
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembly of oppositely charged polyelectrolyte block copolymers containing short thermoresponsive blocks†
I. A.
van Hees
 a,
P. J. M.
Swinkels
b,
R. G.
Fokkink
a,
A. H.
Velders
c,
I. K.
Voets
d,
J.
van der Gucht
a and
M.
Kamperman
*e
a,
P. J. M.
Swinkels
b,
R. G.
Fokkink
a,
A. H.
Velders
c,
I. K.
Voets
d,
J.
van der Gucht
a and
M.
Kamperman
*e
aPhysical Chemistry and Soft Matter, Wageningen University and Research, Stippeneng 4, 6708 WE Wageningen, The Netherlands
bInstitute of Physics, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, the Netherlands
cLaboratory of BioNanoTechnology, Wageningen University and Research, Bornse Weilanden 9, 6708 WG, Wageningen, The Netherlands
dDepartment of Chemical Engineering and Chemistry, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands
ePolymer Science, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: marleen.kamperman@rug.nl
First published on 9th April 2019
Abstract
The assembly of oppositely charged block copolymers, containing small thermoresponsive moieties, was investigated as a function of salt concentration and temperature. Aqueous solutions of poly-[N-isopropylacrylamide]-b-poly[dimethylaminoethyl methacrylate] (NIPAM44-b-DMAEMA216) and PNIPAM-b-poly[acrylic acid]-b-PNIPAM (NIPAM35-b-AA200-b-NIPAM35) were mixed in equal charge stoichiometry, and analysed by light scattering (LS), NMR spectroscopy and small angle X-ray scattering (SAXS). At room temperature, two different micelle morphologies were found at different salt concentrations. At NaCl concentrations below 0.75 M, complex coacervate core micelles (C3M) with a PNIPAM corona were formed as a result of interpolyelectrolyte complexation. At NaCl concentrations exceeding 0.75 M, the C3M micelles inverted into PNIPAM cored micelles (PCM), containing a water soluble polyelectrolyte corona. This behavior is ascribed to the salt concentration dependence of both the lower critical solution temperature (LCST) of PNIPAM, and the complex coacervation. Above 0.75 M NaCl, the PNIPAM blocks are insoluble in water at room temperature, while complexation between the polyelectrolytes is prevented because of charge screening by the salt. Upon increasing the temperature, both types of micelles display a cloud point temperature (Tcp), despite the small thermoresponsive blocks, and aggregate into hydrogels. These hydrogels consist of a complexed polyelectrolyte matrix with microphase separated PNIPAM domains. Controlling the morphology and aggregation of temperature sensitive polyelectrolytes can be an important tool for drug delivery systems, or the application and hardening of underwater glues.
Introduction
Complex coacervation is a liquid–liquid phase separation that occurs when two oppositely charged polyelectrolyte solutions are mixed, resulting in two coexisting phases: (1) the complex coacervate, a water-insoluble polyelectrolyte phase, and (2) a dilute solvent phase.1 Complex coacervates display unique characteristics, such as a low interfacial tension and a high water content, while being water-insoluble.2,3 This combination of properties makes complex coacervates interesting for many applications, such as underwater adhesives or encapsulants.4–8The properties of complex coacervates depend on several parameters, including the chemical nature of polyelectrolytes, salt concentration, and in the case of weak polyelectrolytes, the pH. These parameters influence not only the water content, but also the interaction strength and mobility of the polyelectrolytes in the complex.3,9 Complex coacervate core micelles (C3M) (also known as poly-ion complex (PIC) micelles, block ionomer complex (BIC) micelles, or interpolyelectrolyte complex (IPEC) micelles) can be formed when water-soluble blocks are connected to the polyelectrolytes. These water-soluble blocks will form a stabilizing corona around the water-insoluble complex coacervate core of the C3M.9–11
A special class of C3Ms are stimuli responsive micelles, which are promising systems for sensors or controlled delivery systems, and many reports describe C3Ms that are responsive to ionic strength, pH or temperature.9,12–20 Most of these C3Ms are designed to be stable in solution, which is important for applications such as drug delivery. Therefore, large stabilizing blocks with a minimum block length of 30 mol% are typically used to form the corona.13–18 However, for applications such as adhesives, the formation of a dense solid phase is needed and smaller temperature responsive blocks may be preferred to obtain a different morphology.
