
Tools and strategies of systems metabolic engineering for the development of microbial cell factories for chemical production
Yoo-Sung
Ko†
 a,
Je Woong
Kim†
a,
Jong An
Lee
a,
Taehee
Han
a,
Gi Bae
Kim
a,
Jeong Eum
Park
a and
Sang Yup
Lee
*abc
a,
Je Woong
Kim†
a,
Jong An
Lee
a,
Taehee
Han
a,
Gi Bae
Kim
a,
Jeong Eum
Park
a and
Sang Yup
Lee
*abc
aMetabolic and Biomolecular Engineering National Research Laboratory, Systems Metabolic Engineering and Systems Healthcare (SMESH) Cross-Generation Collaborative Laboratory, Department of Chemical and Biomolecular Engineering (BK21 Plus Program), Institute for the BioCentury, Korea Advanced Institute of Science and Technology (KAIST), 291 Daehak-ro, Yuseong-gu, Daejeon, 34141, Republic of Korea. E-mail: leesy@kaist.ac.kr
bBioinformatics Research Center, KAIST, 291 Daehak-ro, Yuseong-gu, Daejeon, 34141, Republic of Korea
cBioProcess Engineering Research Center, KAIST, 291 Daehak-ro, Yuseong-gu, Daejeon, 34141, Republic of Korea
First published on 22nd June 2020
Abstract
Sustainable production of chemicals from renewable non-food biomass has become a promising alternative to overcome environmental issues caused by our heavy dependence on fossil resources. Systems metabolic engineering, which integrates traditional metabolic engineering with systems biology, synthetic biology, and evolutionary engineering, is enabling the development of microbial cell factories capable of efficiently producing a myriad of chemicals and materials including biofuels, bulk and fine chemicals, polymers, amino acids, natural products and drugs. In this paper, many tools and strategies of systems metabolic engineering, including in silico genome-scale metabolic simulation, sophisticated enzyme engineering, optimal gene expression modulation, in vivo biosensors, de novo pathway design, and genomic engineering, employed for developing microbial cell factories are reviewed. Also, detailed procedures of systems metabolic engineering used to develop microbial strains producing chemicals and materials are showcased. Finally, future challenges and perspectives in further advancing systems metabolic engineering and establishing biorefineries are discussed.
 Yoo-Sung Ko | Yoo-Sung Ko received his BS in Chemical and Biomolecular Engineering from Korea Advanced Institute of Science and Technology (KAIST) and currently is an Integrated MS/PhD candidate in Professor Sang Yup Lee's Lab (Metabolic & Biomolecular Engineering National Research Laboratory) at KAIST. His research focuses on the production of industrial chemicals using metabolically engineered bacteria through systems metabolic engineering. |
 Je Woong Kim | Je Woong Kim received his BS in Chemical and Biomolecular Engineering from Korea Advanced Institute of Science and Technology (KAIST) and currently is an Integrated MS/PhD candidate in Professor Sang Yup Lee's Lab (Metabolic & Biomolecular Engineering National Research Laboratory) at KAIST. His research focuses on the efficient production of industrial bulk chemicals through metabolic engineering. |
 Sang Yup Lee | Sang Yup Lee is a distinguished professor at the Department of Chemical and Biomolecular Engineering, Korea Advanced Institute of Science and Technology (KAIST). He is currently the Dean of KAIST Institutes, Director of BioProcess Engineering Research Center, and Director of Bioinformatics Research Center. His research interests are metabolic engineering, systems and synthetic biology and biotechnology, industrial biotechnology, and nanobiotechnology. |
Key learning points(1) Systems metabolic engineering, which integrates traditional metabolic engineering with systems biology, synthetic biology, and evolutionary engineering, accelerates the development of efficient microbial cell factories.(2) Systems metabolic engineering considers upstream (raw material preparation), midstream (fermentation), and downstream (recovery and purification) processes together when developing microbial strains. (3) Through the integration of the tools and strategies of systems biology, genome-wide scale prediction of cellular status, including metabolic fluxes, is possible. (4) The use of synthetic biology tools allows the design and construction of enzymes and pathways for the production of more diverse chemicals and materials. (5) Evolutionary engineering allows enzymes, regulatory proteins, metabolic pathways, and/or entire cells to evolve to possess desired functions, characteristics, and/or phenotypes. |
1. Introduction
Numerous chemicals and materials around us are produced from fossil resources through petrochemical refinery processes, and such petrochemical products have brought unprecedented prosperity to humankind. However, we are now facing serious threats from the excessive use of fossil resources causing a climate crisis and environmental problems. Furthermore, fossil resources are not infinite, and will ultimately be depleted. To cope with these problems, it is essential to establish a sustainable chemical industry capable of producing chemicals and materials from renewable non-edible biomass as a raw material. For the bio-based production of chemicals and materials, microorganisms are most often employed as they are ethically sound and can be securely cultured in a closed tank (e.g., a fermentor). Microbial production of chemicals and materials is typically performed under mild conditions such as relatively low temperature and pressure without using toxic solvents or metal catalysts, adding another set of advantages. According to a recent report by Market Research Future in 2019, the global market size of bio-based chemicals is estimated to grow with a CAGR of 10.47% to reach USD 97.2 billion by 2023.For convenience, a biorefinery can be divided into four processes: the first upstream process that converts renewable biomass into fermentable carbohydrates, the second upstream process that develops a microbial strain capable of efficiently producing a desired product, the midstream process that cultivates microorganisms and produces a chemical or material of interest, and the downstream process that recovers and purifies the desired product. Although none of these four processes is unimportant, the second upstream process of developing microbial strains is the most important as it determines the overall efficiency of converting raw material to a product. Since microorganisms isolated from nature are not optimized for the production of our desired product, their performance needs to be enhanced. This is where metabolic engineering comes into play. Metabolic engineering can be defined as the purposeful modification of cellular networks to achieve defined objectives.1
These objectives include enhanced production of a desired chemical or material, production of a novel chemical or material that the wild-type strain has no metabolic capability to biosynthesize, and degradation of a toxic or undesirable chemical or material.2 Also, metabolic engineering can be performed to allow microorganisms to utilize the least expensive carbon substrates available at the place of fermentation operation. Furthermore, metabolic engineering is performed to reduce the production of byproducts, which consequently facilitates the downstream processes and increases the yield of the product.
Over the past three decades, metabolic engineering has allowed the development of many different microbial strains for the production of chemicals and materials. However, earlier metabolic engineering research required a large amount of manpower, time, and cost to develop industrially competitive microbial strains. More recently, systems metabolic engineering has been established through integrating traditional metabolic engineering with the tools and strategies of systems biology, synthetic biology, and evolutionary engineering for more efficient and rapid development of microbial cell factories. When developing microbial strains by systems metabolic engineering (e.g., the second upstream process), it is extremely important to consider the first upstream, midstream, and downstream processes together for the overall optimization of the entire process (Fig. 1).3 The advent of systems metabolic engineering promoted the development of high-performance strains producing various bioproducts, including bulk chemicals, fine chemicals, polymers and materials, biofuels, and natural products (Fig. 1). Several of them, including lactic acid, polylactic acid (PLA), polyhydroxyalkanoates (PHAs), succinic acid, ethanol, butanol, isobutanol, 1,3-propanediol (1,3-PDO), 1,4-butanediol (1,4-BDO), isoprene, farnesene, artemisinin, and various amino acids, have been commercialized or are close to commercialization.4 Also, some biologically produced chemicals can serve as platform chemicals and be further converted into many other valuable derivatives or polymers by integrating with chemical processes. A comprehensive map visualizing the strategies and pathways for the production of bio-based chemicals through biological and/or chemical reactions has recently been presented.4
 | ||
| Fig. 1 Overall strategies and procedures of systems metabolic engineering for the bio-based production of chemicals and materials. Before building such cell factories, a target product (bulk chemical, fine chemical, polymer, biofuel, or natural product) to be produced is selected. Also, the most appropriate raw material (carbon substrate) based on the availability and price is selected. Then, a host strain for the efficient production of the target product is chosen. The biosynthetic metabolic pathway for the target chemical production is constructed and introduced into the host strain. Using the advanced tools of systems biology and synthetic biology, the metabolic fluxes toward the target chemical are optimized to maximize the production performance indices including the titer, yield, and productivity. At an appropriate stage, adaptive laboratory evolution is performed to obtain an improved strain having a desired phenotype under selection pressure. During the pathway construction, directed evolution is performed to develop enzymes with new or improved functionalities by iterative rounds of mutagenesis and selection. Various chemicals (shown on the right) can be produced by the strains developed by applying systems metabolic engineering. During strain development, lab-scale (0.5–30 L) fermentations are performed. Once the desired performance indices are obtained in lab-scale fermentation, pilot-scale and/or demoplant-scale fermentations are performed to see if the strain developed is exhibiting the desired performance. If problems are observed, additional systems metabolic engineering and process optimization are performed to address such problems. Finally, the target products are recovered from the fermentation broth and purified with high yield. Abbreviations are: 1,3-PDO, 1,3-propanediol; 1,4-DAB, 1,4-diaminobutane; 1,5-DAP, 1,5-diaminopentane; 3-HP, 3-hydroxypropionic acid; FAEE, fatty acid ethyl ester; FAME, fatty acid methyl ester; LA, lactic acid; MANT, methylanthranilic acid; MEG, monoethylene glycol; P3HB, poly(3-hydroxybutyric acid); P(3HB-co-PhLA), poly(3-hydroxybutyric-co-phenyllactic acid); PLA, polylactic acid; PLGA, poly(lactic-co-glycolic acid); P(LA-co-4HB), poly(lactic-co-4-hydroxybutyric acid); SA, succinic acid; TPA, terephthalic acid. | ||
In this paper, we review the detailed procedure of systems metabolic engineering for the development of microbial cell factories. The general strategies of systems metabolic engineering we previously proposed3 were used as a guideline for a detailed description of the procedure. Also, the tools and strategies of systems metabolic engineering are reviewed together with a detailed explanation of their basic principles and practical application examples. Furthermore, several examples of developing microbial strains capable of producing chemicals and materials are showcased to demonstrate how microbial cell factories are successfully developed in practice. To help readers to better understand the complete procedure of systems metabolic engineering, we detailed a case study on developing a process for microbial production of succinic acid (Fig. 2).5 Last but not least, future challenges in further advancing systems metabolic engineering and perspectives on successfully establishing biorefineries are discussed.
 | ||
| Fig. 2 The procedural flowcharts of systems metabolic engineering for the development of (a) Escherichia coli and (b) Mannheimia succiniciproducens strains for the production of succinic acid (SA), a four-carbon dicarboxylic acid widely used as a monomer and a precursor for industrial commodity chemicals, as an example. This flowchart illustrates how systems metabolic engineering strategies were applied for the development of SA overproducing microbial strains. The systems metabolic engineering strategies used are described in a sequential order together with the actual decisions made or experiments performed. In detail, SA was selected as the target product (1). First, E. coli was chosen as a host strain due to the availability of various genetic engineering tools and its best understood metabolism (2). Since E. coli is a native SA producer, introduction of a heterologous metabolic pathway was not necessary. To increase the SA production, genes responsible for byproduct synthesis (pflB and ldhA) were deleted (3). Furthermore, the mdh gene was overexpressed to improve cell growth and SA production (4). However, the dual-phase fermentation (the aerobic cell growth phase followed by the anaerobic SA production phase) of the developed E. coli strain did not produce SA to a high enough level (5, 6). To further increase SA production, evolutionary engineering was performed to mutate the chromosomal ptsG gene (7). When the Rhizobium etli pyc gene was additionally overexpressed (8), the final engineered E. coli strain produced SA to a high titer by fed-batch fermentation (9). However, the specific productivity achieved was not high and several byproducts were co-produced. Thus, another native SA overproducer, M. succiniciproducens, isolated from the rumen of Korean cows, was selected as a new host strain (10). It was necessary to first develop genetic engineering tools including plasmid construction and gene deletion tools. As the metabolism of M. succiniciproducens was unknown, cultivation experiments under various conditions were performed. Then, whole-genome sequencing was performed (11). Although genome sequencing is a routine job nowadays, it was not so 16 years ago and was quite expensive when this sequencing was done; in fact, the genome sequence of M. succiniciproducens became the first ever full genome sequence reported from Korea. Analysis of the full genome sequence allowed better understanding of its metabolic characteristics through genome-scale metabolic network reconstruction and also metabolic flux analyses during fermentation (12, 13). Based on these results, the byproduct forming pathways were blocked by deleting the ldhA, pflB, and pta and ackA genes (14). However, in silico metabolic flux analysis suggested that restoring the pflB gene was beneficial for the enhanced production of SA (15, 16). A chemically defined medium was also developed by analyzing the genome-based auxotrophies (17). Glycerol was chosen as an auxiliary carbon source since it supported enhanced production of SA through providing twice the amount of reducing equivalents (NADH) compared with 1/2 glucose. Fed-batch fermentation of the engineered M. succiniciproducens strain in a chemically defined medium successfully produced SA with high titer, yield, and productivity, from glucose and glycerol as carbon sources (18). Then, in silico genome-scale analysis predicted the use of glycerol and sucrose as the optimal dual carbon sources. To relieve the carbon catabolite repression in M. succiniciproducens, the mechanism of glycerol utilization in the presence of sucrose was studied, and as a result the fruA gene was additionally deleted or E. coli glpK was overexpressed (19). Bioprocess engineering studies were also performed. Using a membrane cell recycling bioreactor (MCRB) system, the final engineered M. succiniciproducens strain produced homo-SA with the highest productivity reported to date using sucrose and glycerol as carbon sources (20, 21). Homo-SA produced by fermentation of metabolically engineered M. succiniciproducens allowed simple and economically feasible recovery and purification of SA from the fermentation broth (22). Abbreviations are: ackA, acetate kinase; fruA, fructose phosphotransferase; glpK, glycerol kinase; ldhA, lactate dehydrogenase; MCRB, membrane cell recycling bioreactor; mdh, malate dehydrogenase; pflB, pyruvate formate lyase; pta, phosphoate acetyltransferase; ptsG, glucose phosphotransferase; pyc, pyruvate carboxylase. | ||
