
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLocus-patterned sequence oriented enrichment for multi-dimensional gene analysis†
Yue
Zhao
a,
Xiaoxing
Fang
a,
Feng
Chen
a,
Min
Bai
a,
Chunhai
Fan
b and
Yongxi
Zhao
 *a
*a
aInstitute of Analytical Chemistry and Instrument for Life Science, Key Laboratory of Biomedical Information Engineering of Ministry of Education, School of Life Science and Technology, Xi'an Jiaotong University, Xianning West Road, Xi'an, Shaanxi 710049, P. R. China. E-mail: yxzhao@mail.xjtu.edu.cn
bSchool of Chemistry and Chemical Engineering, Institute of Molecular Medicine, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai 200240, P. R. China
First published on 22nd July 2019
Abstract
Multi-dimensional gene analysis provides in-depth insights into gene sequence, locus variations and molecular abundance, whereas it is vulnerable to the perturbation of complex reaction networks and always compromises on the discrimination of analogous sequences. Here, we present a sequence oriented enrichment method patterned by the prescribed locus without crosstalk between concurrent reactions. Energetically favourable structures of nucleic acid probes are theoretically derived and oriented to a specific gene locus. We designed a pair of universal probes for multiple conserved loci to avoid side reactions from undesired interactions among increased probe sets. Furthermore, competitive probes were customized to sink analogues for differentiating the reaction equilibrium and kinetics of sequence enrichment from the target, so variant loci can be synchronously identified with nucleotide-level resolution. Thus, the gene locus guides sequence enrichment and combinatorial signals to create unique codes, which provides access to multidimensional and precise information for gene decoding.
Introduction
Gene heterogeneity is a widespread biological property due to the stochastic and variable nature of molecular evolution.1,2 In-depth analysis of gene sequence, locus variations and molecular abundance is pivotal to understanding the essence of gene heterogeneity.3 Advances in next-generation sequencing technologies have paved the way for whole-genome analysis, yet the random sequence enrichment induced biased coverage impedes the access to trace variants. So the sequencing depth necessary to quantitatively detect rare events is substantially higher and always results in higher consumption of sequencing resources.4,5 Although digital polymerase chain reactions are developed for absolute quantification of individual molecules, the modest specificity and limited resolution may not be sufficient for homologous analysis.6–8 Toward that end, considerable efforts are focused on the discrimination of single-nucleotide variants (SNVs) during multi-dimensional gene analysis.9–11 DNA strand branch migration provides opportunities to recognize SNVs, relying on the specificity of Watson–Crick hybridization between different dynamic DNA complexes at some step of strand displacement workflow.12–15 However, the effects are impaired by the small thermodynamic penalty from a single base mismatch and are always vulnerable to environmental perturbation.16 Polymerase-assisted locus identification is based on allele-specific amplification, which shows a preference to matched primer–template complexes rather than the mismatched.17 The essential concept of a competitive sequestration mechanism is to design a scavenger probe, which acts as a sink to consume spurious analogues and improve the target specificity.18,19 All the above methods exhibit discrimination among SNVs, yet leakage signals from undesired molecular interactions or crosstalks among multiplexed reactions always lead to equivocal results, which are scarcely to offer valid and precise information for multi-dimensional gene analysis.20,21Here we present a sequence oriented enrichment method, which is exclusively patterned by the gene locus of interest. A couple of hairpin probes are designed to indicate the multiple conserved locus, by which sequence information of all variants can be enriched simultaneously. Furthermore, a competitive mechanism was introduced to identify the variant locus. The sequential reaction paths from the reaction equilibrium of DNA strand branch migration to the kinetics of sequence enrichment are coupled and tightly governed, thus the variant locus with nucleotide variations can be distinguished. To be specific, we predicted the energetically favourable structure of probes and oriented the reaction equilibrium of strand exchange between targets and analogues towards opposite directions. Subsequently, the thermodynamic discrimination could be further expanded by the kinetic efficiency of sequence enrichment. Then various loci output signals with distinct patterns which can be encoded as unique codes for gene decoding.22,23
The principle of our approach is illustrated in Fig. 1. Two sets of standard loop-mediated isothermal amplification (LAMP) primers are involved to amplify gene 1 and gene 2. Then a couple of universal probes are designed with hairpin structures to improve the recognition specificity to the conserved loci of multiple genes. So the common sequence of all variants can be synchronously enriched and generate corresponding fluorescence signals without crosstalk from parallel reactions or increased probe sets. Variant probes, consisting of F–Q (duplex strands respectively labelled with fluorescence F and quench Q) and S (scavenger for sinking analogues), utilize the competitive mechanism to regulate the reaction equilibrium among S, F–Q, F and S–Q. The negative Gibbs energy change (ΔG < 0) favours the DNA branch migration and promotes the strand exchange between F–Q, S and T, yielding numerous F–T complexes. Thereout, T enables the previous reaction equilibrium to shift forward with an obvious fluorescence response. Then F perfectly annealing to T spontaneously works as a candidate primer to initiate the sequence enrichment with high efficiency. In contrast, A with nucleotide variations possesses base-pair deficiency (marked as a red bulge) and energetic weakness (ΔG > 0) for migration to F–A, thus are inclined to form S–A. Ascribed to the consumption of S, the reaction equilibrium is driven to shift backward and signals are reduced in turn. Even if bits of F–A escape from the thermodynamic differentiation, their roles as mismatched primers can be readily precluded via kinetic discrimination. With universal probes to recognize common sequences of the conserved locus and variant probes to identify nucleotides within the variant locus, diverse genes create combinatorial fluorescence signals with multiple colours. Thus, signals of distinct patterns can be recorded as unique codes, providing multidimensional and precise information for gene sequence, locus variation and quantification analysis.
