
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multi-component supramolecular fibers with elastomeric properties and controlled drug release†
Matilde
Putti
ab,
Tristan
Mes
c,
Jingyi
Huang
ab,
Anton W.
Bosman
c and
Patricia Y. W.
Dankers
 *ab
*ab
aEindhoven University of Technology, The Netherlands. E-mail: p.y.w.dankers@tue.nl
bInstitute for Complex Molecular Systems, Eindhoven, The Netherlands
cSupraPolix BV, The Netherlands
First published on 25th October 2019
Abstract
Supramolecular materials based on hydrogen bonding ureido-pyrimidinones (UPy) are highly versatile substrates for tissue engineering, as they provide a platform in which specific functions can be introduced in a modular fashion by means of components with matching supramolecular motifs. In this work, a core–shell fiber mesh is generated by coaxial electrospinning of a robust elastomeric UPy-poly(hexamethylene carbonate) (UPy-PC) core with a hydrophilic shell of poly(ethylene glycol) (UPy-PEG), which is exploited to confer drug release properties to the load-bearing core. The effect of PEG chain length and supramolecular crosslink density on mechanical properties and drug elution profiles is investigated. Hydrated UPy-PC/UPy-PEG meshes containing 30 mol% of UPy-PEG have a Young's modulus matching that of UPy-PC meshes of approximately 0.5 MPa, and elongation at break of 600%. Drug release experiments with low molecular weight drugs encapsulated in the UPy-PEG shell during electrospinning reveal a combined role of drug and matrix hydrophilicity on the elution profile. Our results indicate that a hydrophobic drug is retained in the UPy-PEG shell for several days with a maximum drug release of 56 ± 8% after 14 days, a highly water soluble drug undergoes burst release within one day, and the UPy-modification of a highly water soluble compound increases its retention in the UPy-PEG shell up to multiple weeks. Taken together, our results indicate that the proposed multi-component system is a drug delivery vehicle of excellent versatility for applications requiring strong and durable materials.
Introduction
Requirements for the latest generation of biomedical materials have gradually shifted towards multi-functionality. Well-known examples that have reached clinical applications are materials that incorporate drugs into matrices originally chosen for their mechanical and degradation properties (e.g. drug-eluting stents or growth factor-loaded wound dressings).1,2 Current options are rather simple systems with limited control over properties, lack of flexibility in design and limited responsiveness. Clearly, there is a need for material platforms that can host a complexity of functions and allow for fine-tuning of host–material interactions, as these are cumulated in supramolecular materials.Advances in supramolecular chemistry have revolutionized the fields of biomaterials with the introduction of life-like complexity, a feature that allows material to closely mimic the natural biological environment. Electrostatic interactions, host–guest interactions and hydrogen bonds are common non-covalent interactions at the base of supramolecular biomaterials and their ability to self-assemble. Exemplary self-assembling biomaterials are based on nature-inspired molecules forming β-sheets, cylindrical or helical structures. These supramolecular biomaterials are composed by peptoids and peptide amphiphiles, silk, elastin, carbohydrate-derivatives and nucleotides. Another class of supramolecular materials owes its dynamic nature to synthetic building blocks such as cucurbiturils, cyclodextrines, calixarenes, benzene-1,3,5-carboxamides and ureido-pyrimidinones (UPy).3–5 The UPy is a self-complementary unit that can dimerize via quadruple hydrogen bonds in a strong yet reversible manner.6,7 UPy-based systems are highly valued as biomaterials because of their versatility, which boosted their biomedical application as solid materials, hydrogels and nanoparticles.8,9 Hydrogen-bonded thermoplastic elastomers are designed to have load-bearing properties, and the polymeric backbone can be either end-functionalized with UPy groups or chain-extended with UPy-units in the main chain. Bifunctional UPy-elastomers represent a well-defined substrate for fundamental studies of modular surface modification,10,11 whereas chain-extended UPy-polymers have reached in vivo applications due to the outstanding combination of mechanical properties, processability and synthetic accessibility.12–15 Similarly, UPy-hydrogelators have been reported in both bifunctional and chain-extended molecular designs. The former constitute a highly dynamic transient network that has been exploited for the release of growth factors, anti-cancer drugs, therapeutic siRNA and MRI-contrast agents.16–20 The chain-extended UPy-hydrogel based on poly(ethylene glycol) (UPy-PEG) reported by Guo et al. exhibited shape memory behavior after more than 300% strain while incorporating a water weight fraction up to 0.85. Furthermore, a UPy-PEG/gelatin blend was processable with electrospinning and the resulting mesh supported growth of a healthy monolayer of kidney epithelial cells.21,22
Co-axial electrospinning makes use of double or triple23 concentric nozzles for fabricating core–shell fibers, thereby being identified as an excellent processing technique for the creation of porous networks with hybrid material properties24,25 and encapsulation of active substances.26–28 Co-axial electrospinning can be used to process solutions which would not be suitable for single-fluid electrospinning, thereby expanding the range of materials processable into fibers.23 Furthermore, the core–shell geometry of the fibers is instrumental in drug delivery applications, as it allows confinement and control over the release kinetic of substances loaded both in the core or in the shell.29
Herein, we demonstrate the use of coaxial electrospinning to fabricate multi-component supramolecular fibers, thereby combining drug release and load-bearing properties into a core–shell fiber mesh. Chain-extended UPy-poly(hexamethylene carbonate) (UPy-PC) serves as elastomeric core material, while the chain-extended UPy-PEG component confers water swelling an drug eluting properties as shell material (Fig. 1). The multi-block chain-extended UPy-PEG employed as shell material has a length of the PEG block of 600, 1000 and 1500 g mol−1 (named UPy-PEG600, UPy-PEG1000 and UPy-PEG1500 respectively), in order to study the relationship between the hydrophilic polymer structure and the functionality of the elastomer–hydrogel hybrid construct. After demonstrating the feasibility of co-axial electrospinning with the described chain-extended UPy-PEG polymers, the mechanical properties of hybrid UPy-PC/UPy-PEG electrospun meshes are characterized, and an application of the core–shell fibers as a drug release platform is described. The release properties are evaluated using small molecule drugs. Eventually, the supramolecular nature of the proposed system is exploited to tailor drug–matrix affinity with a UPy-modified model molecule. This strategy allowed us to achieve controlled drug release, thereby implementing additional therapeutic properties into an elastomeric porous construct for biomedical implants.
