
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMeasuring human carboxylesterase 2 activity in pancreatic cancer patient-derived xenografts using a ratiometric fluorescent chemosensor†
Karishma
Kailass
a,
Oleg
Sadovski
a,
Michela
Capello
b,
Ya'an
Kang
c,
Jason B.
Fleming
d,
Samir M.
Hanash
b and
Andrew A.
Beharry
 *a
*a
aDepartment of Chemical and Physical Sciences, University of Toronto Mississauga, Mississauga, ON L5L 1C6, Canada. E-mail: andrew.beharry@utoronto.ca
bDepartment of Clinical Cancer Prevention, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
cDepartment of Surgical Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
dDepartment of Gastrointestinal Oncology, H. Lee Moffitt Cancer Center, Tampa, FL, USA
First published on 29th July 2019
Abstract
Irinotecan-based therapy is a common treatment for pancreatic cancer. To elicit its anticancer activity, the drug requires first the hydrolysis action of the enzyme human carboxylesterase 2 (hCES2). It has been established that pancreatic cancer patients have various levels of hCES2, whereby patients having low levels respond poorer to Irinotecan than patients with higher levels, suggesting that hCES2 can be used to predict response. However, current methods that measure hCES2 activity are inaccurate, complex or lengthy, thus being incompatible for use in a clinical setting. Here, we developed a small molecule ratiometric fluorescent chemosensor that accurately measures hCES2 activity in a single-step within complex mixtures. Our chemosensor is highly selective for hCES2 over hCES1, cell permeable and can measure hCES2 activity in pancreatic cancer patient-derived xenografts. Given the simplicity, accuracy and tissue compatibility of our assay, we anticipate our chemosensor can be used to predict patient response to Irinotecan-based therapy.
Introduction
Carboxylesterases (CE) are a family of conserved α,β-fold intracellular hydrolases that catalyze the hydrolysis and transesterification of a variety of exogenous substrates containing esters, amides, carbamates and thioester groups.1 Humans have two major CE known as human carboxylesterase 1 (hCES1) and human carboxylesterase 2 (hCES2), which both act as phase-I drug metabolizing enzymes but differ in tissue distribution and substrate specificity.1 Importantly, the activity of CE, particularly hCES2 (expressed mainly in gastrointestinal tract, minor expression in liver), was found to be responsible for the activation of several anticancer agent prodrugs.2 For example, the prodrug Gemcitabine requires hCES2-mediated hydrolysis before it can be phosphorylated to the active triphosphate form that ultimately inhibits DNA synthesis.3,4 The prodrug pentyl PBABC-Doxaz is first activated by hCES2 to the anthracycline antitumour drug, Doxazolidine, a DNA cross-linking agent used to kill cancer cells.5,6 Finally, the chemotherapeutic prodrug Irinotecan, is converted to its active metabolite SN-38 by hCES2 to inhibit the DNA repair enzyme topoisomerase 1.7Given the importance of hCES2 in facilitating the conversion of prodrugs to drugs, a major factor governing anticancer drug efficacy has been the level of hCES2 activity found in cancers and patients. Most notably, hCES2 has been used as an indicator of response to Irinotecan treatment against neuroblastoma, metastatic colorectal cancer, and non-small cell lung cancer.8–10 In pancreatic ductal adenocarcinoma (PDAC) patients, hCES2 expression and activity was found to be a major contributor to Irinotecan sensitivity when administered in the chemotherapeutic cocktail, FOLFIRINOX.11 Thus, by using hCES2 as a predictive marker in biopsied pancreatic tissues, a patient's response towards drugs like Irinotecan can be determined before therapy to help guide the choice of the most appropriate therapeutic drug for that individual. Moreover, by continuously monitoring hCES2 activity over the course of therapy, any acquired resistance to Irinotecan can be caught early and the treatment plans can be adjusted accordingly. To achieve these goals in the clinic, a hCES2 assay that is simple, accurate and can be used in high throughput formats is highly warranted.
The gold standard to measure hCES2 activity uses para-nitrophenyl acetate (p-NPA), whereby active CEs will hydrolyze the acetate group, producing para-nitrophenol and an absorbance increase at 405 nm. Although direct in activity, the assay can only be used after the cells are lysed, and is non-selective for measuring hCES2 activity (e.g. active hCES1 will also produce a signal). Other methods, such as immunoblotting, mass spectrometry, liquid chromatography, and proteomic techniques are often used, although they are costlier, labor intensive, non-selective, and inaccurate as they often correlate hCES2 RNA or protein expression levels with enzymatic activity.12–15 Fluorescent chemosensors are an attractive alternative to conventional methods due to their simplicity, low detection limit, non-invasive nature, and potential for real-time cellular/tissue applications. Perhaps the most common chemosensor for hCES2 is fluorescein diacetate, which is non-fluorescent in its spirocyclized form, but becomes fluorescent upon hCES2 mediated hydrolysis of the acetate groups to produce fluorescein.16 Although this probe has been used to measure hCES2 activity in cells, it suffers from high false positives due to background hydrolysis in the cellular media and false negatives due to quenching of fluorescein from certain cellular components.16,17 Several other fluorescent probes have been designed that contain a fluorophore conjugated via an ester linkage with a benzoyl moiety.18–21 These probes were found to be selective for hCES2, but were sensitive to environmental factors (e.g. temperature and pH), producing substantial background signal when introduced in cells. To overcome these issues, several ratiometric fluorescent probes were constructed to more accurately quantify hCES2 activity in living cells.20–24 However, these probes either have aqueous solubility issues, absorb and emit at short wavelengths, suffer from photostability issues or are poorly selective for hCES2 over hCES1.21,25 Moreover, the use of these probes in patient-derived xenografts has yet to be demonstrated.
To address this, we describe the design, synthesis and characterization of a fluorescent chemosensor that responds to hCES2 activity ratiometrically in long wavelength emission channels (i.e. yellow and red), and with high selectivity over its related family member, hCES1. Our probe exhibits high water solubility, photostability and cell permeability, and can be used to quantify hCES2 activity in cultured cancer cells as well as in pancreatic cancer patient-derived tissue samples.
Results and discussion
Design, synthesis and evaluation of fluorogenic hCES2 probes
To design probes that can report on hCES2 activity, we focused on developing a strategy for coupling a fluorescence change to the CE hydrolysis reaction. With this in mind, we designed two probes containing the pyruval (1) or benzoyl (2) group conjugated to the dye, 6-amino-1-phenalenone (3). Although the photophysical properties of 3 have not been thoroughly characterized,26 it was selected since it contains an exocyclic amine in conjugation with the pi system of the dye. We reasoned that conversion of the amine to an amide via ligation to a pyruval or benzyl moiety would cause a blue shift in the absorption spectrum, and a red-shift (i.e. recovering the properties of the parent dye) upon hCES2-mediated hydrolysis of the amide bond. Compound 3 was synthesized according to established methods27,28 (see ESI, Scheme 2†), and then coupled with pyruvyl chloride (see ESI, Scheme 3†), or benzoyl chloride (see ESI, Scheme 4†), to yield probes 1 and 2, respectively.The photophysical properties and stability of probes 1 and 2, and compound 3 in the absence of hCES2 were first investigated and are summarized in Table 1. As expected, probes 1 and 2 exhibited blue shifts in their absorption spectra compared to the parent dye, 3. Interestingly, conversion of the amine to an amide did not diminish its fluorescence, but rather resulted in a blue-shift towards the yellow spectrum region with overall similar brightness to 3. To determine aqueous stability, each probe was excited at their corresponding excitation maxima and the fluorescence emission at 605 nm was monitored as a function of time. We reasoned that any potential instability would be due to the presence of the aryl amide linkage, which could undergo background, uncatalyzed hydrolysis to generate the parent dye, 3. We found that probe 1 exhibited a measurable increase in fluorescence at 605 nm when incubated under physiological conditions (PBS pH 7.4, 37 °C), however probe 2 showed no change in fluorescence under the same conditions (Fig. S1†). We hypothesized the difference in stability may be due to the electron-withdrawing carbonyl functionality neighboring the carbonyl of the amide bond in probe 1, which is replaced by an electron donating phenyl moiety in probe 2 making the carbonyl carbon less electrophilic. Thus, given its superior aqueous stability, enzymatic and cellular studies were conducted using probe 2.
