
Insights into isothiourea-catalyzed asymmetric [3 + 3] annulation of α,β-unsaturated aryl esters with 2-acylbenzazoles: mechanism, origin of stereoselectivity and switchable chemoselectivity†
Congcong
Wang
 a,
Shi-Jun
Li
b,
Qiao-Chu
Zhang
b,
Donghui
Wei
*b and
Lina
Ding
*a
a,
Shi-Jun
Li
b,
Qiao-Chu
Zhang
b,
Donghui
Wei
*b and
Lina
Ding
*a
aCollaborative Innovation Center of New Drug Research and Safety Evaluation, Henan Province, Key Laboratory of Technology of Drug Preparation (Zhengzhou University), Ministry of Education of China, Key Laboratory of Henan Province for Drug Quality and Evaluation, School of Pharmaceutical Sciences, Zhengzhou University, Zhengzhou 450001, P. R. China. E-mail: dinglina123@126.com
bCollege of Chemistry, and Institute of Green Catalysis, Zhengzhou University, 100 Science Avenue, Zhengzhou 450001, Henan, P. R. China. E-mail: donghuiwei@zzu.edu.cn
First published on 29th April 2020
Abstract
Recently, isothiourea-catalyzed asymmetric [3 + 3] annulation reactions of α,β-unsaturated aryl esters with 2-acylbenzothiazole (or 2-acylbenzoxazole) were reported with switchable chemoselectivity to form either dihydropyridone or dihydropyranone, but predicting the origin of chemoselectivity and stereoselectivity is still challenging in these kinds of reactions. Herein, density functional theory (DFT) was used to study the general mechanism and explore the origin of stereoselectivity and chemoselectivity in these reactions. The calculated results show that three stages including adsorption, [3 + 3] annulation and dissociation are involved in the reaction, and the C–C bond formation involved in [3 + 3] annulation is a key step that determines both chemoselectivity and stereoselectivity. The origin of stereoselectivity was further investigated by analysis of distortion and non-covalent interactions (NCI), and the C–H⋯O interaction between the chiral substituents of the catalyst and the carbonyl oxygen on 2-acylbenzazoles contributes greatly to the stereoselectivity. In addition, the switchable chemoselectivity associated with the competitive [3 + 3] cyclizations for formation of N- and O-heterocyclic compounds can be predicted by using local nucleophilic Parr function (Pk−) and nucleophilic atom energy (Ea−) analysis. This work provides guidance for the rational design of potential catalysts for such highly stereoselective reactions with switchable chemoselectivities.
Introduction
Functionalized six-membered ring compounds such as pyridone and pyranone are important building blocks for synthesis of pharmaceutical drugs and natural products.1 Catalytic stereoselective access to these kinds of ring compounds has thus attracted considerable research interest. Various kinds of efficient pathways were reported to synthesize these cyclic compounds, such as the common [4 + 2] cycloaddition and the emerging [3 + 3] cyclization reaction. For these synthesis processes, the use of a suitable catalyst will result in a convenient reaction associated with mild conditions and high selectivity.2 Noteworthily, the employment of Lewis base organocatalysts,3 including cinchona alkaloid derivatives,4N-heterocyclic carbenes,5 phosphine,6 and isothiourea,7 was found to be an ideal choice for promoting various kinds of asymmetric syntheses of those N- and O-heterocyclic compounds.8More recently, the use of chiral isothioureas9 has become more and more popular in enantioselective synthesis of six-membered ring compounds. Notably, Smith et al. widely extended their applications to build many complex compounds with high levels of regio-, chemo- and stereo-control capabilities. Besides isothiourea-catalyzed [4 + 2] cyclization reactions for formation of various six-membered ring compounds,10 Smith's group reported the isothiourea-catalyzed [3 + 3] cyclization reaction of 2-acylbenzoxazoles with homo-anhydrides to form dihydropyranone and dihydropyridinone (Scheme 1a).11,12 Moreover, Smith et al. reported the isothiourea-catalyzed formal [3 + 3] cyclization reaction of 2-acylbenzoxazole with α,β-unsaturated aryl esters to generate dihydropyranone and dihydropyridinone, which has good stereoselectivity and chemoselectivity (Scheme 1b).13 However, the origin of the chemoselectivity and stereoselectivity in these kinds of isothiourea-catalyzed [3 + 3] cyclization reactions remains unclear so far. As can be seen in Scheme 1a, the product is exclusively dihydropyranone when the X atom is an oxygen atom, but the main product would be dihydropyridinone when the X atom is a sulfur atom, and the yield ratio of dihydropyridinone![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dihydropyranone is 85:15 in the experiment.11 Similarly, as shown in Scheme 1b, similar products would be obtained and chemoselectivity can be switchable by using different substrates with X = S or O atoms.
:dihydropyranone is 85:15 in the experiment.11 Similarly, as shown in Scheme 1b, similar products would be obtained and chemoselectivity can be switchable by using different substrates with X = S or O atoms.
 | ||
| Scheme 1 Isothiourea-catalysed [3 + 3] annulation reaction of (a) homoanhydrides with 2-acylbenzazole derivatives and (b) α,β-unsaturated aryl esters with 2-acylbenzazole. | ||
Although there are a lot of valuable insights in these excellent experiments, some issues still need to be further studied and explored in theory: (1) how is an acyl ammonium precursor converted from an α,β-unsaturated aryl ester? (2) What is the role of the leaving group ArO−? (3) How does the deprotonation proceed to produce the corresponding enolate intermediate? (4) How do protonic media assist [1,3]-proton transfer? (5) What is the origin of high stereoselectivity? (6) What role does the isothiourea catalyst play? To the best of our knowledge, few theoretical studies on the isothiourea-catalyzed asymmetric [3 + 3] annulation of α,β-unsaturated aryl esters (or homoanhydrides) with 2-acylbenzazoles have been reported so far,11 and the general principle on the origin of the switchable chemoselectivities and selectivities should be highly desirable to explore them systematically. These questions and our continuous interest in organocatalysis14–16 encourage us to perform this computational study, in which the reactions of 3-trifluoromethyl-1-(2,4,6-trichlorophenoxy)-2-acetyl (denoted as R1, Scheme 1b) with 2-benzoxazole-1-acetophenone (denoted as R2O/S with X = O/S, Scheme 1b) leading to dihydropyridone and dihydropyranone in the presence of isothiourea (denoted as Cat) were selected as the research object. All of the calculations were conducted at the M06-2X-D3/6-311++G(2df, 2pd)/IEF-PCMTHF//M06-2X17,18/6-31G(d, p)/IEF-PCMTHF19,20 level in the Gaussian 09 program,21 and more computational details are provided in the ESI.† This theoretical study aims to explore the exact map for general mechanisms of such reactions, and thus provides valuable clues for guiding experimental design.
Results and discussion
Mechanism for the [3 + 3] cyclization
As shown in Scheme 2, we suggested and studied the possible mechanisms of [3 + 3] cyclization of an α,β-unsaturated aryl ester with 2-acylbenzothiazole catalyzed by isothiourea, and Fig. 1 shows the relative free energy profiles of the entire catalytic reaction, in which the free energy of R1 + Cat was set to 0.0 kcal mol−1. The entire catalytic cycle comprises four reaction steps, and we would elaborate on the specific mechanism and free energy change of each step as follows. | ||
| Scheme 2 The competitive reaction mechanisms for the isothiourea-catalyzed [3 + 3] annulation of an α,β-unsaturated aryl ester with 2-acylbenzothiazole. | ||
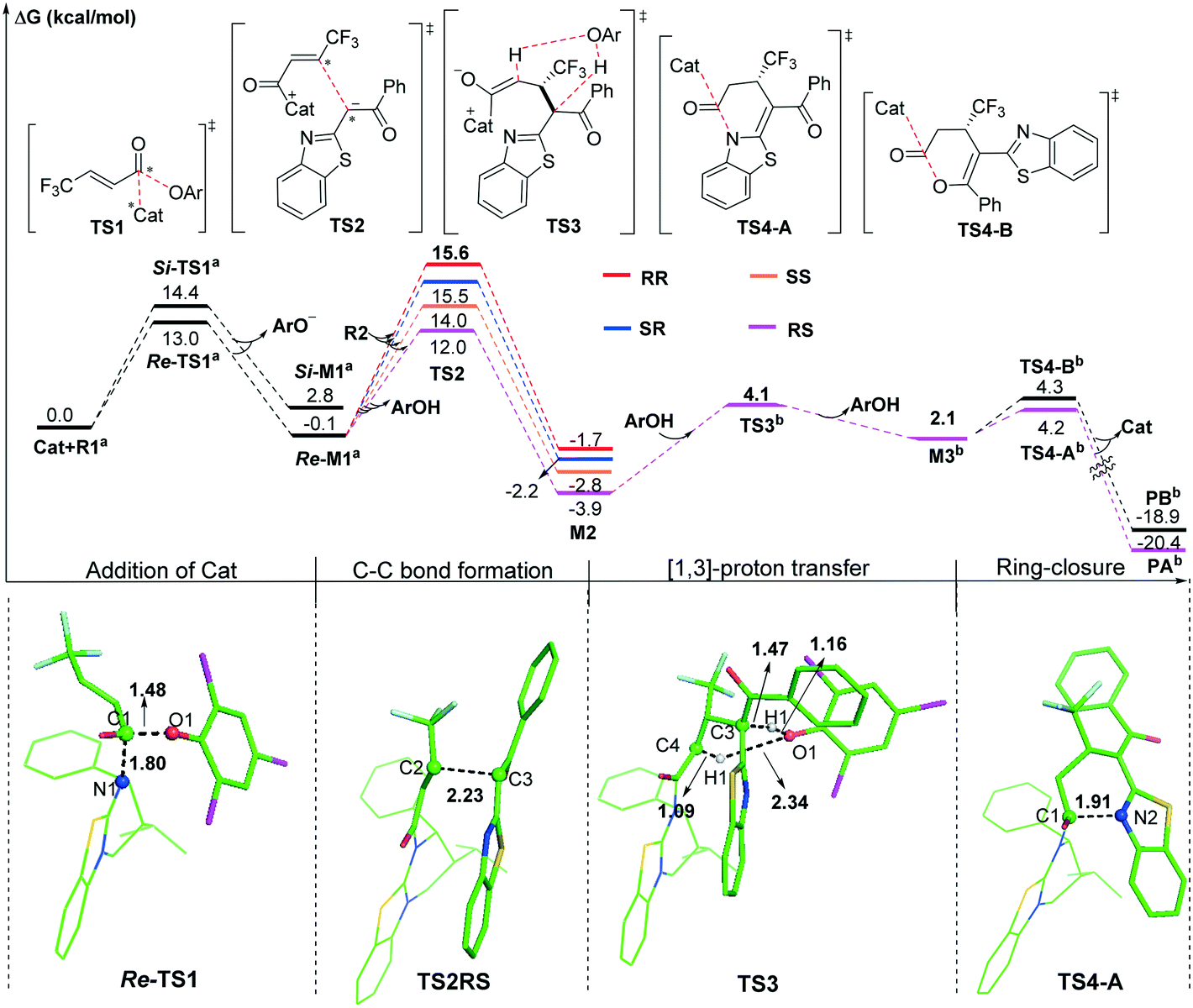 | ||
| Fig. 1 The relative Gibbs free energy profiles of the isothiourea-catalyzed [3 + 3] annulation reaction. (the superscript “a” represents adding the free energy of pre-R2, the superscript “b” represents the S configuration, and distances are in angstroms). | ||
The first step is initiated with nucleophilic attack on the carbonyl carbon of reactant R1 from either the Si or Re face of the carbonyl group of R1 by the imine nitrogen atom of Catvia transition state Si- or Re-TS1 to generate intermediate Si- or Re-M1 (Fig. S1 of the ESI†). For convenience, the unchanged chirality of the catalyst has not been included in the names of the following stationary points including the diastereomer intermediates and transition states. It should be noted that the chirality of the catalyst was not mentioned in the compound names. The Si- or Re-before the compound names corresponds to the Si- or Re-face of reactant R1, and the same signs for the associated Si- or Re-M1 were retained. As seen from Fig. 1, the energy barrier of the pathway associated with attacking the Re surface of the carbonyl group of reactant R1 through transition state Re-TS1 (13.0 kcal mol−1) is lower than that viaSi-TS1 (14.4 kcal mol−1). In addition, the energy of intermediate Re-M1 (−0.1 kcal mol−1) is also lower than that of Si-M1 (2.8 kcal mol−1). Therefore, the pathway associated with Re-M1 should be more energetically favourable in both kinetics and thermodynamics, so we only considered this pathway in the following steps.
The second step is Michael addition to M1 by nucleophile R2, which is generated via the α-C–H deprotonation process of reactant precursor pre-R2 in the presence of ArO− through transition state TS1′ (7.4 kcal mol−1, Fig. S2 of the ESI†). It should be noted that a molecule of ArOH can be generated to assist the following [1,3]-proton transfer. In this step, α-C of R2 attacks the activated α-C of intermediate M1 to form intermediate M2via transition state TS2. As shown in Scheme 1, there are two nucleophilic sites on R2, namely α-C and the oxygen atom of the carbonyl group. In order to explore which atom is easier to attack to form intermediate M1, local nucleophilic (Pk−) and electrophilic (Pk+) Parr function analysis22 was performed for R2, and the calculated results indicated that nucleophilicity of α-C is much greater than that of the carbonyl oxygen atom (Table S1 of the ESI†). Furthermore, we analysed the global reaction index (GRI23) to understand the actual role of the isothiourea catalyst. As shown in Table S2 of the ESI,† the ArO− can be nucleophilically substituted by isothiourea, which makes the substrate more electrophilic to facilitate the reaction with nucleophile R2.
As shown in Fig. 2, four different reaction modes would lead to the corresponding diastereoisomer transition states, i.e.TS2SS, TS2RS, TS2RR and TS2SR. According to Fig. 1, the energy barriers via transition states TS2SS, TS2RS, TS2RR and TS2SR are 14.0, 12.0, 15.6 and 15.5 kcal mol−1, and the relative energies of the corresponding intermediates M2SS, M2RS, M2RR and M2SR are −2.8, −3.9, −1.7 and −2.2 kcal mol−1, respectively. These data indicate that the reaction pathway for generating intermediate M2RSviaTS2RS requires much lower energy than the other three pathways, so the reaction pattern for generating the RS isomer is more advantageous. Noteworthily, we considered and studied multiple conformations for transition state TS2 to make sure that the selected conformations of the diastereoisomers are the lowest energy conformations, and more details can be found in Table S3 in the ESI.†
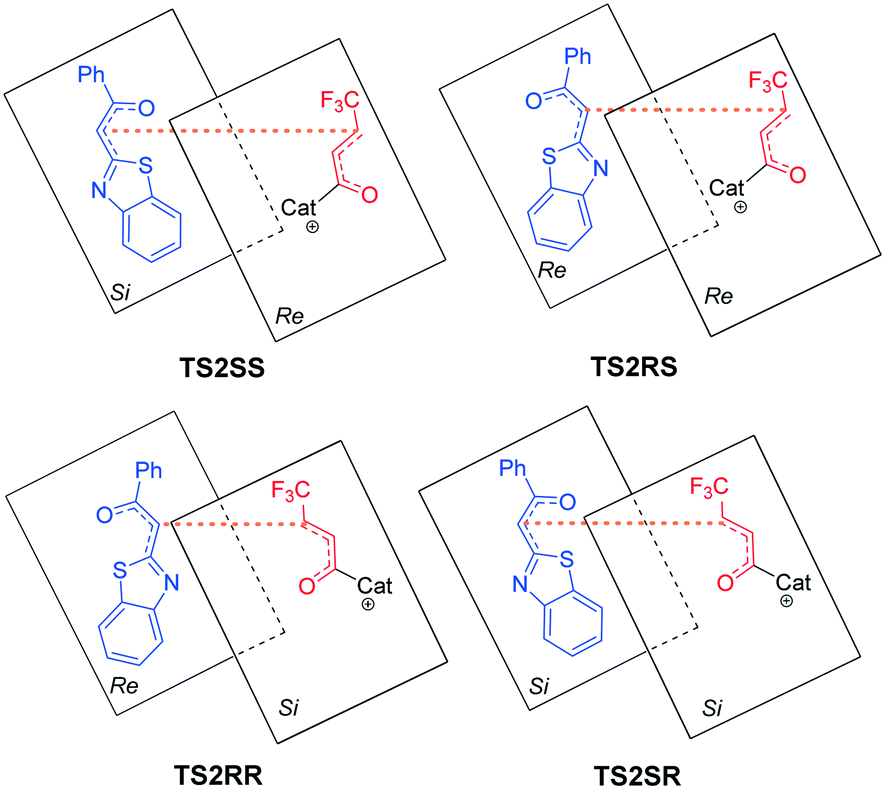 | ||
| Fig. 2 Stereochemistry involved in transition states TS2SS, TS2RS, TS2RR and TS2SR. | ||
In third step, two possible pathways, including in situ generated ArOH and i-Pr2NEt·H+ assisted [1,3]-proton transfer via transition states TS3 (with an energy barrier of 2.5 kcal mol−1, Fig. 1) and TS3′ (with an energy barrier of 23.2 kcal mol−1, Fig. S3 of the ESI†), were considered and investigated. Obviously, the ArOH assisted [1,3]-proton transfer has a much lower energy barrier and should be the main pathway in the reaction.
The final step is ring closure coupled with dissociation of Cat for formation of either product PA or PBvia transition state TS4-A or TS4-B (Scheme 2), separately. As seen from Fig. 1, the energy barrier via transition state TS4-A (4.2 kcal mol−1) is slightly lower than that via transition state TS4-B (4.3 kcal mol−1), and the relative energy of product PA is also lower than that of product PB, which is in agreement with the product ratio (PA:PB = 54:42) obtained in the experiment. In contrast, for X = O shown in Fig. S4 of the ESI,† the energy barrier via transition state TS4-B′ (4.5 kcal mol−1) is much lower than that via transition state TS4-A′ (11.3 kcal mol−1), the relative energy of product PB′ (−12.8 kcal mol−1) is higher than that of product PA′ (−19.7 kcal mol−1), and PA′ should be the kinetically-unfavoured but thermodynamically-favoured product, which is consistent with the main product being changed to PA′ under heating conditions. The calculated results are consistent with the observed switchable chemoselectivity in the experiment, and this interesting phenomenon encouraged us to further explore the origin of chemoselectivity as follows.24
Origin of stereoselectivity
According to the energy barrier diagram (Fig. 1) and above discussed in step 2, the Michael addition was identified as the stereoselectivity-determining step, and the reaction pathway associated with the RS configuration has the lowest energy. The energy difference between TS2RS and TS2SR is 3.5 kcal mol−1, which corresponds to 99% ee in theory, and this is close to the 94% ee reported in experiment. In order to explore the origin of stereoselectivity, the distortion/interaction energy25 and non-covalent interaction (NCI) analyses were performed as follows.Based on Houk's definition,25,26 the energy difference between the reaction partners (M1 and R2) and the corresponding segments separated from each transition state (ΔE‡dist_M1/ΔE‡dist_R2) is the distortion energy, and the total distortion energy (ΔE‡dist_total) is the sum of ΔE‡dist_M1 and ΔE‡dist_R2. The weak interaction ΔE‡int is the difference between ΔE‡ and ΔE‡dist_total. As summarized in Table 1, although the transition state TS2RS has the largest distortion energy (15.45 kcal mol−1), it also has the largest interaction energy (−38.63 kcal mol−1), indicating that the weak interactions contribute significantly for its favourability. Therefore, we further performed non-covalent interaction (NCI) analysis on transition states TS2SS, TS2RS, TS2RR and TS2SR to discover those interactions. Several kinds of interactions have been identified by using NCI analysis, i.e. lp⋯π interaction, C–H⋯O interaction, C–H⋯S interaction, and C–H⋯π interaction. Noteworthily, the lone pair⋯π (lp⋯π) interaction is similar to the anion⋯π interaction, namely the non-covalent bonding association between a neutral electron-rich atom and an electron-poor π ring.27 As depicted in Fig. 3, there are one lp⋯π interaction (2.97 Å) between carbonyl oxygen and the five-membered ring and one lp⋯π interaction (3.31 Å) between the sulfur atom and the six-membered ring in TS2SS; one lp⋯π interaction (2.94 Å) between the carbonyl oxygen and the five-membered ring, one C–H⋯S interaction (2.91 Å) and one strong C–H⋯O interaction (2.34 Å) in TS2RS; one lp⋯π interaction (2.98 Å) between the carbonyl oxygen and the five-membered ring and one lp⋯π interaction (3.17 Å) between the sulfur atom and the six-membered ring in TS2RR, one C–H⋯π interaction (2.45 Å), one C–H⋯S interaction (2.99 Å) and one C–H⋯O interaction (2.59 Å) in TS2SR. Therefore, the stronger C–H⋯O interaction between R2 and the chiral center of the catalyst in transition state TS2RS should be the key to determine the stereoselectivity of the reaction.
| SP | ΔΔG‡ | ΔE‡dist_R2 | ΔE‡dist_M1 | ΔE‡dist_total | ΔE‡int |
|---|---|---|---|---|---|
| TS2SS | 1.93 | 4.32 | 8.67 | 12.99 | −33.20 |
| TS2RS | 0.00 | 4.69 | 11.01 | 15.70 | −37.27 |
| TS2RR | 3.52 | 2.49 | 6.92 | 9.41 | −28.38 |
| TS2SR | 3.48 | 1.68 | 7.89 | 9.57 | −26.74 |
 | ||
| Fig. 3 Stereochemistry involved in transition states TS2SS, TS2RS, TS2RR and TS2SR. | ||
Prediction of chemoselectivity
Inspired by Smith's excellent contribution,11,28–30 we think that it should be interesting to explore the origin of the switchable chemoselectivity. Noteworthily, the number of 1,5-S⋯O interactions could be used for explaining the chemoselectivity involved in this kind of reaction depicted in Fig. 4a. However, as shown in Fig. 4b, the two chemoselective transition states have only one 1,5-S⋯O interaction, so the switchable chemoselectivity cannot be well explained so far.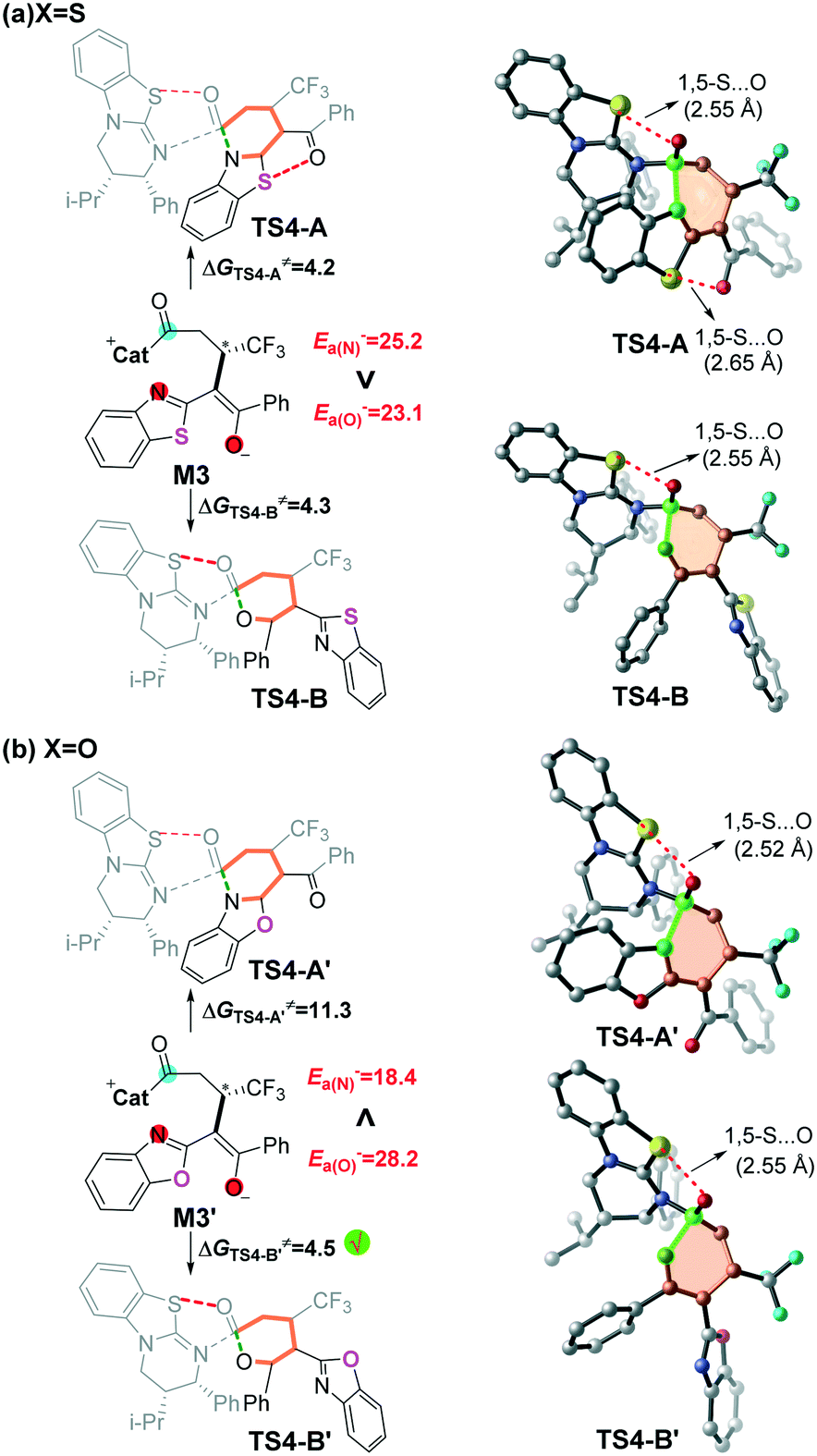 | ||
| Fig. 4 Nucleophilic atom energy (Ea−, kcal mol−1) of N and O atoms in intermediates M3 and M3′. | ||
In order to predict the chemoselectivity associated with [3 + 3] cyclization products PA or PB by using a general principle, we determined the global nucleophilic index (N (ref. 23d and 31 = EHOMO (M3, M3′) − EHOMO (TCNE)) of intermediate M3 or M3′ and local nucleophilicity (Pk−), and performed nucleophilic atom energy (Ea− = Pk− × N) analyses on the nucleophilic nitrogen and oxygen atoms in intermediate M3 or M3′ (Table S4 of the ESI†). The computed results (Fig. 4) show that the nucleophilicity of the nitrogen atom (Ea− = 25.2 kcal mol−1) is slightly higher than that of the oxygen atom (Ea− = 23.1 kcal mol−1) in intermediate M3. While the nucleophilicity of the nitrogen atom (Ea− = 18.4 kcal mol−1) is significantly lower than that of the oxygen atom (Ea− = 28.2 kcal mol−1) in intermediate M3′. The higher nucleophilic atom energy represents the stronger nucleophilic activity, and thus leads to a lower energy barrier. Therefore, we can successfully predict the switchable chemoselectivity by performing single-point energy calculations according to the above two local reactivity indexes.
Conclusions
In this work, density functional theory (DFT) calculations were carried out to study the possible reaction mechanisms and origin of selectivities of the reaction between α,β-unsaturated aryl esters and 2-acylbenzazole (X = O/S) to generate dihydropyridone and dihydropyranone under isothiourea organocatalysis. The calculated results reveal that the reaction includes four reaction steps, including complexation of α,β-unsaturated aryl esters using the isothiourea catalyst, Michael addition, ArOH assisted [1,3]-proton transfer, and the ring-closure process coupled with dissociation of the catalyst. The Michael addition step (i.e. C–C bond formation) was identified as the stereoselectivity-determining step. Analysis of the distortion/interaction and non-covalent interaction (NCI) shows that the C–H⋯O interaction formed between the two chiral centers on the catalyst with the carbonyl oxygen on 2-acylbenzothiazole has a significant influence on the stereoselectivity. In addition, analysis of a newly suggested local activity index (i.e. nucleophilic atom energy Ea−) and nucleophilic Parr function (Pk−) was performed to predict the origin of the switchable chemoselectivity in these kinds of reactions. Therefore, this work should be helpful in understanding the detailed reaction mechanism, role of the isothiourea organocatalyst, and origin of both stereoselectivity and chemoselectivity, and thus provides useful clues for rational design of more efficient organocatalyzed [3 + 3] cyclization reactions with high stereoselectivity and special chemoselectivity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the Science and Technology Innovation Talents of Henan Provincial Education Department (19IRTSTHN001) and the National Natural Science Foundation of China (No. 21773214, 21303167, and 21403200).Notes and references
- C. G. Hui, F. Chen, F. Pu and J. Xu, Nat. Rev. Chem., 2019, 3, 85–107 CrossRef.
- J. P. Page and T. V. RajanBabu, J. Am. Chem. Soc., 2012, 134, 6556–6559 CrossRef CAS PubMed.
- C. Guo, M. Fleige, D. Janssen-Meller, C. G. Daniliuc and F. Glorius, Nat. Chem., 2015, 7, 842–847 CrossRef CAS PubMed.
- (a) L. Jiang and Y. C. Chen, Catal. Sci. Technol., 2011, 1, 354 RSC; (b) P. Melchiorre, Angew. Chem., Int. Ed., 2012, 51, 9748 CrossRef CAS PubMed.
- (a) H. U. Vora, P. Wheeler and T. Rovis, Adv. Synth. Catal., 2012, 354, 1617 CrossRef CAS PubMed; (b) S. J. Ryan, L. Candish and D. W. Lupton, Chem. Soc. Rev., 2013, 42, 4906 RSC.
- W. Wang, Y. Wang, L. J. Zheng, Y. Qiao and D. H. Wei, ChemistrySelect, 2017, 2, 5266–5273 CrossRef CAS.
- T. H. West, D. M. Walden, J. E. Taylor, A. C. Brueckner, R. C. Johnston, P. H. Y. Cheong, G. C. L. Jones and A. D. Smith, J. Am. Chem. Soc., 2017, 139, 4366–4375 CrossRef CAS PubMed.
- (a) D. G. Stark, L. C. Morrill, P.-P. Yeh, A. M. Z. Slawin, T. J. C. ORiordan and A. D. Smith, Angew. Chem., Int. Ed., 2013, 52, 11642–11646 CrossRef CAS PubMed; (b) S. L. Wang, J. Izquierdo, C. R. Escrich and M. A. Pericàs, ACS Catal., 2017, 7, 2780–2785 CrossRef CAS.
- B. V. Birman and X. Li, Org. Lett., 2006, 8, 1351–1354 CrossRef PubMed.
- S. L. Wang, J. Izquierdo, C. Rodriguez-Escrich and M. A. Pericàs, ACS Catal., 2017, 7, 2780 CrossRef CAS.
- E. R. T. Robinson, D. M. Walden, C. Fallan, M. D. Greenhalgh, P. H.-Y. Cheong and A. D. Smith, Chem. Sci., 2016, 7, 6919–6927 RSC.
- For a review on isothiourea catalysis see: (a) J. Merad, J.-M. Pons, O. Chuzel and C. Bressy, Eur. J. Org. Chem., 2016, 2016, 5589–5610 CrossRef CAS; (b) C. Joannesse, C. P. Johnson, C. Concellon, C. Simal, D. Philp and A. D. Smith, Angew. Chem., Int. Ed., 2009, 48, 8914–8918 CrossRef CAS PubMed; (c) C. A. Leverett, V. C. Purohit and D. Romo, Angew. Chem., Int. Ed., 2010, 49, 9479–9483 CrossRef CAS PubMed; (d) D. Belmessieri, L. C. Morill, C. Simal, A. M. Z. Slawin and A. D. Smith, J. Am. Chem. Soc., 2011, 133, 2714–2720 CrossRef CAS PubMed.
- M. D. Greenhalgh, S. Qu, A. M. Z. Slawin and A. D. Smith, Chem. Sci., 2018, 9, 4909 RSC.
- (a) Y. Y. Wang, D. H. Wei, Y. Wang, W. J. Zhang and M. S. Tang, ACS Catal., 2016, 6, 279–289 CrossRef CAS; (b) S. Li, Z. W. Tang, Y. Wang, D. Wang, Z. L. Wang, C. X. Yu, T. J. Li, D. H. Wei and C. S. Yao, Org. Lett., 2019, 21, 1306–1310 CrossRef CAS PubMed.
- (a) X. K. Guo, L. B. Zhang, D. H. Wei and J. L. Niu, Chem. Sci., 2015, 6, 7059–7071 RSC; (b) K. Sun, S. J. Li, X. L. Chen, Y. Liu, X. Q. Huang, D. H. Wei, L. B. Qu, Y. F. Zhao and B. Yu, Chem. Commun., 2019, 55, 2861–2864 RSC.
- (a) D. H. Wei, B. L. Lei, M. S. Tang and C.-G. Zhan, J. Am. Chem. Soc., 2012, 134, 10436–10450 CrossRef CAS PubMed; (b) D. H. Wei, X. Q. Huang, Y. Qiao, J. J. Rao, L. Wang, F. Liao and C.-G. Zhan, ACS Catal., 2017, 7, 4623–4636 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 106, 5151 CrossRef CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- (a) L. R. Domingo, P. Pérez and J. A. Sáez, RSC Adv., 2013, 3, 1486–1494 RSC; (b) L. J. Zheng, Y. Wang, D. H. Wei and Y. Qiao, Chem. – Asian J., 2016, 11, 3046–3054 CrossRef CAS PubMed.
- (a) R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS; (b) W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef; (c) L. J. Sham and W. Kohn, Phys. Rev., 1966, 145, 561 CrossRef CAS; (d) L. R. Domingo, E. Chamorro and P. Pérez, Eur. J. Org. Chem., 2009, 2009, 3036–3044 CrossRef; (e) L. R. Domingo and P. Pérez, Org. Biomol. Chem., 2013, 11, 4350–4358 RSC; (f) L. R. Domingo, J. A. Saéz, R. J. Zaragozá and M. Arnó, J. Org. Chem., 2008, 73, 8791–8799 CrossRef CAS PubMed; (g) D. Yepes, J. S. Murray, P. Pérez, L. R. Domingo, P. Politzer and P. Jaque, Phys. Chem. Chem. Phys., 2014, 16, 6726–6734 RSC.
- (a) N. Liu, Y. F. Xie, C. Wang, S. J. Li, D. H. Wei and Bin Dai, ACS Catal., 2018, 8, 9945–9957 CrossRef CAS; (b) Q. Q. Shi, W. Wang, Y. Wang, Y. Lan, C. S. Yao and D. H. Wei, Org. Chem. Front., 2019, 6, 2692 RSC; (c) Q. C. Zhang, X. Li, X. H. Wang, S. J. Li, L. B. Qu, Y. Lan and D. H. Wei, Org. Chem. Front., 2019, 6, 679–687 RSC.
- (a) R. Gordillo and K. N. Houk, J. Am. Chem. Soc., 2006, 128, 3543–3553 CrossRef CAS PubMed; (b) C. D. Anderson, T. Dudding, R. Gordillo and K. N. Houk, Org. Lett., 2008, 10, 2749–2752 CrossRef CAS PubMed; (c) O. Gutierrez, R. G. Iafe and K. N. Houk, Org. Lett., 2009, 11, 4298–4301 CrossRef CAS PubMed; (d) M. A. M. Capozzi, C. Centrone, G. Fracchiolla, F. Naso and C. Cardellicchio, Eur. J. Org. Chem., 2011, 4327–4334 CrossRef CAS.
- F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS PubMed.
- (a) D. Quinoñero, C. Garau, C. Rotger, A. Frontera, P. Ballester, A. Costa and P. M. Deya, Angew. Chem., Int. Ed., 2002, 41, 3389–3392 CrossRef; (b) T. J. Mooibroek, P. Gamez and J. Reedijk, CrystEngComm, 2008, 10, 1501–1515 RSC.
- C. Shu, H. L. Liu, A. M. Z. Slawin, C. Carpenter-Warrena and A. D. Smith, Chem. Sci., 2020, 11, 241–247 RSC.
- T. H. West, D. M. Walden, J. E. Taylor, A. C. Brueckner, R. C. Johnston, P. H. Y. Cheong, G. C. L. Jones and A. D. Smith, J. Am. Chem. Soc., 2017, 139, 4366–4375 CrossRef CAS PubMed.
- D. J. B. Antúnez, M. D. Greenhalgh, A. C. Brueckner, D. M. Walden, P. E. Rodríguez, P. Roberts, B. G. Young, T. H. West, A. M. Z. Slawin, P. H. Y. Cheong and A. D. Smith, Chem. Sci., 2019, 10, 6162–6173 RSC.
- (a) L. R. Domingo, E. Chamorro and P. Perez, J. Phys. Chem. A, 2008, 112, 4046–4053 CrossRef CAS PubMed; (b) L. R. Domingo and P. Perez, Org. Biomol. Chem., 2011, 9, 7168–7175 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details; the DFT-calculated geometries, energies, and frequencies for all stationary points along the reactions studied. See DOI: 10.1039/d0cy00295j |
| This journal is © The Royal Society of Chemistry 2020 |