In this article, we study the assembly of oppositely charged block copolymers with short temperature-sensitive blocks. The system is composed of two block-copolymers, poly-[N-isopropylacrylamide]-b-poly[dimethylaminoethyl methacrylate] (PNIPAM-b-PDMAEMA) and PNIPAM-b-poly[acrylic acid]-b-PNIPAM (PNIPAM-b-PAA-b-PNIPAM) (Fig. 1). Both PDMAEMA and PAA are weak polyelectrolytes that are positively and negatively charged at neutral pH, respectively, therewith enabling complex coacervation.1,3 Thermo-responsiveness is introduced into the system by means of PNIPAM, which is a well-explored polymer displaying a lower critical solution temperature (LCST).21 Below the LCST, PNIPAM is water-soluble, while above the LCST, the polymer chain collapses and PNIPAM becomes water-insoluble.22 The LCST of PNIPAM in aqueous solution is about 32 °C, but varies with molecular weight, salt concentration, and block length ratio when copolymerized.22–25
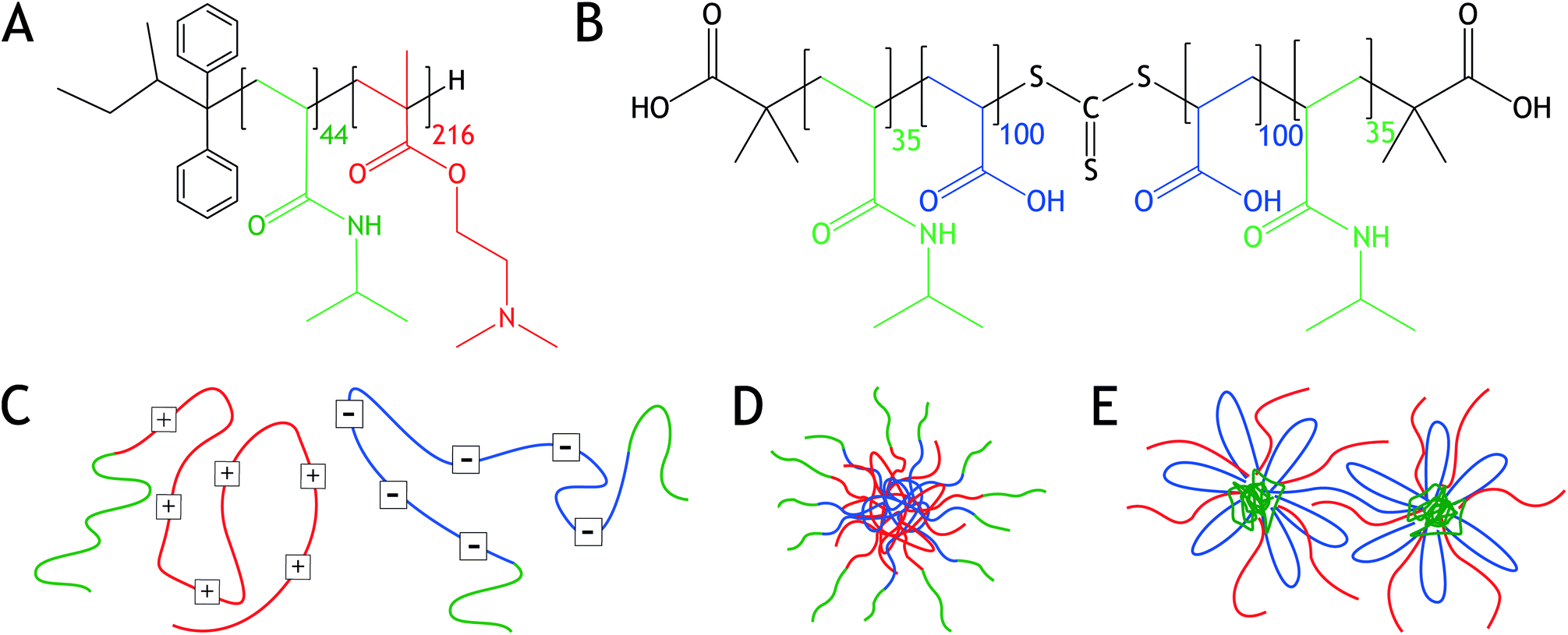 | ||
| Fig. 1 Chemical structures of (a) cationic PNIPAM-b-PDMAEMA, which was synthesized by anionic polymerization, and (b) PNIPAM-b-PAA-b-PNIPAM, which was synthesized by reversible addition fragmentation chain transfer (RAFT) polymerization. Schematic representations of (c) PNIPAM-b-PDMAEMA and PNIPAM-b-PAA-b-PNIPAM, (d) a C3M, and (e) PCMs. The red block represents PDMAEMA, the blue block PAA and the green blocks PNIPAM. | ||
We study both the influence of salt concentration and temperature on the morphology of the block copolymer mixtures. Analysis is performed using light scattering (LS), nuclear magnetic resonance spectroscopy (NMR), and small angle X-ray scattering (SAXS). We show that at low salt concentrations, C3Ms with a PNIPAM corona are formed, Fig. 1. However, sufficiently elevated salt concentrations turn the C3Ms inside out, leading to PNIPAM-cored micelles (PCMs) with a water-soluble polyelectrolyte corona. Upon temperature increase, both C3Ms and PCMs aggregate and form hydrogels, displaying a salt concentration dependent cloud point temperature (Tcp).
Experimental
Materials
1,4-Dioxane (99.8%), NIPAM (97%), azobisisobutyronitrile (AIBN) (98%), tert-butyl acrylate (98%, 10–20 ppm monomethyl ether hydroquinone inhibitor), aluminium oxide (neutral, Brockmann I) and hydrochloric acid (37%, RG) were purchased from Sigma Aldrich and used as received, unless mentioned otherwise. Sodium chloride (>99.5%) was bought from Acros Organics. Methanol (HPLC grade), dichloromethane (DCM) (AR), diethyl ether (AR), 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) (AR) and n-hexane (HPLC grade) were purchased from Biosolve and used as received, unless mentioned otherwise. Sodium hydroxide solution (TitriPUR, 0.1 M) was bought from Merck Chemicals. PNIPAM-b-PDMAEMA was purchased from Polymer Source, Mn 38.2 kDa and PDI 1.05, ESI Fig. 1.† The chain transfer agent S,S′-bis(α,α′-dimethyl-α′′-acetic acid) trithiocarbonate (BDAT) was synthesized using a previously described method.26,27Triblock synthesis and characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :R:i as 80:1:0.2, [m] 0.8 M). The reactants were dissolved and the mixture was purged with nitrogen for 60 minutes. The polymerization took place at 70 °C for 85 minutes. Subsequently, the reaction was quenched by exposure to air and rapid cooling. The resulting polymer was purified by precipitation in diethyl ether. The final product was dried under vacuum. 1H-NMR (400 MHz, D2O, ESI Fig. 2†): δ 1.05 (s, 6H), 1.36–2.1 (m, 3H), 3.80 (s, 1H), 6.24 (s, 1H). GPC: Mn 7.6 kDa and PDI 1.26.
