
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesulfonative photoredox alkylation of N-heteroaryl sulfones – an acid-free approach for substituted heteroarene synthesis†
Zheng-Jun
Wang‡
 ab,
Shuai
Zheng‡
a,
Jennifer K.
Matsui
a,
Zhipeng
Lu
a and
Gary A.
Molander
*a
ab,
Shuai
Zheng‡
a,
Jennifer K.
Matsui
a,
Zhipeng
Lu
a and
Gary A.
Molander
*a
aRoy and Diana Vagelos Laboratories, Department of Chemistry, University of Pennsylvania, 231 South 34th Street, Philadelphia, Pennsylvania 19104-6323, USA. E-mail: gmolandr@sas.upenn.edu
bState Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, Hunan University, Changsha 41000, China
First published on 14th March 2019
Abstract
Minisci-type alkylation of electron-deficient heteroarenes has been a pivotal technique for medicinal chemists in the synthesis of drug-like molecules. However, such transformations usually require harsh conditions (e.g., strong acids, stoichiometric amount of oxidants, elevated temperatures, etc.). Herein, by utilizing photoredox catalysis, a highly-selective alkylation method using heteroaryl sulfones has been developed that can be carried out under acid-free and redox-neutral conditions. Because of these mild conditions, challenging yet privileged structures, such as monosaccharides and unprotected secondary amines, can be installed.
Introduction
N-Heteroarenes are among the most prevalent motifs in drug-like compounds.1 Notably, by 2014, over 84% of FDA-approved drugs contain at least one nitrogen, with 60% containing nitrogen heterocycles.2 Of the reported structures, pyrimidines are a prominent moiety, appearing in structures from nucleic acids to hundreds of bioactive molecules (Fig. 1).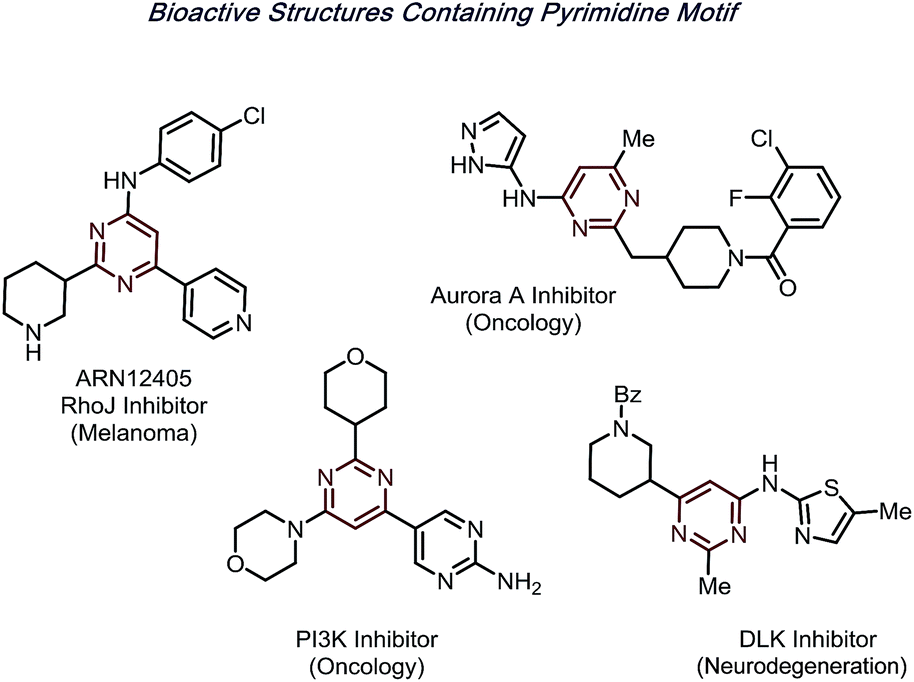 | ||
| Fig. 1 Bioactive molecules with pyrimidine cores. | ||
Significant progress has been made toward functionalization of such useful structures in the past few decades. One representative protocol is the Minisci reaction,3–5 wherein nucleophilic radicals attack activated, electron-deficient heterocycles. Due to the recent renaissance of radical chemistry, various radical precursors can now be generated selectively via photoredox6–10 or electrochemical catalysis,11–13 creating an expansive toolbox for building molecular complexity in synthetic chemistry.