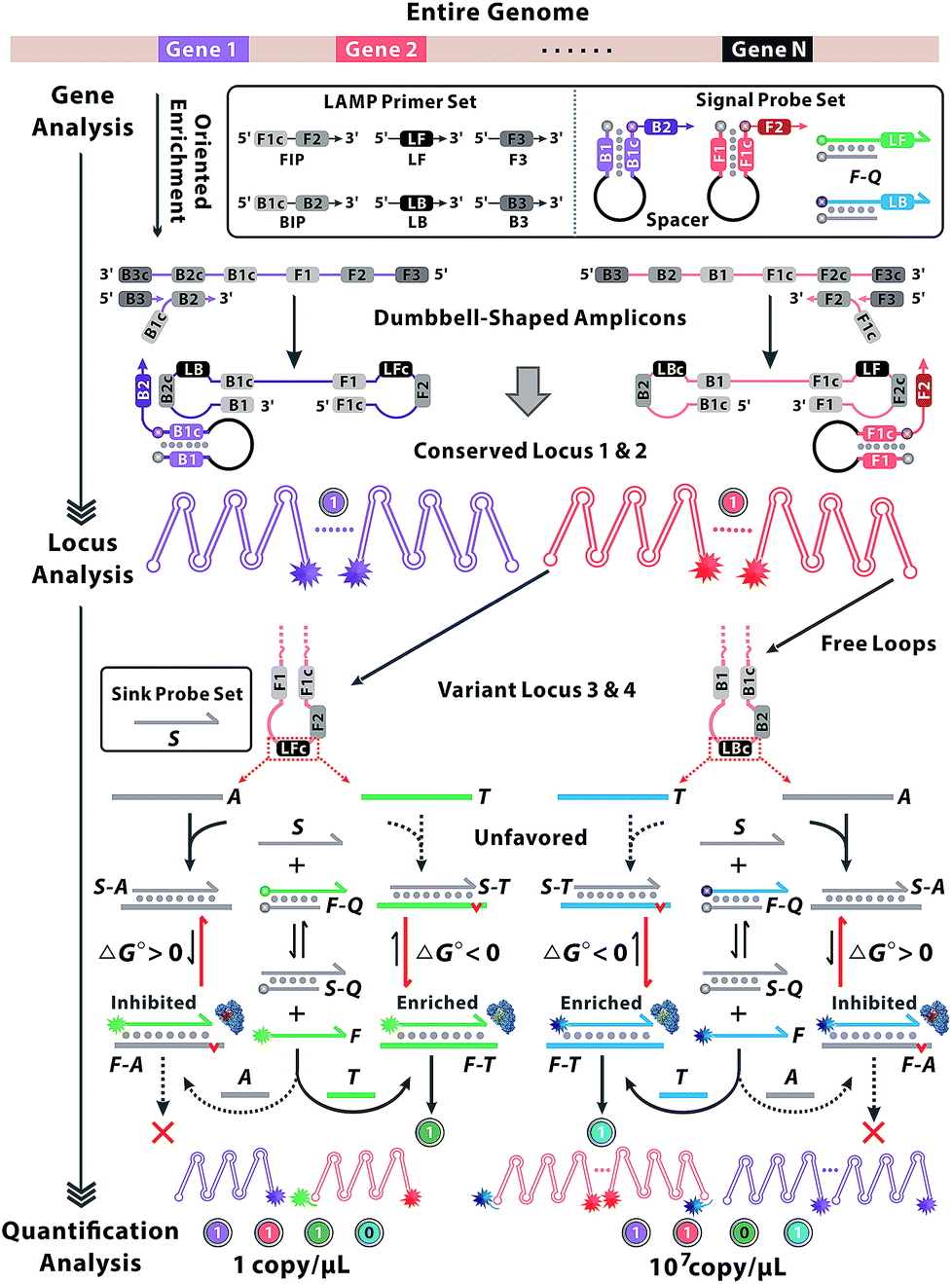 | ||
| Fig. 1 Working principle of locus-patterned sequence oriented enrichment for multi-dimensional gene analysis. Two sets of standard LAMP primers are involved to amplify gene 1 and gene 2. So loci 1 and 2 within the conversed region can be enriched independently and indicated by the spacer-linked signal probes. Loci 3 and 4 represent two variant loci and can be discriminated by nucleotide variations within the free loop region of the amplified concatemers during the enrichment of locus 2. Signal probes labelled with fluorescence and quench tags (F–Q) are competitive with scavenger probes (S) in the reaction equilibrium. Targets (T) are denoted in bold blue or green and analogues (A) are represented in grey. The mismatched base pairing is marked as a red bulge. The involvement of A or T shifts the reaction equilibrium toward opposite directions, and appears as a distinct discrimination of Gibbs energy change (ΔG) in strand migration. Subsequently, polymerase shows a preference to matched primer (yellow in the crystal structures of the enzyme) rather than the mismatched (red in the crystal structures of the enzyme), by which corresponding signal probes could be illuminated during the locus-oriented sequence enrichment. Thus, various loci (conserved loci 1 & 2 and variant loci 3 & 4) impart fluorescence signals of multiple colours and generate 4-bit binary codes for gene sequence, variant locus and quantification analysis. | ||
Results and discussion
Theoretical prediction and experimental verification
The thermodynamics of the oriented sequence enrichment path is rationally modulated via prediction of probe configuration and regulation of reaction equilibrium to improve the discrimination among variant loci. We chose hepatitis B virus (HBV) as the model due to its global hazards and numerous variants. Since variations between close homologues bring dramatic phenotypic consequences, different variants of HBV have been identified as genotypes according to the variant locus. Each type shares conserved sequences over the whole genome of about 92%, so the variable locus within the recognition sequences is always a SNV.24–26 Through alignment of HBV genome sequences from the National Center for Biotechnology Information (NCBI) nucleotide sequence database (GenBank), we chose the conserved loci of open reading frames encoding the viral surface envelope (S gene) and core capsid (C gene) proteins for universal probes. Comparison of complete genome sequences reveals genetic clusters to be classified as six types A–F, corresponding with their variant loci in the S gene (Fig. S1†). Since B and C are the predominant HBV genotypes in the Asia-Pacific region.26 we investigated our method on the discrimination between type B and type C with partial HBV gene fragments cloned into plasmid as the template (Fig. S2–S4†). As presented in Fig. 2, we supply a competitive mechanism with F–Q and S, wherein both T (type B) and A (type C) have the potential to hybridize with the two probes. S is manifested to suppress hybridization of A to F as S is complementary to A. Owing to the intrinsic superiority of the single strand over the duplex strand for hybridization with the other, A will perfectly hybridize with S rather than F–Q. Although T could also be masked by S even with mismatched base-pairings, the branch migration of F is energetically favorable (ΔG° = −0.71 kcal mol−1), being driven by the higher stability of the final duplex. Notably, when A binds to S, there are no free