
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe rapid microwave-assisted hydrothermal synthesis of NASICON-structured Na3V2O2x(PO4)2F3−2x (0 < x ≤ 1) cathode materials for Na-ion batteries†
Daria Burovaa,
Iaroslava Shakhova *a,
Polina Morozovaa,
Anna Iarchuka,
Oleg A. Drozhzhinab,
Marina G. Rozovab,
S. Praneethac,
Vadivel Muruganc,
Jean-Marie Tarascond and
Artem M. Abakumova
*a,
Polina Morozovaa,
Anna Iarchuka,
Oleg A. Drozhzhinab,
Marina G. Rozovab,
S. Praneethac,
Vadivel Muruganc,
Jean-Marie Tarascond and
Artem M. Abakumova
aCenter for Energy Science and Technology, Skolkovo Institute of Science and Technology, Nobel Str. 3, 143026 Moscow, Russia. E-mail: y.shakhova@skoltech.ru; Tel: +7 495 280 14 81 ext. 3244
bChemical Department, Moscow State University, Leninskie Gory, 1, 119234 Moscow, Russia
cAdvanced Functional Nanostructured Materials Laboratory, Centre for Nanoscience and Technology, Madanjeet School of Green Energy Technologies, Pondicherry University (A Central University), Kalapet, Puducherry 605014, India
dCollège de France, Chimie du Solide et de l'Energie, UMR 8260, 11 Place Marcelin Berthelot, 75231 Paris Cedex 05, France
First published on 20th June 2019
Abstract
NASICON-structured Na3V2O2x(PO4)2F3−2x (0 < x ≤ 1) solid solutions have been prepared using a microwave-assisted hydrothermal (MW-HT) technique. Well-crystallized phases were obtained for x = 1 and 0.4 by reacting V2O5, NH4H2PO4, and NaF precursors at temperatures as low as 180–200 °C for less than 15 min. Various available and inexpensive reducing agents were used to control the vanadium oxidation state and final product morphology. The vanadium oxidation state and O/F ratios were assessed using electron energy loss spectroscopy and infrared spectroscopy. According to electron diffraction and powder X-ray diffraction, the Na3V2O2x(PO4)2F3−2x solid solutions crystallized in a metastable disordered I4/mmm structure (a = 6.38643(4) Å, c = 10.62375(8) Å for Na3V2O2(PO4)2F and a = 6.39455(5) Å, c = 10.6988(2) Å for Na3V2O0.8(PO4)2F2.2). With respect to electrochemical Na+ (de)insertion as positive electrodes (cathodes) for Na-ion batteries, the as-synthesized materials displayed two sloping plateaus upon charge and discharge, centered near 3.5–3.6 V and 4.0–4.1 V vs. Na+/Na, respectively, with a reversible capacity of ∼110 mA h g−1. The application of a conducting carbon coating through the surface polymerization of dopamine with subsequent annealing at 500 °C improved both the rate capability (∼55 mA h g−1 at a discharge rate of 10C) and capacity retention (∼93% after 50 cycles at a discharge rate of C/2).
Introduction
The rapid growth in global interest in renewable and sustainable energy, including solar, wind, and tidal power, has promoted research on high-performance, environmentally friendly, and cost-effective electrical energy storage systems.1 Na-ion batteries are being considered as good candidates for a new generation of rechargeable energy storage devices owing to the natural abundance of sodium in the earth's crust compared with lithium resources, which are scarce and distributed non-uniformly worldwide. Therefore, although Na-ion batteries cannot yet compete with the already mature Li-ion technology in terms of energy density and power, they are promising cost-wise.2 Materials with the NASICON-type structure are known for their high Na+ ionic conductivity, and are undergoing extensive study as positive electrodes (cathodes) for Na(Li)-ion batteries. Among NASICONs, Na3V2O2x(PO4)2F3−2x (0 ≤ x ≤ 1) solid solutions have demonstrated high energy densities of ∼350 W h kg−1.3,4 Na3V2O2x(PO4)2F3−2x can be synthesized using various methods, including solid state reactions from VPO4 and VOPO4 precursors,5–7 and carbothermal,8,9 sol–gel,10 and spray-drying11 techniques. However, in all cases, high-temperature treatment at 650–800 °C under an inert gas flow for between several hours and 24 h is needed. Hydrothermal/solvothermal synthesis is an energy-saving alternative to high-temperature annealing, allowing the reaction to be conducted under significantly milder conditions (<200 °C). This method has been used to prepare Na3V2O2x(PO4)2F3−2x from VPO4 as precursor, which was obtained from a conventional high temperature solid state reaction.12 Therefore, energy-intensive high-temperature treatment cannot be avoided. The one-step hydrothermal synthesis of Na3V2O2x(PO4)2F3−2x has been realized at temperatures as low as 120–170 °C,4,13–17 but with the disadvantages of requiring long treatment durations (9–60 h) and/or expensive and poorly accessible reagents. As the cathode material contributes to up to 30% of the total cell cost,2 developing sustainable resource- and energy-saving green processes for cathode material production is vitally important for making metal-ion battery technology commercially competitive.18 This inspired us to explore the microwave-assisted hydrothermal (MW-HT) synthesis of electrode materials. Microwave radiation, which directly transfers energy to the reacting species, can shorten reaction processes from several hours to several minutes, making this technique among the most energy- and cost-effective.19–21 Herein, we report that Na3V2O2x(PO4)2F3−2x solid solutions with different O/F ratio can be prepared rapidly (<15 min) in a single step at low temperatures (180–220 °C) from inexpensive and easily available precursor materials based on pentavalent vanadium, and appropriately selected reducing agents, using the MW-HT technique. We also show that selecting an appropriate reducing agent can be used to tune the V oxidation state and O/F ratio in the final product, along with the morphology, to obtain Na3V2O2x(PO4)2F3−2x powders with a sustained reversible capacity of 110 mA h g−1.Experimental
Commercially available V2O5 (Sigma Aldrich), NaF (RusKhim), and NH4H2PO4 (Sigma Aldrich) were used as V, Na, F, and P sources. As V(V) had to be reduced to V(III)/V(IV) during the synthesis, different reducing agents were tested, namely, oxalic acid (C2H2O4, RusKhim), citric acid (C6H8O7, RusKhim), hydrazine chloride (N2H4·HCl, RusKhim), hydrazine sulfate (N2H4·H2SO4, RusKhim), and sodium borohydride (NaBH4, Sigma Aldrich). Initial materials V2O5, NaF, and NH4H2PO4 were mixed in stoichiometric amounts, and reducing agent was added in double excess with respect to V2O5 to reduce all V(V) to V(III) (Table S1†). Detailed synthesis conditions are listed in Table S1.† Next, deionized water (10 mL) was added to the initial precursors and the solution was magnetically stirred at 65–70 °C for 10–20 min while adding NH3·H2O (1 mL, 25 wt%). A 5 mL aliquot of the obtained homogeneous solution was transferred to a 10 mL glass vessel and placed into a microwave hydrothermal reactor (Anton Paar 400) for treatment under the conditions listed in Table S1† with constant stirring. Scale-up of the microwave synthesis was conducted using an Anton Paar Multiwave PRO reactor (Pondicherry University, India). The V, Na, F, and P sources were mixed in stoichiometric amounts and oxalic acid was added in double excess with respect to V2O5. The overall precursor concentration was increased by 25%. Deionized water (20 mL) was added to the precursor mixture followed by magnetic stirring at 65–70 °C for 10–20 min while adding NH3·H2O (2 mL, 25 wt%). The homogeneous solution was divided into two portions (11 mL each), transferred into two 60 mL glass vessels, placed in the reactor, and treated under the conditions listed in Table S1† (sample #6). The final solid products were centrifuged and washed with deionized water several times and air-dried prior to characterization of the phase purity and composition.To obtain a carbon coating, the material synthesized with oxalic acid as reducing agent was mixed with glucose (8 wt%) in a mortar. The mixture was annealed at 600 °C during 1 h under argon flow. In another carbon-coating procedure, a suspension of Na3V2(PO4)2O2F (412 mg) in Tris–HCl buffer (10 mL, pH 8.5) was mixed with dopamine (DOPA) hydrochloride (10 wt%). The obtained solution was stirred for 24 h followed by removal of the Na3V2(PO4)2O2F/DOPA precipitate by centrifugation. The precipitate was washed with deionized water several times and dried at 75 °C for 24 h. DOPA-coated Na3V2(PO4)2O2F particles were annealed at 500 °C for 3 h under an argon atmosphere to achieve polydopamine carbonization.
The phase purity was determined by X-ray powder diffraction (XRPD) using a Huber G670 Guinier camera (CoKα1 radiation (λ = 1.78892 Å), curved Ge(111) monochromator, image plate detector) and Rigaku Ultima IV diffractometer (radiation CuKα1 (λ = 1.54056 Å), CuKα2 (λ = 1.54433 Å)). PXRD data for the Rietveld refinement were collected on a STOE STADI-P diffractometer (CoKα1 radiation (λ = 1.78892 Å), curved Ge(111) monochromator, linear PSD) and a Huber G670 Guinier camera. The LeBail profile fitting and Rietveld refinement were performed using JANA2006 software.22
The particle morphology was investigated using a Helios NanoLab 660 scanning electron microscope equipped with an EDAX energy dispersive X-ray (EDX) spectrometer. Electron diffraction (ED) patterns were obtained using an FEI Tecnai G2 transmission electron microscope operating at 200 kV. Electron energy loss spectroscopy (EELS) was performed in scanning transmission (STEM) mode using an FEI Titan G3 transmission electron microscope operated at 120 kV and equipped with a monochromator and Gatan Enfinium ER spectrometer. The energy resolution, measured from the full width at half maximum of the zero loss peak, was 0.175 eV. Samples for TEM investigation were prepared by crushing and mixing the powder with ethanol, followed by deposition of the suspension onto a Cu-supported holey carbon grid.
