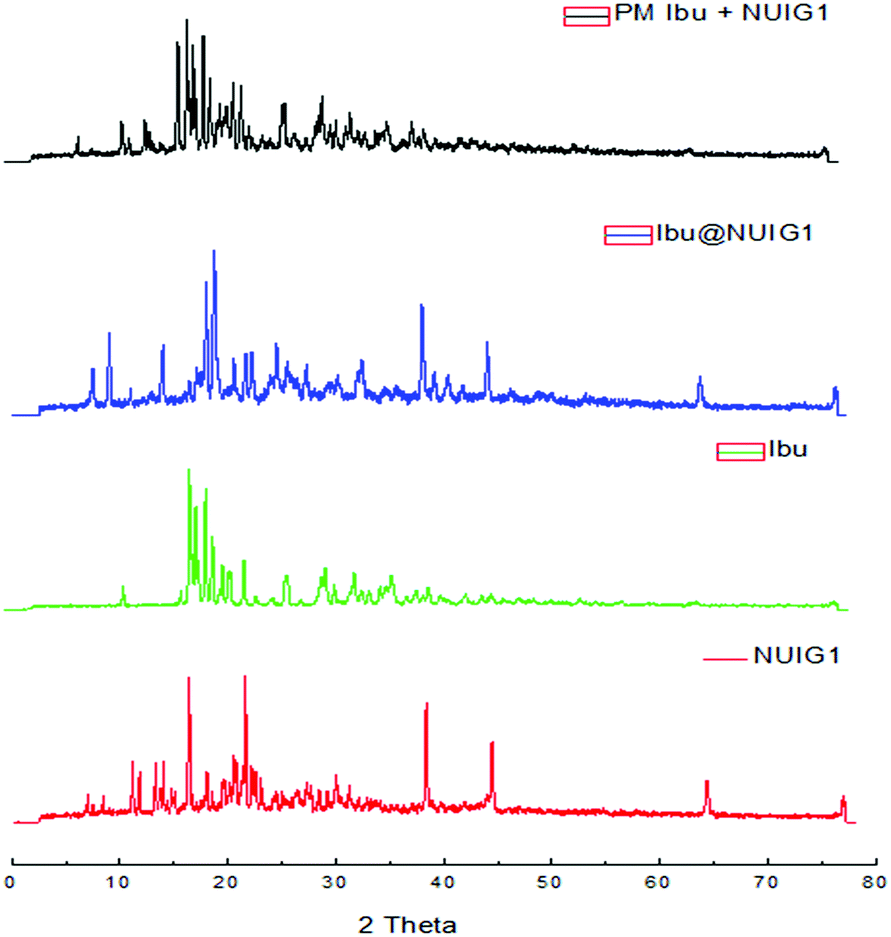 | ||
| Fig. 4 XRPD patterns of NUIG1, Ibu, Ibu@NUIG1, and a physical mixture of NUIG1 and Ibu. | ||
The encapsulation rate then slows down slightly before it reaches a plateau after 144 hours. The initial low drug uptake rate, followed by a sharp increase, is related to the breathing effect in NUIG1, indicating that the pore expansion is initiated after the encapsulation of a small amount of Ibu over the first 40 hours.
To confirm the unprecedently high drug adsorption capacity of NUIG1 and gain insight into the interactions between NUIG1 framework and Ibu, thermal stability (Fig. S4, ESI†) and solid state NMR studies were pursued. Solid-state NMR has proven to be able to provide significant information on the local environments of host systems such as MOFs, zeolites and covalent organic frameworks, and subsequently probe the host–guest interaction of those systems, ranging from adsorbed CO2 to encapsulated drug molecules.41,42
The 1H → 13C CP spectra are shown in Fig. 5. There is a distinct difference in the chemical shifts due to the carboxyl functional groups of the samples, with deprotonated COO− (173 ppm) being observed in the unloaded MOF, whilst the protonated –COOH is seen in crystalline ibuprofen (183 ppm).43
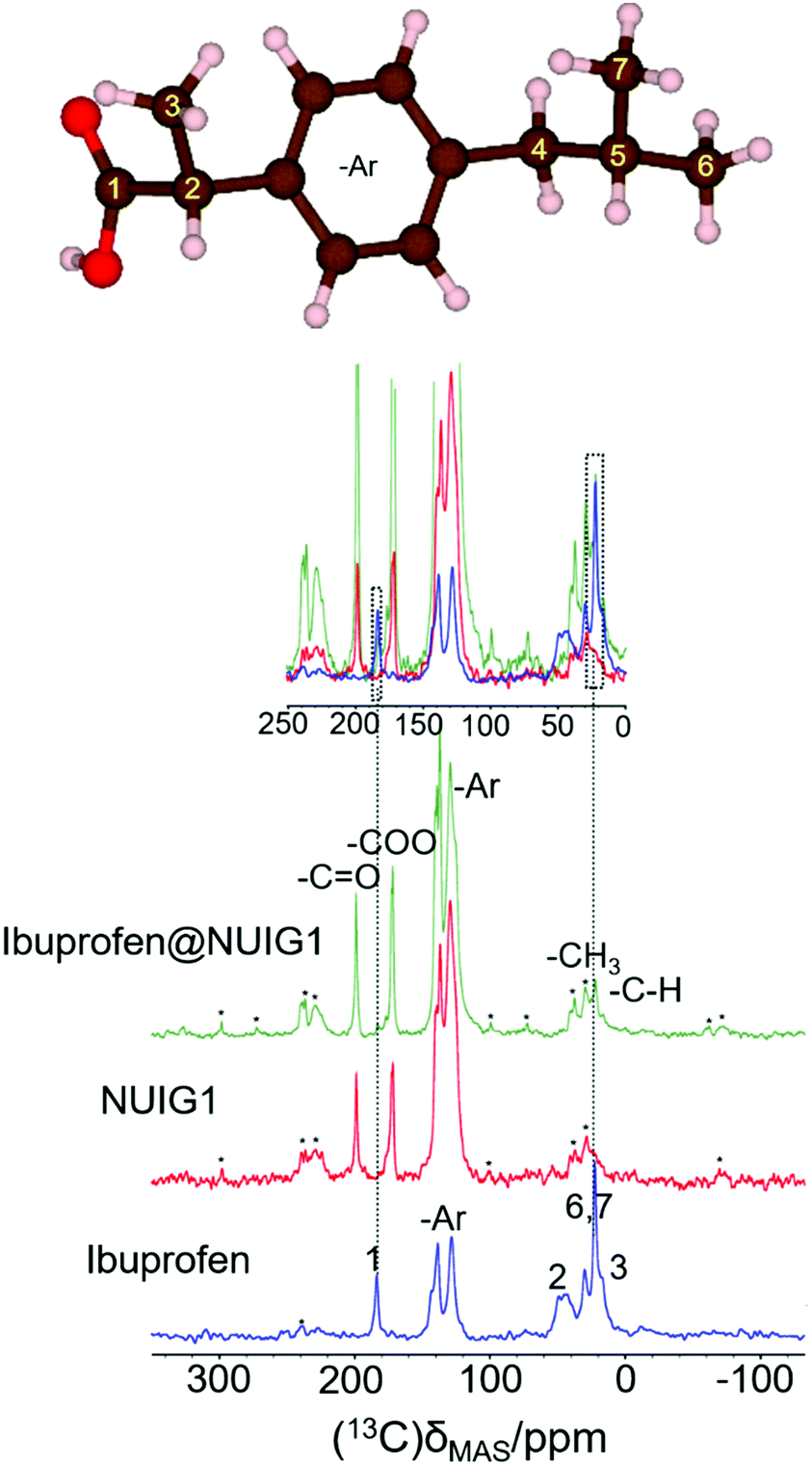 | ||
| Fig. 5 (top): The ibuprofen molecule, reproduced with VESTA.42d (bottom) The 1-D 1H → 13C cross polarization spectra for Ibu (blue), pristine NUIG1 (red), and Ibu@NUIG1 (green). Inset: The expanded spectral regions, highlighting the aliphatic and carboxylic species that are present in both the Ibuprofen and the Ibu loaded MOF. Spinning sidebands are labelled with asterisks. | ||
Both resonances are seen in the spectrum of the loaded MOF, with a very low intensity COOH resonance being slightly shifted to 181.5 ppm. Additionally, peaks at 21.8 ppm and 16.3 ppm, corresponding to the aliphatic region of Ibuprofen, are also visible in the spectrum of the loaded NU1G (but not for the unloaded MOF), whose correlation with the respective proton shift at 0.9 ppm is further manifested in the HETCOR experiment (Fig. S5, ESI†). The presence of both sets of resonances in the spectrum of ibu@NUIG1 indicate that ibuprofen is indeed taken up into the MOF. The comparatively small change in chemical shift of the carboxyl resonance of ibuprofen on adsorption suggests that there is no deprotonation of the ibuprofen, and no covalent bonding to the MOF network, suggesting that the adsorption mechanism is predominantly physisorption.42b