2. Selection of target products and carbon substrates
Before the construction of microbial cell factories, the chemicals and materials to be produced should be carefully decided in response to demands from the market and society. Recent advances in the fields of computational biology and synthetic biology have facilitated the design of novel and efficient metabolic pathways for desired chemicals, consequently resulting in the expansion of the spectrum of bio-based products. Obviously, chemicals that currently have high demand or high potential with promising applications in the near future are often the most desired production targets. Also, these chemicals and materials to be produced can be so far unknown ones in addition to the known ones. Bioproducts can be typically classified into five categories: bulk chemicals, fine chemicals (including drugs), polymers and materials, biofuels, and natural products (Fig. 3).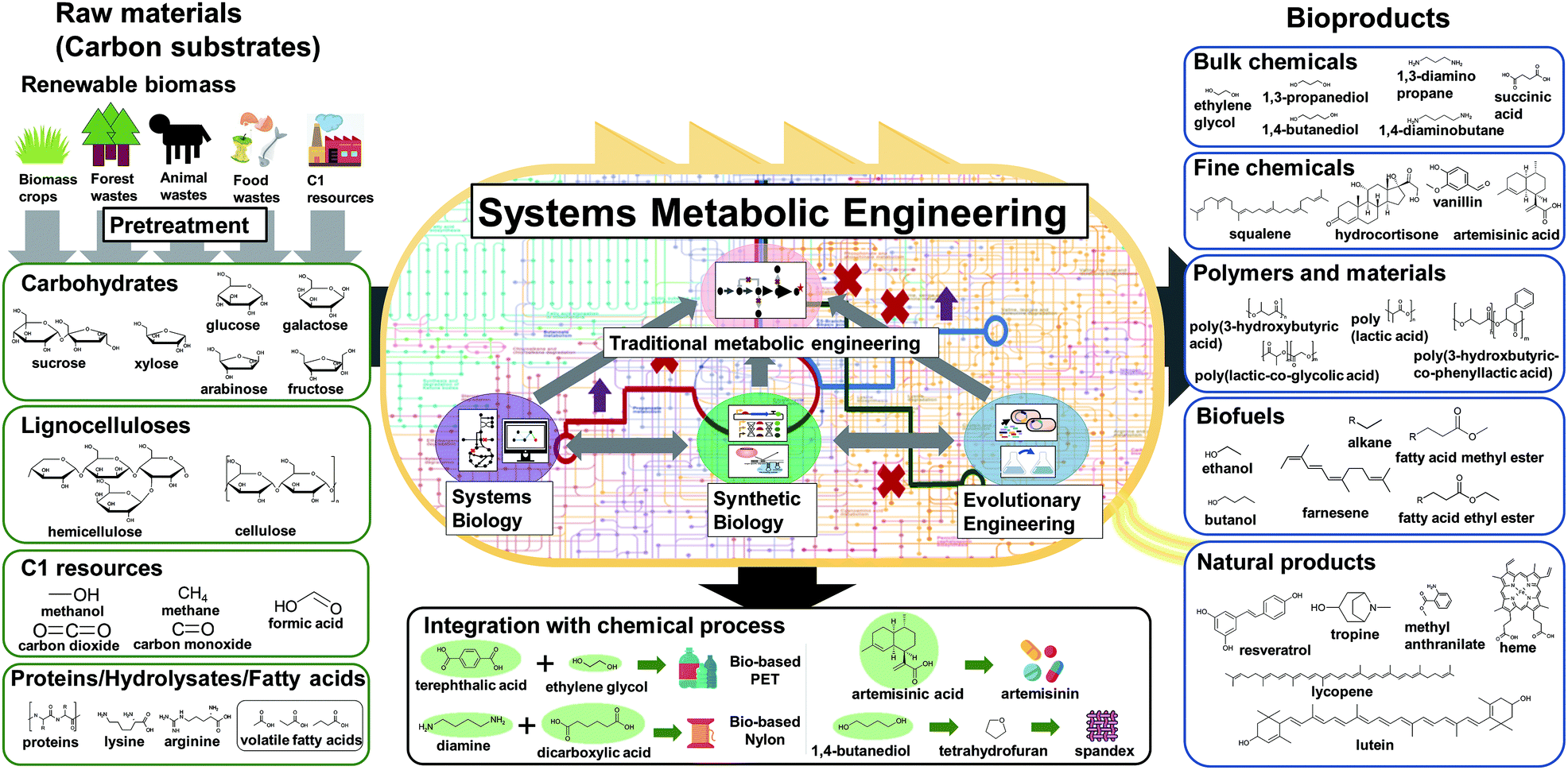 | ||
| Fig. 3 Raw materials and types of bioproducts to be produced by metabolically engineered microorganisms. Non-edible biomass and more preferably waste biomass, including carbohydrates, lignocelluloses, protein hydrolysates, and fatty acids derived from biomass crops, forest/animal waste, and food waste, can be used as raw materials for the production of chemicals and materials. Also, one carbon gases, such as carbon dioxide, carbon monoxide, and methane, can be used as carbon sources. Although not covered in detail here, the preparation of fermentable substrates through the pretreatment of biomass (e.g., the first upstream process) is important in overall biorefinery economics. Systems metabolic engineering, which integrates tools and strategies of systems biology, synthetic biology, and evolutionary engineering with traditional metabolic engineering, enables the successful development of efficient microbial cell factories. Various types of bioproducts including bulk chemicals, fine chemicals, polymers and materials, biofuels, and natural products can be produced using efficient microbial cell factories developed by systems metabolic engineering. Some of the biologically produced platform chemicals (shaded green circles) can be further converted into valuable derivatives by integrating with chemical processes (middle bottom box). | ||
Bulk chemicals refer to commodity chemicals that are produced in large quantities, and thus typically at a lower price than fine chemicals. Although achieving three key performance indices (product titer, yield, and productivity) as high as possible is a common objective of all biorefineries, it is more critical for bulk chemicals due to the high competition with petrochemicals. These indices are tightly linked to the overall production costs, which include the substrate costs (including the cost of the carbon source), fermentation and other operation costs, and separation and purification costs. With the aid of newly emerged engineering tools and strategies, production of several bulk chemicals by metabolically engineered microorganisms has reached near the theoretical maximum yield with high titer and productivity.4 For example, the successful commercialization of microbially produced 1,3-PDO, which is important as a monomer for the synthesis of polytrimethylene terephthalate, has been demonstrated.6 In this study, an engineered Escherichia coli strain capable of producing 135 g L−1 of 1,3-PDO from glucose with a productivity of 3.5 g L−1 h−1 and a yield of 0.51 g g−1 glucose (81% of the theoretical maximum yield) was developed. Compared with the petrochemical process, bio-based 1,3-PDO production exhibited 42% less energy consumption with 56% reduced greenhouse gas emission.6
Fine chemicals are those chemicals produced in smaller amounts than bulk chemicals, and are usually sold at a higher price. Thus, they have high potential for successful commercialization if they can be produced biologically with reasonably high efficiencies. However, due to the inherent limitation in metabolic capabilities, for example, the lack of the corresponding biosynthetic enzymes and pathways, there have only been a few fine chemicals produced at the industrial level.2 Such a limitation is being quickly resolved with advances in metabolic engineering, and an increasing number of microbial strains capable of producing various fine chemicals are under development.4 One of the successful examples is the development of a microbial strain producing artemisinic acid, an anti-malarial drug precursor, which will be detailed in Section 4.1.
Bio-based polymers have recently received great attention to replace, at least some, current petroleum-based plastics. Among them, PHAs, which are natural polyesters produced and accumulated in various microorganisms, have attracted much attention as they possess similar material properties to synthetic commodity plastics as well as biodegradability and biocompatibility. Through the application of systems metabolic engineering, the portfolio of biopolyesters that can be produced by engineered microorganisms has been expanded to cover non-natural polymers such as PLA and poly(lactic-co-glycolic acid), and various copolymers with natural PHAs. Readers can consult a recent review paper7 and many excellent papers cited therein.
Biofuels refer to fuels produced from renewable resources by biological and/or chemical processes, but those produced biologically are only considered in this paper. Biofuels can be categorized based on the key metabolic pathways involved: ethanol pathways, keto acid pathways, isoprenoid pathways, CoA-dependent reverse β-oxidation pathways, and fatty acid biosynthetic pathways.4 Beyond bioethanol, currently the most prevalent biofuel, a diverse range of higher alcohols and hydrocarbon fuels have been produced by metabolically engineered microorganisms. Bio-based hydrocarbon fuels are of particular interest due to their high-energy contents and superior fuel properties, and thus can substitute gasoline, diesel, and jet-fuel depending on the carbon chain lengths. However, it will not be possible (and in fact not desirable) to replace large amounts of petroleum-derived liquid fuels with biofuels considering the availability of non-food biomass. More realistically, biofuels can be blended with fossil-based liquid fuels to contribute to reducing greenhouse gas emissions.4 So far, microbial production of these hydrocarbon biofuels by employing well-known microorganisms such as E. coli and Saccharomyces cerevisiae has been pursued, but the production performance indices were rather low. More recently, oleaginous microorganisms are increasingly utilized as alternative host strains for the production of hydrocarbon biofuels, which will be described in Section 3.
Natural products are chemicals produced by living organisms in nature, and are widely used in our daily lives as food additives, nutraceuticals, and cosmetic ingredients. Natural products can be classified based on their structures as terpenoids, phenylpropanoids, polyketides, and alkaloids; there are also other natural products that do not belong to any of these four categories.8 Most natural products have so far been isolated through extraction from natural resources, including plants or animals. However, their production has been rather limited due to the poor yields of extraction, which consequently result in insufficient supply and high costs. Several natural products can be produced through chemical synthetic routes. However, such a chemical process is unfavorable when multistep reactions are involved, and generates stereoisomers or intermediates under harsh operating conditions. Thus, the heterologous production of natural products using metabolically engineered microorganisms has been increasingly implemented as it allows the production of enantiomerically pure compounds under benign conditions. Metabolic engineering strategies for the production of a variety of natural products with associated examples are reviewed in a recent paper.8
After the selection of a target product, the raw materials or carbon substrates to be utilized should be carefully selected. Currently, sucrose from sugarcane or sugar beet, glucose in the form of starch hydrolysate, and glycerol obtained as a byproduct of biodiesel production are the most commonly used carbon substrates. Due to the increasing recognition of the food shortage issue, there is a clear vision in the field for using non-edible and waste substrates, including lignocellulosics of non-edible plants such as rapidly growing biomass crops, crop residues, wood, and forest waste. Also, animal waste and food waste are increasingly considered as raw materials. The pretreatment of these raw materials into fermentable carbohydrates (e.g., the first upstream process) is crucial for the overall economics of biorefineries. Obviously, a carbon substrate with a stable supply at low cost should be used. The choice of carbon substrate also affects the selection of the host strain as each microorganism prefers different carbon substrates; of course, systems metabolic engineering can solve this problem of carbon substrate preference, as will be described later. Recently, C1 resources such as methane, carbon monoxide, carbon dioxide, and formic acid (which can be considered a liquid form of carbon dioxide) are emerging as new carbon substrates as well.
3. Selection of a host strain
Recruiting reliable microbial host strains is crucial for ensuring the efficient production of desired chemicals. Among numerous microorganisms, E. coli and S. cerevisiae have been dominantly employed as host strains due to our better understanding of their metabolisms and the availability of gene and genome engineering tools.4 Due to recent advances in engineering tools facilitating the efficient manipulation of various microorganisms, including those less studied ones, we are no longer confined to E. coli or S. cerevisiae for the production of target chemicals and can employ microorganisms with inherently better metabolic characteristics toward the target products. It is obvious that pathogenic microorganisms of any sort should not be considered as production hosts. In general, one of the following three cases can be initially considered in selecting the production host: a microorganism that naturally overproduces the target product, a microorganism that produces the target product with low efficiency, and a microorganism that does not produce the target product. Different metabolic engineering strategies are employed for each of these three cases.For the efficient production of chemicals and materials, natural overproducers are obviously the preferred host strains as they already have evolved to possess a high tolerance to and strong metabolic fluxes toward the target products. Some examples include: Corynebacterium glutamicum for amino acids; Clostridium sp. for butanol; Rhodococcus opacus and Yarrowia lipolytica for oleochemicals; and Mannheimia succiniciproducens for succinic acid. These overproducers can be employed for the production of other related chemicals through metabolic engineering. For example, an industrial L-lysine producing C. glutamicum PKC strain was used as a chassis strain for the production of cadaverine, which is a monomer for polyamides.9L-Lysine can be converted into cadaverine efficiently through the direct decarboxylation reaction by L-lysine decarboxylase (LDC). By integrating the E. coli ldcC gene into the C. glutamicum genome with concomitant disruption of L-lysine exporter (LysE), an engineered C. glutamicum strain produced 103.8 g L−1 of cadaverine with a productivity and yield of 1.47 g L−1 h−1 and 0.36 g g−1 glucose, respectively, by fed-batch fermentation of glucose. The microbially produced cadaverine was purified and chemically polymerized with sebacic acid, which can be produced from plant oil, to synthesize nylon 510, a commodity plastic with high temperature resistance.9 Thus, it is possible to produce fully bio-based nylon 510 and also other bio-based nylons in a similar way.
Fatty acids and their derivatives including biodiesel are an important family of products, and have mostly been produced from plant oils and animal fats. However, plant oils such as palm oil that have been used for biodiesel production are in conflict with our consumption as a food. Thus, there has been much interest in producing fatty acids and derivatives from carbohydrates that can be obtained from lignocellulosics. There have been many attempts to engineer E. coli and S. cerevisiae to produce fatty acids and derivatives, but the final titers, yields, and productivities were rather low. Oleaginous microorganisms, which accumulate large amounts of triacylglycerols (TAGs) inside the cell under nitrogen-limiting conditions, are ideal hosts for the production of lipid-based chemicals. For example, the wild-type R. opacus PD630 strain produced 82.9 g L−1 of TAGs from glucose by optimization of the fed-batch fermentation conditions without any gene manipulation.10 For the production of valuable derivatives from TAGs, R. opacus PD630 was further metabolically engineered to produce free fatty acids (FFAs), fatty acid ethyl esters (FAEEs), and long chain hydrocarbons (LCHCs). More specifically, when the genes encoding TAG lipase and lipase-specific foldase were overexpressed and the genes encoding several major acyl-CoA synthetases were deleted, an engineered R. opacus strain produced 50.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) g L−1 of FFAs from glucose by one-step direct fermentation.10 For the production of FAEEs, an R. opacus strain was engineered by disrupting several key acyl-CoA dehydrogenases and overexpressing the genes encoding native acyl-CoA synthetases. The subsequent introduction of heterologous aldehyde/alcohol dehydrogenase and wax ester synthase into this engineered R. opacus strain led to the production of 21.3 g L−1 of FAEEs from glucose.10 LCHC production was also achieved by overexpressing the genes encoding acyl-CoA reductase, acyl-CoA synthetase, aldehyde deformylating oxygenase, TAG lipase, and lipase-specific foldase while disrupting several major acyl-CoA dehydrogenases and alkane-1-monooxygenase. Fed-batch fermentation of the resulting strain produced 5.2 g L−1 of LCHCs from glucose.10
g L−1 of FFAs from glucose by one-step direct fermentation.10 For the production of FAEEs, an R. opacus strain was engineered by disrupting several key acyl-CoA dehydrogenases and overexpressing the genes encoding native acyl-CoA synthetases. The subsequent introduction of heterologous aldehyde/alcohol dehydrogenase and wax ester synthase into this engineered R. opacus strain led to the production of 21.3 g L−1 of FAEEs from glucose.10 LCHC production was also achieved by overexpressing the genes encoding acyl-CoA reductase, acyl-CoA synthetase, aldehyde deformylating oxygenase, TAG lipase, and lipase-specific foldase while disrupting several major acyl-CoA dehydrogenases and alkane-1-monooxygenase. Fed-batch fermentation of the resulting strain produced 5.2 g L−1 of LCHCs from glucose.10
If the target products are for food and pharmaceutical applications, the use of ‘generally recognized as safe’ (GRAS) microorganisms should be considered to cope with public concerns on safety issues. Bacillus subtilis, Lactobacillus sp., Pseudomonas putida KT2440, and S. cerevisiae are representative well-explored GRAS strains. Beyond typical mesophilic microorganisms often employed, thermophilic or halophilic microorganisms can be advantageous as host strains at they can grow and survive at a high cultivation temperature or high salt concentration, which consequently prevents contamination during fermentation. Recently, microorganisms such as cyanobacteria and methanotrophs have been spotlighted as next-generation host strains, as they can utilize C1 carbon sources as substrates.