single-stranded domains, rendering S–A inert and unable to react with any other single strand. Thus, A is prevented from binding to F and causes negligible fluorescence signal leakage (ΔG° = 6.02 kcal mol−1). However, the basic reaction path relying on simplex strand displacement has limited power for the specific identification of T and A due to the small energy difference. According to the van't Hoff equation, ΔG of a locus-oriented reaction path is given byΔGT = ΔG0FT + ΔG0SQ − ΔG0ST − ΔG0FQ + RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnQ lnQ | (1) |
| ΔGA = ΔG0FA + ΔG0SQ − ΔG0SA − ΔG0FQ + RTlnQ | (2) |
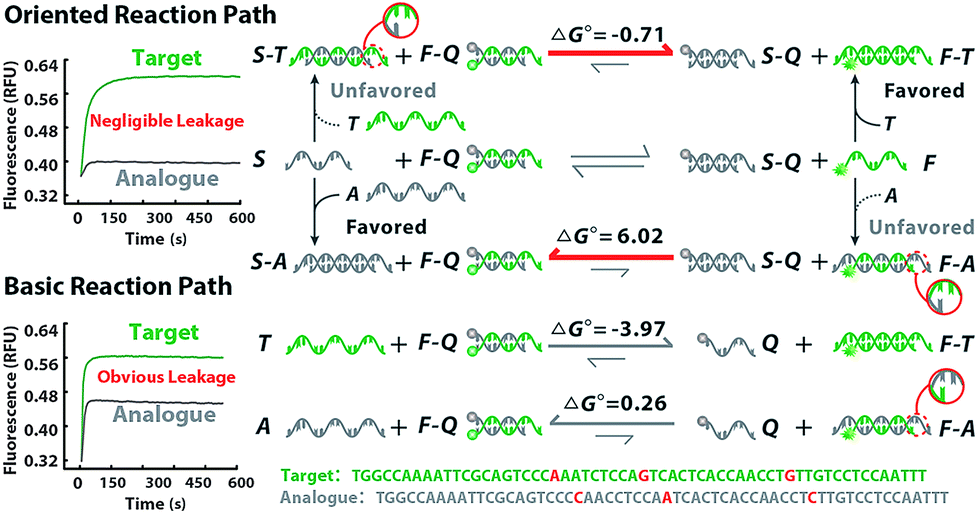 | ||
| Fig. 2 Theoretical prediction and experimental verification of the thermodynamic discrimination via the oriented sequence enrichment reaction path and basic reaction path. The targets (T) and analogues (A) are denoted in green and grey, respectively. The bold arrow in red indicates the direction of equilibrium shift. The mismatched base pairing at the variant locus is circled to zoom in. The real-time fluorescence signals of strand migration in the oriented reaction and basic reaction are presented, in which the green line represents T and the grey line represents A. | ||
Principle and evaluation of locus-oriented sequence enrichment
Subsequently, F perfectly annealing to T spontaneously works as a candidate primer to initiate sequence enrichment with high kinetic efficiency. Even if bits of F–A leakages escape from thermodynamic obstacles, the gap of amplification efficiency with orders of magnitude between the paired primer and distal mismatched primer can be eliminated via reaction kinetics discrimination (Fig. S6 and S7†). Utilizing six primers to recognize eight distinct regions on the target gene, LAMP is appropriate for locus-oriented sequence enrichment.27–30 We chose four loci for gene heterogeneity analysis, and the detailed workflow of locus-patterned sequence oriented enrichment is shown in Fig. S8.† As shown in Fig. 3A, the spacer-linked (denoted as a black loop) hairpin probe labelled with Texas Red (highlighted in purple) is designed to recognize the conserved locus 1 of the C gene and Hex (highlighted in red) corresponds to locus 2 of the S gene. The fluorescence responses are independently recoded in channel 1 (Ch. 1) and Ch. 2, which are potent indications of conserved sequence identification and general for all variants. The hairpin probes can anneal to the conserved locus, initiate extension and subsequently generate a new template for other primers to bind. The new-synthesized strand will displace the original stem region of hairpin probes, generate fluorescence and be blocked by the reserved spacer. With the loop-back feature predesigned in hairpin probes, their extension products readily form cauliflower-like structures for self-primed DNA synthesis to create abundant sequence enrichment and numerous fluorescence signals. Meanwhile, the other two probe sets are involved in discriminating the variant locus. The double-stranded probe labelled with FAM (highlighted in green) is specific to type B and Cy5 (highlighted in blue) is for type C. Once strand branch migration occurs, obvious fluorescence signal can be obtained from Ch. 3 and Ch. 4, respectively. Then the fluorophore-labelled strand, which matches the target perfectly, recruits polymerase and works as a new primer to spontaneously initiate new parallel amplification pathways. To evaluate the oriented ability of our method, we designed a proof-of-principle ‘locus-mixing’ experiment with 1 μg type B and 1 μg type C. As we are unable to differentiate the real-time fluorescence signal source from a homogeneous solution, the spatially resolved fluorescence images at the end-point of reactions are recorded with a confocal fluorescence microscope for direct display of the oriented sequence enrichment. As shown in Fig. 3B, fluorescence spots detected in the same positions between Ch. 2 and Ch. 4 indicate the sequence enrichment of type C, corresponding with the coexisting conserved locus 2 and variant locus 4. In contrast, no colocalization or crosstalk is observed between Ch. 3 and Ch. 4, which primarily proves the nucleotide-level resolution of sequence enrichment (Fig. 3C). Inspired by the algorithm used in fluorescence in situ hybridization, we evaluated the cross-correlation between multichannel images in the same way.31 From the cross-correlation results in Fig. 3 and S9,† we further confirmed that the sequence enrichment is patterned by the gene locus.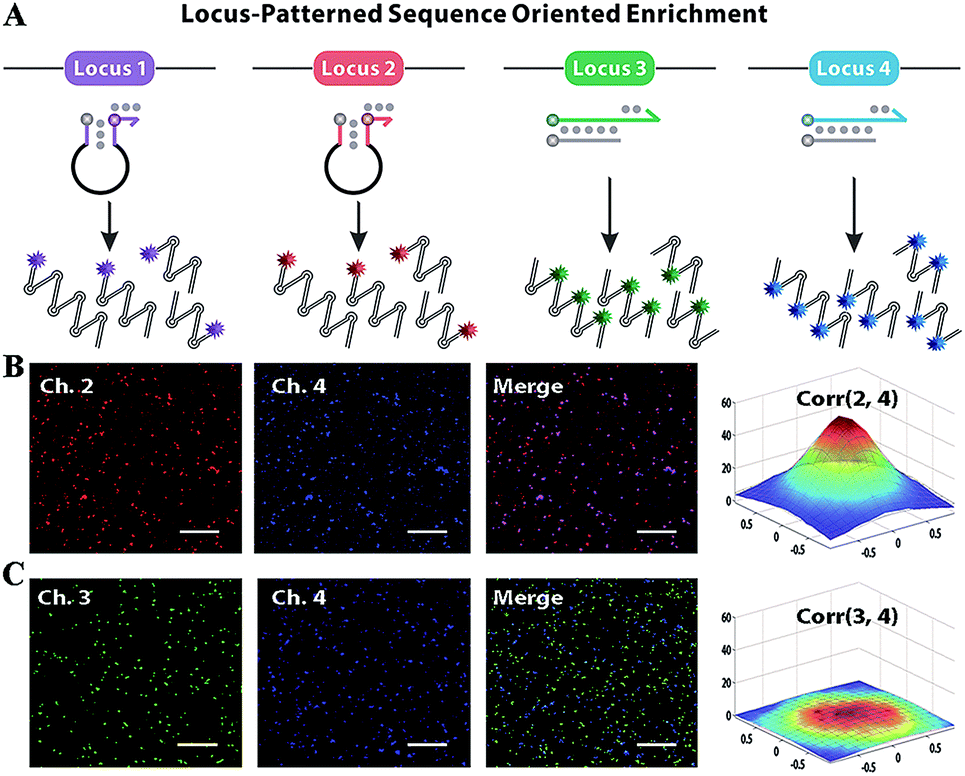 | ||
| Fig. 3 (A) The principle of locus-patterned sequence oriented enrichment. Four loci coloured in purple, red, green and blue correspond with the probes labelled with different fluorescent tags (Texas Red, Hex, FAM and Cy5), the signals of which can be recorded in channel (Ch.) 1, 2, 3 and 4. (B) Confocal fluorescence image of locus-patterned sequence enrichment with 1 μg type B and 1 μg type C template. The signals from the conserved locus (Ch. 2) and variant locus (Ch. 4) show obvious positive correlations, corresponding with the sequence enrichment of type C template. (C) The pair-wise correlations between signals from different variant loci (Ch. 3 and Ch. 4) are negligible. Corr is the abbreviation of correlations and the number in brackets denotes the pairwise channels. Scale bar = 20 μm. | ||
Heterogeneous gene decoding
Heterogeneous gene alterations impart unique colour combinations of fluorescence responses and can be encoded as binary codes corresponding to their gene locus. The fluorescence signals are monitored real-time (Fig. S10†) and recorded in the heat maps (Fig. 4). Then some crucial parameters are modulated to evaluate the performance of all parallel channels (Fig. S11–S14†). Considering the different fluorescence quantum yields and enrichment efficiency of each channel, fluorescence intensity is normalized and rational thresholds are set to transform signals to binary codes. The conserved locus of the C gene exhibits a prominent fluorescence signal in Ch. 1 and can be described as (1,0,0,0). The conserved locus of the S gene with diverse variants generates different signal combinations patterned by their variant locus. As a result, type B presents synchronous signals in Ch. 2 and Ch. 3, so as to be recorded as (0,1,1,0), whereas type C with distinctive signals in Ch. 2 and Ch. 4 corresponds with the code (0,1,0,1). Thereout, heterogeneous genes with different loci containing different sequence and nucleotide information can also be clearly defined and encoded as (1,1,1,0), (1,1,0,1), (0,1,1,1) and (1,1,1,1). Compared with the well-known allele specific-PCR, the proposed method offers an improved discrimination to nucleotide variations, especially for identifying trace targets from analogues of orders of magnitude excess (Fig. S15†). | ||