To obtain a better understanding of the mechanism, 23Na MAS NMR experiments were carried out. 23Na is a quadrupolar nucleus (spin 3/2), whose quadrupolar charge moment interacts with the electric field gradient of the local electronic environment. The one- dimensional 23Na MAS NMR spectrum of the unloaded MOF (Fig. 6) exhibits a resonance broadened by the quadrupolar interaction centred on −20 ppm, with two sharp peaks likely due to sodium salt impurities. The TOP-DQF-STMAS spectra (Fig. 6) clearly show that this broad feature is actually two distinct resonances, due to the two different sodium sites present in the structures of NUIG1. These sodium sites lack the distinctive features of the second order quadrupolar lineshapes, which indicates a structural disorder that induces a distribution of quadrupolar coupling constants. The TOP-DQF-STMAS spectrum of Ibu@NUIG1 (Fig. S6, ESI†) is largely unchanged, and the two sets of resonances yield chemical shifts and quadrupolar products that are the same within experimental error (Table S2, ESI†), which suggests that the sodium sites are not extensively affected by the presence of Ibuprofen. This result corroborates with the full hexacoordination of both Na sites and confirms that physisorption is the dominant mechanism of adsorption of Ibu by NUIG1.
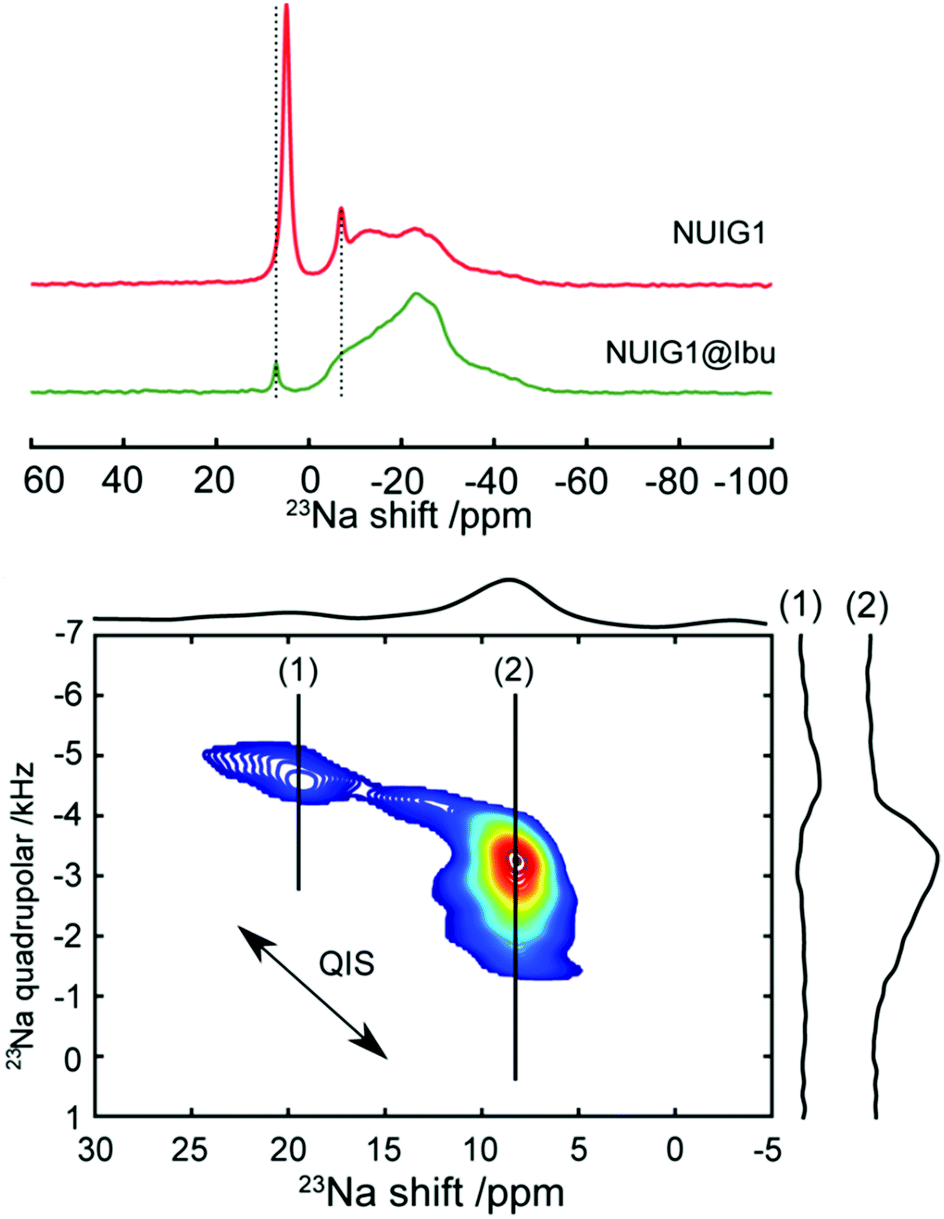 | ||
| Fig. 6 Top: 23Na solid-state NMR spectrum of unloaded (red) and loaded (green) MOF. Bottom: DQF-TOP-STMAS 23Na spectrum of the loaded MOF. Two separate resonances (1) and (2) can be identified. Horizontal extracted slices of these resonances are also represented. The arrow indicates the direction of the second order quadrupolar induced shift (QIS), which accounts for the small differences in resonance position between the one-dimensional spectra (top) and the two-dimensional spectrum (bottom). | ||
The drug release performance NUIG1 was also studied to H2O and PBS at 37 °C in order to simulate physiological conditions. 50 mg of the loaded Ibu@NUIG1 was immersed in the corresponding solution and left under stirring; at certain time intervals a sample from the suspension was removed and analysed using UV-vis. Fig. S7 (ESI†) shows the Ibu release kinetics in both H2O and PBS. In the first case, the maximum amount of the released Ibu is 73% with the release being completed within 50 hours; 40% of the drug is released over the first 35 hours, after which an increase in the release rate being observed. The remaining 33% of the drug is released during the next 15 hours. On the other hand, a faster release is observed in PBS conditions with the 30% drug loss being evident in the first 5 hours. A plateau is reached after 24 hours, where the 55% of the drug has been released.