Despite the significant advances,14–16 limitations are still present. Because of the requirement for harsh oxidants and elevated temperatures,5 Minisci reactions carried out via traditional approaches suffer from diminished regioselectivity and functional group tolerance, attributed to the electronics of the heteroarene. Recently, the employment of photoredox catalysis in the Minisci reaction has provided much milder conditions,15,17–23 thus significantly improving its utility. However, the inherent requirement for acidic conditions hampers functional group tolerance and thus the potential for late-stage functionalization.
A few efforts have been made to overcome these challenges, one of which being homolytic aromatic substitution (HAS) of pre-functionalized heteroarenes. Traditionally, this concept has been underdeveloped in alkylation reactions, with the few extant examples utilizing harsh conditions,24–26 or stoichiometric radical initiators (e.g., AIBN, Bu3SnH, etc.).27,28 In a seminal report by MacMillan and coworkers, α-aminoalkyl species were used in the generation of nucleophilic radicals for HAS of chloro-substituted heteroaromatic compounds.29 Although various heteroarenes served as suitable partners, acceptable selectivity was difficult to achieve when polychlorinated compounds were applied. Kamijo and co-workers reported an alternative strategy,30 wherein heteroaromatic sulfones were used. In this study, benzophenone acted as a photosensitizer to generate radicals from α-alkoxy- or α-aminoalkanes. Using this protocol, selective alkylation of sulfones was achieved in the presence of chlorides. Although readily available, UV light must be applied to excite benzophenone. Notably, in both of these studies, the reactions were limited to stabilized α-alkoxyalkyl- or α-aminoalkyl radicals. With this in mind, alkyl bis(catecholato)silicates7 