FT-IR spectra (average of 64 scans with 4 cm−1 resolution) were obtained using a standalone FT-IR microscope LUMOS (Bruker) equipped with a liquid-N2-cooled MCT detector. A ZnSe disk was used as the substrate for powder samples. Raman spectra were recorded using a DXRxi Raman Imaging Microscope (Thermo Scientific) equipped with a 532 nm laser (power at sample plane, 1 mW) and full-range grating (range, 75–3400 cm−1; spectral dispersion, 2 cm−1).
Electrochemical measurements were conducted in two-electrode cells using a potentiostat-galvanostat (Biologic VMP-3, EC-Lab software) at room temperature. Galvanostatic cycling was performed in the potential range 2.5–4.3 V vs. Na/Na+ at different scanning rates (C/10, C/3, 1C, 3C, and 10C). The working electrode was prepared by mixing active material Na3V2O2x(PO4)2F3−2x with conductive carbon black Super-P and polyvinylidene fluoride (PVDF) as binder in a 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15:15 weight ratio. First, carbon black and the active material were mixed in a mortar with a small amount of acetone to obtain better homogeneity. PVDF was then mixed rapidly with N-methyl-2-pyrrolidone and the dry mixture of active material and carbon super black was added to form the final slurry. This slurry was applied to Al foil using the “doctor blade” technique with a thickness of 150 μm. The obtained electrodes were rolled at room temperature, punched into disks with a diameter of 16 mm and dried at 100 °C for 8 h under dynamic vacuum. Two-electrode cells were assembled in an MBraun glovebox under an Ar atmosphere. Metallic Na was used as a negative electrode and was separated from the Na3V2O2x(PO4)2F3−2x positive electrode by borosilicate glass fiber soaked with electrolyte. The electrolytes used were 1 M NaClO4 (NaPF6) solutions in (a) propylene carbonate (PC), (b) a mixture of propylene carbonate and ethylene carbonate (PC:EC), (c) a mixture of propylene carbonate ethylene carbonate and fluoroethylene carbonate (5 wt%) (PC:EC:FEC), and (d) a mixture of ethylene carbonate and diethyl carbonate (EC:DEC) with a 1:1 weight ratio.
:15:15 weight ratio. First, carbon black and the active material were mixed in a mortar with a small amount of acetone to obtain better homogeneity. PVDF was then mixed rapidly with N-methyl-2-pyrrolidone and the dry mixture of active material and carbon super black was added to form the final slurry. This slurry was applied to Al foil using the “doctor blade” technique with a thickness of 150 μm. The obtained electrodes were rolled at room temperature, punched into disks with a diameter of 16 mm and dried at 100 °C for 8 h under dynamic vacuum. Two-electrode cells were assembled in an MBraun glovebox under an Ar atmosphere. Metallic Na was used as a negative electrode and was separated from the Na3V2O2x(PO4)2F3−2x positive electrode by borosilicate glass fiber soaked with electrolyte. The electrolytes used were 1 M NaClO4 (NaPF6) solutions in (a) propylene carbonate (PC), (b) a mixture of propylene carbonate and ethylene carbonate (PC:EC), (c) a mixture of propylene carbonate ethylene carbonate and fluoroethylene carbonate (5 wt%) (PC:EC:FEC), and (d) a mixture of ethylene carbonate and diethyl carbonate (EC:DEC) with a 1:1 weight ratio.
Synchrotron X-ray powder diffraction (SXPD) in the operando regime was performed at Swiss Norwegian Beamlines (SNBL) BM01 and BM31 of the European Synchrotron Radiation Facility (ESRF, Grenoble, France). The original electrochemical cell with single-crystal sapphire X-ray windows was used.23 Experiments were conducted in a low-intensity beam mode (∼40 mA, 4 × 10 filling mode). The PILATUS@SNBL diffractometer was used for SXPD studies (wavelength, 0.68987 Å).24 2D diffraction data from the Pilatus 2 M detector were processed using the SNBL Toolbox and BUBBLE software. The time of data acquisition was 10 s per pattern. Patterns were refined using FullProf software.25
Results and discussion
Synthesis and crystal structures
Samples obtained with oxalic acid or equimolar mixtures of oxalic acid with hydrazine sulfate, citric acid, and sodium borohydride as reducing agents were light-green powders containing a well-crystalized single phase, according to PXRD. Regardless of the reducing agent used, the Bragg peaks for the solid products were indexed to a body-centered tetragonal lattice with nearly identical unit cell parameters of a = 6.3915(2) Å and c = 10.6289(5) Å (see Table S1,† ESI†) and unit cell volumes (434.21(4)–435.06(1) Å3, except for samples #11 and #12 synthesized with the hydrazine chloride–oxalic acid mixture) indicating that the chemical composition of the product remained almost identical. These unit cell parameters were typical of Na3V2O2(PO4)2F with the NASICON-type structure.6,13,16,26,27 Nevertheless, other studies have reported the primitive aP = aI√2, cP = cI tetragonal supercell for Na3V2O2(PO4)2F,5,12,15 and a more systematic crystallographic investigation revealed that the primitive supercell was indeed stable at room temperature, while the smaller I-centered tetragonal subcell was characteristic of the high-temperature polymorph of Na3V2O2(PO4)2F (stable above ∼500 K).28 To verify the unit cell of the Na3V2O2(PO4)2F phase prepared by the MW-HT method, we performed electron diffraction analysis. All reflections in the ED patterns (Fig. 1) were indexed to the unit cell determined from the PXRD data and the hkl: h + k + l = 2n reflection condition corroborated the I4/mmm space group.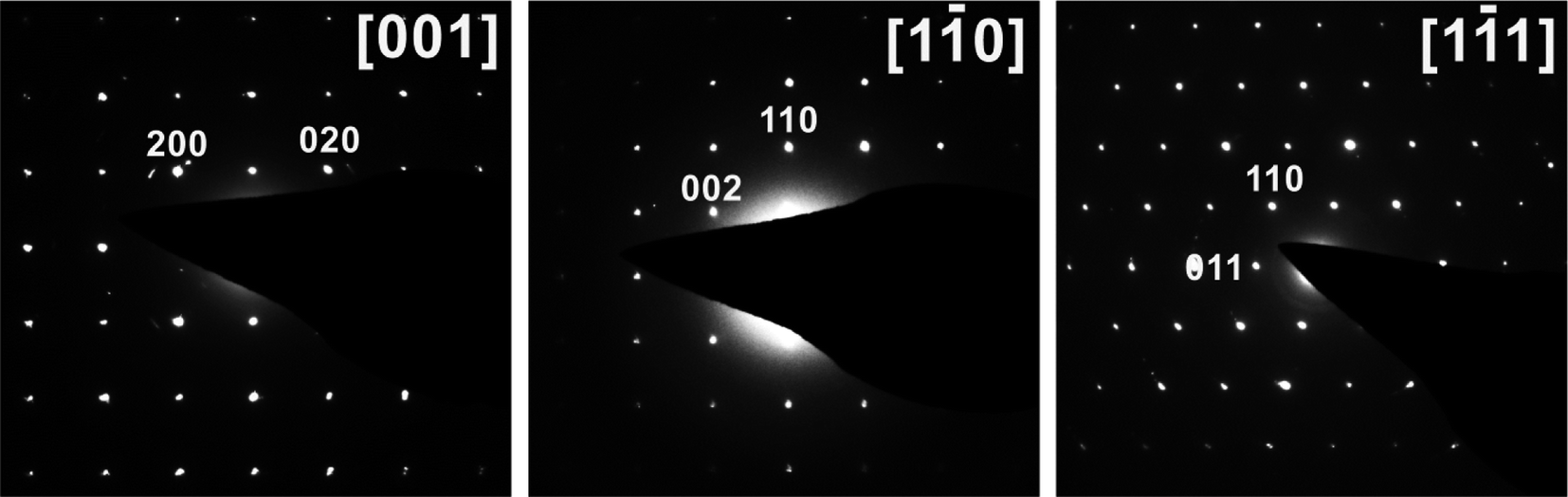 | ||
| Fig. 1 SAED patterns of Na3V2O2(PO4)2F synthesized with oxalic acid indexed on an I-centered tetragonal lattice. | ||
Therefore, the structure of the high-temperature Na3V2O2(PO4)2F phase was selected as the initial structure model for the Rietveld refinement from the PXRD data. This implied pronounced static disorder of the Na atoms over the 8h and 16l positions of the I4/mmm space group. The independent refinement of the occupancy factors for these two positions resulted in a phase composition of Na3.02(7)V2O2(PO4)2F, and the final refinement was performed by restricting the sum of occupancy factors to be equal to 3Na atoms per formula unit. The Na3.02(7)V2O2(PO4)2F structure was registered in the Cambridge Crystallographic Data Centre (CCDC) with CSD 1904096. The refinement parameters, atomic coordinates and main interatomic distances are listed in Tables S2, S3 and S4†, respectively. The experimental, calculated, and difference PXRD profiles are shown in Fig. 2. Notably, carbon coating of the pristine Na3V2O2(PO4)2F phase by dopamine did not affect the crystal structure, and the PXRD pattern of the coated material was easily indexed to the I4/mmm space group (Fig. S1†).