:R:i as 380:1:0.2, [m] 3.9 M). The reactants were dissolved and the mixture was purged with nitrogen for 60 minutes. The polymerization took place for 55 minutes at 70 °C. The reaction was quenched by exposure to air and rapid cooling. The polymer was purified by precipitation in a cold methanol/water mixture, 3/1 v/v. A dry product was obtained by redissolving in minimal DCM and subsequent drying under vacuum. 1H-NMR (400 MHz, CDCl3): δ 1.11 (s, 6H, (CH2)2 isopropyl), 1.4–1.5 (s, 9H, (CH3)3tert-butyl), 1.6–2.2 (m, backbone), 3.99 (s, 1H, CH isopropyl). 13C-NMR (400 MHz, MeOD): δ 22.38 ((CH3)2 isopropyl), 28.07 ((CH3)3tert-butyl), 36–40 (backbone), 41.9 (CH isopropyl), 42.38 (backbone), 174.12 ((C
:R:i as 80:1:0.2, [m] 0.8 M). The reactants were dissolved and the mixture was purged with nitrogen for 60 minutes. The polymerization took place at 70 °C for 85 minutes. Subsequently, the reaction was quenched by exposure to air and rapid cooling. The resulting polymer was purified by precipitation in diethyl ether. The final product was dried under vacuum. 1H-NMR (400 MHz, D2O, ESI Fig. 2†): δ 1.05 (s, 6H), 1.36–2.1 (m, 3H), 3.80 (s, 1H), 6.24 (s, 1H). GPC: Mn 7.6 kDa and PDI 1.26.
:R:i as 380:1:0.2, [m] 3.9 M). The reactants were dissolved and the mixture was purged with nitrogen for 60 minutes. The polymerization took place for 55 minutes at 70 °C. The reaction was quenched by exposure to air and rapid cooling. The polymer was purified by precipitation in a cold methanol/water mixture, 3/1 v/v. A dry product was obtained by redissolving in minimal DCM and subsequent drying under vacuum. 1H-NMR (400 MHz, CDCl3): δ 1.11 (s, 6H, (CH2)2 isopropyl), 1.4–1.5 (s, 9H, (CH3)3tert-butyl), 1.6–2.2 (m, backbone), 3.99 (s, 1H, CH isopropyl). 13C-NMR (400 MHz, MeOD): δ 22.38 ((CH3)2 isopropyl), 28.07 ((CH3)3tert-butyl), 36–40 (backbone), 41.9 (CH isopropyl), 42.38 (backbone), 174.12 ((C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)N acrylamide).
O)N acrylamide).
The resulting tert-butyl acrylate copolymer was deprotected by dissolving in HFIP containing 0.12 M hydrochloric acid and the mixture was stirred for 3 hours.28 The sample was dried under vacuum and redissolved in water, by adding a 0.1 M sodium hydroxide solution. The polymer solution was centrifuged to remove any insoluble by-products of the deprotection and was further purified by dialysis. The final product was obtained after freeze drying. 1H-NMR (400 MHz, MeOD, ESI Fig. 3†): δ 1.16 (s, 6H, (CH3)2 isopropyl), 1.4–2.3 (m, backbone), 3.97 (s, 1H, CH isopropyl). 13C-NMR (400 MHz, D2O, ESI Fig. 4†): δ 21.65 ((CH3)2 isopropyl), 35–40 (backbone), 41.8 (CH isopropyl), 44.80 (backbone), 175.4 ((CO)N acrylamide), 183.4 ((CO)OH carboxylic acid). GPC: Mn 26.8 kDa and PDI 1.59.
Methods
LS was used to determine the cloud point temperature Tcp. In this article, the Tcp of the assemblies is defined as the temperature where the scattering intensity has doubled compared to its value at 17 °C for 0.75 M NaCl, and 25 °C for all other samples. The data used for this determination are shown in ESI Fig. 8.†
θ), where 2θ represents the observation angle. Silver behenate was used for the calibration of the beam centre and the q-range. Samples were contained in 2 mm quartz capillaries (Hilgenberg GmbH, Germany) and the temperature was controlled using a Julabo heating circulator. The two-dimensional SAXS patterns were azimuthally averaged to obtain one-dimensional SAXS profiles.
Results and discussion
NaCl concentrations up to 0.75 M
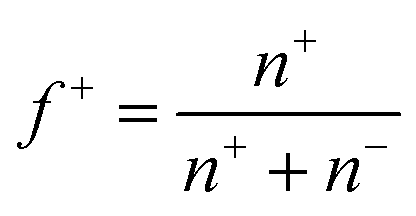 , was set at 0.5 (ESI Fig. 7†) by adding equal amounts of DMAEMA and AA monomers, which should approximately result in a net zero charge at pH 6.5. To verify this, zetapotential measurements were performed, and the ratio between the monomers was adjusted when needed. The samples were investigated using light scattering while increasing the temperature (ESI Fig. 8†). At room temperature, a single phase is observed, while at elevated temperatures, an aggregated system is found. To show the transition between these systems, a summary of the LS data is given in Fig. 2. In the single phase system at salt concentrations below 0.75 M, most often objects with well-defined and monodisperse hydrodynamic radii are observed at room temperature (ESI Fig. 9†). CONTIN analysis revealed a monomodal decorrelation curve, suggesting the assembly of the polymers into well-defined objects, such as C3Ms.
, was set at 0.5 (ESI Fig. 7†) by adding equal amounts of DMAEMA and AA monomers, which should approximately result in a net zero charge at pH 6.5. To verify this, zetapotential measurements were performed, and the ratio between the monomers was adjusted when needed. The samples were investigated using light scattering while increasing the temperature (ESI Fig. 8†). At room temperature, a single phase is observed, while at elevated temperatures, an aggregated system is found. To show the transition between these systems, a summary of the LS data is given in Fig. 2. In the single phase system at salt concentrations below 0.75 M, most often objects with well-defined and monodisperse hydrodynamic radii are observed at room temperature (ESI Fig. 9†). CONTIN analysis revealed a monomodal decorrelation curve, suggesting the assembly of the polymers into well-defined objects, such as C3Ms.