and 4-alkyl-1,4-dihydropyridines (DHPs)8 were envisioned to serve as more versatile radical precursors under visible light photoredox conditions, with the potential to alkylate polysubstituted heteroaryl sulfones selectively (Scheme 1). Herein, an acid- and oxidant-free photoredox-mediated alkylation strategy of N-heteroaryl sulfones is described, which can be conducted with excellent selectivity and functional group tolerance.
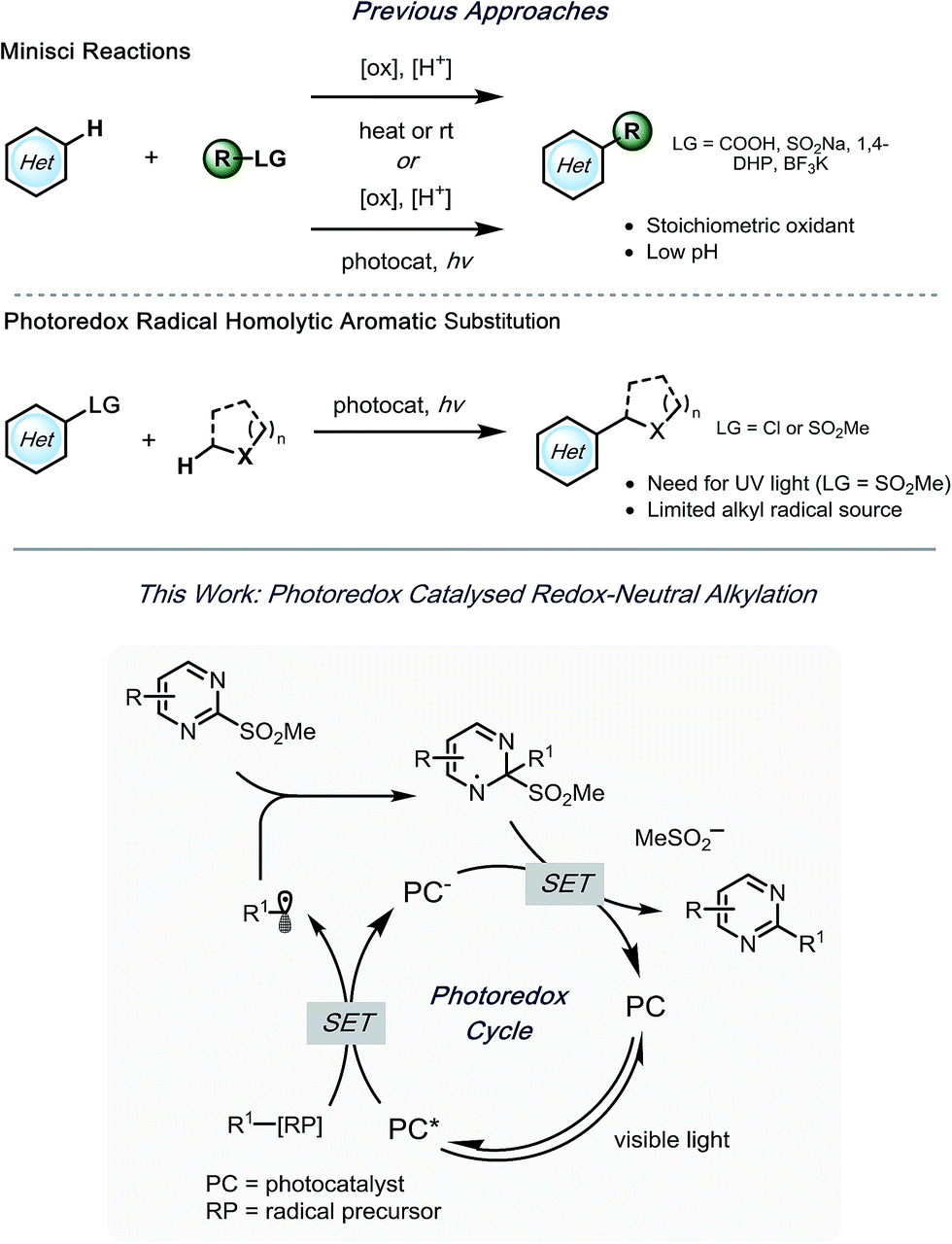 | ||
| Scheme 1 Reported precedents and envisioned transformation. | ||
Results and discussion
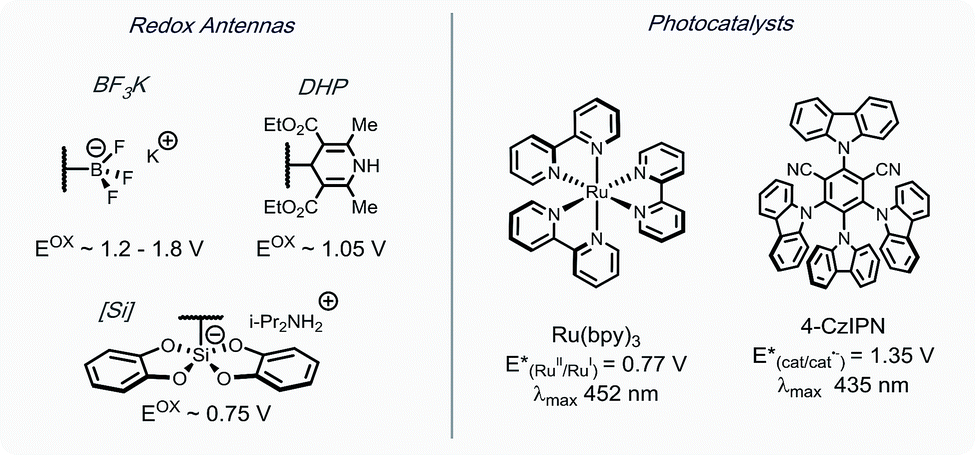
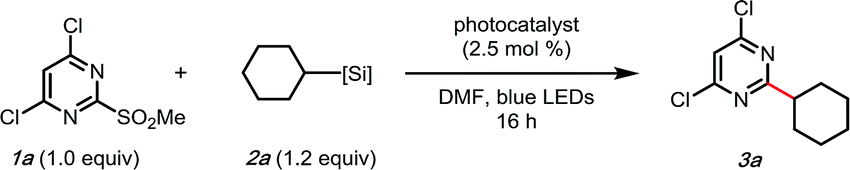
As outlined in Scheme 1, a single-electron transfer (SET) event was envisioned to generate alkyl radicals from the radical precursor. These radicals could engage heteroaromatic sulfones, with subsequent sulfinate extrusion leading to a recovery of aromaticity, simultaneously turning over the catalytic cycle by oxidation of the reduced photocatalyst. Because of their low oxidation potentials (Ered = +0.4 to 0.7 V vs. SCE),7 an investigation was undertaken using alkyl bis(catecholato)silicates as radical precursors. The initial attempt of using [Ru(bpy)3][PF6]2, whose photoexcited state is sufficient to oxidize (Ered = +0.77 V vs. SCE) alkylsilicates,7,31 provided a 78% isolated yield (entry 4, Table 1). In an attempt to achieve metal-free catalysis, we then investigated the applicability of different organophotocatalysts (entries 1–3, Table 1). Unfortunately, although the desired product was observed, the yield was not satisfactory. Various solvents were screened, and strong, polar aprotic solvents generally showed better activity, with DMF as the optimal solvent, while the diminished solubility of alkylsilicates in MeCN hampered the reaction (entries 4–6, Table 1).7,32 Finally, to examine the compatibility of