4. Pathway construction
4.1. Introduction of heterologous pathways to non-native producers
As explained in Section 3, recruiting a native overproducer as a host strain is advantageous for the efficient production of desired chemicals. However, the number of natural host strains capable of overproducing target products is often limited. In such cases, target chemicals can be produced by establishing the biosynthetic pathways from native producers and/or introducing created pathways into a non-native host strain (Fig. 4).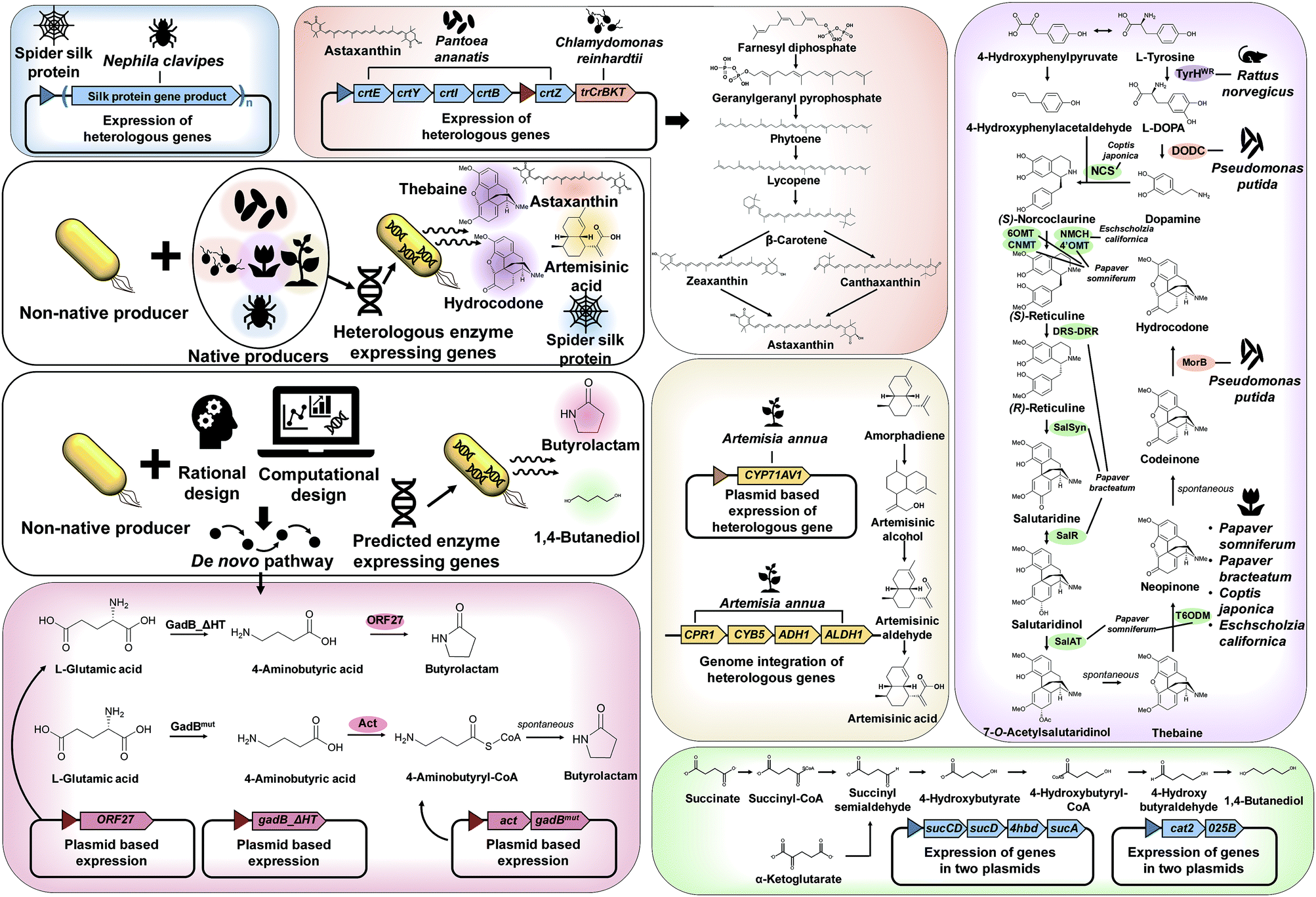 | ||
| Fig. 4 General strategies and examples of metabolic pathway construction. When the biosynthetic pathway for the target chemical is known, a suitable heterologous pathway from native producers can be established in a non-native (model) producer. For example, for the production of spider silk, astaxanthin, artemisinic acid, thebaine, and hydrocodone, the genes encoding the required heterologous enzymes were expressed in E. coli or S. cerevisiae. If the enzymes and metabolic pathways are not known, de novo biosynthetic pathways are designed by rational approaches or by using pathway prediction tools. For the production of butyrolactam and 1,4-butanediol, newly designed de novo pathways were established in an E. coli strain. The key pathways and genes/enzymes involved are shown for the production of astaxanthin, artemisinic acid, thebaine, hydrocodone, butyrolactam, and 1,4-butanediol. Abbreviations are: 025B, aldehyde dehydrogenase; 4hbd, 4-hydroxybutyrate dehydrogenase; 4′OMT, 3′-hydroxy-N-methylcoclurine 4′-O-methyltransferase; 6OMT, norcoclaurine 6-O-methyltransferase; act, β-alanine CoA transferase; ADH1, putative alcohol dehydrogenase; ALDH1, aldehyde dehydrogenase; cat2, 4-hydroxybutyryl-CoA transferase; CNMT, coclaurine N-methyltransferase; CPR1, amorphadine oxidase; crtB, 15-cis-phytoene synthase; crtE, geranyl diphosphate synthase; crtI, phytoene desaturase; crtY, lycopene beta-cyclase; crtZ, beta-carotene 3-hydroxylase; CYB5, amorphadine oxidase; CYP71AV1, amorphadine oxidase; DODC, 3,4-dihydroxyphenylalanine decarboxylase; DRS-DRR, 1,2-dehydroreticuline synthase-1,2-dehydroreticuline reductase; gadB_ΔHT, mutant L-glutamate decarboxylase; gadBmut, mutant L-glutamate decarboxylase; MorB, morphinone reductase; NCS, (S)-norcoclaurine synthase; NMCH, N-methylcoclaurine hydroxylase; ORF27, butyrolactam synthase; SalAT, salutaridinol 7-O-acetyltransferase; SalR, salutaridine reductase; SalSyn, salutaridine synthase; sucA, α-ketoglutarate dehydrogenase; sucCD, succinyl-CoA synthase; sucD, succinyl-CoA synthetase; T6ODM, thebaine 6-O-demethylase; trCrBKT, truncated β-carotene ketolase; TyrHWR, L-tyrosine hydroxylase. | ||
The first example is microbial production of astaxanthin, which has a wide range of applications in the pharmaceutical and cosmetic industry, due to its excellent antioxidant property. Recently, astaxanthin was successfully produced by a metabolically engineered E. coli strain (Fig. 4).11 A series of heterologous genes, including crtEYIBZ from Pantoea ananatis and trCrBKT from Chlamydomonas reinhardtii, were introduced into the E. coli strain to establish the astaxanthin biosynthetic pathway. Additional engineering strategies, including fusion of E. coli signal peptides and solubility enhancing tags to trCrBKT and overexpression of the native genes (ispDF) based on in silico metabolic flux analysis, were implemented to enhance astaxanthin production. Fed-batch fermentation of the final engineered E. coli strain produced 432.82 mg L−1 (equivalent to 7.12 mg g DCW−1) of astaxanthin from glycerol.11
Another great example of establishing heterologous pathways is production of artemisinic acid, an anti-malarial drug precursor, in S. cerevisiae (Fig. 4).12 For this, genes from Artemisia annua that complete the artemisinic acid biosynthetic pathway were discovered first. Three-step conversion of amorphadiene to artemisinic acid was found to be catalyzed by amorphadiene oxidase (CYP71AV1, CPR1, and CYB5), artemisinic aldehyde dehydrogenase (ALDH1), and alcohol dehydrogenase (ADH1). Then, an amorphadiene overproducing S. cerevisiae strain was engineered to down-regulate squalene synthase (ERG9) expression under the copper regulated CTR3 promoter, which increased the amorphadiene concentration. When the heterologous pathway was introduced into an engineered S. cerevisiae strain, 25 g L−1 of artemisinic acid was produced from a mixed glucose/ethanol feed in fed-batch fermentation.12 The biologically produced artemisinic acid was subsequently converted to anti-malarial drug artemisinin through a chemical process.12
In addition, direct fermentative production of opioids, a medicine for pain relief, was first demonstrated by metabolically engineered S. cerevisiae (Fig. 4).13 In this study, many enzymes from several different biological sources, including plants, mammals, bacteria, and yeast, were introduced into the S. cerevisiae strain for the construction of thebaine and also hydrocodone biosynthetic pathways. Here a bioinformatic tool for enzyme discovery and protein engineering played key roles in enhancing the enzyme solubility, which otherwise was a big problem. The final engineered S. cerevisiae strain could produce 6.4 and 0.3 μg L−1 of thebaine and hydrocodone, respectively, from glycerol.13
Other than chemicals, enhanced production of recombinant proteins is also possible by systems metabolic engineering. As an example, spider silk protein was produced by metabolically engineered E. coli (Fig. 4).14 Due to its outstanding mechanical properties, spider silk has been considered as a promising material for various industrial applications. To produce spider silk protein, the gene encoding recombinant spidroid I (MaSp1) protein, the main component of spider silk proteins from Nephila clavipes, was expressed in an E. coli strain. However, the expression of spider silk protein in large quantities was limited as spider silk gene expression is known to be notoriously difficult due to the glycine-rich (40–45%) nature of spider silk protein with highly repetitive sequences of high GC content (∼70%). Thus, additional metabolic engineering strategies based on comparative proteomic analysis were applied for supplying sufficient glycine and glycyl-tRNA. High cell density cultivation of the final engineered E. coli strain produced 2.7 g L−1 of recombinant spider silk protein.14
4.2. Development of de novo pathways
As discussed in Section 4.1, the design, construction, and establishment of heterologous pathways in a non-native production host strain can facilitate the efficient production of target chemicals. Unfortunately, however, enzymes and metabolic pathways for many target chemicals are not available. In such cases, de novo pathways should be designed by developing (or creating) new enzymes, e.g., engineering known enzymes that catalyze similar reactions by protein engineering and directed evolution (Fig. 4).One such example of non-natural chemicals produced through de novo pathway design and construction is lactam production by engineered E. coli. Lactams are important industrial chemicals that can be directly polymerized into polyamides. As there are no known natural biosynthetic pathways for the production of lactams, a de novo pathway had to be developed for the direct fermentative production of lactams. Recently, two research groups separately reported the development of lactam producing E. coli strains through different biosynthetic pathways (Fig. 4).15,16 In one study, a newly found butyrolactam synthase (ORF27) from Streptomyces aizunensis was employed for the ring-closing dehydration of 4-aminobutyric acid to butyrolactam via an ATP dependent mechanism. By expressing the genes encoding mutant glutamate decarboxylase and ORF27, the recombinant E. coli strain produced 1.1 g L−1 of butyrolactam from 9 g L−1 of L-glutamate.15 In another study, a platform for the direct production of four-, five- and six-carbon lactams via a β-alanine-CoA transferase (Act) route was developed. Act is a key enzyme that allows CoA activation of ω-amino acids followed by spontaneous cyclization to lactams. When the act gene was overexpressed in different E. coli strains engineered to produce 4-aminobutyric acid, 5-aminolevulinic acid, and 6-aminocaproic acid, the final engineered strains could produce 54.1 g L−1 of butyrolactam, 1.2 g L−1 of valerolactam from glucose and 79.6 μg L−1 of caprolactam, respectively, from glycerol by fed-batch fermentation.16
Rational identification of suitable enzymes is not an easy task. Computer-aided pathway prediction tools can be utilized to predict novel biosynthetic pathways for various target chemicals.2 For example, BNICE, which allows the discovery of novel metabolic pathways using generalized reaction rules, was applied for the prediction of promiscuous enzymes responsible for converting syngas to monoethylene glycol. As a result, seven different previously unknown biological routes to monoethylene glycol were identified, and are waiting to be tested in practice.17 In addition, several prediction tools such as DESHARKY, GEM-Path, RetroRules, and RetroPath are also available for metabolic pathway design. In general, these prediction tools propose potential pathways and rank them based on different factors employed in each framework to provide the most feasible route for the biosynthesis of desired chemicals. Some of the predicted engineering strategies might not lead to the successful production of target products, but we should not complain as this situation will change with our increasing knowledge of metabolism.
One great example demonstrating the successful application of a pathway prediction tool in metabolic engineering is the development of an E. coli strain capable of producing 1,4-BDO, which is a major commodity chemical used in the manufacture of polyurethanes including spandex and is produced from petroleum (Fig. 4).18 There had been no known organism that naturally produces 1,4-BDO. Thus, SimPheny BioPathway Predictor software was applied to elucidate all possible biosynthetic routes to 1,4-BDO from key central metabolites such as acetyl-CoA, L-glutamate, succinyl-CoA, and α-ketoglutarate. Assessment of the best pathway based on several criteria, including the theoretical maximum yield, pathway length, number of non-native and novel steps, and thermodynamic feasibility, revealed that two synthetic pathways via succinyl-CoA and α-ketoglutarate are most suitable for 1,4-BDO production (Fig. 4). After the construction and introduction of the proposed 1,4-BDO biosynthetic pathway into E. coli, the 1,4-BDO production was further optimized by deleting knockout targets suggested by OptKnock simulation (an in silico prediction algorithm for identifying gene knockout targets, which will be detailed in Section 6.1). Fed-batch fermentation of the final engineered E. coli strain resulted in the production of 18 g L−1 of 1,4-BDO from glucose.18 This process was further improved and commercialized by Genomatica (USA) in collaboration with Novamont (Italy), producing 30000 tons of bio-based 1,4-BDO per year.
There has been emerging interest in metabolic engineering of microorganisms for the utilization of C1 resources as a raw material. The reconstructed tetrahydrofolate (THF) cycle coupled with the reverse glycine cleavage reaction or Calvin–Benson–Bassham (CBB) cycle has been heterologously established in E. coli to confer the capability of assimilating CO2 and formic acid; formic acid can be considered as a liquid form of CO2. In the most recent study, the ftl, fch, and mtd genes from Methylobacterium extorquens were integrated into the E. coli genome for the construction of the reconstructed THF cycle.19 Also, the glycine cleavage operon was overexpressed. Further metabolic engineering to increase pyruvate synthesis from glycine and the reducing power allowed the engineered E. coli to grow on CO2 and formic acid as sole carbon sources, but at a low growth rate (a doubling time of 65–80 h). To increase the growth rate, adaptive laboratory evolution (ALE) was performed to improve the growth rate (a doubling time of <10 h).19
5. Evolutionary engineering
As described in the above section, systems metabolic engineering has enabled the construction of many different microbial cell factories capable of producing valuable natural and non-natural chemicals. However, the development of high-performing strains is still challenging, since the characteristics of the metabolic, regulatory, and signaling networks of host strains are not yet fully elucidated. Genotype-to-phenotype relationships were found to be much more complicated than previously thought (e.g., the so-called classical “one-gene/one-enzyme/one-function” paradigm), which consequently resulted in the rational engineering of microbial strains lagging behind efforts. Even for well-known microorganisms such as E. coli or S. cerevisiae, it is difficult to precisely predict their cellular behaviors in response to environmental or genetic perturbations. Thus, many iterative studies should be carried out to discover the optimal strain and process designs by simultaneously considering factors such as cell growth, product/substrate toxicity, performance indices (titer, yield, and productivity), byproduct formation, and culture conditions (e.g., culture medium, temperature, pH, oxygen transfer, and carbon source).When one is stuck with difficulties of further strain improvement by rational design, evolutionary engineering can be considered. Evolutionary engineering has emerged to compensate for the lack of comprehensive understanding of host strains. The key principle of evolutionary engineering is rapidly evolving the production strain to possess desired cellular and metabolic phenotypes by mimicking the natural evolution process, but much more rapidly by applying constant or increasing selection pressure. Evolutionary engineering is complementary to rational metabolic engineering to improve the performance of microbial cell factories, and is becoming more powerful through integration with automated cell culturing and monitoring systems as well as advanced DNA sequencing technology and multi-omics analysis. In this section, tools and strategies in two main fields of evolutionary engineering, adaptive laboratory evolution (ALE) and directed evolution, are described. Also, recent advances in high-throughput screening (HTS) methods are discussed.