| ε (M−1 cm−1) | λ ex (nm) | Φ F | λ em (nm) | Brightness (M−1 cm−1) | Stability in PBS | |
|---|---|---|---|---|---|---|
| 3 (AP) | 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 736 736 |
548 | 0.20 ± 0.01 | 605 | 2547 ± 150 | Yes |
| Probe 2 (Benz-AP) | 8880 | 413 | 0.39 ± 0.1 | 562 | 3500 ± 900 | Yes |
| Probe 1 (Pyr-AP) | 8872 | 413 | 0.16 ± 0.03 | 550 | 1420 ± 200 | No |
To determine if probe 2 can undergo hCES2 catalyzed hydrolysis, we first monitored its UV-Vis spectrum as a function of time. Upon addition of hCES2 (110 nM) we observed a decrease in absorbance at 413 nm followed by an increase at 548 nm, with a final spectrum resembling that of compound 3 (Fig. S2†). Given that probe 2 has weak absorbance at 548 nm, we reasoned that excitation at 548 nm (where 3 would have maximum absorbance) would lead to an increase in fluorescence as 3 is produced. Indeed, we found that addition of 110 nM hCES2 led to a 40-fold increase in fluorescence emission at 605 nm, with a fluorescence spectrum matching that of the expected product, 3 (Fig. 1), which was further validated by HPLC analysis of the reaction mixture (Fig. S3†). Similarly, exciting 2 at its maximum absorbance at 413 nm resulted in a decrease in emission at 562 nm upon addition of hCES2 (Fig. 1). Similar fluorescence responses were also observed as low as 5 nM hCES2 with an expected slower rate (Fig. S4†). Lastly, probe 2 and compound 3 were found to be photostable with constant irradiation for 1 hour and soluble in PBS up to 75 μM and 140 μM, respectively (Fig. S5†).
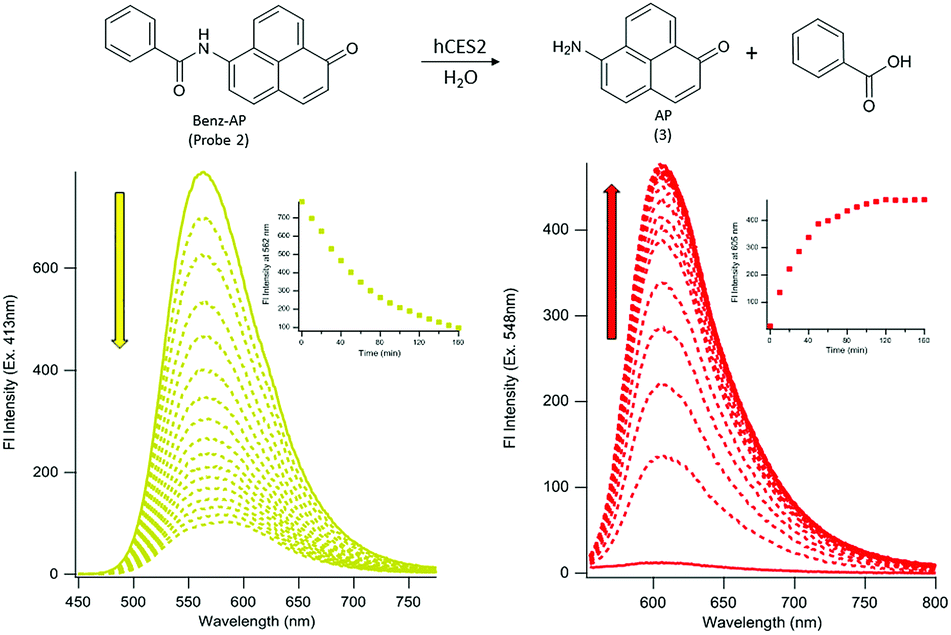 | ||
| Fig. 1 Ratiometric fluorescence response of probe 2 for hCES2 activity. Top, scheme of probe 2 upon hCES2 hydrolysis of the aryl amide bond produces the dye 3 and benzoic acid as a by-product. Bottom, time-dependent fluorescence emission spectra upon excitation at 413 nm (left) and excitation at 548 nm (right) results in a decrease and increase in yellow and red fluorescence, respectively, upon reaction with hCES2 (insets, plot of emission maxima as a function of time). Scans are taken at 10 minute intervals at 37 °C. Probe 2 = 11 μM, hCES2 = 110 nM. | ||
To validate the fluorescence response is in fact due to hCES2 activity, we pre-incubated hCES2 with general CE covalent inhibitor bis(4-nitrophenyl)phosphate (BNPP) or a hCES2-specific non-covalent inhibitor loperamide (LPA) followed by addition of probe 2. We observed an overall lower fluorescence increase at 605 nm (Fig. 2 and S6†) suggesting that our probe is in fact responding to hCES2 activity and that the fluorescence response observed is due to hCES2 catalyzed hydrolysis of the aryl amide bond to produce 3 (Fig. 1). Lastly, we determined if probe 2 is selective for hCES2 over other major human carboxylesterase isoforms, mainly hCES1, which have been shown to act on similar substrates.23 Although active on p-NPA (Fig. S7†), addition of hCES1 (110 nM) to probe 2 led to no significant increase in fluorescence emission at 605 nm (Fig. 2, S8 and S9†). Further increasing the concentration of hCES1 up to 440 nM also did not produce any product (Fig. S9†). These results suggest probe 2 is highly selective for hCES2 over hCES1 with a measured Km and kcat of 6.6 ± 3.7 μM and 0.26 ± 0.04 s−1, respectively (Fig. S10†). Finally, probe 2 tested against other hydrolases also did not produce a significant fluorescent response (Fig. 2).
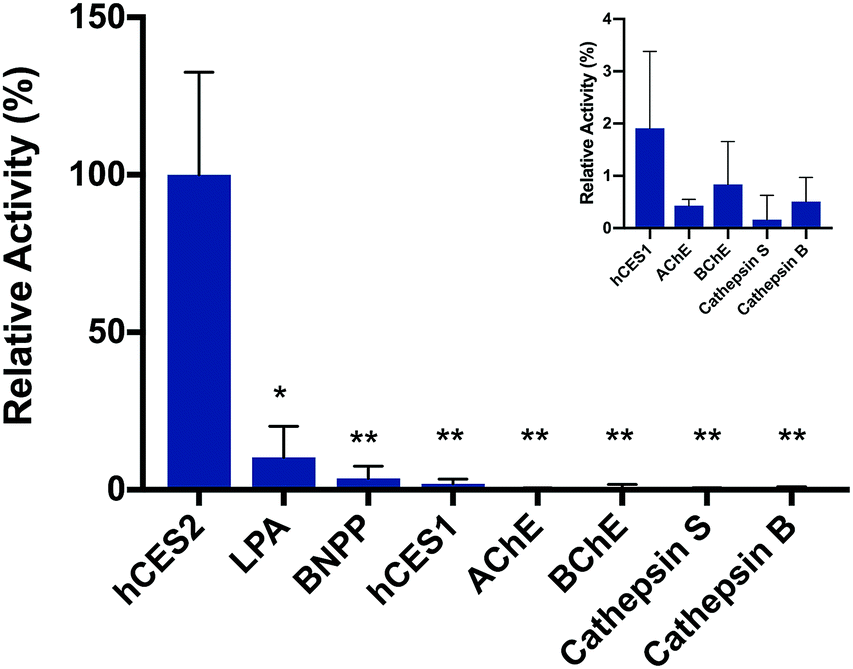 | ||
| Fig. 2 Probe 2 selectivity for hCES2 and its response to CE inhibitors. Incubation of probe 2 with 110 nM of either hCES1, AChE, BChE, Cathepsin S or Cathepsin B resulted in no fluorescence increase. Pre-incubation of hCES2 with the inhibitors LPA or BNPP resulted in a substantial reduction in overall fluorescence compared to the positive control. Fluorescence at 605 nm (Ex. 548 nm) was measured after 1 hour at 37 °C. The positive control (“hCES2”) was normalized to 100% activity. Experiments were performed in triplicates. Statistical significance is calculated using unpaired t test compared with hCES2 (**p < 0.01, *p < 0.05). LPA = loperamide, BNPP = bis(4-nitrophenyl)phosphate, AChE = acetylcholinesterase, BChE = butyrylcholinesterase. | ||
hCES2 activity in pancreatic cancer cells