different radical precursors, 4-cyclohexyl-1,4-dihydropyridine (DHP) and cyclohexyltrifluoroborate were also used under modified conditions (entries 7–8, Table 1).8,33 The alkyl DHP exhibited a comparable yield using the 4CzIPN organophotocatalyst (Ered = +1.35 V vs. SCE), while a much lower yield was observed with the RBF3K salt.|
|
|||
|---|---|---|---|
| Entry | Photocatalyst | Deviation from standard conditions | Yieldb (%) |
| a 1a (0.1 mmol, 1.0 equiv.), 2a, (0.12 mmol, 1.2 equiv.), photocatalyst (2.5 mol%), DMF (1 mL, 0.1 M) at rt under blue LED irradiation. b Isolated yield. c Acetone as the solvent. d MeCN as the solvent. | |||
| 1 | Eosin Y | None | 26 |
| 2 | 4CzIPN | None | 14 |
| 3 | Rhodamine 6G | None | 0 |
| 4 | [Ru(bpy) 3 ][PF 6 ] 2 | None | 78 |
| 5 | [Ru(bpy)3][PF6]2 | DMSO | 52 |
| 6 | [Ru(bpy)3][PF6]2 | MeCN | 0 |
| 7 | 4CzIPN | Cy-DHP instead of Cy-[Si] | 75 |
| 8 | 4CzIPN | Cy-BF3K instead of Cy-[Si]d | 15 |
| 9 | na | No photocatalyst | 0 |
| 10 | [Ru(bpy)3][PF6]2 | Open to air | 14 |
| 11 | [Ru(bpy)3][PF6]2 | No light | 24 |
|
|||
With suitable conditions in hand, the substrate scope was investigated. As shown in Table 2, various alkylsilicates were applied toward the alkylation of several heteroaryl sulfones. Both secondary (3a and 3b) and primary alkyl radicals (3c–h) were installed with moderate to good yields. Different functional groups, such as alkenes (3d), esters (3e), perfluoroethers (3f, 3j, and 3m) and an alkyl chloride (3i) were accommodated. Notably, an unprotected secondary amine survived the reaction with a relatively good yield (3g), which would be otherwise challenging under previously reported Minisci conditions.15 The electron-rich pyrrole group (3h) was challenging to install, likely because of its propensity to polymerize,32 but the desired product was still isolated, albeit with a compromised yield.