| Fig. 4 The real-time fluorescence signals obtained from locus-patterned sequence enrichment of heterogeneous genes and the corresponding pseudo-colour images at the end point. The colour bands from bottom to top correspond with signals obtained from Ch. 1, 2, 3 and 4. (A) Blank without template, (B) conserved locus of the C gene, (C) conserved locus of the S gene with variant locus of type B, (D) conserved locus of the S gene with variant locus of type C, (E) conserved locus of C and S genes with variant locus of type B, (F) conserved locus of C and S genes with variant locus of type C, (G) conserved locus of the S gene with variant locus of type B and type C, (H) conserved locus of C and S genes with variant locus of type B and type C. 1 μg of each standard template with a specific locus was involved and reactions were incubated at 65 °C for 3 h, with fluorescence read steps at intervals of 2 min. FI is the abbreviation of fluorescence intensity. | ||
Clinical sample analysis
To further characterize the molecular abundance, we investigated the proposed method on a standard template (Fig. S16†) and next sought to verify the quantitative performances in clinical samples. Serum from 24 HBV patients and 8 healthy volunteers generate binary codes (Fig. S17†). In theory, uninfected samples can't display any signal in the locus-patterned sequence oriented enrichment, so as to be coded as (0,0,0,0). Infected samples of any variant process the same conserved sequence locus, so they can create codes with at least 2 bits occupied by 1, which is clearly separated from negative samples of the healthy. As presented in Fig. 5, 32 samples are divided into two groups via sequence decoding of the first two bits. Then with nucleotide decoding for the variant locus, all infected samples can be further identified as type B, type C and other types, which has been subsequently validated by sequencing (Fig. S18†). With redundant data acquired from multiple channels, the methods provide abundant information to quantify molecules even with low-abundance (Fig. S19 and S20†), which are consistent with the results of clinical diagnosis.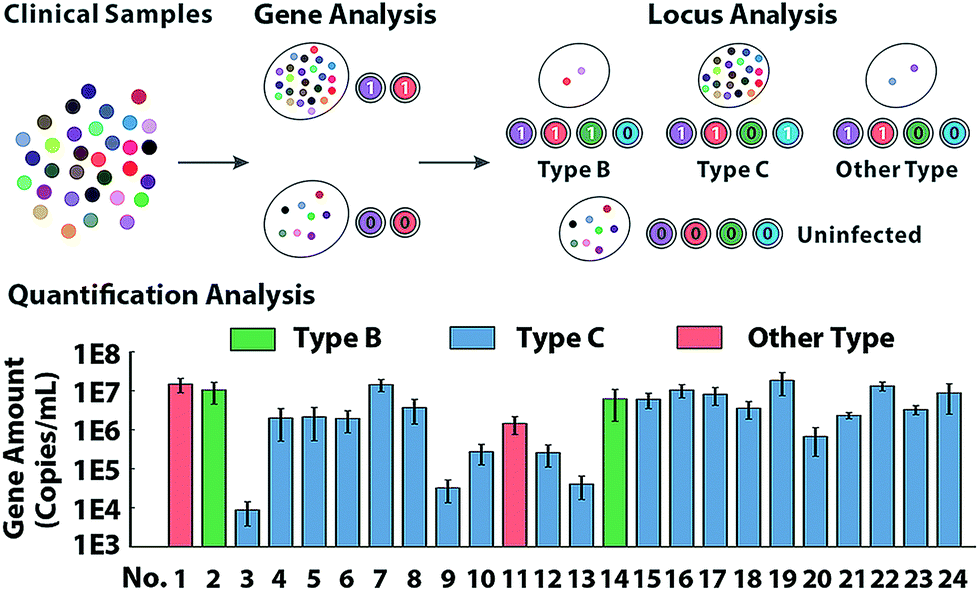 | ||
| Fig. 5 The analysis of HBV gene in clinical samples from 24 patients and 8 healthy volunteers. The HBV samples are discriminated from the healthy samples via conserved gene sequence analysis. Then with variant locus discrimination, the infected samples can be classified into three groups: 2 cases type B HBV, 20 cases type C HBV and 2 cases of other types, which are further verified by sequencing. The molecular abundance obtained from quantification analysis indicates the copy number of HBV gene in 1 mL blood of patients. The error bar indicates the standard error from three independent experiments. | ||
Conclusions
In conclusion, we have successfully demonstrated locus-patterned sequence oriented enrichment, providing a precise and multidimensional approach to heterogeneous gene analysis. The oriented probes are coupled and governed to simultaneously recognize the conserved and variant loci of genes. Thus the reaction equilibrium and kinetics of sequence enrichment reaction networks are exclusively oriented with a nucleotide-level resolution. Heterogeneous gene alterations evoke multiple colour combinations of fluorescence responses in distinct patterns and can be encoded as binary codes corresponding to their locus information. Overall, this method proves to be a prospective approach for gene decoding, promising an available avenue for high-throughput gene analysis and may also be adapted to build a comprehensive molecular map in future studies.Experimental section
DNA strand branch migration reaction
A set of fragments from the S gene are applied as the target (T) or analogue (A) to investigate the thermodynamics and equilibrium of the reaction. The sequence from type B (LF template of type B) is defined as T and the sequence from type C (LF template of type C) simulates A. The forward loop primer for type C (HBVS LFC) serves as the scavenger (S). Fluorescence labelled loop primers HBVS LFB FAM and quencher labelled primers LF Block BHQ2 form a complex signal probe.The basic reaction was performed in 30 μL volumes in thin-walled PCR strip tubes. 30 nM T or A and 300 nM signal probes were added into the reaction mixture, which had a final composition of 1× ThermoPol reaction buffer (20 mM Tris–HCl, 10 mM KCl, 10 mM (NH4)2SO4, 2 mM MgSO4 and 0.1% Triton X-100, pH 8.8) supplemented with an additional 4 mM MgSO4 (final 6 mM MgSO4) and 1 M betaine. Reactions were incubated at a constant temperature of 65 °C, with fluorescence read steps at intervals of 10 s.
The locus-oriented reaction was performed in 30 μL volumes in thin-walled PCR strip tubes. 30 nM T or A, 300 nM signal probes and 30 nM S were added into the reaction mixture, which had a final composition of 1× ThermoPol reaction buffer (20 mM Tris–HCl, 10 mM KCl, 10 mM (NH4)2SO4, 2 mM MgSO4 and 0.1% Triton X-100, pH 8.8) supplemented with an additional 4 mM MgSO4 (final 6 mM MgSO4) and 1 M betaine. Reactions were incubated at a constant temperature of 65 °C, with fluorescence read steps at intervals of 10 s.
Rate effects of distal mismatches on primer extension
The kinetic efficiency of primer extension was monitored by real-time quantitation of the synthesized double stranded DNA stained with a high-resolution melting (HRM) fluorescent dye, SYTOX Orange. Since changes in fluorescence are directly proportional to DNA synthesis, quantitative measurements are obtained by calibrating the fluorescence signal against synthesized standards. We investigated the extension ability of matched and distal mismatched primers. HRM dyes were firstly confirmed for a linear dose-dependent response to the yield of duplex DNA strands. The average rate of DNA synthesis is measured by units of nucleotides per minute (nt per min). The DF of average extension efficiency between T and A can be defined as the ratio of DNA synthesis.Measurements were performed in 30 μL volumes in thin-walled PCR strip tubes that contained 30 nM primer (HBVS LFB or HBVS LFC), 30 nM template (LF template of type B), 1× Thermopol buffer (20 mM Tris–HCl, 10 mM KCl, 10 mM (NH4)2SO4, 2 mM MgSO4 and 0.1% Triton X-100, pH 8.8) supplemented with an additional 4 mM MgSO4 (final 6 mM MgSO4), 30 μM each dNTP, 500 nM SYTOX Orange and 1 M betaine. 1 U Bst 2.0 WarmStart® DNA polymerase was added and reactions were incubated at a constant temperature of 65 °C, with fluorescence read steps at intervals of 5 s.
Nucleic acid sequence enrichment reaction
Primers were designed using the PrimerExplorer V5 program at the website (http://primerexplorer.jp/e/) and used in the amounts typically recommended for classical LAMP.For the C gene: 0.4 μM each for outer primers HBVC F3 and HBVC B3; 1.2 μM each for inner primers HBVC FIP (F1–F2) and HBVC BIP (B1–B2); 0.2 μM fluorescence labelled inner primers HBVC BIP Tex, and 0.8 μM each for loop primers HBVC LF and HBVC LB.
For the S gene (type B): 0.4 μM each for outer primers HBVS F3 and HBVS B3B; 1.2 μM each for inner primers HBVS FIP and HBVS BIPB; 0.2 μM fluorescence labelled inner primers HBVS FIP Hex; 0.8 μM loop primers HBVS LBB; 0.4 μM fluorescence labelled loop primers HBVS LFB FAM; and 0.4 μM quencher labelled primers LF Block BHQ2.