5.1. Adaptive laboratory evolution
ALE is the process of screening and selecting strains having beneficial mutations through culturing strains under desired selection pressure for a prolonged period (Fig. 5). When cells are cultured under selection pressure for generations, cells that possess mutations giving competitively advantageous phenotypes will outgrow others. ALE is commonly performed in many biotechnological applications: (1) improving the carbon source utilization, (2) increasing the tolerance to products or metabolic intermediates, (3) increasing the product titer, yield, and productivity, (4) optimizing cell growth, and (5) identifying unidentified biological mechanisms.20 | ||
| Fig. 5 Adaptive laboratory evolution (ALE) of strains. During ALE, a microorganism is cultivated under desired selection pressure for prolonged periods until the evolved strain possesses beneficial mutations. The evolved strains possessing desired phenotypes are isolated and their genome sequences determined. Based on the genome sequencing data, reverse engineering is performed in the parental strain to reveal which genomic mutations are responsible for phenotypic improvement. For example, a strain evolved through ALE to become highly tolerant to a target chemical can produce a chemical of interest to a much high titer. Sometimes, the production strain itself can be evolved and used directly without reverse engineering. | ||
Recently, ALE has been successfully applied to rewire the unbalanced cellular metabolism of a genome-reduced E. coli strain.21 Theoretically, a microorganism having a minimal genome containing only essential genes and desired genotypes might be beneficial for more rapid growth and more efficient production of a desired product with reduced byproduct formation. A genome-reduced MS56 strain was constructed by deleting 1.1 million base pairs of the genome of the wild-type MG1655 strain, while retaining the genes related to cell growth. Unexpectedly, however, the MS56 strain showed a much lower growth rate than the wild-type strain in a glucose minimal medium. To restore cellular fitness, ALE was performed for 807 generations in M9 minimal medium, which allowed isolation of the eMS57 strain showing a cell growth rate comparable to the wild-type. Multi-omics analysis of eMS57 revealed that the evolved strain had remodeled transcriptional and translational profiles to recover unbalanced metabolism, especially exhibiting a decreased translational buffering capacity compared to MS65.21
Recently, a high-performance FFA producer was developed by implementing ALE to completely reprogram the metabolism of S. cerevisiae.22 Due to its strong inherent flux to ethanol, the production of target chemicals by metabolically engineered S. cerevisiae is often limited. Thus, the central carbon flux in S. cerevisiae should be reprogrammed to ensure the efficient production of target products. After establishing an efficient FFA biosynthetic pathway, S. cerevisiae was engineered to balance the supply of metabolites and cofactors for FFA biosynthesis, including acetyl-CoA, NADPH, and ATP, by overexpressing the gene encoding ATP citrate lyase (ACL), up-regulating the pentose phosphate pathway (PPP), and down-regulating the TCA cycle. After the abolishment of the ethanol production pathway by deleting pyruvate decarboxylase (PDC1/5/6), ALE was performed for about 200 generations to restore cell growth on glucose. Whole genome sequencing of isolated mutants showing higher cell growth rates revealed that a key mutation in pyruvate kinase (PYK1) resulted in the down-regulation of the glycolytic flux. Additionally, it was found that increased expression of an alternative pyruvate kinase isoform (PYK2) enabled cell growth on glucose without ethanol production. Fermentation of the final engineered S. cerevisiae strain produced 33.4 g L−1 of extracellular FFA, which is a 400% improvement compared with the starting strain. This study demonstrates the successful combination of rational metabolic engineering and ALE.22
Furthermore, ALE has been successfully applied to increase the tolerance to inhibitory substrates or products for the enhanced production of target chemicals. For example, microbial production of L-serine, a promising platform chemical for the production of pharmaceuticals and cosmetic ingredients, was hindered by its negative impacts on peptidoglycan synthesis and cell division.23 By performing ALE on an engineered E. coli strain lacking L-serine degradation pathways (sdaA, sdaB, tdcG, and glyA) for 45 days, an evolved strain that could tolerate 100 g L−1 of L-serine was isolated. This is remarkable as the wild-type MG1655 cannot tolerate more than 50 g L−1 of L-serine. Whole genome sequencing of evolved populations showing a positive phenotype revealed that key mutations in a potential L-serine binding site of homoserine dehydrogenase, which is responsible for branched amino acid synthesis, resulted in the enhanced tolerance to L-serine. When the native genes responsible for L-serine biosynthesis, including serA, serC, and serB, were overexpressed, fed-batch cultures of the parental strain before ALE and the evolved L-serine tolerant E. coli strain produced 8.3 and 37.3 g L−1 of L-serine, respectively, from glucose.23 The L-serine titer achieved is comparable to the highest titer obtained with an engineered C. glutamicum (42.6 g L−1 of L-serine from sucrose), demonstrating the power of ALE in metabolic engineering. Readers can consult an excellent review paper20 for more details and examples of recent ALE studies.
5.2. Directed evolution
As enzymes are catalysts in metabolic reactions inside the cell, metabolic engineers are always interested in optimizing their activities, stability, and substrate/product specificities and selectivities. It is possible to rationally redesign enzymes if their catalytic mechanisms and the relationship between the protein structure, sequence, and function are relatively well understood. However, such information is not always available, and the complexity of the structure/function relationship in enzymes limits the general application of rational design. Directed evolution allows the development of enzymes with new or improved functionalities by applying desired selection pressure without a comprehensive understanding of their structures or characteristics (Fig. 6). It involves iterative mutagenesis of an enzyme and selection of an evolved enzyme with improved function and activity through screening the library. Directed evolution is routinely used in protein engineering to enhance the catalytic activity, substrate/product specificity, thermal stability, and enantioselectivity of enzymes (Fig. 6).24 Directed evolution is performed on a gene encoding a protein (including enzymes) or a series of genes encoding multiple enzymes, while ALE is carried out on an entire microorganism aiming at genome-wide mutations.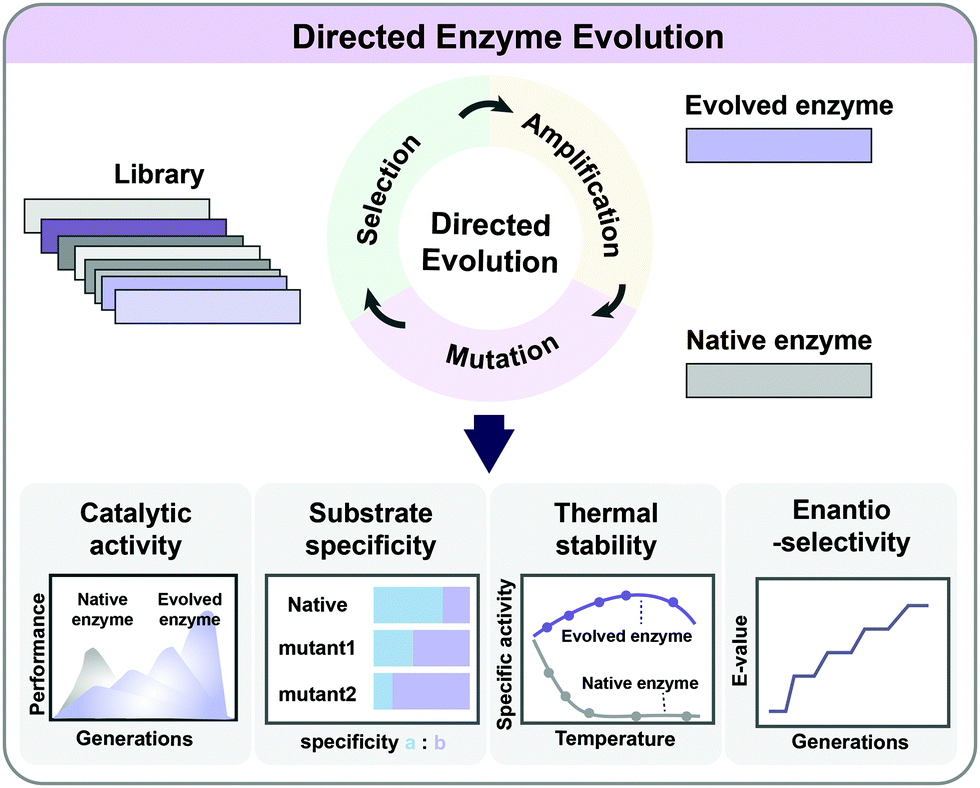 | ||
| Fig. 6 Directed evolution of enzymes. Through iterative cycles of mutation, selection and amplification, evolved enzymes with new or improved functionalities can be obtained. Directed evolution is commonly applied in protein engineering to improve the catalytic activity, substrate specificity, thermal stability, and enantioselectivity. | ||
The mutant library of enzymes during directed evolution can be generated by random mutagenesis or targeted mutagenesis. Random mutagenesis is an efficient way of increasing genetically diverse populations when information on the enzyme structure or key residues is not available. The simplest method of random mutagenesis is error-prone PCR (ep-PCR), which employs a proofreading activity-deficient DNA polymerase for PCR amplification to generate mutations throughout the gene encoding the target enzyme of interest. For example, isoprene synthase (ISPS) with 3.8-fold enhanced catalytic activity was successfully screened from a library generated by ep-PCR.25 In the isoprene biosynthetic pathway, the insufficient activity of ISPS converting dimethylallyl diphosphate (DMAPP) to isoprene is responsible for the limited production of isoprene. An engineered S. cerevisiae strain expressing the evolved ispS gene produced 3.7 g L−1 of isoprene by fed-batch fermentation using glucose.25 Other random mutagenesis methods, including DNA shuffling, incremental truncation for the creation of hybrid enzymes (ITCHY), and the staggered extension process (StEP), can be employed for the construction of mutant libraries.24
Although powerful, random mutagenesis has a big disadvantage of the need for generating a huge library of mutant enzymes, containing mostly unimproved mutants. Targeted mutagenesis (or focused mutagenesis) can maximize the possibility of generating a smaller library containing beneficial mutants by introducing mutations into only specific sites that are likely to determine the desired characteristic. In recent years, structural information on various enzymes has been increasingly available. Through combined bioinformatic and structural analyses of enzymes, core parts within the protein or de novo proteins can be identified to design a ‘small but smart’ library. For example, an aldehyde dehydrogenase from Cupriavidus necator (GabD4), which converts 3-hydroxypropionaldehyde (3-HPA) to 3-hydroxypropionic acid (3-HP), was evolved by applying targeted mutagenesis.26 To enhance its catalytic activity, potential engineering sites were predicted by homology modeling, and mutations were introduced into the corresponding amino acid residues by site-directed mutagenesis (SDM). Fed-batch culture of an engineered E. coli expressing the gene encoding the mutant GabD4 (E209Q/E269Q) having 1.4-fold enhanced activity on 3-HPA produced 71.9 g L−1 of 3-HP from glucose and glycerol as carbon sources.26
5.3. High-throughput screening
When enzyme assay or strain screening is possible through changes in color, fluorescence, size, higher cell density, or other easily observable characteristics, the desired mutant can be easily acquired using various techniques such as fluorescence-activated cell sorting (FACS), colorimetric assays, spectrophotometer or microfluidic sorting devices. However, such easily screenable characteristics are not available for many engineered enzymes and strains. In such cases, a significant bottleneck in evolutionary engineering is the lack of proper screening methods to select improved variants from a population of strains or enzymes. Thus, selection of an enzyme or strain with desired characteristics is often limited by the use of slow and low-throughput techniques such as high-performance liquid chromatography (HPLC), gas chromatography, or mass spectrometry. When there are no intuitive screening methods available, synthetic biology strategies can be applied to address this issue by developing new screening systems that expand the spectrum of traits that can be selected and screened.Biosensors that can monitor cellular metabolism in vivo are promising for HTS applications. Genetically encoded biosensors, which interact with intracellular metabolites and generate readable output, can be combined with HTS devices such as FACS to enable the rapid screening of the desired phenotype. Two types of biosensors are routinely used for facilitating the screening/selection process in metabolic engineering: transcription factor (TF)-based biosensors and RNA-based biosensors (Fig. 7). TFs have been explored to construct biosensors as they can recognize a variety of metabolites with high specificities. When the target molecule of interest interacts with the TF binding site, TFs respond as transcriptional activators or repressors to mediate the expression of the reporter gene to make an output signal in proportion to the target metabolite concentration. Various types of TF-based biosensors capable of screening metabolites, including L-valine, malonyl-CoA, glucaric acid, succinic acid, naringenin, and NADPH, have been developed.27
 | ||
| Fig. 7 High-throughput screening (HTS) of an enzyme or strain library. Libraries of enzymes or microbial strains generated by mutagenesis or evolution are screened by various HTS methods. Two major types of biosensor-based HTS methods, transcription factor-based biosensors and RNA-based biosensors, are commonly applied to efficiently select improved variants. Abbreviations are: FACS, fluorescence-activated cell sorting; RBS, ribosome binding site; RNAP, RNA polymerase; TF, transcription factor. | ||
In RNA-mediated biosensing systems, both transcription and translation of the target gene are regulated by changing the mRNA secondary structure (Fig. 7). Riboswitches are the most common RNA-based biosensor where the ribosome binding site (RBS) of mRNA is blocked from translation when the target molecule is missing. When the target molecule binds to the riboswitch, a conformational change of secondary RNA structures occurs to initiate the translation of reporter genes. Various synthetic riboswitches have been developed for sensing many different metabolites, such as L-lysine, L-tryptophan, theophylline, and thiamine pyrophosphate.27
6. Pathway optimization
After the construction of metabolic pathways for the biosynthesis of a target product, the metabolic flux toward the target chemical should be optimized to maximize the production. To achieve this, various systems biology strategies, including the construction and simulation of in silico genome-scale metabolic models (GEMs) and multi-omics analyses, have been developed to comprehensively understand the complex metabolic network and identify further engineering targets. In addition, recent advances in synthetic biology tools have facilitated the fine-tuned expression of the target genes in a more efficient and controllable manner, resulting in the rapid construction of high-performing strains. In this section, we discuss how tools and strategies from systems and synthetic biology can be applied for the optimization of the metabolic pathway fluxes.6.1. Pathway optimization by genome-scale metabolic simulation
GEMs are a mathematical representation of gene–protein-reaction (GPR) associations within an organism, and have played an essential role in understanding the whole metabolism of a host strain through simulation.28 Enzymes, metabolic reactions, and corresponding genes are assembled for the construction of a metabolic network of the microorganism. The metabolic reactions are expressed as equations which satisfy the mass balance of the chemical reactions according to the stoichiometry of metabolites. Therefore, the assembly of the reactions forms a linear system that enables engineers to easily analyze the mass actions of the metabolites at the systems level. Analyzing the mass action of metabolites in the form of metabolic flux at the genome-scale describes the metabolism of a microorganism, which can provide metabolic engineering strategies.First, a GEM specific to a microorganism of interest should be reconstructed (Fig. 8a). GEM reconstruction starts with the annotation of the genome sequence. To extract metabolic reactions from the genes, annotation of metabolic functions is conducted for the proteins encoded by the genes. With the aid of various annotation tools and available biochemical databases, the aforementioned procedures became easier. The retrieved reaction data are used to set up the mass- and charge-balanced reaction equations considering reaction stoichiometry, reaction reversibility, enzyme localization, and GPR associations, among others. Detailed instructions for the manual reconstruction of GEMs are well described in a protocol paper.29 More recently, automated tools are available for the reconstruction of draft GEMs.30 Fully or partially automated reconstruction procedures allow engineers to avoid tedious and labor-intensive steps in manual reconstruction processes. With the increasing availability of genomic data of various organisms, the convenient reconstruction procedure makes engineers utilize not only GEMs of model organisms, but also less studied microorganisms of specific interest. Even though thousands of GEMs have been reconstructed, many of them remain in draft versions, which need further refinements or modifications to precisely predict metabolic characteristics through manual curation and updates. As this is also a tedious step, several computational methods have been developed to upgrade those draft models by (semi-)automatic refinements and assessments.30 Altogether, the ever-increasing availability of GEMs serves as an important tool for analyzing the metabolic traits of microorganisms at the systems level.