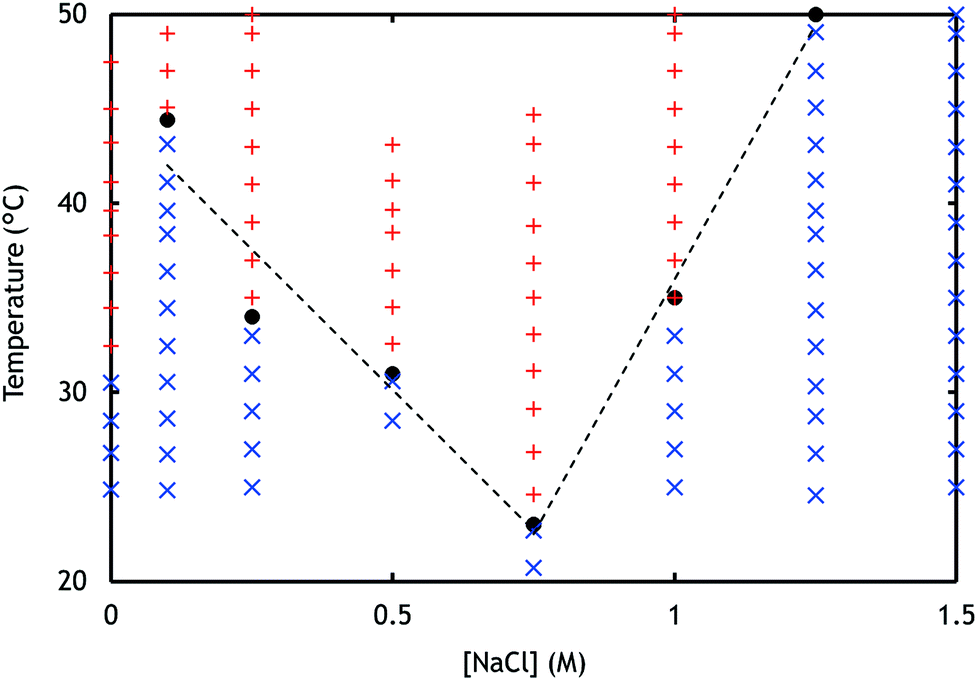 | ||
| Fig. 2 Light scattering was used to determine the Tcp of samples containing PNIPAM-b-PDMAEMA and PNIPAM-b-PAA-b-PNIPAM with varying concentrations of NaCl. The results were used to create a phase diagram; the blue crosses represent a one phase system, and the red plusses represent an aggregated system. The dotted lines represent the phase boundary that was deduced from the observed Tcps (black dots). | ||
The assembly of the polymers into C3Ms can be explained by the interactions between the polyelectrolytes and the solubility of PNIPAM under these conditions. Mixed homopolymers of PDMAEMA and PAA, of block lengths comparable to the electrolyte blocks used in this research, form complexes below the critical salt concentration (cs) of approximately 1.1 M NaCl.3 This means that below 0.75 M NaCl, complex coacervation between the PDMAEMA and PAA blocks can occur. PNIPAM, on the other hand, is soluble at room temperature and at salt concentrations of 0.75 M NaCl and below. Regarding the well-defined objects observed in LS, the PNIPAM blocks are able to solubilize the complexed polyelectrolytes, resulting in C3Ms with a PNIPAM corona.
From LS, an estimated size for the C3Ms can be obtained. For 0.5 M NaCl, an Rh of 31 nm was observed. This value is similar to values that were reported before.9,13 By using LS, Voets et al. observed C3Ms with an apparent Rh of 31.3 ± 0.9 nm that were composed of poly(N-methyl-2-vinyl pyridinium iodide)-b-poly(ethylene oxide), PM2VP38-b-PEO211, and PAA55-b-PNIPAM88. Park et al. observed micelles upon mixing poly(2-isopropyl-2-oxazoline)-b-poly(lysine), PiPrOx45-b-P(Lys)85, PiPrOx-b-poly(aspartic acid), and PiPrOx45-b-P(Asp)76, with an apparent Rh of 22.6 nm by LS.
Upon sufficient temperature increase, the aggregation of the micelles from solution was observed as is indicated by a sudden increase in the radius, scattering intensity, and/or polydispersity in the LS data (ESI Fig. 8 and 10†).17 In Fig. 2, the determined Tcp (black dots) are shown for all the measured salt concentrations. It was observed that below 0.75 M NaCl, the Tcp decreases with increasing salt concentration. This trend can be attributed to the salt concentration dependent solubility of PNIPAM. PNIPAM solubility decreases when salt concentrations increase, which is expressed by a decreasing LCST and thus collapsing of the polymer chain at lower temperatures (ESI Fig. 21†).23,29 A similar behaviour was observed not only for PNIPAM containing micelles but also for micelles containing different temperature-responsive blocks.13 After sufficient cooling of the samples, aggregates disappeared again and radii similar to the sizes observed before heating were measured, demonstrating the reversibility of the system.
The Tcp observed at 0 M NaCl deviates from the tendency of increasing Tcp with decreasing salt concentration. Without added salt, oppositely charged polyelectrolytes strongly interact immediately upon mixing, through electrostatic bonds with long relaxation times. This most likely results in a heterogeneous and kinetically trapped system. The system may not have enough mobility to arrange in stable micelles, which in turn will result in a lower Tcp.