Cytotoxicity studies
The MCF-7 (breast cancer) cell line was used to assess the toxicity of NUIG1 and Ibu@NUIG1. The cytotoxicity was determined using the MTT assay revealing that both NUIG1 and Ibu@NUIG1 are non-toxic (cell viability > 70%, Fig. S8, ESI†). This is not surprising as previously reported ZnII MOFs have been proven to be non-toxic, whereas ibuprofen does not show anticancer activity against breast cancer cells,44a but it has been found to show a chemopreventive effect against the development of breast cancer, only under regular use.44bMetal uptake studies
The outstanding drug delivery performance of NUIG1 prompted us to study its metal adsorption potential. The metal encapsulation studies were carried out by soaking activated crystals of NUIG1 into aqueous solutions of Co2+, Cu2+ and Ni2, respectively. Immediately, the MOF crystals underwent a color change from white (NUIG1) to purple (Co@NUIG1), blue (Cu@NUIG1), or green (Ni@NUIG1), Fig. S9 (ESI†); the colour change is directly related to the adsorbed metal; this property can be used for the naked-eye detection of a range of metal ions. The metal encapsulation was initially investigated by batch studies using UV-vis spectroscopy, (Fig. S10, ESI†). The maximum loading capacity obtained for Co2+, Cu2+ and Ni2+ were 6% wt (equivalent to 67 mg Co2+ per g NUIG1), 8.3% wt (equivalent to 81 mg Cu2+ per g NUIG1), and 3.8% wt (equivalent to 37 mg Ni2+ per g NUIG1), respectively.The metal adsorption by NUIG1 exhibits fast kinetics; there is a smooth increase in the adsorption capacity over time for the three metals, which after 20 min reaches a plateau (Fig. 7, top). There is no metal adsorption after 20 min. In order to get a better insight into the metal adsorption mechanism, the experimental kinetic data were fitted to a theoretical model;45 pseudo-first order and pseudo-second order kinetic models were used according to eqn (2) and (3), respectively.
| ln(qe − qt) = lnqe − k1t | (2) |
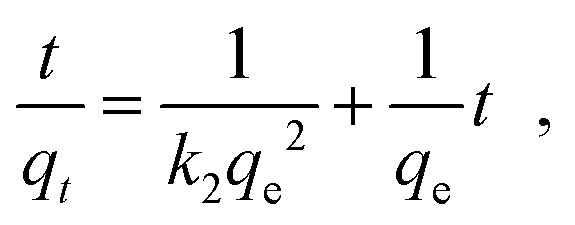 | (3) |
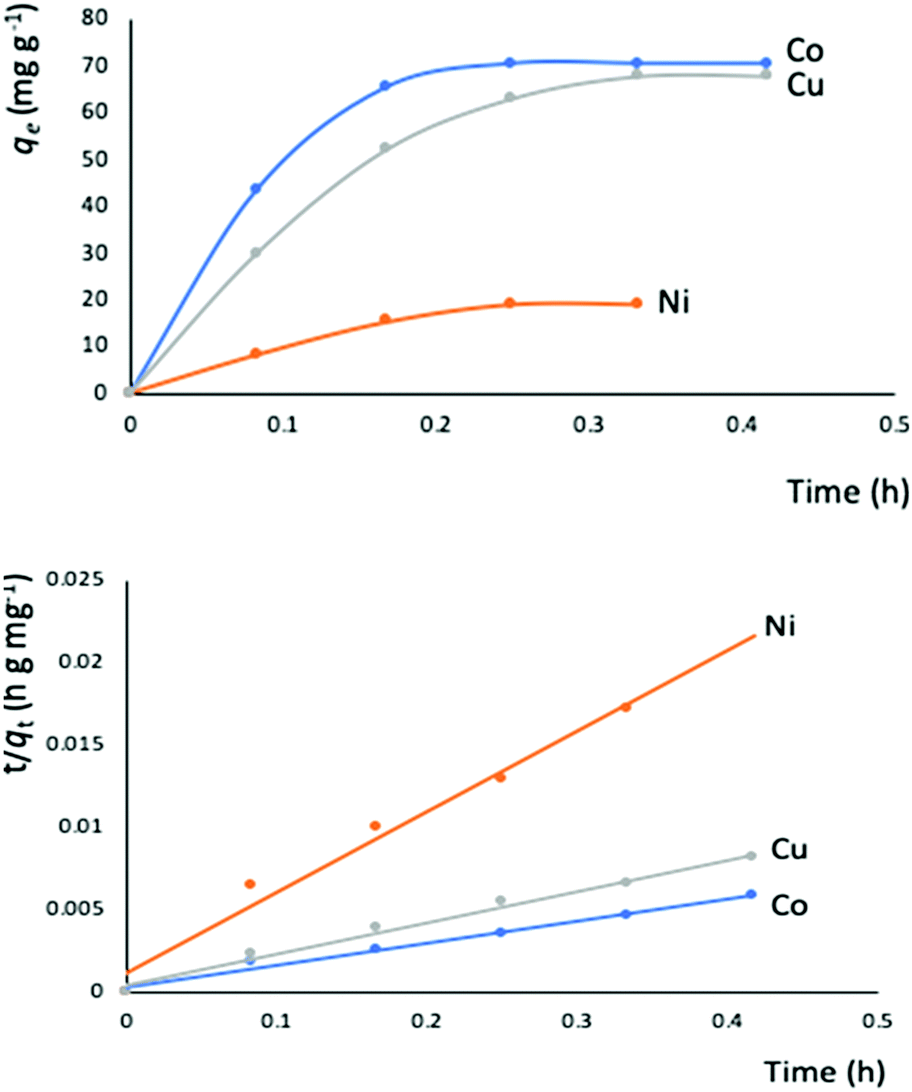 | ||
| Fig. 7 Top: Metal adsorption capacity (mg g−1) versus time (h) plot. The solid lines is a guide to the eye; bottom: simulation of the experimental data to the pseudo-second order kinetic model. The solid lines represent the fitting of the data. | ||
The metal adsorption equilibrium data are plotted in Fig. 8. The best description of the data is provided by the Langmuir model (Fig. S11, ESI†),46 considering a monolayer adsorption with a finite number of homogeneous and equivalent active sites (eqn (4)):
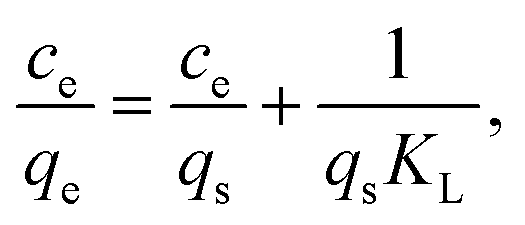 | (4) |
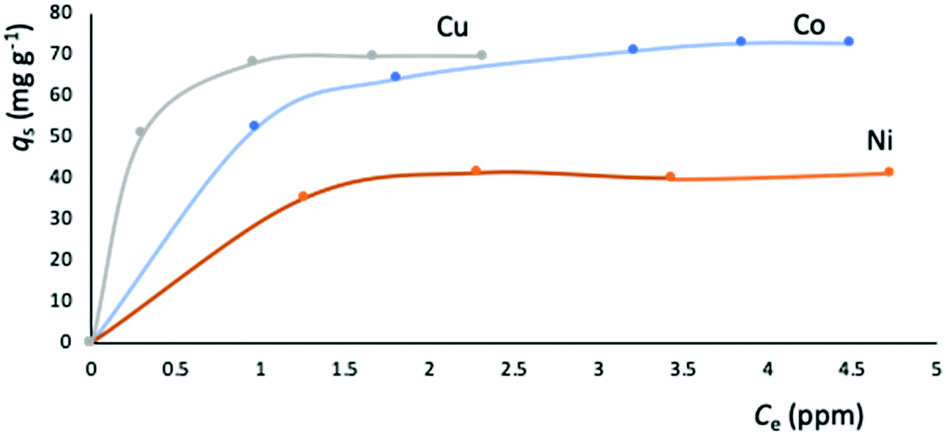 | ||
| Fig. 8 Equilibrium data for the metal adsorption by NUIG1 (contact time: 30 min). The solid lines are a guide to the eye. | ||
| Metal ion | q s (mg g−1) | K L (104 L mol−1) | R 2 |
|---|---|---|---|
| Co2+ | 76.92 | 1.9696 | 0.9979 |
| Cu2+ | 70.42 | 20.2860 | 0.9956 |
| Ni2+ | 42.55 | 5.0000 | 0.9985 |
The metal adsorption capacity of NUIG1 was further investigated by EDX, ICP, TGA and SEM studies. Fig. 9 shows the EDX spectra of NUIG1, Co@NUIG1, Cu@NUIG1, and Ni@NUIG1 samples. Zn, Na, C and O are detected in the four samples, with the spectra of the M@NUIG1 (M = Co, Cu, Ni) displaying one additional peak corresponding to the metal ion that has been adsorbed, hence confirming the metal uptake. This was further confirmed by ICP studies, from which it was found that the maximum % wt M content in the M@NUIG1 samples is 5.0%Co (in Co@NUIG1), 6.8%Cu (in Cu@NUIG1), and 3.8%Ni (in Ni@NUIG1); this is in good agreement with the results from the UV-vis studies.