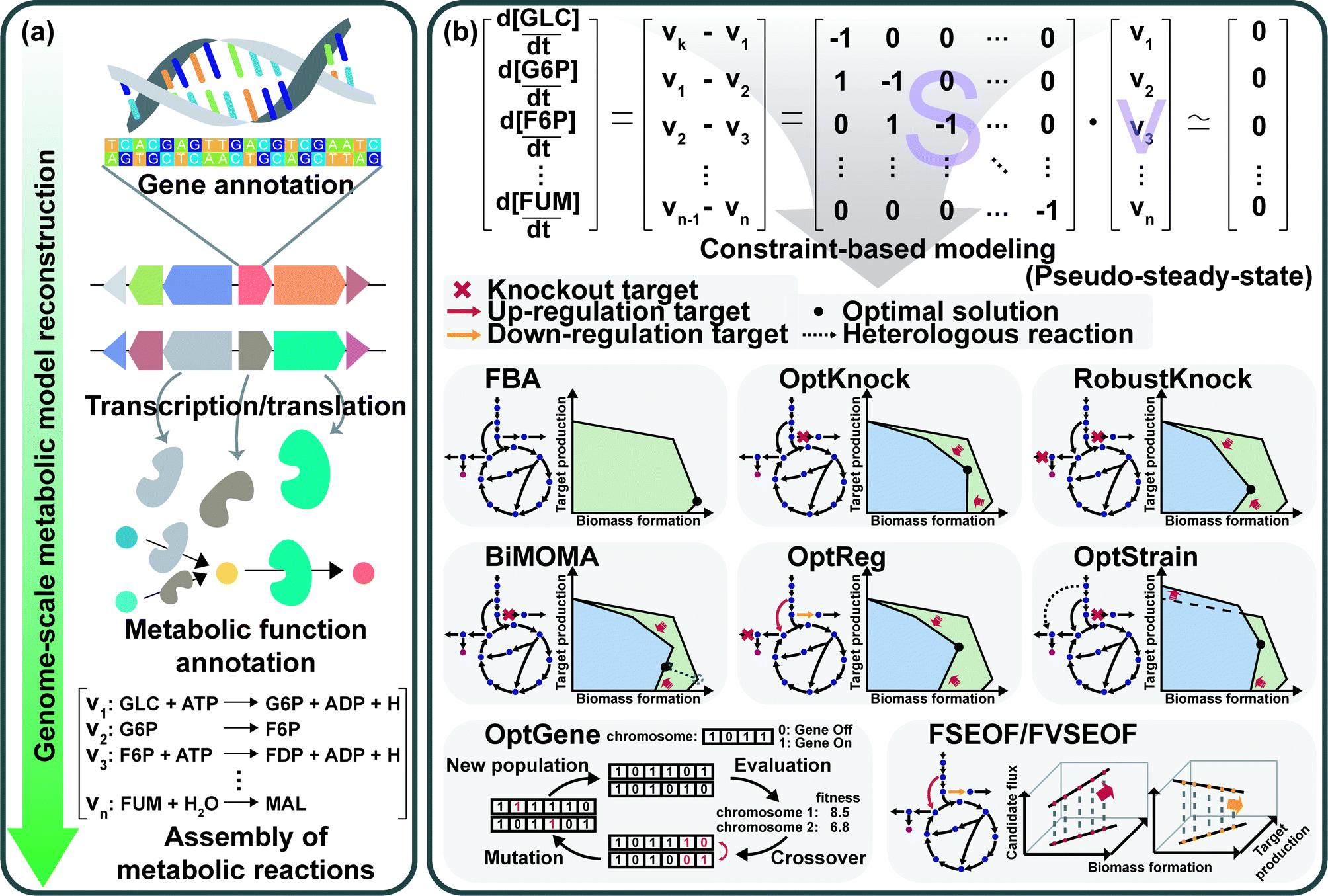 | ||
| Fig. 8 Reconstruction and applications of genome-scale metabolic models (GEMs). (a) A procedure to reconstruct a GEM. Starting with the genome sequence, annotation of genes, extraction of metabolic functions, and assembly of the metabolic functions in the mass- and charge-balanced reaction equations are performed. (b) Constraint-based modeling (CBM) approaches to perform simulations of the reconstructed metabolic network. A number of in silico genome-scale simulation algorithms including OptKnock, RobustKnock, BiMOMA, OptReg, OptStrain, OptGene, and FSEOF/FVSEOF have been developed for the prediction of knockout or amplification candidate genes to improve the production of a desired chemical. In the phenotypic phase planes, which show the achievable range of target chemical production rate versus biomass formation rate, the shaded green area and blue area represent the solution space of the wild-type and engineered strains, respectively. Abbreviations are: CBM, constraint-based modeling; FBA, flux balance analysis; GEM, genome-scale metabolic model. Abbreviated metabolites are: G6P, D-glucose 6-phosphate; GLC, D-glucose; F6P, D-fructose 6-phosphate; FDP, D-fructose 1,6-bisphosphate; FUM, fumarate; MAL, L-malate. | ||
The constraint-based modeling (CBM) approach has been employed for decades to interpret the reconstructed metabolic network.28 CBM accounts for metabolic flux distributions under various physicochemical constraints governed by thermodynamics, the pseudo-steady-state assumption, and even the culture conditions (Fig. 8b). Flux balance analysis (FBA), one of the most basic CBM methods, allows calculation of metabolic fluxes of all the reactions in the GEM through linear programming-based optimization under exponential growth conditions. In the exponential growth phase, balanced growth occurs without the accumulation of metabolites. Thus, the pseudo-steady-state assumption can be applied to make the differential mass balance equations into linear equations. Since the system is underdetermined (e.g., the number of fluxes to calculate is greater than the number of mass balance equations), FBA uses linear programming-based optimization of a biological objective function, such as maximizing the biomass formation rate, maximizing the product formation rate, maximizing the use of ATP, minimizing the byproduct formation rate, or other objectives of interest, together with other constraints such as upper and lower bounds of each flux. Among various objective functions, maximizing biomass production is the most frequently used one. Due to the underdetermined nature of the system, alternative solutions can be generated depending on how the simulation is done. To address this problem, several algorithms, such as flux variability analysis (FVA) and parsimonious flux balance analysis (pFBA), have been developed.28 FVA considers alternative flux distributions by analyzing possible flux ranges of all reactions while satisfying a base type value of an objective function. pFBA calculates flux distributions by minimizing the sum of all fluxes in the model while fulfilling an objective function.
GEMs can also be used for determining the alterations of metabolic fluxes upon various environmental or genetic perturbations. Most importantly, simulation of GEMs can be used to identify gene manipulation targets for metabolic engineering. Experimentally constructing all the potentially interesting strains having combinatorial gene knockouts and amplifications requires tremendous time, effort, and costs. Thus, in silico genome-scale metabolic simulations can significantly reduce the time, effort, and costs needed for constructing optimal engineered strains. For example, gene knockout targets can be identified by simulation of the GEM during which the metabolic flux is set to zero when the gene (enzyme) responsible for that reaction is knocked out. Single and multiple gene knockouts can be simulated until one finds an optimal knockout strain that shows desired metabolic flux distributions.
Several sophisticated algorithms have been developed for gene knockout and amplification simulations. The details of the methods and algorithms can be found in a review paper28 and references cited therein. Minimization of metabolic adjustment (MOMA) is quadratic programming, which minimizes changes in fluxes from a flux distribution of the wild-type strain upon genetic perturbation.31 MOMA was successfully applied to predict knockout candidate genes for enhancing lycopene production in metabolically engineered E. coli strains.32 OptKnock predicts gene knockout targets through bi-level mixed integer linear programming (MILP) that couples biomass formation with target chemical production (Fig. 8b).33 Similar algorithms have also been developed: RobustKnock for finding knockout reaction targets making the target chemical as an obligatory byproduct, BiMOMA for finding knockout reaction targets while satisfying the MOMA constraint, OptReg for finding reaction targets to be up- or down-regulated from the basal steady-state fluxes, and OptStrain for finding target reactions to be added and to be deleted (Fig. 8b). OptGene employs a genetic algorithm to identify knockout target genes by iteratively performing generation of the chromosome population, crossover among the chromosomes, evaluation of the generated population, and mutation on the chromosomes (Fig. 8b). Flux scanning based on enforced objective flux (FSEOF) and flux variability scanning based on enforced objective flux (FVSEOF) identify target reactions to be up-regulated (or down-regulated) that have positive (or negative) correlations with enforced objective reactions (Fig. 8b). In addition to these algorithms, various simulation methods are also available: flux response analysis that analyzes the response of an objective flux upon varying metabolic fluxes, flux-sum analysis that analyzes the turnover rates of metabolites, and elementary flux mode analysis that decomposes the metabolic network into the simplest functional vectors.
Recently, GEMs have served as a computational platform to accommodate massive data sets, including transcriptome, proteome, metabolome, and fluxome, for more comprehensive understanding of complex metabolism and identifying further metabolic engineering strategies. Various algorithms have been developed for the integration of the ever-increasing amount of massive omics data with GEMs.28 Also, the network coverage of GEMs has been expanded to deal with more biological processes: the genome-scale model of metabolism and macromolecular expression (ME-model)34 accounting for metabolism with gene expression and the whole-cell model35 accounting for all annotated gene functions. These models are expected to innovate systems metabolic engineering one step further.
6.2. Pathway optimization by using plasmids
A plasmid is a small circular extrachromosomal DNA that can replicate independently from the chromosome. Plasmids have been widely employed in metabolic engineering for the expression of target genes. The plasmid-based expression system is easy to manipulate and can be used to transform a host strain to display desired phenotypes. However, metabolic burdens imposed by plasmids often retard cell growth, leading to low productivity of producing strains. Thus, it is important to select a plasmid having an appropriate copy number, and consequently an appropriate gene dosage effect. Also, the promoter, terminator, and RBS of the gene expression cassette in the plasmid need to be carefully designed to achieve optimal gene expression levels (Fig. 9).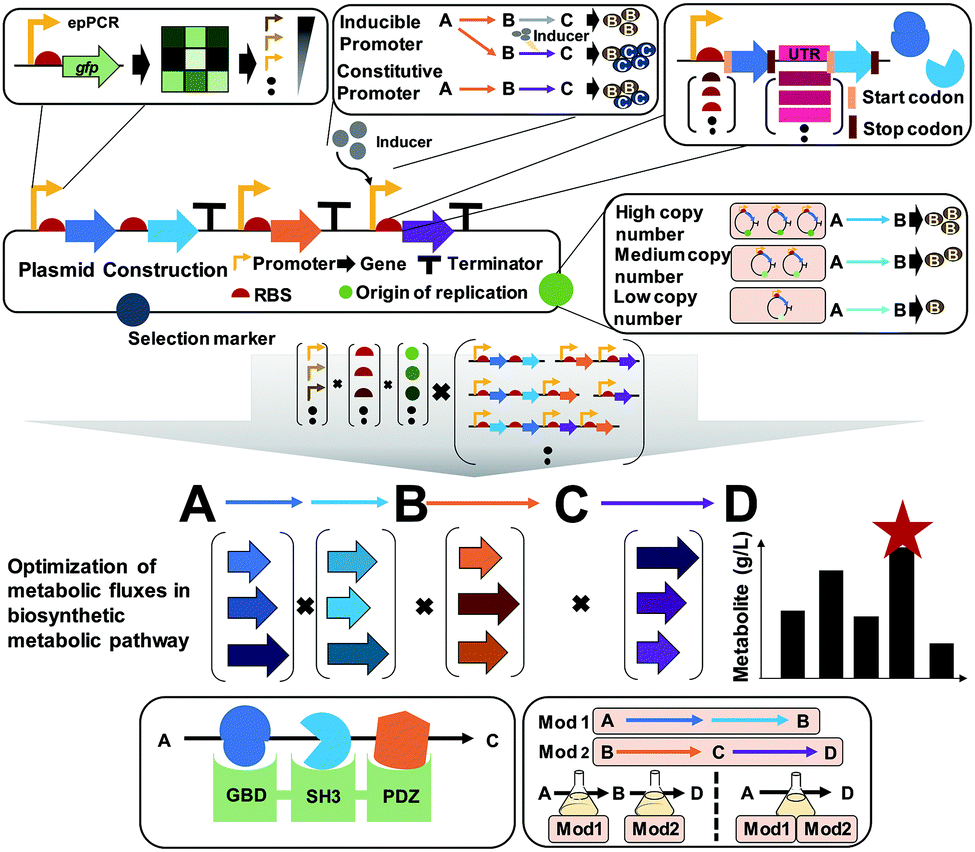 | ||
| Fig. 9 Plasmid-based gene expression for metabolic pathway flux optimization. A plasmid generally comprises the origin of replication, selection markers, and the promoter, a ribosome binding site (RBS), and a terminator for a target gene to be expressed. Multiple genes can also be expressed using a single promoter or multiple promoters. Various engineering tools and strategies have been developed for optimizing the individual components in a plasmid. Combinatorial optimization of all components in the plasmid construction should be performed for the balanced expression of multiple genes within complex metabolic pathways. Also, substrate channeling through tethering enzymes or using synthetic scaffolds can enhance metabolic fluxes toward a desired chemical. For the optimization of metabolic fluxes in long metabolic pathways, pathway modularization can be applied using at least two production strains, followed by culturing them together or sequentially. Abbreviations are: ep-PCR, error-prone PCR; Mod, module; UTR, untranslated region. | ||
Plasmids can have different copy numbers per cell depending on the type of origin of replication (ORI): low (e.g., pSC101 having ∼5 copies per cell), medium- (e.g., pBR322 having ∼20 copies per cell), and high-copy plasmids (e.g., pUC9 having ∼600 copies per cell) (Fig. 9). When more than one plasmid needs to be introduced into the host strain, their ORIs should be compatible to ensure their maintenance, i.e., plasmids belonging to different incompatibility groups need to be used. One might think that the use of high-copy plasmids will be good to enhance the metabolic fluxes by having more enzymes, which is sometimes true but in other cases not true. Differently from overexpressing a gene for the production of a recombinant protein as a product of interest, metabolic engineering requires the optimal and finely-tuned expression of a series of genes encoding enzymes for achieving the highest possible titer, yield and productivity. The use of high-copy plasmids sometimes causes side effects such as metabolic burden with cell growth retardation, formation of inclusion bodies (of enzymes), and accumulation of toxic intermediates, consequently resulting in poor production performance. In such a case, one can use low- or medium-copy plasmids or integrate the constructed biosynthetic pathway into a host chromosome (this will be explained in Section 6.4).
A promoter is a DNA sequence where RNA polymerase binds to initiate the transcription of the target genes. Promoters can be either inducible or constitutive. A constitutive promoter is an unregulated promoter that allows continuous transcription of a target gene. An inducible promoter is only active upon induction with chemical agents, temperature, or light (Fig. 9). Among these, chemically inducible promoters are the most widely utilized. Obviously, the use of expensive chemical inducers such as isopropyl β-D-1-thiogalactopyranoside (IPTG) for gene expression is not desirable for industrial-scale production. In industry, constitutive promoters are preferred whenever possible. The specific sequence of the promoter defines the rate of transcription, which is called the promoter strength. For the precise control of gene expression, various synthetic promoters of different strengths have been developed by employing strategies such as ep-PCR, SDM, hybrid promoters, randomization of the non-conserved region, and design of synthetic transcriptional factor binding sites. Recently, promoters facilitating the stable expression of target genes regardless of any environmental or genetic perturbations were developed by engineering an incoherent feedforward loop (iFFL) using a transcription activator-like effector (TALE; which will be described in Section 6.4). These iFFL-stabilized promoters exhibited constant gene expression levels in various plasmids having different copy numbers.36 Terminator, a nucleic acid sequence located at the end of a gene or operon, plays an important role in controlling the gene expression by mediating transcriptional termination and triggering the release of mRNAs. The use of the right terminator is also important to ensure the stable transcription of genes to the desired levels.