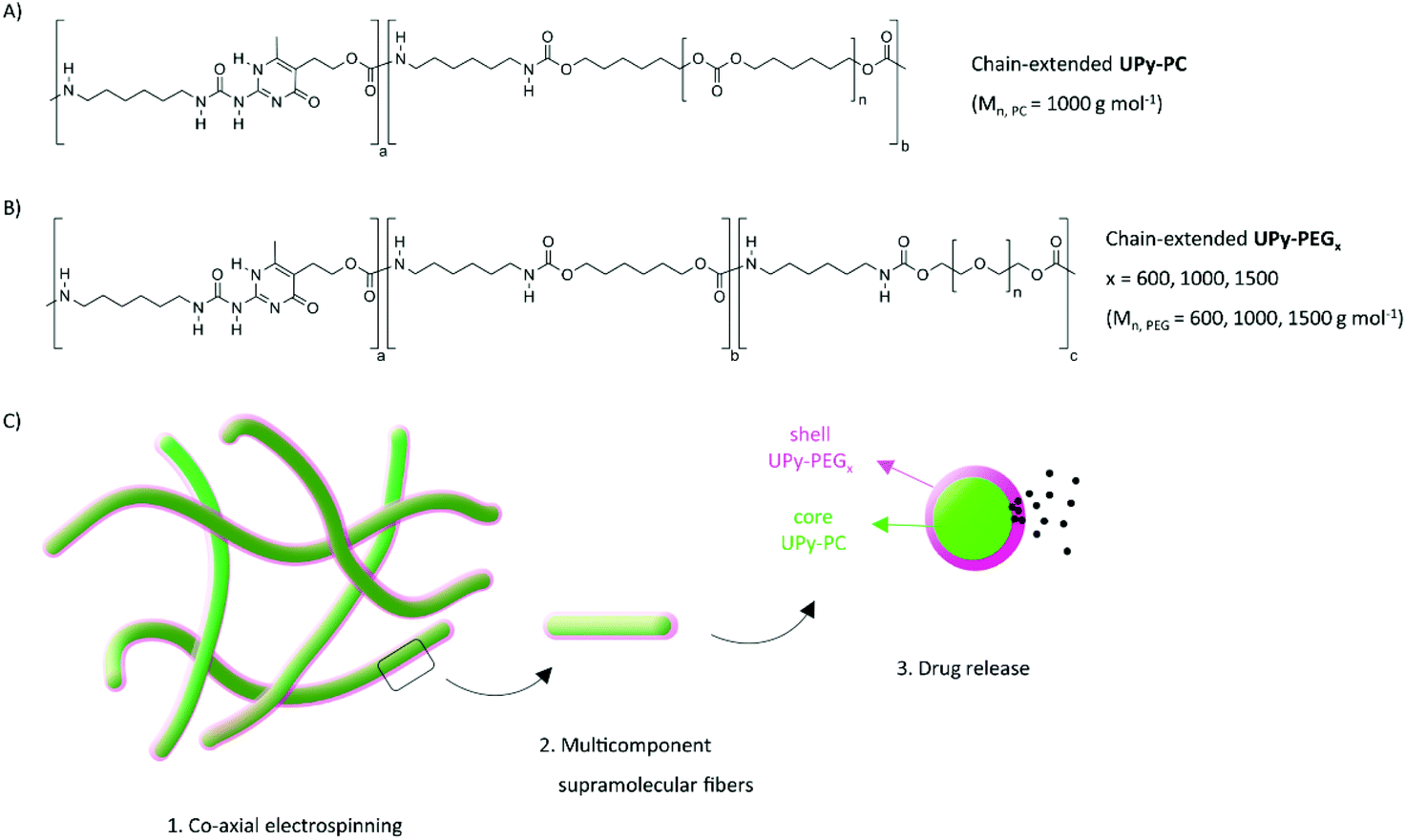 | ||
| Fig. 1 Chain-extended polymers used in this study and schematic representation of the core–shell fibers produced by co-axial electrospinning. Chemical structure of (A) UPy-PC made of PC blocks with Mn, PC = 1000 g mol−1; (B) UPy-PEG made of PEG blocks with Mn, PEG = 600, 1000, 1500 g mol−1. (C) Concept of fabrication of multicomponent supramolecular fibers with supramolecular cross-links and exemplary application as drug delivery system (green core: UPy-PC; magenta shell: UPy-PEGx). | ||
Experimental
Materials
Chain-extended UPy-modified poly(hexamethylene carbonate) (UPy-PC) with PC chain of 1 kg mol−1 (Mn: 19.4 kg mol−1, Mw: 47.7 kg mol−1) and chain-extended UPy-modified poly(ethylene glycol) (UPy-PEGx) were developed by SupraPolix BV (The Netherlands). The UPy-PEG polymers contain PEG chains of 600, 1000 and 1500 g mol−1, namely UPy-PEG600 (Mn: 15.4 kg mol−1, Mw: 30.0 kg mol−1), UPy-PEG1000 (Mn: 20.6 kg mol−1, Mw: 61.7 kg mol−1) and UPy-PEG1500 (Mn: 14.4 kg mol−1, Mw: 29.9 kg mol−1), respectively. General materials and instrumentations are described in the ESI.†Synthesis of rhodamine B-piperazine-amide (RBP)
Rhodamine B piperazine amide was prepared as described previously in literature.30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CH2Cl2/MeOH solution. The combined filtrate was concentrated and the residue was dissolved in CH2Cl2, filtered to remove insoluble salts, and concentrated again. The resulting glassy solid was then partitioned between dilute aqueous NaHCO3 and EtOAc. After isolation, the aqueous layer was washed with 3 additional portions of EtOAc to remove residual starting material. The retained aqueous layer was saturated with NaCl, acidified with 1 M aqueous HCl, and then extracted with multiple portions of 2:1 iPrOH/CH2Cl2, until a faint pink color persisted. The combined organic layers were then dried over Na2SO4, filtered, and concentrated under reduced pressure. The glassy purple solid was dissolved in a minimal amount of MeOH and precipitated by dropwise addition to a large volume of Et2O. The product was collected by filtration as a dark purple solid 0.4 g, (65%): 1H NMR (500 MHz, CD3OD): δ 7.76–7.80 (m, 3), 7.51–7.54 (m, 1), 7.26–7.27 (d, 2, J = 9.5), 7.09–7.11 (dd, 2, J = 2.5, 10.0), 6.97–6.98 (d, 2, J = 2.5), 3.64–3.74 (m, 12), 3.12 (br s, 4), 1.28–1.33 (t, 12, J = 7.5). 13C NMR (100 MHz, CD3OD) δ 169.48, 159.32, 157.28, 156.82, 135.75, 133.08, 132.54, 131.97, 131.63, 131.50, 129.04, 115.67, 114.87, 97.51, 47.05, 45.59, 44.34, 13.07. LC-MS (ESI), calc. m/z 511.31, found: [M + H]+, 511.33 (Scheme S1†).
:1 CH2Cl2/MeOH solution. The combined filtrate was concentrated and the residue was dissolved in CH2Cl2, filtered to remove insoluble salts, and concentrated again. The resulting glassy solid was then partitioned between dilute aqueous NaHCO3 and EtOAc. After isolation, the aqueous layer was washed with 3 additional portions of EtOAc to remove residual starting material. The retained aqueous layer was saturated with NaCl, acidified with 1 M aqueous HCl, and then extracted with multiple portions of 2:1 iPrOH/CH2Cl2, until a faint pink color persisted. The combined organic layers were then dried over Na2SO4, filtered, and concentrated under reduced pressure. The glassy purple solid was dissolved in a minimal amount of MeOH and precipitated by dropwise addition to a large volume of Et2O. The product was collected by filtration as a dark purple solid 0.4 g, (65%): 1H NMR (500 MHz, CD3OD): δ 7.76–7.80 (m, 3), 7.51–7.54 (m, 1), 7.26–7.27 (d, 2, J = 9.5), 7.09–7.11 (dd, 2, J = 2.5, 10.0), 6.97–6.98 (d, 2, J = 2.5), 3.64–3.74 (m, 12), 3.12 (br s, 4), 1.28–1.33 (t, 12, J = 7.5). 13C NMR (100 MHz, CD3OD) δ 169.48, 159.32, 157.28, 156.82, 135.75, 133.08, 132.54, 131.97, 131.63, 131.50, 129.04, 115.67, 114.87, 97.51, 47.05, 45.59, 44.34, 13.07. LC-MS (ESI), calc. m/z 511.31, found: [M + H]+, 511.33 (Scheme S1†).
Synthesis of UPy-carboxylic acid
UPy-carboxylic acid (UPy-C6-U-C12-Ut-OEG12-COOH) was synthesized similar to previous methods.31Synthesis of UPy-rhodamine B-piperazine (UPy-RBP)
UPy-C6-U-C12-Ut-OEG12-COOH (10 mg, 0.01 mmol) was dissolved in DMF (1 mL) and HATU (5.1 mg, 0.13 mmol) and pyridine (0.11 mL, 0.13 mmol) were added. The solution was stirred for 30 minutes under argon. Thereafter, rhodamine B piperazine amide (11.7 mg, 0.02 mmol) dissolved in 1 mL DMF was added. The reaction mixture was stirred overnight and subsequently poured into 2% FA water solution and centrifuged (2×). Eluting over silica with FA/MeOH/CHCl3 1:5:94 afforded UPy-rhodamine B-piperazine (1.28 mg, 87%) as a pink solid. 1H NMR (399 MHz, CDCl3) δ 7.71–7.72 (m, 2H), 7.56–7.57 (m, 1H), 7.37–7.40 (m, 1H), 7.22–7.25 (d, 2H, J = 9.6), 6.91 (d, 2H, J = 2.0), 6.75–6.81 (br s, 2H), 5.86 (s, 1H), 4.99 (s, 3 NH), 4.20 (t, 2H), 3.65 (m, 56H), 3.42–3.46 (q, 8H, J = 7.2), 3.14 (m, 8H), 2.61 (t, 2H), 2.19 (s, 3H), 1.23–1.49 (m, 40H) (Fig. S3†). 13C NMR (100 MHz, CDCl3) δ 187.26, 187.24, 169.62, 159.32, 157.28, 156.82, 156.48, 139.45, 135.75, 133.08, 132.20, 130.54, 130.39, 127.80, 114.18, 106.52, 96.37, 75.60, 70.52, 69.67, 67.36, 66.70, 63.74, 46.16, 41.46, 40.66, 39.19, 33.53, 31.14, 30.92, 30.15, 29.92, 29.37, 29.28, 29.20, 28.90, 26.73, 26.59, 25.91, 25.61, 12.61. LC-MS (ESI), calc. m/z 1631.00, found: [M + H]+, 1631.83, [M + 2H]2+, 816.25 (Scheme S2†).
Preparation and characterization of cast films
For preparation of polymer films, UPy-PC, UPy-PEG600, UPy-PEG1000, UPy-PEG1500 were dissolved in HFIP at concentration of 25 mg mL−1. Drop cast films were prepared by distributing 50 μL of solution on 13 mm ∅ glass coverslip. The HFIP was evaporated overnight in vacuo at 40 °C. For cell culture experiments, samples were sterilized with UV light for 10 minutes. Water contact angle (WCA) measurements on drop cast films were performed on an OCA 30 system from Dataphysics using SCA20 software. A 5 μL drop of deionized water was placed in three different regions of three different samples. Images were captured immediately and 5 seconds after placement of the water drop. Water contact angles were determined from the recorded images.Cell culture
Mouse NIH/3T3 fibroblasts (ECACC 93061524) were cultured in complete medium consisting of Dulbecco's Modified Eagle Medium (DMEM, Gibco) supplemented with 5 v% heat inactivated fetal bovine serum (Invitrogen) and 1 v% penicillin–streptomycin solution (Invitrogen), at 37 °C and 5% CO2 in a humidified atmosphere, passaged at 80% confluency and seeded at a concentration of 0.6 × 105 cells per cm2 on drop cast films. For fluorescent staining, cells seeded on cast films (n = 3) were first washed with phosphate buffered saline (PBS), fixated in 3.7% formaldehyde (Merck) for 10 minutes, washed with PBS, and permeabilized with 0.5% Triton X-100 (Merck) for 15 minutes. Subsequently, cells were washed with PBS and incubated with actin labelling (phalloidin-Atto488) in the dark for 45 minutes, followed by nuclei labelling by incubation with 0.1 μg mL−1 4′,6′-diamidino-2-phenylindole (DAPI) in PBS for 5 minutes. Finally, samples were washed and mounted on cover glasses with Mowiol (Sigma). Samples were imaged with a Zeiss Axiovert 200M epifluorescence microscope. Cell's viability was assessed on cells cultured on cast films for 24 hours (n = 4) by XTT in vitro Toxicology Assay kit (Sigma-Aldrich) following manufacturer's instructions.Fabrication of core–shell fibers
Electrospinning was carried out in a climate-controlled EC-CLI electrospinning apparatus equipped with a co-axial nozzle EM-CAX from IME Technologies (Waalre, NL). UPy-PC was electrospun as core solution at 70 mg mL−1 in CHCl3/HFIP 9/1 v/v. UPy-PEGs were electrospun as shell solution at 80 or 100 mg mL−1 in CHCl3/HFIP 9/1 v/v for UPy-PEG1500 or CHCl3/HFIP/MeOH 9/0.9/0.1 v/v for UPy-PEG1000 and UPy-PEG600. Polymer solutions were stirred overnight before loading in a syringe connected to the co-axial nozzle. The core solution was fed at a constant flow rate of 40 μL min−1. For tuning the composition of core–shell fibers, the flow of shell solution was varied between 5 and 20 μL min−1. The applied voltage of 13 kV with a tip–collector distance of 15 cm generated fibers collected on a rotating (100 rpm) cylindrical collector covered in aluminum foil. After electrospinning, the meshes were dried overnight in vacuo.Scanning electron microscopy
Scanning Electron Microscopy (SEM) was performed using FEI Quanta 600 and Xt Microscope Control software. Mesh samples were mounted on a metal stub by using double sided carbon tape. The samples were visualized under high vacuum with an acceleration voltage of 1 kV and a working distance of 10 mm. The fiber diameters were determined from multiple high magnification images using ImageJ software.Visualization of core–shell morphology
In order to visualize the core–shell morphology of the fibers, electrospinning solutions of UPy-PC and UPy-PEG were labelled with 0.01 wt% of fluorescein (Sigma-Aldrich) and rhodamine B (Sigma-Aldrich), respectively, from stock solutions in HFIP. After electrospinning, the dry-mounted scaffolds were imaged by confocal microscopy with a 63×/1.4 Oil immersion objective. The dyes were excited with Ar (488 nm, 10% power) and HeNe (543 nm, 30% power) lasers and imaged in a multitrack configuration, detecting the emission of fluorescein with a 500–500 nm band-pass filter and the emission of rhodamine with a 560 nm low-pass filter on a Zeiss LSM500 confocal microscope.32Fiber composition analysis and swelling properties
The ratio between UPy-PC and UPy-PEG in electrospun meshes was determined by 1H-NMR in a Varian Mercury Vx 400 MHz (Bruker) in d6-DMSO. For all three UPy-PEGs, each combination of flow rate and solution concentration was electrospun twice and the compositions from the two independent electrospinning sessions were analyzed and averaged. To assess the dependence of the swelling properties on the PEG chain length in UPy-PEG, the material was incubated in water at 37 °C for 30 minutes, and excess water was removed with a tissue. Subsequently, the masses of the material before and after swelling were compared.Fabrication of core–shell fibers with drugs in the UPy-PEG shell
For release studies, co-axial electrospinning was performed with shell solutions of UPy-PEG at 100 mg mL−1 with flow of 15 μL min−1, in order to obtained meshes with comparable UPy-PEG content of approximately 30 mol%, as determined by 1H-NMR. Pirfenidone (Bio-Connect BV), SB431542 (Tocris), rhodamine B-piperazine (RhodBP) and UPy-rhodamine B-piperazine (UPy-RhodBP) were added to the UPy-PEG shell solution at concentration of 0.1 wt% from stock solutions in HFIP.Mechanical properties
Electrospun hybrid meshes (n = 4) containing approximately 30 mol% UPy-PEG and one UPy-PC control mesh were cut into rectangular strips of 0.5 × 3 cm, the extremities were secured to the clamps of a EMT Criterion tensile tester, equipped with a 5000 N load cell. A pre-load of 0.5 N was applied and samples were loaded until break over a test length of 10 mm with loading speed of 1 mm min−1. Elongation at break, ultimate tensile stress (UTS) and Young's modulus were calculated from the loading curves of independent electrospun samples and averaged.Drug release studies
Mesh samples were cut with an 8 mm ∅ biopsy punch and incubated in PBS at 37 °C under shaking at 600 rpm. The supernatant was collected at each time point, and the amount of release drug was measured on a microplate reader and drug concentration was extrapolated from the linear calibration curves. Pirfenidone content in the supernatant was measured by reading absorbance at λ = 317 nm, SB431542 content was measured by fluorescence intensity of the supernatant with λex = 360 nm and λem = 590 nm. The concentration of RhodBP and UPy-RhodBP in supernatant samples was measured by fluorescence intensity with λex = 530 nm and λem = 590 nm.Results and discussion
In order to control the range of swelling properties and hydrophilicity, the chain-extended UPy-PEG polymers used here have different length of the PEG block (UPy-PEGx with x = 600, 1000, 1500, corresponding to Mn,PEG = 600, 1000, 1500 g mol−1). Hydrophilic poly(ethylene glycol) and hydrophobic poly(hexamethylene carbonate) oligomers were chain extended with UPy-moieties comprising alkyl spacers. The resulting UPy-PC and UPy-PEGx polymers exhibited molecular weights within a small range of 14–20 kDa as assessed with SEC (Table S1†). The thermal behavior of UPy-PC and UPy-PEGx was studied by differential scanning calorimetry (DSC). UPy-PC exhibited a glass transition temperature Tg of −33 °C and a melting temperature Tm of 123 °C (ΔHm = 1.6 J g−1) which is attributed to the formation of UPy-nanofibrous domains in the material.33 In the context of the proposed application, these results confirm that UPy-PC is an amorphous polymer presenting UPy-nanoaggregates at body temperature of 37 °C. UPy-PEGx exhibit a similar thermal behavior to UPy-PC with glass transition temperatures Tg corresponding to −48 °C, −47 °C, −54 °C for UPy-PEG600, UPy-PEG1000 and UPy-PEG1500, respectively. However, differently from UPy-PC, UPy-PEGx show two pronounced melting temperatures, indicating that UPy-PEGx are semi-crystalline polymers presenting both crystalline domains of PEG (showing phase transition around Tm of 60 °C) and UPy-assemblies, whereby the latter are responsible for the phase transition at higher temperatures. Nevertheless, the PEG crystalline domains are unlikely to have any influence on the properties of the material as they are plasticized by water when used in the swollen state, for example when in contact with body fluids. Bulk films of UPy-PEG variants underwent tensile test in both dry and wet conditions, and indeed, the measured elastic modulus (E) of the bulk material decreased significantly from 99, 15 and 20 MPa in the dry state, to 35, 15 and 8 MPa in the swollen state for UPy-PEG600, UPy-PEG1000 and UPy-PEG1500, respectively (Table S2, Fig. S1†). Consistently, ultimate tensile strength and elongation at break of UPy-PEGx decreases significantly upon hydration (Table S2, Fig. S1†). This result is in agreement with previous research on similar polymers which showed that upon water swelling of the chain-extended UPy-PEG, no melting transition related to the PEG crystalline domains is detected any longer due to hydration of the network.21The water contact angle (WCA) of UPy-PC measured immediately after deposition of a water drop was 77 ± 1°, and the WCA value decreased to 64 ± 1°, 63 ± 1° and 23 ± 6° for polymers with PEG length of 600, 1000 and 1500 g mol−1, respectively (Fig. 2A, full circles). The measured WCA value after 5 seconds from deposition of the drop remained unvaried for UPy-PC, while it decreased slightly with respect to the initial value for UPy-PEG600 and UPy-PEG1000 to 57 ± 2° and 50 ± 1° respectively, as expected. The most hydrophilic was found to be UPy-PEG1500, which exhibits complete spreading of the water drop over the film surface (Fig. 2A, empty circles). The swelling behavior of the UPy-PEGx was compared with that of the control UPy-PC by recording the mass gain after immersion in water at 37 °C for 30 minutes. As anticipated, UPy-PC only gained 6 ± 5% of its original weight, while the increase in mass after water uptake was 109 ± 4% for UPy-PEG600, 137 ± 20% for UPy-PEG1000 and 200 ± 11% for UPy-PEG1500 (Fig. 2B). PEG is commonly exploited in material science for the creation of antifouling and cell repellent surfaces,34,35 therefore the adhesion of 3T3 cells was investigated in response to UPy-PC and UPy-PEGx films. Adhering cells with elongated morphology were observed on UPy-PC and UPy-PEG600. Cell adhesion decreased slightly on UPy-PEG1000 and UPy-PEG1500, although it was not compromised (Fig. 2D). Cell viability is found to be similar between UPy-PC and UPy-PEGx, demonstrating that any diminished degree of cell adhesion and spread morphology is likely due to the surface properties of the material and not to cytotoxic effects (Fig. 2C). Co-axial electrospinning makes use of concentric nozzles for fabricating core–shell structures, thereby being identified as an excellent processing technique for the creation of porous networks with hybrid material properties25,32 and encapsulation of active substances.26–28 In this study, coaxial electrospinning was carried out with an inner UPy-PC solution and an outer UPy-PEG solution. The two polymer solutions were labelled with fluorescent dyes to distinguish the core from the shell with confocal fluorescence microscopy. A variable degree of separation between the core UPy-PC fiber and the exterior UPy-PEG shell was detected in all observed samples, thereby confirming the formation of bi-layered fibers (Fig. 3). Important parameters in the co-axial electrospinning working process have been identified in the Taylor's cone shape and dimensions and the length of the linear jet.36 To ascertain whether processing conditions can be used to control the final mesh morphology and composition, the effect of concentration and flow rate of the outer polymer solution was studied systematically. Fluid flow rate has been reported as an important tunable parameter in the fabrication of drug-loaded core–shell fibers which correlated eventually with the drug release profile.37 The concentration and flow rate of the inner UPy-PC solution were kept constant at 70 mg mL−1 and 40 μL min−1, respectively. As a general trend, higher flow rates of the outer shell solution led to a higher degree of connected or fused fibers, especially for UPy-PC/UPy-PEG600 (Fig. 3A) and UPy-PC/UPy-PEG1500 fibers (Fig. 3E). Furthermore, increasing flow rate of the shell solution appears to increase the thickness of the UPy-PEG shell in fibers of UPy-PC/UPy-PEG1000 (Fig. 3C). The concentration of the shell polymer solution seems to have no significant effect on fiber aspect. In line with expectations, the analysis of UPy-PC/UPy-PEG ratio confirmed that increasing UPy-PEG solution flow rate increases its molar ratio in the electrospun mesh (Fig. 3B, D and F). With a flow rate of 5 μL min−1 the relative amount of UPy-PEG in the scaffold was approximately 10 mol%, and it increased linearly up to approximately 40 mol% with a flow rate of 20 μL min−1. Contrarily to expectations, the lower concentration of shell solution appears to yield a higher UPy-PEG content at the same flow rate in most of the explored conditions. SEM micrographs of the fibers are available in Fig. S2.† Taken together, these analyses confirm that a core–shell structure can be created by coaxial electrospinning of UPy-PC and UPy-PEG solutions for all the three variants of UPy-PEG employed in this study. Furthermore, it is possible to adjust the composition of the fiber mesh mainly by tuning the flow rate of the shell polymer solution.
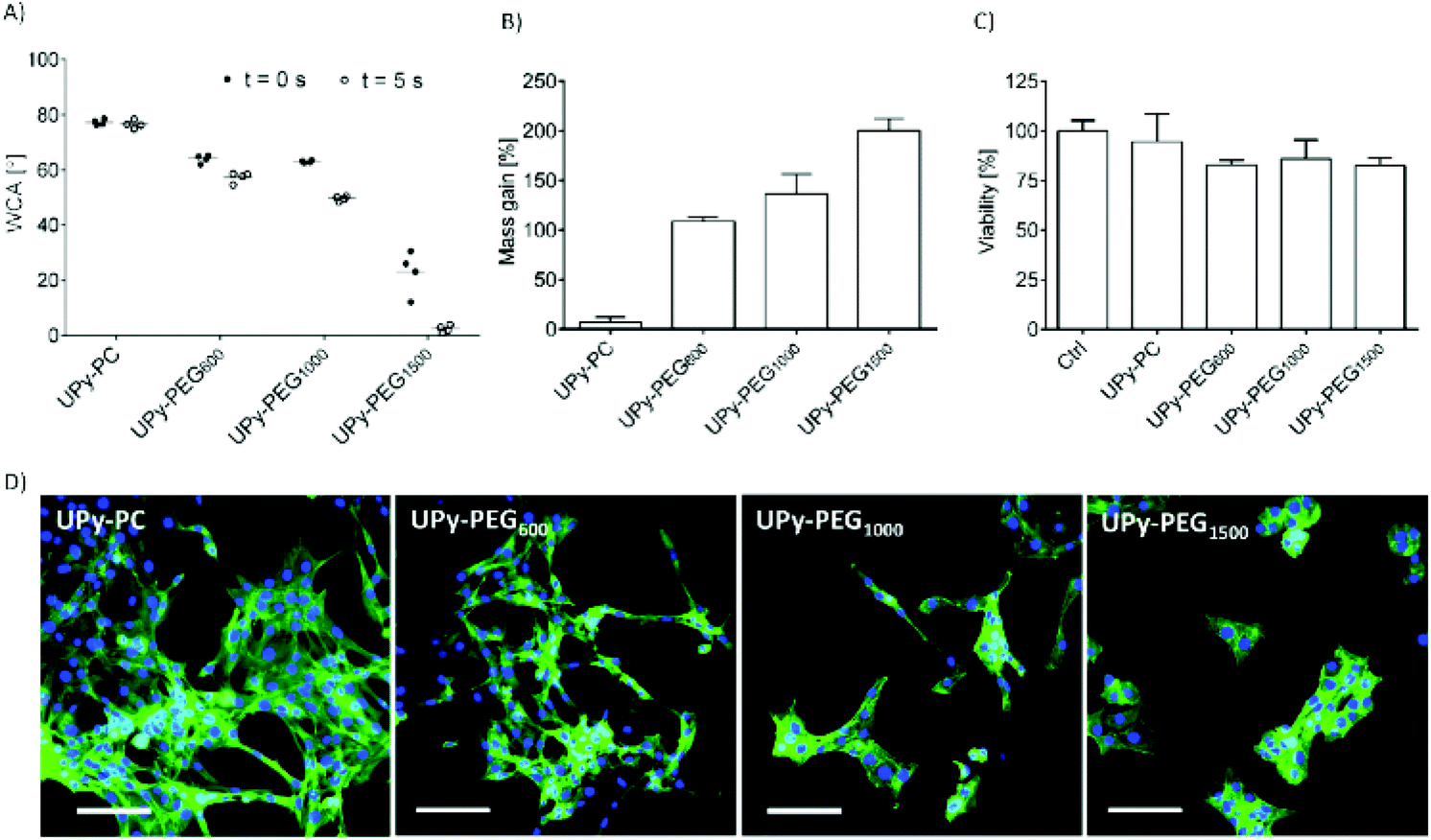 | ||
| Fig. 2 Comparison of surface and swelling properties of UPy-PEG600, 1000, 1500 with those of elastomeric UPy-PC. (A) Water contact angle measured immediately after drop deposition (full circles) and after 5 seconds (empty circles). (B) Water uptake at 37 °C measured after removal of excess water as mass gain with respect to original mass. (C) Representative fluorescence micrographs of adhesion and morphology of 3T3 mouse fibroblasts cultured on cast films for 48 hours. Actin cytoskeleton is stained with phalloidin (green) and nuclei with DAPI (blue). Scale bar represents 100 μm. (D) Viability of 3T3 fibroblasts cultured on cast films for 48 hours. Control condition represents cells cultured on glass surface. | ||
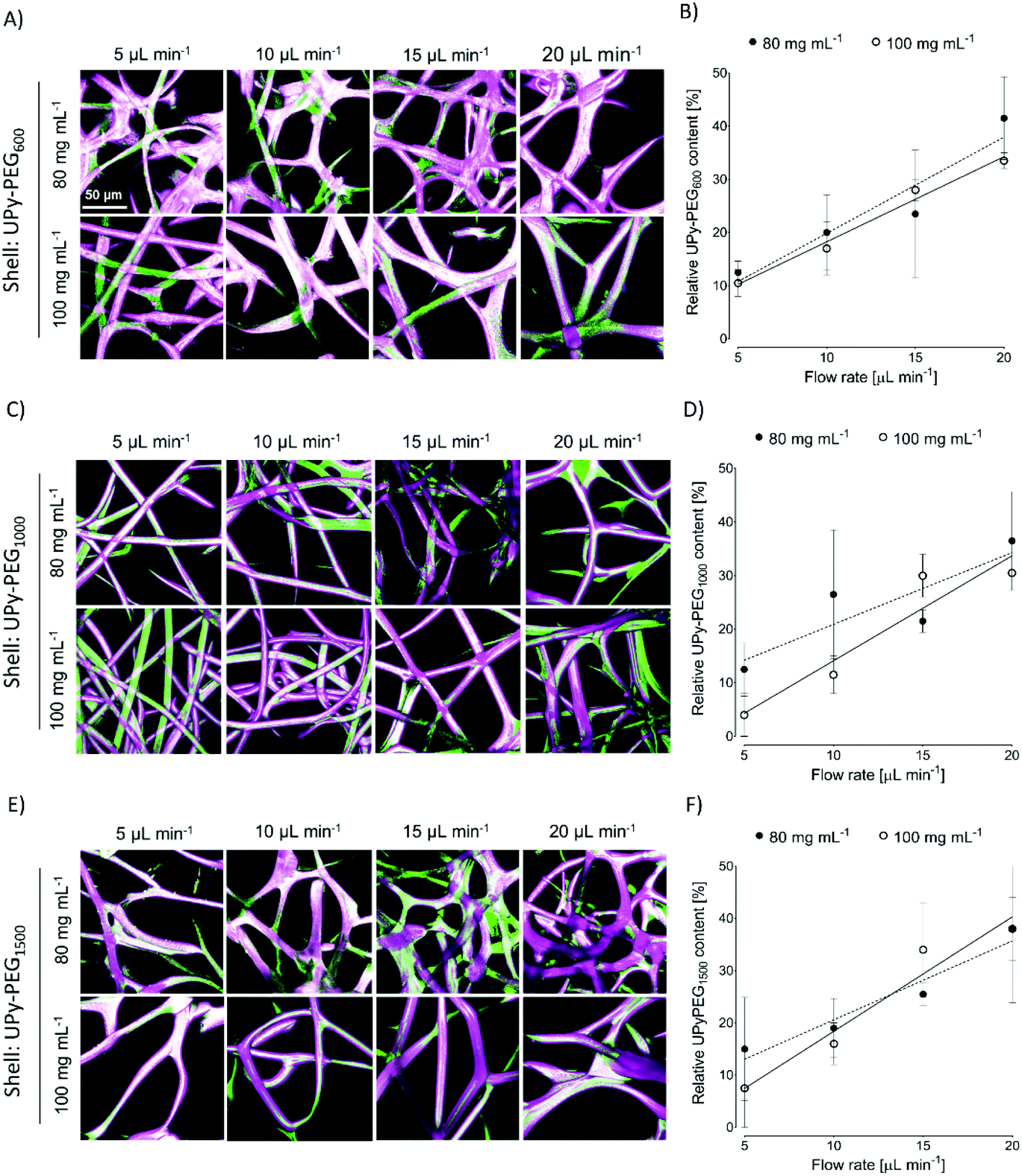 | ||
| Fig. 3 Fabrication of core–shell electrospun fibers. (A, C and E) Maximum intensity projection fluorescence micrographs of core–shell fibers fabricated with a UPy-PC core solution labelled with fluorescein (green) and a shell UPy-PEG solution labeled with rhodamine-B (magenta). The flow rate and concentration of the shell solution were varied between 5 and 20 μL min−1 and 80 and 100 mg mL−1. (B, D and F) Relative UPy-PEG content as a measure for mesh composition with varying flow rate and solution concentration: 80 mg mL−1, full circle and 100 mg mL−1, empty circle; linear fit by least squares method, dashed and continuous line respectively. Scale bar is valid for all the images and represents 50 μm. | ||
In order to be able to compare the effect of different soft block length of UPy-PEG on the resulting mesh properties, the following characterizations and application studies were carried out using hybrid meshes all having a UPy-PC/UPy-PEG molar ratio of approximately 7/3. The desired composition was obtained by electrospinning a shell solution concentration of 100 mg mL−1 with flow rate of 15 μL min−1, in accordance with the results illustrated in Fig. 3, and an inner core UPy-PC solution at 100 mg mL−1 with flow rate of 40 μL min−1. This combination of processing parameters allowed us to obtain smooth and homogeneous fibers with an average diameter of 16 ± 2 μm for UPy-PC/UPy-PEG600, 14 ± 2 μm for UPy-PC/UPy-PEG1000 and 13 ± 2 μm for UPy-PC/UPy-PEG1500 (Fig. 4A-left). The core–shell morphology was present in all the three hybrid meshes (Fig. 4A, right). The swelling behavior was studied for the hybrid meshes with 30 mol% UPy-PEG in water at 37 °C, confirming that all UPy-PC/UPy-PEG meshes are capable of water uptake in the form of an electrospun mesh as well (Fig. 4B).
 | ||
| Fig. 4 Morphology and swelling of core–shell electrospun fibers. (A) SEM micrographs (left) and zoom of maximum intensity projection fluorescence micrograph (right) of non-swollen fibers. UPy-PC (green) form the core, and UPy-PEG600, 1000, 1500 (magenta) forms the shell layer. Hybrid fibers contain 30 mol% of UPy-PEG. (B) Water uptake at 37 °C measured after removal of excess water as mass gain with respect to original mass. Scale bars of SEM images correspond to 100 μm, scale bars of fluorescence micrographs represent 10 μm. | ||
The thermal properties of electrospun fibers were investigated by analyzing the first heating run in DSC experiments (Table S3†). Electrospun UPy-PC meshes have two melting peaks at 58 °C and 93 °C, with enthalpies of 4.3 J g−1 and 1.9 J g−1, respectively. Two melting endotherms are present in the first heating run of bulk samples of UPy-PC as well, at 66 °C and 125 °C, which can be attributed to incipient PC crystallization and UPy-domains, respectively, while the sample is left at room temperature. However, PC semi-crystalline domains are present in minor amount and they are likely to have negligible effects on the overall polymer properties. Core–shell fibers composed by UPy-PC and 30 mol% UPy-PEG600 and UPy-PEG1000 show two melting endotherms, similarly to electrospun UPy-PC, while meshes of UPy-PC/UPy-PEG1500 have three melting peaks. The presence of three melting peaks is found in the first heating run for bulk UPy-PEG1500 as well, suggesting that the longer PEG chain is capable of forming multiple types of ordered structures.
Core–shell meshes of UPy-PC/UPy-PEG were subjected to tensile tests in dry and swollen state to investigate the effect of a swollen shell layer on the mechanical properties of the meshes. Single-component UPy-PC meshes exhibit classic thermoplastic elastomer behavior in the dry state, with elongation at break reaching up to 708 ± 80% (Fig. 5A and E). Hybrid meshes of UPy-PC/UPy-PEG exhibit lower elongation at break and higher modulus in the dry state with respect to UPy-PC meshes (Fig. 5A). The elongation at break is reduced to 515 ± 49% and 490 ± 32% for UPy-PC/UPy-PEG600 and UPy-PC/UPy-PEG1000 meshes, respectively (Fig. 5E, left). The elastic modulus increases from 0.8 ± 0.2 MPa in UPy-PC meshes to 1.3 ± 0.1 MPa and 1.4 ± 0.1 MPa for UPy-PC/UPy-PEG600 and UPy-PC/UPy-PEG1000 meshes, respectively (Fig. 5C, left). This change in properties is likely due to the contribution of a degree of crystallinity present in the UPy-PEG component which confers stiffness and brittleness into the electrospun construct. In the case of UPy-PC/UPy-PEG1500 meshes, the decrease in elongation at break is dramatic and is accompanied by a pronounced drop in ultimate tensile strength (UTS) (Fig. 5D, left). We can speculate that the observed weakness of the mesh containing UPy-PEG1500 comes from the fact that this polymer contains the least supramolecular junctions and alkyl spacers among the investigated variants of UPy-PEG, therefore the applied load is mostly focused on the brittle semi-crystalline PEG segments.
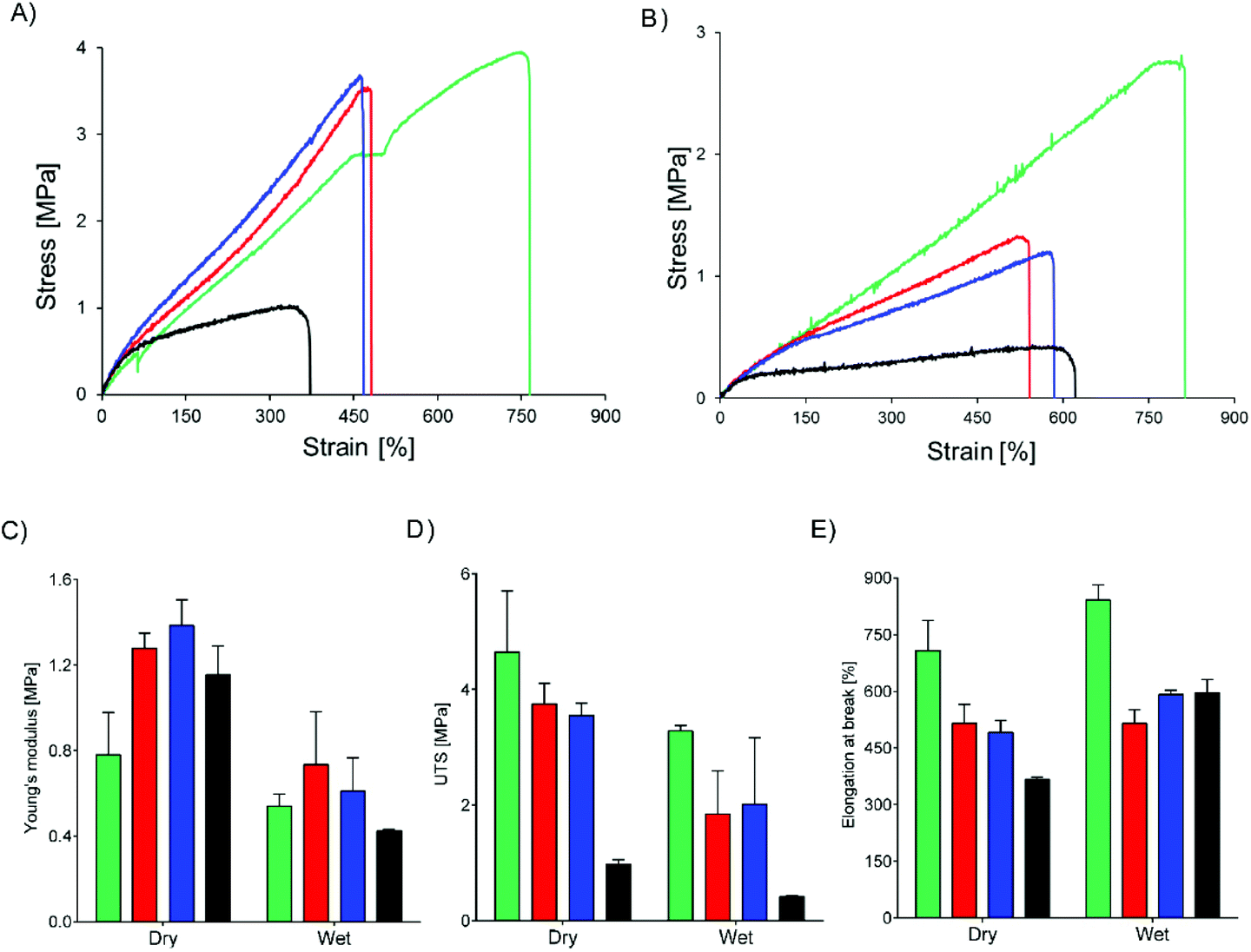 | ||
| Fig. 5 Mechanical properties of core–shell meshes in dry and wet conditions. Core polymer is UPy-PC (green), shell polymers are UPy-PEG600 (red), UPy-PEG1000 (blue), UPy-PEG1500 (black). Representative stress–strain curves from uniaxial tensile test performed on meshes in the dry state (A) or swollen in water (B). Young's modulus (C), ultimate tensile strength (UTS) (D) and elongation at break (E) calculated from the stress–strain curves. | ||
Materials for biomedical implants are generally used in contact with body fluids, therefore it is relevant to study the mechanical behavior of the hybrid meshes after water uptake. As expected, the tensile properties of UPy-PC fiber meshes do not significantly change after the material has been exposed to water (Fig. 5B). For the hybrid meshes, a decrease in Young's modulus is observed to match the modulus of UPy-PC (Fig. 5C, right), which might be a highly desired characteristics in applications in which the properties of UPy-PC were designed specifically to ensure mechanical compliance is required of the material with the surrounding environment. The UTS of hybrid meshes in the swollen state decreases compared to the dry state, consistently with the increase in length of the PEG soft block (Fig. 5D, right), suggesting that swelling of the hydrogel network releases part of the entanglements present both at the molecular level and at fiber level. After water uptake these physical constraints are likely to become weaker, allowing sliding of fibers over each other upon application of tensile load. This hypothesis is supported by the observation that, for hybrid UPy-PC/UPy-PEG meshes, the elongation at break increases in the wet state with respect to the dry state (Fig. 5E, right). Overall, these results suggest that the introduction of 30 mol% UPy-PEG in electrospun UPy-PC meshes preserves the elastomeric properties of the core material.
The core–shell UPy-PC/UPy-PEG fibers were evaluated as a delivery system for small molecule drugs. The drugs were chosen based on their highly different water solubility, but similar polarity. Pirfenidone is a water soluble, FDA-approved substance with anti-fibrotic and anti-inflammatory properties.38 SB431542 is a potent TGF-β pathway inhibitor with low water solubility which was found to prevent scar formation in glaucoma surgery39 and improve long-term patency of tissue engineered vascular grafts.40 The water soluble pirfenidone diffused out of the UPy-PEG shell of the co-axial electrospun fibers almost instantaneously, reaching 80–100% of release within the first hours of incubation of the scaffold in PBS at 37 °C (Fig. 6A). No difference between the UPy-PEG variants was observed. This finding is consistent with those of Pape et al.,17 who reported full release of pirfenidone within one day from a bifunctional UPy-PEG hydrogel. On the contrary, the lipophilic drug SB431542 was released gradually from the outer layer of core–shell fibers over several days and in a UPy-PEG variant-dependent manner (Fig. 6B). After 2 weeks of incubation in PBS at 37 °C, the maximum release of SB431542 was 12 ± 2%, 30 ± 2% and 56 ± 9% when encapsulated in UPy-PEG600, UPy-PEG1000 and UPy-PEG1500, respectively. By design, there is no interaction between the compound SB431542 and the UPy-PEG. However, the chain-extended polymer contains hydrophobic supramolecular linkers and alkyl spacers which are relatively more abundant in the UPy-PEG600 variant, with shorter PEG chains, than in the more hydrophilic UPy-PEG1500. It is proposed that the relatively more hydrophobic nature of UPy-PEG with shorter PEG chains is at the origin of the improved retention of hydrophobic drug SB431542, as it provides a matrix which has greater affinity with the drug than the aqueous environment.
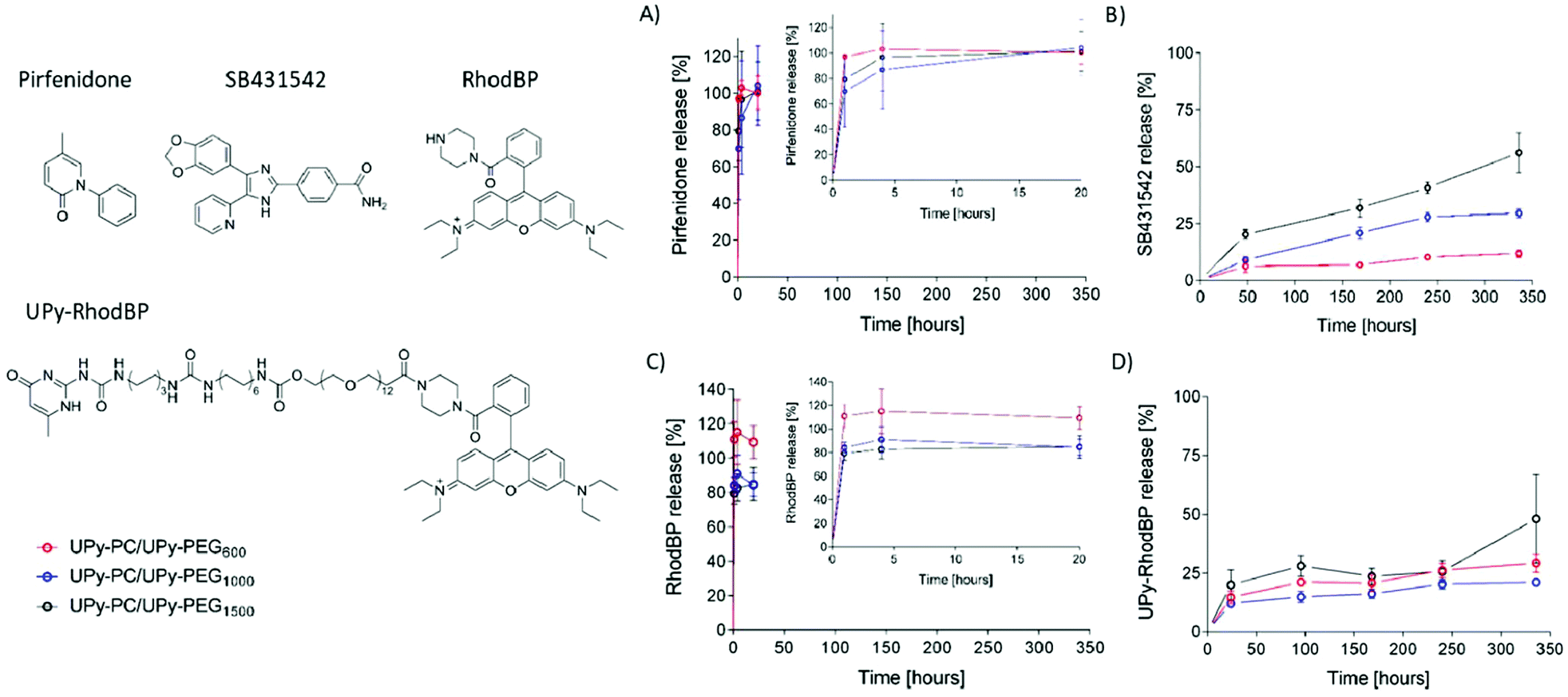 | ||
| Fig. 6 Release of small molecule drugs from core–shell fibers. Chemical structures of pirfenidone, SB431542, rhodamine B-piperazine (RhodBP) and its UPy-modified version (UPy-RhodBP). Release curves over a period of 14 days (336 hours) of pirfenidone (inset contains release over a period of 20 hours) (A); SB431542 (B), RhodBP (inset contains release over a period of 20 hours) (C) and UPy-RhodBP (D). Release from core–shell fibers with shell layer composed of UPy-PEG600 (red), UPy-PEG1000 (blue) and UPy-PEG1500 (black) in PBS at 37 °C. | ||
Drug–matrix affinity is an important determinant of molecule's retention in the carrier for both large and small drug molecules, and it is a target of many strategies for tuning release kinetics from hydrogel systems.18,41–43 Our material system based on strong and reversible supramolecular interactions presents the unique advantage of modularity. Therefore, we designed and synthesized a UPy-functionalized rhodamine B-piperazine dye (UPy-RhodBP), which is positively charged and highly water soluble, but at the same time has specific supramolecular interactions with the UPy-PEG matrix. As expected, complete release of the also highly water-soluble unmodified RhodBP from the UPy-PEG shell of hybrid fibers occurs within the first hour of exposure to aqueous environment, regardless of the UPy-PEG variant employed for encapsulation (Fig. 6C). Instead, the UPy-modified compound UPy-RhodBP is eluted slowly, with only 15–20% release from the fiber mesh over a period of two weeks (Fig. 6D). Interestingly, there seems to be only a minor difference in release between the three UPy-PEG shell polymers, indicating that the strong UPy–UPy interactions are dominant in determining the retention of UPy-RhodBP over the other features of the polymer matrix. With these results, we demonstrate that the simple UPy-modification of a model water soluble molecule is effective in increasing drug–matrix affinity, thereby achieving long-term retention of a compound which would otherwise undergo burst release. The release behavior presented in Fig. 6D is a mere proof-of-concept implementation of slow release induced by supramolecular drug–matrix interactions, therefore the UPy-RhodBP molecule has been designed to be incorporated in the UPy-PEG with strong and specific supramolecular interactions. For applications requiring an intermediate release rate, a rational molecular design can be of help in tuning the elution rate.
Conclusions
In this work, supramolecular poly(hexamethylene carbonate) and poly(ethylene glycol) containing UPy groups in the polymer chain have been processed into core–shell fibers to create a multi-functional robust mesh with water swelling and drug eluting properties. The molecular design of chain-extended UPy-PEG, combining relatively low molecular weight PEG segments separated by UPy units, allows the preparation of hydrophilic yet processable and cell-adhesive polymers. The introduction of UPy-PEG shell has an impact on elasticity and strength of the hybrid construct, yet the elastic modulus of the monocomponent UPy-PC mesh is preserved. The multicomponent UPy-PC/UPy-PEG system proved itself suitable as a drug delivery vehicle for small molecule drugs. A hydrophobic model drug exhibited sustained release up to 14 days of incubation. Highly hydrophilic compounds underwent burst release, but a simple modification with a UPy-moiety allowed the creation of drug–matrix interaction, thereby significantly increasing drug retention. In conclusion, the supramolecular core–shell fiber system based on UPy groups presented here provides an excellent platform for the creation of multi-component systems encompassing tunable strength and drug release.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the European Research Council (FP7/2007–2013) Grant Agreement 604514 and ERC Grant Agreement 308045, and the Ministry of Education, Culture and Science (Gravity Program 024.001.03).Notes and references
- E. Im and M. K. Hong, Expert Rev. Cardiovasc. Ther., 2016, 14, 87–104 CrossRef CAS.
- L. I. F. Moura, A. M. A. Dias, E. Carvalho and H. C. De Sousa, Acta Biomater., 2013, 9, 7093–7114 CrossRef CAS.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS.
- M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Nat. Mater., 2015, 15, 13–26 CrossRef.
- Self-Assembling Biomaterials: Molecular design, Characterization and Applications in Biology and Medicine, ed. H. S. Azevedo and R. M. P. da Silva, Woodhead Publishing, 2018 Search PubMed.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. Folmer, J. H. Hirschberg, R. F. Lange, J. K. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS.
- R. P. Sijbesma, E. W. Meijer and A. W. Bosman, Mater. Today, 2004, 34–39 Search PubMed.
- O. J. G. M. Goor, S. I. S. Hendrikse, P. Y. W. Dankers and E. W. Meijer, Chem. Soc. Rev., 2017, 46, 6621–6637 RSC.
- M. H. Bakker, R. E. Kieltyka, L. Albertazzi and P. Y. W. Dankers, RSC Adv., 2016, 6, 110600–110603 RSC.
- P. Y. W. Dankers, M. C. Harmsen, L. A. Brouwer, M. J. A. Van Luyn and E. W. Meijer, Nat. Mater., 2005, 4, 568–574 CrossRef CAS.
- O. J. G. M. Goor, H. M. Keizer, A. L. Bruinen, M. G. J. Schmitz, R. M. Versteegen, H. M. Janssen, R. M. A. Heeren and P. Y. W. Dankers, Adv. Mater., 2017, 29, 1–8 CrossRef.
- L. A. Bockeria, O. Svanidze, A. Kim, K. Shatalov, V. Makarenko, M. Cox and T. Carrel, J. Thorac. Cardiovasc. Surg., 2017, 153, 1542–1550 CrossRef.
- L. Hympanova, M. G. M. C. Mori da Cunha, R. Rynkevic, R. A. Wach, A. K. Olejnik, P. Y. W. Dankers, B. Arts, T. Mes, A. W. Bosman, M. Albersen and J. Deprest, J. Mech. Behav. Biomed. Mater., 2018, 88, 431–441 CrossRef CAS.
- P. W. Serruys, Y. Miyazaki, A. Katsikis, M. Abdelghani, M. B. Leon, R. Virmani, T. Carrel, M. Cox, Y. Onuma and O. I. I. Soliman, EuroIntervention, 2017, 13, AA68–AA77 CrossRef PubMed.
- D. E. P. Muylaert, G. C. van Almen, H. Talacua, J. O. Fledderus, J. Kluin, S. I. S. Hendrikse, J. L. J. van Dongen, E. Sijbesma, A. W. Bosman, T. Mes, S. H. Thakkar, A. I. P. M. Smits, C. V. C. Bouten, P. Y. W. Dankers and M. C. Verhaar, Biomaterials, 2016, 76, 187–195 CrossRef CAS.
- P. Y. W. Dankers, T. M. Hermans, T. W. Baughman, Y. Kamikawa, R. E. Kieltyka, M. M. C. Bastings, H. M. Janssen, N. A. J. M. Sommerdijk, A. Larsen, M. J. A. Van Luyn, A. W. Bosman, E. R. Popa, G. Fytas and E. W. Meijer, Adv. Mater., 2012, 24, 2703–2709 CrossRef CAS.
- A. C. H. Pape, M. H. Bakker, C. C. S. Tseng, M. M. C. Bastings, S. Koudstaal, P. Agostoni, S. A. J. Chamuleau and P. Y. W. Dankers, J. Visualized Exp., 2015, 100, 1–8 Search PubMed.
- M. H. Bakker, M. Grillaud, D. J. Wu, P. P. K. H. Fransen, I. H. de Hingh and P. Y. W. Dankers, Macromol. Rapid Commun., 2018, 39, 1–6 CrossRef.
- M. H. Bakker, E. van Rooij and P. Y. W. Dankers, Chem. – Asian J., 2018, 13, 3501–3508 CrossRef CAS.
- M. H. Bakker, C. C. S. Tseng, H. M. Keizer, P. R. Seevinck, H. M. Janssen, F. J. Van Slochteren, S. A. J. Chamuleau and P. Y. W. Dankers, Adv. Healthcare Mater., 2018, 7, 1–8 Search PubMed.
- M. Guo, L. M. Pitet, H. M. Wyss, M. Vos, P. Y. W. Dankers and E. W. Meijer, J. Am. Chem. Soc., 2014, 136, 6969–6977 CrossRef CAS.
- B. B. Mollet, S. Spaans, P. G. Fard, N. A. M. Bax, C. V. C. Bouten and P. Y. W. Dankers, Macromol. Biosci., 2017, 17, 1–8 CrossRef.
- Y. Yang, W. Li, D. G. Yu, G. Wang, G. R. Williams and Z. Zhang, Carbohydr. Polym., 2019, 203, 228–237 CrossRef CAS.
- A. Saraf, G. Lozier, A. Haesslein, F. K. Kasper, R. M. Raphael, L. S. Baggett and A. G. Mikos, Tissue Eng., Part C, 2009, 15, 333–344 CrossRef CAS PubMed.
- P. McClellan and W. J. Landis, BioRes. Open Access, 2016, 5, 212–227 CrossRef CAS.
- C. Wang, K. W. Yan, Y. D. Lin and P. C. H. Hsieh, Macromolecules, 2010, 43, 6389–6397 CrossRef CAS.
- S. J. Song, Y. C. Shin, S. E. Kim, I. K. Kwon, J. H. Lee, S. H. Hyon, D. W. Han and B. Kim, Sci. Rep., 2018, 8, 1–11 CrossRef.
- Y. Su, Q. Su, W. Liu, M. Lim, J. R. Venugopal, X. Mo, S. Ramakrishna, S. S. Al-Deyab and M. El-Newehy, Acta Biomater., 2012, 8, 763–771 CrossRef CAS.
- M. Jin, D. G. Yu, X. Wang, C. F. G. C. Geraldes, G. R. Williams and S. W. A. Bligh, Adv. Healthcare Mater., 2016, 5, 977–985 CrossRef CAS.
- T. Nguyen and M. B. Francis, Org. Lett., 2003, 5, 3245–3248 CrossRef CAS.
- I. De Feijter, O. J. G. M. Goor, S. I. S. Hendrikse, M. Comellas-Aragonès, S. H. M. Söntjens, S. Zaccaria, P. P. K. H. Fransen, J. W. Peeters, L. G. Milroy and P. Y. W. Dankers, Synlett, 2015, 26, 2707–2713 CrossRef CAS.
- A. Saraf, G. Lozier, A. Haesslein, F. K. Kasper, R. M. Raphael, L. S. Baggett and A. G. Mikos, Tissue Eng., Part C, 2009, 15, 333–344 CrossRef CAS.
- M. Hutin, E. Burakowska-Meise, W. P. J. Appel, P. Y. W. Dankers and E. W. Meijer, Macromolecules, 2013, 46, 8528–8537 CrossRef CAS.
- A. C. H. Pape, B. D. Ippel and P. Y. W. Dankers, Langmuir, 2017, 33, 4076–4082 CrossRef CAS.
- B. D. Ippel, H. M. Keizer and P. Y. W. Dankers, Adv. Funct. Mater., 2019, 29, 1–11 CrossRef.
- H. Zhou, Z. Shi, X. Wan, H. Fang, D.-G. Yu, X. Chen and P. Liu, Nanomaterials, 2019, 9, 843–857 CrossRef CAS.
- Y. Yang, T. Zhu, Z. Liu, M. Luo, D. Yu and S. W. A. Bligh, Int. J. Pharm., 2019, 569, 118634–118641 CrossRef CAS.
- C. Aravena, G. Labarca, C. Venegas, A. Arenas and G. Rada, PLoS One, 2015, 10, 1–15 Search PubMed.
- Y. qin Xiao, K. Liu, J. F. Shen, G. T. Xu and W. Ye, Invest. Ophthalmol. Visual Sci., 2009, 50, 1698–1706 CrossRef.
- Y. U. Lee, J. De Dios Ruiz-Rosado, N. Mahler, C. A. Best, S. Tara, T. Yi, T. Shoji, T. Sugiura, A. Y. Lee, F. Robledo-Avila, N. Hibino, J. S. Pober, T. Shinoka, S. Partida-Sanchez and C. K. Breuer, FASEB J., 2016, 30, 2627–2636 CrossRef CAS.
- K. Vulic and M. S. Shoichet, Biomacromolecules, 2014, 15, 3867–3880 CrossRef CAS.
- M. Qiao, D. Chen, X. Ma and Y. Liu, Int. J. Pharm., 2005, 294, 103–112 CrossRef CAS.
- A. G. Cheetham, R. W. Chakroun, W. Ma and H. Cui, Chem. Soc. Rev., 2017, 46, 6638–6663 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9bm01241a |
| This journal is © The Royal Society of Chemistry 2020 |