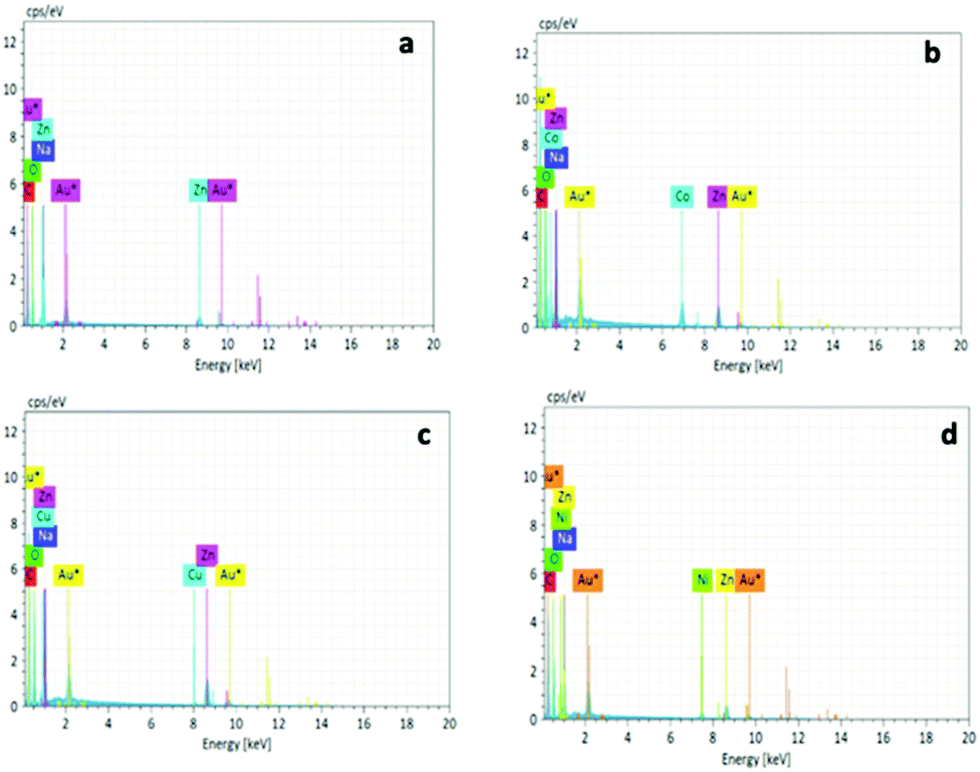 | ||
| Fig. 9 EDX spectra of NUIG1 (a), Co@NUIG1 (b), Cu@NUIG1 (c), and Ni@NUIG1 (d). | ||
The M@NUIG1 samples are decomposed in two steps (Fig. S12, ESI†), with the step at 450 °C corresponding to the framework breakdown. The additional drop at 250 °C is evidence of the absorbed metal, with the mass loss at this temperature being less than 10%, in accordance with the % wt M content in the M@NUIG1 species that has been determined by the UV-vis and ICP studies.
The SEM images of the four MOF species are depicted in Fig. 10. NUIG1 comprises a significant number of parallel nanosheets. It is noteworthy that, upon metal encapsulation, sponge-like nanospheres are formed, with the ratio of the coexistent nanosheets: nanospheres depending on the kind of the metal ion that has been adsorbed. In particular, the number of nanospheres is high in Co@NUIG1, and considerably lower in the case of Cu@NUIG1 and Ni@NUIG1, whose morphology is very similar to that of NUIG1.
 | ||
| Fig. 10 The SEM images of NUIG1, Co@NUIG1, Cu@NUIG1, and Ni@NUIG1. | ||
The regeneration of NUIG1 by M@NUIG1 is not feasible possibly due to the strong interactions between the metal ions and the framework, as suggested by the kinetic studies. Several attempts were performed, that included washing of M@NUIG1 with a concentrated aqueous solution of EDTA or 1 M HCl. While the guest metal was removed in all cases, XRPD showed degradation of the framework.
Magnetism studies
The magnetism properties of polycrystalline M@NUIG1 samples were measured as a means to confirm the metal uptake capacity of NUIG1, and also preform an initial investigation of the potential of this technique to be used for the development of novel MOF-based sensors. Note that NUIG1 is diamagnetic, hence any paramagnetic component would be due to the presence of paramagnetic metal ions into its pores. Direct current magnetic susceptibility measurements were carried out for Co@NUIG1, Ni@NUIG1 and Cu@NUIG1 in the 2–300 K temperature range (Fig. 11).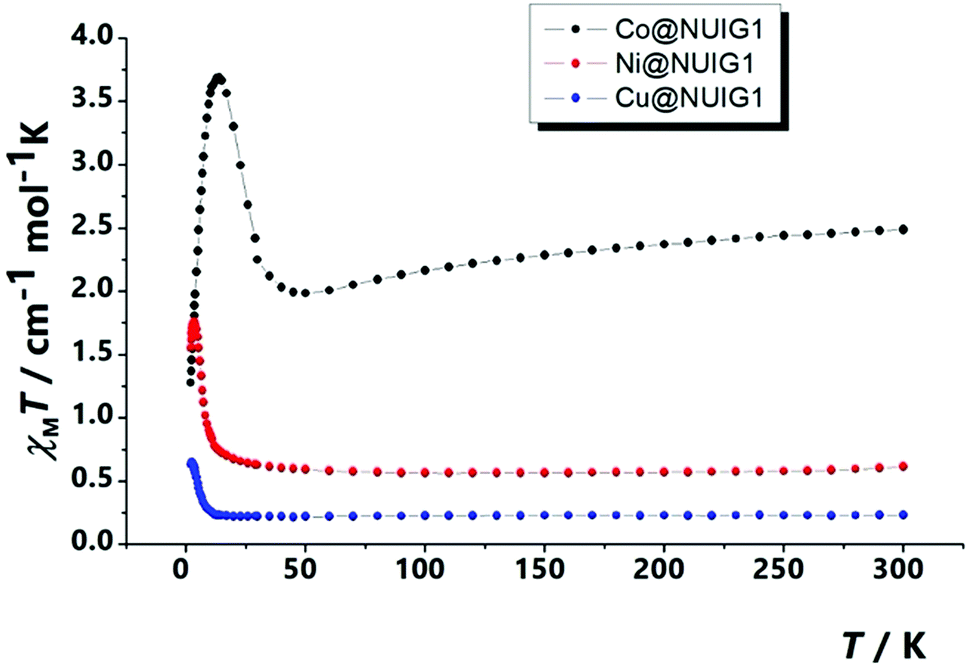 | ||
| Fig. 11 χ M T vs. T plot for the three M@NUIG1 species. | ||
A closer inspection in Fig. 11 reveals that the presence of the metal ions into the NUIG1 pores affects the magnetic properties of the MOF with the observed χMT value depending on the kind and the amount of the encapsulated metal ion; this is a very desirable property for the magnetic sensing of environmentally hazardous species. The values of the χMT products at room temperature indicate that the %M w/w uptake is in good agreement with the values derived from the previous characterization techniques: 0.6 cm−1 mol−1 K for Ni with a g = 2.30; 0.25 cm−1 mol−1 K for Cu, with a g = 2; 2.3 for CoII which is slightly higher than the expected value taking into account the anisotropy of the CoII ion (g = 2.45). The χMT value remains essentially stable until low temperature indicating weak interactions between the metal ions. There is an increase at the χMT at lower temperatures which can be attributed to a ferromagnetic coupling between the cations. The decay of the curve at very low temperatures for the Cu and Ni cations can be attributed to intramolecular interactions, whereas in the case of the Co analogue this is due to the presence of axial zero field splitting. The ferromagnetic response of the three complexes evidences that the cations are not isolated inside the pores, forming polynuclear aggregates that according the low temperature data should be of medium/low nuclearity.
Looking at the static magnetic properties of Co@NUIG1 and Ni@NUIG1, we decided to investigate the dynamic magnetic properties and the slow relaxation of the magnetization potential. Both aggregates present clear out-of-phase peaks (Fig. S13, ESI†), which are observed at relatively high temperature for the Ni case. In both cases, there are frequency dependent χM′′ signals in absence of an external dc external field, which indicates an SMM behaviour for the complexes.
Computational studies
The BET surface area for NUIG1 was calculated by simulating the N2 adsorption; the N2 uptake isotherm was calculated at 77 K (Fig. 12, top) and the data were analysed as previously reported in the literature.47 The pressure limit, where the Nitrogen monolayer is formed, was determined by the V·(P0 − P) (V = Volumetric uptake) vs. P graph. After defining the pressure limit, the BET surface area for NUIG1 was calculated from the slope of the diagram in Fig. 12, bottom, and was found to be 1040 m2 g−1. The void volume for NUIG1 was also determined by using the helium void volume method48 and was found to be 44%, resulting to a gravimetric pore volume of 0.45 cm3 g−1. This is in perfect agreement with the solvent accessible area as calculated by PLATON.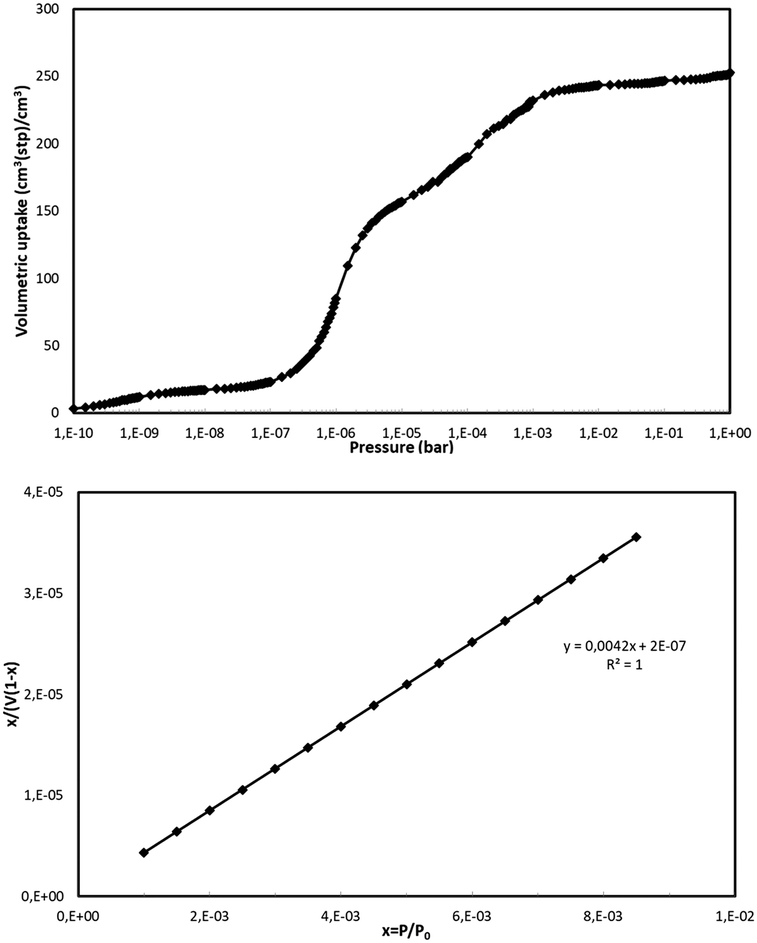 | ||
| Fig. 12 N2 volumetric uptake isotherm at 77 K for the calculation of the BET surface area of NUIG1 (top), and plot of the selected linear region that that satisfies the consistency criterion (bottom).48 | ||
NUIG1 was also tested for its ability to adsorb nitric oxide. Nitric oxide is known for its therapeutic properties, including among others its relaxing effect on smooth muscle dilating blood vessels, especially in the lungs, while recent reports show that NO might play an important role in the treatment of COVID-19;49 as such, the development of materials that can adsorb and deliver NO to the cells in a controlled way is very important. The isotherm (Fig. 13, top) was from GCMC simulations at 121 K and for pressures up to P0. To the best of our best knowledge, there are no similar studies at 121 K In literature. In contrast there are some studies at room temperature. For this reason and for the sake of comparison of our results with results from similar studies we also performed similar simulations at 298 K. The gravimetric uptake is presented in Fig. 13, bottom; the gravimetric uptake is 1.3 mmol g−1 and 6 mmol g−1 at 1 and 10 bar, respectively. These values are significantly higher than the corresponding values of ca. 0.5 mmol g−1 and 3.6 mmol g−1 for the top performing materials,50a which were studied at the same temperature. This value for the top performing material50b at 1 bar is ca. 1 mol kg−1 but this study was at 313 K. Fig. S14 (ESI†) shows the isosteric heat of adsorption for NO as taken from the simulations at 298 K. At low loading this value is ca. 20 kJ mol−1. This value is also very high showing the strong interaction between fluid molecules and MOF framework and is consistent with the high adsorption capacity of the material. This is also consistent with the fact that this value is considerably higher than the largest values taken from the top performing materials of the two studies referred earlier which are 10 and 20 kJ mol−1, respectively.
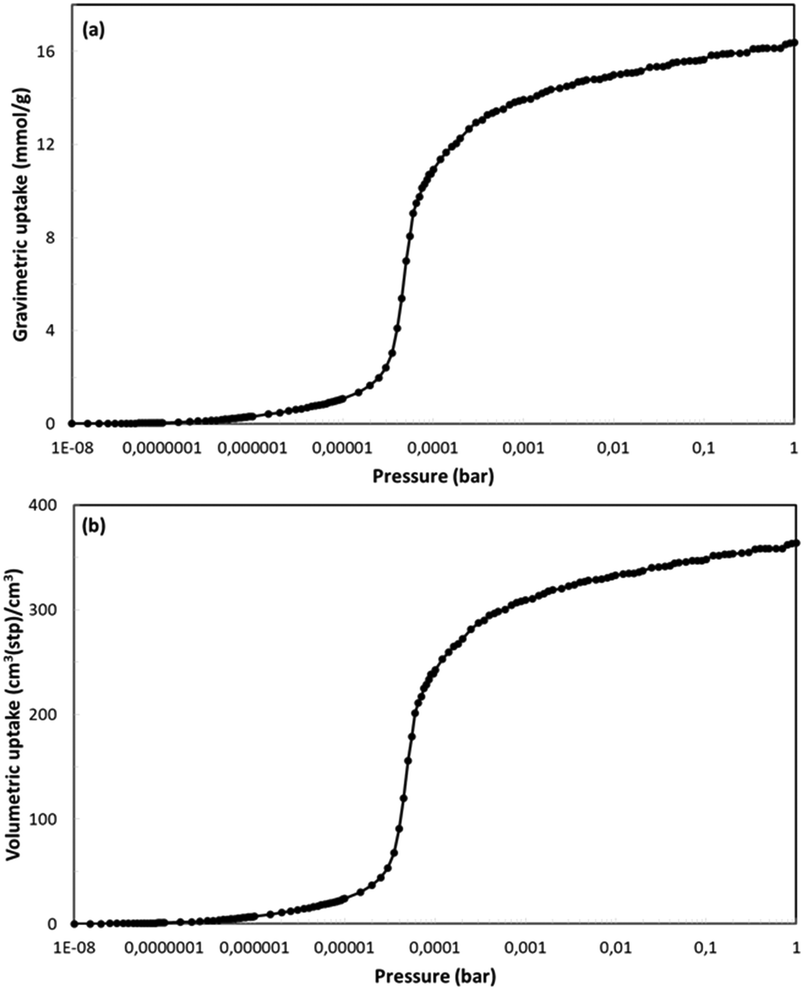 | ||
| Fig. 13 Gravimetric (top) and volumetric (bottom) uptake isotherms for NO at 121 K. | ||
Similar Monte Carlo simulations were conducted for ibuprofen to find the maximum uptake by the MOF. The simulations were done at various fugacity values, instead of pressure, since it is not possible to apply the equation of state for ibuprofen. The calculations were performed considering that there is an almost proportional dependence of the adsorbed amount of drug on the gravimetric pore volume of the material.51 Literature data were used in order to produce this linear dependence of the uptake on the pore volume (Fig. S15, ESI†).52 According to this, the maximum calculated gravimetric loading is 230 mg drug per g NUIG1, which is significantly lower than the maximum drug uptake that has been experimentally determined. There are several possible explanations for this including the fact that the breathing property is not considered in the theoretical model, as well as to possible structural distortions that the MOF undergoes during activation, e.g. it is likely that not only the solvents that occupy the pores of the MOF are removed, but also the terminally ligated solvents, which will result in a considerable expansion of the MOF's pores, and hence its drug uptake capacity increases. Another possible reason is that a percentage of the amount of the drug is likely adsorbed by the external surface of the MOF and does not enter in to its pores.
Conclusions
The discovery of new materials with improved performance in environmental and biomedical applications, including drug delivery, and adsorption and removal of hazardous species from aqueous systems is very important. Mixed metal MOFs, although they display desirable features for these applications, have not been systematically investigated so far. NUIG1 is a new addition to the relatively small family of mixed-metal MOFs, being a rare example of such a species whose structure is based on rod SBUs. It is non-toxic and displays a new network topology. NUIG1 exhibits an outstanding performance in Ibu and NO adsorption capacity. The mechanism of adsorption was studied in detail by solid-state NMR, which revealed that the dominant mechanism is physisorption. Furthermore, NUIG1 possesses a considerable metal uptake capacity for Co, Ni and Cu. Dc magnetic susceptibility studies revealed that the magnetic properties of the M@NUIG1 species change significantly according to the kind and the amount of the encapsulated metal ion.This work establishes the mixed-metal MOFs as excellent candidates in important biomedical and environmental applications. It also reveals that magnetism can be a powerful technique for the detection of environmentally hazardous chemicals, and this can be useful especially for species that do not affect the photoluminescence properties or the colour of a compound, hence they cannot be detected by the commonly used sensors. Further work is currently in progress towards this direction and will be reported in due course. The isolation of analogues of NUIG1 with paramagnetic metal ions instead of Zn is also of special interest in order to investigate the impact on the adsorption capacity and the magnetism-based sensing properties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been funded by the Science Foundation Ireland. W. P., J. P. C., and A. J. P. were supported by the Swedish Research Council (project no. 2016-03441).References
- (a) H.-C. J. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415 RSC; (b) M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O’Keeffe and O. Yaghi, Acc. Chem. Res., 2001, 34, 319 CrossRef PubMed; (c) S. Yuan, L. Feng, K. Wang, J. Pang, M. Bosch, C. Lollar, Y. Sun, J. Qin, X. Yang, P. Zhang, Q. Wang, L. Zou, Y. Zhang, L. Zhang, Y. Fang, J. Li and H.-C. Zhou, Adv. Mater., 2018, 37, 1704303 CrossRef PubMed.
- (a) H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed; (b) A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and A. R. Fischer, Chem. Soc. Rev., 2014, 43, 6062 RSC.
- N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. Yaghi, Science, 2003, 300, 1127 CrossRef PubMed.
- (a) M. Kurmoo, Chem. Soc. Rev., 2009, 38, 1353 RSC; (b) J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC.
- (a) P. Kumar, A. Deep and K.-H. Kim, TrAC, Trends Anal. Chem., 2015, 73, 39 CrossRef; (b) P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J.-S. Chang, Y. K. Hwang, V. Marsaud, P.-N. Bories, L. Cynober, S. Gil, G. Ferey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172 CrossRef CAS PubMed.
- P. Horcajada, C. Serre, M. Vallet-Regí, M. Sebban, F. Taulelle and G. Férey, Angew. Chem., Int. Ed., 2006, 45, 5974 CrossRef CAS.
- (a) P. Horcajada, C. Serre, G. Maurin, N. A. Ramsahye, F. Balas, M. Vallet-Regí, M. Sebban, F. Taulelle and G. Férey, J. Am. Chem. Soc., 2008, 130, 6774 CrossRef CAS; (b) A. C. McKinlay, R. E. Morris, P. Horcajada, G. Férey, R. Gref, P. Couvreur and C. Serre, Angew. Chem., Int. Ed., 2010, 49, 6260 CrossRef CAS.
- (a) Y. Han, W. Liu, J. Huang, S. Qiu, H. Zhong, D. Liu and J. Liu, Pharmaceutics, 2018, 10, 271 CrossRef CAS; (b) C. He, K. Lu, D. Liu and W. Lin, J. Am. Chem. Soc., 2014, 136, 5181 CrossRef CAS PubMed.
- (a) C.-Y. Sun, C. Qin, X.-L. Wang, G.-S. Yang, K.-Z. Shao, Y.-Q. Lan, Z.-M. Su, P. Huang, C.-G. Wang and E.-B. Wang, Dalton Trans., 2012, 41, 6906 RSC; (b) I. A. Lazarov and R. S. Forgan, Coord. Chem. Rev., 2019, 380, 230 CrossRef.
- (a) L. Sercombe, T. Veerati, F. Moheimani, S. Y. Wu, A. K. Sood and S. Hua, Front. Pharmacol., 2015, 6, 286 Search PubMed; (b) W. Xu, P. Ling and A. Zhang, J. Drug Delivery, 2013, 340315 Search PubMed.
- (a) E. Abbasi, S. F. Aval, A. Akbarzadeh, M. Milani, H. T. Nasrabadi, S. W. Joo, Y. Hanifehpour, K. Nejati-Koshki and R. Pashaei-Asl, Nanoscale Res. Lett., 2014, 9, 247 CrossRef PubMed; (b) W. H. De Jong and P. J. A. Borm, Int. J. Nanomed., 2008, 3, 133 CrossRef CAS PubMed; (c) A. Bianco, K. Kostarelos and M. Prato, Curr. Opin. Chem. Biol., 2005, 9, 674 CrossRef CAS PubMed.
- (a) P. Zrazhevskiy, M. Sena and X. Gao, Chem. Soc. Rev., 2010, 39, 4326 RSC; (b) A. Z. Wilczewska, K. Niemirowicz, K. H. Markiewicz and H. Car, Pharmacol. Rep., 2012, 64, 1020 CrossRef CAS PubMed.
- (a) M. Hoop, C. F. Walde, R. Riccò, F. Mushtaq, A. Terzopoulou, X.-Z. Chen, A. J. deMello, C. J. Doonan, P. Falcaro, B. J. Nelson, J. Puigmartí-Luis and S. Pané, Appl. Mater. Today, 2018, 11, 13 CrossRef; (b) K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O’Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186 CrossRef CAS.
- (a) M. Witman, S. Ling, A. Gladysiak, K. C. Stylianou, B. Smit, B. Slater and M. J. Haranczyk, J. Phys. Chem. C, 2017, 121, 1171 CrossRef CAS PubMed; (b) X. Huang and C. S. O. Brazel, J. Controlled Release, 2001, 73, 121 CrossRef CAS.
- (a) K. Roth Stefaniak, C. C. Epley, J. J. Novak, M. L. McAndrew, H. D. Cornell, J. Zhu, D. K. McDaniel, J. L. Davis, I. C. Allen, A. J. Morris and T. Z. Grove, Chem. Commun., 2018, 54, 7617 RSC; (b) H. Zheng, Y. Zhang, L. Liu, W. Wan, P. Guo, A. M. Nyström and X. Zou, J. Am. Chem. Soc., 2016, 138, 962 CrossRef CAS.
- Z. Zou, S. Li, D. He, X. He, K. Wang, L. Li, X. Yang and H. Li, J. Mater. Chem. B, 2017, 5, 2126 RSC.
- F. Ke, Y.-P. Yuan, L.-G. Qiu, Y.-H. Shen, A.-J. Xie, J.-F. Zhu, X.-Y. Tian and L.-D. Zhang, J. Mater. Chem., 2011, 21, 3843 RSC.
- (a) R. Ricco, M. J. Styles and P. Falcaro, Metal-Organic Frameworks (MOFs) for Environmental Applications, ed. S. K Ghosh, Elsevier, 2019, pp. 383–426 Search PubMed; (b) H. Molavi, A. Hakimian, A. Shojaei and M. Raeiszadeh, Appl. Surf. Sci., 2018, 445, 424 CrossRef CAS.
- (a) P. A. Kobielska, A. J. Howarth, O. K. Farha and S. Nayak, Coord. Chem. Rev., 2018, 358, 92 CrossRef CAS; (b) K. Vellingiri, J. E. Szulejko, P. Kumar, E. E. Kwon, K.-H. Kim, A. Deep, D. W. Boukhvalov and R. J. C. Brown, Sci. Rep., 2016, 6, 27813 CrossRef CAS.
- (a) I. Ahmed and S. H. Jhung, J. Hazard. Mater., 2016, 301, 259 CrossRef CAS; (b) P. W. Seo, B. N. Bhadra, I. Ahmed, N. A. Khan and S. H. Jhung, Sci. Rep., 2016, 6, 34462 CrossRef CAS; (c) M. S. Khan, M. Khalid and M. Shahid, Mater. Adv., 2020 10.1039/d0ma00291g.
- (a) J. Yang, B. Hou, J. Wang, B. Tian, J. Bi, N. Wang, X. Li and X. Huang, Nanomaterials, 2019, 9, 424 CrossRef CAS PubMed; (b) E. Zanin, J. Scapinello, M. de Oliveira, C. L. Rambo, F. Franscescon, L. Freitas, J. M. M. de Mello, M. A. Fiori, J. V. Oliveira and J. Magro, Process Saf. Environ. Prot., 2017, 105, 194 CrossRef CAS.
- (a) E. M. Dias and C. Petit, J. Mater. Chem. A, 2015, 3, 22484 RSC; (b) L. Zhao, M. R. Azhar, X. Li, X. Duan, H. Sun, S. Wang and X. Fang, J. Colloid Interface Sci., 2019, 542, 421 CrossRef CAS; (c) J. Barreto, M. D. G. Xavier, R. P. P. L. Ribeiro, D. Martins, I. A. A. C. Esteves, M. Branco, T. Tirolien, J. P. B. Mota and G. Bonfait, J. Chem. Eng. Data, 2019, 64, 5407 CrossRef CAS; (d) S. Ramanayaka, M. Vithanage, A. Sarmah, T. An, K.-H. Kim and Y. Sik Ok, RSC Adv., 2019, 9, 34359 RSC.
- W. P. Lustig, S. Mukherjee, N. D. Rudd, A. V. Desai, J. Li and S. K. Ghosh, Chem. Soc. Rev., 2017, 46, 3242 RSC.
- C.-H. Chen, X.-S. Wang, L. Li, Y.-B. Huang and R. Cao, Dalton Trans., 2018, 47, 3452 RSC.
- S. Abednatanzi, P. Gohari Derakhshandeh, H. Depauw, F.-X. Coudert, H. Vrielinck, P. Van Der Voort and K. Leus, Chem. Soc. Rev., 2019, 48, 2535 RSC.
- (a) M. Y. Masoomi, A. Morsali, A. Dhakshinamoorthy and H. Garcia, Angew. Chem., Int. Ed., 2019, 58, 15188 CrossRef CAS PubMed; (b) Q.-G. Zhai, X. Bu, C. Mao, X. Zhao and P. Feng, J. Am. Chem. Soc., 2016, 138(8), 2524 CrossRef CAS PubMed.
- (a) H. J. Park and M. P. Suh, Chem. – Eur. J., 2008, 14, 8812 CrossRef CAS; (b) Y. Ge, N.-Y. Li, W. Ma, D. Liu and X.-Y. Tang, CrystEngComm, 2014, 16, 10949 RSC; (c) H. J. Park and M. P. Suh, Chem. Commun., 2010, 46, 610 RSC; (d) V. Bon, I. Senkonsva, D. Wallacher, D. M. Toebbens, I. Zizak, R. Feyerharm, U. Mueller and S. Kaskel, Inorg. Chem., 2014, 53, 1513 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef.