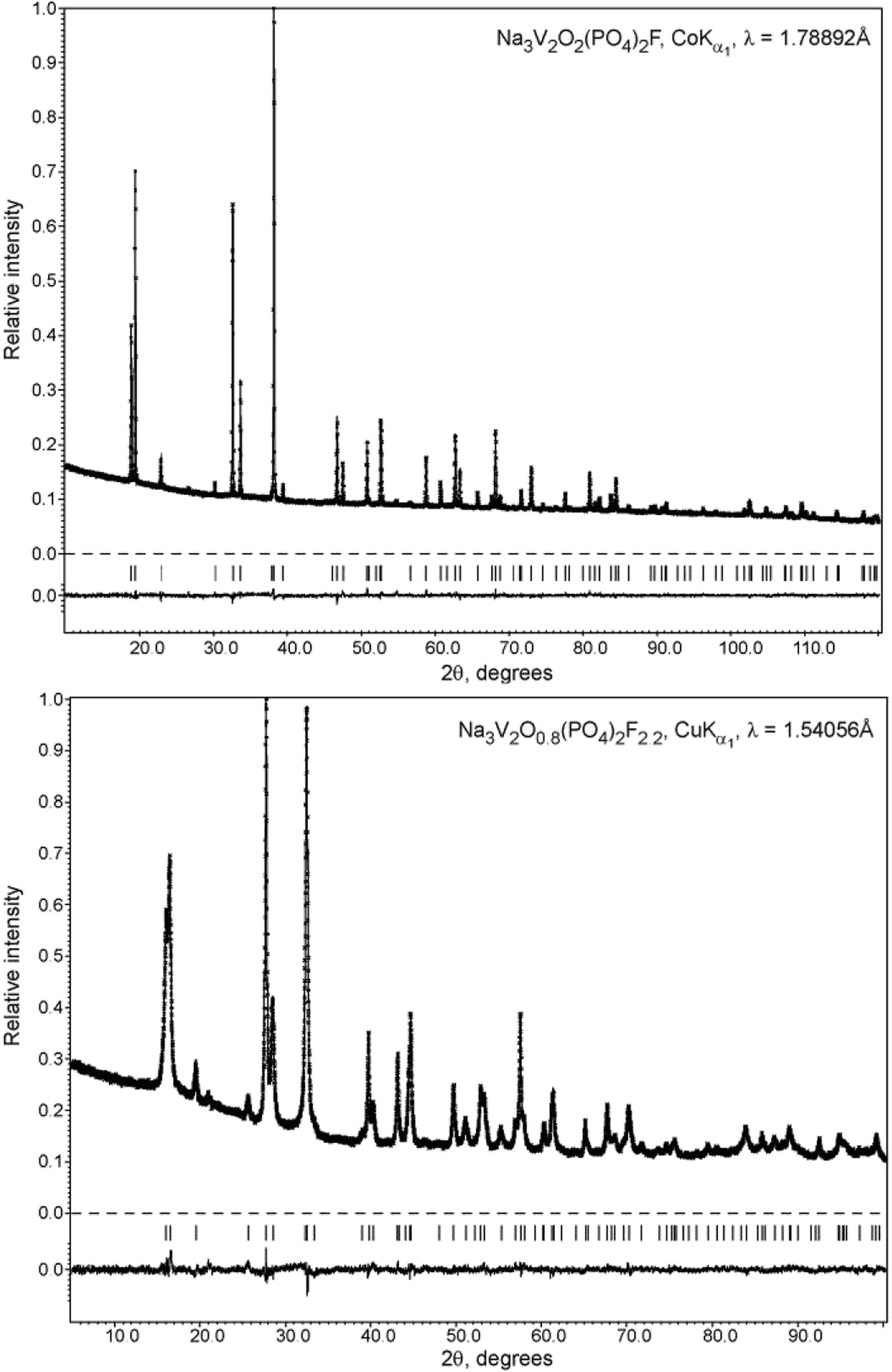 | ||
| Fig. 2 Experimental, calculated, and difference PXRD profiles for Na3V2O2(PO4)2F and Na3V2O0.8(PO4)2F2.2 after Rietveld refinement. | ||
The coordination environment of vanadium atoms in the refined Na3V2O2(PO4)2F structure indicated that V was in the +4 oxidation state. The VO5F octahedron was formed by four long equatorial V–O1 bonds (2.002 Å), one long apical V–F1 bond (2.119 Å), and one very short apical V–O2 bond (1.624 Å). The length of this short V–O2 separation was typical of so-called “vanadyl” bonds intrinsic to V4+ cations in octahedral coordination environments.29 Indeed, bond valence sum (BVS) calculations provided a formal vanadium oxidation state of +4.04(2) (Table S4†), which confirmed the O/F ratio in the Na3V2O2(PO4)2F formula. In addition to the BVS method, electron energy loss spectroscopy (EELS) was used to estimate the vanadium oxidation state. The quantitative approach based on the L3/L2 intensity ratio of the V core loss edge for materials with V and O was hampered by overlapping of the O–K edge (532 eV) with the continuum region of the V-L2,3 edges (521 and 513 eV, respectively).30 Therefore, we restricted the analysis to a comparison with the EELS spectra of vanadium oxo- and fluorophosphates KVOPO4 (V4+) and KVPO4F (V3+) with crystal chemistry similar to that of the Na3V2O2x(PO4)2F3−2x solid solutions (namely, the octahedral coordination environment for V and part of O linked into PO4 tetrahedra).31,32 The typical V-L2,3 edge in Na3V2O2(PO4)2F closely resembled the V-L2,3 edge in KVOPO4 (Fig. 3), confirming that the vast majority of V was in the +4 oxidation state, although the contribution to the L3 edge peak at ∼517.8 eV indicated that fractional V3+ was also present. Spatial STEM-EELS mapping of V4+ and V3+ showed that the interior part of the crystallites comprised V4+ (in agreement with the Rietveld refinement results), while the surface was more reduced and enriched with V3+ (Fig. S2†).
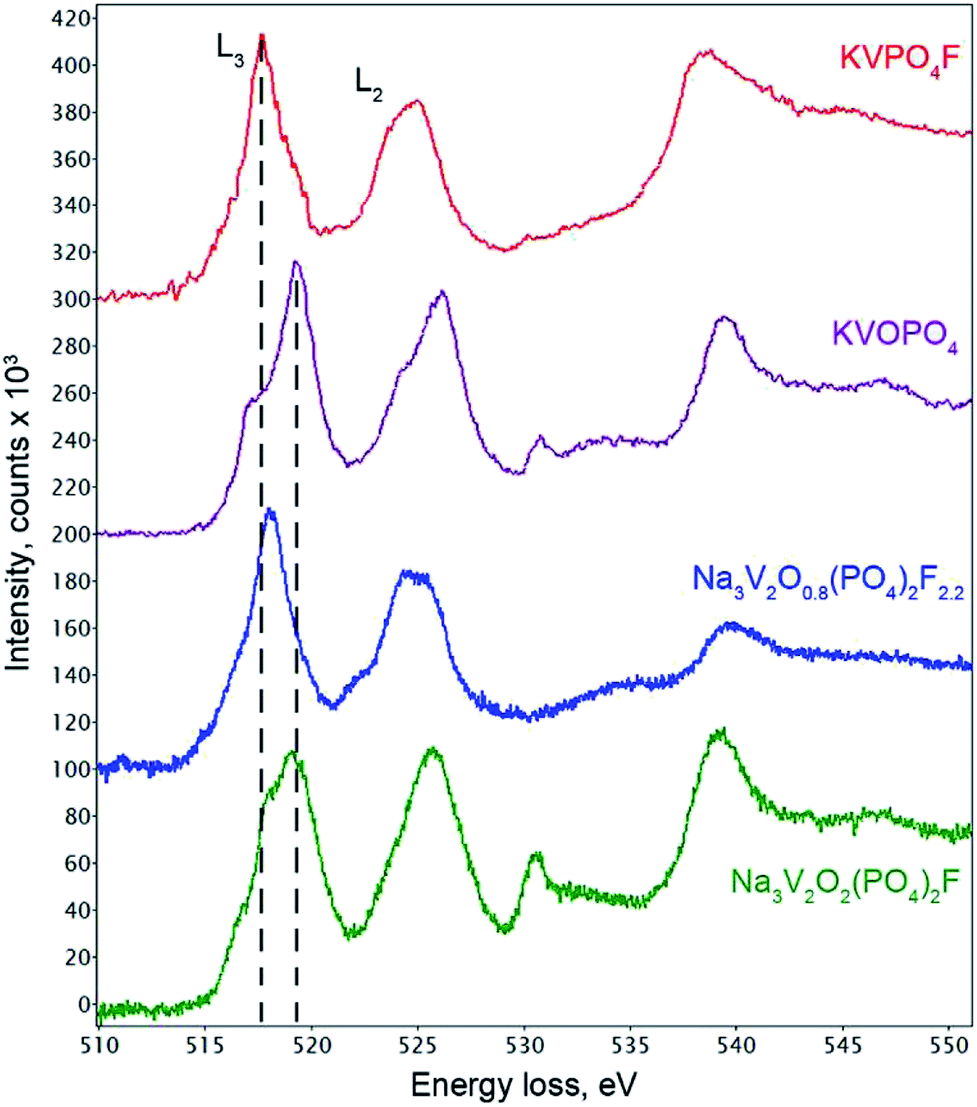 | ||
| Fig. 3 EELS spectra of Na3V2O2(PO4)2F synthesized with oxalic acid and Na3V2O0.8(PO4)2F2.2 synthesized with the hydrazine chloride–oxalic acid mixture, along with reference spectra of KVOPO4 and KVPO4F. | ||
The unit cell volume of the material obtained with an equimolar mixture of oxalic acid and hydrazine chloride was noticeably larger (437.74(2) Å3) compared to that obtained with other reducing agents (Table S1†). This indicated the formation of a Na3V2O2x(PO4)2F3−2x solid solution with more reduced vanadium and a higher fluorine content, in accordance with the ionic radii of V3+ (0.64 Å) and V4+ (0.58 Å) in an octahedral coordination environment.33 As the PXRD pattern of this phase corresponded well to the disordered I4/mmm model, the Rietveld refinement was performed according to the scheme adopted for the Na3V2O2(PO4)2F phase. As the vanadium oxidation state estimated using the BVS method was +3.393(7), the O/F content was set accordingly to satisfy the Na3V2O0.8(PO4)2F2.2 formula. The Na3V2O0.8(PO4)2F2.2 phase was registered in the Cambridge Crystallographic Data Centre (CCDC) with CSD 1904025. Excess fluorine was placed into the O2 position, as demonstrates by the BVS of 1.36, which was significantly smaller than the formal oxygen valence of −2. The V-L2,3 edge in the Na3V2O0.8(PO4)2F2.2 EELS spectrum was very similar to that of KVPO4F (V3+) (Fig. 3), but was shifted towards a higher energy loss by 0.4 eV, which is characteristic of a mixed +3/+4 vanadium oxidation state.30 The refinement parameters, atomic coordinates, and main interatomic distances are listed in Tables S2, S3 and S4,† respectively. The experimental, calculated, and difference PXRD profiles are shown in Fig. 2.
Compared with the strongly distorted VO5F octahedron in Na3V2O2(PO4)2F, the V(O,F)6 octahedron in Na3V2O0.8(PO4)2F2.2 became more isotropic, with the V–O1 and V–F1 bonds becoming shorter and V–O2 bond elongating from 1.624 Å to 1.748 Å. The octahedral distortion parameter, 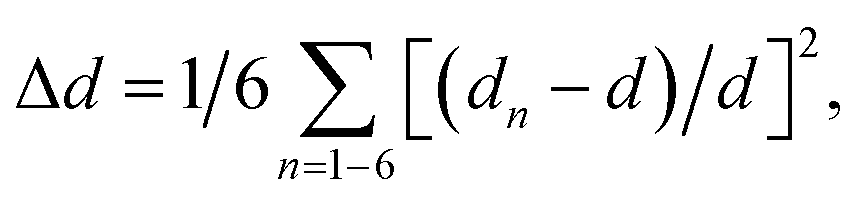 where dn is an individual V–(O,F) bond length and d is an average V–(O,F) bond length,34 decreased from 6.3 × 10−3 for Na3V2O2(PO4)2F to 2.5 × 10−3 in Na3V2O0.8(PO4)2F2.2. This strongly indicated that the formal oxidation state of vanadium approached +3 and the vanadyl bond was suppressed, with the upper limit for the V4+–O vanadyl bond considered to be 1.74 Å.29
where dn is an individual V–(O,F) bond length and d is an average V–(O,F) bond length,34 decreased from 6.3 × 10−3 for Na3V2O2(PO4)2F to 2.5 × 10−3 in Na3V2O0.8(PO4)2F2.2. This strongly indicated that the formal oxidation state of vanadium approached +3 and the vanadyl bond was suppressed, with the upper limit for the V4+–O vanadyl bond considered to be 1.74 Å.29
The Na3V2O2(PO4)2F phase being prepared in its high-temperature disordered form, rather than in the thermodynamically stable room temperature form with partial Na-vacancy ordering and a larger unit cell volume, was attributed to the low temperature of MW-HT treatment and extremely short synthesis time of 5–30 min. These conditions precluded migration of the Na+ cations and did not allow structure relaxation from the metastable high-temperature polymorph to the thermodynamically stable room-temperature structure.
Finally, regarding scaled-up material production, some synthesis experiments were conducted by increasing the solution concentration by a factor of 1.25 and the amount of simultaneously treated solution by a factor of 2, while retaining the developed treatment procedure. Overall, independent of both the reactor size and excess of oxalic acid, the routine preparation of 3 g batches of single phase Na3V2O2(PO4)2F material with the same unit cell parameters and unit cell volume as the samples obtained using smaller quantities was feasible (Table S1 and Fig. S3†).
No impurity peaks were detected in the Raman spectra of Na3V2O2x(PO4)2F3−2x materials synthesized using different reducing agents (Fig. S4†), indicating that a pure NVPOF phase was easily synthesized regardless of the reducing agent used. In general, all spectra were characterized by the presence of distinct bands at 940 and 1045 cm−1, which corresponded to the symmetric P–O stretching vibration and anti-symmetric stretching bands of PO43−.35 Meanwhile, detailed analysis of the Raman spectra was complicated due to bands from VO6 overlapping with bands from PO43−.36 Notably, some negligible differences in the band intensity were observed for samples with hydrazine chloride. Therefore, FT-IR spectroscopy was used to clearly compare the samples synthesized with various reducing agents. As expected, the FT-IR spectra showed differences between samples obtained using oxalic or citric acids and that synthesized using an equimolar mixture of hydrazine chloride and oxalic acid (Fig. 4). Interpretation of the FT-IR spectra was somewhat difficult owing to overlap of the absorption bands for PO43− symmetric stretching vibrations at ∼900–1200 cm−1, V–O and V–F single bond stretching vibrations at 900–950 cm−1, and double “vanadyl” V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond vibrations near 1000 cm−1.6,15,37–39 According to a systematic IR study of the Na3V2O2x(PO4)2F3−2x solid solutions,37 the narrow bands at 912 cm−1 and 954 cm−1 can be tentatively ascribed to V–O and V–F vibrations, respectively. For the Na3V2O2(PO4)2F phase obtained with oxalic or citric acids, the V–F band had a much lower strength compared with that of the V–O band according to the low fluorine content. However, in the Na3V2O0.8(PO4)2F2.2 material prepared with hydrazine chloride and oxalic acid, the strength of this band was increased significantly (Fig. 4). This was generally considered as a signature of increasing fluorine content in the Na3V2O2x(PO4)2F3−2x formula.15,37 An interesting feature of the FT-IR spectrum of Na3V2O0.8(PO4)2F2.2 is a splitting of the broad band at ∼900–1100 cm−1, which consists of the overlapped contributions of the PO43− and “vanadyl” VO bond stretching vibrations. A short “vanadyl” VO bond with a V–O distance of 1.624 Å in the Na3V2O2(PO4)2F phase resulted in a strong absorption band near 1000 cm−1. However, this band was shifted to a lower frequency due to lengthening of this bond to 1.748 Å in Na3V2O0.8(PO4)2F2.2, which enlarged the separation between the PO43− and VO bands. Some extra peaks also appeared in the IR spectrum of Na3V2O0.8(PO4)2F2.2, which were attributed to the presence of water and trace ammonium, but in general the FT-IR study corroborated a higher fluorine content and suppressed “vanadyl” bonding in Na3V2O0.8(PO4)2F2.2.
O bond vibrations near 1000 cm−1.6,15,37–39 According to a systematic IR study of the Na3V2O2x(PO4)2F3−2x solid solutions,37 the narrow bands at 912 cm−1 and 954 cm−1 can be tentatively ascribed to V–O and V–F vibrations, respectively. For the Na3V2O2(PO4)2F phase obtained with oxalic or citric acids, the V–F band had a much lower strength compared with that of the V–O band according to the low fluorine content. However, in the Na3V2O0.8(PO4)2F2.2 material prepared with hydrazine chloride and oxalic acid, the strength of this band was increased significantly (Fig. 4). This was generally considered as a signature of increasing fluorine content in the Na3V2O2x(PO4)2F3−2x formula.15,37 An interesting feature of the FT-IR spectrum of Na3V2O0.8(PO4)2F2.2 is a splitting of the broad band at ∼900–1100 cm−1, which consists of the overlapped contributions of the PO43− and “vanadyl” VO bond stretching vibrations. A short “vanadyl” VO bond with a V–O distance of 1.624 Å in the Na3V2O2(PO4)2F phase resulted in a strong absorption band near 1000 cm−1. However, this band was shifted to a lower frequency due to lengthening of this bond to 1.748 Å in Na3V2O0.8(PO4)2F2.2, which enlarged the separation between the PO43− and VO bands. Some extra peaks also appeared in the IR spectrum of Na3V2O0.8(PO4)2F2.2, which were attributed to the presence of water and trace ammonium, but in general the FT-IR study corroborated a higher fluorine content and suppressed “vanadyl” bonding in Na3V2O0.8(PO4)2F2.2.
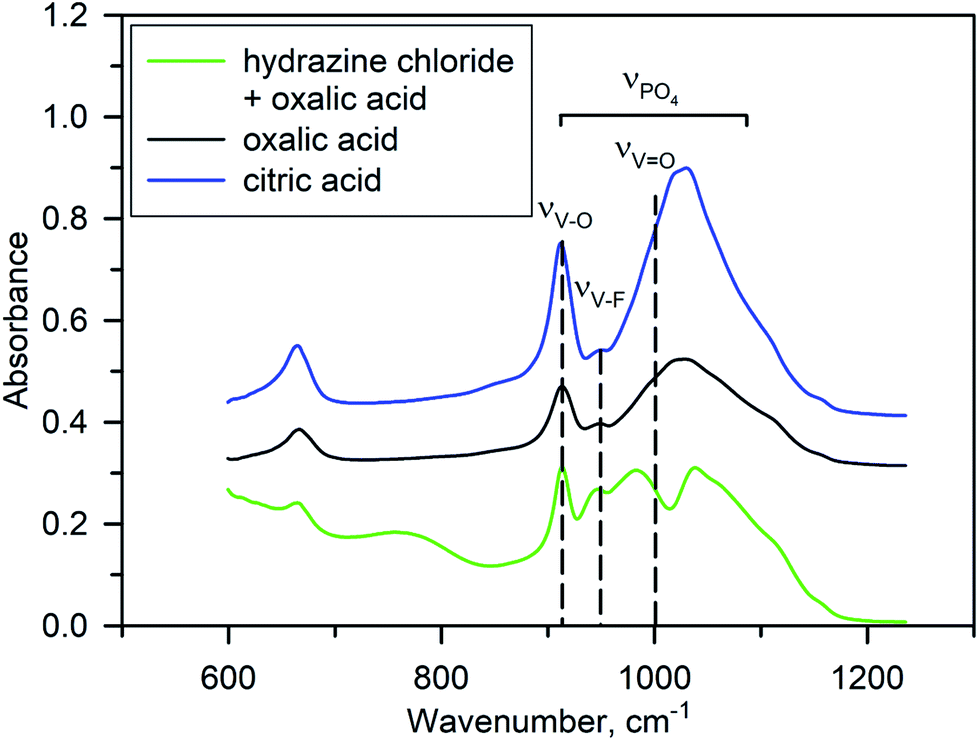 | ||
| Fig. 4 FT-IR spectra of Na3V2O2(PO4)2F prepared with citric acid (blue) and oxalic acid (black), and Na3V2O0.8(PO4)2F2.2 synthesized with the hydrazine chloride–oxalic acid mixture (green). Inset, enlarged part of the spectra with contributions from PO43− symmetric stretching vibrations at ∼900–1200 cm−1, V–O and V–F single bond stretching vibrations at 900–950 cm−1, and double “vanadyl” VO bond vibrations near 1000 cm−1. | ||
The Na3V2O2x(PO4)2F3−2x materials can be synthesized using different methods, such as solid state,5–9 sol–gel,10 or hydrothermal4,13,17 techniques. The main disadvantage of the former two techniques is that energy-consuming high-temperature annealing is always required as the last step of the synthesis. A high annealing temperature causes coarsening of the particles that may compromise rate performance and cycling stability.40 Furthermore, solid-state and sol–gel routes comprised multiple steps, which are time consuming and demand advanced parameter control throughout. The main advantages of the hydrothermal/solvothermal methods are a significant decrease in the synthesis temperature to <150 °C17 and their flexibility regarding the synthetic parameters and reagents used.37 These methods provide more scope for controlling morphology and size of particles. For example, using Na2HPO4 as a phosphorus source results in the formation of nanoflowers with a diameter of 8 μm and thickness of 4 μm, while using NH4H2PO4 leads to a cube-like morphology and particles with sizes less than 1 μm.14,27 However, treatment time is a limiting parameter of hydrothermal synthesis, with up to 60 h perhaps required to afford the final product.13 The long synthesis duration is closely related with the requirement to heat the solution homogeneously, while heating occurs from the “outside” to “inside”. Simultaneously, a new synthesis method was recently proposed that combined hydrothermal synthesis with microwaves.19 The effect of microwaves significantly decreased synthesis time from hours to minutes. Furthermore, the direction of heating was changed (from “inside” to “outside”) compared with conventional hydrothermal synthesis, leading to homogeneous heat distribution in the solution and the formation of particles with narrow size distributions. The morphology and size of particles can be easily controlled using various reducing agents and by changing the synthesis parameters (temperature and time), as clearly demonstrated in the present study for Na3V2O2x(PO4)2F3−2x materials.
Morphology
The effect of various reducing agents on the morphology of the Na3V2O2x(PO4)2F3−2x materials was examined by scanning electron microscopy (Fig. 5). Significant morphological changes were observed. First, a nucleation-growth process lead to the formation of well-defined cubic crystallites with salient edges when oxalic acid (Fig. 5a and b) or an equimolar mixture of hydrazine sulfate/oxalic acid were used as reducing agents. For the latter (Fig. 5c and d), once the cubes reached nearly ∼5 μm in size, the growing process stopped, most likely due to surface energy minimization between the cube faces and edges, at the expense of agglomeration. Despite the similar morphology of samples synthesized using oxalic acid and an equimolar mixture of hydrazine sulfate/oxalic acid, the particle surface area was significantly different, reaching 3.54 m2 g−1 and 24.81 m2 g−1, respectively. A similar morphology was obtained with sodium borohydride as the reducing agent (not shown in Fig. 5). In contrast, using citric acid drastically changed the morphology, resulting in agglomerates containing rhombus-like plates together with rod-like particles (Fig. 5e). The plate thickness was around 0.5 μm, while the rod diameter was around 1 μm (Fig. 5f). Finally, an equimolar mixture of hydrazine chloride and oxalic acid led to the formation of fibrous particles with average transverse sizes of less than 0.5 μm and a particle surface area of 44.28 m2 g−1 (Fig. 5g and h). To further explore the structural relationship between the precursor and the end phase, semi in situ analytical measurements (IR, PXRD) are currently being designed. | ||
| Fig. 5 SEM images of Na3V2O2x(PO4)2F3−2x produced by microwave-assisted hydrothermal synthesis using different reducing agents: (a and b) oxalic acid; (c and d) an equimolar mixture of hydrazine sulfate with oxalic acid; (e and f) citric acid; and (g and h) an equimolar mixture of hydrazine chloride with oxalic acid. | ||
Electrochemical performance
Numerous batches of Na3V2O2x(PO4)2F3−2x powders, with different particle sizes and morphologies depending on the reducing agents used, were tested for their electrochemical performance. A two-electrode custom-designed cell hardware, comprising metallic Na as the counter electrode, was used to perform the measurements. Data was collected in galvanostatic charge–discharge (GCD) cycling mode at a rate of C/10 (1 Na+ in 1 h) and over a potential window range of 2.5–4.3 V vs. Na+/Na. Measurements were repeated twice prior to being reported. For conciseness, the collected data are solely reported for the 1st and 10th cycles, together with the derivative (dQ/dV) curves for various Na3V2O2x(PO4)2F3−2x samples. A common feature, regardless of the reducing agent (Fig. 6, S5 and S6†), was the presence of two redox activities at around 4.00 V and 3.55 V, which can be visualized by semi-plateaus in the V-capacity curves and peaks in the derivative plots. These pseudo-plateaus, similar to those already reported for partially oxygen substituted Na3V2O2x(PO4)2F3−2x samples, contrasted with the well-defined plateaus observed for pure Na3V2(PO4)2F3. Upon first charge, the material obtained with oxalic acid delivered its full theoretical capacity of ∼130 mA h g−1 (Fig. 6a). However, a significant irreversible capacity between the first charge and discharge culminated at ∼35% for samples prepared with oxalic acid (Fig. 6a), and was reduced to 18% for samples prepared with the equimolar hydrazine sulfate–oxalic acid mixture (Fig. 6d). This then switched back to 30% when an equimolar hydrazine chloride–oxalic acid mixture was used (Fig. 6e). The irreversible capacity completely disappeared after 10 cycles regardless of the reducing agent used (Fig. 6a, d, g, S5a and S6a†). These large irreversible capacities were higher than those for samples prepared using the ceramic process, implying that the remaining surficial groups inherent to the solution process were most likely irreversibly oxidized during the charging process. This hypothesis was confirmed by the 15% decrease in irreversible capacity observed when the sample was annealed for a 1 h at 600 °C with a carbon source (Fig. 7a). Therefore, this sample displayed poor capacity retention upon cycling (Fig. 7c and S7a†). Pleasingly, the capacity retention of the sample was dramatically improved by simply coating the pristine phase with dopamine followed by annealing at 500 °C for 3 h (Fig. 7d, f and S7b†). This treatment produced a uniform carbon coating with thickness less than 10 nm (Fig. S1b and c†), which was also beneficial to the material performance at a higher discharge rate (Fig. 7e). The capacity decreased by nearly 50% (∼110 mA h g−1 to 55 mA h g−1) when going from C/10 to 10C, compared with nearly 60% for the sample annealed with glucose (Fig. 7b). Qualitative dependence was observed between particle morphology and rate capability, which decreased concomitantly with increasing particle size observed by SEM, which, in turn, depended on the reducing agent used. This trend was illustrated by discharge capacities at the 10C rate of ∼22 mA h g−1 (Fig. 6b), ∼35 mA h g−1 (Fig. 6e), and ∼55 mA h g−1 (Fig. 6h) for the samples prepared with oxalic acid (cubes of >5 μm in size, Fig. 5a and b), hydrazine sulfate–oxalic acid (cubes of ∼2–5 μm in size, Fig. 5c and d), and hydrazine chloride–oxalic acid (fibrous particles with transverse sizes of <0.5 μm, Fig. 5e and f), respectively. Furthermore, an increase in current density led to pseudo-plateau formation being suppressed, most probably owing to the suppression of Na+ ion ordering. Briefly, among all samples synthesized, that prepared with oxalic acid as reducing agent, coated with dopamine, and annealed at 500 °C for 3 h showed the highest first discharge capacity at a current density of C/2 (92 mA h g−1), retaining a capacity of ∼82 mA h g−1 after 50 cycles (Fig. S7b†).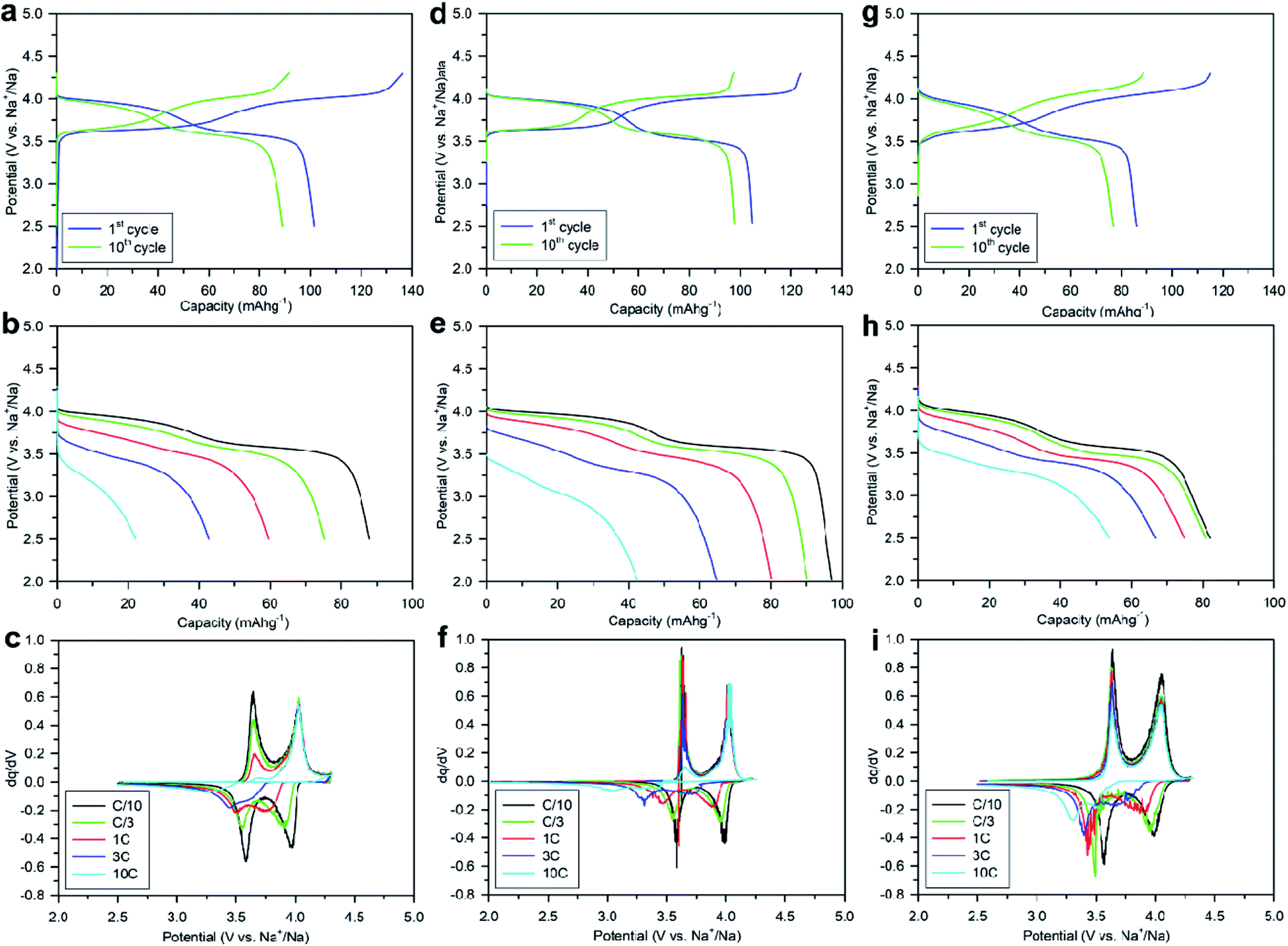 | ||
| Fig. 6 (a–c) Electrochemical performance of Na3V2O2(PO4)2F obtained with oxalic acid, (d–f) an equimolar mixture of hydrazine sulfate with oxalic acid, and (g–i) an equimolar mixture of hydrazine chloride with oxalic acid as reducing agents, in the potential range 2.5–4.3 V vs. Na+/Na at room temperature: (a, d and g) galvanostatic charge–discharge profiles at a C/10 current density; (b, e and h) galvanostatic discharge profiles at different current densities, and (c, f and i) derivative curves (dQ/dV). | ||
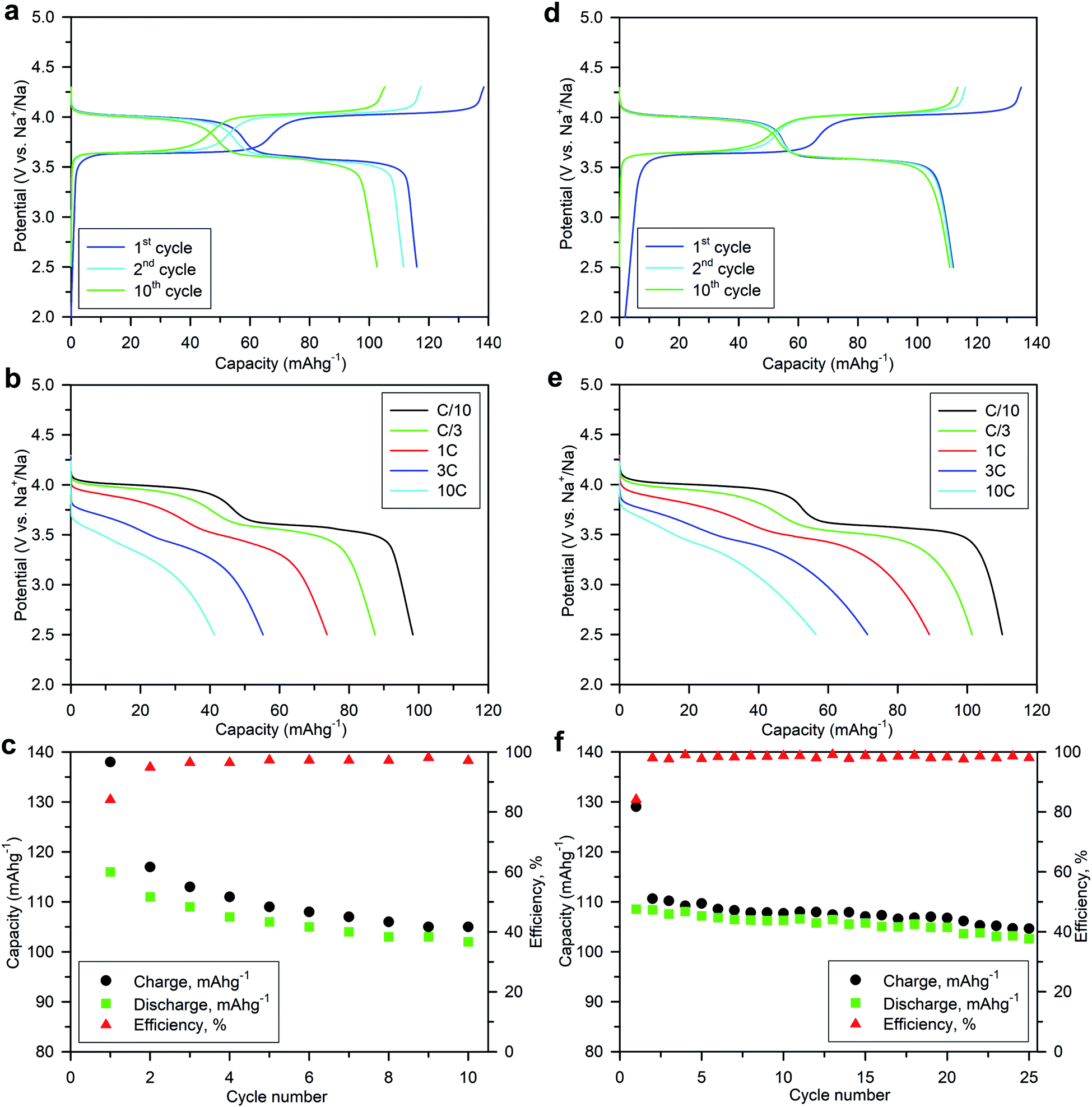 | ||
| Fig. 7 Electrochemical performance of Na3V2O2(PO4)2F (a–c) annealed with glucose (8 wt%) at 600 °C for 1 h, and (d–f) coated with dopamine for 24 h and annealed at 500 °C for 3 h, over the potential range 2.5–4.3 V vs. Na+/Na at room temperature: (a and d) galvanostatic charge–discharge profiles at a C/10 current density; (b and e) galvanostatic discharge profiles at different current densities; (c and f) capacity retention and coulombic efficiency at a C/10 current density. | ||
The rate capability is another figure of merit used to compare electrode materials. Fig. 6b, d, e, 7b and e show a decrease in the delivered capacity with increasing current density for all samples. To better convey the differences between samples, we constructed a Ragone plot (Fig. 8). From this, the best power rate performance was obtained by the sample prepared from oxalic acid and carbon-coated with dopamine, which delivered more than 50% of its initial capacity a rate of 10C (in 6 min).
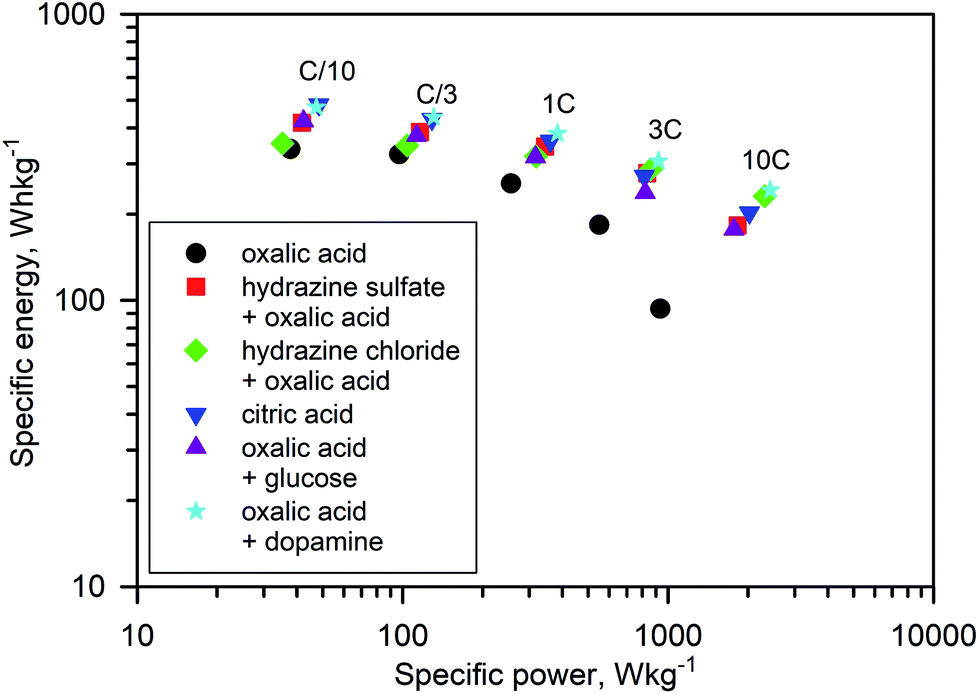 | ||
| Fig. 8 Ragone plot showing the electrochemical performance of the Na3V2O2x(PO4)2F3−2x materials at current densities ranging from C/10 to 10C. | ||
To further explore the origin of this irreversible capacity at the first charge, we performed SXPD experiments in the operando regime for two charge–discharge cycles at a current density of C/5 for the sample synthesized with an equimolar hydrazine sulfate–oxalic acid mixture as the reducing agent. The general view of the SXPD data (Fig. 9a and S8†), and the results of the Rietveld refinement (Fig. 9b–e) of the selected patterns showed that the Na3V2O2(PO4)2F material was characterized by a combination of two-phase and single-phase (de)intercalation mechanisms. In agreement with the previous operando data on Na3V2O2(PO4)2F,16,41 the first charge proceeded through a very short single-phase region at the beginning of the first charge. A new phase with similar crystallographic symmetry and unit cell parameters then appeared, and desodiation followed a two-phase mechanism until the extraction of 1Na. Notably, the unit cell parameters of both phases varied within this region (corresponding to the low voltage plateau at ≈3.6 V). Further desodiation proceeded via Na3−xV2O2(PO4)2F solid solution (denoted as NδVPOF in Fig. 9) until 2Na were extracted. At this fully charged state, the occupancy of the Na2 position dropped to zero, and the occupancy of the Na1 positions decreased by ∼56%. Most discharge also followed a single-phase mechanism, and the initial fully “sodiated” phase reappeared at the end, but with a mass fraction not exceeding 50%. The second partially “sodiated” phase has Na2.2V2O2(PO4)2F refined composition where the occupancy of the Na1 position amounts to only ∼75% of its initial value, whereas the occupancy of the Na2 position gets ∼40% higher. This redistribution was in agreement with the conjecture that (de)intercalation occurs by moving Na from/in the Na1 position through the Na2 position.12 Regarding the lattice parameters, desodiation results in of the unit cell in the ab plane and its expansion along the c axis for both phases. Sodiation was accompanied by reverse variation in the unit cell parameters. The total unit cell volume change was 2.2% at first charge and only 1.5% at discharge. The second cycle was symmetric relative to the discharge process. The Na occupancy refinements, along with lattice parameter variations, indicated that the irreversible capacity observed at the first desodiation of Na3V2O2(PO4)2F was related to internal structural changes (most probably associated with redistribution of the sodium atoms among the Na1 and Na2 positions). However, the surface contributions, as inferred from the electrochemical data, should also play a role, because EELS indicated the presence of V3+ at the edges of the Na3V2O2(PO4)2F crystallites (Fig. S2†), and this reduced surface layer can be oxidized irreversibly during the first charge.
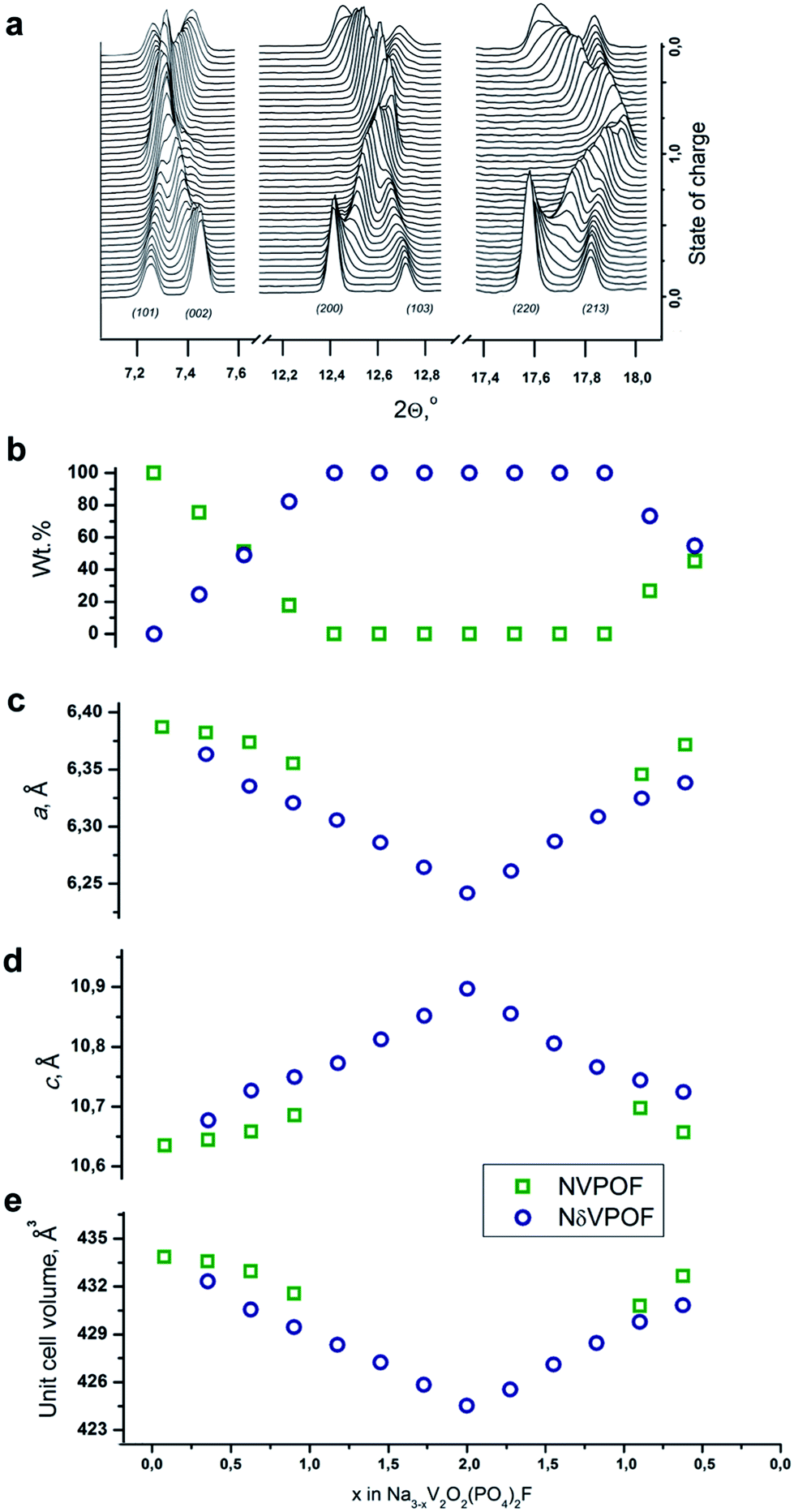 | ||
| Fig. 9 Results of operando SXPD experiments on Na3V2O2(PO4)2F: (a) evolution of the selected regions of the SXPD patterns; (b) mass fraction; and (c–e) unit cell parameters of sodiated (NVPOF) and desodiated (NδVPOF) phases. | ||
Conclusions
The microwave-assisted hydrothermal synthesis of NASICON-structured Na3V2O2x(PO4)2F3−2x solid solutions has been developed, providing an economically viable, energy-saving, and environmentally friendly pathway toward this important family of cathode materials for Na-ion batteries. Microwave-assisted hydrothermal treatment provided a well-crystallized material at temperatures as low as 180–200 °C that required an exceptionally short preparation time of 5–15 min, providing significant energy saving compared with conventional synthesis techniques. The precursor materials were readily available, including inexpensive salts, reducing agents, and V2O5 as the vanadium source, which allowed intermediate stages for V5+ conversion to V3+/V4+ or the preparation of precursors containing reduced forms of vanadium to be avoided. This method was based solely on aqueous solutions without organic solvents, which contributed to the environmental friendliness. When applied to the Na3V2O2x(PO4)2F3−2x solid solutions, this microwave-assisted hydrothermal method appeared to be very flexible, allowing for control of the vanadium oxidation state and O/F content, as well as the microstructure of the material, by varying the nature of the reducing agent. This method also seems to be easily scalable, as demonstrated by the preparation of larger batches in larger-volume reactors by the direct transfer of the synthesis conditions developed in a 10 mL reactor vessel. The electrochemical behavior of the Na3V2O2x(PO4)2F3−2x solid solutions prepared with the microwave-assisted hydrothermal technique was similar to that reported for these materials synthesized with other methods [see ref. 42 and references therein]. The samples prepared using citric acid, hydrazine sulfate/oxalic acid, and hydrazine chloride/oxalic acid as reducing agents demonstrated two sloping plateaus on charge and discharge centered at near 3.5–3.6 V and 4.0–4.1 V vs. Na+/Na with a reversible capacity of ∼100–110 mA h g−1 at a rate of C/10, as previously reported for Na3V2O2x(PO4)2F3−2x solid solutions.5,13,15,35,43 The application of a conducting carbon coating through the surface polymerization of dopamine with subsequent annealing at 500 °C improved both the rate capability (∼55 mA h g−1 at discharge rate of 10C) and capacity retention (∼93% after 50 cycles at a discharge rate of C/2). Considering that a main advantage of Na-ion batteries compared with their Li-ion counterparts is their reduced cost, the developed synthesis method might significantly contribute to the competitiveness of Na-ion energy storage technology.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Russian Foundation of Basic Research (grant 16-53-48011) and the Department of Science and Technology, Government of India (under grant No. DST/INT/RFBR/IDIR/P-09/2016/G). Dr Iaroslava Shakhova would like to thank Dr S. Ryazantcev for his help with Raman and FT-IR spectroscopy experiments and discussion of the experimental data.References
- K. Liao, S. Chen, H. Wei, J. Fan, Q. Xu and Y. Min, Micropores of pure nanographite spheres for long cycle life and high-rate lithium-sulfur batteries, J. Mater. Chem. A, 2018, 6, 23062–23070 RSC.
- C. Vaalma, D. Buchholz, M. Weil and S. Passerini, A cost and resource analysis of sodium-ion batteries, Nat. Rev. Mater., 2018, 3, 18013 CrossRef.
- R. A. Shakoor, D.-H. Seo, H. Kim, Y.-U. Park, J. Kim, S.-W. Kim, H. Gwon, S. Leec and K. Kang, A combined first principles and experimental study on Na3V2(PO4)2F3 for rechargeable Na batteries, J. Mater. Chem., 2012, 22, 20535–20541 RSC.
- P. Serras, V. Palomares, P. Kubiak, L. Lezama and T. Rojo, Enhanced electrochemical performance of vanadyl (IV) Na3(VO)2(PO4)2F by ex situ carbon coating, Electrochem. Commun., 2013, 34, 344–347 CrossRef CAS.
- Y.-U. Park, D.-H. Seo, H. Kim, J. Kim, S. Lee, B. Kim and K. Kang, A Family of High-Performance Cathode Materials for Na-ion Batteries, Na3(VO1−xPO4)2F1+2x (0 ≤ x ≤ 1): Combined First-Principles and Experimental Study, Adv. Funct. Mater., 2014, 24, 4603–4614 CrossRef CAS.
- F. Sauvage, E. Quarez, J.-M. Tarascon and E. Baudrin, Crystal structure and electrochemical properties vs. Na+ of the sodium fluorophosphate Na1.5VOPO4F0.5, Solid State Sci., 2006, 8, 1215–1221 CrossRef CAS.
- R. K. B. Gover, A. Bryan, P. Burns and J. Barker, The electrochemical insertion properties of sodium vanadium fluorophosphate, Na3V2(PO4)2F3, Solid State Ionics, 2006, 177, 1495–1500 CrossRef CAS.
- R. Väli, P. Möller and A. Jänes, Synthesis and Characterization of Na3V2(PO4)2F3 Based Cathode Material for Sodium Ion Batteries, ECS Trans., 2015, 69(39), 27–36 CrossRef.
- W. Song, X. Ji, Z. Wu, Y. Zhu, F. Li, Y. Yao and C. E. Banks, Multifunctional dual Na3V2(PO4)2F3 cathode for both lithium-ion and sodium-ion batteries, RSC Adv., 2014, 4, 11375–11383 RSC.
- T. Jiang, G. Chen, A. Li, C. Wang and Y. Wei, Sol–gel preparation and electrochemical properties of Na3V2(PO4)2F3/C composite cathode material for lithium ion batteries, J. Alloys Compd., 2009, 478, 604–607 CrossRef CAS.
- N. Eshraghi, S. Caes, A. Mahmoud, R. Cloots, B. Vertruyen and F. Boschini, Sodium vanadium (III) fluorophosphate/carbon nanotubes composite (NVPF/CNT) prepared by spray-drying: good electrochemical performance thanks to well-dispersed CNT network within NVPF particles, Electrochim. Acta, 2017, 228, 319–324 CrossRef CAS.
- P. Serras, V. Palomares, J. Alonso, N. Sharma, J. M. López del Amo, P. Kubiak, M. L. Fdez-Gubieda and T. Rojo, Electrochemical Na Extraction/Insertion of Na3V2O2x(PO4)2F3−2x, Chem. Mater., 2013, 25, 4917–4925 CrossRef CAS.
- M. Xu, P. Xiao, S. Stauffer, J. Song, G. Henkelman and J. B. Goodenough, Theoretical and Experimental Study of Vanadium-Based Fluorophosphate Cathodes for Rechargeable Batteries, Chem. Mater., 2014, 26, 3089–3097 CrossRef CAS.
- Y. Qi, L. Mu, J. Zhao, Y.-S. Hu, H. Liu and S. Dai, pH-regulative synthesis of Na3(VPO4)2F3 nanoflowers and their improved Na cycling stability, J. Mater. Chem. A, 2016, 4, 7178–7184 RSC.
- J. Zhao, L. Mu, Y. Qi, Y.-S. Hu, H. Liu and S. Dai, A phase-transfer assisted solvo-thermal strategy for low-temperature synthesis of Na3(VO1−xPO4)2F1+2x cathodes for sodium-ion batteries, Chem. Commun., 2015, 51, 7160–7163 RSC.
- J.-Z. Guo, P.-F. Wang, X.-L. Wu, X.-H. Zhang, Q. Yan, H. Chen, J.-P. Zhang and Y.-G. Guo, High-Energy/Power and Low-Temperature Cathode for Sodium-Ion Batteries: In Situ XRD Study and Superior Full-Cell Performance, Adv. Mater., 2017, 1701968 CrossRef PubMed.
- Y. Cai, X. Cao, Z. Luo, G. Fang, F. Liu, J. Zhou, A. Pan and S. Liang, Caging Na3V2(PO4)2F3 Microcubes in Cross-Linked Graphene Enabling Ultrafast Sodium Storage and Long-Term Cycling, Adv. Sci., 2018, 1800680 CrossRef.
- D. Larcher and J.-M. Tarascon, Towards greener and more sustainable batteries for electrical energy storage, Nat. Chem., 2015, 7, 19–29 CrossRef CAS PubMed.
- S. Komarneni, R. Roy and Q. H. Li, Microwave-hydrothermal synthesis of ceramic powders, Mater. Res. Bull., 1992, 27, 1393–1405 CrossRef CAS.
- C. E. Alarcón-Suesca and T. Nilges, Phospho-olivines as cathode materials for lithium ion battery: trends on microwave synthesis, J. Phys.: Conf. Ser., 2014, 480, 012018 CrossRef.
- A. Vadivel Murugan, T. Muraliganth and A. Manthiram, Comparison of Microwave Assisted Solvothermal and Hydrothermal Syntheses of LiFePO4/C Nanocomposite Cathodes for Lithium Ion Batteries, J. Phys. Chem. C, 2008, 112, 14665–14671 CrossRef.
- V. Petricek, M. Dusek and L. Palatinus, Crystallographic Computing System JANA2006: general features, Z. Kristallogr., 2014, 229, 345–352 CAS.
- O. A. Drozhzhin, I. V. Tereshchenko, H. Emerich, E. V. Antipov, A. M. Abakumov and D. Chernyshov, An Electrochemical Cell with Sapphire Windows for Operando Synchrotron X-Ray Powder Diffraction and Spectroscopy Studies of High-Power and High-Voltage Electrodes for Metal-Ion Batteries, J. Synchrotron Radiat., 2018, 25(2), 468–472 CrossRef CAS PubMed.
- V. Dyadkin, P. Pattison, V. Dmitriev and D. Chernyshov, A New Multipurpose Diffractometer PILATUS@SNBL, J. Synchrotron Radiat., 2016, 23(3), 825–829 CrossRef CAS PubMed.
- J. Rodríguez-Carvajal, Recent Developments of the Program FULLPROF, Newsletter of the Commission for Powder Diffraction of the IUCr, 2001, 26, 12–19 Search PubMed.
- W. Massa, O. V. Yakubovich and O. V. Dimitrova, Crystal structure of a new sodium vanadyl(IV) fluoride phosphate Na3{V2O2F[PO4]2}, Solid State Sci., 2002, 4, 495–501 CrossRef CAS.
- H. Jin, J. Dong, E. Uchaker, Q. Zhang, X. Zhou, S. Hou, J. Lic and G. Cao, Three dimensional architecture of carbon wrapped multilayer Na3V2O2(PO4)2F nanocubes embedded in graphene for improved sodium ion batteries, J. Mater. Chem. A, 2015, 3, 17563–17568 RSC.
- A. A. Tsirlin, R. Nath, A. M. Abakumov, Y. Furukawa, D. C. Johnston, M. Hemmida, H.-A. Krug von Nidda, A. Loidl, C. Geibel and H. Rosner, Phase separation and frustrated square lattice magnetism of Na1.5VOPO4F0.5, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 014429 CrossRef.
- M. Schindler, F. C. Hawthorne and W. H. Baur, Crystal Chemical Aspects of Vanadium: Polyhedral Geometries, Characteristic Bond Valences, and Polymerization of (VOn) Polyhedra, Chem. Mater., 2000, 12, 1248–1259 CrossRef CAS.
- H. Tan, J. Verbeeck, A. Abakumov and G. Van Tendeloo, Oxidation state and chemical shift investigation in transition metal oxides by EELS, Ultramicroscopy, 2012, 116, 24–33 CrossRef CAS.
- S. S. Fedotov, N. R. Khasanova, A. S. Samarin, O. A. Drozhzhin, D. Batuk, O. M. Karakulina, J. Hadermann, A. M. Abakumov and E. V. Antipov, AVPO4F (A = Li, K): A 4 V Cathode Material for High-Power Rechargeable Batteries, Chem. Mater., 2016, 28, 411–415 CrossRef CAS.
- K. Chihara, A. Katogi, K. Kubota and S. Komaba, KVPO4F and KVOPO4 toward 4 volt-class potassium-ion batteries, Chem. Commun., 2017, 53, 5208–5211 RSC.
- R. D. Shannon, Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- I. D. Brown and R. D. Shannon, Empirical Bond-Strength Bond-Length Curves for Oxides, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1973, 29, 266–282 CrossRef CAS.
- H. Chen, H. Mi, L. Sun, P. Zhang and Y. Li, One-step synthesis of 3D-sandwiched Na3V2(PO4)2O2F@rGO composites as cathode material for high-rate sodium-ion batteries, ChemElectroChem, 2018, 5, 2593–2599 CrossRef CAS.
- R. L. Frost, K. L. Erickson, M. L. Weier and O. Carmody, Raman and infrared spectroscopy of selected vanadates, Spectrochim. Acta, Part A, 2005, 61, 829–834 CrossRef PubMed.
- Y. Qi, L. Mu, J. Zhao, Y.-S. Hu, H. Liu and S. Dai, Superior Na-Storage Performance of Low-Temperature-Synthesized Na3(VO1-xPO4)2F1+2x (0≤x≤1) Nanoparticles for Na-Ion Batteries, Angew. Chem., Int. Ed., 2015, 54, 9911–9916 CrossRef CAS PubMed.
- L. D. Frederickson and D. M. Hausen, Infrared Spectra-Structure Correlation Study of Vanadium-Oxygen Compounds, Anal. Chem., 1963, 35, 818–827 CrossRef CAS.
- W. Jastrzebski, M. Sitarz, M. Rokita and K. Bułat, Infrared spectroscopy of different phosphates structures, Spectrochim. Acta, Part A, 2011, 79, 722–727 CrossRef CAS PubMed.
- C. Zhu, C. Wu, C.-C. Chen, P. Kopold, P. A. van Aken, J. Maier and Y. Yu, High Power–high energy Na3V2(PO4)2F3 sodium cathode: investigation of transport parameters, rational design and realization, Chem. Mater., 2017, 29, 5207–5215 CrossRef CAS.
- V. Palomares, P. Serras, H. E. A. Brand, T. Rojo and N. Sharma, Structural evolution of mixed valent (V3+/V4+) and V4+ sodium vanadium fluorophosphates as cathodes in sodium-ion batteries: comparisons, overcharging and mid-term cycling, J. Mater. Chem. A, 2015, 3, 23017–23027 RSC.
- P. Serras, V. Palomares and T. Rojo, High-Voltage Cathodes for Na-Ion Batteries: Sodium–Vanadium Fluorophosphates, in Alkali-ion Batteries, ed. D. Yang, Intech Open, 2016, ch. 8, pp. 155–172 Search PubMed.
- P. R. Kumar, Y. H. Jung, C. H. Lim and D. K. Kim, Na3V2O2x(PO4)2F3-2x: a stable and high-voltage cathode material for aqueous sodium-ion batteries with high energy density, J. Mater. Chem. A, 2015, 3, 6271–6275 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02257k |
| This journal is © The Royal Society of Chemistry 2019 |