An RBS is a sequence of nucleotides in mRNA, which engages a ribosome for initiating translation. Manipulation of the translation initiation rate (TIR) by engineering the RBS is an efficient strategy to achieve desired expression levels of genes encoding the target enzymes. A number of synthetic RBS sequences have been developed by employing several computational tools such as UTR designer, RBS designer, RBS calculator, and RBS library calculator (Fig. 9). These modeling tools generally predict the TIRs of RBSs and help design a library of RBSs giving diverse expression levels by changing a few sequences of the RBS. Recently, an algorithm RedLibs was developed to generate smart RBS libraries with small and user-specific sizes that uniformly cover the entire accessible TIR space in a linear manner.37 These smart RBS libraries can minimize experimental effort while providing high TIR coverage. The degenerate RBS libraries generated in silico were applied for the production of violacein in E. coli. During the violacein biosynthesis from L-tryptophan, the formation of a byproduct, deoxyviolacein, is inevitable due to the the presence of a branched pathway. Thus, the expression levels of the vioC, vioD, and vioE genes involved in the violacein biosynthetic pathway were modulated with the optimal RBSs selected from the smart RBS library. As a result, an engineered E. coli strain showed a 1.35-fold increase in the violacein fraction with 91% purity.37
The construction and optimization of a desired biosynthetic pathway are not the end. In a multi-step conversion pathway typically needed in metabolic engineering, some intermediate metabolites can accumulate and cause problems. In such a case, substrate channeling can be applied to enhance metabolic fluxes toward a target product by directly fusing the enzymes in cascade reactions or tethering through the use of synthetic scaffolds (Fig. 9). Substrate channeling allows spatial colocalization of enzymes and thus efficient enzyme-to-enzyme substrate transfer to biosynthesize a target product through decreased substrate diffusion, reduced metabolic burden, low levels of accumulation of toxic intermediates, and increased levels of local intermediates to the corresponding enzymes. A similar approach of enzyme compartmentalization can be employed. For further details on substrate channeling systems and compartmentalization, readers can consult many papers cited in a review paper.38
When quite a long biosynthetic pathway is required for the production of a desired product, it will be difficult to construct a strain expressing all these long pathway genes, causing severe metabolic burden. In such a case, the metabolic pathway can be divided into several modules to effectively balance the metabolic fluxes (Fig. 9). This approach requires construction of at least two production strains with optimally distributed pathway modules. Then, stepwise fermentation and/or co-culture of these strains can allow production of a desired product through multi-step metabolic reactions with a reduced metabolic burden.
6.3. Pathway optimization by using regulatory RNAs
Regulatory RNAs have been increasingly used as key modulators for controlling gene expression by interacting with DNA, RNA, protein, or metabolite. Among regulatory RNAs, trans-acting RNAs, which can recognize specific target DNA or RNA sequence by the base-pairing principle, are widely used due to their modular, tunable, and designable characteristics. Various synthetic biology tools using trans-acting RNAs have been developed for enabling genome-wide engineering and fine-tuning of gene expression levels. Among them, several representative tools, including synthetic small regulatory RNA (sRNA), RNA interference (RNAi), antisense RNA (asRNA), and small transcription activating RNAs (STARs), are discussed in this section.The sRNA system comprises a target-specific sequence and a scaffold sequence (Fig. 10a). The target-specific sequence allows sRNA to bind to the translation initiation region of the target mRNA. The scaffold sequence recruits an RNA chaperone Hfq for the regulatory activity (Fig. 10a). The sRNA system can be computationally designed to achieve a desirable level of knockdown of the target gene expression on a genome-wide scale. The sRNA system was applied to increase the production of tyrosine and cadaverine in engineered E. coli strains. By knocking down the expression of the tyrR (encoding tyrosine repressor) and csrA (encoding carbon-storage regulator, which regulates genes involved in glycolysis) genes using sRNAs in a tyrosine producing recombinant E. coli strain, the tyrosine production increased to 21.9 g L−1 in fed-batch fermentation. Similarly, knocking down the expression of the murE gene encoding UDP-N-acetylmuramoyl-L-alanyl-D-glutamate:meso-diaminopimelate ligase in a cadaverine producing recombinant E. coli strain allowed increased cadaverine production to 12.6 g L−1 in fed-batch fermentation.39 Through the application of the sRNA system, which allows rapid, easy, modular, portable, multiplex, and fine-tunable knockdown of the expression of desired genes, it was possible to enhance the production of various chemicals in E. coli including 1,4-diaminobutane, violacein, indigo, and 1,3-diaminopropane. Furthermore, the sRNA system was successfully applied to engineer Clostridium acetobutylicum, which is one of the most challenging host strains to genetically manipulate. Thus, the sRNA platform technology is expected to facilitate metabolic engineering of various microorganisms which have not been explored as host strains due to the lack of proper genetic engineering tools.
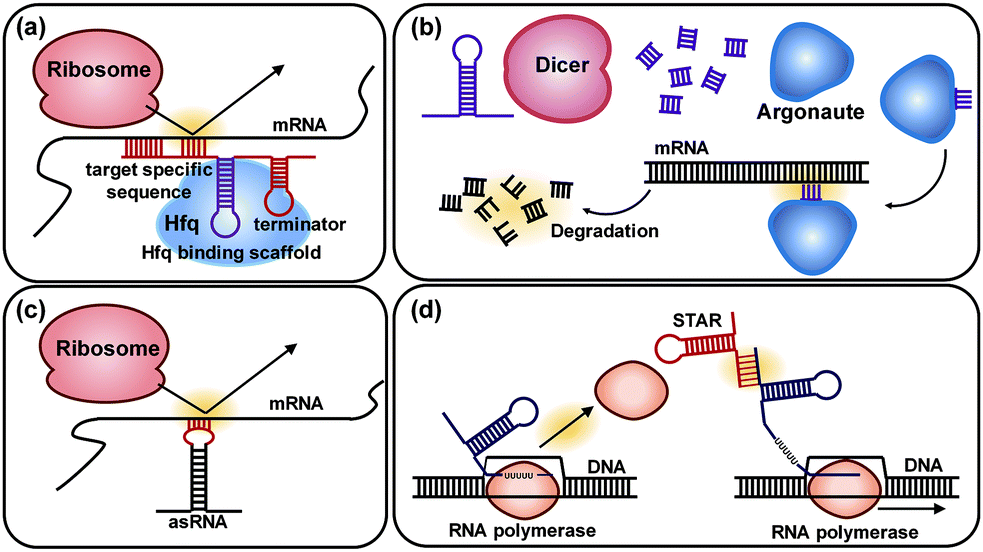 | ||
| Fig. 10 Regulatory RNAs as tools for metabolic pathway flux optimization. (a) The synthetic small regulatory RNA (sRNA) system comprises a target-specific sequence that binds to the translation initiation region and a scaffold sequence that recruits the Hfq protein for regulation. When sRNA is bound to the target mRNA, ribosomes can no longer bind to translate the mRNA; thus, the sRNA system allows translation-level gene expression modulation. The binding strength of sRNA can be designed by precalculation for modulating the translation-repression level. (b) The RNA interference (RNAi) system comprises small interfering RNA (siRNA), a dicer protein, and argonaute. Once siRNA is generated from degradation of foreign double-stranded RNA by the dicer protein, an argonaute guided by siRNA cleaves the target mRNA. (c) Antisense RNA (asRNA) having a complementary sequence to the target mRNA for gene expression regulation. A single-stranded RNA binds to the target mRNA, preventing translation by ribosomes. (d) The small transcription activating RNAs (STARs) system comprises an asRNA that is specific to the anti-terminator sequence for up-regulation of gene expression. The STAR asRNAs bind to the 5′ stem of terminator hairpin structures to activate transcription. | ||
RNAi is a representative gene silencing system in eukaryotes. In the RNAi system, a dicer protein degrades a heterologous double-stranded RNA to a small interfering RNA, which guides an RNA-induced silencing complex protein (RISC) to the target mRNA. The RISC protein has an essential catalytic component called argonaute that can cleave the target mRNA (Fig. 10b). The RNAi system was further combined with an evolutionary engineering strategy, resulting in RNAi-assisted genome evolution (RAGE) that enables RNAi-assisted genome-scale evolution of S. cerevisiae. As a proof-of-concept, RAGE was applied to develop an acetic acid-tolerant S. cerevisiae strain, and three synergistic knockdown target genes that confer improved tolerance to acetic acid were found. The mutant strain obtained through RAGE showed a 20-fold increase in cell growth on 0.9% (v/v) acetic acid compared with the parental strain.40
An asRNA is a single-stranded RNA that has a complementary sequence to the target mRNA. When the asRNA is bound to the target mRNA, translation is blocked because the ribosome can no longer bind to the RBS (Fig. 10c). Compared with sRNA and RNAi, the use of asRNAs has been limited since the gene silencing mechanism of asRNA is not fully understood yet. A few studies have demonstrated the application of the asRNA system for the enhanced production of chemicals such as butanol, resveratrol, naringenin, and 4-hydroxycoumarin.
Other than down-regulation of gene expression using the above RNA-based tools, up-regulation of the target genes is also possible by RNA. The expression activation tool named small transcription activating RNAs (STARs) uses synthetic sRNAs that bind to the upstream region of a target gene to prevent the formation of intrinsic terminator hairpin structures and activate transcription (Fig. 10d).41 Although the STARs system is not available for up-regulating chromosomal genes yet, it can be used as a new tool in the construction of transcriptional logic gates in synthetic gene circuits.
6.4. Pathway optimization by genome engineering
As discussed in Section 6.2, applying a plasmid-based system is generally effective and convenient in expressing genes involved in the biosynthesis of a target product. However, the use of plasmids has some problems including metabolic burden, plasmid instability, and expensive cost of selection markers. To address these problems, direct chromosomal genome engineering tools, including deletion, insertion, repression, overexpression, and mutations in nucleotide sequences, have been developed. In this section, such genome engineering tools with associated examples for metabolic pathway construction and optimization are described.Recombineering, recombination-mediated genetic engineering, is one of the most commonly utilized genome engineering tools. In the past, RecABCD system-based homologous recombination was the most widely used recombineering method. However, due to its low efficiency, the λ Red recombination method was applied for recombineering with a higher recombination efficiency of linear double-stranded DNA (dsDNA). Genes of interest can be efficiently integrated into the chromosome or genes of interest in the chromosome can be knocked out by using the λ Red recombination method. In most cases, a selection marker (e.g., an antibiotic resistance gene) is used during the integration, and thus needs to be removed. In combination with the λ Red recombination method, site-specific recombination systems including Cre-lox and Flippase-Flippase Recombinase Target (Flp-FRT) are often used for this purpose (Fig. 11a). The Cre-lox recombination method utilizes recombinase Cre and its specific locus of crossover loxP site derived from bacteriophage P1. This recombination mediated by the Cre protein catalyzes the in vivo site-specific excision (also integration and inversion) of DNA sequences flanked by two loxP recognition sites. Similar to Cre-lox recombination, Flp-FRT recombination utilizes the Flp recombinase derived from S. cerevisiae that recognizes a pair of FRT sequences flanking a genomic region of interest. The λ Red recombination method with site-specific recombination is one of the most popular recombineering methods for the deletion of chromosomal genes by a one-step inactivation method (Fig. 11a).
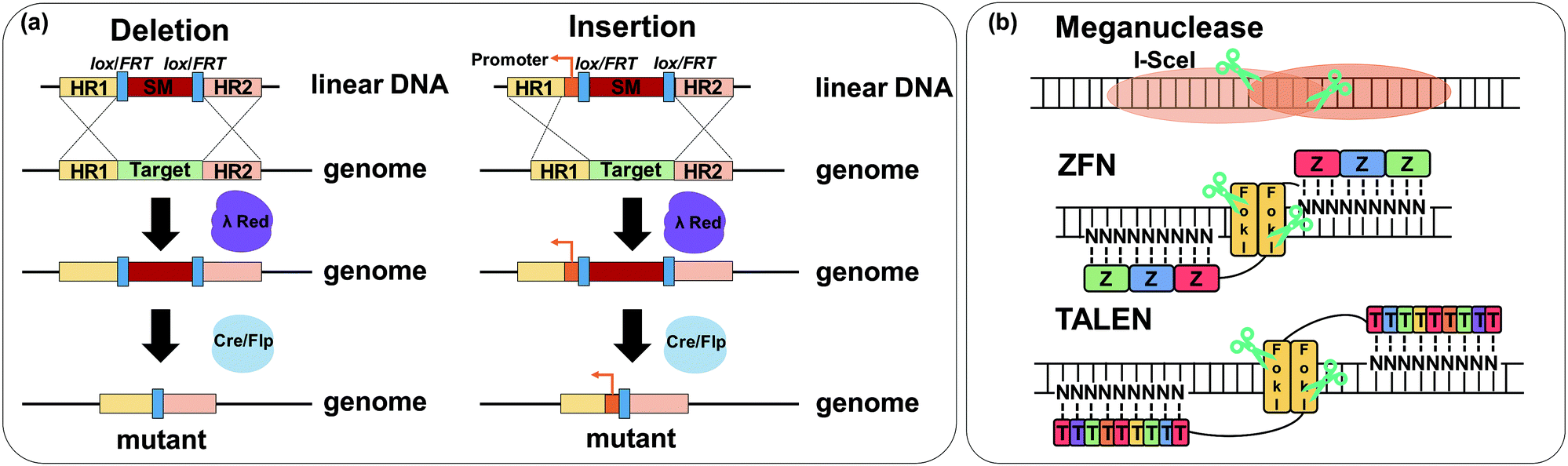 | ||
| Fig. 11 Genome engineering strategies including recombineering mediated by λ Red recombination with site-specific recombination and genome engineering for the targeted cleavage of the genomic region. (a) λ Red-based site-specific recombination. By applying two recombination systems, site-specific recombineering such as gene deletion or insertion can be performed. (b) Meganucleases, zinc finger nucleases (ZFNs), and transcription activator-like effector nucleases (TALENs) for genome engineering. A meganuclease recognizes 12–40 base pairs (bps) for cleavage. A ZFN comprises zinc finger protein (ZFP) domains for DNA binding through the recognition of a 3 bp DNA sequence and restriction of endonuclease FokI. Similar to ZFNs, a TALEN comprises a TALE for DNA binding through the recognition of only a single bp and FokI. N represents any nucleotide. Abbreviations are: HR, homology region; SM, selection marker. | ||
A meganuclease, also known as a homing endonuclease, recognizes and cleaves the sites of 12 to 40 base pairs (Fig. 11b). After the first discovery of I-SceI meganuclease from S. cerevisiae, other meganucleases from different hosts including Chlamydomonas reinhardtii and Desulfurococcus mobilis were reported. The characteristic of the meganuclease having a long recognition site led to its scarce appearance in the genome, which makes direct genome engineering challenging. Thus, to target and cleave the genomic region of interest more specifically, a fusion endonuclease called zinc finger nuclease (ZFN) was developed (Fig. 11b).42 By engineering and combining domains of zinc finger protein and restriction endonuclease FokI, a genomic region of interest was targeted more specifically with two ZFNs. However, each zinc finger protein recognizes three base pairs at once, requiring at least a library size of 64 modules to cover all types of base pairs. This difficulty together with a high chance of off-target effects led to the development of transcription activator-like effector nucleases (TALENs) as an alternative nuclease by combining FokI and TALEs (Fig. 11b).43 TALEs were first discovered in plant pathogen Xanthomonas spp. and are composed of 33 to 35 amino acid repeats. Each repeat of the TALE was reported to have specific binding to a single nucleotide giving a small size of the library (e.g., for four types of base pairs, only four corresponding types of repeats are necessary for binding).
Clustered regularly interspaced short palindromic repeats (CRISPR) along with the CRISPR-associated protein (Cas) system (CRISPR/Cas), originating from adaptive immune systems of microorganisms to cleave foreign invaders (e.g., bacteriophages), has recently been receiving much interest as a genome engineering tool since it enables rapid, simple, and robust engineering compared with other conventional genome engineering tools. The CRISPR-Cas system is useful for engineering even so called difficult-to-engineer strains. Among various types of CRISPR/Cas systems, the CRISPR/Cas9 system derived from Streptococcus pyogenes, which belongs to type II and class 2, has been the most widely employed in genome editing applications (Fig. 12a). The detailed mechanisms and other types of CRISPR/Cas systems can be found in an excellent review paper44 and papers cited therein.
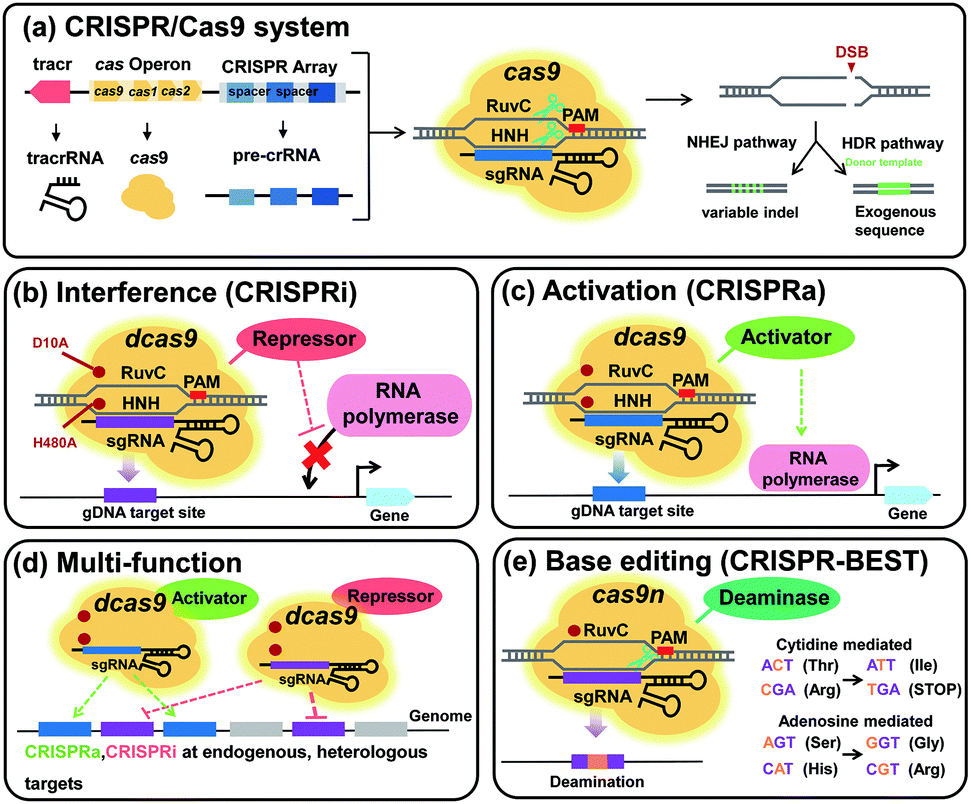 | ||
| Fig. 12 CRISPR/Cas9 systems for genome engineering. (a) Mechanism of CRISPR/Cas9 genome editing. The effector nuclease Cas9 is guided to the target DNA with the help of an RNA duplex comprising CRISPR RNA (crRNA) and trans-activating CRISPR RNA (tracrRNA). Cas9 scans for the presence of an NGG sequence at the end of the protospacer, called the protospacer-adjacent motif (PAM), to determine the target sites to be cleaved. Cas9 contains two nuclease domains each breaking a different DNA strand. The HNH domain breaks the complementary sequence of the crRNA and the RuvC domain breaks the non-complementary sequence of the crRNA. Double strand breaks (DSBs) induced by Cas9 are recovered by one of the two endogenous DNA repairing systems: error-prone based non-homologous end joining (NHEJ) giving random mutations or homology-directed repair (HDR) with a required repair donor template for precisely targeted mutation. (b) CRISPR interference (CRISPRi) employing dCas9 to repress gene expression. dCas9 is a catalytically inactive or dead Cas9 protein having D10A and H480A mutations, resulting in a lack of endonuclease activity. The CRISPR/dCas9 system physically blocks the binding of RNA polymerase, and thus transcription initiation and elongation are blocked, resulting in down-regulation of target gene expression. (c) CRISPR activation (CRISPRa) to activate gene expression. The CRISPR/dCas9 complex combined with a transcriptional activator domain binds to the upstream promoter region for up-regulating the target gene expression. (d) A multi-functional CRISPR/Cas9 system combining CRISPRa and CRISPRi enables multiple gene expression regulations, activation and repression, simultaneously. (e) CRISPR-Base Editing SysTem (CRISPR-BEST) to edit single nucleotides on the chromosome. A fusion protein combining Cas9 nickase (Cas9n) having a D10A mutation and specific deaminase performs the specific conversion of one nucleotide in the DNA sequence. Cytidine-mediated base editing allows conversion of cytidine (C) to thymidine (T), while adenosine-mediated base editing converts adenosine (A) to guanosine (G). | ||
After the Cas9-guide RNA complex introduces double strand breaks (DSBs) to the target DNA sequence, either homology-directed repair (HDR) or non-homologous end joining (NHEJ) machinery is recruited to repair the DSBs (Fig. 12a). During HDR, which is the dominant route of DSB repair in prokaryotes, an additional homologous DNA fragment (i.e., donor template DNA) is required as a sequence template that directs the repairing. In contrast, NHEJ, which is dominant in eukaryotes and mostly absent in prokaryotes, is independent of a homologous DNA template and randomly inserts or deletes DNA base pairs at the DSB site, introducing a frameshift mutation to the target gene at a 67% chance.
In addition, the CRISPR/Cas9 system has been repurposed to knock down (or repress) and activate (or overexpress) the target chromosomal genes. In these repurposed CRISPR/Cas9 systems, a catalytically inactive or dead Cas9 (dCas9) protein having D10A and H840A mutations in S. pyogenes Cas9 and lacking in endonuclease activity is utilized. CRISPR interference (CRISPRi) utilizes the dCas9-guide RNA ribonucleoprotein (RNP) complex that can specifically bind to the target DNA sequences and interfere with initiation or elongation of transcription by RNA polymerase, leading to repression of the target genes (Fig. 12b). Recently, CRISPRi was applied to increase the production of β-amyrin, a pentacyclic triterpenoid compound, using an engineered S. cerevisiae strain.45 Using a previously developed β-amyrin producing S. cerevisiae strain, seven genes involved in competing pathways (ADH1, ADH4, ADH5, and ADH6 in ethanol production pathways, CIT2 and MLS2 in the peroxisomal acetyl-CoA consumption pathway, and ERG7 in the ergosterol synthesis pathway) were simultaneously repressed using CRISPRi. Fed-batch fermentation of the final engineered S. cerevisiae strain produced 156.7 mg L−1 of β-amyrin, the highest titer reported in yeast.45 To activate the expression level of the target genes, dCas9 has been fused to a transcriptional activator and guided to upstream of the promoter, leading to transcription activation (CRISPRa) (Fig. 12c). CRISPRa has been actively employed in eukaryotes due to the availability of various transcriptional activators, while its more wide application in prokaroytes is to be seen.
During the optimal design and construction of metabolic pathways, the number of genes to engineer increases. Examination of the synergistic effect of combinatorial engineering on improving the phenotype or production performance requires sequential engineering of numerous genes, which is time-consuming and labor-intensive. To solve this problem, the CRISPR/Cas9 system has been exploited to simultaneously engineer multiple target genes (Fig. 12d). For example, an orthogonal tri-functional CRISPR system that enables simultaneous transcriptional activation, interference, and gene deletion (CRISPR-AID) was developed by optimizing the hybrid of CRISPRa, CRISPRi, and gene deletion.46 As a proof-of-concept, this CRISPR-AID system was applied in a β-carotene producing S. cerevisiae strain and enhanced its production by 3-fold by the deletion of ROX1 (encoding a stress-responsive transcriptional regulator), overexpression of HMG1 (encoding a rate-limiting enzyme of the mevalonate pathway), and repression of ERG9 (an essential gene at the branching point of the β-carotene biosynthesis and endogenous sterol biosynthesis) simultaneously.46
Also, targeted base editing technologies have been developed by combining Cas9 nickase (Cas9n) and base deaminases (Fig. 12e). While the Cas9n protein having a D10A mutation introduces a single strand break (SSB) to the target site and facilitates DNA repair, either cytidine deaminase or adenosine deaminase converts cytidine (C) to thymidine (T) or adenosine (A) to guanosine (G), respectively (Fig. 12e). Recently, a highly efficient DSB-free CRISPR-Base Editing SysTem (CRISPR-BEST) with single nucleotide resolution for actinomycetes was developed by employing a cytidine deaminase (CRIPSR-cBEST) and an adenosine deaminase (CRISPR-aBEST).47 CRISPR-BEST could be used to introduce mutations on the genomes of non-model actinomycetes, Streptomyces collinus Tü 365 and Streptomyces griseofuscus DSM 40191 strains, with high efficiencies and low off-target effects.47
More recently, a novel “search-and-replace” Cas9-based system with high efficiency and versatility named prime editing was developed. This precise genome editing was established without introducing DSBs or donor templates.48 In this system, the single guide RNA was modified to prime editing guide RNA (pegRNA) by extending its 3′ end with a sequence complementary to the non-target strand of the target DNA and an additional nucleotide sequence that directs genome editing. In addition, Cas9n was fused to reverse transcriptase (rT). As a result, the Cas9n-rT-pegRNA RNP complex introduces an SSB to the non-target strand of the target gene, and the 3′ end of the pegRNA complementarily binds to the cleaved non-target strand. Subsequently, rT fused to the Cas9n extends the 3′ end of the cleaved non-target strand using the complementarily bound 3′ end region of the pegRNA. The resulting single-stranded DNA flap serves as a template for DNA repair machineries recruited to the SSB site of the target gene. With prime editing, efficient and precise genome editing including all 12 possible base-to-base conversions, insertions, deletions, and their combinations was conducted with low off-target effects. Thus, prime editing is expected to be increasingly employed in modifying genomes of a number of different cell lines in an unprecedentedly efficient manner.48
7. Scale-up fermentation
The above sections described various tools and strategies employed for the development of overproducer strains. During and after strain development, lab-scale fermentation is performed to validate the performance of the strain and to find optimal fermentation conditions. After confirmation of their production performance in the lab-scale fermentor, pilot-scale (and sometimes demoplant-scale) fermentations need to be performed to find the optimal production conditions and also to see whether the developed strain can be used for the large-scale production of the desired product without unexpected problems (Fig. 1). In general, a bioprocess developed using a lab-scale fermentor (0.5–30 L) is first translated into a pilot-scale fermentor (30–3000 L) to assess the fermentation performance in the scaled-down version of a much larger production fermentor. If needed, fermentations can be performed in a demoplant-scale (3000–20000 L) fermentor not only for further optimization of the bioprocess, but also for actual production of a desired bioproduct for marketing and sales purposes, which can lower the risk of building and operating a costly full-scale (20000–2000000 L) production fermentor. If one encounters problems in pilot-scale fermentation reproducing the high performance of the lab-scale fermentation, the developed strain needs to undergo additional metabolic engineering to address these problems; this is why it is important to perform systems metabolic engineering (the second upstream process) by considering the raw materials (the first upstream process), fermentation (midstream process) and recovery and purification (downstream process). Some of the common things to be considered during the scale-up are described below.
Minimizing manufacturing costs without affecting the fermentation performance is considered as a priority in scale-up fermentation. It is obvious that the use of expensive ingredients, such as inducers for gene expression and antibiotics for maintaining plasmid stability and preventing contamination, is not possible in large-scale fermentations. As described earlier, one of the most commonly used methods is to integrate all the biosynthetic pathways into the genome and express the corresponding genes under constitutive promoters of desired strength. If the desired performance is not achieved through the chromosomal integration and the use of plasmid-based expression is needed, several different antibiotic-free plasmid systems, such as a toxin/antitoxin system, metabolism-based system, and operator repressor titration system, can be applied instead. It should also be noted that the number of generations (e.g., the number of cell divisions) in fed-batch fermentation, the industrially preferred standard mode of operation, is not that high; starting with the initial optical density (OD) of 0.1–0.3 to the final OD of 100–300 needs only 9–12 generations. Sometimes, it can be confirmed a priori that plasmids are maintained stably during the entire fermentation without selection pressure (e.g., addition of antibiotics). Nonetheless, one does not want the large-scale fermentation to fail due to the high costs associated with it, and thus it is preferred to make the strain stable as described above. Impurities present in inexpensive raw carbon sources or industrial-grade medium constituents often lead to the accumulation of inhibitors and non-fermentable components in the fermentor, which can consequently cause cell growth retardation or even contamination. In such a case, ALE can be performed to increase the tolerance level.
In scale-up fermentation, the flow patterns inside the large bioreactor will differ from the lab-scale bioreactor due to its different physical properties. If homogeneous mixing is not available, gradients of feed and oxygen concentrations inside the bioreactor can cause reduced cell growth and increased production of byproducts such as formic acid, lactic acid, and succinic acid.3 Computational fluid dynamics, which can predict mixing behaviors or shear stress on cell morphology under the specific fermentation conditions, can be employed to develop sophisticatedly designed impellers enabling efficient oxygen transfer and mixing of substrates without disrupting cells.
Another main challenge in scale-up fermentation is maintaining the genomic stability of high-performing strains. In some cases, low- or non-producing populations can emerge when they fail to tolerate metabolic burden or product toxicity.3 The emergence of escape variants is crucial in the context of industrial fermentation as they reduce the product titer, yield, productivity, and quality. While tools for completely preventing the emergence of sub-performing populations are unavailable, synthetic control circuits, which regulate cellular metabolism in response to extracellular and intracellular perturbations, might be employed for mitigating genetic heterogeneity problems.
8. Downstream process
The downstream process to recover a target chemical from the culture broth and purify it with high yield and purity is an essential finishing step of any commercial fermentation process (Fig. 13). It should be noted that the cost of separation and purification generally accounts for 20–50% of the total production cost. Thus, the development of an economically-feasible and energy-efficient downstream process is essential for successful commercialization of bioprocesses. | ||
| Fig. 13 Overview of the downstream recovery and purification processes. To purify the extracellular products, cells are first removed by separating them from the fermentation broth by centrifugation or filtration. For the intracellular products, cells collected by centrifugation or filtration are disrupted using a bead mill or French press. After the solution containing a target product is obtained, various separation and purification technologies are employed. Some representative methods include equilibrium-based (adsorption and distillation), membrane-based (reverse osmosis, microfiltration, ultrafiltration, and electrodialysis), affinity-based (adsorption, ion exchange, and chromatography), solid–liquid separation (filtration, crystallization, and precipitation), and liquid–liquid separation (solvent extraction and aqueous two-phase extraction). These methods are combinatorially used depending on the type and characteristics of a product, the presence of contaminating compounds, and several other factors including the targeted purity of a product. | ||
After the successful scale-up fermentation to the full-scale fermentor, various recovery and purification techniques can be applied for the efficient recovery of target products; of course, the recovery and purification processes are being developed together with lab-scale fermentation instead of waiting until the scale-up fermentation studies are finished. For the purification of extracellular products, the first step is separation of cells from the fermentation broth by centrifugation or filtration. For intracellular products, cells collected by centrifugation or filtration are disrupted by a bead mill or French press. Once a solution containing a desired product is obtained, several different recovery and purification techniques can be employed depending on the characteristics of the product-containing solution. These techniques can be broadly classified based on their basic separation principles: equilibrium-based separation (e.g., absorption, distillation, and liquid–liquid extraction), affinity-based separation (e.g., adsorption, ion-exchange, and chromatography), membrane separation (e.g., reverse osmosis, microfiltration, ultrafiltration, and electrodialysis), solid–liquid separation (e.g., conventional filtration, direct crystallization, and precipitation), and liquid–liquid extraction (e.g., solvent extraction and aqueous two-phase extraction), among others (Fig. 13). Again, strain development through systems metabolic engineering can contribute significantly to lowering the costs of the downstream process through reducing the byproduct production, which also increases the yield of the desired product.