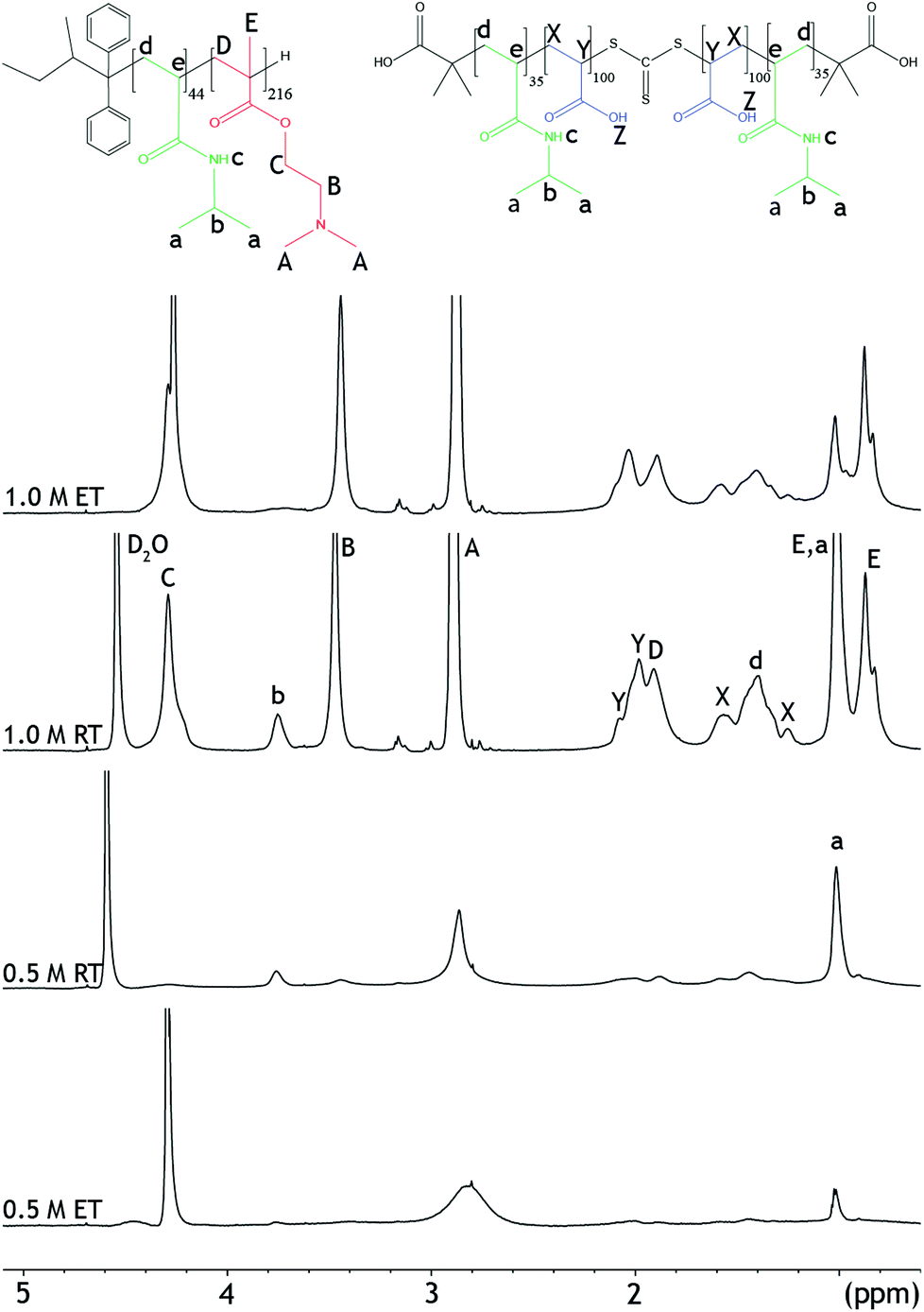 | ||
| Fig. 3 1H-NMR spectra of mixtures of PNIPAM-b-PDMAEMA and PNIPAM-b-PAA-b-PNIPAM in 0.5 M and 1.0 NaCl, at 27 °C (RT), and 67 °C (ET). | ||
NOESY-NMR is a 2D-NMR technique that establishes correlations between chemical moieties that are in close proximity through space, i.e. within 1 nm distance from each other. Therefore, this NMR technique can also be used to verify the proposed micelle morphologies.14 ESI Fig. 15† depicts the NOESY spectrum at 0.5 M NaCl. A cross-peak between the PDMAEMA and PAA (2.1;2.9) can be observed in the spectrum at room temperature.
Furthermore, cross-peaks can be observed between peaks belonging to the same polymer block, e.g. for PNIPAM peak b at 3.9 ppm. In NOESY spectra obtained from unmixed polymer solutions, cross-peaks can only be observed between PNIPAM peaks, or only between polyelectrolyte peaks (ESI Fig. 19 and 20†). Altogether, the data show that PAA and PDMAEMA are close in space when the polymers are mixed. Therefore, both 1D and 2D NMR techniques indicate the presence of C3Ms, with a water insoluble complex coacervate core and a hydrated PNIPAM corona at low salt concentration and temperature.
Additionally, NMR experiments were performed at elevated temperatures to investigate whether the aggregation of the micelles leads to differences in the morphology between the samples at different salt concentrations. At 67 °C, the peak splitting and peak intensities of the PDMAEMA (peaks A and B), and the PNIPAM (peaks a and b) decreased even further, Fig. 3. This reflects the decreased solubility of the micelles at elevated temperatures. In NOESY, cross-peaks that were present between DMAEMA and PAA at room temperature disappeared after increasing the temperature (ESI Fig. 16†). Both observations can be explained by the reduced solubility of the polymers resulting from the aggregation, which reduces visibility in NMR. After cooling the samples back to room temperature, similar 1H spectra could be obtained as before heating, showing the reversibility of the system (ESI Fig. 13†).