Given the selective ratiometric fluorescent response of probe 2 for hCES2, we asked whether it can be used to measure hCES2 activity in cultured cancer cells. Previous work by Capello et al. have shown that hCES2 activity in pancreatic cancers is a crucial determinant for the effectiveness of the anti-cancer drug, Irinotecan.11 To test whether the new probes might address this, we first incubated probe 2 (20 μM) with the pancreatic cell line, SU.86.86, known to contain no detectable amounts of hCES2 by Western blot (Fig. 3).11 After a 3 hour incubation, cells were washed and imaged in the yellow channel for probe 2 and the red channel for the expected hCES2 product, 3.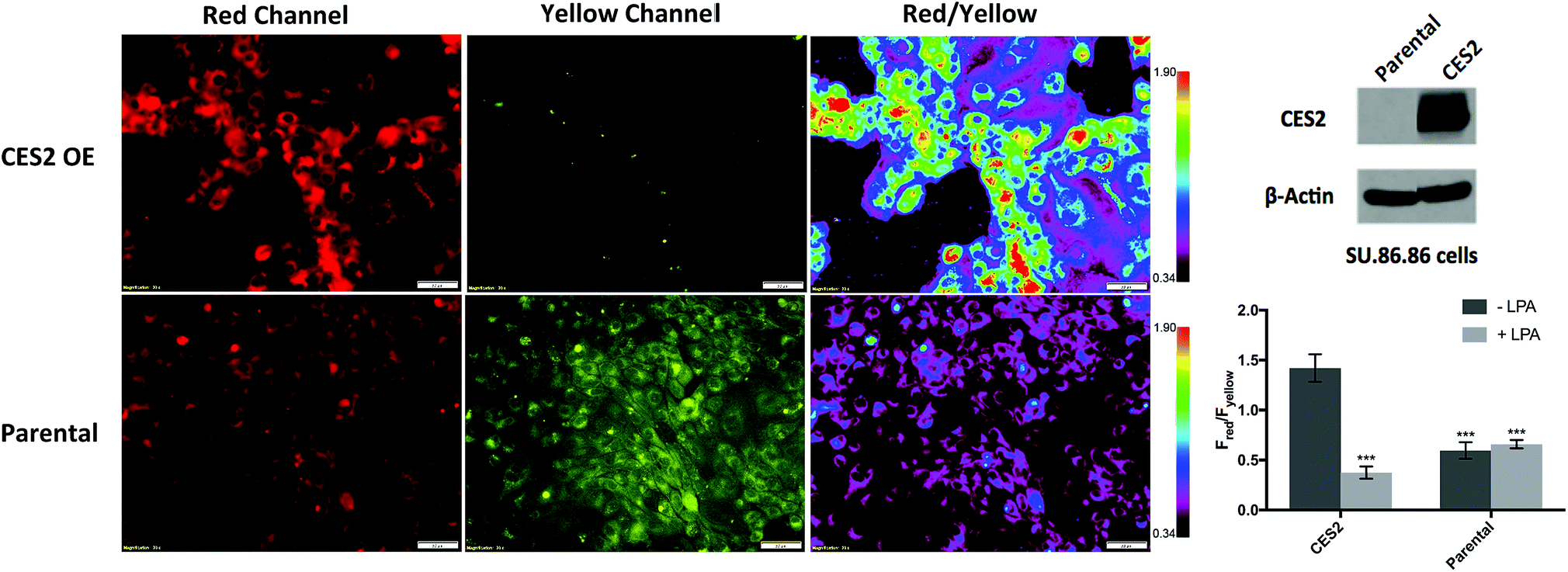 | ||
| Fig. 3 Measuring hCES2 activity in pancreatic cancer cells. Probe 2 (20 μM) was incubated for 3 hours with the pancreatic cancer cell line, SU.86.86. Top row, probe 2 with SU.86.86 cells containing a vector overexpressing CES2 (CES2 OE) displayed strong red over yellow fluorescence, indicating high conversion of probe 2 to product 3. Bottom row, probe 2 with parental SU.86.86 showed weak red fluorescence but high yellow fluorescence indicative of poor conversion of probe 2 to 3. Far top right, Western blot showing hCES2 expression levels. Far bottom right, bar graph quantifying the ratio of red over yellow fluorescence for CES2 OE and parental. Light gray bars indicate pretreatment with the CES2 inhibitor, LPA, dark gray bars are no pretreatment (images in Fig. S9†). Ratios are an average of 3 regions in an 8-chambered well plate. Magnification is 20×. Scale bars = 50 μm. Statistical significance is calculated using unpaired t test compared with CES2 (***p < 0.001). | ||
As shown in Fig. 3 (Fig. S11†), yellow fluorescence appeared to be diffuse in the cytosol with no fluorescence detected in the nucleus. Switching to the red channel revealed low red fluorescence relative to the yellow channel (Fred/Fyellow = 0.60 ± 0.080), suggesting that most of probe 2 is present relative to the product dye, 3. Next, we added probe 2 to SU.86.86 cells that contain a vector overexpressing hCES2 (CES2 OE) (Fig. 3).11 Incubation of probe 2 under the same conditions, we now observed high red fluorescence and low yellow fluorescence (Fred/Fyellow = 1.4 ± 0.13), indicating a significant portion of probe 2 has been converted to 3 (Fig. 3 and S11†). To control for any effects a vector could have on our fluorescence signals, we also incubated probe 2 with parental SU.86.86 containing an empty vector (“control”). As seen in Fig. S12,† control and parental cell lines yielded similar fluorescence ratios. To ensure the difference between CES2 OE and parental is due to the action of hCES2, we pre-incubated CES2 OE cells with the hCES2 specific LPA inhibitor, followed by incubation of probe 2 for 3 hours. We observed a significant decrease in the Fred/Fyellow ratio (Fred/Fyellow = 0.37 ± 0.060) compared to cells untreated with LPA suggesting cellular hCES2 activity is responsible for the conversion of probe 2 to compound 3 (Fig. 3 and S11†). Treating the parental SU.86.86 with LPA did not result in a lower ratio suggesting there is no significant level of active hCES2, consistent with previous Western blots.11 In addition to ratiometric imaging, we also performed single red channel measurements (no washing), which yielded similar results (Fig. S13†). Lastly, we found that incubation of our probe for 3 hours up to 20 μM did not cause any cell toxicity (Fig. S14†).
hCES2 activity in pancreatic cancer patient-derived xenografts
Next, we determined whether probe 2 can be used to measure hCES2 activity in pancreatic cancer tissue samples. We first obtained control vector and CES2 OE SU.86.86 orthotopic xenograft tumors as frozen tissue slices (10 μm in thickness) on microscope slides. We added 100 μL of probe 2 directly on the area containing the tissue, then captured red channel fluorescence readings every 30 min. In both tissue samples, we observed a time dependent increase in red fluorescence, however, the CES2 OE orthotopic tissues showed an overall larger fluorescence increase after 3 hours (p < 0.0001) (Fig. 4A). After 3 hours, tissues were washed 3× with PBS to capture fluorescence from both red and yellow emission channels. The average fluorescence intensity was collected at 3 hours at 12 different regions of a given tissue sample, then their five-number summary distribution was plotted to construct a box and whisker plot (Fig. 4B). Consistent with cell culture experiments, the CES2 OE SU.86.86 tissues displayed higher red/yellow ratio (median Fred/Fyellow = 2.71) compared with control vector tissues (median Fred/Fyellow = 0.44; p < 0.0001). We further performed H&E staining which indeed confirmed the presence of cancerous tissue (Fig. S15†).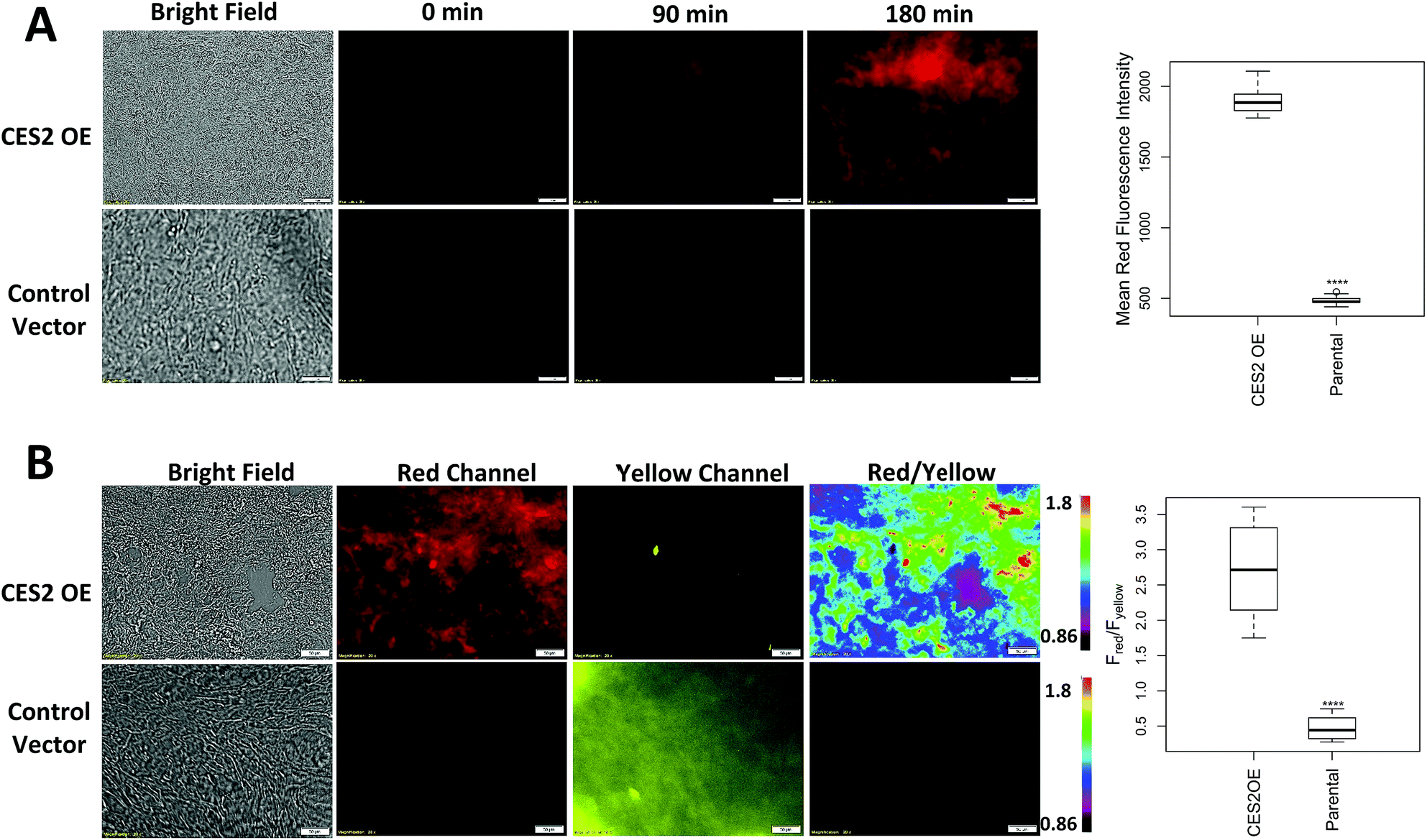 | ||
| Fig. 4 Detecting hCES2 activity in orthotopic SU.86.86 xenografts. A few drops of probe 2 (20 μM) was added to a SU.86.86 frozen tissue slice (10 μm in thickness) on a microscope slide and imaged. (A) Single channel red fluorescence as a function of time whereby probe 2 added to SU.86.86 CES2 OE tissues (top row) led to larger time dependent increase in fluorescence, compared to control vector SU.86.86 tissues (bottom row). (B) Ratiometric images obtained after 3 hour incubation. Tissues were washed 3 × with PBS to obtain images in the yellow channel. A higher red/yellow ratio was observed for CES2 OE orthotopic xenografts compared to the respective control. Average fluorescence was collected at 12 different regions of the tissues at 3 hours to construct a box plot (far right). Magnification is 20×. Scale bars = 50 μm. Statistical significance is calculated using the Mann–Whitney–Wilcoxon test compared to CES2 OE (****p < 0.0001). | ||
Lastly, we obtained patient-derived xenografts (PDX) which have previously served as reliable models for predicting anticancer efficacy of therapeutic agents.29–31 We applied probe 2 to tissue samples from two pancreatic cancer patients (PDX112 and PDX121) and captured ratiometric images following the same procedure we established for orthotopic xenografts. Fig. 5 shows PDX112 displaying a higher red/yellow emission ratio (median Fred/Fyellow = 0.98) compared to PDX121 (median Fred/Fyellow = 0.34; p < 0.0001). H&E staining confirmed the presence of cancerous tissue (Fig. S15†) while Western blots confirmed the presence of CES2, with significantly higher levels found in PDX112 over PDX121 (Fig. 5). Although PDX121 shows no detectable expression, fluorescence reveals some degree of hCES2 activity consistent with what we observed with parental SU.86.86 also containing no detectable hCES2 expression and with previous reports.11 Single channel measurements also showed a higher median fluorescence for PDX112 over PDX121, however there is increased variability in PDX121 signals due to a relatively larger interquartile range compared to ratiometric measurements (Fig. S16†). Finally, it is worth noting that addition of probe 2 to mostly stroma or necrotic tissues produced low ratiometric signals (Fig. S17†) suggesting some CES2 activity consistent with previous reports.6
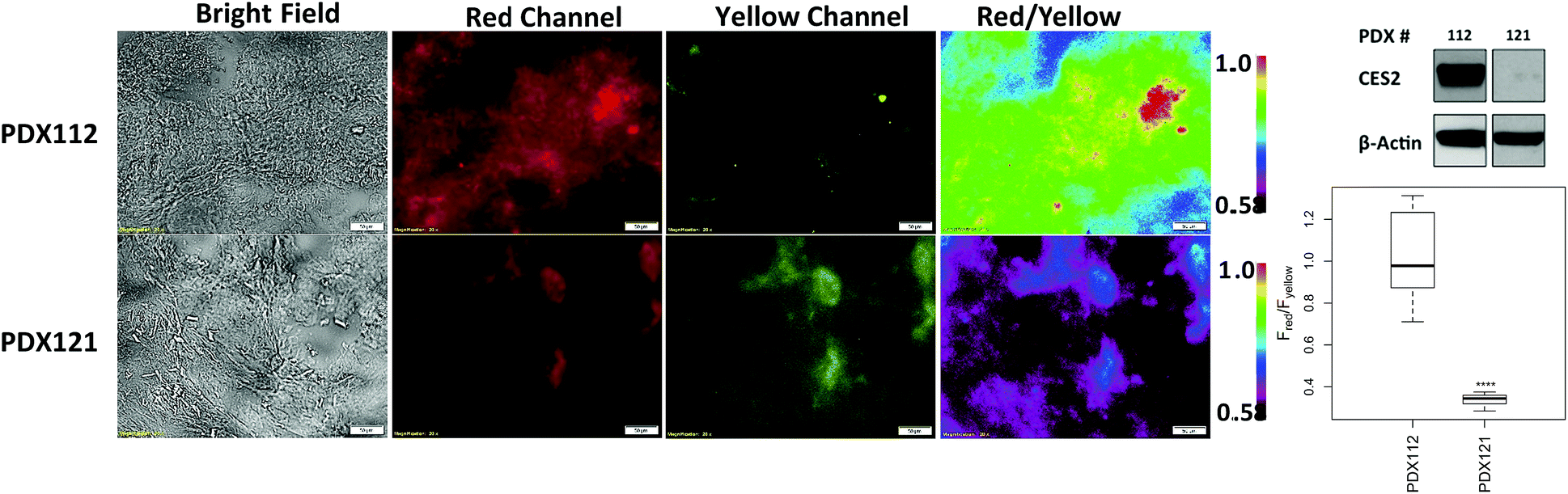 | ||
| Fig. 5 Detecting hCES2 activity in patient-derived xenografts. A few drops of probe 2 (20 μM) was added to a tissue slice (10 μm in thickness) on a microscope slide and ratiometric images were acquired for after 3 hours. Far top right, Western blot showing hCES2 expression levels. Far bottom right, average fluorescence collected at 12 different regions of the tissues at 3 hours to construct a box plot. Magnification is 20×. Scale bars = 50 μM. Statistical significance is calculated by the Mann–Whitney–Wilcoxon test (****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05). | ||
We have developed a small-molecule ratiometric fluorescent chemosensor that can report on the activity of hCES2 in pancreatic cancer cells and in patient-derived xenografts. Although other hCES2 fluorescent chemosensors have been reported, probe 2 described here overcomes several limitations that have hindered previous hCES2 chemosensors from providing accurate and practical measurements of hCES2 activity.18–22,25,32–34 First, probe 2 can be readily synthesized following the facile coupling of 3 to benzoyl chloride and demonstrates enhanced selectivity for hCES2 over hCES1 to the extent where Michaelis–Menten parameters for hCES1 could not be measured.21,24,33,34 Second, our sensor demonstrates improved solubility in aqueous media, with a maximal use of 0.5% organic solvent as opposed to 50% organic solvent used in other reported probes.18–22,25 Thirdly, in addition to being a ratiometric chemosensor, probe 2 also has the advantage of monitoring at longer wavelength emission channels (i.e. red and yellow) compared to previous sensors, thereby minimizing interference from auto-fluorescence and improving tissue penetration depth.18,19,32 Furthermore, excitation of our sensor in the visible region reduces photodamage compared to several other hCES2 fluorescent probes excited in the UV range (<400 nm).20,25,32 Overall, these properties of probe 2 render it by far superior to other reported hCES2 probes.
The gold standard for measuring cellular hCES2 activity is the colorimetric p-NPA assay, which requires cell lysis prior to measurements. The act of lysis not only adds additional steps, but can also lead to deactivation of enzymes in the process. Moreover, p-NPA is not selective for hCES2 and so the presence of related esterases such as hCES1 would lead to an overestimation of hCES2 activity. In contrast to p-NPA, probe 2 undergoes a ratiometric fluorescence response highly selective for hCES2 over hCES1 activity. We show that probe 2 is cell permeable and can readily respond to levels of hCES2 activity in live pancreatic cancer cells in just a few hours. Given the simplicity and accuracy of our assay, we envision that probe 2 can be used in conjunction with flow cytometry to map hCES2 activity across various pancreatic cancer cell lines to make stronger correlations between activity and Irinotecan potency, as was previously generated using p-NPA assay. Probe 2 may also be used to discover new connections between hCES2 activity and anticancer drug resistance in a variety of other cancer cell lines in more of a throughput manner compared to other methods. Finally, given the change in fluorescence against known existing CE inhibitors such as BNPP and LPA, probe 2 may also be used to discover new inhibitors or activators of hCES2 directly in its native cellular environment.
Lastly, we demonstrate for the first time the use of a fluorescent chemosensor for detecting hCES2 activity in patient-derived xenografts. Previous work by Capello et al. found that hCES2 expression is highly variable amongst pancreatic cancer patients.11 They further demonstrated that the potency of the anti-cancer drug Irinotecan is highly dependent on the active hCES2. In our work, we showed that probe 2 can detect hCES2 activity within intact orthotopic SU.86.86 xenografts. Consistent with cells in culture, probe 2 was capable of reporting the expected high level of hCES2 in CES2 OE tissues compared to control vector tissues. Moreover, adding probe 2 to PDX samples revealed hCES2 activity consistent with expression levels via Western blots. Unfortunately, only one patient (PDX112) out of the two had been treated with FOLFIRINOX, and so we could not determine how the measured hCES2 activity correlates with survival rates. Given the simplicity of the assay demonstrated here, it seems feasible that probe 2 can be used to measure hCES2 activity on a large number of patient tissues whereby their clinical response to Irinotecan had been recorded. We then envision that one could determine a threshold for hCES2 activity that is required to elicit certain survival rates. Such data may even first be obtained in high throughput on PDXs that have been treated with anti-cancer drugs, using recently developed live tissue sensitivity assays.35 Finally, given that ratiometric signals were also observed in stroma and necrotic tissue, we note that fluorescent chemosensing should be complemented by H&E staining to rule out false positives/negatives. Future work will also aim to apply probe 2 directly to fresh biopsied tissue samples, where, by predicting hCES2 activity, could improve pancreatic cancer patient stratification for Irinotecan-based therapy.
Conclusions
We have developed a small molecule ratiometric fluorescent chemosensor that directly measures hCES2 activity. Probe 2 is readily synthesized, soluble in aqueous media, photostable, and can selectively monitor hCES2 activity at long wavelength emissions (red and yellow). Probe 2 can be used to measure hCES2 activity in live pancreatic cancer cells with no cytotoxicity at active concentrations. We also show for the first time the use of a fluorescent chemosensor for detecting hCES2 in pancreatic cancer patient-derived xenografts. The developed assay raises the possibility of predicting patient response to Irinotecan-based therapy in a high throughput clinical setting.Experimental section
General information
Chemicals and solvents were purchased from either Fisher Scientific or Sigma-Aldrich unless otherwise stated and were used without further purification. All reactions were conducted under argon environment and in oven-dried glassware. Reactions were monitored by silica gel TLC plates and were viewed with a low intensity short/long UV wavelength lamp. 1H and 13C NMR spectra were taken on Bruker 400 MHz NMR spectrometer. HRMS were acquired from the AIMS Mass Spectrometry Laboratory at the Department of Chemistry, University of Toronto. High performance liquid chromatography (HPLC) was performed on a LC-20AT Shimadzu liquid chromatograph, equipped with an SPD-M20A VD diode array detector, a CBM-20A VP system controller, and a C18 semi-prep column. Human CES2 (expressed in HEK293 cells, C-terminal His Tag) was purchased from Sino Biological Incorporated (10380-H08H). Human CES1 (expressed in E. coli, N-terminal His tag) was purchased from Abbexa Limited (abx168930). All stock solutions of probe 1, probe 2, and 3 were prepared by dissolving each sample in 1:1 ACN/HyClone Cell Culture Grade Water. Stock solutions of bis(4-nitrophenyl)phosphate (BNPP) and loperamide (LPA) were prepared by dissolving each inhibitor in DMSO (Molecular Probes™ DMSO, Anhydrous). Absorbance spectra were obtained on a Shimadzu UV-1800 UV Spectrophotometer. Fluorescence emission spectra were obtained on a Shimadzu RF-6000 Spectro Fluorophotometer.
Synthesis of phenalenone (1H-phenalen-1-one)
The synthesis of phenalenone was carried out as previously described.27 Aluminum chloride (40 g, 0.3 mol) and naphthalene (16.9 g, 0.1 mol) were added to anhydrous DCM (100 mL). The mixture was cooled to 0 °C. Cinnamoyl chloride (13 g, 0.1 mol) was added at 0 °C and the reaction was stirred at room temperature overnight. The mixture was then refluxed for 3 h, cooled with iced hydrochloric acid, and filtered. The filtrate was extracted with chloroform. The solids were repeatedly boiled with hot chloroform (3 × 200 mL) and filtered until the filtrate became colourless. All organic extracts were combined, dried over anhydrous sodium sulfate, and subjected to a rotary evaporator to give a yellow solid (54%). The crude product was further purified by column chromatography (petroleum ether/ethyl acetate 10:1). 1H-NMR (400 MHz, CDCl3): δ = 8.63 (d, J = 7.34 Hz, 1H), 8.20 (d, J = 8.07 Hz, 1H), 8.02 (d, J = 8.44 Hz, 1H), 7.66–7.82 (m, 3H), 7.59 (t, J = 7.70, 1H), 6.74 (d, J = 9.90 Hz, 1H). 13C-NMR (101 MHz, CDCl3): δ = 185.71, 141.83, 134.95, 132.17, 131.97, 131.42, 130.40, 129.48, 129.25, 127.84, 127.55, 127.15, 126.67. ESI-MS (m/z): [M + H]+ calculated for C13H8O 181.06 Da, found 181.02 Da.
Synthesis of 6-amino-1-phenalenone (3; 6-amino-1H-phenalen-1-one; AP)
The synthesis of 6-amino-1-phenalenone was carried out as previously described, with some modifications.28 To a solution of phenalenone (5 g, 0.03 mol) in sulphuric acid was added a dropwise mixture of nitric acid and sulphuric acid. The reaction mixture was stirred for 3 h at room temperature. Water was added and the mixture was extracted with dichloromethane. The combined extracts were dried with Na2SO4, filtered and concentrated. The crude mixture was dissolved in dioxene at room temperature. Tin chloride (25 g, 0.11 mol) and acetic acid were slowly added and stirred for 2 days. The reaction mixture was neutralized by sodium hydroxide, and extracted with ethyl acetate. The extract was purified by column chromatography (ethyl acetate/hexanes to 100% ethyl acetate) to yield a bright red solid (15%). 1H NMR (400 MHz, MeOD): δ = 8.65 (d, J = 6.24 Hz, 1H), 8.60 (d, J = 6.97 Hz, 1H), 7.79–7.61 (m, 4H), 6.72 (d, J = 8.07 Hz, 1H), 6.46 (d, J = 9.17 Hz, 1H). 13C NMR (101 MHz, MeOD): δ = 186.69, 160.43, 155.47, 145.25, 139.46, 132.96, 131.25, 131.15, 129.59, 125.94, 122.03, 117.03, 109.84. DART-MS (m/z): [M + H]+ calculated for C13H9NO 195.0684 Da, found 196.07598 Da.Synthesis of probe 1 (2-oxo-N-(1-oxo-1H-phenalen-6-yl)propanamide)
To a solution of pyruvic acid (50 μL, 0.705 mmol) in anhydrous DCM (250 μL), oxalyl chloride (75 μL, 0.886 mmol) was added at room temperature followed by a few drops of anhydrous DMF. After stirring at room temperature for 30 min, the mixture was concentrated by evaporation, resulting in pyruvyl chloride as an oil. Sodium hydride (5.1 mg, 0.127 mmol) was added to a mixture of AP (8 mg, 0.041 mmol) in DMF (750 μL) and was stirred for 20 min. The mixture was then added dropwise to the pyruvyl chloride oil and was stirred overnight. The solvent was removed in vacuo and the residual solid was purified by flash silica chromatography (100% DCM to DCM/MeOH 9:1). The compound was further purified by HPLC (gradient of 5 to 100% water/ACN with 0.1% formic acid over 35 min) to afford probe 1 as a yellow solid (46%). 1H-NMR (400 MHz, CDCl3): δ = 8.69–8.71 (d, J = 7.3 Hz, 1H), 8.45–8.47 (d, J = 7.9 Hz, 1H), 8.28–8.30 (d, J = 8.9 Hz, 1H), 7.87–7.91 (t, 1H), 7.78–7.80 (d, J = 7.9 Hz, 1H), 7.72–7.75 (d, J = 9.7 Hz, 1H), 6.70–6.73 (d, J = 9.7 Hz, 1H), 2.67 (s, 3H). 13C-NMR (100 MHz, CDCl3): δ = 192.43, 171.33, 157.66, 143.07, 133.31, 130.06, 128.39, 124.72, 112.04, 111.93, 22.91, 1.03. HRMS (m/z): [M + H]+ calculated for C16H11NO3 266.07 Da, found 266.08 Da.