For the S gene (type C): 0.4 μM each for outer primers HBVS F3 and HBVS B3C; 1.2 μM each for inner primers HBVS FIP and HBVS BIPC; 0.2 μM fluorescence labelled inner primers HBVS FIP Hex; 0.8 μM loop primers HBVS LFC; 0.4 μM fluorescence labelled loop primers HBVS LBC Cy5; and 0.4 μM quencher labelled primers LB Block BHQ2.
Sequence enrichment reactions were performed in 30 μL volumes in thin-walled PCR strip tubes. Various combinations of primers with different templates were added into the reaction mixture, which had a final composition of 1× ThermoPol reaction buffer (20 mM Tris–HCl, 10 mM KCl, 10 mM (NH4)2SO4, 2 mM MgSO4 and 0.1% Triton X-100, pH 8.8) supplemented with an additional 4 mM MgSO4 (final 6 mM MgSO4), 0.8 mM each dNTP and 1 M betaine. Mixtures were heated to 95 °C for 5 min to assist genome degeneration and primer annealing, followed by chilling on ice for 2 min. Then 8 U Bst 2.0 WarmStart® DNA polymerase was added to initiate the LAMP reaction. Reactions were incubated at a constant temperature of 65 °C for 3 h, with fluorescence read steps at intervals of 2 min.
Allele specific-PCR reactions were performed in 20 μL volumes in thin-walled PCR strip tubes. 250 nM primers with different templates were added into the reaction mixture, which had a final composition of 1× Standard Taq reaction buffer (10 mM Tris–HCl, 50 mM KCl, 1.5 mM MgCl2, pH 8.3). Then 1 U Hot Start Taq DNA polymerase, and 50 μM each dNTP and Sybr Green I (0.4×) were added. Mixtures were heated to 95 °C for 5 min to assist genome degeneration and primer annealing, followed by 45 cycles with 15 s denaturation at 95 °C, 30 s annealing at 55 °C and 15 s extension at 65 °C.
Confocal fluorescence analysis
Sequence enrichment reactions were incubated at a constant temperature of 65 °C for 3 h followed by heat-inactivation for 20 min at 80 °C. Then the reaction mixture was added to the cover glass and the fluorescence images of the time series model were obtained using a commercial high-resolution laser confocal microscope (TCS SP8 STED 3X, Leica). Paired lasers (523, 633 nm) and (488, 633 nm) were synchronously used as the excitation sources.Image correlation evaluation
Slight modifications are made to the MATLAB source codes of maincorrfishsimulation in ref. 31. Raw images are provided for two channels within the same field of view. Then the cross-correlation result provides common spots between images and presents as a three-dimensional Gaussian fit curve.Real-time fluorescence signal normalization
Considering the different enrichment efficiencies of multiple reaction paths and fluorescence quantum yields of diverse fluorescent molecules, fluorescence intensities of four channels need to be normalized and common thresholds values set to identify whether the result is negative (0) or positive (1). The intensity plateau of each channel is defined as 100%, so all fluorescence signals can be transformed to the percentage of saturated signal and normalized via a uniform standard.Sample preparation
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of China [No. 21705124, 31671013 and 21874105]; the China Postdoctoral Science Foundation [No. 2017M613102 and 2018T111032] and the “Young Talent Support Plan” of Xi'an Jiaotong University. Generous thanks to the Instrumental Analysis Center of Xi'an Jiaotong University for the confocal fluorescence imaging. We also would like to thank Dr Lei Zhang of the Department of Clinical Laboratory in The Second Affiliated Hospital of Xi'an Jiaotong University for experimental support on clinical samples.References
- S. Mamlouk, L. H. Childs, D. Aust, D. Heim, F. Melching, C. Oliveira, T. Wolf, P. Durek, D. Schumacher, H. Blaker, M. von Winterfeld, B. Gastl, K. Mohr, A. Menne, S. Zeugner, T. Redmer, D. Lenze, S. Tierling, M. Mobs, W. Weichert, G. Folprecht, E. Blanc, D. Beule, R. Schafer, M. Morkel, F. Klauschen, U. Leser and C. Sers, Nat. Commun., 2017, 8, 12 CrossRef PubMed.
- K. M. Turner, V. Deshpande, D. Beyter, T. Koga, J. Rusert, C. Lee, B. Li, K. Arden, B. Ren, D. A. Nathanson, H. I. Kornblum, M. D. Taylor, S. Kaushal, W. K. Cavenee, R. Wechsler-Reya, F. B. Furnari, S. R. Vandenberg, P. N. Rao, G. M. Wahl, V. Bafna and P. S. Mischel, Nature, 2017, 543, 122–125 CrossRef CAS PubMed.
- C. M. O'Keefe, T. R. Pisanic II, H. Zec, M. J. Overman, J. G. Herman and T.-H. Wang, Sci. Adv., 2018, 4, eaat6459 CrossRef PubMed.
- A. M. Streets, X. N. Zhang, C. Cao, Y. H. Pang, X. L. Wu, L. Xiong, L. Yang, Y. S. Fu, L. Zhao, F. C. Tang and Y. Y. Huang, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7048–7053 CrossRef CAS PubMed.
- Z. T. Chen, W. X. Zhou, S. Qiao, L. Kang, H. F. Duan, X. S. Xie and Y. Y. Huang, Nat. Biotechnol., 2017, 35, 1170–1178 CrossRef CAS PubMed.
- X. R. Li, D. F. Zhang, H. M. Zhang, Z. C. Guan, Y. L. Song, R. C. Liu, Z. Zhu and C. Y. Yang, Anal. Chem., 2018, 90, 2570–2577 CrossRef CAS PubMed.
- H. Tian, Y. Y. Sun, C. H. Liu, X. R. Duan, W. Tang and Z. P. Li, Anal. Chem., 2016, 88, 11384–11389 CrossRef CAS PubMed.
- Z. Zhu and C. Y. J. Yang, Acc. Chem. Res., 2017, 50, 22–31 CrossRef CAS PubMed.
- M. G. Mohsen, D. Ji and E. T. Kool, Chem. Sci., 2019, 10, 3264–3270 RSC.
- Z.-L. Hu, M.-Y. Li, S.-C. Liu, Y.-L. Ying and Y.-T. Long, Chem. Sci., 2019, 10, 354–358 RSC.
- S. Hu, W. Tang, Y. Zhao, N. Li and F. Liu, Chem. Sci., 2017, 8, 1021–1026 RSC.
- L. R. Wu, J. S. Wang, J. Z. Fang, E. R. Evans, A. Pinto, I. Pekker, R. Boykin, C. Ngouenet, P. J. Webster, J. Beechem and D. Y. Zhang, Nat. Methods, 2015, 12, 1191–1196 CrossRef CAS PubMed.
- M. X. Li, C. H. Xu, N. Zhang, G. S. Qian, W. Zhao, J. J. Xu and H. Y. Chen, ACS Nano, 2018, 12, 3341–3350 CrossRef CAS PubMed.
- M. X. You, Y. F. Lyu, D. Han, L. P. Qiu, Q. L. Liu, T. Chen, C. S. Wu, L. Peng, L. Q. Zhang, G. Bao and W. H. Tan, Nat. Nanotechnol., 2017, 12, 453–459 CrossRef CAS PubMed.
- D. Y. Zhang and E. Winfree, J. Am. Chem. Soc., 2009, 131, 17303–17314 CrossRef CAS PubMed.
- L. R. Wu, S. X. Chen, Y. L. Wu, A. A. Patel and D. Y. Zhang, Nat. Biomed. Eng., 2017, 1, 714–723 CrossRef CAS PubMed.
- P. Chen, D. Pan, C. H. Fan, J. H. Chen, K. Huang, D. F. Wang, H. L. Zhang, Y. Li, G. Y. Feng, P. J. Liang, L. He and Y. Y. Shi, Nat. Nanotechnol., 2011, 6, 639–644 CrossRef CAS PubMed.
- J. S. Wang and D. Y. Zhang, Nat. Chem., 2015, 7, 545–553 CrossRef CAS PubMed.
- S. X. Chen and G. Seelig, J. Am. Chem. Soc., 2016, 138, 5076–5086 CrossRef CAS PubMed.
- X. Sun, B. Wei, Y. Guo, S. Xiao, X. Li, D. Yao, X. Yin, S. Liu and H. Liang, J. Am. Chem. Soc., 2018, 140, 9979–9985 CrossRef CAS PubMed.
- Y. Zhao, F. Chen, J. Qin, J. Wei, W. H. Wu and Y. X. Zhao, Chem. Sci., 2018, 9, 392–397 RSC.
- J. J. Zhang and Y. Lu, Angew. Chem., Int. Ed., 2018, 57, 9702–9706 CrossRef CAS PubMed.
- N. R. Y. Ho, G. S. Lim, N. R. Sundah, D. N. Lim, T. P. Loh and H. L. Shao, Nat. Commun., 2018, 9, 11 CrossRef PubMed.
- J. Kim, M. J. Biondi, J. J. Feld and W. C. W. Chan, ACS Nano, 2016, 10, 4742–4753 CrossRef CAS PubMed.
- M. Mizokami, T. Nakano, E. Orito, Y. Tanaka, H. Sakugawa, M. Mukaide and B. H. Robertson, FEBS Lett., 1999, 450, 66–71 CrossRef CAS PubMed.
- Y. Miyakawa and M. Mizokami, Intervirology, 2003, 46, 329–338 CrossRef CAS PubMed.
- T. Notomi, H. Okayama, H. Masubuchi, T. Yonekawa, K. Watanabe, N. Amino and T. Hase, Nucleic Acids Res., 2000, 28, e63 CrossRef CAS PubMed.
- Y. S. Jiang, S. Bhadra, B. Li, Y. R. Wu, J. N. Milligan and A. D. Ellington, Anal. Chem., 2015, 87, 3314–3320 CrossRef CAS PubMed.
- Y. S. Jiang, T. E. Riedel, J. A. Popoola, B. R. Morrow, S. Cai, A. D. Ellington and S. Bhadra, Water Res., 2018, 131, 186–195 CrossRef CAS PubMed.
- Y. Tang, B. Lu, Z. Zhu and B. Li, Chem. Sci., 2018, 9, 760–769 RSC.
- A. F. Coskun and L. Cai, Nat. Methods, 2016, 13, 657–660 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02496d |
| This journal is © The Royal Society of Chemistry 2019 |