- P. McArdle, K. Gilligan, D. Cunningham, R. Dark and M. Mahon, CrystEngComm, 2004, 6, 303 RSC.
- P. Van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194 CrossRef.
- (a) Z. Gan,, J. Am. Chem. Soc., 2000, 122, 3242 CrossRef; (b) J. P. Carvalho, A. Jaworski, M. Brady and A. J. Pell, Magn. Reson. Chem., 2020 Search PubMed , in press; (c) B. M. Fung, A. K. Khitrin and K. Ermolaev, J. Magn. Reson., 2000, 142, 97 CrossRef CAS; (d) P. Caravatti, L. Braunschweiler and R. R. Ernst, Chem. Phys. Lett., 1983, 100, 305 CrossRef CAS.
- E. Tylianakis and G. E. Froudakis, J. Comput. Theor. Nanosci., 2009, 6, 335 CrossRef.
- (a) D.-Y. Peng and D. B. Robinson, Ind. Eng. Chem. Fundam., 1976, 15, 59 CrossRef CAS; (b) S. L. Mayo, B. D. Olafson and W. A. Goddard, J. Phys. Chem., 1990, 94, 8897 CrossRef CAS.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. M. Breneman and K. B. Wiberg, J. Comput. Chem., 1990, 11, 361 CrossRef CAS.
- (a) J. J. Potoff and J. I. Siepmann, AIChE J., 2001, 47, 1676 CrossRef CAS; (b) J. Yang, Y. Ren, A. Tian and H. Sun, J. Phys. Chem. B, 2000, 104, 4951 CrossRef CAS; (c) M. C. Bernini, D. Fairen-Jimenez, M. Pasinetti, A. J. Ramirez-Pastor and R. Q. Snurr, J. Mater. Chem. B, 2014, 2, 766 RSC.
- (a) V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576 CrossRef CAS; (b) M. O'Keeffe, M. A. Peskov, S. J. Ramsden and O. M. Yaghi, Acc. Chem. Res., 2008, 41, 1782 CrossRef; (c) E. V. Alexandrov, V. A. Blatov, A. V. Kochetkov and D. M. Proserpio, CrystEngComm, 2011, 13, 3947 RSC.
- A. Schoedel, M. Li, D. Li, M. O’Keefe and O. Yaghi, Chem. Rev., 2016, 116, 12466 CrossRef CAS.
- M. Alhamami, H. Doan and C.-H. Cheng, Materials, 2014, 7, 3198 CrossRef.
- F. Nouar, T. Devic, H. Chevreau, N. Guillou, E. Gibson, G. Clet, M. Daturi, A. Vimont, J. M. Grenèche, M. I. Breeze, R. I. Walton, P. L. Llewellyne and C. Serre, Chem. Commun., 2012, 48, 10237 RSC.
- (a) S. Devautour-Vinot, G. Maurin, C. Serre, P. Horcajada, D. Paula da Cunha, V. Guillerm, E. de Souza Costa, F. Taulelle and C. Martineau, Chem. Mater., 2012, 24, 2168 CrossRef CAS; (b) M. Castro, P. Losch, W. Park, M. Haouas, F. Taulelle, C. Loerbroks, G. Brabants, E. Breynaert, C. E. A. Kirschhock, R. Ryoo and W. Schmidt, Chem. Mater., 2018, 30, 2676 CrossRef CAS; (c) S.-Y. Ding, M. Dong, Y.-W. Wang, Y.-T. Chen, H.-Z. Wang, C.-Y. Su and W. Wang, J. Am. Chem. Soc., 2016, 138, 3031 CrossRef CAS PubMed.
- (a) P. Rzepka, Z. Bacsik, A. J. Pell, N. Hedin and A. A. Jaworski, J. Phys. Chem. C, 2019, 123, 21497 CrossRef CAS; (b) N. Liédana, A. Galve, C. Rubio, C. Téllez and J. Coronas, ACS Appl. Mater. Interfaces, 2012, 4, 5016 CrossRef PubMed; (c) N. Rasenack and B. W. Müller, Drug Dev. Ind. Pharm., 2002, 28, 1077 CrossRef CAS PubMed; (d) K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272 CrossRef CAS.
- T. Azaïs, C. Tourné-Péteilh, F. Aussenac, N. Baccile, C. Coelho, J.-M. Devoisselle and F. Babonneau, Chem. Mater., 2006, 18, 6382 CrossRef.
- (a) S. Rojas, T. Devic and P. Horcajada, J. Mater. Chem. B, 2017, 14, 2560 RSC; (b) R. E. Harris, R. T. Chlebowski, R. D. Jackson, D. J. Frid, J. L. Ascenseo, G. Anderson, A. Loar, R. J. Rodabough, E. White and A. McTiernan, Cancer Res., 2003, 63, 6096 CAS.
- (a) S. Lagergren, Zur Theorie der Sogenannten Absorption Gelöster Stoffe, PA Norstedt & Söner, Stockholm, Sweden, 1898, pp. 1–39 Search PubMed; (b) Y. S. Ho and G. Mckay, Process Biochem., 1999, 34, 451 CrossRef CAS; (c) K.-D. Zhang, F.-C. Tsai, N. Ma, Y. Xia, H.-L. Liu, X.-Q. Zhan, X.-Y. Yu, X.-Z. Zeng, T. Jiang, D. Shi and C.-J. Chang, Materials, 2017, 10, 205 CrossRef PubMed.
- (a) I. Langmuir, J. Am. Chem. Soc., 1915, 38, 102 Search PubMed; (b) N. Ayawei, A. N. Ebelegi and D. Wankasi, J. Chem., 2017, 3039817 Search PubMed.
- K. S. Walton and R. Q. Snurr, J. Am. Chem. Soc., 2007, 129, 8552 CrossRef CAS PubMed.
- M. A. Lyers and P. A. Monson, Langmuir, 2002, 18, 10261 CrossRef.
- (a) J. Kobayashi and I. Murata, Ann. Intensive Care, 2020, 10, 61 CrossRef PubMed; (b) J.-L. Burgaud, E. Ongini and P. Del Soldato, Ann. N. Y. Acad. Sci., 2002, 962, 360 CrossRef CAS.
- (a) L. Wang, L. Wang, J. Zhao and T. Yan, J. Appl. Phys., 2012, 111, 112628 CrossRef; (b) W. Sun, L.-C. Lin, X. Peng and B. Smit, AIChE J., 2014, 60, 2314 CrossRef CAS.
- I. Erucar and S. Keskin, Ind. Eng. Chem. Res., 2016, 55, 1929 CrossRef CAS.
- (a) M. C. Bernini, D. Fairen-Jimenez, M. Pasinetti, A. J. Ramirez-Pastor and R. Q. Snurr, J. Mater. Chem. B, 2014, 2, 766 RSC; (b) R. Babarao and J. J. Jiang, J. Phys. Chem. C, 2009, 113, 18287 CrossRef CAS; (c) B. Liu, Y. Lian, Z. Li and G. Chen, Acta Chim. Sin., 2014, 72, 942 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental data, graphs and tables with fitting parameters for the adsorption studies; thermal analysis graphs. CCDC 2008513. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ma00450b |
| This journal is © The Royal Society of Chemistry 2020 |