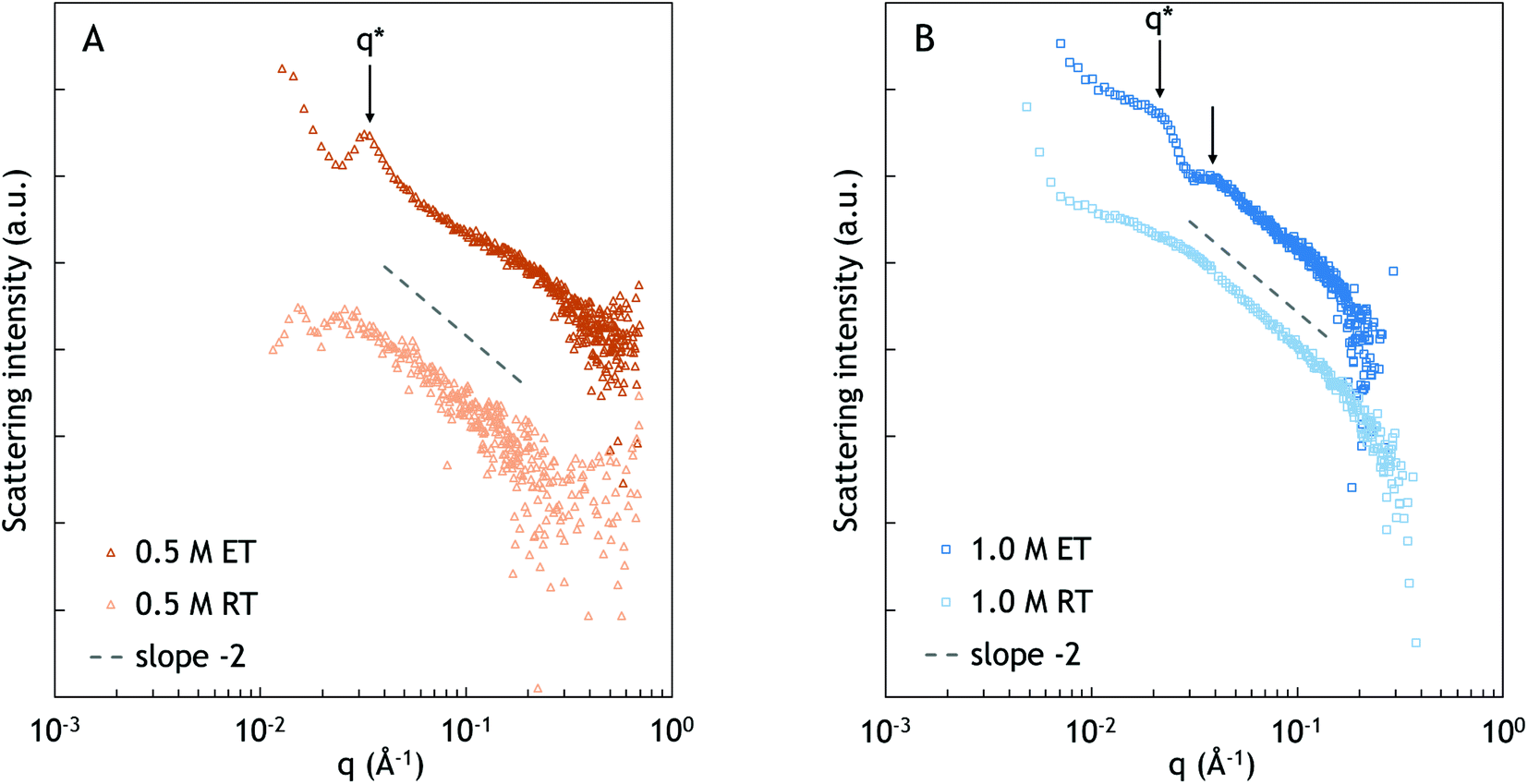 | ||
| Fig. 4 SAXS spectra of PNIPAM-b-PDMAEMA and PNIPAM-b-PAA-b-PNIPAM in 0.5 M (A) and 1.0 M (B) NaCl at 27 °C (RT) and 67 °C (ET). As a guide to the eye, a dashed line is displayed to indicate a slope of −2. The presented data were corrected for solvent contributions and plotted on a log–log scale. Scattering intensities of the ET data were adjusted for clear display. | ||
The SAXS spectra at an elevated temperature exhibit more morphological features compared to the spectra at room temperature. At 67 °C, a peak appears at 0.032 Å−1, corresponding to a characteristic distance of approximately 20 nm. It is plausible that the observed distance corresponds to the typical distance between PNIPAM and complex coacervate domains. However, the lack of higher order peaks suggests that there is no specific long range arrangement of the domains.
To obtain monodisperse micelle solutions, the equilibration of the samples is a key factor. Immediately after the preparation of the 0.5 M NaCl samples, small pieces with a gel like structure appeared in the 0.5 M NaCl solutions and dissolved over time. The dissolution of these solid pieces could be sped up by refrigerating, which is also reported by Park et al.13 Furthermore, for elevated polymer concentrations at room temperature, micelle solutions at low salt concentrations were not fully stable and after one day, sediments of a complex coacervate coexisted with a dilute phase which contained micelles. This observation can be explained by the short PNIPAM chains that are likely not able to completely stabilize the micelles in solutions with higher polymer concentrations, resulting in aggregation and sedimentation. A similar behaviour was observed by De Santis et al. who investigated C3Ms with different sizes of water-soluble blocks.17 Micelles composed of block copolymers with the smallest stabilizing blocks formed aggregates, while the other micelles did not. Bayati et al. also observed the aggregation of PNIPAM-containing micelles but underlined that PNIPAM can not only aggregate above the LCST, but also below the LCST due to weak hydrophobic interactions, leading to aggregation as well.14 Furthermore, they observed that increasing polymer concentrations resulted in increased aggregation, similar to our findings.
