
Photocatalytic hydrogen evolution over nickel cobalt bimetallic phosphate anchored graphitic carbon nitrides by regulation of the d-band electronic structure†
 *ac
*ac
Abstract
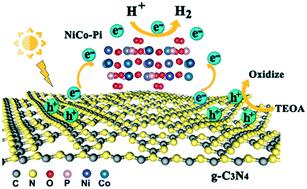
Non-precious metal co-catalysts with high activity and stability are extremely desirable for economically viable photocatalytic molecular hydrogen (H2) evolution. Herein, nickel cobalt phosphate (NiCo–Pi) was introduced into graphitic carbon nitride layers (g-C3N4) via a sonication-assisted ion intercalation method as a substitute for noble metal co-catalysts. Under visible light irradiation, NiCo–Pi/g-C3N4 (Ni/Co molar ratio of 4 : 5) exhibited the highest photocatalytic activity (ca. 10 184 μmol h−1 g−1) and stability for H2 evolution. Synchrotron radiation X-ray absorption spectroscopy (XAS) indicated that NiCo–Pi is closely bound to g-C3N4via covalent binding, which accelerates electron transport. Moreover, the unoccupied d-orbital in NiCo–Pi causes the surface to strongly adsorb atomic hydrogen (*H). Theoretically, density functional theory (DFT) calculations demonstrated that the d-band center position of NiCo–Pi is relocated upon adjusting the Ni/Co molar ratio, which changes the adsorption energy of NiCo–Pi toward intermediate state *H. This work provides new insights for exploring the role of the bimetallic composition in non-noble co-catalysts for highly efficient H2 evolution.
 Please wait while we load your content...
Please wait while we load your content...