|
|
|---|
| a Reaction conditions: alkylsilicate (0.6 mmol, 1.2 equiv.), heteroaryl sulfone (0.5 mmol, 1.0 equiv.) [Ru(bpy)3][PF6]2 (0.0125 mmol, 2.5 mol%), DMF (5 mL, 0.1 M). |

|
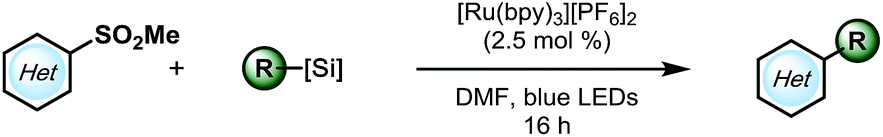
Challenges emerged in moving to the scope of heteroaryl methyl sulfones. Different heteroaromatic ring systems (3l and 3m), and different substitution patterns, such as mono-chloro substitution, 4-substituted sulfones (3j), and mono-methoxy substitution (3k), are tolerable, but the yields were heavily compromised. Even though diminished SNAr reactivity would be expected with more electron-rich systems, previous reports have shown that little activation of the pyrimidyl backbone is required. This led to a hypothesis that it might be the stability of the alkylsilicates that caused the problem.30
With a higher oxidation potential (∼+1.05 V vs. SCE)8 and better stability, alkyl 1,4-dihydropyridines (DHPs) seemed to be a promising potential solution to this problem. Indeed, our initial screening had shown only a slightly lower yield using these partners (Table 1, entry 7). Moreover, with the application of the DHP derivatives, the inexpensive organophotocatalyst, 4CzIPN, could be used. Furthermore, the DHP partners are derived from commodity chemicals (alkyl aldehydes, primary alcohols) that are much more abundant than the silicates, making this transformation infinitely more appealing from the viewpoint of structural diversity. In the event, using cyclohexyl DHP (5a) under optimal conditions, the desired product was isolated in a slightly higher yield than that with the corresponding silicate (3a).
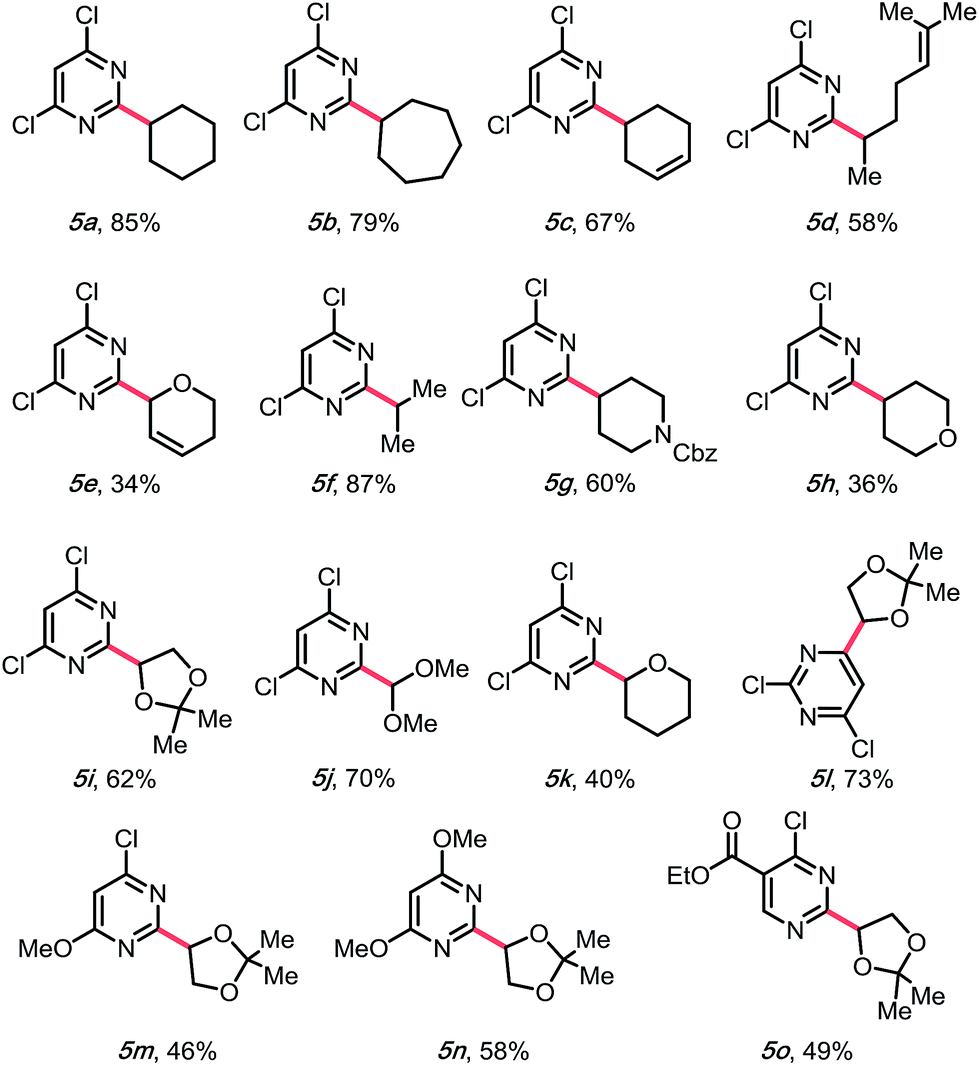
Encouraged by this result, we moved on to explore the scope with diverse DHPs (Table 3). Fortunately, a general improvement of yield was observed, with transformations ranging from 34% to 87%. In addition to their compatibility with olefins (5c–e), various nitrogen- or oxygen-containing saturated heterocycles were installed, including dioxolanes (5i and 5l–5o), dihydro- and tetrahydropyrans (5e, 5h, and 5k), and a protected piperidine (5g), which are of interest in medicinal chemistry. Notably, the dimethoxymethylene group was also compatible (5j), providing a 70% yield, rendering a convenient way of installing an aldehyde on the pyrimidyl backbone. Of note, various substitution patterns on the pyrimidyl backbones, including 6-sulfonyl (5l), mono- and di-methoxy (5m, 5n) and ethyl ester (5o) have been achieved with yields ranging from 46 to 73%. With multiple functionalization handles on the products, the substrates would appear valuable for rapid synthesis of diverse heteroaromatic structures.
|
|
|---|
| a Reaction conditions: 4-alkyl-1,4-dihydropyridine (0.6 mmol, 1.2 equiv.), heteroaryl sulfone (0.5 mmol, 1.0 equiv.), 2,4,5,6-tetra-(9H-carbazol-9-yl)isophthalonitrile (4CzIPN) (0.0125 mmol, 2.5 mol%), acetone (5 mL, 0.1 M). |
|
To demonstrate the utility of the developed method further, we attempted to apply a saccharide-derived DHP to this transformation, as these scaffolds are of some value in drug discovery (Scheme 2).34–37 Gratifyingly, not only could 5p be prepared in good yield (54%, Scheme 2), but the diastereoselectivity was also excellent (dr > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). It is noteworthy that when a previously developed Minisci-type transformation introduced by our group was used to access this target structure,14 no desired product was observed.
:1). It is noteworthy that when a previously developed Minisci-type transformation introduced by our group was used to access this target structure,14 no desired product was observed.
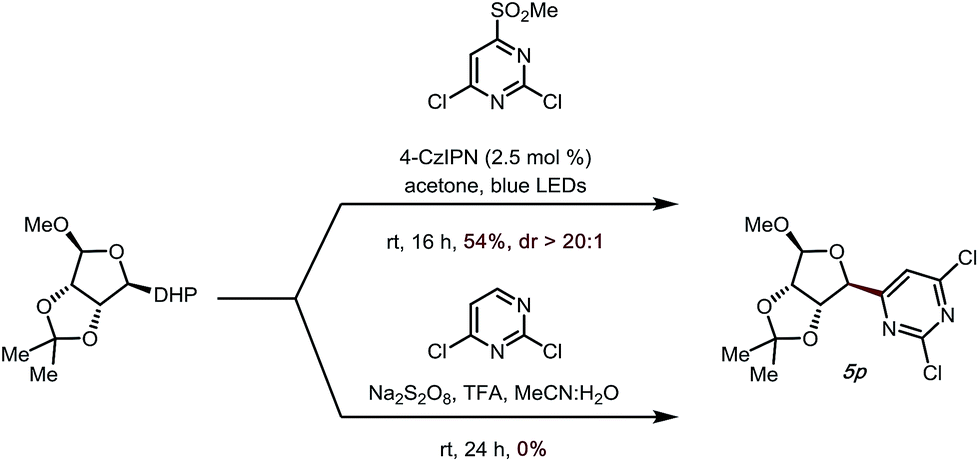 | ||
| Scheme 2 Heteroaryl alkylation with sugar DHPs. | ||
Several investigations support the proposed mechanistic reaction pathway. Control experiments (entries 9–11, Table 1) showed the necessity of light and photocatalyst. Stern–Volmer fluorescence quenching studies of 4CzIPN showed no quenching with 4,6-dichloro-2-(methylsulfonyl)pyrimidine, while an efficient dynamic quenching was observed using cyclohexyl DHP as the quencher, with KSV = (9.3 ± 0.6) × 102 M−1, largely disfavoring the possibility of a photoactivating sulfone species. To further confirm the unlikelihood of heteroaryl sulfone reduction, we carried out a cyclic voltammetry study of 4,6-dichloro-2-(methylsulfonyl)pyrimidine. The measured potential was −1.68 V vs. SCE (see ESI†), implying that the formation of alkyl radical via oxidation of the alkyl DHP was significantly more favorable.
Conclusions
In conclusion, a selective photoredox-catalyzed alkylation reaction of N-heteroaryl sulfones has been developed. To highlight the advantages of the acid- and oxidant-free conditions, several challenging motifs have been installed on the medicinally valuable heteroaromatic systems – in particular, monosaccharide motifs and unprotected amines. With excellent selectivity and several functionalization handles accommodated on the backbone, this method may be of value in accelerating the synthesis of drug-like molecules.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors are grateful for the financial support provided by NIGMS (R01 GM 113878). We thank the NIH (S10 OD011980) for supporting the University of Pennsylvania (UPenn) Merck Center for High Throughput Experimentation, which funded the equipment used in screening efforts. Z.-J. Wang acknowledges the financial support from the Chinese Scholarship Council. J. K. M. was supported by a Bristol-Myers Squibb Graduate Fellowship. We thank Dr Charles W. Ross, III (UPenn) for help in HRMS data acquisition.Notes and references
- M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Chem. Biol., 2010, 14, 347–361 CrossRef CAS PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- F. Minisci, R. Bernardi, F. Bertini, R. Galli and M. Perchinummo, Tetrahedron, 1971, 27, 3575–3579 CrossRef CAS.
- F. Minisci, E. Vismara and F. Fontana, J. Org. Chem., 1989, 54, 5224–5227 CrossRef CAS.
- M. A. J. Duncton, MedChemComm, 2011, 2, 1135–1161 RSC.
- J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433–436 CrossRef CAS PubMed.
- M. Jouffroy, D. N. Primer and G. A. Molander, J. Am. Chem. Soc., 2016, 138, 475–478 CrossRef CAS PubMed.
- Á. Gutiérrez-Bonet, J. C. Tellis, J. K. Matsui, B. A. Vara and G. A. Molander, ACS Catal., 2016, 6, 8004–8008 CrossRef PubMed.
- J. K. Matsui, S. B. Lang, D. R. Heitz and G. A. Molander, ACS Catal., 2017, 7, 2563–2575 CrossRef CAS PubMed.
- J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Acc. Chem. Res., 2016, 49, 1911–1923 CrossRef CAS PubMed.