NaCl concentrations from 0.75 M and above
With LS, polydisperse structures are observed. As a control, angle dependent LS was performed on samples containing 1.25 M NaCl and 0.01 M charged groups. These measurements resulted in a linear relationship between the decay rate Γ and q2, which only slightly deviated at high q. The linear decay indicates the presence of spherical particles at elevated salt concentrations, instead of, for example, cylindrical objects. The triblock PNIPAM-b-PAA-b-PNIPAM can form bridges between the different micelles, as is schematically shown in Fig. 1e. The presence of interconnected micelles at higher polymer concentrations could be an explanation for the polydisperse structures observed with LS.
For PCMs, aggregation is observed with increasing Tcp for higher salt concentrations, Fig. 2, while at lower salt concentrations, a decrease in Tcp was observed. The aggregation of PCMs is caused by the reoccurrence of complexation of the polyelectrolytes, which can be explained by two phenomena. Firstly, complex coacervation is most likely entropically driven, thus temperature dependent.31 Secondly, by increasing the temperature, the cs,cr also increases. When cs,cr exceeds the salt concentration of the sample, complex coacervates can be formed. For higher salt concentrations, higher temperatures are needed to exceed the cs,cr. This behaviour was observed before for PAA/PDMAEMA homopolymers and in addition to other homopolymer couples, such as poly(trimethyl amino ethyl methacrylate) (PTMAEMA) and poly(sulphopropyl methacrylate) (PSPMA).31 The influence of the LCST of PDMAEMA on the aggregation at a high salt concentration is considered unlikely, as charges on the polyelectrolyte prevent LCST behaviour. At this pH, the PDMAEMA is charged, and the charges cannot be screened by PAA due to the high salt concentration. Increasing Tcp with increasing NaCl concentrations is therefore likely the result of increased polyelectrolyte solubility, and a complex coacervation-driven aggregation of the PCMs.
With LS, different tendencies in the radii can be observed with increasing temperature, between the samples at low and high salt concentrations, ESI Fig. 8.† Below 1.0 M NaCl, the radius steeply increases with increasing temperature, while at a high salt concentration, first a decrease in the radius is observed, followed by an increase. This tendency might be explained by a collapse of either the core or the corona preceding aggregation.32 As a result, the scattering intensity would increase while the micellar radius decreases, as is observed at 1.0 M NaCl.
Equal to samples containing less than 0.75 M NaCl, sedimentation was observed for the samples prepared above 0.75 M NaCl. However, the sedimentation only occurred after a couple of days. The polyelectrolyte blocks of the PCMs are much larger than the insoluble PNIPAM blocks under these conditions and therefore more efficiently stabilize the micelles without aggregating. Therefore, higher polymer concentrations could also be obtained without visible aggregation within a day.
At 67 °C, the intensity of the peaks has slightly decreased and the proton peak of the PNIPAM has even disappeared, Fig. 3. This indicates a reduced solubility at temperatures above the LCST. Also at this salt concentration, the temperature transition is a reversible process, ESI Fig. 14.† Furthermore, cross-peaks between PDMAEMA and PAA, 2.1;2.9 ppm, appear in the NOESY spectra when the temperature is increased, ESI Fig. 18.† The appearance of the cross-peaks indicates reoccurring complex coacervation at elevated temperatures.33
At elevated temperature, two broad peaks can be observed at approximately 0.021 and 0.037 Å−1, as is indicated with the black arrows in Fig. 4B. Similar to 0.5 M NaCl, the peaks likely originate from a typical distance between PNIPAM and complex coacervate domains. The broad higher order peak at q ≈ 0.037 Å−1 indicates ordering over longer distances. However, the shape and order of the domains cannot be determined from the spectrum. From q*, the characteristic distance between the domains is calculated as approximately 30 nm.34–37 Compared to the sample at 0.5 M NaCl, the characteristic distance is larger, which could be related to the difference in the salt concentration, and thus the difference in the water content, as was also observed in the peak splitting in NOESY-NMR. The sample with a high salt concentration has weaker interpolyelectrolyte interactions, leading to a looser structure and lower polymer concentration, and thus larger distances between the PNIPAM domains, as was also found by Krogstad et al.3,34
Conclusions
By mixing oppositely charged polyelectrolytes functionalized with PNIPAM, different micelle morphologies could be obtained at room temperature. At low salt concentrations, C3Ms were observed with a PNIPAM corona and a polyelectrolyte core. At sufficiently high salt concentrations, the micelles turned inside out, into PCMs with a water-soluble polyelectrolyte corona and a PNIPAM core. Both micelles displayed a salt dependent Tcp, leading to aggregation and sedimentation in aqueous solution. The resulting concentrated phase contains domains of PNIPAM and complex coacervate with a salt concentration dependent separation distance. The ability to adjust the morphology and solubility of the micelles can be an important tool to apply complex coacervates as drug delivery vehicles, or as underwater or medical adhesives.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank Dr Merve Mocan for providing the RAFT agent BDAT. Ing. Marco Hendrix is thanked for his help while operating the SAXS equipment at TU/e ICMS, Eindhoven, The Netherlands. Dr Pieter de Waard is thankfully acknowledged for his help with the NMR measurements at the magnetic resonance facility MAGNEFY in Wageningen, The Netherlands. This research was financially supported by the Netherlands Organization for Scientific Research (NWO) via a Vidi innovational research grant.References
- E. Spruijt, M. A. C. Stuart and J. van der Gucht, Macromolecules, 2013, 46, 1633–1641 CrossRef CAS.
- J. van der Gucht, E. Spruijt, M. Lemmers and M. A. C. Stuart, J. Colloid Interface Sci., 2011, 361, 407–422 CrossRef CAS PubMed.
- E. Spruijt, A. H. Westphal, J. W. Borst, M. A. C. Stuart and J. van der Gucht, Macromolecules, 2010, 43, 6476–6484 CrossRef CAS.
- R. J. Stewart, C. S. Wang and H. Shao, Adv. Colloid Interface Sci., 2011, 167, 85–93 CrossRef CAS PubMed.
- S. Kaur, G. M. Weerasekare and R. J. Stewart, ACS Appl. Mater. Interfaces, 2011, 3, 941–944 CrossRef CAS PubMed.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2012, 64, 18–23 CrossRef.
- D. Priftis and M. Tirrell, Soft Matter, 2012, 8, 9396–9405 RSC.
- A. H. Hofman, I. A. van Hees, J. Yang and M. Kamperman, Adv. Mater., 2018, 30, 1704640 CrossRef PubMed.
- I. K. Voets, P. M. Moll, A. Aqil, C. Jerome, C. Detrembleur, P. de Waard, A. de Keizer and M. A. C. Stuart, J. Phys. Chem. B, 2008, 112, 10833–10840 CrossRef CAS PubMed.
- A. Harada and K. Kataoka, Macromolecules, 1995, 28, 5294–5299 CrossRef CAS.
- D. V. Pergushov, A. H. E. Muller and F. H. Schacher, Chem. Soc. Rev., 2012, 41, 6888–6901 RSC.
- N. Rapoport, Prog. Polym. Sci., 2007, 32, 962–990 CrossRef CAS.
- J. S. Park, Y. Akiyama, Y. Yamasaki and K. Kataoka, Langmuir, 2007, 23, 138–146 CrossRef CAS PubMed.
- S. Bayati, K. E. Bergquist, K. Z. Zhu, B. Nystrom, J. S. Pedersen, L. Galantini and K. Schillen, J. Polym. Sci., Part B: Polym. Phys., 2017, 55, 1457–1469 CrossRef CAS.
- G. Masci, S. De Santis and C. Cametti, J. Phys. Chem. B, 2011, 115, 2196–2204 CrossRef CAS PubMed.
- H. Dautzenberg, Y. B. Gao and M. Hahn, Langmuir, 2000, 16, 9070–9081 CrossRef CAS.
- S. De Santis, R. D. Ladogana, M. Diociaiuti and G. Masci, Macromolecules, 2010, 43, 1992–2001 CrossRef CAS.
- C. Dahling, G. Lotze, H. Mori, D. V. Pergushov and F. A. Plamper, J. Phys. Chem. B, 2017, 121, 6739–6748 CrossRef PubMed.
- J. Li, W. D. He, N. He, S. C. Han, X. L. Sun, L. Y. Li and B. Y. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1450–1462 CrossRef CAS.
- M. Kamimura, J. O. Kim, A. V. Kabanov, T. K. Bronich and Y. Nagasaki, J. Controlled Release, 2012, 160, 486–494 CrossRef CAS PubMed.
- M. A. Ward and T. K. Georgiou, Polymers, 2011, 3, 1215–1242 CrossRef CAS.
- K. J. Krzyminski, M. Jasionowski and A. Gutowska, Polym. Int., 2008, 57, 592–604 CrossRef CAS.
- T. Patel, G. Ghosh, S. Yusa and P. Bahadur, J. Dispersion Sci. Technol., 2011, 32, 1111–1118 CrossRef CAS.
- J. Adelsberger, A. Kulkarni, A. Jain, W. N. Wang, A. M. Bivigou-Koumba, P. Busch, V. Pipich, O. Holderer, T. Hellweg, A. Laschewsky, P. Muller-Buschbaum and C. M. Papadakis, Macromolecules, 2010, 43, 2490–2501 CrossRef CAS.
- H. Feil, Y. H. Bae, J. Feijen and S. W. Kim, Macromolecules, 1993, 26, 2496–2500 CrossRef CAS.
- M. Cetintas, J. de Grooth, A. H. Hofman, H. M. van der Kooij, K. Loos, W. M. de Vos and M. Kamperman, Polym. Chem., 2017, 8, 2235–2243 RSC.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS.
- A. D. Filippov, I. A. van Hees, R. Fokkink, I. K. Voets and M. Kamperman, Macromolecules, 2018, 51, 8316–8323 CrossRef CAS PubMed.
- Y. J. Zhang, S. Furyk, D. E. Bergbreiter and P. S. Cremer, J. Am. Chem. Soc., 2005, 127, 14505–14510 CrossRef CAS PubMed.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- E. Spruijt, Strength, structure and stability of polyelectrolyte complex coacervates, Wageningen University, Wageningen, 2012 Search PubMed.
- C. M. Li, N. J. Buurma, I. Haq, C. Turner, S. P. Armes, V. Castelletto, I. W. Hamley and A. L. Lewis, Langmuir, 2005, 21, 11026–11033 CrossRef CAS PubMed.
- H. Wu, J. M. Ting, O. Werba, S. Q. Meng and M. V. Tirrell, J. Chem. Phys., 2018, 149, 163330 CrossRef PubMed.
- D. V. Krogstad, N. A. Lynd, S. H. Choi, J. M. Spruell, C. J. Hawker, E. J. Kramer and M. V. Tirrell, Macromolecules, 2013, 46, 1512–1518 CrossRef CAS.
- A. V. Ruzette, S. Tence-Girault, L. Leibler, F. Chauvin, D. Bertin, O. Guerret and P. Gerard, Macromolecules, 2006, 39, 5804–5814 CrossRef CAS.
- J. K. Kim, H. H. Lee, S. Sakurai, S. Aida, J. Masamoto, S. Nomura, Y. Kitagawa and Y. Suda, Macromolecules, 1999, 32, 6707–6717 CrossRef CAS.
- F. M. Abuzaina, A. J. Patel, S. Mochrie, S. Narayanan, A. Sandy, B. A. Garetz and N. P. Balsara, Macromolecules, 2005, 38, 7090–7097 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9py00250b |
| This journal is © The Royal Society of Chemistry 2019 |