9. Conclusion and future challenges
As described above, systems metabolic engineering has become an essential strategy for the development of high-performance microbial strains capable of producing a desired chemical or material with high titer, yield, and productivity. However, many challenges still remain to be addressed to firmly establish biorefineries through systems metabolic engineering.First, development of microbial strains that can efficiently utilize all the components derived from non-edible biomass (see Section 2) is of the utmost importance to avoid the so called food versus fuel issue. To fully utilize these massive non-edible substrates, strain construction through metabolic engineering should be accompanied by the development of cost-effective, eco-friendly, and highly-efficient pretreatment processes of deconstructing the raw materials into fermentable carbohydrates. In particular, we should pay more attention to those carbon sources derived from lignocellulosics, animal waste, food waste, and C1 carbon sources.
Second, further advances need to be made in systems metabolic engineering tools and strategies, including in particular enzyme and pathway design. Currently, one of the major obstacles of systems metabolic engineering is to engineer or even create enzymes for desired reactions. To address this problem, protein and pathway design tools that facilitate the development of a suitable enzyme and pathway required for the production of natural and non-natural chemicals are needed. Recently, an enzyme capable of forming a carbon–silicon bond to produce organosilicon compounds by evolving cytochrome C from Rhodothermus marinus has been reported,49 which suggested that our imagination might be a limitation in innovative development. As more and more enzymes having new and novel catalytic functions are discovered or developed, the portfolio of bio-based chemicals that can be produced by microbial fermentation will be drastically expanded by performing systems metabolic engineering. Also, systems metabolic engineering is now integrating machine learning to better utilize exploding volumes of bio big data. Machine-learning techniques can be applied for the prediction of novel biosynthetic pathways and functions of unknown proteins, optimization of gene expression levels, and ideal fermentation conditions. The introduction of machine learning-based tools into systems metabolic engineering is expected to dramatically reduce the time and cost of developing strains and bioprocesses by minimizing the time and effort of repeated experiments, especially through integration with advanced robotic systems for automatic cloning, transformation, strain characterization and selection, mini-scale fermentation, and even downstream processes.
Third, the spectrum of microorganisms to be employed for production needs to be more diversified. Microorganisms have survived over billions of years through evolution and acquired their unique characteristics under particular environments. Thus, it is obviously advantageous if we can properly select a microorganism for a particular product of interest, as explained in Section 3. One good example is succinic acid production by engineered M. succiniciproducens isolated from the rumen of Korean cows. Succinic acid requires carboxylation of a three carbon metabolite (e.g., phosphoenolpyruvate or pyruvate) using carbon dioxide as one carbon donor. Thus, we searched for a bacterium in the rumen based on the finding that the gas phase of the rumen is rich in carbon dioxide. Indeed, M. succiniciproducens capable of efficiently producing succinic acid was isolated. Systems metabolic engineering of M. succiniciproducens allowed production of succinic acid with the highest overall performance indices. Another example is the use of halophilic microorganisms, which can reduce the use of freshwater in industrial-scale fermentation. For the production of organic acids and dicarboxylic acids, the use of low-pH tolerant microorganisms will be beneficial for facilitating the downstream process. These microorganisms never or less exploited before require development of genetic engineering tools to perform metabolic engineering. Also, investigations on their safety and impacts on the environment and human health should be made together with the regulatory bodies.
Last but not least, all these advances should be accompanied by market development, e.g., penetration into the existing market or establishing a new market for bioproducts to replace current petroleum-based chemistry. Even though efficient microbial cell factories efficiently producing bioproducts have been developed, they will be useless if no one is willing to use these bioproducts due to the lack of advantages with respect to price and/or function. One example is 2,3-butanediol (2,3-BDO), which can have three stereoisomers, (2R, 3R), (2S, 3S), or (2R, 3S), and is useful in various applications in polymer, agriculture, pharmaceuticals, and cosmetics areas. However, there was no 2,3-BDO market because it was difficult to chemically produce optically pure 2,3-BDO from fossil resources. Since optically pure 2,3-BDO can now be produced by using metabolically engineered microorganisms, new applications in agriculture and cosmetics are being developed.50 This example clearly demonstrates good opportunities for new applications of bioproducts, in addition to their potential to replace petroleum-based chemicals and materials.
Taken together, systems metabolic engineering will play increasingly important roles in developing microbial cell factories for the environmentally-friendly production of bulk chemicals, fine chemicals, fuels, drugs, functional compounds, and polymers and materials. With further advances in systems metabolic engineering, more competitive microbial cell factories will be developed. Also, the range of products that can be biologically produced will expand. In some cases with difficulties in finding a biological conversion method, one does not have to give up. Instead, combined biological and chemical methods can be employed to more efficiently produce products of interest; thus, the collaboration between metabolic engineers and chemists will have a greater impact. It is hoped that the key principles and strategies of systems metabolic engineering described in this paper will be helpful for researchers who are interested in sustainable bio-based production of chemicals and materials.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the C1 Gas Refinery Program (NRF-2016M3D3A1A01913250) through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT. This work was further supported by Hanwha Chemical through the KAIST-Hanwha Chemical Future Technology Institute. We thank Dr Kyeong Rok Choi for helpful discussion on genome engineering tools. We also thank Dr Won Jun Kim and Dr Jung Ho Ahn for kind advice on the genome-scale metabolic simulation and succinic acid case study, respectively.References
- G. Stephanopoulos, A. A. Aristidou and J. H. Nielsen, Metabolic Engineering: Principles and Methodologies, Academic Press, San Diego, 1998 Search PubMed.
- J. Nielsen and J. D. Keasling, Cell, 2016, 164, 1185–1197 CrossRef CAS PubMed.
- S. Y. Lee and H. U. Kim, Nat. Biotechnol., 2015, 33, 1061–1072 CrossRef CAS PubMed.
- S. Y. Lee, H. U. Kim, T. U. Chae, J. S. Cho, J. W. Kim, J. H. Shin, D. I. Kim, Y.-S. Ko, W. D. Jang and Y.-S. Jang, Nat. Catal., 2019, 2, 942–944 CrossRef CAS.
- J. H. Ahn, Y. S. Jang and S. Y. Lee, Curr. Opin. Biotechnol., 2016, 42, 54–66 CrossRef CAS PubMed.
- C. E. Nakamura and G. M. Whited, Curr. Opin. Biotechnol., 2003, 14, 454–459 CrossRef CAS PubMed.
- S. Y. Choi, M. N. Rhie, H. T. Kim, J. C. Joo, I. J. Cho, J. Son, S. Y. Jo, Y. J. Sohn, K. A. Baritugo, J. Pyo, Y. Lee, S. Y. Lee and S. J. Park, Metab. Eng., 2019, 58, 47–81 CrossRef PubMed.
- S. Y. Park, D. Yang, S. H. Ha and S. Y. Lee, Adv. Biosyst., 2018, 2, 1700190 CrossRef.
- H. T. Kim, K.-A. Baritugo, Y. H. Oh, S. M. Hyun, T. U. Khang, K. H. Kang, S. H. Jung, B. K. Song, K. Park, I.-K. Kim, M. O. Lee, Y. Kam, Y. T. Hwang, S. J. Park and J. C. Joo, ACS Sustainable Chem. Eng., 2018, 6, 5296–5305 CrossRef CAS.
- H. M. Kim, T. U. Chae, S. Y. Choi, W. J. Kim and S. Y. Lee, Nat. Chem. Biol., 2019, 15, 721–729 CrossRef CAS PubMed.
- S. Y. Park, R. M. Binkley, W. J. Kim, M. H. Lee and S. Y. Lee, Metab. Eng., 2018, 49, 105–115 CrossRef CAS PubMed.
- C. J. Paddon, P. J. Westfall, D. J. Pitera, K. Benjamin, K. Fisher, D. McPhee, M. D. Leavell, A. Tai, A. Main, D. Eng, D. R. Polichuk, K. H. Teoh, D. W. Reed, T. Treynor, J. Lenihan, M. Fleck, S. Bajad, G. Dang, D. Dengrove, D. Diola, G. Dorin, K. W. Ellens, S. Fickes, J. Galazzo, S. P. Gaucher, T. Geistlinger, R. Henry, M. Hepp, T. Horning, T. Iqbal, H. Jiang, L. Kizer, B. Lieu, D. Melis, N. Moss, R. Regentin, S. Secrest, H. Tsuruta, R. Vazquez, L. F. Westblade, L. Xu, M. Yu, Y. Zhang, L. Zhao, J. Lievense, P. S. Covello, J. D. Keasling, K. K. Reiling, N. S. Renninger and J. D. Newman, Nature, 2013, 496, 528–532 CrossRef CAS PubMed.
- S. Galanie, K. Thodey, I. J. Trenchard, M. Filsinger Interrante and C. D. Smolke, Science, 2015, 349, 1095–1100 CrossRef CAS PubMed.
- X. X. Xia, Z. G. Qian, C. S. Ki, Y. H. Park, D. L. Kaplan and S. Y. Lee, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14059–14063 CrossRef CAS PubMed.
- J. Zhang, E. Kao, G. Wang, E. E. K. Baidoo, M. Chen and J. D. Keasling, Metab. Eng. Commun., 2016, 3, 1–7 CrossRef CAS PubMed.
- T. U. Chae, Y. S. Ko, K. S. Hwang and S. Y. Lee, Metab. Eng., 2017, 41, 82–91 CrossRef CAS PubMed.
- M. A. Islam, N. Hadadi, M. Ataman, V. Hatzimanikatis and G. Stephanopoulos, Metab. Eng., 2017, 41, 173–181 CrossRef CAS PubMed.
- H. Yim, R. Haselbeck, W. Niu, C. Pujol-Baxley, A. Burgard, J. Boldt, J. Khandurina, J. D. Trawick, R. E. Osterhout, R. Stephen, J. Estadilla, S. Teisan, H. B. Schreyer, S. Andrae, T. H. Yang, S. Y. Lee, M. J. Burk and S. Van Dien, Nat. Chem. Biol., 2011, 7, 445–452 CrossRef CAS PubMed.
- S. Kim, S. N. Lindner, S. Aslan, O. Yishai, S. Wenk, K. Schann and A. Bar-Even, Nat. Chem. Biol., 2020, 16, 538–545 CrossRef CAS PubMed.
- T. E. Sandberg, M. J. Salazar, L. L. Weng, B. O. Palsson and A. M. Feist, Metab. Eng., 2019, 56, 1–16 CrossRef CAS PubMed.
- D. Choe, J. H. Lee, M. Yoo, S. Hwang, B. H. Sung, S. Cho, B. Palsson, S. C. Kim and B. K. Cho, Nat. Commun., 2019, 10, 935 CrossRef PubMed.
- T. Yu, Y. J. Zhou, M. Huang, Q. Liu, R. Pereira, F. David and J. Nielsen, Cell, 2018, 174, 1549–1558 CrossRef CAS PubMed.
- H. Mundhada, J. M. Seoane, K. Schneider, A. Koza, H. B. Christensen, T. Klein, P. V. Phaneuf, M. Herrgard, A. M. Feist and A. T. Nielsen, Metab. Eng., 2017, 39, 141–150 CrossRef CAS PubMed.
- M. C. Bassalo, R. Liu and R. T. Gill, Curr. Opin. Biotechnol., 2016, 39, 126–133 CrossRef CAS PubMed.
- F. Wang, X. Lv, W. Xie, P. Zhou, Y. Zhu, Z. Yao, C. Yang, X. Yang, L. Ye and H. Yu, Metab. Eng., 2017, 39, 257–266 CrossRef CAS.
- H. S. Chu, Y. S. Kim, C. M. Lee, J. H. Lee, W. S. Jung, J. H. Ahn, S. H. Song, I. S. Choi and K. M. Cho, Biotechnol. Bioeng., 2015, 112, 356–364 CrossRef CAS.
- T. C. Williams, I. S. Pretorius and I. T. Paulsen, Trends Biotechnol., 2016, 34, 371–381 CrossRef CAS PubMed.
- N. E. Lewis, H. Nagarajan and B. O. Palsson, Nat. Rev. Microbiol., 2012, 10, 291–305 CrossRef CAS.
- I. Thiele and B. O. Palsson, Nat. Protoc., 2010, 5, 93–121 CrossRef CAS PubMed.
- S. N. Mendoza, B. G. Olivier, D. Molenaar and B. Teusink, Genome Biol., 2019, 20, 158 CrossRef PubMed.
- D. Segre, D. Vitkup and G. M. Church, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15112–15117 CrossRef CAS.
- H. Alper, Y. S. Jin, J. F. Moxley and G. Stephanopoulos, Metab. Eng., 2005, 7, 155–164 CrossRef CAS PubMed.
- A. P. Burgard, P. Pharkya and C. D. Maranas, Biotechnol. Bioeng., 2003, 84, 647–657 CrossRef CAS PubMed.
- E. J. O'Brien, J. A. Lerman, R. L. Chang, D. R. Hyduke and B. O. Palsson, Mol. Syst. Biol., 2013, 9, 693 CrossRef PubMed.
- J. R. Karr, J. C. Sanghvi, D. N. Macklin, M. V. Gutschow, J. M. Jacobs, B. Bolival, Jr., N. Assad-Garcia, J. I. Glass and M. W. Covert, Cell, 2012, 150, 389–401 CrossRef CAS PubMed.
- T. H. Segall-Shapiro, E. D. Sontag and C. A. Voigt, Nat. Biotechnol., 2018, 36, 352–358 CrossRef CAS PubMed.
- M. Jeschek, D. Gerngross and S. Panke, Nat. Commun., 2016, 7, 11163 CrossRef CAS PubMed.
- T. Obata, Curr. Opin. Biotechnol., 2019, 64, 55–61 CrossRef PubMed.
- D. Na, S. M. Yoo, H. Chung, H. Park, J. H. Park and S. Y. Lee, Nat. Biotechnol., 2013, 31, 170–174 CrossRef CAS PubMed.
- T. Si, Y. Luo, Z. Bao and H. Zhao, ACS Synth. Biol., 2015, 4, 283–291 CrossRef CAS PubMed.
- J. Chappell, M. K. Takahashi and J. B. Lucks, Nat. Chem. Biol., 2015, 11, 214–220 CrossRef CAS PubMed.
- Y. G. Kim, J. Cha and S. Chandrasegaran, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 1156–1160 CrossRef CAS PubMed.
- M. Christian, T. Cermak, E. L. Doyle, C. Schmidt, F. Zhang, A. Hummel, A. J. Bogdanove and D. F. Voytas, Genetics, 2010, 186, 757–761 CrossRef CAS PubMed.
- G. J. Knott and J. A. Doudna, Science, 2018, 361, 866–869 CrossRef CAS.
- J. Ni, G. Zhang, L. Qin, J. Li and C. Li, Synth. Syst. Biotechnol., 2019, 4, 79–85 CrossRef PubMed.
- J. Lian, M. HamediRad, S. Hu and H. Zhao, Nat. Commun., 2017, 8, 1688 CrossRef PubMed.
- Y. Tong, C. M. Whitford, H. L. Robertsen, K. Blin, T. S. Jorgensen, A. K. Klitgaard, T. Gren, X. Jiang, T. Weber and S. Y. Lee, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 20366–20375 CrossRef CAS.
- A. V. Anzalone, P. B. Randolph, J. R. Davis, A. A. Sousa, L. W. Koblan, J. M. Levy, P. J. Chen, C. Wilson, G. A. Newby, A. Raguram and D. R. Liu, Nature, 2019, 576, 149–157 CrossRef CAS PubMed.
- S. B. Kan, R. D. Lewis, K. Chen and F. H. Arnold, Science, 2016, 354, 1048–1051 CrossRef CAS PubMed.
- C. W. Song, J. M. Park, S. C. Chung, S. Y. Lee and H. Song, J. Ind. Microbiol. Biotechnol., 2019, 46, 1583–1601 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |