
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nano-inclusion in one step: spontaneous ice-templating of porous hierarchical nanocomposites for selective hydrogen release†
Simon
Champet
a,
Jan
van den Berg
ab,
Robert
Szczesny
 c,
Agata
Godula-Jopek
de and
Duncan H.
Gregory
*a
c,
Agata
Godula-Jopek
de and
Duncan H.
Gregory
*a
aWestCHEM, University of Glasgow, School of Chemistry, University Avenue G128QQ Glasgow, UK. E-mail: Duncan.Gregory@glasgow.ac.uk
bHumboldt Universität zu Berlin, Institut für Chemie, Brook-Taylor Strasse 2, 12489 Berlin, Germany
cNicolaus Copernicus University in Toruń, Faculty of Chemistry, Gagarina 7, 87-100 Toruń, Poland
dAirbus Group Innovations, XCXDI, 81663 Munich, Germany
eInstitute of Chemical Engineering, Polish Academy of Sciences, 44100 Gliwice, Poland
First published on 7th January 2019
Abstract
3-Dimensional porous scaffold materials can be fabricated by ice templating sheets of graphene oxide (GO) or partially reduced graphene oxide (rGO). Aqueous suspensions of GO (or rGO) can be cast into monoliths or formed as beads on cooling and the solid matrices then fashioned with either laminar or radial porosity as a result. Further, ammonia borane (AB) can be integrated into the hierarchical structures in situ in a one-step process without the requirement of melt infiltration or solution impregnation techniques. Compared to AB itself, the ensuing self-assembled beads release hydrogen at a reduced onset temperature and without volume expansion on heating, suppressing the release of diborane, borazine and ammonia. Pre-reduction of the GO matrix material (to rGO) eliminates CO/CO2 release from the composites.
Introduction
Carbon has an illustrious recent history of use as a porous support and matrix material.1–3 Disordered activated carbons have inherently high surface areas after processing and micro- and mesoporous carbons can be prepared by an array of chemical templating techniques.3 Porous carbons themselves not only exhibit many useful chemical and physical properties as a function of their microstructure, but can also act as hosts for “active” guests with specific functionality. Simple routes to prepare porous (functionalised) carbon scaffolds with characteristic internal structures are extremely attractive in the design of new and improved composite materials.Hydrogen has huge potential as a sustainable alternative to fossil fuels, but its adoption has been frustrated by the challenge of its storage. Solid state storage could provide a remedy if performance requirements can be met.4–7 Ammonia borane (AB) contains 19.6 wt% hydrogen (ca. 13 wt% is readily accessible).8–10 However, its decomposition also releases deleterious gaseous by-products.8–15 The formation of nanocomposites with porous hosts is one way to limit these by-products without the use of noble metal catalysts and can also improve performance in a number of other ways.6,16–20 Commonly solution impregnation is required to support AB in porous structures. Diffusion of AB through the pores of the host (carbon, metal–organic frameworks (MOFs) or silica) can be challenging, time consuming and usually requires organic solvents.10,16–18,20,21 Inhomogeneous and/or impure materials can result.
Here we describe a fresh approach to nanocomposite fabrication, first designing a porous host material from graphene oxide (GO) and subsequently engineering nanocomposites with AB in a simple one-step process otherwise using only water. Nanocomposite formation occurs in situ and requires no impregnation or infiltration. Aqueous self-assembly is driven by the growth of ice crystals during the templating process. Moreover, the pre-reduction of GO (to partially reduced graphene oxide (rGO)) eliminates CO and CO2 release during AB thermal decomposition, reducing the weight of the host matrix further and ensuring COx-free hydrogen is released.
Results and discussion
It has been suggested that AB and GO sheets can form sandwich-like structures when AB is impregnated into GO in THF.15 Similar multi-layers are the base components of the hierarchical nanocomposite architectures we observe here. The self-assembly that occurs during the ice-templating of a GO(-AB) suspension in water is similar to the process exploited in the processing of ceramics;22 ice crystals grow and compress the suspended particles together. The crystal growth, and thus the skeletal structure, can be mediated by control of the temperature gradient. Moreover, the shape of the porous material can be controlled (Fig. S1†).23 The use of a mould in contact with a coolant leads to a material of prescribed shape (e.g. block monolith) with laminar porosity (in the direction of the temperature gradient; Fig. S2†), whereas dropping the suspension into liquid nitrogen, for example, leads to beads with radial porosity (where the spherical surface is cooler than the core; Fig. S3†). In our process, the GO sheets (ca. 3.5 nm and <6 GO layers thick by atomic force microscopy (AFM; Fig. S4†)) or sandwich-like AB–GO structures organise to form the internal monolith or bead structure (Fig. 1). GO beads are typically 2.8 ± 0.2 mm in diameter, have a density of 8.3 ± 0.3 kg m−3 and a specific surface area of 33.5 ± 3.1 m2 g−1 (from BET calculations). The highest order layers in the hierarchical internal structure are ca. 5 μm thick (Fig. 1c). The internal lamellar structure is consistent with the self-assembly process of GO sheets into 3D forms.24 | ||
| Fig. 1 (a) Schematic of the assembly of AB–GO composite beads where sandwich-like GO/AB layers rearrange radially during ice-templating; SEM images of the radial structure of: (b), (c) GO and (d) AB–GO composite. | ||
FT-IR spectra (Fig. S5†) and thermogravimetric-differential thermal analysis with evolved gas mass spectrometry (TG-DTA-MS) profiles demonstrate the presence of AB across a range of composites with AB![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :GO weight ratios varying from 1:5 to 7:1. Scanning transmission electron microscopy (STEM) confirmed the assimilation of AB within both the GO (Fig. S6†) and pre-reduced rGO scaffolds (Fig. 2a; see below). The largest AB clusters observed were ca. 300 nm across and scanning electron microscopy (SEM) reveals the presence of a dispersion of AB clusters throughout the thickness of the solids (Fig. S7†). Interestingly, powder X-ray diffraction (PXD) patterns comparing ice-templated GO, as-received AB and AB–GO nanocomposites showed that whereas AB peak positions remained unchanged, the (001) GO reflection was shifted to lower 2θ in nanocomposites, suggesting expansion of the GO interlayer spacing and intercalation of AB between GO layers at the atomic scale (Fig. 2b). Similar expansion phenomena occur when gases or polymers are intercalated between GO layers.25,26
:GO weight ratios varying from 1:5 to 7:1. Scanning transmission electron microscopy (STEM) confirmed the assimilation of AB within both the GO (Fig. S6†) and pre-reduced rGO scaffolds (Fig. 2a; see below). The largest AB clusters observed were ca. 300 nm across and scanning electron microscopy (SEM) reveals the presence of a dispersion of AB clusters throughout the thickness of the solids (Fig. S7†). Interestingly, powder X-ray diffraction (PXD) patterns comparing ice-templated GO, as-received AB and AB–GO nanocomposites showed that whereas AB peak positions remained unchanged, the (001) GO reflection was shifted to lower 2θ in nanocomposites, suggesting expansion of the GO interlayer spacing and intercalation of AB between GO layers at the atomic scale (Fig. 2b). Similar expansion phenomena occur when gases or polymers are intercalated between GO layers.25,26
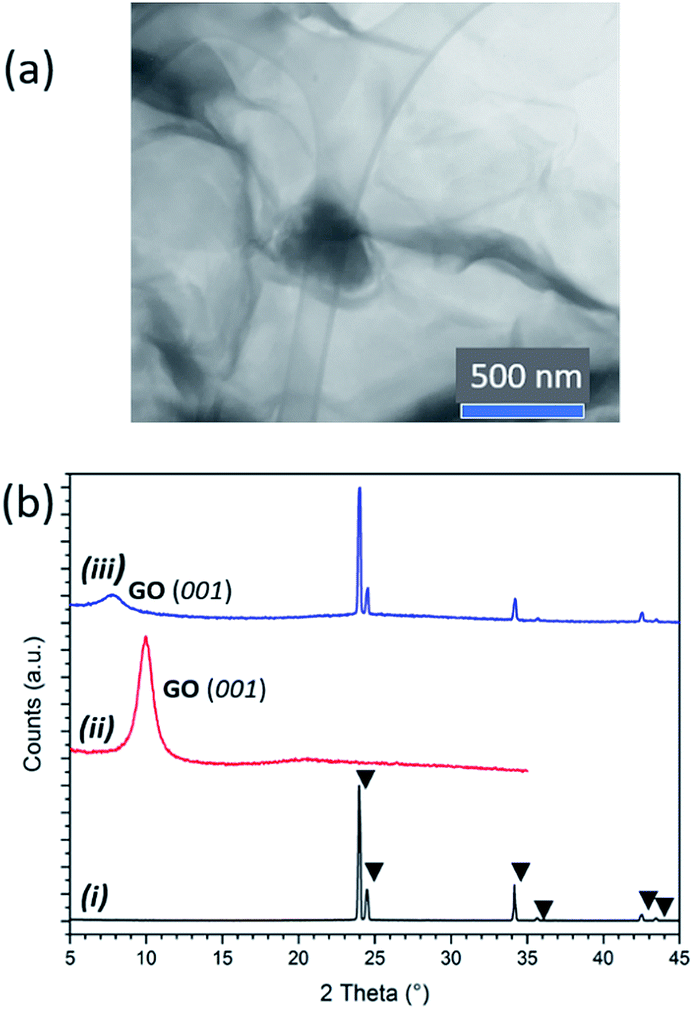 | ||
| Fig. 2 (a) STEM image of 1:2 AB:rGO composite showing a larger cluster of AB among rGO sheets; (b) PXD patterns of: (i) as-received AB, (ii) ice-templated GO beads; (iii) 5:4 AB:GO nanocomposite. AB reflections are indicated by triangles. | ||
In fact, at low AB:GO ratios only the shifted GO reflection is visible in the PXD pattern (e.g. 1:5), but as the ratio increases (e.g. to 3:4) the AB reflections become more apparent, the GO reflection shifts further to lower 2θ (Fig. S8†) and AB clusters become more prominent in micrographs. At higher AB:GO ratios there is no further shift of the GO reflection in the diffractograms and the AB clusters in SEM images grow in size. These data suggest that AB is therefore present across length scales, within both the crystal structure and the microstructure of the GO hosts, first intercalating between GO layers and subsequently enmeshed as particles in the matrix as the AB:GO ratio increases.
The formation of nanocomposites has profound effects on the AB thermal decomposition. Pristine AB presents an endotherm at 112 °C incorporating melting, an exotherm 10 °C higher (1st decomposition step) and a further exotherm at 156 °C (2nd decomposition step; Fig. S9†). AB has an average mass loss of 61 ± 2% by the end of the second step (250 °C) forming polymeric (NHBH)n and releasing hydrogen, borazine, diborane and ammonia, beginning with hydrogen evolution at ca. 90 °C (Fig. 3a), matching literature data.10,27 This gas release during the AB polymerisation step leads to a documented foam formation and volume expansion. The gaseous by-products are not only toxic, but also poison fuel cell catalysts.6,8–11,14 By contrast, no foaming is observed in the composites and the matrix structure remains intact during thermal decomposition. Hence the GO scaffold mitigates against volume increases. Correspondingly, there is no “recoil” effect observed in the thermogravimetric (TG) curves of the composites near the melting point. GO beads themselves lose ca. 34 wt% up to 250 °C (Fig. S10†). For both the GO matrix and the composites, most of the mass change arises from loss of CO2 (and CO).
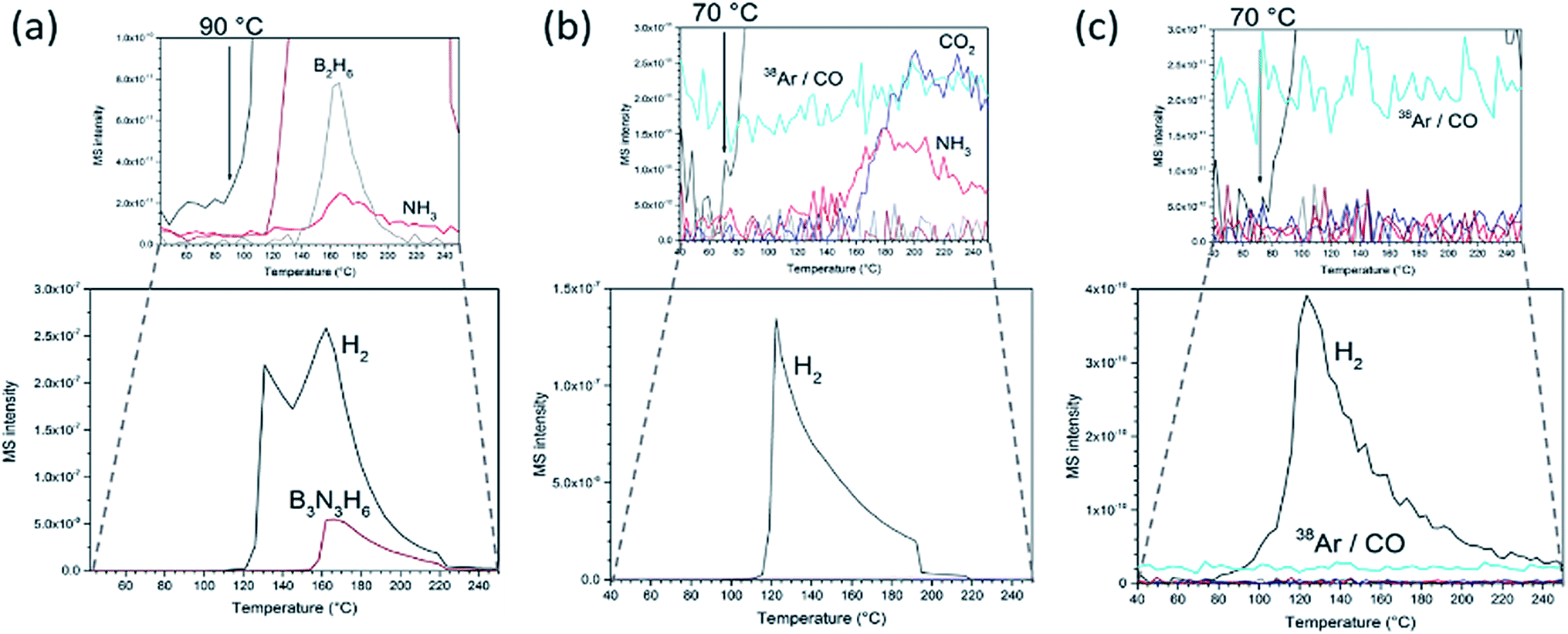 | ||
| Fig. 3 Evolved gas mass spectra vs. temperature (RT – 250 °C) of: (a) as-received AB; (b) 1:1 AB:GO composite and (c) 1:2 AB:rGO composite. 38Ar is present at approximately constant partial pressure in the TG-DTA-MS gas stream in all experiments. | ||
The evolution of the deleterious AB decomposition by-products is significantly diminished in the composites and hydrogen is first detected at 70 °C (20 °C lower than in AB alone; Fig. 3b). In fact, all composites with an AB:GO weight ratio ≤ 1 release no detectable borazine or diborane below 250 °C. Similarly, ammonia release is reduced by 1–2 orders of magnitude to trace levels as compared to pristine AB. Interestingly, unlike AB itself, the composite shows no evidence of a second decomposition step (the exotherm for AB at 156 °C). The data imply that both the thermodynamics and kinetics of decomposition are altered on incorporating AB within the 3D GO matrix. Including that associated with COx from the matrix, the mass loss below 250 °C is ca. 22.5% for an optimised 1:1 AB:GO composite. This corresponds to a release of ca. 5.5 wt% H2 in the 1:1 composite and a gravimetric capacity of 11 wt% for the active material. This would be consistent with 84% of the expected hydrogen release for AB itself at similar temperature (13.1 wt% H2; 2 equiv. of H2).
The performance of the composites can be improved considerably by pre-treating the GO (250 °C/Ar(g)) prior to templating to produce partially reduced graphene oxide (rGO). The reduction (as performed by TG-DTA-MS to constant sample mass) leads to evolution of CO2 (and trace CO) (Fig. S11, S12†). FTIR spectra show that C–O bands gradually diminish during reduction (Fig. S13, S14†). Subsequent TG-DTA-MS of rGO beads shows no further mass loss or gas emission up to 250 °C and use of the pre-treated rGO to make AB composites subsequently yields no CO2 or CO during decomposition.
Most significantly, the use of an rGO matrix provides materials which release hydrogen of even higher purity on heating. In addition to the absence of borazine and diborane, no ammonia is detected in the evolved gas (Fig. 3c). Hence replacing GO by rGO in the nanocomposites removes all the deleterious gaseous by-products from AB decomposition (in addition to the COx emissions from the matrix itself). Previously it has proved difficult to eliminate ammonia by impregnating AB in carbon matrices. Promising recent work has indicated that ammonia emission can be reduced significantly by building carbon scaffolds with a narrow microporous pore distribution.28 Alternatively, its suppression is enhanced by confining AB in MOF matrices rather than carbon (where the former supply metal centres to which N donor atoms can coordinate).16 Otherwise, the only alternative is to employ filters such as metal halides as part of a storage system which abstracts ammonia from hydrogen downstream.27,29
Given that thermal analyses indicate that not only the kinetics but also the thermodynamics of AB decomposition are altered in AB–(r)GO nanocomposites, then the decomposition reaction pathways must differ significantly from AB itself. This would suggest that gaseous by-product species are not simply physically trapped by the matrix and previous evidence suggests that AB follows alternative reaction paths when confined in certain porous structures.10,18 From experimental and theoretical studies it has been proposed that AB infiltrated into porous silica,21,30,31 or activated carbons,10,20,27 can undergo acid–base reactions with surface hydroxyl groups evolving H2 and tethering the remainder of the AB molecule to the surface for subsequent reactions with other AB molecules or other surface groups. The formation of strong B–O bonds at the matrix surface prevents the evolution of borazine and diborane observed in free AB. A range of hydrogen- and oxygen-containing functionalities exist on the surface of GO and are likely to behave similarly.15 The presence of B–O bands in IR spectra of the dehydrogenated r(GO)–AB nanocomposites would support this premise with Lewis acidic B bound to O functionalities (e.g. carboxyl-, epoxy-) at the nanocomposite surface and surface hydroxyl-groups providing alternative routes to dehydrogenation by combination with Hδ− from –BH3.
One of the historical challenges associated with nanoconfinement of AB is that impregnation/infiltration is dependent on AB diffusion through the pores of the porous material. This diffusion is in turn dependent on the solvent system and the accessibility of pores in the internal structure. Homogeneous dispersions of active materials in composites can be difficult to control and reproduce.32–35 This is one of the advantages in producing AB–(r)GO composites in situ in a single step. The direct fabrication of a 3D composite structure avoids impregnation/infiltration and such issues of AB diffusion (Fig. 1). We are exploring ways to exert more precise control over the bead dimensions, shape and internal porosity and, for example, nebulisation of the (r)GO and AB–(r)GO suspensions at the ice templating stage results in matrices of even lower density and higher porosity (Fig. S15†) with beads that can be tuned in diameter to sub-micron levels. We are also investigating routes to regenerate the composites, both by recycling the active AB components and by re-using the GO matrix. The preservation of the (r)GO skeleton after hydrogen release and the retention of the dehydrogenated active material within (Fig. S16–S18†), facilitates the recycling of composites (although, if necessary, they could be simply reconstructed in a further ice templating step). Among several mooted schemes for AB regeneration, one of the simplest to implement involves reaction of the partially-spent AB product, polyborazylene with hydrazine solvothermally (at 313 K) using ammonia.36 This is one feasible route to return to the charged AB–(r)GO nanocomposite without compromising the microstructure. In principle, the (r)GO ice-templating technique should be readily scalable and composite monoliths of prescribed size and shape should be within reach. Moreover, the technique is not restricted to AB as an active material. Other components with a vast array of different chemical and physical properties could be integrated to make new functional and/or chemically-active composite materials.
Experimental
Full experimental details are provided in the ESI.† GO was synthesised following Tour's method.37 The washed GO dispersion was concentrated to 20 mg mL−1 and sonicated (Elma S30 Elmasonic bath). For GO–AB composites, AB (Sigma, 97%) was added to the GO suspension at this point in the process. The suspension was stirred at RT for 24 h. To prepare beads, drops of the concentrate were added to liquid nitrogen. The frozen beads were freeze dried, isolated and retained for analyses. The thermal behaviour of the templated materials was evaluated by TG-DTA-MS under argon (Netzsch STA 409 PC and Hiden Analytical mass spectrometer). Microstructure was investigated by SEM (Philips XL30 microscope with Oxford Instruments INCA500 detector and Quanta 3D FEG microscope), AFM (Bruker Dimension ICON, scanasyst mode) and STEM (Quanta 3D FEG instrument, with copper grid). The density of the composites was measured via helium pycnometry using multiple room temperature isotherms from vacuum to ≥4 bar (IGA; Hiden Isochema). Specific surface area was determined by BET analysis (Quantachrome Evo instrument with Quadrawin software). Purity, structure and bonding were evaluated by PXD (PANalytical X'Pert Pro and Bruker D8 advance diffractometers; Cu Kα and Cu Kα1 radiation respectively) and vibrational spectroscopy (FTIR and Raman using Shimadzu IRAffinity-1S and Horiba LabRAM HR instruments respectively).Conclusion
Ice templating techniques can be utilised to freeze cast monoliths or beads with either laminar or radial porosity respectively from aqueous suspensions of (reduced) graphene oxide (r)GO sheets. Ammonia borane (AB) can be incorporated in situ within the porous scaffolds by following an otherwise identical process when AB is dissolved in the initial (r)GO suspension. The facile one-step process entirely overcomes the requirements of melt infiltration or solution impregnation in achieving a nanoconfined composite. The self-assembled hierarchical beads offer significant performance advantages for hydrogen release when compared to AB itself. Hydrogen is evolved at a reduced onset temperature and without the characteristic volume expansion characteristic of AB on heating. The GO matrix suppresses the release of deleterious diborane, borazine and ammonia AB decomposition by-products and by pre-reducing the GO matrix material (to rGO), unwanted CO/CO2 release from the composite is eliminated.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. H. G. thanks Airbus Group Innovations for funding a studentship for S. C.References
- J. Lee, J. Kim and T. Hyeon, Recent progress in the synthesis of porous carbon materials, Adv. Mater., 2006, 18, 2073–2094 CrossRef CAS.
- R. White, Porous Carbon Materials from Sustainable Precursors, Royal Society of Chemistry, Cambridge, 2015 Search PubMed.
- Y. Xia, Z. Yang and R. Mokaya, in Porous Materials, John Wiley & Sons, Ltd, Chichester, UK, 2010, pp. 217–264 Search PubMed.
- T. K. Mandal and D. H. Gregory, Hydrogen: A future energy vector for sustainable development, Proc. Inst. Mech. Eng., Part C, 2010, 224, 539–558 CrossRef.
- J. Yang, A. Sudik, C. Wolverton and D. J. Siegel, High capacity hydrogen storage materials: attributes for automotive applications and techniques for materials discovery, Chem. Soc. Rev., 2010, 39, 656–675 RSC.
- H. Reardon, J. M. Hanlon, R. W. Hughes, A. Godula-Jopek, T. K. Mandal and D. H. Gregory, Emerging concepts in solid-state hydrogen storage: the role of nanomaterials design, Energy Environ. Sci., 2012, 5, 5951 RSC.
- D. J. Durbin and C. Malardier-Jugroot, Review of hydrogen storage techniques for on board vehicle applications, Int. J. Hydrogen Energy, 2013, 38, 14595–14617 CrossRef CAS.
- B. Peng and J. Chen, Ammonia borane as an efficient and lightweight hydrogen storage medium, Energy Environ. Sci., 2008, 1, 479–483 CAS.
- A. Karkamkar, C. Aardahl and T. Autrey, Recent Developments on Hydrogen Release from Ammonia-Borane, Mater. Matters, 2007, 2, 6–9 CAS.
- U. B. Demirci, Ammonia borane, a material with exceptional properties for chemical hydrogen storage, Int. J. Hydrogen Energy, 2016, 42, 9978–10013 CrossRef.
- B. L. Davis, D. a. Dixon, E. B. Garner, J. C. Gordon, M. H. Matus, B. Scott and F. H. Stephens, Efficient regeneration of partially spent ammonia borane fuel, Angew. Chem., Int. Ed., 2009, 48, 6812–6816 CrossRef CAS PubMed.
- M. Diwan, V. Diakov, E. Shafirovich and A. Varma, Noncatalytic hydrothermolysis of ammonia borane, Int. J. Hydrogen Energy, 2008, 33, 1135–1141 CrossRef CAS.
- C.-C. Chou, D.-J. Lee and B.-H. Chen, Hydrogen production from hydrolysis of ammonia borane with limited water supply, Int. J. Hydrogen Energy, 2012, 37, 15681–15690 CrossRef CAS.
- K. Chang, E. Kim, P. F. Weck and D. Tomanek, Nanoconfinement effects on the reversibility of hydrogen storage in ammonia borane: A first-principles study, J. Chem. Phys., 2011, 134, 214501 CrossRef PubMed.
- Z. Tang, H. Chen, X. Chen, L. Wu and X. Yu, Graphene oxide based recyclable dehydrogenation of ammonia borane within a hybrid nanostructure, J. Am. Chem. Soc., 2012, 134, 5464–5467 CrossRef CAS PubMed.
- Z. Li, G. Zhu, G. Lu, S. Qiu and X. Yao, Ammonia Borane Confined by a Metal–Organic Framework for Chemical Hydrogen Storage: Enhancing Kinetics and Eliminating Ammonia, JACS Communication, 2015, 132, 1490–1491 CrossRef PubMed.
- L. Zhang, G. Xia, Y. Ge, C. Wang, Z. Guo, X. Li and X. Yu, Ammonia borane confined by nitrogen-containing carbon nanotubes: enhanced dehydrogenation properties originating from synergetic catalysis and nanoconfinement, J. Mater. Chem. A, 2015, 3, 20494–20499 RSC.
- M. A. Wahab, H. Zhao and X. D. Yao, Nano-confined ammonia borane for chemical hydrogen storage, Front. Chem. Sci. Eng., 2012, 6, 27–33 CrossRef CAS.
- Y. Peng, T. Ben, Y. Jia, D. Yang, H. Zhao, S. Qiu and X. Yao, Dehydrogenation of ammonia borane confined by low-density porous aromatic framework, J. Phys. Chem. C, 2012, 116, 25694–25700 CrossRef CAS.
- G. Moussa, S. Bernard, U. B. Demirci, R. Chiriac and P. Miele, Room-temperature hydrogen release from activated carbon-confined ammonia borane, Int. J. Hydrogen Energy, 2012, 37, 13437–13445 CrossRef CAS.
- M. Rueda, L. M. Sanz-Moral, J. J. Segovia and Á. Martín, Improvement of the kinetics of hydrogen release from ammonia borane confined in silica aerogel, Microporous Mesoporous Mater., 2017, 237, 189–200 CrossRef CAS.
- S. Deville, Freeze-Casting of Porous Ceramics: A Review of Current Achievements and Issues, Adv. Eng. Mater., 2008, 10, 155–169 CrossRef CAS.
- Ammonia borane confinement in graphene oxide 3D structures, US Pat., App. No. 15/862,905, (filing date Jan. 10, 2017), 17150852–6, 2017.
- L. Qiu, J. Z. Liu, S. L. Y. Chang, Y. Wu and D. Li, Biomimetic superelastic graphene-based cellular monoliths, Nat. Commun., 2012, 3, 1241 CrossRef PubMed.
- D. Kim, D. W. Kim, H.-K. Lim, J. Jeon, H. Kim, H.-T. Jung and H. Lee, Intercalation of Gas Molecules in Graphene Oxide Interlayer: The Role of Water, J. Phys. Chem. C, 2014, 118, 11142–11148 CrossRef CAS.
- K. Zhang, L. L. Zhang, X. S. Zhao and J. Wu, Graphene/Polyaniline Nanofiber Composites as Supercapacitor Electrodes, Chem. Mater., 2010, 22, 1392–1401 CrossRef CAS.
- L. Bravo Diaz, J. M. Hanlon, M. Bielewski, A. Milewska and D. H. Gregory, Ammonia borane-based nanocomposites as solid state hydrogen stores for portable power applications, Energy Technol., 2018, 6, 583–594 CrossRef CAS.
- Z. Yang, D. Zhou, B. Chen, Z. Liu, Q. Xia, Y. Zhu and Y. Xia, Improved hydrogen release from ammonia borane confined in microporous carbon with narrow pore size distribution, J. Mater. Chem. A, 2017, 5, 15395 RSC.
- J. Breternitz, Y. E. Vilk, E. Giraud, H. Reardon, T. K. A. Hoang, A. Godula-Jopek and D. H. Gregory, Facile Uptake and Release of Ammonia by Nickel Halide Ammines, ChemSusChem, 2016, 9, 1312–1321 CrossRef CAS PubMed.
- T. Lu, PhD thesis, James Cook University, 2010.
- V. I. Simagina, N. V. Vernikovskaya, O. V. Komova, N. L. Kayl, O. V. Netskina and G. V. Odegova, Experimental and modeling study of ammonia borane-based hydrogen storage systems, Chem. Eng. J., 2017, 329, 156–164 CrossRef CAS.
- K. P. De Jong, Synthesis of solid catalysts, Wiley-VCH, 2009 Search PubMed.
- D. S. Clark, J. E. Bailey and D. D. Do, A mathematical model for restricted diffusion effects on macromolecule impregnation in porous supports, Biotechnol. Bioeng., 1985, 27, 208–213 CrossRef CAS PubMed.
- M. Börnhorst, P. Walzel, A. Rahimi, A. Kharaghani, E. Tsotsas, N. Nestle, A. Besser, F. Kleine Jäger and T. Metzger, Influence of pore structure and impregnation–drying conditions on the solid distribution in porous support materials, Drying Technol., 2016, 34, 1964–1978 CrossRef.
- Heterogeneous catalysis: fundamentals and applications, ed. J. R. H. Ross, Elsevier, Amsterdam, 2012 Search PubMed.
- A. D. Sutton, A. K. Burrell, D. A. Dixon, E. B. Garner III, J. C. Gordon, T. Nakagawa, K. C. Ott, J. P. Robinson and M. Vasiliu, Regeneration of ammonia borane spent fuel by direct reaction with hydrazine and liquid ammonia, Science, 2011, 331, 1426–1429 CrossRef CAS PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, Improved synthesis of graphene oxide, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Online library or from the author. See DOI: 10.1039/c8se00526e |
| This journal is © The Royal Society of Chemistry 2019 |