Synthesis of probe 2 (N-(1-oxo-1H-phenalen-6-yl)benzamide)
Sodium hydride (5.1 mg, 0.127 mmol) was added to a mixture of AP (8 mg, 0.041 mmol) in DMF (750 μL) and stirred for 20 min. After dropwise addition of benzoyl chloride (82 μL, 0.699 mmol), the mixture was stirred overnight and the solvent was removed under reduced pressure. The crude product was purified by flash silica chromatography (100% DCM to DCM/MeOH 9:1) and further purified by HPLC (gradient of 5 to 100% water/ACN with 0.1% formic acid over 35 min), resulting in a yellow solid (52%). 1H NMR (400 MHz, CDCl3): δ = 8.72–8.73 (d, 1H), 8.52 (s, 1H), 8.41–8.43 (d, J = 7.8 Hz, 1H), 8.31–8.34 (d, 1H), 8.03–8.05 (d, 2H), 7.88–7.90 (t, 1H), 7.83–7.85 (d, J = 8.0 Hz, 1H), 7.77–7.79 (d, J = 9.8 Hz, 1H), 7.66–7.68 (d, 1H), 7.62–7.64 (d, 2H). 13C-NMR (100 MHz, CDCl3): δ = 171.21, 162.39, 153.14, 142.20, 134.56, 132.49, 131.94, 130.97, 130.16, 127.36, 126.00, 125.90, 113.16, 112.51. HRMS (m/z): [M + H]+ calculated for C16H11NO3 300.09, found 300.1020.
hCES2 UV-Vis assays
Enzyme assays were performed in a 1 cm path length, 60 μL quartz cuvette. Stock solutions of probes were diluted in DPBS containing magnesium chloride and calcium chloride to a final concentration of 11 μM. Enzyme was added to yield a final concentration of 110 nM. To confirm hCES1 was active, 110 nM hCES1 was added to 2 mM p-NPA in DPBS at 37 °C and the UV-Vis spectra was measured in 30 s intervals.HPLC analysis of enzyme reaction
Probe 2, 3, and the hCES2 reaction with probe 2 were analyzed by HPLC. Samples of purified probe 2 and 3 were prepared to 110 μM in ACN/0.1% formic acid. The hCES2 reaction was prepared by incubating 11 μM probe 2 with 110 nM hCES2 in DPBS at 37 °C for 3 h. The reaction mixture was concentrated 10× to achieve 110 μM product and dissolved in ACN/0.1% formic acid for HPLC analysis. HPLC was performed on a LC-20AT Shimadzu liquid chromatograph, equipped with an SPD-M20A VD diode array detector, a CBM-20A VP system controller, and an ACE Equivalence 5 C18 column (250 × 3 mm ID). The mobile phase consisted of gradient of 5–100% water/ACN with 0.1% formic acid over 35 min using a flow rate of 0.2 mL min−1.Water solubility
Stock solutions of probe 2 and compound 3 were prepared in 50% ACN/50% water. Dilutions were performed to yield solutions ranging in concentration from 10 nM to 142 μM, with the concentration of ACN maintained at less than 1%. UV-Vis spectra were taken in DPBS in a 60 μL quartz cuvette and absorbance at λmax (413 nm for probe 2, 548 nm for 3) was plotted against concentration. The maximum linear response was used to quantify the solubility limit.hCES2 fluorescence assays
Enzyme assays were performed using a 1 cm path length, 60 μL quartz cuvette. Stock solutions of probe were diluted in DPBS containing magnesium chloride and calcium chloride to a final concentration of 11 μM. Enzyme was added to a final concentration of 110 nM and the fluorescence intensity was measured at 605 nm with 1 s intervals at a scan speed of 600 nm min−1 and 3 nm excitation and emission bandwidths at 37 °C. Emission spectra were collected before and after the time course at excitation wavelengths of each probe and 3. Ratiometric fluorescent properties were measured by collecting emission spectra from 0 to 160 min in 10 min intervals at excitation wavelengths of 413 nm and 548 nm.hCES2 fluorescence inhibition assays and selectivity
For inhibition assays, 1 μM BNPP or LPA was incubated with 110 nM CES2 at 37 °C in a DPBS/1% DMSO solution for 30 min. 11 μM probe 2 was then added to the solution and fluorescence emission at 605 nm was measured. For selectivity assays, 110 nM enzyme was added to 11 μM probe 2 in DPBS in a 60 μL quartz cuvette and fluorescence emission at 605 nm was measured for 1 h at 37 °C.Fluorescence quantum yield
The fluorescence quantum yields of probe 1 and probe 2 were determined using the fluorophore perylene (ΦF = 0.94 in cyclohexane) as a standard due to overlap between the absorption spectra at the 413 nm excitation band of the probes.36 Separate solutions of perylene in cyclohexane and probe 1 or probe 2 in DPBS were prepared to yield an absorbance of 0.05 at 366 nm. Emission spectra of each sample were taken with excitation at 366 nm (1.5 nm bandwidth) and emission was measured over the 376–800 nm range (1 nm emission bandwidth) at a 60 nm min−1 scan speed. The emission spectra were integrated by Riemann sums at 1 nm intervals of the midpoint of the fluorescence intensity values from 376 to 700 nm. To determine the fluorescence quantum yield of 3, the fluorophore rhodamine B (ΦF = 0.77 in ethanol) was used as a standard due to its similar absorption profile.37 Emission spectra solutions of rhodamine B in ethanol and 3 in DPBS with fluorescence values between 0.04 and 0.05 at 542 nm were measured with excitation at 542 nm (1.5 nm bandwidth) from 552 nm to 800 nm (1 nm bandwidth). The Riemann sum of the fluorescence values at the midpoint of 1 nm intervals from 552 nm to 710 nm was taken as the integration of the emission spectra. Fluorescence quantum yields were calculated according to the following equation:| Φsample = (Φstandard)(Isample/Istandard)(Astandard/Asample)(nsample2/nstandard2) |
Molar extinction coefficient
3 (1.0 mg) was dissolved in 1:1 ACN/water (1.00 mL) to make a stock solution. The slope of absorbance measurements of a series of dilutions of the stock solution was taken to determine the molar extinction coefficient according to the Beer–Lambert law. To determine the molar extinction coefficient of the probes, 0.6 μL of probe was diluted in DPBS to a total volume of 60 μL and an absorbance spectrum was obtained. 110 nM CES2 was added and absorbance spectra were recorded in 10 min intervals. The concentration of 3 upon the completion of the reaction was calculated. Assuming that this concentration is equal to the concentration of probe before the addition of probe, the molar extinction coefficient was determined by the Beer–Lambert law.
Michaelis–Menten enzyme kinetics assays
55 nM CES2 was added to various solutions of probe 2 (0–66 μM) prepared in DPBS buffer. Fluorescence intensity was collected at 605 nm (548 nm excitation) with 1 s intervals for 10 min at 37 °C. Fluorescence intensity rates were converted to rates of product formation (nM min−1) by a fluorescence calibration curve (fluorescence intensity versus concentration of AP produced (μM)) generated from the reaction of 66 μM probe 2 with 55 nM CES2, assuming linearity. Michaelis–Menten kinetic parameters (Km) were calculated by nonlinear regression analysis of the Michaelis–Menten equation (GraphPad Prism) by fitting to the model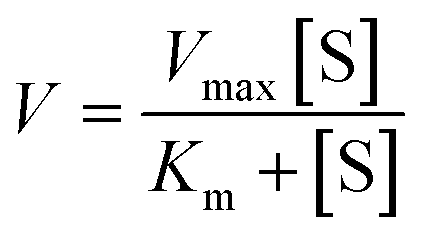 , where V represents enzyme velocity, Vmax represents maximal enzyme velocity, [S] represents substrate concentration, and Km represents substrate concentration required to achieve half of the maximal enzyme velocity. kcat was determined by the equation
, where V represents enzyme velocity, Vmax represents maximal enzyme velocity, [S] represents substrate concentration, and Km represents substrate concentration required to achieve half of the maximal enzyme velocity. kcat was determined by the equation 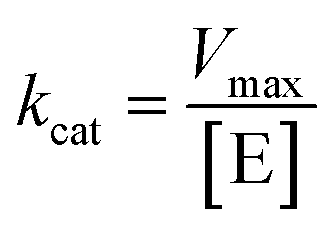 , where kcat represents the turnover number and [E] represents the concentration of enzyme.
, where kcat represents the turnover number and [E] represents the concentration of enzyme.
Cell culture
Parental SU.86.86 cells were purchased from ATCC. Manipulated SU.86.86 cell lines were provided by Dr Samir Hanash from MD Anderson Cancer Center and were cultured as previously described.11 For overexpression, CES2 was cloned into pLenti-C-Myc-DDK-IRESPuro (OriGene, Rockville, MD) vector, an empty vector was used as a control. Lentiviral infections were performed using 293LTV cells (Cell Biolabs, Inc., San Diego, CA) as producers of viral supernatants. 293LTV cells were grown on 10 cm plates to 70% confluence and cotransfected with lentiviral DNA and the helper vectors pCMVΔ8.74 and pVSV-G (Clontech, Mountain View, CA) using Lipofectamine 3000 (Life Technologies), according to the manufacturer's instructions. The medium was harvested 48 h post-transfection, filtered through a 0.45 μm filter and used to cross-transduce cancer cells in the presence of 8 μg mL−1 Polybrene (Sigma-Aldrich). Subsequently clones were selected by puromycin. All cells were maintained in a 5% CO2 atmosphere at 37 °C. SU.86.86 cells were grown in Roswell Park Memorial Institute (RPMI) 1640 Medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin/antimycotic (complete growth medium).Microscopy imaging
All fluorescence microscopy images and corresponding brightfield images were obtained on an Olympus IX73 microscope. All probe 2 cell images were obtained using a filter cube consisting of 380–420 nm excitation bandpass filter, a 484 nm dichroic mirror, and a 513–604 nm emission bandpass filter. Cell images of 3 were acquired using a filter cube comprising 503–557 nm excitation bandpass filter, a 580 nm dichroic mirror, and a 600–700 nm emission bandpass filter. All cells were plated in 8-chambered wells (Lab-Tek® Chambered #1.0 Borosilicate Coverglass System) at a density of 5 × 104 cells per well one day before imaging. The cells were washed once with DPBS, and were incubated in Opti-MEM with or without 100 μM LPA for 1 h. 20 μM probe 2 was then diluted into the appropriate wells and the cells were incubated for 3 h. The cells were washed five times in DPBS and were imaged in DPBS. All images were analysed using Olympus CellSens Dimension V1.18. H&E images were acquired from a Nikon Eclipse 50i Microscope with a Nikon DS-Ri1 camera and NIS-Elements BR3.0 software.Cell viability assays
SU.86.86 Parental, IRES control vector and CES2 overexpressing cells were each seeded at a density of 5000 cells per well in 48-well plates (Thermo Scientific Nunclon™ Delta Surface) and were incubated in supplemented RPMI 1640 Medium overnight at 37 °C in 5% CO2 atmosphere. The media was discarded; cells were washed once with DPBS, replaced with solutions of probe 2 or 3 in complete growth media at different concentrations (0–20 μM), and were incubated for 3 h. The media was discarded; the cells were washed once with DPBS, and were replaced with 250 μL of a 0.5 mg mL−1 solution of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) in complete RPMI. Following 2 h of incubation, the solution was removed and replaced with 250 μL DMSO to dissolve the produced formazan. Absorbance values were then read by a Tecan Infinite M1000 plate reader. Cell viability (CV) was determined by CV = Asample/Acontrol × 100%, where Asample and Acontrol are the absorbance values of the experimental group (treated with probe or AP) and control group (not treated with probe or AP) at 560 nm respectively, with cell viability of the control group adjusted to 100%.Western blot analysis
Cell pellets were lysed in RIPA buffer. Tumor tissues were cut into small pieces and disrupted with a Dounce homogenizer in RIPA buffer at 4 °C for use in all procedures. The lysates were then separated by SDS–PAGE and subjected to Western blotting. The following antibodies were used: CES2 (Sigma-Aldrich, HPA018897), and β-actin (Sigma-Aldrich).Animal studies, tissue preparation and imaging
Orthotopic and patient-derived xenografts were provided by Dr Samir Hanash and Dr Jason Fleming from MD Anderson Cancer Center. Animal experiment protocols were reviewed and approved by The University of Texas MD Anderson Cancer Center IRB and in accordance with the Guidelines for the Care and Use of Laboratory Animals published by the NIH (Bethesda, MD). Heterotopic engraftment of patient tumors into immunodeficient mice and expansion of direct xenograft tumors were performed as previously described.38,39 Briefly, excised patient tumor tissues were obtained at surgery at The University of Texas MD Anderson Cancer Center and mechanically minced into fragments (<1 mm). Five tumor fragments were individually placed into formed tissue pockets. Once tumors reached 1.2 cm in greatest diameter, the mice were sacrificed and the tumors dissected from mouse subcutaneous tissue under sterile conditions. Approximately one-third of the tumor was prepared for paraffin embedment, one-third was cut into quadrants and placed in liquid nitrogen for future study (F1), and other one-third was injected into NOD/SCID mice again to generate additional generations (F2, F3) of direct xenograft tumors applied in this study. For orthotopic xenograft models, a total of 106 viable cells in 50 μL of complete growth medium with 50% growth factor-reduced Matrigel were injected into the pancreas of 4 to 6 week-old female nude mice; a total of five mice were used for each experimental condition. The condition of mice was monitored two times weekly over a period of 5 weeks. As the mice tumor burden increased, mice were euthanized immediately upon becoming moribund, as required by our institutional animal care guidelines. All tissues were harvested and flash frozen with liquid nitrogen, then stored at −80 °C until needed. Using a cryotome, the harvested tissues were cut into 10 μm thick slices and placed in glass microscope slides. These were kept frozen until ready for the assessment of CES2 activity. All slides were imaged before addition of probe 2 to determine normal tissue fluorescence. Slides were incubated with 100 μL of 20 μM probe 2 at 37 °C for 3 h. Slides were then imaged for red fluorescence measurements. Ratiometric measurements were taken after washing three times with DPBS. H&E staining was performed by established protocols.40Statistical analysis
All data in bar graphs are expressed as mean ± standard deviation of triplicate measurements. Statistical differences were determined using a Student's t-test via GraphPad Prism 7.00 for Mac OS X (GraphPad Software, La Jolla California, USA), with p < 0.05 considered statistically significant. All box-and-whisker plots of mean fluorescence intensity were designed using R Studio (version 3.4.4), with boxes indicating the first and third quartiles, and bolded horizontal lines in the boxes representing the median value. Upper whiskers represent the maximum and lower whiskers represent the minimum values. All cell and tissue fluorescence data have normal cell or tissue fluorescence for each channel (fluorescence with no probe) subtracted respectively. To determine statistical differences in median values, Mann–Whitney–Wilcoxon rank sum test was performed using R studio, with p < 0.05 considered statistically significant.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
A. A. B. acknowledges support from Natural Sciences and Engineering Research Council of Canada (NSERC Discovery).Notes and references
- S. C. Laizure, V. Herring, Z. Hu, K. Witbrodt and R. B. Parker, Pharmacotherapy, 2013, 33, 210–222 CrossRef CAS PubMed.
- D. Wang, L. Zou, Q. Jin, J. Hou, G. Ge and L. Yang, Human carboxylesterases: a comprehensive review, Acta Pharm. Sin. B, 2018, 8, 699–712 CrossRef PubMed.
- S. E. Pratt, S. Durland-Busbice, R. L. Shepard, K. Heinz-Taheny, P. W. Iversen and A. H. Dantzig, Clin. Cancer Res., 2013, 19, 1159–1168 CrossRef CAS PubMed.
- L. de Sousa Cavalcante and G. Monteiro, Eur. J. Pharmacol., 2014, 741, 8–16 CrossRef CAS PubMed.
- B. L. Barthel, R. C. Torres, J. L. Hyatt, C. C. Edwards, M. J. Hatfield, P. M. Potter and T. H. Koch, J. Med. Chem., 2008, 51, 298–304 CrossRef CAS PubMed.
- B. L. Barthel, Z. Zhang, D. L. Rudnicki, C. D. Coldren, M. Polinkovsky, H. Sun, G. G. Koch, D. C. F. Chan and T. H. Koch, J. Med. Chem., 2009, 52, 7678–7688 CrossRef CAS PubMed.
- V. Charasson, R. Bellott, D. Meynard, M. Longy, P. Gorry and J. Robert, Clin. Pharmacol. Ther., 2004, 76, 528–535 CrossRef CAS PubMed.
- K. Uchida, K. Otake, K. Tanaka, K. Hashimoto, S. Saigusa, K. Matsushita, Y. Koike, M. Inoue, M. Ueeda, Y. Okugawa, Y. Inoue, Y. Mohri and M. Kusunoki, J. Pediatr. Surg., 2013, 48, 502–509 CrossRef PubMed.
- C. Shaojun, H. Li, H. Haixin and L. Guisheng, Cancer Biol. Ther., 2018, 19, 153–159 CrossRef PubMed.
- K. Ohtsuka, S. Inoue, M. Kameyama, A. Kanetoshi, T. Fujimoto, K. Takaoka, Y. Araya and A. Shida, Lung Cancer, 2003, 41, 187–198 CrossRef PubMed.
- M. Capello, M. Lee, H. Wang, L. Babel, M. H. Katz, J. B. Fleming, A. Maitra, H. Wang, W. Tian, A. Taguchi and S. M. Hanash, J. Natl. Cancer Inst., 2015, 107, djv132 CrossRef PubMed.
- M. K. Ross, A. Boraziani, R. Wang, J. A. Crow and S. Xie, Arch. Biochem. Biophys., 2012, 522, 44–56 CrossRef CAS PubMed.
- Y. Sato, A. Miyashita, T. Iwatsubo and T. Usui, Drug Metab. Dispos., 2012, 40, 1389–1396 CrossRef CAS PubMed.
- S. K. Quinney, S. P. Sanghani, W. I. Davis, T. D. Hurley, Z. Sun, D. J. Murry and W. F. Bosron, J. Pharmacol. Exp. Ther., 2005, 313, 1011–1016 CrossRef CAS PubMed.
- H. J. Zhu, D. I. Appel, Y. Jiang and J. S. Markowitz, Drug Metab. Dispos., 2009, 37, 1819–1825 CrossRef CAS PubMed.
- J. Wang, E. T. Williams, J. Bourgea, Y. N. Wong and C. J. Patten, Drug Metab. Dispos., 2011, 39, 1329–1333 CrossRef CAS PubMed.
- J. M. Clarke, M. R. Gillings, N. Altavilla and A. J. Beattie, J. Microbiol. Methods, 2001, 46, 261–267 CrossRef CAS PubMed.
- L. Feng, Z.-M. Liu, L. Xu, J. Ning, J. Hou, G.-B. Ge, J.-N. Cui and L. Yang, Chem. Commun., 2014, 50, 14519–14522 RSC.
- Q. Jin, L. Feng, D.-D. Wang, J.-J. Wu, J. Hou, Z.-R. Dai, S.-G. Sun, J.-Y. Wang, G.-B. Ge, J.-N. Cui and L. Yang, Biosens. Bioelectron., 2016, 83, 193–199 CrossRef CAS PubMed.
- Z. M. Liu, L. Feng, J. Hou, X. Lv, J. Ning, G. B. Ge, K. W. Wang, J. N. Cui and L. Yang, Sens. Actuators, B, 2014, 205, 151–157 CrossRef CAS.
- S. J. Park, Y. J. Kim, J. S. Kang, L. Y. Kim, K. S. Choi and H. M. Kim, Anal. Chem., 2018, 90, 9465–9471 CrossRef CAS PubMed.
- Q. Jin, L. Feng, D. D. Wang, Z. R. Dai, P. Wang, L. W. Zou, Z. H. Liu, J. Y. Wang, Y. Yu, G. B. Ge, J.-N. Cui and L. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 28474–28481 CrossRef CAS PubMed.
- Z. M. Liu, L. Feng, G. B. Ge, X. Lv, J. Hou, Y. F. Cao, J. N. Cui and L. Yang, Biosens. Bioelectron., 2014, 57, 30–35 CrossRef CAS PubMed.
- S. J. Park, H. W. Lee, H. R. Kim, C. Kang and H. M. Kim, Chem. Sci., 2016, 7, 3703–3709 RSC.
- L. Feng, Z.-M. Liu, J. Hou, X. Lv, J. Ning, G. B. Ge, J. N. Cui and L. Yang, Biosens. Bioelectron., 2015, 65, 9–15 CrossRef CAS PubMed.
- T. Bezrodna, A. Negrviko, Y. Bezrodnyi and L. Kosyanchuk, J. Mol. Liq., 2018, 267, 89–95 CrossRef CAS.
- Y. N. Zhang, Y. A. Feng, Z. Li and X. S. Shao, Chin. Chem. Lett., 2017, 28, 1228–1231 CrossRef CAS.
- M. B. Freijo, A. López-Arencibia, J. E. Piñero, G. McNaughton-Smith and T. Abad-Grillo, Eur. J. Med. Chem., 2018, 143, 1312–1324 CrossRef CAS PubMed.
- G. Bousquet, J. P. Feugeas, L. Ferreira, L. Vercellino, N. Jourdan, P. Bertheau, C. de Bazelaire, E. Barranger and A. Janin, Breast Cancer Res., 2014, 16, 401 CrossRef PubMed.
- M. Hidalgo, E. Bruckheimer, N. V. Rajeshkumar, I. Garrido-Laguna, E. De Oliviera, B. Rubio-Viqueira, S. Strawn, M. J. Wick, J. Martell and D. Sidransky, Mol. Cancer Ther., 2011, 10, 1311–1316 CrossRef CAS PubMed.
- M. P. Morelli, E. Calvo, E. Ordoñez, M. J. Wick, B. R. Viqueira, P. P. Lopez-Casas, E. Bruckheimer, A. Calles-Blanco, D. Sidransky and M. Hidalgo, J. Clin. Oncol., 2012, 30, e45–e48 CrossRef CAS PubMed.
- K. Yoshioka, T. Komatsu, A. Nakada, J. Onagi, Y. Kuriki, M. Kawaguchi, T. Terai, T. Ueno, K. Hanaoka, T. Nagano and Y. Urano, J. Am. Chem. Soc., 2015, 137, 12187–12190 CrossRef CAS PubMed.
- Y. Wu, S. Huang, F. Zeng, J. Wang, C. Yu, J. Huang, H. Xie and S. Wu, Chem. Commun., 2015, 51, 12791–12794 RSC.
- Y. Zhang, W. Chen, D. Feng, W. Shi, X. Li and H. Ma, Analyst, 2012, 137, 716–721 RSC.
- D. Roife, B. Dai, Y. Kang, M. V. Rios Perez, M. Pratt, X. Li and J. B. Fleming, Clin. Cancer Res., 2016, 22, 6021–6030 CrossRef CAS PubMed.
- A. M. Brouwer, Pure Appl. Chem., 2011, 83, 2213–2228 CAS.
- K. Rurack, in Standardization and Quality Assurance in Fluorescence Measurements, ed. U. Resch-Genger, Springer-Verlag, Berlin Heidelburg, Germany, 2008, pp. 101–146 Search PubMed.
- M. P. Kim, D. B. Evans, H. Wang, J. L. Abbruzzese, J. B. Fleming and G. E. Gallick, Nat. Protoc., 2009, 4, 1670–1680 CrossRef CAS PubMed.
- M. P. Kim, M. J. Truty, W. Choi, Y. Kang, X. Chopin-Lally, G. E. Gallick, H. Wang, D. J. McConkey, R. Hwang, C. Logsdon, J. Abbruzzesse and J. B. Fleming, Ann. Surg. Oncol., 2012, 19, S395–S403 CrossRef PubMed.
- A. H. Fischer, K. A. Jacobson, J. Rose and R. Zeller, Cold Spring Harb. Protoc., 2008, prot4986 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc00283a |
| This journal is © The Royal Society of Chemistry 2019 |