- A. G. O'Brien, A. Maruyama, Y. Inokuma, M. Fujita, P. S. Baran and D. G. Blackmond, Angew. Chem., Int. Ed., 2014, 53, 11868–11871 CrossRef PubMed.
- Q.-Q. Wang, K. Xu, Y.-Y. Jiang, Y.-G. Liu, B.-G. Sun and C.-C. Zeng, Org. Lett., 2017, 19, 5517–5520 CrossRef CAS PubMed.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS.
- G. A. Molander, V. Colombel and V. A. Braz, Org. Lett., 2011, 13, 1852–1855 CrossRef CAS PubMed.
- J. K. Matsui, D. N. Primer and G. A. Molander, Chem. Sci., 2017, 8, 3512–3522 RSC.
- Á. Gutiérrez-Bonet, C. Remeur, J. K. Matsui and G. A. Molander, J. Am. Chem. Soc., 2017, 139, 12251–12258 CrossRef PubMed.
- G.-X. Li, C. A. Morales-Rivera, Y. Wang, F. Gao, G. He, P. Liu and G. Chen, Chem. Sci., 2016, 7, 6407–6412 RSC.
- W.-M. Cheng, R. Shang and Y. Fu, ACS Catal., 2017, 7, 907–911 CrossRef CAS.
- T. McCallum and L. Barriault, Chem. Sci., 2016, 7, 4754–4758 RSC.
- D. A. DiRocco, K. Dykstra, S. Krska, P. Vachal, D. V. Conway and M. Tudge, Angew. Chem., Int. Ed., 2014, 53, 4802–4806 CrossRef CAS PubMed.
- J. Jin and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2015, 54, 1565–1569 CrossRef CAS PubMed.
- J. Jin and D. W. C. MacMillan, Nature, 2015, 525, 87–90 CrossRef CAS PubMed.
- D. A. Nagib and D. W. C. MacMillan, Nature, 2011, 480, 224–228 CrossRef CAS PubMed.
- A. Shifman, N. Palani and S. Hoz, Angew. Chem., Int. Ed., 2000, 39, 944–945 CrossRef CAS PubMed.
- N. Palani, K. Jayaprakash and S. Hoz, J. Org. Chem., 2003, 68, 4388–4391 CrossRef CAS PubMed.
- Z. Zhang, Z. Yin, J. F. Kadow, N. A. Meanwell and T. Wang, J. Org. Chem., 2004, 69, 1360–1363 CrossRef CAS PubMed.
- W. R. Bowman and J. M. D. Storey, Chem. Soc. Rev., 2007, 36, 1803–1822 RSC.
- M. Gurry and F. Aldabbagh, Org. Biomol. Chem., 2016, 14, 3849–3862 RSC.
- C. K. Prier and D. W. C. MacMillan, Chem. Sci., 2014, 5, 4173–4178 RSC.
- S. Kamijo, K. Kamijo and T. Murafuji, J. Org. Chem., 2017, 82, 2664–2671 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- S. Zheng, D. N. Primer and G. A. Molander, ACS Catal., 2017, 7, 7957–7961 CrossRef CAS PubMed.
- D. N. Primer, I. Karakaya, J. C. Tellis and G. A. Molander, J. Am. Chem. Soc., 2015, 137, 2195–2198 CrossRef CAS PubMed.
- T. Bililign, B. R. Griffith and J. S. Thorson, Nat. Prod. Rep., 2005, 22, 742–760 RSC.
- R. W. Gantt, P. Peltier-Pain and J. S. Thorson, Nat. Prod. Rep., 2011, 28, 1811–1853 RSC.
- É. Bokor, S. Kun, D. Goyard, M. Tóth, J.-P. Praly, S. Vidal and L. Somsák, Chem. Rev., 2017, 117, 1687–1764 CrossRef PubMed.
- A. Doumoulin, J. K. Matsui, Á. Gutiérrez-Bonet and G. A. Molander, Angew. Chem., Int. Ed., 2018, 57, 6614–6618 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc00776h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |