
Lipids: chemical tools for their synthesis, modification, and analysis
Judith
Flores†
a,
Brittany M.
White†
 b,
Roberto J.
Brea
a,
Jeremy M.
Baskin
*b and
Neal K.
Devaraj
*a
b,
Roberto J.
Brea
a,
Jeremy M.
Baskin
*b and
Neal K.
Devaraj
*a
aDepartment of Chemistry and Biochemistry, University of California, San Diego, La Jolla, CA 92093, USA. E-mail: ndevaraj@ucsd.edu
bDepartment of Chemistry and Chemical Biology, Cornell University, Ithaca, NY 14853, USA. E-mail: jeremy.baskin@cornell.edu
First published on 17th June 2020
Abstract
Lipids remain one of the most enigmatic classes of biological molecules. Whereas lipids are well known to form basic units of membrane structure and energy storage, deciphering the exact roles and biological interactions of distinct lipid species has proven elusive. How these building blocks are synthesized, trafficked, and stored are also questions that require closer inspection. This tutorial review covers recent advances on the preparation, derivatization, and analysis of lipids. In particular, we describe several chemical approaches that form part of a powerful toolbox for controlling and characterizing lipid structure. We believe these tools will be helpful in numerous applications, including the study of lipid–protein interactions and the development of novel drug delivery systems.
 Judith Flores | Judith Flores received her BS in Biochemistry from California State University, San Marcos in 2017. Under the supervision of Prof. Kambiz Hamadani, she conducted biophysical research focusing on protein kinetics. She then began her doctoral studies at University of California, San Diego in Prof. Neal K. Devaraj's laboratory in 2017. She currently focuses on studying membrane protein integration in biomimetic lipid membranes. |
 Brittany M. White | Brittany M. White received her PhD in Chemistry (2018) from the University of Oregon under the guidance of Prof. Ramesh Jasti, synthesizing functionalized cycloparaphenylenes as novel biocompatible fluorescent probes and organic materials. Afterwards, she moved to Cornell University to carry out postdoctoral studies with Prof. Jeremy M. Baskin as a NIH postdoctoral fellow. Her current research focuses on developing metabolic and fluorescent probes to image the dynamics of de novo phospholipid biosynthesis. |
 Roberto J. Brea | Roberto J. Brea received his PhD in Chemistry (2013) from the University of Santiago de Compostela (Spain), under the supervision of Prof. Juan R. Granja, working on the design, synthesis and potential applications of supramolecular nanosystems based on α,γ-cyclic peptides. Afterwards, he moved to the University of California, San Diego (USA) to carry out postdoctoral studies with Prof. Neal K. Devaraj as a Human Frontier Science Program (Cross-Disciplinary) fellow. Since 2018, he has worked as an Assistant Project Scientist at University of California, San Diego (USA). His current research focuses on the design and preparation of self-assembled non-natural cellular systems. |
 Jeremy M. Baskin | Jeremy M. Baskin received his PhD in Chemistry (2009) from the University of California, Berkeley under the guidance of Prof. Carolyn R. Bertozzi. Then, he moved to the Yale School of Medicine, where he was a postdoctoral fellow in the group of Prof. Pietro De Camilli and developed a passion for membrane biology and lipid metabolism. Afterwards, he joined the Department of Chemistry and Chemical Biology and Weill Institute for Cell and Molecular Biology at Cornell University (2015). His research laboratory operates at the interface of chemical biology tool development and mechanistic cell biology to understand important roles of lipids in fundamental and disease-related cellular signaling. He is the recipient of the 2020 Walter A. Shaw Young Investigator Award in Lipid Research (ASBMB). |
 Neal K. Devaraj | Neal K. Devaraj earned his PhD in chemistry (2007) from Stanford University under the mentorship of Prof. James Collman and Christopher Chidsey. After a postdoctoral fellowship with Prof. Ralph Weissleder at the Harvard Medical School, he joined the faculty of Chemistry and Biochemistry at the University of California, San Diego. A major research thrust of his lab is understanding how non-living matter, such as simple organic molecules, can assemble to form life. Along these lines, he has developed approaches for the in situ synthesis of synthetic cell membranes by using selective reactions to “stitch” together lipid fragments. His work has been recognized by the 2017 ACS Award in Pure Chemistry, being selected as the 2018 Blavatnik National Laureate in Chemistry, and the 2019 Eli Lilly Award in Biological Chemistry. |
Key learning points1. Importance of chemical tools in the control of lipid properties.2. Synthetic methodologies for the construction and modification of lipids. 3. Recent approaches to monitor and analyze lipids. 4. Study of interactions between lipids and other biomolecules. 5. Lipid tools for the development of novel applications. |
1. Introduction
Lipids are fundamental building blocks of cells that play key roles in signaling, energy storage, and membrane formation. Lipid species are highly diverse in structure, and their distribution varies between organisms. This immense diversity arises from the biosynthesis of various combinations of these building blocks and results in a wide range of functional implications. Despite their significant relevance and essential functions, lipids remain less studied than equally important biomolecules, such as proteins and nucleic acids. One reason why lipids have been difficult to study is the relative lack of techniques to both interrogate their manipulation and visualize them. Moreover, given the high structural diversity of lipid families, it is not possible to accommodate all classes with common methods of extraction, purification, characterization, and analysis. Therefore, much of what we know about lipids has come from studying synthetic membranes with specific lipid compositions. These model membranes consist of very few lipid species, which enables a better understanding of the specific properties and functions of the constituted lipids. Over the last few decades, lipid-based probes have become more prevalent as powerful tools in chemical and synthetic biology and novel chemical techniques have been described for studying lipids both in vitro and in vivo.In this tutorial review, we cover recently developed technologies employed for chemical synthesis and modifications of lipids in vitro and in live cells, as well as explore their associated biological applications. These topics include chemoselective strategies for in situ formation of lipids, the design of chemical probes for lipid imaging and mass spectrometry analysis, the study of lipid–protein interactions, and the construction of lipid-based drug delivery systems. Applications of the discussed topics include origin of life studies, developing cancer drug delivery systems, and understanding the fundamental cell biology of lipid biosynthesis, trafficking, and signaling. Throughout, we highlight techniques that, collectively, constitute a toolbox to control lipid structure.
2. Synthesis of membrane-forming lipids
A major goal of biomimetic membrane chemistry is to develop methods to synthesize cell-like membranes in the absence of enzymes. The general strategy is to find synthetic routes to produce membrane-forming lipids from simple non-membrane forming precursors, all in the presence of water, which is the solvent in which lipid self-assembly takes place. Given the importance of mimicking biological systems, several research groups have recently explored chemoselective coupling reactions for the de novo generation of synthetic membranes. These robust methodologies have allowed for building a diverse toolbox to synthesize lipids chemically. The evolution of bilayer membrane formation by in situ lipid synthesis has been set forth through the efforts of several groups. For instance, pioneering work by Zepik et al. demonstrated that chemical reactions that form a sulfur bridge between surfactant precursors led to vesicle-forming amphiphiles that resembled naturally occurring phospholipids.1 In this section, we highlight the most prominent strategies that have been employed for in situ formation of lipids capable of driving self-assembly of biomimetic membranes.2.1. Copper-catalyzed click chemistry
Budin and Devaraj demonstrated the use of a copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction for the efficient synthesis of phospholipids,2 paving the way for further bioorthogonal reactions to be employed for lipid synthesis. In this study, a simple alkyne-functionalized lysolipid and an oleyl azide were coupled using a copper(I) catalyst, which afforded a biomimetic triazole-containing phospholipid. Remarkably, the CuAAC approach has been utilized to design a self-reproducing system that can drive the repeated synthesis and growth of phospholipid membranes (Fig. 1).3 The regeneration of membrane-bound autocatalysts continuously induces the formation of triazole phospholipids, mimicking natural membrane generation. Employing a similar biomimetic approach, Enomoto et al. added spatiotemporal control to phospholipid synthesis using an intramolecular photoinduced electron transfer to generate copper(I) from a photosensitizer dyad, triggering the CuAAC reaction and the subsequent in situ phospholipid membrane formation.4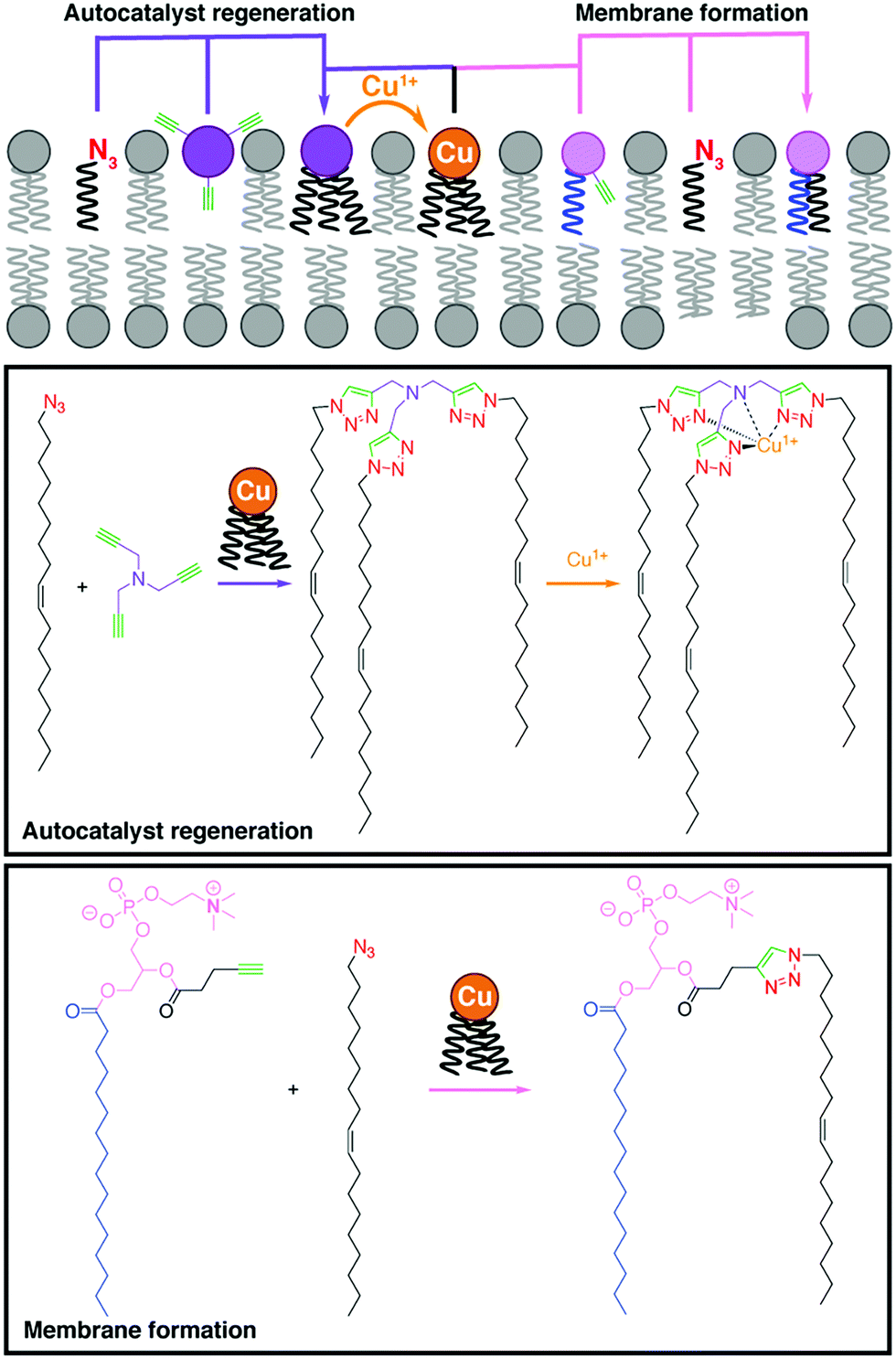 | ||
| Fig. 1 Self-reproducing system that uses autocatalysis to drive triazole-containing phospholipid synthesis and membrane growth by using a CuAAC reaction.3 | ||
2.2. Native chemical ligation (NCL)
NCL is one of the most popular tools for the synthesis and derivatization of large peptides, small proteins, and nucleic acids. Recently, Brea et al. used NCL to couple long-chain acyl thioesters to cysteine-functionalized lysolipids in a highly specific and chemoselective way to form the corresponding phospholipids.5 These lipids are capable of self-assembly into vesicles that can grow to several microns in diameter. NCL versatility was further employed for the incorporation of functional integral membrane proteins into biomimetic membranes (Fig. 2A).6 For instance, Brea et al. developed an in situ lipid synthesis for protein reconstitution technology (INSYRT) for the construction of adenosine A2A receptor (A2AR) proteoliposomes (Fig. 2).6 INSYRT proceeds in the absence of dialysis and/or detergent absorbents, which make it an excellent tool for the assimilation of G protein-coupled receptors (GPCRs) into synthetic liposomes.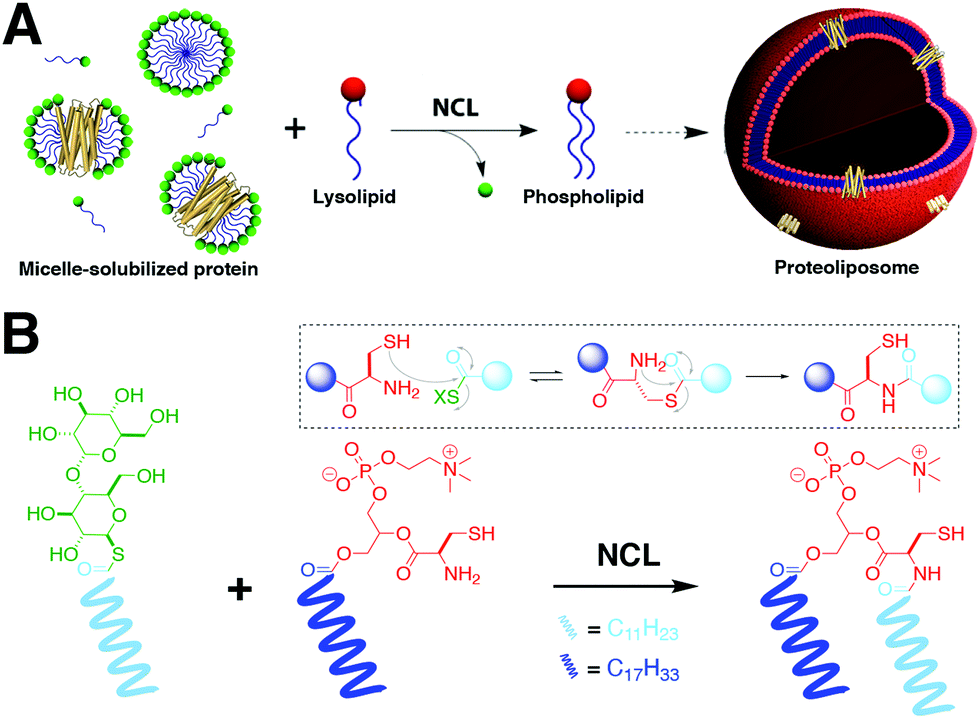 | ||
| Fig. 2 In situ lipid synthesis for protein reconstitution technology (INSYRT).6 (A) Schematic representation of the NCL-based phospholipid membrane formation with embedded transmembrane proteins. The protein is initially solubilized with a synthetic surfactant to form micelle-solubilized protein complexes. Addition of the complementary reactive precursor and subsequent NCL results in the formation of the desired proteoliposomes. (B) Synthesis of phospholipids by NCL between acyl maltose thioesters and cysteine-functionalized lysophospholipids. | ||
The Devaraj group has also shown that a reversible NCL reaction could be used to achieve in situ remodeling of both lipid acyl chains and head groups, triggering changes in vesicle spatial organization, composition, and morphology.7 Application of this novel approach allowed the construction of dynamic membranes that can spontaneously generate and subsequently modulate their physical and chemical properties by using non-enzymatic chemoselective reactions.
2.3. Dynamic imine chemistry
Another relevant chemoselective strategy for the in situ formation of lipids is the combination of aldehyde and amine amphiphiles to form an imine linkage. One of the most attractive features of this reaction is its reversibility depending on environmental factors, which is advantageous when engineering synthetic cells for the purpose of adding dynamic features. Although the ligation is reversible, Sugawara showed that combination of two single chains containing the necessary moieties afforded a kinetically stable product.8 Advantageously, this reaction results in the release of a negatively charged molecule, which aids to in situ formation of giant vesicles.Continuing the efforts to develop vesicles that resemble simple cell-like features, Matsuo et al. showed an imine based self-reproducing protocell formed from a synthetic oleyl phospholipid (V− ole).9 The unsaturation of the lipid facilitated the fluidity of the structure, resulting in the budding and replication of the vesicles by the continuous addition of the phospholipid precursor. Furthermore, the reported vesicles can serve as microreactors, as shown by the encapsulation and amplification of DNA. Similarly, Seoane et al. used the reversibility of the imine formation approach to develop liposomes that respond to external stimuli, and showed controlled release of encapsulated cargo.10
2.4. Traceless ceramide ligation (TCL)
Most of the previously described methods have mainly focused on phospholipid formation. Analogous strategies have been developed for in situ synthesis of other relevant lipids, such as ceramides. Ceramides are a class of sphingolipids that are abundant in the cell membrane. In addition to their well-known function as structural elements, ceramides also participate in a variety of cellular processes, including apoptosis, cell growth arrest, gene regulation, and differentiation. Interestingly, their role in tumor suppression has led to investigation of ceramide delivery as a cancer therapeutic. Given their complex formation and importance in different biological processes, Rudd et al. successfully developed a straightforward method, termed traceless ceramide ligation (TCL), for synthesizing natural ceramides in live cells. TCL uses fatty acid salicylaldehyde esters and sphingosine as precursors followed by incubation with human or mouse cells, leading to in situ formation of ceramides.11 Additionally, in order to obtain spatiotemporal control, photocaged coumarin sphingosine precursors were synthesized and tested to reveal that uncaging led to TCL (Fig. 3A). Consequently, they also demonstrated that the newly synthesized ceramides triggered apoptosis in the tested cells (Fig. 3B) and remarkably revealed previously unreported saturation-dependent apoptotic effects. Following this simple methodology, a diverse group of relevant lipids could be synthesized in living cells and evoke a therapeutic response.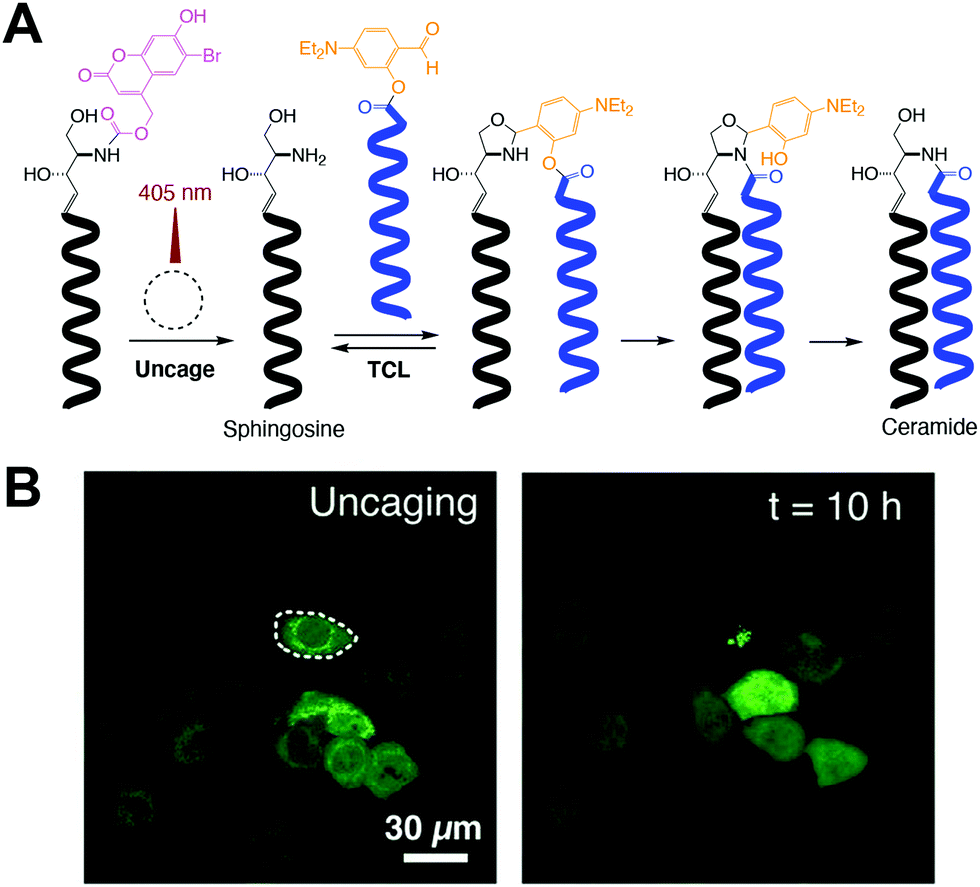 | ||
| Fig. 3 Photocontrolled traceless ceramide ligation (TCL).11 (A) Photocaged sphingosine is irradiated with 405 nm laser light to produce sphingosine and subsequently trigger TCL with a fatty acid salicylaldehyde ester, which affords the corresponding ceramide. (B) Cellular apoptosis mediated by spatiotemporally controlled TCL. One specific GFP-marked HeLa cell was exposed to 10 s of 405 nm light (left), observing its cell death after 10 h of incubation (right) [adapted with permission from A. K. Rudd and N. K. Devaraj, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 7485–7490].11 | ||
2.5. Other ligations
Histidine ligation (HL) has also been employed to form phospholipids in situ.12 Thus, the catalytic role of the imidazole ring of a histidine-containing lysolipid was used to drive its coupling to a long-chain acyl thioester, which afforded a new class of biomimetic phospholipids. These synthetic lipids have physical properties like their natural counterparts and spontaneously self-assemble into micron-sized vesicles. The chemoselectivity, high reactivity, and biocompatibility of the HL approach make it a powerful tool for the efficient encapsulation of relevant biomolecules, such as proteins.Zhao and coworkers also reported the formation of novel synthetic phospholipids using a two-step coupling consisting of a sulfur transfer and a subsequent [2,3]-sigmatropic rearrangement.13 The ligation afforded an ortho-sulfiliminyl phenol product by combining an oxyactamide moiety with a thiophthalimide group. Fluorescent microscopy dictated that these phospholipids formed biomimetic vesicles.
3. Imaging of cellular lipids
Key parts of the chemical toolkit for understanding biological functions of lipids are small-molecule probes for imaging the subcellular locations and dynamics of different classes of lipids within intact cells.14 For the most common imaging modality, fluorescence, we divide the discussion into fully synthetic lipid analogs (Section 3.1) and analogs of lipid biosynthetic precursors that can metabolically label lipids (Section 3.2). Further, Raman spectroscopy imaging has emerged as an alternate modality that enables imaging of lipid analogs with very small tags (Section 3.3).3.1. Fluorescence imaging with synthetic phospholipids
Unnatural, synthetic lipid analogs equipped with either a fluorophore or a bioorthogonal handle have been incorporated into cellular membranes. The increased size and hydrophobicity of lipid-fluorophore derivatives can substantially change the properties of the lipid, and thus the two-step, bioorthogonal approach has gained favor in recent years. In this way, lipid derivatives with smaller structural perturbations can be incorporated into membranes to mimic the localization and trafficking patterns of native lipids, prior to fluorophore tagging.This concept was demonstrated with a synthetic analog of phosphatidic acid (PA) containing terminal alkyne or cyclooctyne groups within the acyl tails. Subsequent fluorophore tagging via CuAAC or the strain-promoted azide–alkyne cycloaddition (SPAAC)15 in fixed or live cells, respectively, revealed lipid fluorescence distributed within several intracellular membranes (Fig. 4A). By contrast, analogous lipid-fluorophore conjugates distributed differently within the cell, highlighting the utility of a two-step labeling. Live-cell imaging of the cyclooctyne–PA analog using SPAAC revealed fluorescently labeled membranes, as a pioneering example of the chemical tagging of lipids in living systems.
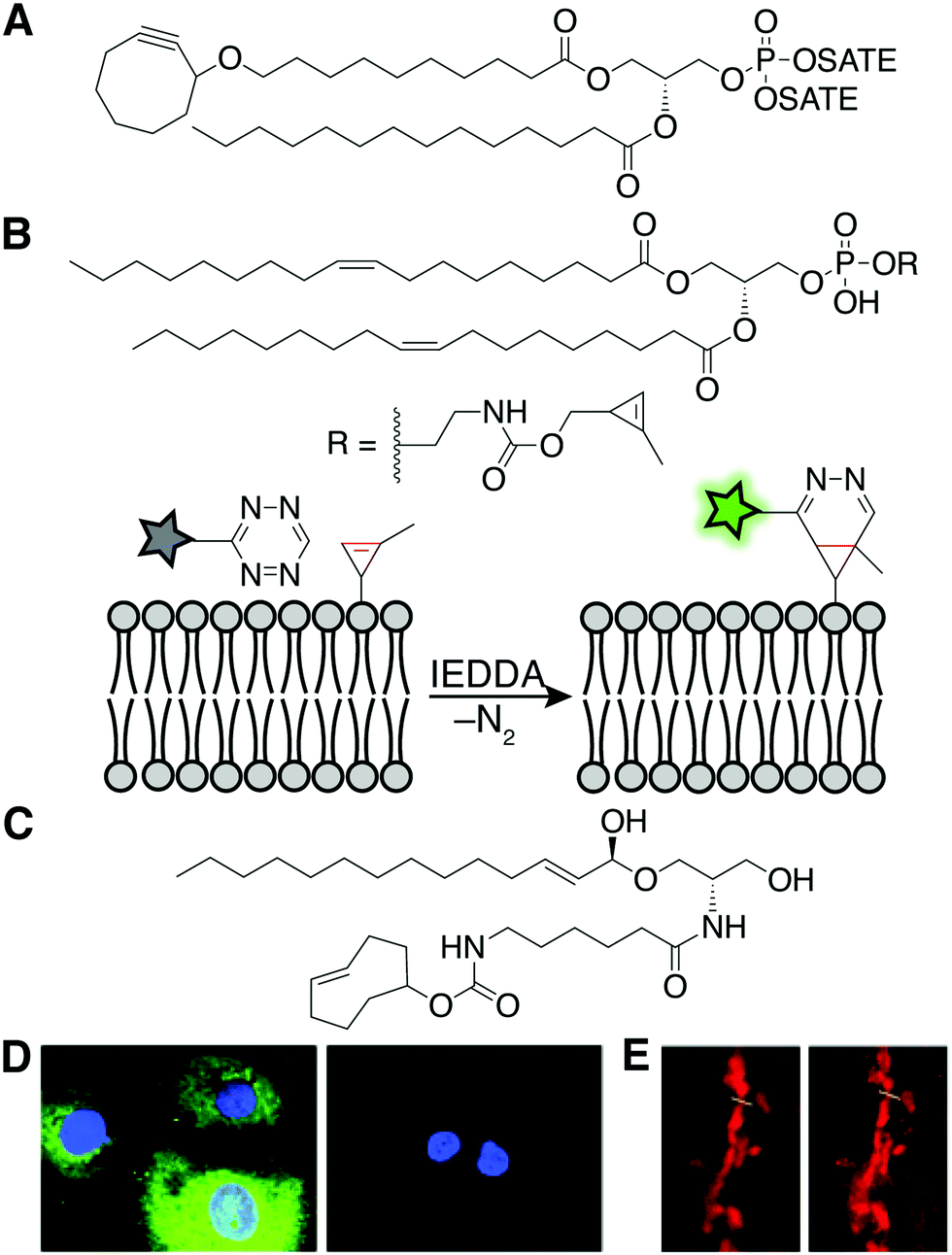 | ||
| Fig. 4 Synthetic lipids for fluorescent, live-cell imaging of lipids. (A) The chemical structure of the cyclooctyne–PA mimic for fluorescent imaging in live cells.15 (B) A phospholipid with a methyl cyclopropene headgroup enables live-cell imaging after IEDDA tagging with a tetrazine fluorophore.16 (C) The chemical structure of the trans-cyclooctene–ceramide conjugate for superresolution imaging of the Golgi.17 (D) Fluorescence microscopy after labeling with probes described in (B). Fluorescence is only observed in the presence (left) and not the absence (right) of the cyclopropene phospholipid [adapted from ref. 16 with permission from John Wiley & Sons, copyright 2012].16 (E) Confocal (left) and stimulated emission depletion superresolution (STED) (right) microscopy imaging of the Golgi complex enabled by a trans-cyclooctene-containing ceramide and IEDDA tagging [adapted from ref. 17 with permission from John Wiley & Sons, copyright 2014].17 | ||
More recently, the rapid and fluorogenic inverse electron-demand Diels–Alder (IEDDA) bioorthogonal reaction between strained alkenes and tetrazines has inspired a new generation of lipid imaging probes. For example, a phospholipid analog whose head group bears a methyl cyclopropene, the smallest stable strained alkene, can label multiple intracellular organelle membranes and be visualized following IEDDA tagging with a fluorophore (Fig. 4B and D).16 As well, a synthetic ceramide analog bearing a trans-cyclooctene (TCO)-functionalized acyl chain (Cer-TCO) targets to the Golgi and enables live-cell, superresolution imaging of this organelle and dynamic vesicle traffic emanating from it, following IEDDA tagging with a silicon rhodamine–tetrazine fluorophore (Fig. 4C and E).17 Together, these and other synthetic, bioorthogonally tagged lipids enable fluorescent labeling of the bulk of intracellular, organelle membranes.
3.2. Fluorescence imaging with metabolic reporters
As a complement to bulk dosing of cells with synthetic lipid analogs, delivery of bioorthogonally tagged biosynthetic precursors enables the metabolic labeling of specific lipid classes as a function of flux through their biosynthetic pathway. These precursors can be either modified head groups or fatty acyl tails. Head group analogs exhibit the advantage of selectivity, i.e., incorporation into, typically, a single class of lipids. By contrast, bioorthogonal fatty acids are incorporated not only into many types of lipids but, as discussed in detail in Section 4.2, also hijack protein posttranslational lipidation pathways. Here we focus on two sets of functionalized head group mimics for visualizing two contrasting phospholipid classes, phosphatidylcholines (PC) and phosphatidic acids (PA).PC is the most abundant eukaryotic phospholipid, and tools to monitor its biosynthesis and distribution serve as a reasonable proxy for cellular membrane biogenesis. De novo PC biosynthesis proceeds via the Kennedy pathway, wherein choline is activated as a nucleotide analog (CDP-choline) and then coupled to diacylglycerol (DAG) to generate PC. Both alkynyl and azido analogs of choline can traverse this pathway, in certain scenarios (e.g., with the alkyne-containing propargylcholine, ProCho) enabling replacement of up to 50% of natural PC with the bioorthogonal analog.18,19 It is worth noting that these choline analogs also tag other choline-containing lipid classes, notably the abundant sphingomyelin and the rarer ether PCs. Following tagging using an appropriate clickable fluorophore, these probes enable visualization of organelle locations for both the biosynthesis [endoplasmic reticulum (ER)] and trafficking destinations (e.g., Golgi, endolysosomes, mitochondria, plasma membrane, etc.) of choline-containing phospholipids in both cultured cells and even animals (Fig. 5A and C).
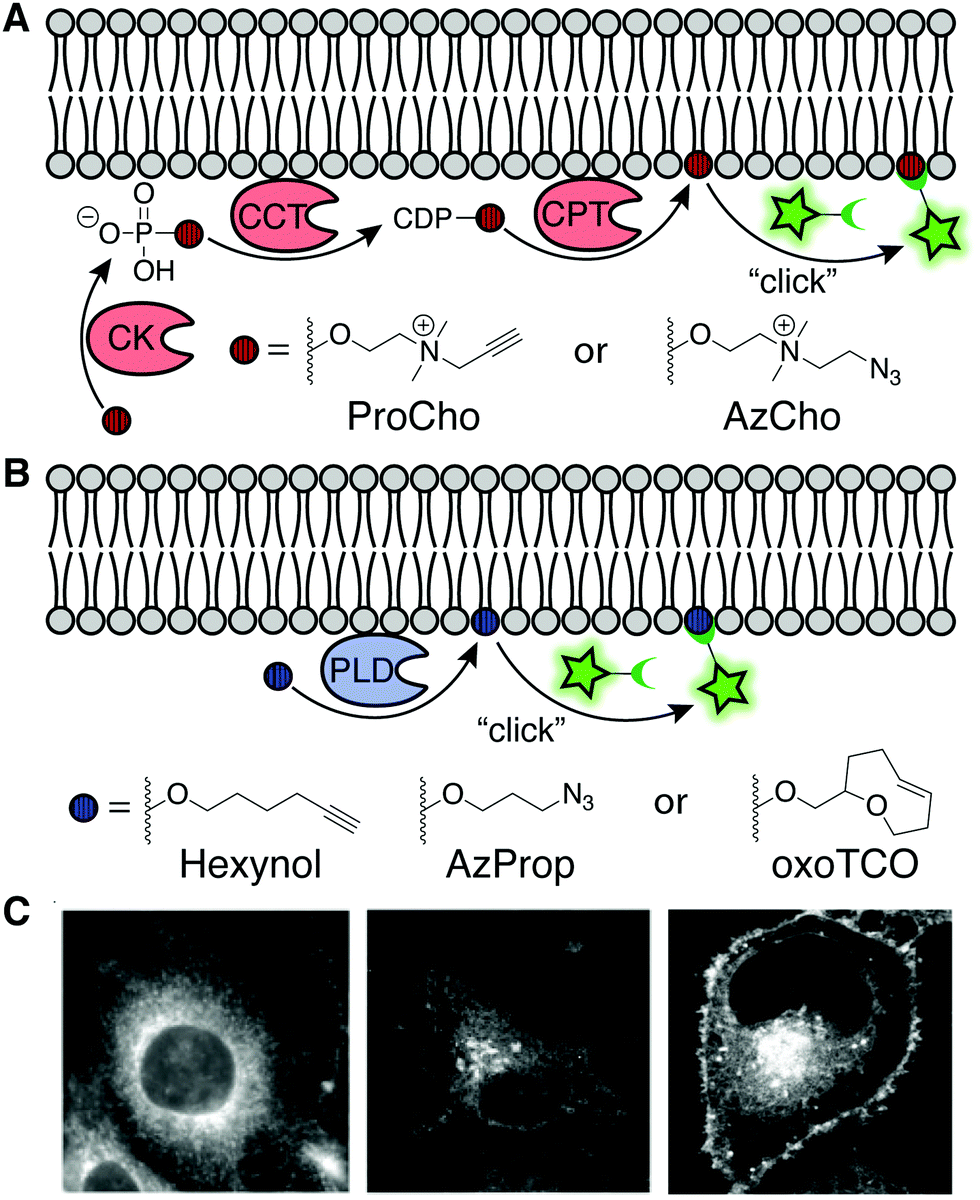 | ||
| Fig. 5 Enzymatic labeling of specific lipid classes. (A) Analogs of choline, ProCho and AzCho, are incorporated via the Kennedy pathway of PC biosynthesis [CK: choline kinase, CCT: phosphocholine cytidylyltransferase, CPT: cholinephosphotransferase]. CuAAC, SPAAC, or Staudinger ligation of the tagged lipids enable fluorescent imaging.22 (B) IMPACT labeling with non-natural primary alcohols report on phospholipase D (PLD) activity. Fluorophore conjugation via CuAAC, SPAAC, or IEDDA labels pools of PA generated by PLD. (C) Fluorescence imaging of membranes using ProCho after CuAAC tagging (left) [adapted with permission from C. Y. Jao, M. Roth, R. Welti and A. Salic, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15332–15337],18 and localization of PLD activity via IMPACT with AzProp and SPAAC (middle) [adapted with permission from https://pubs.acs.org/doi/10.1021/acscentsci.7b00222. Further permissions for the material excerpted should be directed to the American Chemical Society],21 and RT-IMPACT with oxoTCO after IEDDA tagging (right) [adapted with permission from D. Liang, K. Wu, R. Tei, T. W. Bumpus, J. Ye and J. M. Baskin, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 15453–15462].23 | ||
In addition to important structural roles, PC also serves as a major biosynthetic source of the lipid second messenger PA via a hydrolysis reaction catalyzed by phospholipase D (PLD) enzymes. In turn, PA engages many effector proteins on different intracellular membranes to modulate a host of signal transduction pathways, and thus its levels are highly regulated. To visualize sites of PLD-mediated PA synthesis, the Baskin lab took advantage of the ability of PLDs to accept primary alcohols such as ethanol, in lieu of water, in a transphosphatidylation reaction, to generate unnatural phosphatidyl alcohol lipids whose abundance and structure report on PA synthesis by PLDs. Several short, aliphatic alkynyl and azido primary alcohols are effective PLD substrates. Following CuAAC or SPAAC-mediated click chemistry tagging either in lysate or in intact cells, PLD activity can be read out by LC-MS, fluorescence-coupled HPLC, flow cytometry, or fluorescence microscopy in a method termed IMPACT (Imaging Phospholipase D Activity with Clickable Alcohols via Transphosphatidylation; Fig. 5B and C).20,21 Importantly, IMPACT is an activity-based imaging approach, revealing distinct information – sites of activated PLD enzymes – from total cellular pools of PA or of PLDs.
Interestingly, the Baskin lab found that ProCho, which can report on PC biosynthesis via the Kennedy pathway as discussed above, is also a PLD substrate, and by tuning labeling time and cellular stimulation, this single chemical probe can be coaxed to report on different lipid biosynthetic pathways.22
A drawback of live-cell, SPAAC-based IMPACT is the relatively long labeling time, which allows for the fluorescent lipid reporters to traffic away from their site of synthesis via diffusion and transport to other organelle membranes. To accurately reveal subcellular localizations of PLD-mediated PA synthesis, the Baskin lab developed oxo-trans-cyclooctene (oxoTCO) alcohol reagents that, remarkably despite their size, are accepted as PLD substrates. These reporters enable visualization with the rapid and fluorogenic IEDDA reaction, in a variant termed real-time (RT)-IMPACT.23 Using RT-IMPACT, differential locations of PLD activity were observed as elicited by distinct upstream stimuli (e.g., GPCR and receptor tyrosine kinase activation). Additionally, tracking of the RT-IMPACT-derived lipids on the second-to-minute timescale holds promise to reveal mechanisms of rapid, non-vesicular lipid transport pathways between organelles (Fig. 5B and C). Lastly, both IMPACT variants served as indispensable tools in the design and validation by the Baskin lab of a light-controlled, or optogenetic, PLD (optoPLD), which provides a complementary method for generating, rather than visualizing, organelle-specific PA pools for understanding the requirements for local PA synthesis in driving cell signaling pathways.24
3.3. Stimulated Raman spectroscopy imaging
Benefits of using bioorthogonal tagging and fluorescence microscopy for visualizing lipids include the wide availability of probes and microscopy systems. A drawback is the relatively large size of the chemical modification to the lipid structure represented by the contrast agent, which can lead to different biophysical properties and localization patterns from the natural lipid. Stimulated Raman spectroscopy (SRS) imaging represents an alternate modality wherein a functional group as small as a bond (e.g., C–C triple bond, C–D single bond, both of which are located in biologically silent regions of the Raman spectrum) can serve as the contrast agent, minimizing these problems. This approach enables real-time imaging of various classes of lipids bearing alkynes, diynes and carbon-deuterium bonds in living systems to uncover biological phenomena invisible to fluorescence microscopy. SRS imaging can reveal PC using minimally perturbing tags, such as alkynyl or deuterated choline analogs. For example, the alkyne in ProCho enables generation and SRS imaging of metabolically labeled PC in hippocampal neurons.25 Due to the positively charged nitrogen in ProCho, the vibrational mode of alkynyl PC is spectroscopically distinct from other alkyne-functionalized biomolecules, enabling multiplex imaging of different alkyne-tagged species.SRS imaging has also been leveraged to study disordered and ordered regions in the ER. Detection of deuterated palmitic (saturated) and oleic (monounsaturated) acids revealed striking micron-sized patches of slow lateral diffusion in the ER membrane unobservable with larger, more perturbing fluorescent lipid probes (Fig. 6A and B). These domains, not previously known to occur in the ER, were induced by palmitate and attenuated by oleate, revealing a potential role of this unsaturated fatty acid in overcoming cytotoxic effects of palmitate-induced ER microdomain formation.26
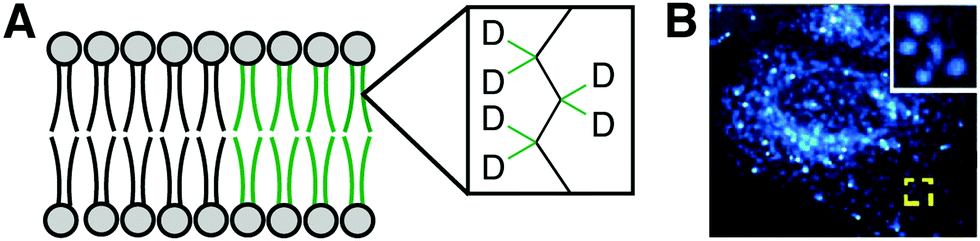 | ||
| Fig. 6 SRS lipid imaging. (A) Deuterated palmitate and oleate incorporated into lipid acyl chains. (B) SRS imaging of C–D single bond stretches in cells treated with palmitate-d31 reveals patched of slow lateral diffusion in the ER [adapted with permission from Y. Shen, Z. Zhao, L. Zhang, L. Shi, S. Shahriar, R. B. Chan, G. Di Paolo and W. Min, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 13394–13399].26 | ||
4. Probing protein lipidation and protein–lipid interactions
In addition to playing critical structural and signaling roles within membranes, lipids also interact covalently and non-covalently with proteins, modifying their localization and function. Chemical tools have been instrumental in revealing the important biological roles of lipid modifications to the proteome.4.1. Bioorthogonal probes for protein prenylation
A major form of protein lipidation is posttranslational modification (PTM) of cysteine residues within a short C-terminal recognition motif by isoprenoid groups (e.g., farnesyl, geranylgeranyl) to give thioethers in a process generally known as prenylation. The prenylation enzymes, farnesyltransferases (FTases) and geranylgeranyl transferases (GGTases), exhibit impressive substrate plasticity for the lipid substrate yet specificity for the protein substrate, enabling the incorporation of bioorthogonal lipid analogs into bona fide prenylated proteins.27,28 In particular, azido analogs of precursors to farnesyl pyrophosphate and geranylgeranyl pyrophosphate, the lipid substrates for FTases and GGTases, respectively, have been used to detect and identify prenylated proteins. These studies have revealed unexpected targets of prenylation, including proteins with an alternative “CGGDD” motif in addition to the canonical “CAAX” motif (Fig. 7A).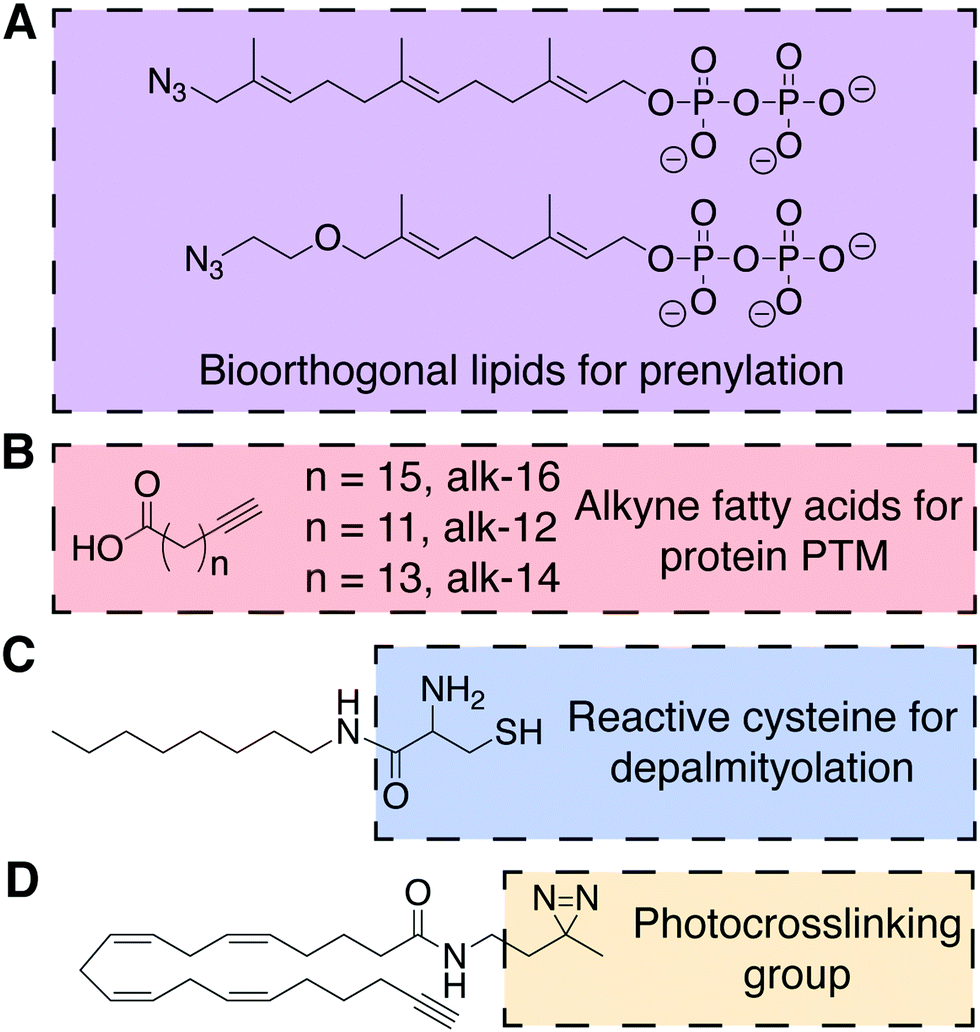 | ||
| Fig. 7 Chemical structures of probes for the posttranslational lipidation of proteins.27–31 (A) Azido isoprenoids identify sites of prenylation in the proteome. (B) Fatty acids with terminal alkynes to study protein lipidation. (C) A substrate for amphiphile-mediated depalmitoylation (AMD) native chemical ligation. (D) Example of a bifunctional lipid containing a photocrosslinkable diazirine functional group and terminal alkyne. | ||
4.2. Chemical approaches to study protein fatty acylation
Proteins can also be lipidated by covalent attachment of saturated and unsaturated fatty acids to several nucleophilic residues, including cysteine, lysine, N-terminal glycine, and serine. Like prenylation, these forms of lipidation, collectively termed fatty acylation, can impact protein localization and function. Due to the chemical nature of the modifications (i.e., thioester, ester, and amide) and the dynamic activities of conjugating and deconjugating enzymes, in certain instances, fatty acylation can be added and removed rapidly. Protein fatty acylation is catalyzed by families of acyltransferases that use the appropriate acyl-coenzyme A (acyl-CoA) conjugate as the lipid substrate to selectively lipidate proteins. These transferases, as well as upstream acyl-CoA synthetases, are permissive of unnatural substrates, enabling both azide- and alkyne-functionalized free fatty acids to serve as metabolic probes for this modification. As mentioned earlier, such bioorthogonal fatty acids also metabolically label de novo glycerolipids and sphingolipids, but biochemical separation of proteomes from lipidomes enables the utility of these fatty acid probes for selectively probing certain forms of protein fatty acylation. In particular, alkynyl fatty acids are optimal probes due to the minimal structural change from natural fatty acids and favorable selectivity of CuAAC tagging with azido detection probes.Notably, bioorthogonal analogs of the 16-carbon fatty acid palmitic acid can metabolically label protein S-acylation, including alkyne-16 (alk-16, Fig. 7B). This PTM most commonly involves palmitoylation of cysteine residues as thioesters by one of several DHHC family palmitoyltransferases and is removed by thioesterases. Though alk-16 also labels N- and O-acylated proteins, S-acylation can be differentiated due to the hydroxylamine sensitivity of thioester linkages, facilitating biochemical analysis.32 Alk-16 has also been used to characterize the O-acylation of Wnt3a, an important protein in embryonic development and stem cell differentiation, by the acyltransferase porcupine (PORCN).29 This chemical probe enabled identification of the palmitoylated serine residue in Wnt3a and, coupled with a FRET assay, the subcellular location and secretion pathway of palmitoylated Wnt3a.
As a complement to metabolic probes for studying the formation of S-palmitoylation, the Devaraj group reported a method for chemoselective depalmitoylation in live cells. This approach, termed amphiphile-mediated depalmitoylation (AMD), capitalizes upon the principle of NCL, wherein unnatural 1,2-aminothiol-functionalized lipids can capture the acyl group from S-acylated proteins (Fig. 7C).30 This chemical depalmitoylation approach has therapeutic potential to rescue loss of palmitoyl-protein thioesterase (PPT1) activity, as occurs in a rare fetal disease, infantile neuronal ceroid lipofuscinosis, which features inactivating mutations in the PPT1 gene.
A shorter alkynyl fatty acid (alk-12, Fig. 7B) mimics the 14-carbon myristic acid, which is co-translationally attached as an amide to N-terminal glycine residues by N-myristoyltransferase (NMT). A notable application of alk-12 revealed key roles for N-myristoylation during infection by the malaria parasite Plasmodium falciparum and supported the idea of NMT inhibition as a potential anti-malarial therapy.31
An alternative form of amide-based lipidation is the fatty acylation of lysine residues. Studies using yet another alkynyl fatty acid (alk-14, Fig. 7B) have, surprisingly, indicated that the abundance and frequency of lysine fatty acylation rivals that of the more extensively studied S-palmitoylation.33 In particular, alk-14 revealed that certain Ras superfamily small GTPases exhibit lysine fatty acylation, and that this modification occurs differentially in closely related paralogs within this family (e.g., the Ras-like proto-oncogenes A and B). Though the identity of acyltransferases that catalyze lysine fatty acylation remains unknown, this study elucidated the NAD-dependent class III histone deacetylase SIRT2 as the deacylase that removes this posttranslational modification from RalB.
4.3. Photocrosslinking lipids for trapping protein–lipid interactions
Bilayer lipids exhibit both stable and transient interactions with integral and peripheral membrane proteins, but it is challenging to detect and study these native interactions in vivo or in model systems. Lipids with photocrosslinking groups such as diazirene, phenyl azide, or benzophenone can enable the covalent trapping of protein–lipid interactions. In particular, a diazirene imposes only a subtle modification to the lipid structure, therefore increasing its popularity in recent years. Inclusion of a bioorthogonal handle (e.g., alkyne, azide) to a photocrosslinking lipid enables enrichment, identification, and characterization of the resultant crosslinked lipid–protein complex. For example, a panel of dual (diazirine and alkyne) functionalized analogs of the polyunsaturated arachidonic acid (Fig. 7D) was used in competitive binding experiments to determine the mechanism of action of an anti-diabetic natural product, KDT501, a lipid-binding molecule to the protein PTGR2.34 More generally, this approach has the potential to limit the need for synthetically challenging derivatization of drugs to uncover their protein targets.5. Lipid modification for the construction of drug delivery systems
Conjugation of lipids to therapeutic molecules has recently undergone explosive growth due to potential drug delivery applications (Fig. 8). The hydrophobic character of lipids enables the enhancement of the drug loading process and increases the stability of the resulting lipid–drug conjugates (LDCs). Lipids can self-associate to form different types of assemblies, including emulsions, liposomes, micelles, and nanoparticles, consequently leading to facilitated drug delivery (Fig. 8A). Additionally, enhanced tumor targeting and reduced off-target toxicity are key features that allow LDCs to be applied in the discovery of anticancer carriers. Therefore, lipids (fatty acids, steroids, phospholipids, glycerides) have been conjugated to an assortment of drug molecules (e.g., doxorubicin, paclitaxel) with different therapeutic applications.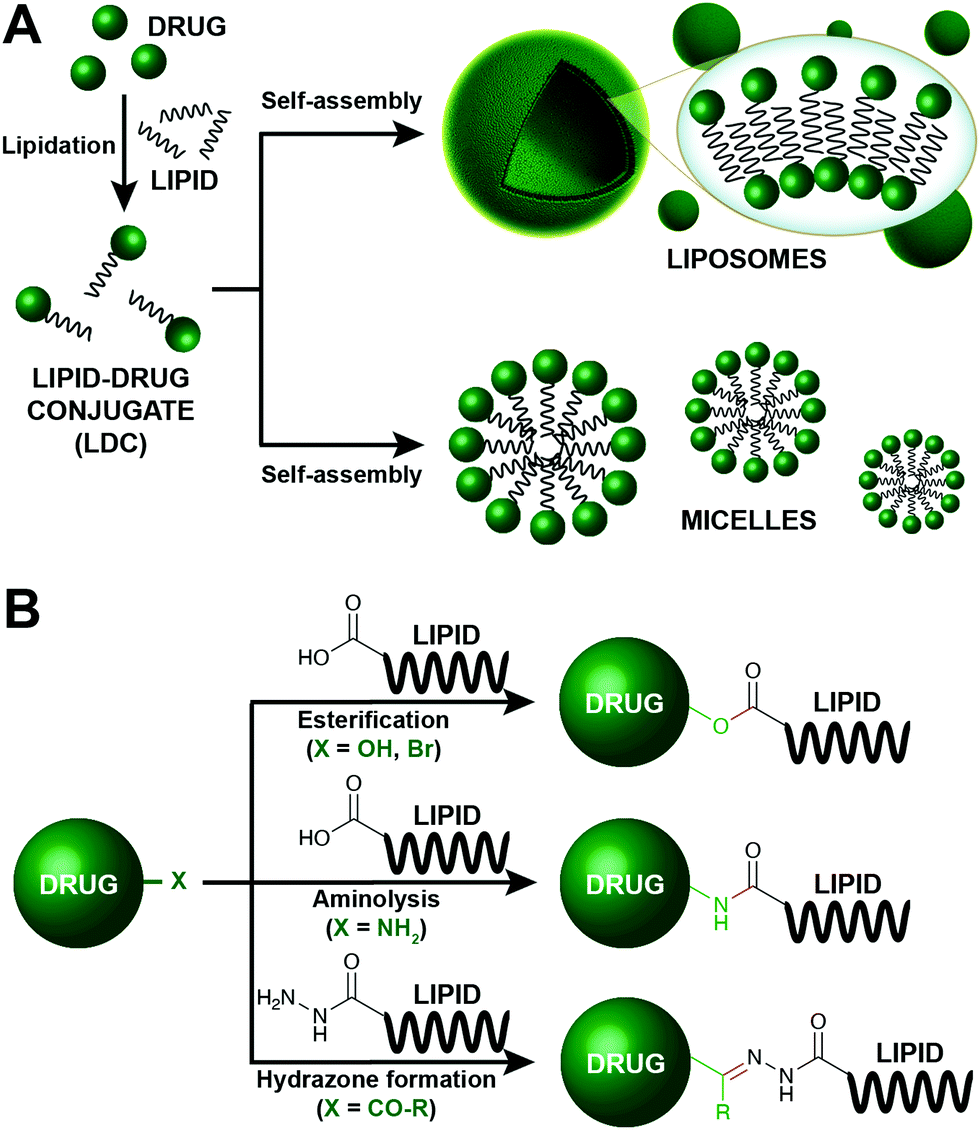 | ||
| Fig. 8 Lipid modification for the construction of enhanced drug carriers.35–40 (A) Schematic representation of the preparation of drug delivery systems based on the lipidation of the corresponding drug. The drug is initially lipidated, affording a lipid–drug conjugate (LDC), which can spontaneously self-assemble into different type of structures, including liposomes and micelles. (B) Common strategies employed for the formation of LDCs. | ||
In this section, we will focus on the prominent chemical strategies to conjugate lipids and drugs (Fig. 8B). Although most LDCs aim to have a therapeutic value, these conjugation methods also highlight the importance of simple bioconjugation reactions. The majority of LDCs are designed to carry prodrugs that become active upon the cleavage of the lipid by an endogenous enzyme. Thus, LDCs depend on broad substrate enzymes such as esterases, amidases, and hydrolases. As such, among the different chemical modifications, esterification, aminolysis and hydrazine formation are especially appealing to create LDCs.
5.1. Esterification
Over the last decade, ester formation has been commonly employed to prepare LDCs (Fig. 8B). This reaction takes advantage of the free carboxylic acid group that is contained in some relevant lipids, such as fatty acids. Specifically, doxorubicin, an anthracycline antibiotic, has been covalently linked to a diverse collection of fatty acids35 and squalenes36 through ester bonds. For instance, Chhikara et al. recently synthesized a wide collection of fatty acyl-doxorubicin conjugates via a succinic acid ester linkage.35 These LDCs have higher cellular uptake and stability in certain cancer cells compared to free doxorubicin. Furthermore, unique squalene–doxorubicin derivatives were prepared by simple nucleophilic displacement-type esterifications. In this particular case, a bromo-functionalized doxorubicin was attached by the free carboxylic acid of squalenoic acid, affording a LDC that self-assembles into elongated nanostructures.36 Remarkably, the squalene assemblies were also observed when the lipid was conjugated with paclitaxel, a relevant chemotherapeutic agent. Subsequent in vivo studies showed comparably lower cytotoxicity than other Food and Drug Administration (FDA) approved paclitaxel conjugates. Thus, the conjugation of lipids to cancer drugs has shown to be promising for decrease of unwanted toxicity.More recently, Callmann and colleagues reported the synthesis of a novel class of paclitaxel-α,ω-dicarboxylic acid conjugates driven by the formation of an ester linkage.37 These derivatives were combined with human serum albumin (HSA) for enabling the efficient delivery of the associated drug. HSA is often used as a carrier drug due to its abundancy in the blood stream. Covalent functionalization of paclitaxel with the corresponding lipid moieties showed increased drug binding to HSA and ultimately higher delivery efficacy. In vivo studies were also carried out by treating xenograft mouse models of human cancer with the ester-based lipid-paclitaxel derivatives, showing high animal survival rates and stalled tumor growths compared to the controls.
5.2. Aminolysis
Amide bond formation has also been extensively explored as a conjugation strategy for the construction of LDCs (Fig. 8B). Using a simple carbodiimide-based chemical methodology, Paliwal et al. conjugated low molecular weight heparin (LMWH) to several fatty acids, such as stearic acid, palmitic acid, and myristic acid.38 The approach took advantage of a standard coupling reaction between the free amine group of LMWH and the carboxylic group of the corresponding fatty acid, which allowed the formation of a stable amide bond. All of the resulting derivatives showed increased bioavailability of the drug. Additionally, they employed a vesicle-forming phospholipid, phosphatidylcholine (PC), for encapsulation purposes. Overall, the fatty acid protected the drug from biodegradation and showed higher absorbance in in vivo studies.Amido-based lipid-doxorubicin conjugates have been formed by covalently linking the DNA-interacting chemotherapeutic drug with D-α-tocopherol succinate via a standard aminolysis reaction.39 In this particular case, the corresponding LDC self-assembles into well-defined nanocarriers, further demonstrating the advantages of using lipids as tools for therapeutics. Characterization of the lipid vehicles suggested that this conjugation methodology facilitates the formation of small nanostructures, which are perfect candidates for tumor drug delivery.
5.3. Hydrazone formation
The formation of hydrazones is routinely employed as a straightforward and versatile bioconjugation strategy. Moreover, the hydrazone bond presents particular interest due to its pH sensitive characteristics. The robustness of the linkage at physiological conditions and its amenability to disassemble in mildly acidic aqueous solutions renders them extremely attractive for applications in LDC-based drug delivery systems (Fig. 8B). Thus, the resulting carriers are stable in storage conditions but can readily release the conjugated drug for endosome uptake (acidic conditions).Over the last few years, several studies have reported the synthesis of pH-sensitive lipid conjugated doxorubicin through hydrazone bond formation. For instance, Li et al. prepared a new class of hydrazine-based palmitic acid–doxorubicin conjugates, which self-assemble into discrete micelles.40 Micelle formation guarantees increase stability and avoids drug precipitation, while hydrazone linkage facilitates the release of free doxorubicin inside cancer cells. These LDCs may see application as novel micelle-based cancer nanomedicines.
6. Light-controlled lipids
In a native, cellular setting, signaling lipids often exert their functions with a high degree of spatial and temporal control. Consequently, bulk dosing of bioactive lipids to cells can lead to ectopic localizations and not faithfully recapitulate signaling. Synthetic lipid analogs delivered to cells in an OFF state but possessing chemical functionality to enable reversible or irreversible switching to an ON state, typically using light stimulation, can afford high spatiotemporal control of lipid signaling. These chemical tools are useful for fundamental mechanistic studies as well as translational, clinical applications.6.1. Photoswitchable lipids
Photoswitchable lipids are a class of synthetic lipids that can exist in either an ON or OFF state dependent upon the conformation of a lipophilic, photosensitive azobenzene functional group within one of the acyl tails. trans-Azobenzenes can be isomerized to the cis form by exposure to UV light (365 nm), and blue light (460 nm) or thermal relaxation allows return to the trans isomer. The two isomers have very different shapes, and in many instances, azobenzenes within acyl tails enable a bioactive lipid to only engage its targets with high affinity in one of the two isomeric forms. For example, analogs of lipophilic ligands for the vanilloid receptor (TRPV1), which comprise azobenzene fatty acids linked to an aromatic head group, enabled the reversible, light-induced control of signaling through this ligand-activated ion channel, which regulates body temperature and pain transduction in the nervous system.41This photolipid strategy has also been applied to other lipid classes for various applications. For example, azobenzene-bearing ceramides (ACes) were developed to study liquid-ordered (Lo) and liquid-disordered (Ld) microdomains within supported lipid bilayers (SLBs) (Fig. 9A).42 Notably, the trans and cis forms of the ACes mimic the structures of saturated and unsaturated lipids respectively, making the photoconversion of ACes a method for reversible control of membrane physical properties.
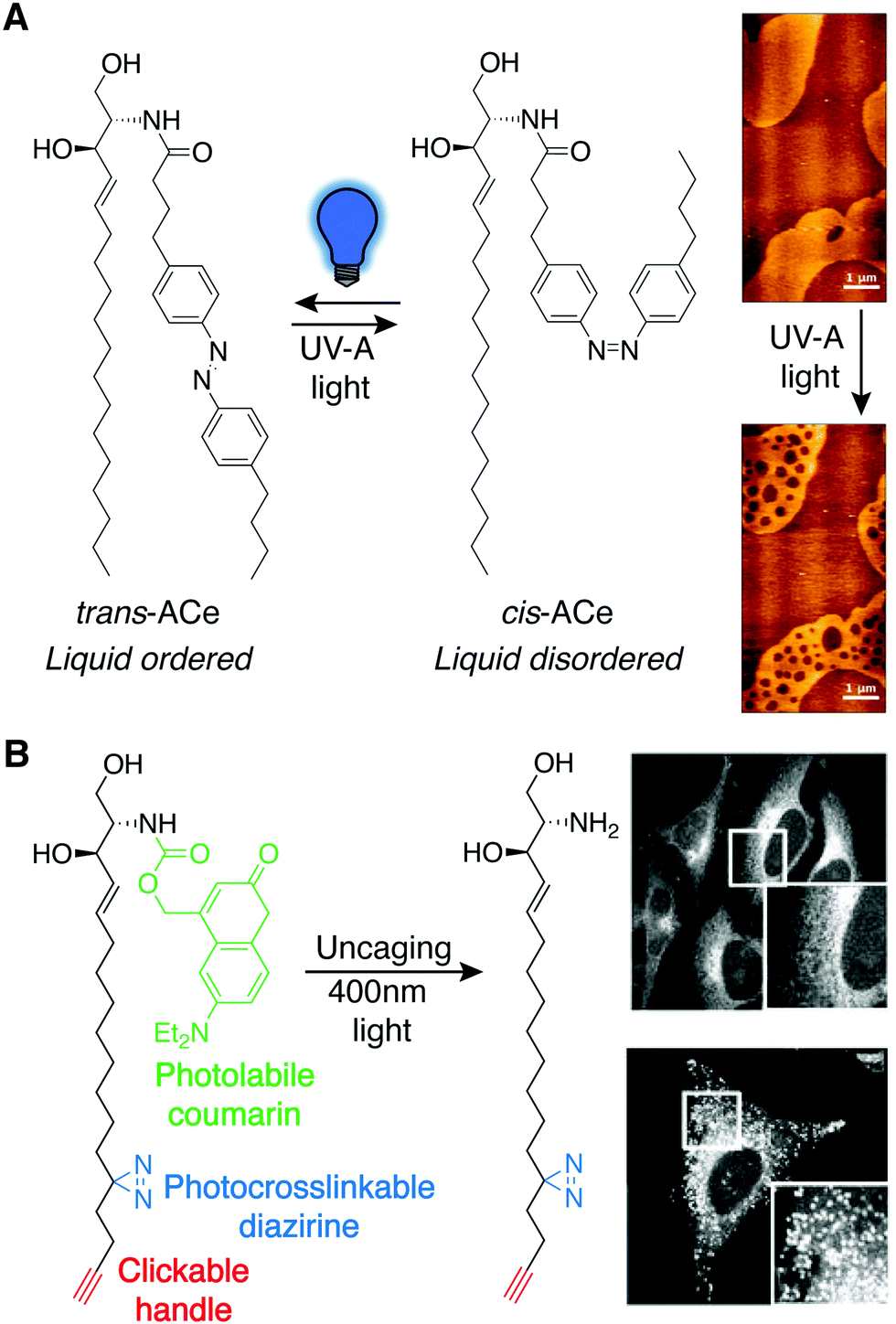 | ||
| Fig. 9 Photoswitchable lipids. (A) Azobenzene-bearing ceramides (ACes) that induce liquid-ordered (Lo) membranes as the trans isomer are converted to the liquid-disordered-favoring (Ld) cis isomer with UV-A light (365 nm), demonstrated by AFM imaging of ACe containing supported lipid bilayers (right) [adapted with permission from J. A. Frank, H. G. Franquelim, P. Schwille and D. Trauner, J. Am. Chem. Soc., 2016, 138, 12981–12986. Copyright 2016 American Chemical Society].42 (B) A trifunctional sphingosine is selectively uncaged with 400 nm light revealing sphingosine localization with fluorescence imaging in control cells (top) and cells where the Niemann-Pick C1 protein (NPC1) was depleted using siRNA (bottom), following photocrosslink-mediated immobilization and CuAAC tagging of the sphingosine probe [adapted with permission from D. Höglinger, A. Nadler, P. Haberkant, J. Kirkpatrick, M. Schifferer, F. Stein, S. Hauke, F. D. Porter and C. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1566–1571].44 | ||
In addition, photoswitchable analogs have been developed of the sphingolipid sphingosine-1-phosphate (S1P), a potent lipid second messenger that engages a family of GPCRs (S1P receptors) to elicit physiological responses in the nervous, immune, and other systems.43 Critically, isomerization of PhotoS1P elicited a isomer-dependent response of S1P receptors, wherein only one isomer of PhotoS1P was able to bind tightly within the receptor binding pocket. Specific effects of the trans and not cis forms of PhotoS1P were seen both in cultured neurons and also in vivo, using changes in pain sensitivity in mice as a readout for S1P receptor engagement by the photolipid.
6.2. Photocaged lipids
A major benefit of photoswitchable lipids is their ability to reversibly control signaling. However, the need to incorporate the abiotic and bulky azobenzene group into what is ultimately a non-native lipid analog makes their design challenging and, sometimes, empirical. By contrast, photocaged lipids enable spatiotemporal control over lipid signaling by the native lipid structure. This feat is accomplished by synthetic installation of a photolabile protecting (or “caging”) group that masks a key part of the lipid. These lipid analogs are irreversible in nature, being delivered to the cell in the OFF state and convertible to the ON state by photo-uncaging.For example, a sphingosine derivative was prepared and equipped with a photocleavable coumarin that contained an additional mitochondrial targeting group that masked the hydroxyl head group of sphingosine.45 After mitochondrial targeting of this inactive lipid, uncaging with UV light liberated the caging and targeting group, revealing free, native sphingosine, enabling temporally resolved analysis of its organelle-specific metabolism to downstream metabolites including S1P. Excitingly, the rates of mitochondrial sphingosine phosphorylation to generate S1P stood in contrast to observed rates of this transformation on the whole-cell level, revealing dramatic differences in metabolic fates and signaling properties of this lipid dependent on subcellular location.
Finally, the photocaging concept has been combined with photocrosslinking and enrichment groups (Section 4.3) to produce trifunctional lipids capable of acute lipid production and identification of lipid–protein interaction partners. For example, trifunctional sphingosine and diacylglycerol probes contain a coumarin photolabile protecting group, a diazirine photocrosslinkable group, and a terminal alkyne for enrichment and live-cell imaging (Fig. 9B).44 These probes have revealed previously unknown scarce or short-lived lipid–protein interactions and enabled imaging of sphingosine accumulation and trafficking during diseases featuring imbalance of sphingolipid and cholesterol metabolism such as Niemann-Pick disease type C (NPC). Thus, the combination of different chemical approaches in the design of lipid probes can serve as a powerful approach to reveal precise biological functions and physiologically relevant behaviors of bioactive lipids within a native, cellular context.
7. Lipid modification for mass spectrometry (MS) analysis
Recent improvements in mass analyzers, ionization methods, and bioinformatic tools have given mass spectrometry (MS) a higher sensitivity, accuracy, and specificity compared to other analytical methods. For these reasons, MS has become a powerful tool for complex lipidome analysis in basic and translational research. Thus, it has been extensively used to understand lipid organization and composition of biological membranes, protein–lipid interactions, and lipid localization. Unfortunately, lipid MS analysis often has associated difficulties in sample preparation, solubility, and identification of sites of lipid unsaturation. Chemical strategies that modify the lipid structure before MS analysis have been key in overcoming these issues, as well as aiding in the comprehension of the lipidome. We will focus on describing these chemical approaches and the manner in which the corresponding lipid modifications aid in MS analysis.7.1. Double bond derivatization
The unsaturation of lipids refers to the number of carbon–carbon double bonds (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) present in the lipid fatty acid tails. The quantity and position of these double bonds can greatly affect the structure and dynamics of lipids. Therefore, accurate examination of double bonds is important in lipidomic studies. Common strategies, such as electrospray ionization (ESI), are not capable of distinguishing double bond positions among a pool of fragments. To combat this problem, several solutions have been offered by simply derivatizing the double bonds before ionization through ozonolysis,46 Paterno-Büchi reactions,47 and ultraviolet photodissociation (UVPD) (Fig. 10).48
C) present in the lipid fatty acid tails. The quantity and position of these double bonds can greatly affect the structure and dynamics of lipids. Therefore, accurate examination of double bonds is important in lipidomic studies. Common strategies, such as electrospray ionization (ESI), are not capable of distinguishing double bond positions among a pool of fragments. To combat this problem, several solutions have been offered by simply derivatizing the double bonds before ionization through ozonolysis,46 Paterno-Büchi reactions,47 and ultraviolet photodissociation (UVPD) (Fig. 10).48
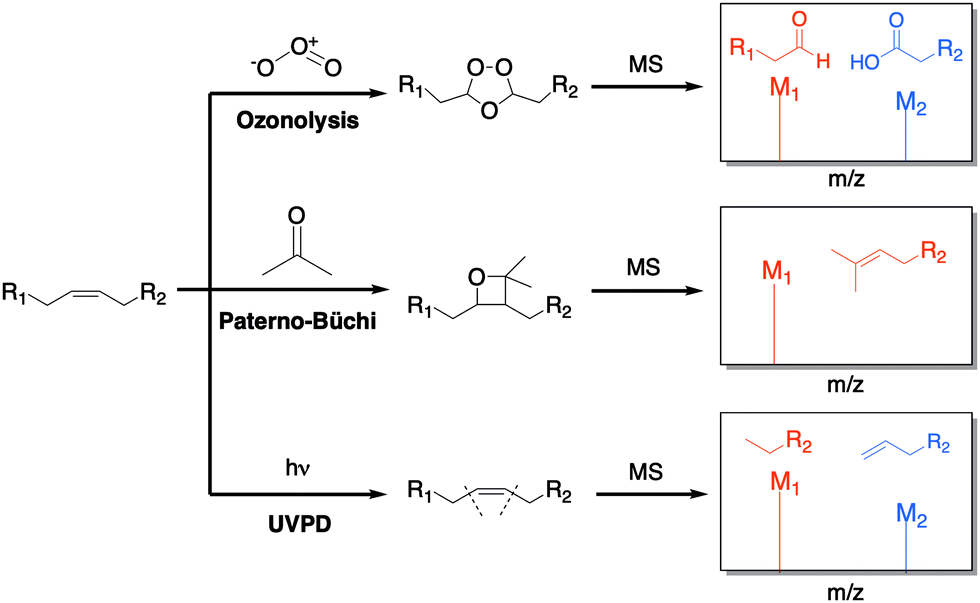 | ||
| Fig. 10 Common strategies for the derivatization of double bonds. The lipid is either treated online or offline, and the double bonds undergo chemical reactions that create distinct fragments, which are then detected in the spectrometer.46–50 | ||
Ozonolysis is one of the most explored methods for double bonds chemical derivatization (Fig. 10). Combined with ESI, it has been a successful way to cleave unsaturated lipids during ionization. In this approach, the sample does not require any prior preparation since the chemical modification occurs during the fragmentation step. Cetraro et al. recently employed a novel approach wherein ozonolysis was coupled with direct analysis in real time (DART) MS to avoid collision-induced dissociation (CID).46 This effectively resulted in the carboxylate and aldehyde ozonolysis products that corresponded to double bond sites. Analysis of lipid extracts from different individuals of the same insect species generated distinct spectra, suggesting population-specific fatty acid signatures can be created and be useful in ecological/biological research.
The Paterno–Büchi reaction is a [2+2] photochemical reaction between a carbonyl and an alkene that affords an oxetane ring (Fig. 10). Ma et al. demonstrated how both Paterno–Büchi and retro Paterno–Büchi reactions could be utilized to elucidate the correct CC position on lipids.47 Following this strategy, an unsaturated lipid [oleic acid, phosphatidylserine (PS) or lysophosphocholine (LPC)] was initially dissolved in an acetone/water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture. Then, the resulting solution was irradiated with a low pressure mercury lamp, followed by nanoelectrospray ionization. The obtained MS fragmentations corresponded to the Paterno–Büchi reaction products. Experiments with lipid mixture samples and yeast lipid extracts indicated that the structures and proper CC diagnosis could be determined in unknown complex mixtures.
:1) mixture. Then, the resulting solution was irradiated with a low pressure mercury lamp, followed by nanoelectrospray ionization. The obtained MS fragmentations corresponded to the Paterno–Büchi reaction products. Experiments with lipid mixture samples and yeast lipid extracts indicated that the structures and proper CC diagnosis could be determined in unknown complex mixtures.
Most recently, Williams and colleagues have described a UVPD method for characterizing both double bonds and sn-positional isomers (the sn-position refers to the carbon in glycerol to which the fatty acid is attached) (Fig. 10).48 This type of lipid modification enables high-throughput applications because it does not rely on reaction times or reactions requiring multiple steps. In this process, the lipid fragments were treated with 193 nm UVPD following static nanospray ionization, which resulted in dissociation at the desired sites. The phospholipid double bond sites and sn-positions were then identified by the resulting fragments. The sn-positions were further investigated by coupling liquid chromatography and peak abundancy. Several lipid extracts from E. coli and bovine liver extracts have also been examined using this strategy.
All of the mentioned approaches have been successfully used to structurally characterize complex lipids, which were challenging to identify in the past due to their small isomeric differences.
7.2. Liposomes
As discussed earlier, liposomes are often used as a simple model for cellular membranes, offering a good platform to study biological membranes. Specifically, liposomes have been routinely employed as an MS tool for studying membrane dynamics, which reveal not only the class of lipids present in the sample but also their proportions. Frick et al. showed that lipids could be introduced into the MS analysis in the form of lipid vesicles, which have a better representation of lipid bilayer composition than extracts that have been treated with harsh organic solvents.49An important goal for MS lipidome analysis is the study of lipid–membrane protein interactions.50 Liposome formation offers a potential “vehicle” for insoluble membrane-bound molecules, facilitating their detection by MS. Using direct infusion to ESI, experiments with liposomes were performed, and gave MS spectra that corresponded to present lipid ratios. Based on lipid mixture results, these liposome methodologies appear to be adequate at detecting low lipid concentrations. Despite such strategies paving the way for relevant MS elucidation, further studies are still needed to better understand liposome roles in lipid–protein dynamics.
8. Conclusions
From answering the most fundamental questions of life to developing cancer therapeutics, lipid-based tools have enabled new avenues of research that were previously not feasible. In this tutorial review, we discussed the recent advances that have made it possible to bridge chemistry and biology together to provide a toolbox for lipid modifications. Simple chemoselective reactions allow for in situ lipid synthesis and conjugation of lipids to other lipids, proteins, fluorophores, and small-molecule drugs. Thus, strategies focused on in situ modifications that give rise to the formation of synthetic cells have potential application in therapeutics, biofuels, biosensors, and drug delivery. Complementary methods that monitor lipid biosynthesis and trafficking dynamics with synthetic tools have revealed valuable insights into fundamental biological signaling pathways. The field is advancing quickly, and there is no doubt that the next decade will offer several exciting emergent methods that will pave the way for a deeper understanding of membrane behavior and lipid biology.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the National Science Foundation (NSF; CHE-1254611, Devaraj; CHE-1749919, Baskin), the Arnold and Mabel Beckman Foundation (Baskin), and the Sloan Research Foundation (Baskin). Judith Flores thanks the NSF for her Graduate Research Fellowship. Brittany M. White acknowledges the National Institutes of Health (NIH, F32GM134632) for her postdoctoral fellowship. Roberto J. Brea thanks the Human Frontier Science Program (HFSP) for his Cross-Disciplinary Fellowship.Notes and references
- H. H. Zepik, P. Walde and T. Ishikawa, Angew. Chem., Int. Ed., 2008, 47, 1323–1325 CrossRef CAS PubMed.
- I. Budin and N. K. Devaraj, J. Am. Chem. Soc., 2012, 134, 751–753 CrossRef CAS PubMed.
- M. D. Hardy, J. Yang, J. Selimkhanov, C. M. Cole, L. S. Tsimring and N. K. Devaraj, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 8187–8192 CrossRef CAS PubMed.
- T. Enomoto, R. J. Brea, A. Bhattacharya and N. K. Devaraj, Langmuir, 2018, 34, 750–755 CrossRef CAS PubMed.
- R. J. Brea, C. M. Cole and N. K. Devaraj, Angew. Chem., Int. Ed., 2014, 53, 14102–14105 CrossRef CAS PubMed.
- R. J. Brea, C. M. Cole, B. R. Lyda, L. Ye, R. S. Prosser, R. K. Sunahara and N. K. Devaraj, J. Am. Chem. Soc., 2017, 139, 3607–3610 CrossRef CAS PubMed.
- R. J. Brea, A. K. Rudd and N. K. Devaraj, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8589–8594 CrossRef CAS PubMed.
- K. Takakura, T. Yamamoto, K. Kurihara, T. Toyota, K. Ohnuma and T. Sugawara, Chem. Commun., 2014, 50, 2190–2192 RSC.
- M. Matsuo, S. Ohyama, K. Sakurai, T. Toyota, K. Suzuki and T. Sugawara, Chem. Phys. Lipids, 2019, 222, 1–7 CrossRef CAS PubMed.
- A. Seoane, R. J. Brea, A. Fuertes, K. A. Podolsky and N. K. Devaraj, J. Am. Chem. Soc., 2018, 140, 8388–8391 CrossRef CAS PubMed.
- A. K. Rudd and N. K. Devaraj, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 7485–7490 CrossRef CAS PubMed.
- R. J. Brea, A. Bhattacharya and N. K. Devaraj, Synlett, 2017, 108–112 CAS.
- F. Xiong, L. Lu, T. Y. Sun, Q. Wu, D. Yan, Y. Chen, X. Zhang, W. Wei, Y. Lu, W. Y. Sun, J. J. Li and J. Zhao, Nat. Commun., 2017, 8, 1–7 CrossRef CAS PubMed.
- T. W. Bumpus and J. M. Baskin, Trends Biochem. Sci., 2018, 43, 970–982 CrossRef CAS PubMed.
- A. B. Neef and C. Schultz, Angew. Chem., Int. Ed., 2009, 48, 1498–1500 CrossRef CAS PubMed.
- J. Yang, J. Šečkutė, C. M. Cole and N. K. Devaraj, Angew. Chem., Int. Ed., 2012, 51, 7476–7479 CrossRef CAS PubMed.
- R. S. Erdmann, H. Takakura, A. D. Thompson, F. Rivera-Molina, E. S. Allgeyer, J. Bewersdorf, D. Toomre and A. Schepartz, Angew. Chem., Int. Ed., 2014, 53, 10242–10246 CrossRef CAS PubMed.
- C. Y. Jao, M. Roth, R. Welti and A. Salic, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15332–15337 CrossRef CAS PubMed.
- C. Y. Jao, M. Roth, R. Welti and A. Salic, ChemBioChem, 2015, 16, 472–476 CrossRef CAS PubMed.
- T. W. Bumpus and J. M. Baskin, Angew. Chem., Int. Ed., 2016, 55, 13155–13158 CrossRef CAS PubMed.
- T. W. Bumpus and J. M. Baskin, ACS Cent. Sci., 2017, 3, 1070–1077 CrossRef CAS PubMed.
- T. W. Bumpus, F. J. Liang and J. M. Baskin, Biochemistry, 2018, 57, 226–230 CrossRef CAS PubMed.
- D. Liang, K. Wu, R. Tei, T. W. Bumpus, J. Ye and J. M. Baskin, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 15453–15462 CrossRef CAS PubMed.
- R. Tei and J. M. Baskin, J. Cell Biol., 2020, 219, e20190713 CrossRef PubMed.
- L. Wei, F. Hu, Y. Shen, Z. Chen, Y. Yu, C. C. Lin, M. C. Wang and W. Min, Nat. Methods, 2014, 11, 410–412 CrossRef CAS PubMed.
- Y. Shen, Z. Zhao, L. Zhang, L. Shi, S. Shahriar, R. B. Chan, G. Di Paolo and W. Min, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 13394–13399 CrossRef CAS PubMed.
- Y. Kho, S. C. Kim, C. Jiang, D. Barma, S. W. Kwon, J. Cheng, J. Jaunbergs, C. Weinbaum, F. Tamanoi, J. Falck, Y. Zhao and P. M. Cuatrecasas, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12479–12484 CrossRef CAS PubMed.
- U. T. T. Nguyen, J. Cramer, J. Gomis, R. Reents, M. Gutierrez-Rodriguez, R. S. Goody, K. Alexandrov and H. Waldmann, ChemBioChem, 2007, 8, 408–423 CrossRef CAS PubMed.
- X. Gao and R. N. Hannoush, Nat. Chem. Biol., 2014, 10, 61–68 CrossRef CAS PubMed.
- A. K. Rudd, R. J. Brea and N. K. Devaraj, J. Am. Chem. Soc., 2018, 140, 17374–17378 CrossRef CAS PubMed.
- M. H. Wright, B. Clough, M. D. Rackham, K. Rangachari, J. A. Brannigan, M. Grainger, D. K. Moss, A. R. Bottrill, W. P. Heal, M. Broncel, R. A. Serwa, D. Brady, D. J. Mann, R. J. Leatherbarrow, R. Tewari, A. J. Wilkinson, A. A. Holder and E. W. Tate, Nat. Chem., 2014, 3, 112–121 CrossRef PubMed.
- E. Thinon, J. P. Fernandez, H. Molina and H. C. Hang, J. Proteome Res., 2018, 17, 1907–1922 CrossRef CAS PubMed.
- N. A. Spiegelman, X. Zhang, H. Jing, J. Cao, I. B. Kotliar, P. Aramsangtienchai, M. Wang, Z. Tong, K. M. Rosch and H. Lin, ACS Chem. Biol., 2019, 14, 2014–2023 CrossRef CAS PubMed.
- K. M. Lum, Y. Sato, B. A. Beyer, W. C. Plaisted, J. L. Anglin, L. L. Lairson and B. F. Cravatt, ACS Chem. Biol., 2017, 12, 2671–2681 CrossRef CAS PubMed.
- B. S. Chhikara, D. Mandal and K. Parang, J. Med. Chem., 2012, 55, 1500–1510 CrossRef CAS PubMed.
- A. Maksimenko, F. Dosio, J. Mougin, A. Ferrero, S. Wack, L. H. Reddy, A. A. Weyn, E. Lepeltier, C. Bourgaux, B. Stella, L. Cattel and P. Couvreur, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 217–226 CrossRef PubMed.
- C. E. Callmann, C. L. M. Leguyader, S. T. Burton, M. P. Thompson, R. Hennis, C. Barback, N. M. Henriksen, W. C. Chan, M. J. Jaremko, J. Yang, A. Garcia, M. D. Burkart, M. K. Gilson, J. D. Momper, P. A. Bertin and N. C. Gianneschi, J. Am. Chem. Soc., 2019, 141, 11765–11769 CrossRef CAS PubMed.
- R. Paliwal, S. R. Paliwal, G. P. Agrawal and S. P. Vyas, Mol. Pharm., 2011, 8, 1314–1321 CrossRef CAS PubMed.
- N. Duhem, F. Danhier, V. Pourcelle, J. M. Schumers, O. Bertrand, C. S. Leduff, S. Hoeppener, U. S. Schubert, J. F. Gohy, J. Marchand-Brynaert and V. Préat, Bioconjugate Chem., 2014, 25, 72–81 CrossRef CAS PubMed.
- F. Li, C. Snow-Davis, C. Du, M. L. Bondarev, M. D. Saulsbury and S. O. Heyliger, J. Visualized Exp., 2016, 2016, 1–8 Search PubMed.
- J. A. Frank, M. Moroni, R. Moshourab, M. Sumser, G. R. Lewin and D. Trauner, Nat. Commun., 2015, 6, 7118 CrossRef PubMed.
- J. A. Frank, H. G. Franquelim, P. Schwille and D. Trauner, J. Am. Chem. Soc., 2016, 138, 12981–12986 CrossRef CAS PubMed.
- J. Morstein, R. Z. Hill, A. J. E. Novak, S. Feng, D. D. Norman, P. C. Donthamsetti, J. A. Frank, T. Harayama, B. M. Williams, A. L. Parrill, G. J. Tigyi, H. Riezman, E. Y. Isacoff, D. M. Bautista and D. Trauner, Nat. Chem. Biol., 2019, 15, 623–631 CrossRef CAS PubMed.
- D. Höglinger, A. Nadler, P. Haberkant, J. Kirkpatrick, M. Schifferer, F. Stein, S. Hauke, F. D. Porter and C. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1566–1571 CrossRef PubMed.
- S. Feng, T. Harayama, S. Montessuit, F. P. A. David, N. Winssinger, J. C. Martinou and H. Riezman, eLife, 2018, 7, e34555 CrossRef PubMed.
- N. Cetraro, R. B. Cody and J. Y. Yew, Analyst, 2019, 144, 5848–5855 RSC.
- X. Ma and Y. Xia, Angew. Chem., Int. Ed., 2014, 53, 2592–2596 CrossRef CAS PubMed.
- P. E. Williams, D. R. Klein, S. M. Greer and J. S. Brodbelt, J. Am. Chem. Soc., 2017, 139, 15681–15690 CrossRef CAS PubMed.
- M. Frick, T. Hofmann, C. Haupt and C. Schmidt, Anal. Bioanal. Chem., 2018, 410, 4253–4258 CrossRef CAS PubMed.
- M. Frick and C. Schmidt, Chem. Phys. Lipids, 2019, 221, 145–157 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |