
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMagic-angle spinning NMR spectroscopy provides insight into the impact of small molecule uptake by G-quartet hydrogels†
G. N.
Manjunatha Reddy‡
 *a,
Gretchen M.
Peters
b,
Ben P.
Tatman
a,
Teena S.
Rajan
c,
Si Min
Kock
c,
Jing
Zhang
c,
Bruno G.
Frenguelli
d,
Jeffery T.
Davis
b,
Andrew
Marsh
*c and
Steven P.
Brown
*a
*a,
Gretchen M.
Peters
b,
Ben P.
Tatman
a,
Teena S.
Rajan
c,
Si Min
Kock
c,
Jing
Zhang
c,
Bruno G.
Frenguelli
d,
Jeffery T.
Davis
b,
Andrew
Marsh
*c and
Steven P.
Brown
*a
aDepartment of Physics, University of Warwick, Coventry CV4 7AL, UK. E-mail: gnm.reddy@univ-lille.fr; S.P.Brown@warwick.ac.uk
bDepartment of Chemistry and Biochemistry, University of Maryland, College Park, Maryland, MD 20742, USA
cDepartment of Chemistry and, University of Warwick, Coventry CV4 7AL, UK. E-mail: a.marsh@warwick.ac.uk
dSchool of Life Sciences, University of Warwick, Coventry CV4 7AL, UK
First published on 5th September 2020
Abstract
Small molecule guests influence the functional properties of supramolecular hydrogels. Molecular-level understanding of sol–gel compositions and structures is challenging due to the lack of long-range order and the inherently heterogeneous sol–gel interface. Here, we employ multinuclear (7Li, 11B, 23Na, 39K and 133Cs) gel-state magic-angle spinning (MAS) NMR spectroscopy to gain insight into the roles of alkali cations and borate anions in the gelation of guanosine (G)-quartets (G4). The MAS NMR spectra of alkali metal ions enabled the cations associated with G-quartets to be distinguished from the free cations. Combined 11B MAS NMR and modeling studies allowed cis and trans configurations of guanosine-borate (GB) diesters to be characterized, thus providing a complementary structural insight that indicates that the GB cis diester favours gelation. In addition, the multinuclear MAS NMR strategy is applied to understand the uptake process of biologically relevant small molecules into GB hydrogels. G4·K+ borate hydrogel can absorb up to 0.3 equivalent of cationic methylene blue (MB) without a significant disruption of the G4 fibrils that make up the gel, whereas the addition of over 0.3 equivalents of MB to the same gel leads to a gel-to-sol transition. By comparison, uptake of heterocyclic molecules such as adenine, cytosine and 1-methylthymine into G4·Na+ borate hydrogels leads to stable and clear gels while increasing the solubility of the nucleobases as compared to the solubility of the same compounds in water. G4·Na+ gel can uptake one equiv. of adenine with minimal disruption to the hydrogel framework, thus enhancing the adenine solubility up to an order of magnitude as compared to water. Multinuclear (1H, 11B and 23Na) MAS NMR analysis and vial inversion tests revealed that the nucleobases are embedded into pores of the solution phase rather than being closely interacting with the G4 fibrils that make up the gel phase. These results indicate that G4 hydrogels have potential applications as carrier systems for small molecule drugs.
Introduction
Self-assembly of small molecules into functional supramolecular assemblies is of great current interest to develop materials for biomedical, environmental remediation and optoelectronic applications.1–6 In particular, biologically relevant molecules such as peptides, nucleobases, starch and polysaccharides have garnered attention to develop functional hydrogels for biomedical and clinical applications.7–11 For example, hydrogel delivery systems can be useful for the controlled uptake and release of drug molecules, so as to achieve therapeutically beneficial outcomes. Upon the addition of drug molecules into the hydrogels, the physicochemical properties of these systems ensure the compatibility and controllability of the drug delivery. Hydrogels differing in their sol–gel composition, archetype, structure and function have been previously developed and used for drug delivery applications.12Guanosine (G) and its derivatives exhibit rich supramolecular chemistry.13–16 The hierarchy of distinct quartet- and ribbon-like structures formed by G-derivatives have been employed to develop stimuli-responsive gels, electrochemical sensors, antiviral gels, synthetic ion channels and liquid crystalline phases.9,14,17–20 Davis and colleagues developed stable and long-lived hydrogels by mixing guanosine and alkali metal borate salts in water (G4·M+ borate hydrogel, Scheme 1).21,22 The mechanism of gelation (Scheme 1) was corroborated by cryogenic transmission electron microscopy (cryo-TEM), rheology, powder X-ray diffraction (PXRD), small-angle neutron scattering (SANS), gel- and solid-state NMR spectroscopy measurements and analyses.21,22 These guanosine borate (GB) hydrogels exhibit unique structural properties that are favourable for absorbing guest species: elongated G4-quartet fibrils of GB gels can bind to G-quadruplex ligands such as thioflavin-T and methylene blue, and allow the cations to exchange between the gel network and solution phase.21,22 In addition, the GB gels can undergo exchange reactions with cis diols, facilitating covalent incorporation of compounds containing 1,2-cis-diols. These beneficial properties associated with GB gels enabled the development of materials for cell growth,23 flexible sensors,19 wound healing,10 and environmental remediation.24 The commonality associated with these functional soft materials is that their bulk properties depend strongly on the molecular interactions at the sol–gel interfaces. Therefore, molecular-level understanding of sol–gel compositions and structures is expected to help with better formulation of such materials, for example, hydrogels for targeted biomedical, environmental remediation and optoelectronics applications.
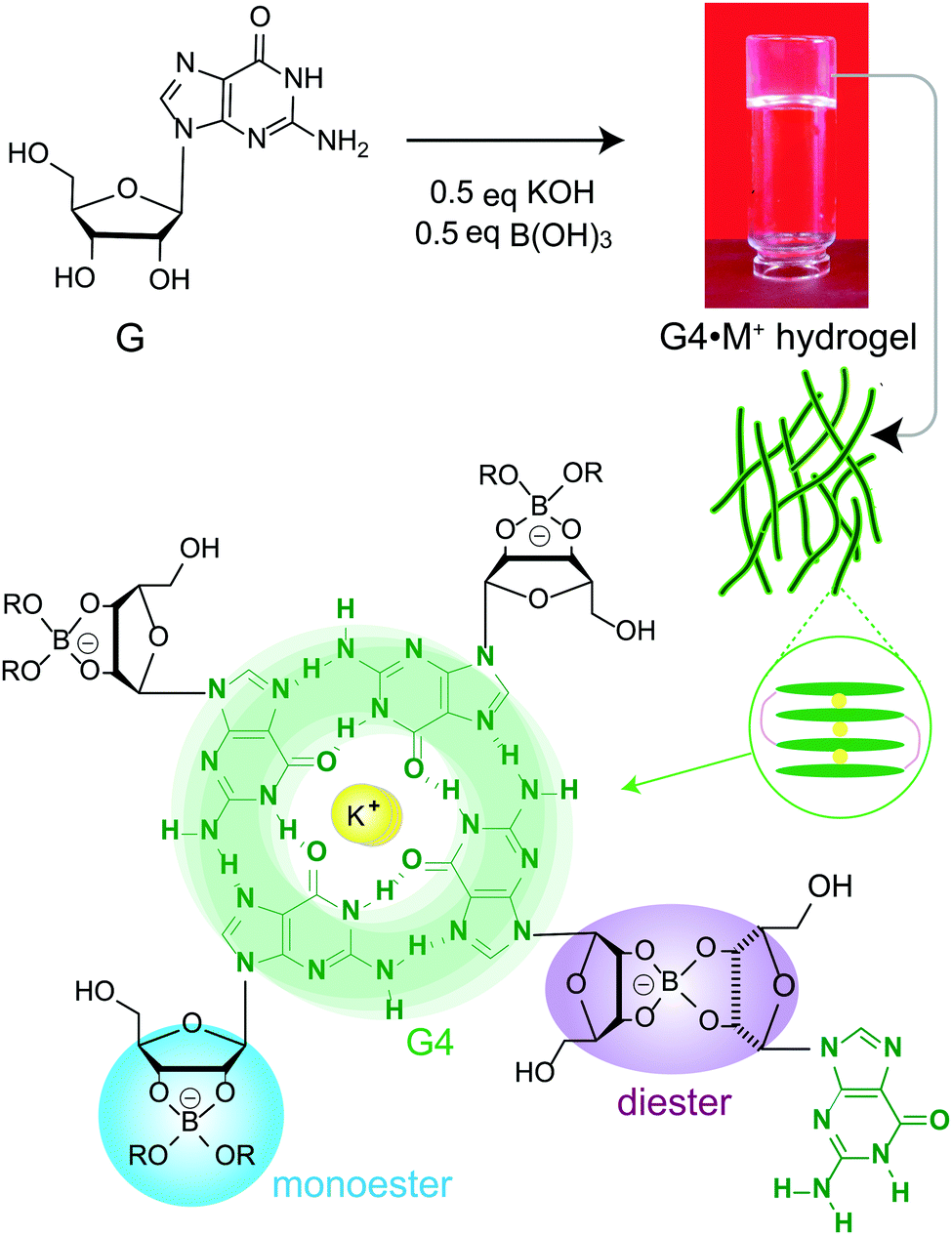 | ||
| Scheme 1 Schematic diagram of the gelation mechanism of G4·M+ (M+ = K+, Na+) hydrogels upon mixing guanosine (G) with metal borate salts. Guanosine borate cis diesters favour gelation by covalently cross-linking stacked G-quartets. | ||
The functional properties of gelator molecules depend strongly on the key structure directing interactions that govern the physicochemical properties of hydrogels as well as the intermolecular interactions between guest molecules and hydrogels. Establishing the structure–function relationships in responsive and dynamic hydrogels represents a challenge. This is due, at least in part, to the compositional and structural heterogeneity associated with the reactive sol–gel interfaces. Stability and integrity of gels is often rationalized by understanding their melting and rheological properties, and insights into the structures of gel networks, gelator molecules and their aggregates at different length scales can be obtained by different characterization techniques such as X-ray scattering, electron microscopy, single particle reconstruction and NMR spectroscopy.6,25–28 In this respect, MAS NMR spectroscopy provides complementary information into the short-range structures and interactions in both sol- and gel domains of soft materials.26,29–33 NMR chemical shifts and dipole–dipole couplings are sensitive to local chemical environments and interatomic distances, which provide information on the host–guest interactions, compositions and structures, and packing interactions in supramolecular assemblies.34–36 For example, recent MAS NMR studies enabled distinct quartet and ribbon-like structures to be elucidated,37–40 and the sol–gel compositions of G4·M+ borate hydrogels to be distinguished and identified.21,22
Here, molecular-level insight into the impact of small molecule uptake by different G4·M+ borate hydrogels (Scheme 1) is obtained and compared. The guest molecules (Scheme 2) are chosen on the basis of their structures and swelling properties into the GB hydrogels. The absorption properties of these small molecules are characterized ex situ by employing solution- and gel-state NMR spectroscopy, and corroborated by vial inversion tests. Notably, multinuclear (7Li, 23Na, 39K and 133Cs) MAS NMR spectroscopy enabled different distributions of cations in sol–gel networks, and their roles in the gelation properties of GB gels, to be distinguished and analyzed. In addition, 1H and 11B MAS NMR has been employed to probe the changes in the local structures of the borate anions that help to hold together the G4 fibrils in GB hydrogels. 11B MAS NMR results are complemented by density functional theory (DFT) calculations, which showed that the GB cis diesters favour the gelation as compared to the GB trans diester and GB monoester components. Emphasis is placed on understanding how changes occur in the sol–gel interfaces upon the addition of cationic dye (methylene blue, MB) and heterocyclic nucleobases such as adenine, cytosine and 1-methylthymine into G4·M+ (M+ = K+, Na+) gels.
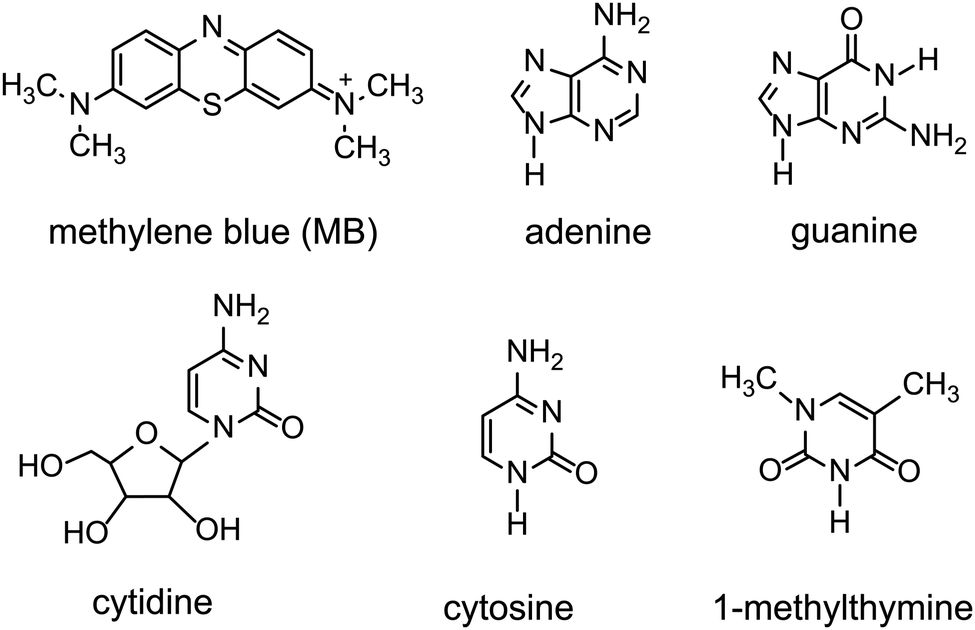 | ||
| Scheme 2 Chemical structures of cationic and neutral molecules added into G4·M+ (M+ = K+, Na) borate gels. Cytidine is used as an external standard to quantify incorporation of adenine into G4·K+ borate gels. | ||
Experimental section
Preparation and addition of small molecules to G4·M+ borate gels
G-Quartet borate hydrogels were prepared according to the procedure described in our previous work.21,22 Guanosine (G·2H2O), boric acid, LiOH, NaOH, KOH and CsOH were purchased from Sigma Aldrich Gillingham UK and used as received. Stock solutions of boric acid (200 mM) and alkali metal hydroxides (200 mM) were prepared in low conductivity Millipore water. To a solution of sonicated guanosine (72 mM) powder in water, half an equivalent of alkali metal borate salt (36 mM) was added. The glass vial containing the milky solution was placed on a hot plate and slowly heated up to 90 °C with continuous stirring until a clear solution was observed. The hot saturated solution was allowed to cool to room temperature. A ‘tap test’ was carried out whereby each gel was given a narrow tap and then inverted to observe whether it remained clear and self-standing. For the studies involving absorption or uptake of small molecules by the hydrogel, the hot saturated solution of G4·K+ hydrogel was pipetted into vials containing different concentrations of methylene blue, and the resulting solutions were manually stirred on a hot plate and then allowed to cool to room temperature. Nucleobase solubility assays were separately carried out for adenine, guanine, cytosine and 1-methylthymine in water, and in G4·K+ and G4·Na+ gels. Saturated solutions of G4·K+ and G4·Na+ gels, prepared either in D2O or Millipore water, were pipetted into vials containing increasing concentration of the nucleobase. Each vial was then reheated and sonicated until the solid powders of nucleobases dissolved and the resultant solution was allowed to cool to room temperature. Solubility of nucleobase molecules was monitored over a week.Characterization
Gels were heated to their sol–gel transition temperature (∼90 °C to ensure a clear solution-like material) to inject them into 4 mm (outer diameter) zirconia rotors containing an inbuilt bottom insert. A procedure for determining melting temperatures (Tm) of different G4·M+ borate gels is described in our previous study.22 The rotors were packed with ∼20 μL of hydrogel using a top insert-screw-lid system and then with a Kel-F cap to ensure the close-fitting. Unless specified, the spinning frequency was 5 kHz in all MAS NMR experiments. 7Li, 23Na, 39K and 133Cs MAS NMR experiments were carried out at 20 T with a Bruker Avance III spectrometer (Larmor frequencies were 1H = 850.2 MHz, 7Li = 330.3 MHz, 23Na = 224.8 MHz, 39K = 39.7 MHz, and 133Cs = 111.5 MHz) and on a double resonance H–X MAS probe. For 7Li, 23Na, 39K and 133Cs MAS NMR, signal averaging was performed over 512, 2048, 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 480 and 4096 co-added transients for a recycle delay of 2, 2, 1 and 5 seconds, respectively. The 7Li, 23Na, 39K and 133Cs chemical shifts were calibrated using aqueous solutions containing 100 mM of LiCl, NaCl, KCl and CsNO3 referenced to 0.0 ppm, respectively. 1D 11B NMR experiments were carried out at different magnetic field strengths: 11.7 T with a Bruker Avance III (1H = 500.1 MHz, 11B = 160.5 MHz), 14.1 T with a Bruker Avance II+ (1H = 599.4 MHz, 11B = 192.3 MHz) and 20.0 T with a Bruker Avance III (1H = 850.2 MHz, 11B = 272.8 MHz) spectrometer equipped with a 4 mm HXY (in double resonance configuration), 4 mm HX and 4 mm HX MAS probe, respectively. The 11B π/2 pulse length was 2.5 μs. Two-Pulse Phase Modulation (TPPM) decoupling41 was applied during the acquisition with a 1H π pulse duration of 5 μs at 11.7 T and 14.1 T, and 6 μs at 20 T, corresponding to a 1H nutation frequency of 100 kHz for 11.7 and 14.1 T, and 85 kHz for 20 T. 11B MAS NMR spectra were acquired by co-adding 2048 transients with a recycle delay of 4 seconds, corresponding to an experimental time of 2 h. The 11B chemical shifts of guanosine borate esters were referenced to NaBH4 at −42.06 ppm as a secondary reference to the primary reference using BF3Et2O at 0.0 ppm.42 For the gels prepared using D2O, one-pulse 1H NMR spectra were recorded using either a Bruker Avance III, 9.4 T (1H, 400 MHz) or a Bruker Avance 11.7 T (1H, 600 MHz) spectrometer. The 1H π/2 pulse lengths were 12.35 μs and 11.4 μs, respectively. Signal averaging was performed using 256 co-added transients with a recycle delay of 1 s, corresponding to a total experimental time of 15 minutes.
480 and 4096 co-added transients for a recycle delay of 2, 2, 1 and 5 seconds, respectively. The 7Li, 23Na, 39K and 133Cs chemical shifts were calibrated using aqueous solutions containing 100 mM of LiCl, NaCl, KCl and CsNO3 referenced to 0.0 ppm, respectively. 1D 11B NMR experiments were carried out at different magnetic field strengths: 11.7 T with a Bruker Avance III (1H = 500.1 MHz, 11B = 160.5 MHz), 14.1 T with a Bruker Avance II+ (1H = 599.4 MHz, 11B = 192.3 MHz) and 20.0 T with a Bruker Avance III (1H = 850.2 MHz, 11B = 272.8 MHz) spectrometer equipped with a 4 mm HXY (in double resonance configuration), 4 mm HX and 4 mm HX MAS probe, respectively. The 11B π/2 pulse length was 2.5 μs. Two-Pulse Phase Modulation (TPPM) decoupling41 was applied during the acquisition with a 1H π pulse duration of 5 μs at 11.7 T and 14.1 T, and 6 μs at 20 T, corresponding to a 1H nutation frequency of 100 kHz for 11.7 and 14.1 T, and 85 kHz for 20 T. 11B MAS NMR spectra were acquired by co-adding 2048 transients with a recycle delay of 4 seconds, corresponding to an experimental time of 2 h. The 11B chemical shifts of guanosine borate esters were referenced to NaBH4 at −42.06 ppm as a secondary reference to the primary reference using BF3Et2O at 0.0 ppm.42 For the gels prepared using D2O, one-pulse 1H NMR spectra were recorded using either a Bruker Avance III, 9.4 T (1H, 400 MHz) or a Bruker Avance 11.7 T (1H, 600 MHz) spectrometer. The 1H π/2 pulse lengths were 12.35 μs and 11.4 μs, respectively. Signal averaging was performed using 256 co-added transients with a recycle delay of 1 s, corresponding to a total experimental time of 15 minutes.
Density functional theory (DFT) calculations
To understand different distributions of 11B chemical shifts of hydrogels, DFT calculations were carried out on a GB monoester, and cis and trans isomers of diesters. All calculations were performed using the Gaussian 09 package.43 The GB mono and dimer structures were optimized to a ground-state equilibrium geometry using the B3LYP functional and aug-cc-pvdz basis set. Using the geometry optimized dimer structure, conformationally different GB dimers were generated by constraining the C–C–C–C dihedral angle between purine and sugar moieties into cis (G moieties facing the same side of borate ester) and trans (G moieties on the opposite sides of the borate ester) conformations. DFT geometry optimized structures of GB monoester, and cis and trans GB dimers were then used to calculate NMR shieldings using the gauge-independent atomic orbital (GIAO) method with B3LYP/6-31G functional and basis set. DFT-calculated 11B shieldings were referenced to the 11B shifts of B2H6 (calculated using the same B3LYP/6-21G functional) included in the Gaussian 09 package.43 DFT-calculated 11B shieldings were plotted against experimental 11B shifts44–46 and regression analysis was carried out by constraining the slope to 1.0. This analysis leads to linear correlation between the experimental and DFT calculated 11B shifts with an intercept of 31.0 ppm. In order to compare with the experimental isotropic 11B shifts shown in the tables, 31.0 ppm should be added to the DFT calculated 11B shieldings.Results and discussion
Roles of cations and borate anions in the gelation of G-borate dimers
We used MAS NMR to separately examine the roles of alkali metal cations and borate anions in the gelation process that gives rise to G4·M+ borate gels. Although boron cross linking of guanosine units in different G-quartet layers apparently enhances the stability of GB gels, the templating cation's size and charge is crucially important in the gelation. In a previous study, we showed that the G4·K+ borate gel exhibited a remarkable stability, relative to G4·M+ gels made with the other alkali metal cations.21,22 To better understand the key role of alkali metal cations in stabilizing these functional hydrogels, a series of G4·M+ gels were prepared with LiB(OH4), NaB(OH4), KB(OH4) and CsB(OH4) salts and characterized using 7Li, 23Na, 39K and 133Cs MAS NMR, respectively (ESI,† Fig. S1). A narrow signal observed at 0 ppm in the 7Li MAS NMR spectrum of the G4·Li+ borate gel indicates the presence of free Li+ ions in a solution-like environment. In contrast, the 133Cs MAS NMR spectrum of a G4·Cs+ gel shows multiple signals, indicating that the Cs+ ions are dispersed into different local chemical environments of this sol–gel material: the relatively broad signals at 50 and 42 ppm originate from different distributions of Cs+ ions in the G4 network (gel) and in the cloudy precipitate that is formed along with the gel, while the narrow signal at 12 ppm is due to free Cs+ ions in the solution phase. Signal integrals obtained from lineshape fitting analysis revealed that 73 ± 6% of the Cs+ was in the gel phase and 27 ± 6% was in solution. In the 39K MAS NMR spectrum of the G4·K+ borate gel (Fig. 1a), partially resolved signals are observed with a broad intensity at 8 ppm, indicative of K+ ions located within the central G4-channel,47 and a relatively narrow signal at ∼0 ppm ascribed to free K+ ions in solution phase. Lineshape fitting analysis of these signals enabled the quantification of K+ ions in the sol and gel phases (the narrow and broad signals in the presented MAS NMR spectra are associated with free cations in the solution-like environment (herein called sol) and cations associated with the G4 network (herein called gel), respectively), which were 53 ± 8% and 47 ± 8%, respectively. To further confirm the assignments, a 39K NMR spectrum was acquired after soaking the G4·K+ borate gel overnight in a solution containing an excess of NaCl (Fig. 1a, inset). The G4·K+ borate gel soaked in 155 mM NaCl solution showed a significant reduction in the intensity of the narrow signal, confirming that the K+ ions in the sol-phase had exchanged with the Na+ ions in the bath solution, while the K+ ions bound to G4 channels are retained due to their stronger binding affinity to G-quartets within the G4·K+ borate gel. Based on these observations, the narrow signal in the 39K spectrum is assigned to the K+ ions in an aqueous environment and the broad signal is assigned to K+ ions in the G4-channel.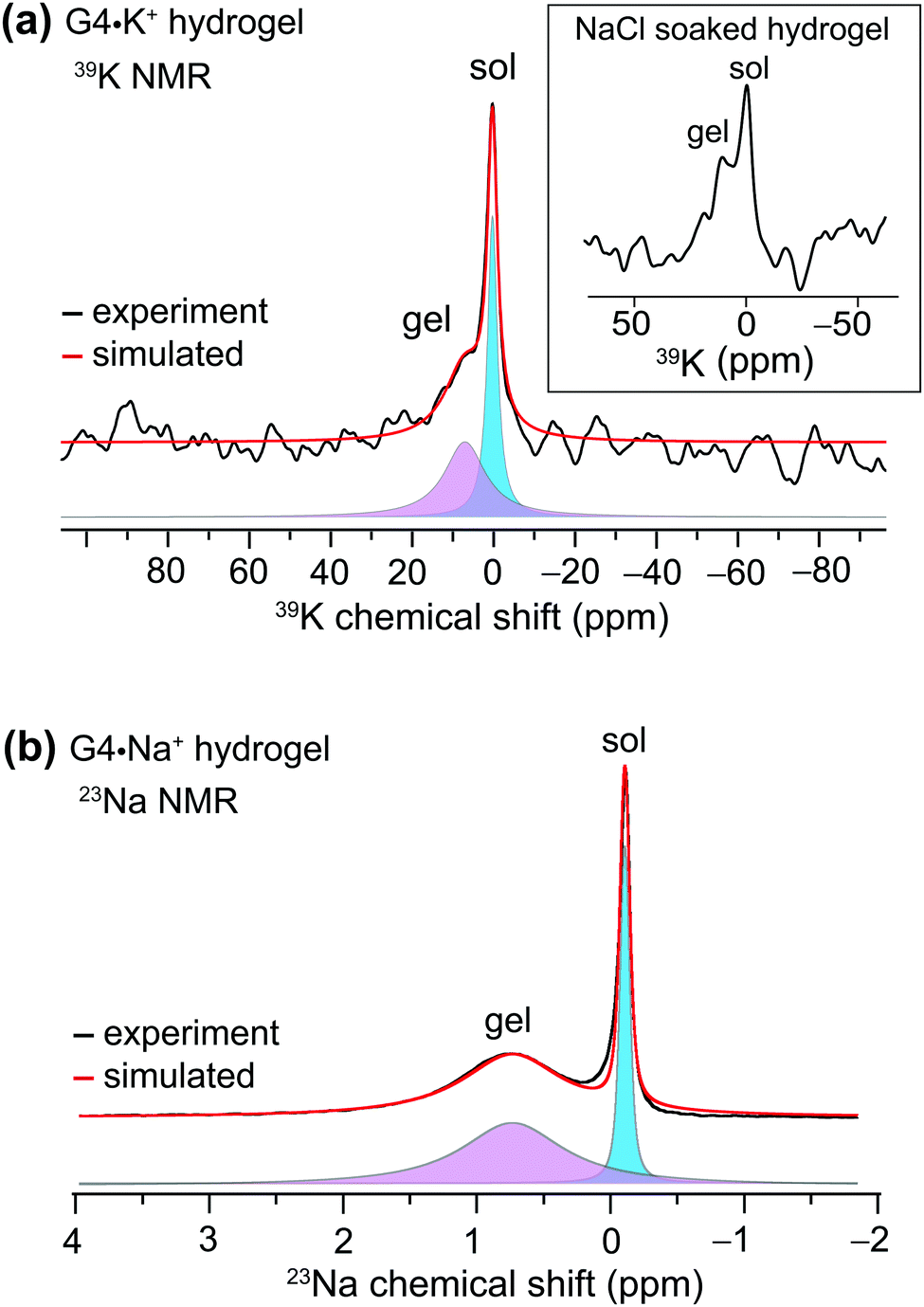 | ||
| Fig. 1 Gel-state 1D MAS NMR spectra of 20 μL G4·M+ hydrogels acquired at 20 T with their deconvolution into broad and narrow components: (a) 39K MAS (5 kHz) NMR spectrum of G4·K+ gel and (b) 23Na MAS (5 kHz) NMR spectrum of G4·Na+ gel. The inset in (a) depicts a 39K MAS NMR spectrum of a G4·K+ borate gel that had been soaked overnight in 155 mM NaCl solution, acquired under the same conditions. The narrow and broad signals originate from the free cations in the solution-like environment (herein called sol) and cations associated with the G4 network (herein called gel), respectively. | ||
Next, we studied the distributions of Na+ cations in the sol- and gel phases of G4·Na+ borate gel. In the 23Na MAS spectrum of G4·Na+ borate gel, two signals are observed (Fig. 1b): the broad signal centered at 0.8 ppm is attributed to the Na+ ions in the G4-channel, and a narrow signal in the vicinity of 0 ppm is assigned to Na+ ions in solution phase. Lineshape analysis of the 23Na signals enabled the quantification of Na+ ions in the sol and gel phases, which were 31 ± 4% and 69 ± 4%, respectively. These results are corroborated by our previous studies, which showed that K+ and Na+ ions stabilize G-quartets as compared to Li+ and Cs+ ions,21,22 and is consistent with the rheological studies, which showed that the G4·K+ gel has a high storage modulus (G′, ∼11 kPa) that is an order of magnitude larger than the loss modulus (G′′), whereas the G4·Li+ gel exhibited a G′ much closer in size to G′′ (Fig. 3, ref. 22). Therefore, based on the results of these above NMR studies and our previous rheology studies, G4·K+ and G4·Na+ gels were chosen for study of the impact of small molecules uptake on the gelation properties.
Complementary insight into the solvent-associated (sol) or G4-network-associated (gel) compositions and structures of G4·M+ borate hydrogels were obtained by analyzing 11B MAS NMR spectra. 11B (spin 3/2, natural abundance 80.5%) MAS NMR has been shown to be a powerful probe for characterizing local boron environments in ion exchange resins, boric acid esters and hydrogels.21,22,42,48–50 The 11B MAS NMR spectra of G4·Na+ and G4·K+ borate gels recorded at 20 T (Fig. 2) showed different 11B shifts, which could be assigned to tetracoordinated borate anions in both the G4 gel network and in a solution-like environment. These experimental shifts (δexpt) are expected to have contributions from the isotropic 11B chemical shifts (δCSiso) and isotropic second order quadrupolar induced shifts (δQiso). Graphical plots of 11B shifts measured at different magnetic fields (11.7 T, 14.1 T and 20 T) as a function of inverse squared Larmor frequency (νL2) are presented in Fig. 2 to the right of the 11B MAS NMR spectra. Similar 11B MAS NMR spectra of G4·Li+ and G4·Cs+ gels are presented in the ESI† (Fig. S2). The isotropic 11B chemical shifts of the mono- and diester signals can be obtained directly from where the extrapolated dashed lines intercept the chemical shift axes (ESI,† Section S2, Table S1). The quadrupolar product (PQ) can be calculated accordingly by the following equation,51–53
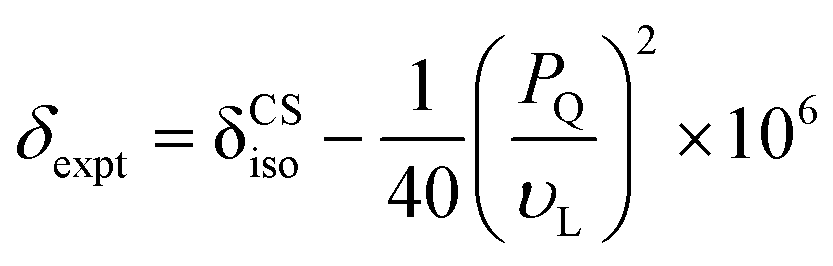 | (1) |
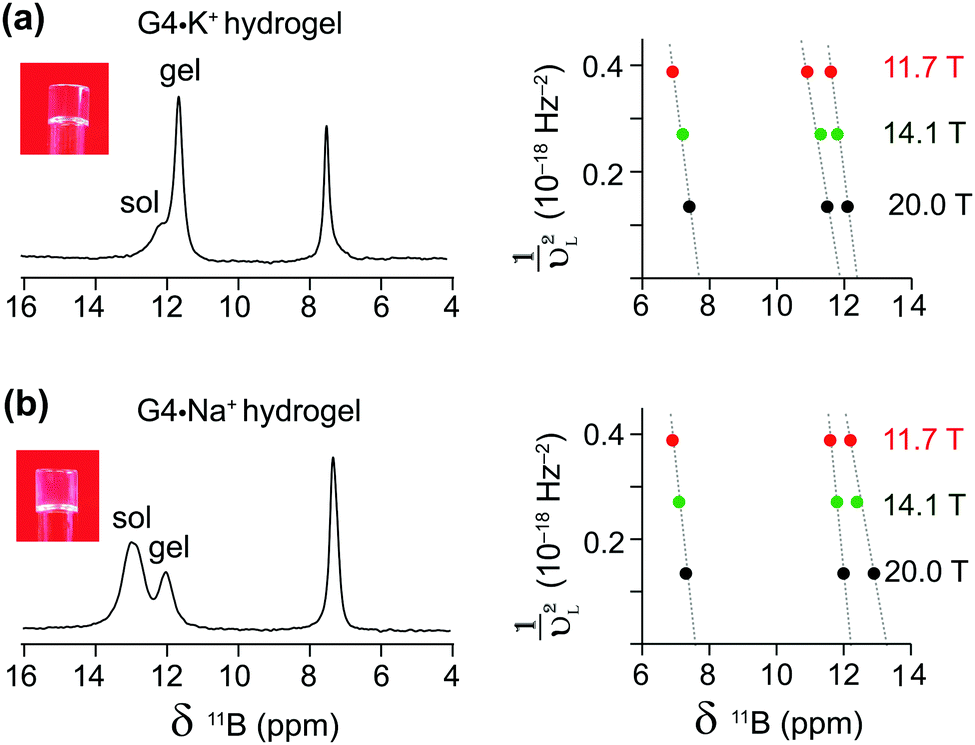 | ||
| Fig. 2 Gel-state 1D 11B MAS (5 kHz) NMR spectra of 20 μL G4·M+ hydrogels acquired at 20 T using 1H decoupling. The experimental 11B NMR shifts of (a) G4·K+ and (b) G4·Na+ gels obtained at different magnetic fields are plotted as a function of the inverse squared Larmor frequency (νL2) for both the GB mono (6–8 ppm) and diester signals (11–14 ppm). Sol and gel 11B signals refer to borate mono- and diesters in a solution-like environment and in a G4 network, respectively. | ||
The δCSiso and PQ values obtained by analyzing the 11B MAS spectra acquired at three different magnetic fields (Table 1) indicate subtle, but detectable, differences in the local boron environments of the G4·Na+ and G4·K+ gels, presumably due to the identity of the countercation. The 11B isotropic chemical shifts and quadrupolar interaction parameters of tetrahedrally coordinated borate anions are in agreement with the analogous tetracoordinated borate anions presented in the literature (δiso in the range of 0.4–12.4 ppm and quadrupolar coupling constants in the range of 0.9–1.7 MHz).49 The 11B isotropic signals at 7.5 and 7.7 ppm were attributed to borate monoesters in the 11B MAS NMR spectra of G4·Na+ and G4·K+ gels, respectively (Fig. 2 and Table 1). In comparison, pairs of signals at 12.2 and 13.2 ppm, and at 11.8 and 12.4 are ascribed to different diastereomeric borate diesters that are in cis and trans conformations with respect to guanosine moieties, respectively, in the 11B NMR spectra of G4·Na+ and G4·K+ gels (Fig. 2 and Table 1). These results are corroborated by the DFT optimized structures of GB diesters and GIAO calculations of 11B isotropic chemical shifts corresponding to cis and trans isomers of borate diesters (ESI,† Fig. S3). A linear correlation is observed between experimental 11B isotropic chemical shifts (shown in Table 1) and DFT-calculated 11B isotropic absolute shielding (ESI,† Fig. S4). These 11B NMR parameters are expected to be influenced by the changes in the boron environments upon the addition of small molecules, as will be studied in the next section. Overall, the MAS NMR studies described above provide us with an excellent method to elucidate the local structures of various G4·M+ borate species (G4-network associated vs. solvent associated) by analyzing the chemicals shifts of both the templating M+ cation and the borate diester as an integral cross-linker in stabilizing these hydrogels. Moreover, as described in the next sections, these multinuclear NMR techniques provide direct insight into the influence that small molecules may have on the structure and properties of the G4·M+ borate gels.
| G4·M+ borate gels | Boron site | 11B NMR shifts (ppm) | P Q (MHz) | δ CSiso (ppm) | ||
|---|---|---|---|---|---|---|
| 11.7 T | 14.1 T | 20 T | ||||
| a Errors on the PQ and δCSiso (ppm) are estimated as ±0.01 MHz and 0.1 ppm, respectively. The values are calculated by the procedure given in the ESI (Section S2). | ||||||
| G4·Na+ gel | Monoester | 6.9 | 7.1 | 7.3 | 0.77 | 7.5 |
| Diester (cis) | 11.6 | 11.8 | 12.0 | 0.76 | 12.2 | |
| Diester (trans) | 12.3 | 12.4 | 12.9 | 0.99 | 13.2 | |
| G4·K+ gel | Monoester | 6.9 | 7.2 | 7.4 | 0.85 | 7.7 |
| Diester (cis) | 11.0 | 11.2 | 11.6 | 0.92 | 11.8 | |
| Diester (trans) | 11.6 | 11.8 | 12.1 | 0.89 | 12.4 | |
The impact of cationic methylene blue (MB) uptake on G·K+ gel
The cationic and planar dye, methylene blue (MB), is well known to bind to G-quadruplex DNA fibrils.54–57 We have previously studied the physical absorption of MB by these anionic GB gels, using both UV-Vis spectroscopy and 11B NMR spectroscopy.21,22 In particular, we found that G4·K+ gels (72 mM in G) were able to quantitatively absorb 12.5 μM MB after soaking the hydrogel in a KCl solution of the dye for 24 h, as determined by UV-Vis spectroscopy.21 Importantly, these low μM concentrations of cationic dyes, including MB, act to strengthen the G4·Li+ gels, as reflected in the increased storage modulus (G′) of the Li+ GB hydrogel in the presence of the dyes.58To gain further insight into the impact of absorption of MB on the structure and stability of the GB gels, high-resolution 1H and 11B MAS NMR spectra of G4·K+ gels (Fig. 3) were compared before and after the incorporation of MB into the gel. The 1H signals at 7.7, 6.8 and 6.2 ppm that correspond to aromatic protons in MB molecules in GB gels are displaced to lower chemical shifts as compared to the analogous 1H chemical shifts of 0.1 mM MB in D2O (7.4, 7.1 and 6.9 ppm).59 These upfield shifts associated with 1H signals of MB in gels (i.e., Δδ = 0.3, 0.3 and 0.7 ppm for the signals at 7.7, 6.8 and 6.2 ppm, respectively) can be attributed to the π–π stacking and CH–π interactions that occur between MB and G4 fibrils. In addition, various binding interactions, such as end stacking, side stacking and parallel stacking of MB molecules to G4-rich DNA, have been previously proposed,55 and those same types of interactions may be relevant here in the context of MB binding to the G4·K+ gels. Also, a priori, ionic interactions between cationic MB and borate anions in the G4·K+ borate gels are certainly important.
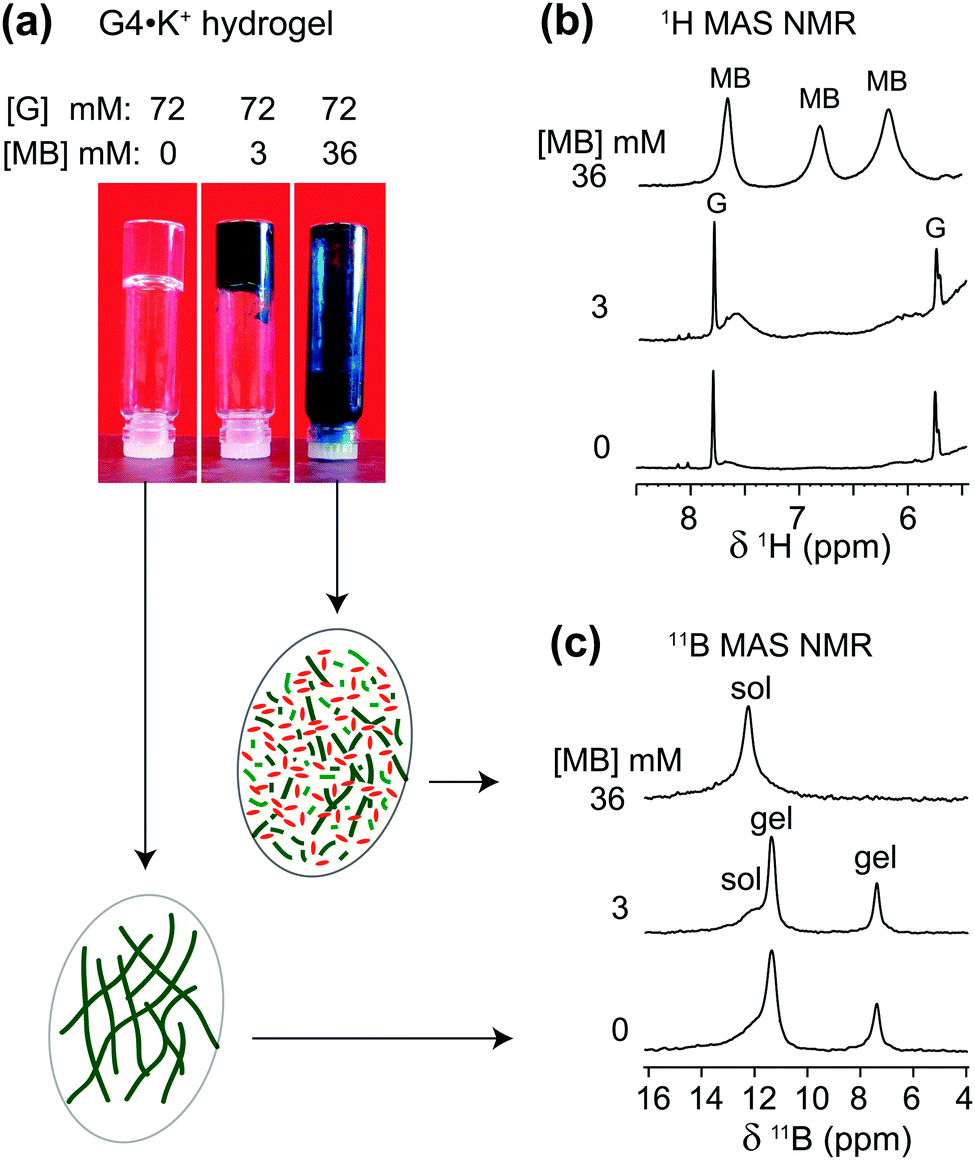 | ||
| Fig. 3 (a) Photographs of 72 mM G4·M+ borate hydrogels taken before and after the incorporation of 3 mM and 36 mM of MB, accompanied by 1D (b) 1H and (c) 11B (20 T, 5 kHz MAS) NMR spectra of G4·K+ borate gels shown in the vials (similar 11B MAS NMR spectra acquired at 11.7 T are presented in ref. 21). Inset figures in ovals represent schematics of G4 fibrils before and after the addition of methylene blue. Sol and gel 11B signals correspond to borate mono- and diesters in a solution-like environment and in the G4 network, respectively. | ||
In the 1H NMR spectrum of a sample that had 3 mM MB added to G4·K+ gels (72 mM in G), relatively broad 1H signals (6.2, 6.8 and 7.6 ppm) are observed for the aromatic protons in MB, indicating that MB is embedded in the gel phase. As illustrated in Fig. 3a, a vial inversion test showed that the G4·K+ gel remained self-standing in the presence of 3 mM MB. In contrast to the broad MB signals for this sample, narrow 1H signals at 5.8 ppm (H1′) and 7.8 ppm (H8) are observed for the free G in the solution phase. This data indicates that MB is sequestered in the gel phase at this concentration. Upon addition of a higher concentration of MB (36 mM) to the gels, a vial inversion test showed a runny solution, indicating that this high concentration of dye had triggered a gel-to-sol transition. Consistent with this phase transition, the broad 1H NMR signals for MB become much narrower and clearly distinguishable (upper spectrum of Fig. 3b). Under these conditions, the G4 fibrils that make up the gel phase are disrupted and the cationic MB molecules are anticipated to be distributed in the vicinities of monomeric GB species.
To investigate whether the uptake of MB influences the local boron chemical environments in the G4·K+ gels, we compared the isotropic 11B chemical shifts and quadrupolar interactions by analyzing 11B NMR spectra acquired at different magnetic fields. In particular, the quadrupolar interactions are expected to be sensitive to the gel-to-sol transition. For example, Wallace et al. have exploited 2H and 14N residual quadrupolar couplings (RQC) to understand the self-assembly process of a dipeptide gelator.60,61 The 11B MAS NMR spectra acquired before and after the addition of MB exhibited different spectral features for different sol–gel ratios, depending on the concentration of added MB. Although similar 11B NMR titrations, monitoring 11B chemical shifts as a function of concentration of added MB, have previously been presented in the ESI of ref. 21, the emphasis in this current study is placed on understanding whether or not the gel-to-sol transition causes significant differences in the local chemical environments of the borate anions, particularly, in the presence of excess of MB (36 mM). It is noteworthy that the 11B signal intensities associated with the GB monoester (7.4 ppm) and the cis isomer of GB diester (11.6 ppm) are reduced upon the addition of 36 mM MB, which indicates that both GB mono- and diesters (cis isomer) enhance the gelation of G4 borate esters in the presence of K+ ions. In contrast, the 11B signal intensity corresponding to the trans isomer of the GB diester (12.4 ppm) increased as a function of the increased concentration of MB, which suggests that the trans GB diester plays a subtle role in the gelation process.18 The isotropic chemical shift δCSiso and quadrupolar product PQ were calculated from the 11B NMR data acquired at two different magnetic field strengths, 11.7 T and 20 T (Table 2).
| [G4·K+gel]:[MB] mM |
Boron site | 11B shifts (ppm) | P Q (MHz) | δ CSiso (ppm) | |
|---|---|---|---|---|---|
| 11.7 T | 20 T | ||||
| a Errors on the PQ and δCSiso (ppm) are estimated as ±0.01 MHz and 0.1 ppm, respectively. The values are calculated by the procedure given in the ESI (Section S2). | |||||
| 72:0 |
Monoester | 6.9 | 7.4 | 0.85 | 7.7 |
| Diester (cis) | 11.0 | 11.6 | 0.92 | 11.8 | |
| Diester (trans) | 11.7 | 12.2 | 0.89 | 12.4 | |
| 72:3 |
Monoester | 7.0 | 7.4 | 0.89 | 7.6 |
| Diester (cis) | 11.0 | 11.6 | 0.92 | 11.9 | |
| Diester (trans) | 11.7 | 12.2 | 0.89 | 12.4 | |
| 72:36 |
Monoester | 7.0 | 7.5 | 0.91 | 7.8 |
| Diester (cis) | 11.2 | 12.2 | 1.22 | 12.7 | |
| Diester (trans) | 11.8 | 12.5 | 1.05 | 12.9 | |
The gel-to-sol transition results in subtle, but detectable, changes in the PQ values of the diester gel signals in the range between 0.85 and 1.2 MHz. While these changes in the PQ values do not confirm the presence of intermolecular interactions between MB and G4·M+ borate gels, they do show that changes occur in local chemical environments of the borate anions upon a gel-to-sol transition, which is consistent with the 1H NMR studies of MB added G4·K+ gels. Together, the visual tip tests and the corresponding 1H and 11B MAS NMR analyses of the different samples shown in Fig. 3 indicate that addition of excess of MB interrupts the G4 quartet borate esters and intermolecular interactions that stabilize the gel. Clearly, gel-state MAS NMR spectroscopy is a valuable method to gauge the impact of how small molecules, such as MB, can influence the sol–gel transition in these G4·M+ borate gels. It is noted that 1H–13C CP-MAS NMR experiments have previously been applied to characterize xerogels and organic phases integrated into mesoporous silica.26,32 However, the low sensitivity of 1H–X (X = 13C, 15N) CP-MAS NMR is a bottleneck, for example, 13C spectra of xerogels have been acquired by signal averaging several hours to days depending on the swelling solvent.26 In the case of GB hydrogels containing ∼2 wt% gel network,22 carrying out a large number of 1H–X (X = 13C, 15N) CP-MAS NMR titration experiments as a function of increasing concentration of guest molecules (Fig. 3) is anticipated to be less suitable, particularly, when dilute concentrations of guest molecules (≤0.1 equiv. of gelator molecules) are involved.
Addition of nucleobases into G·K+ borate hydrogels
To explore the uptake of neutral guest molecules, we next studied the incorporation of nucleosides into G4·M+ borate gels. Indeed, nucleobases such as adenine and closely related heterocycles with antiviral action show relatively poor solubility in water (∼7 mM) and water/additive binary solutions (∼20 mM adenine in the presence of 1 M arginine as an additive),62–64 which poses a challenge for their therapeutic applications. It has been reported that added sugars including ribose65 and especially galactose and lactose66 can enhance the solubility of adenine in water by using CH–π interactions67 that disrupt the base stacking processes that lead to adenine aggregation.68,69 In addition, supramolecular chemistry approaches,70 including using Rebek/Kemp triacid receptors,71 cyclodextrins72,73 and biodegradable polymers74 have been exploited to increase the water solubility of adenine and enhance its transport across biological membranes.To examine the nucleobase solubility in GB gels, we employed adenine concentrations over 30 mM, which is well beyond the limit of its solubility in water (∼7 mM).62–64 While 32 mM adenine precipitates in H2O (Fig. 4, inset photograph), the same concentration of adenine added to a 72 mM G4·K+ borate gel (right vial) shows no precipitation and the hydrogel remains transparent over several days. A solution state 1H NMR spectrum of this G4·K+ borate gel containing adenine indicates that the majority of the nucleobase (∼30.9 mM) remains in the solution-like environment (bottom), i.e., adenine molecules or their aggregates freely diffuse. In comparison to the MB incorporated G4·K+ gel (Fig. 3b), relatively narrow signals are observed in the 1H NMR spectrum of adenine added G4·K+ gels (Fig. 4b), which is consistent with the dissolution of adenine in the solution phase rather than being closely associated with the network that makes up G4·K+ gels. To estimate the adenine content in the solution phase of the G4·K+ gel, we carried out a quantitative 1H NMR analysis. A known concentration of cytidine (32 mM) was dissolved in D2O with a DMSO internal standard (Fig. 4a). The 1H signal integral of two cytidine protons centered at δ = 7.93 ppm was 6.6 with respect to an integral of 1.0 corresponding to the 1H signals of DMSO (δ = 3.09 ppm). Similarly, in a 1H NMR spectrum shown in Fig. 4b, the signal centered at δ = ∼8.1 ppm is representative of adenine's aromatic proton integrated to 3.4 (relative to an integral of 1.0 corresponding to the same 1H signals of DMSO), indicating that ∼31 mM of the adenine was in the solution phase. This suggests that the adenine was introduced into the pores of the gel matrix rather than being closely associated with the boron containing gel. Such minute quantities of DMSO likely do not disrupt the hydrogen bonding networks in hydrogels, yet the gel stability is maintained as confirmed by a vial inversion test. The enhanced adenine solubility in GB gels could be related to several mechanisms and hypotheses proposed in the previous studies; for example, alcohol/water or glycol/water binary mixtures enhance the adenine solubility,62,64 the presence of an amino acid additive such as arginine improves the solubility of adenine in water,63 and also the presence of dideoxynucleosides augments adenine solubility in water.75 In particular, it has been hypothesized that the hydrogen bonding interactions between polar OH groups associated with alcohols and adenine moieties dominate as compared to the base stacking of these latter molecules.62,64 While such noncovalent interactions are anticipated to be relevant in GB gels, the roles of enhanced polarity of the solution phase (due to the borate buffer) of GB gels in enhancing the adenine solubility cannot be ruled out.
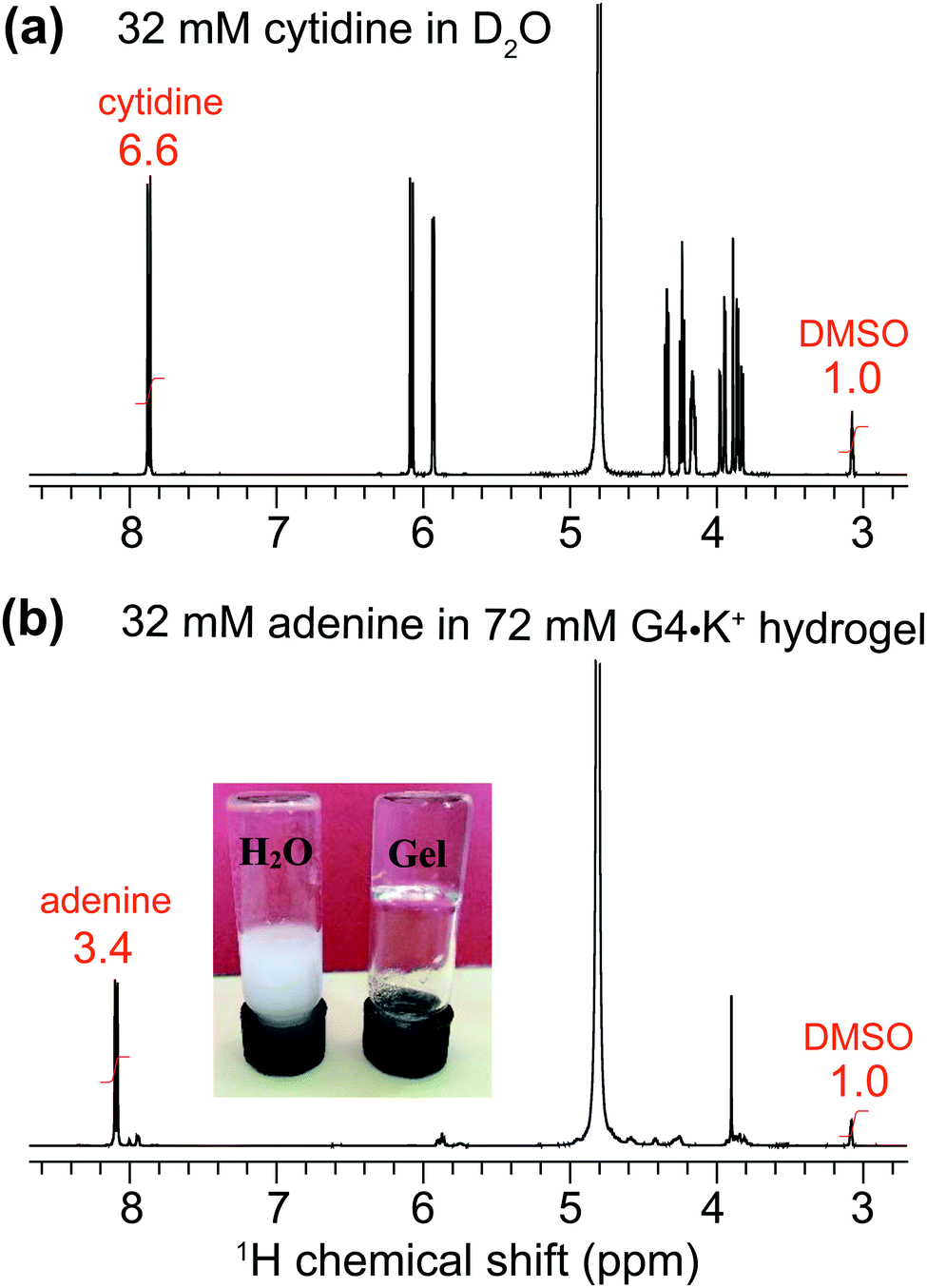 | ||
| Fig. 4 Solution-state 1D 1H (600 MHz) NMR spectrum of (a) 32 mM of cytidine dissolved in D2O using DMSO proton signals (at 3.09 ppm, integral of 1.0) as an internal calibration standard, and (b) of 32 mM adenine incorporated into a G4·K+ gel. The inset image in (b) depicts the dissolution of 32 mM of adenine in H2O (left) and in 72 mM G4·K+ borate gel (right). | ||
Uptake of nucleobases by G4·Na+ borate gels
The enhanced solubility of adenine in G4·K+ gel prompted us to investigate the solubility of different nucleobases in G4·Na+ gel, which did not pose the same cardiotoxicity risks as gels containing potassium ions for one particular adenine delivery application we were envisaging. Hence, in order to examine the solubility of different nucleobases in G4·Na+ borate gel, hot saturated solutions of the G4·Na+ borate gels (72 mM G) were added to the vials containing known concentrations of adenine, guanine, cytosine and 1-methylthymine. The resultant solutions were homogenized by mild agitation and allowed to settle at room temperature. Hydrogel stability was determined by vial inversion tests performed after an hour and after a day (ESI,† Fig. S5–S7). The nucleobase solubility was monitored over a week. These visual observations show that the addition of a relatively low concentration of nucleobases does not influence the gelation properties, however, increased heterocycle loading reduces the gelation properties of the G4·Na+ borate gels (ESI,† Tables S2–S4). The nucleobase solubility is enhanced for adenine, cytosine and 1-methylthymine, although the effect is relatively smaller for cytosine, where there is a tendency for precipitation over time. Notably, transparent gels are observed upon the addition of adenine and 1-methylthymine, meaning that these molecules are situated in the solution phase of the hydrogel. While guanine is notoriously insoluble in water (a cloudy solution was observed when 1 mg of guanine was added to 250 mL water), we found that it can be brought into solution at a low concentration of 0.054 mM by the gels, which was not studied any further.Adenine exhibited remarkable solubility in G4·Na+ hydrogels. After adding 64 mM adenine, the G4·Na+ gel remained stable and clear. The absorption process of adenine is monitored by carrying out solution-state 1H NMR and gel-state 11B and 23Na MAS NMR titration experiments (Fig. 5). The stability and integrity of G4·Na+ gel is reflected in the 1H and 11B NMR spectra, although increased 1H signal (δ = 7.8 ppm) intensities for adenine confirm that there is a rise in the concentration of adenine in the gel. Nevertheless, the lineshape fitting analyses of the gel-state 23Na MAS NMR spectra showed different distributions of Na+ ions in solution and gel regions before and after the addition of adenine. The distribution of Na+ ions content in the adenine added gel is 42 ± 4% (cations associated with the G4 gel network) and 58 ± 4% (free cations in solution) respectively, as compared to 69 ± 4% (gel network) and 41 ± 4% (solution) in the pristine G4·Na+ gel. In addition, the 23Na signal associated with the gel network is displaced to 0.5 ppm, as compared to 0.8 ppm (Δδ = 0.3 ppm) for the same 23Na signal in the pristine gel. These results and analyses confirm that the majority of adenine molecules are located in the pores of the solution phase, and dilute concentrations of adenine molecules are embedded into the gel framework. The impact of increased polarity due to the presence of alkali metal borate salts in the solution phase of the hydrogel on enhanced solubility of adenine cannot be ruled out. The addition of higher concentrations of nucleobases, specifically above 72 mM, reduce the integrity of gels with there being a trend of precipitating out the solute over time.
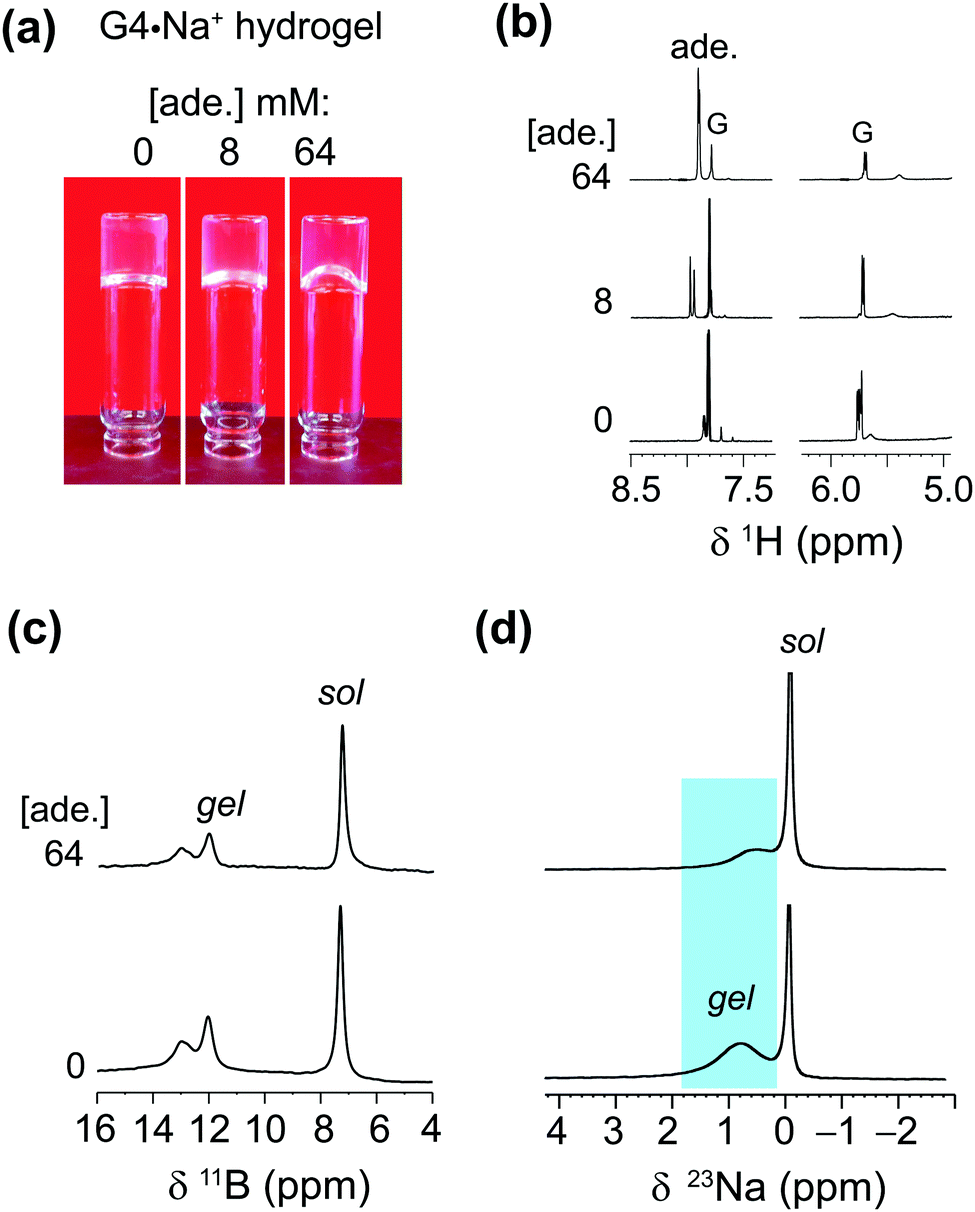 | ||
| Fig. 5 (a) Photographs of 72 mM G4·Na+ borate hydrogels taken before and after the incorporation of 8 mM and 64 mM of adenine. (b) 1H (9.4 T, 400 MHz), (c) 11B (20 T, 5 kHz MAS) and (d) 23Na (20 T, 5 kHz MAS) NMR spectra of G4·Na+ borate gels shown in the inverted vials in (a). Sol and gel signals correspond to free cations or molecules in a solution-like environment and in the G4 network, respectively. | ||
Further insights into the solubility of nucleosides into G4·Na+ borate gels were obtained by monitoring the absorption process of cytosine (Fig. 6). The 1H solution-state NMR titration showed increased cytosine concentration in the solution phase of the gel framework (Fig. 6b), as revealed by the narrow 1H signals at 5.8 and 7.4 ppm. As expected, 11B MAS NMR spectra recorded before and after the incorporation of cytosine showed no significant changes in the spectral patterns (Fig. 6c). These results are corroborated by gel-state 23Na MAS NMR spectroscopy, which showed displacement of the 23Na chemical shift (Δδ = −0.7 ppm) accompanied by changes in the local environments of Na+ ions in the solution phase (Fig. 6d). The signal integrals measured from the lineshape analyses of 23Na MAS NMR spectra confirmed that the distributions of the Na+ ions in solution (69 ± 4%) and G4 gel network (31 ± 4%) before and after the addition of cytosine are the same. From these results and analysis, it could be inferred that cytosine moieties are largely confined within the pores of the gel network and the addition of higher concentrations of over 64 mM cytosine leads to precipitation, which reduces the gel stability. The solubility of 1-methylthymine was however significantly improved from 50 mM to at least 70 mM in G4·Na+ borate gel (ESI,† Table S4). All of these findings reveal that these G4·K+ and G4·Na+ borate gels have the potential to adsorb small molecule drugs in the range between 0.05 and 0.5 equiv. with minimal disruption to the G-quartet gel framework.
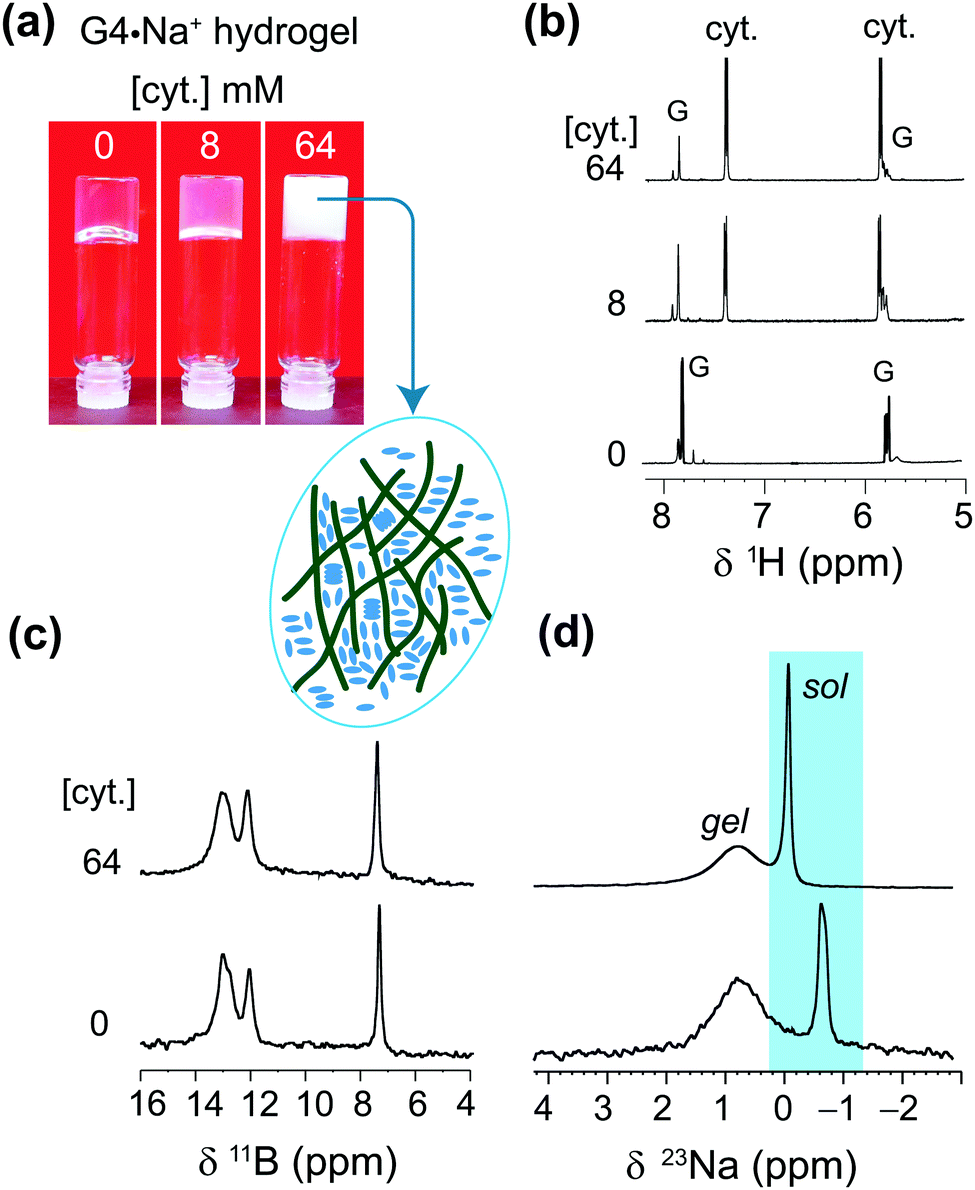 | ||
| Fig. 6 (a) Photographs of 72 mM G4·Na+ borate hydrogels taken before and after the incorporation of 8 mM and 64 mM of cytosine, accompanied by one-pulse (b) 1H (9.4 T, 400 MHz), (c) 11B (20 T, 5 kHz MAS) and (d) 23Na (20 T, 5 kHz MAS) NMR spectra of G·Na+ borate gels shown in the inverted vials (a). The inset figure depicts that how cytosine molecules are aggregated in the pores of the gel framework, which leads to precipitation. Sol and gel signals correspond to free ions or molecules in a solution-like environment and in the G4 network, respectively. | ||
Conclusions
Insights into the roles of alkali cations and borate anions in the gelation of G4 borate hydrogels are obtained by multinuclear (7Li, 11B, 23Na, 39K and 133Cs) MAS NMR spectroscopy in conjunction with DFT modelling. A comparison of 1D MAS NMR spectra of alkali metals enabled free cations versus G-quartet associated alkali metal cations to be distinguished and quantified, which reveals that the K+ and Na+ cations stabilize the borate gel more than gels using Li+ and Cs+ ions. In addition, 11B isotropic chemical shifts and quadrupole interactions were determined by analyzing 1D 11B MAS NMR spectra acquired at different magnetic fields, 11.7 T, 14.1 T and 20 T, enabling the different local bonding environments of borate anions in the G4 network and in solution to be distinguished and identified. A combined 11B MAS NMR and DFT modelling study reveals that the GB cis diesters promote the gelation of G-quartets.Multinuclear MAS NMR is employed to characterize molecular-level interactions between biologically relevant small heterocyclic molecules and G4·M+ (M+ = K+, Na+) hydrogels. While the uptake of up to 0.3 equivalent of cationic methylene blue (i.e., 0.3 equiv. of MB per equiv. of G) yields stable and intact G4·K+ borate gel, the MB concentrations over 0.3 equiv. triggered a gel-to-sol transition caused by the disruption of G4-fibrils that make the gel. The sol–gel transition of MB loaded G4·K+ gel, monitored by ex situ1H and 11B MAS NMR titration experiments, revealed changes in the isotropic 1H and 11B chemical shifts. These changes are corroborated by the vial inversions, which confirm the gel-to-sol transition. The uptake processes of different heterocyclic compounds into G4·K+ and G4·Na+ borate gels were studied and compared. Adenine is readily soluble in G4·Na+ borate gel with adenine concentrations of up to 72 mM, i.e., one equivalent per G (72 mM), although smaller aggregates form when the adenine concentrations exceed 64 mM. Cytosine solubility is enhanced to a more modest 32 mM by the borate gel. The solubility of a pyrimidine, 1-methylthymine, was however significantly improved from 50 mM to at least 70 mM in G4·Na+ borate gel. The uptake process was monitored by solution-state 1H and gel-state 11B and 23Na NMR titration experiments. The 1H NMR results suggest that these heterocyclic nucleobases are incorporated into the solution phase, which are corroborated by the 23Na MAS NMR results.
To summarize, G4-quartet borate gels provide a useful material into which small molecule drugs such as cationic dyes and heterocyclic compounds can be taken up, which we are exploring further as potential delivery agents. Gel-state MAS NMR spectroscopy provides valuable atomic-level insight into the sol–gel compositions and structures, and differentiate local arrangements of order and disorder of as-synthesized gels as well as guest molecules absorbed into the functional gels, which can be correlated and complemented by morphological features and long-range order characterized by conventional bulk techniques such as X-ray scattering and electron microscopy. Although multinuclear 1D MAS NMR is a powerful probe to characterize the GB gels and specific intermolecular interactions between guest molecules and gelator molecules, it can be envisaged that 2D 1H–1H and 1H–X (X = 13C, 11B, 23Na, etc.) correlation NMR spectroscopy in conjunction with other complementary techniques and sophisticated modelling approaches may further help in the determination of packing interactions and the three-dimensional structure of the gel network. However, the complex compositions and structures of GB hydrogels present challenges given the sensitivity and resolution of such MAS NMR techniques, for example, when small quantities of structurally similar guest molecules are added to the hydrogels (i.e., guest molecule concentration lower than 0.1 equiv. of gelator molecule). In this respect, studies of the lowest limit of detection of small molecule guests in GB hydrogels using multinuclear MAS NMR techniques are yet to be examined and compared, but could be a focus of future investigations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding from EPSRC (EP/K003674/1). The UK 850 MHz solid-state NMR Facility used in this research was funded by EPSRC and BBSRC, as well as the University of Warwick including via part funding through Birmingham Science City Advanced Materials Projects 1 and 2 supported by Advantage West Midlands (AWM) and the European Regional Development Fund (ERDF); the Facility Manager, Dr Dinu Iuga, is thanked for experimental assistance. B. P. T. thanks Bruker and the University of Warwick for PhD funding as part of the Warwick Centre for Doctoral Training in Analytical Science. T. S. R. thanks the Warwick Undergraduate Research Scholarship Scheme for funding. S. M. K. and J. Z. thank the Study Abroad Program 2014, NTU Singapore and Xiamen University, China respectively. J. T. D. thanks the Office of Basic Energy Sciences, U.S. Dept. of Energy (DEFG02-98ER14888) for financial support and the NSF (NSF-1726058) for NMR funding. We thank Prof. Sharon E. Ashbrook for insightful discussions on the calculation of quadrupolar parameters. The experimental data for this study are provided as a supporting dataset from WRAP, the Warwick Research Archive Portal at http://wrap.warwick.ac.uk/141042/.References
- B. O. Okesola and D. K. Smith, Chem. Soc. Rev., 2016, 45, 4226–4251 Search PubMed.
- J.-F. Lutz, J.-M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 16024 Search PubMed.
- R. Freeman, M. Han, Z. Álvarez, J. A. Lewis, J. R. Wester, N. Stephanopoulos, M. T. McClendon, C. Lynsky, J. M. Godbe, H. Sangji, E. Luijten and S. I. Stupp, Science, 2018, 362, 808 Search PubMed.
- O. P. Lee, A. T. Yiu, P. M. Beaujuge, C. H. Woo, T. W. Holcombe, J. E. Millstone, J. D. Douglas, M. S. Chen and J. M. J. Fréchet, Adv. Mater., 2011, 23, 5359–5363 Search PubMed.
- A. M. Vibhute, V. Muvvala and K. M. Sureshan, Angew. Chem., Int. Ed., 2016, 55, 7782–7785 Search PubMed.
- R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 Search PubMed.
- X. Shen, J. L. Shamshina, P. Berton, G. Gurau and R. D. Rogers, Green Chem., 2016, 18, 53–75 Search PubMed.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 Search PubMed.
- G. M. Peters and J. T. Davis, Chem. Soc. Rev., 2016, 45, 3188–3206 Search PubMed.
- Q. Tang, T. N. Plank, T. Zhu, H. Yu, Z. Ge, Q. Li, L. Li, J. T. Davis and H. Pei, ACS Appl. Mater. Interfaces, 2019, 11, 19743–19750 Search PubMed.
- M. Li, H. Wang, J. Hu, J. Hu, S. Zhang, Z. Yang, Y. Li and Y. Cheng, Chem. Mater., 2019, 31, 7678–7685 Search PubMed.
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 16071 Search PubMed.
- L. Stefan and D. Monchaud, Nat. Rev. Chem., 2019, 3, 650–668 Search PubMed.
- Y. P. Kumar, R. N. Das, O. M. Schütte, C. Steinem and J. Dash, Nat. Protoc., 2016, 11, 1039 Search PubMed.
- J. T. Davis and G. P. Spada, Chem. Soc. Rev., 2007, 36, 296–313 Search PubMed.
- D. González-Rodríguez, J. L. J. van Dongen, M. Lutz, A. L. Spek, A. P. H. J. Schenning and E. W. Meijer, Nat. Chem., 2009, 1, 151–155 Search PubMed.
- Y. Li, Y. Liu, R. Ma, Y. Xu, Y. Zhang, B. Li, Y. An and L. Shi, ACS Appl. Mater. Interfaces, 2017, 9, 13056–13067 Search PubMed.
- S. Pieraccini, M. Campitiello, F. Carducci, J. T. Davis, P. Mariani and S. Masiero, Org. Biomol. Chem., 2019, 17, 2759–2769 Search PubMed.
- R. Zhong, Q. Tang, S. Wang, H. Zhang, F. Zhang, M. Xiao, T. Man, X. Qu, L. Li, W. Zhang and H. Pei, Adv. Mater., 2018, 30, 1706887 Search PubMed.
- J. Hu, H. Wang, Q. Hu and Y. Cheng, Mater. Chem. Front., 2019, 3, 1323–1327 Search PubMed.
- G. M. Peters, L. P. Skala, T. N. Plank, B. J. Hyman, G. N. Manjunatha Reddy, A. Marsh, S. P. Brown and J. T. Davis, J. Am. Chem. Soc., 2014, 136, 12596–12599 Search PubMed.
- G. M. Peters, L. P. Skala, T. N. Plank, H. Oh, G. N. Manjunatha Reddy, A. Marsh, S. P. Brown, S. R. Raghavan and J. T. Davis, J. Am. Chem. Soc., 2015, 137, 5819–5827 Search PubMed.
- A. Rotaru, G. Pricope, T. N. Plank, L. Clima, E. L. Ursu, M. Pinteala, J. T. Davis and M. Barboiu, Chem. Commun., 2017, 53, 12668–12671 Search PubMed.
- T. N. Plank, L. P. Skala and J. T. Davis, Chem. Commun., 2017, 53, 6235–6238 Search PubMed.
- Molecular Gels: Materials with Self-Assembled Fibrillar Networks, ed. R. G. Weiss and P. Terech, Springer, Dordrecht, 2006 Search PubMed.
- Nonappa, M. Lahtinen, B. Behera, E. Kolehmainen and U. Maitra, Soft Matter, 2010, 6, 1748–1757 Search PubMed.
- V. Noponen, Nonappa, M. Lahtinen, A. Valkonen, H. Salo, E. Kolehmainen and E. Sievänen, Soft Matter, 2010, 6, 3789–3796 Search PubMed.
- Cellulose-Based Superabsorbent Hydrogels, ed. M. Azeera, S. Vaidevi, K. Ruckmani and M. I. H. Mondal, Springer International Publishing, Cham, 2018, pp. 1–24 Search PubMed.
- Nonappa and E. Kolehmainen, Soft Matter, 2016, 12, 6015–6026 Search PubMed.
- Nonappa and E. Kolehmainen, in NMR and MRI of Gels, ed. Y. De Geene, The Royal Society of Chemistry, 2020, pp. 200–227 Search PubMed.
- Y. E. Shapiro, in NMR and MRI of Gels, ed. Y. De Geene, The Royal Society of Chemistry, 2020, pp. 45–88 Search PubMed.
- J. Brus, W. Albrecht, F. Lehmann, J. Geier, J. Czernek, M. Urbanova, L. Kobera and A. Jegorov, Mol. Pharmaceutics, 2017, 14, 2070–2078 Search PubMed.
- B. Escuder, M. Llusar and J. F. Miravet, J. Org. Chem., 2006, 71, 7747–7752 Search PubMed.
- M. R. Hansen, R. Graf and H. W. Spiess, Chem. Rev., 2016, 116, 1272–1308 Search PubMed.
- A. Marchetti, J. Chen, Z. Pang, S. Li, D. Ling, F. Deng and X. Kong, Adv. Mater., 2017, 29, 1605895 Search PubMed.
- M. Siefrid, G. N. M. Reddy, B. F. Chmelka and G. C. Bazan, Nat. Rev. Mater., 2020 DOI:10.1038/s41578-020-00232-5.
- G. N. M. Reddy, A. Huqi, D. Iuga, S. Sakurai, A. Marsh, J. T. Davis, S. Masiero and S. P. Brown, Chem. – Eur. J, 2017, 23, 2235 Search PubMed.
- A. L. Webber, S. Masiero, S. Pieraccini, J. C. Burley, A. S. Tatton, D. Iuga, T. N. Pham, G. P. Spada and S. P. Brown, J. Am. Chem. Soc., 2011, 133, 19777–19795 Search PubMed.
- G. N. M. Reddy, M. Malon, A. Marsh, Y. Nishiyama and S. P. Brown, Anal. Chem., 2016, 88, 11412–11419 Search PubMed.
- C. E. Hughes, G. N. M. Reddy, S. Masiero, S. P. Brown, P. A. Williams and K. D. M. Harris, Chem. Sci., 2017, 8, 3971–3979 Search PubMed.
- A. Khitrin and B. M. Fung, J. Chem. Phys., 2000, 112, 2392 Search PubMed.
- J. W. E. Weiss and D. L. Bryce, J. Phys. Chem. A, 2010, 114, 5119–5131 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision A.02., Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- R. K. Harris, P. Hodgkinson, C. J. Pickard, J. R. Yates and V. Zorin, Magn. Reson. Chem., 2007, 45, S174–S186 Search PubMed.
- G. N. M. Reddy, D. S. Cook, D. Iuga, R. I. Walton, A. Marsh and S. P. Brown, Solid State Nucl. Magn. Reson., 2015, 65, 41–48 Search PubMed.
- G. N. M. Reddy, A. Marsh, J. T. Davis, S. Masiero and S. P. Brown, Cryst. Growth Des., 2015, 15, 5945–5954 Search PubMed.
- G. Wu, Z. Gan, I. C. M. Kwan, J. C. Fettinger and J. T. Davis, J. Am. Chem. Soc., 2011, 133, 19570–19573 Search PubMed.
- M. Hušák, A. Jegorov, J. Rohlíček, A. Fitch, J. Czernek, L. Kobera and J. Brus, Cryst. Growth Des., 2018, 18, 3616–3625 Search PubMed.
- L. Kobera, J. Czernek, M. Streckova, M. Urbanova, S. Abbrent and J. Brus, Macromolecules, 2015, 48, 4874–4881 Search PubMed.
- G. N. M. Reddy, J. A. Gerbec, F. Shimizu and B. F. Chmelka, Langmuir, 2019, 35, 15661–15673 Search PubMed.
- A. Samoson, Chem. Phys. Lett., 1985, 119, 29–32 Search PubMed.
- S. P. Brown and S. Wimperis, J. Magn. Reson., 1997, 128, 42–61 Search PubMed.
- S. P. Brown, S. E. Ashbrook and S. Wimperis, J. Phys. Chem. B, 1999, 103, 812–817 Search PubMed.
- F. Rodríguez-Llansola, J. F. Miravet and B. Escuder, Chem. Commun., 2011, 47, 4706–4708 Search PubMed.
- T. Cao, F.-T. Zhang, L.-Y. Cai, Y.-L. Zhou, N. J. Buurma and X.-X. Zhang, Analyst, 2017, 142, 987–993 Search PubMed.
- D. S.-H. Chan, H. Yang, M. H.-T. Kwan, Z. Cheng, P. Lee, L.-P. Bai, Z.-H. Jiang, C.-Y. Wong, W.-F. Fong, C.-H. Leung and D.-L. Ma, Biochimie, 2011, 93, 1055–1064 Search PubMed.
- F.-T. Zhang, J. Nie, D.-W. Zhang, J.-T. Chen, Y.-L. Zhou and X.-X. Zhang, Anal. Chem., 2014, 86, 9489–9495 Search PubMed.
- G. M. Peters, L. P. Skala and J. T. Davis, J. Am. Chem. Soc., 2016, 138, 134–139 Search PubMed.
- A. Mills, D. Hazafy, J. Parkinson, T. Tuttle and M. G. Hutchings, Dyes Pigm., 2011, 88, 149–155 Search PubMed.
- M. Wallace, J. A. Iggo and D. J. Adams, Soft Matter, 2015, 11, 7739–7747 Search PubMed.
- M. Wallace, A. Z. Cardoso, W. J. Frith, J. A. Iggo and D. J. Adams, Chem. – Eur. J., 2014, 20, 16484–16487 Search PubMed.
- T. T. Herskovits and J. P. Harrington, Biochemistry, 1972, 11, 4800–4811 Search PubMed.
- A. Hirano, H. Tokunaga and M. Tokunaga, Arch. Biochem. Biophys., 2010, 497, 90–96 Search PubMed.
- J. Krzaczkowska, J. Gierszewski and G. Ślósarek, J. Solution Chem., 2004, 33, 395–406 Search PubMed.
- M. G. Sacerdote and J. W. Szostak, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 6004 Search PubMed.
- M. Maresca, A. Derghal, C. Carravagna, S. Dudin and J. Fantini, Phys. Chem. Chem. Phys., 2008, 10, 2792–2800 Search PubMed.
- M. Nishio, Y. Umezawa, J. Fantini and M. S. Weiss, Phys. Chem. Chem. Phys., 2014, 16, 12648–12683 Search PubMed.
- P. O. P. Ts'o, I. S. Melvin and A. C. Olson, J. Am. Chem. Soc., 1963, 85, 1289–1296 Search PubMed.
- P. O. Ts'o, Ann. N. Y. Acad. Sci., 1969, 153, 785–804 Search PubMed.
- G. M. Peters and J. T. Davis, Supramol. Chem., 2014, 26, 286–295 Search PubMed.
- R. K. Castellano, V. Gramlich and F. Diederich, Chemistry, 2002, 8, 118–129 Search PubMed.
- X. Tian-xiang and B. D. Anderson, Int. J. Pharm., 1990, 59, 45–55 Search PubMed.
- K. P. Madhusudanan, S. B. Katti and A. K. Dwivedi, J. Mass Spectrom., 1998, 33, 1017–1022 Search PubMed.
- H. Kuang, S. Wu, Z. Xie, F. Meng, X. Jing and Y. Huang, Biomacromolecules, 2012, 13, 3004–3012 Search PubMed.
- B. D. Anderson, M. B. Wygant, X. Tian-Xiang, W. A. Waugh and V. J. Stella, Int. J. Pharm., 1988, 45, 27–37 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: MAS NMR spectra of alkali metal cations, determination of 11B isotropic chemical shifts and quadrupolar parameters, DFT calculated structures of guanosine borate mono- and diesters, tables of nucleobase solubility assays, and photographs of inverted vials containing guanosine borate gels before and after the incorporation of small molecules. See DOI: 10.1039/d0ma00475h |
| ‡ Present address: Department of Chemistry, University of Lille, CNRS, Centrale Lille Institut, Univ. Artois, UMR 8181-UCCS-Unité de Catalyse et Chimie du Solide, F-59000, Villeneuve d’Ascq, France. |
| This journal is © The Royal Society of Chemistry 2020 |