
Photoinduced C–H bond fission in prototypical organic molecules and radicals
Michael N. R.
Ashfold
 *a,
Rebecca A.
Ingle†
a,
Tolga N. V.
Karsili
b and
Jingsong
Zhang
c
*a,
Rebecca A.
Ingle†
a,
Tolga N. V.
Karsili
b and
Jingsong
Zhang
c
aSchool of Chemistry, University of Bristol, Bristol, BS8 1TS, UK. E-mail: mike.ashfold@bristol.ac.uk
bDepartment of Chemistry, University of Louisiana, Lafayette, LA 70504, USA
cDepartment of Chemistry, University of California at Riverside, Riverside, California 92521, USA
First published on 16th January 2019
Abstract
Recent experimental and computational advances have heralded huge progress in the range and the detail of the database pertaining to photoinduced C–H bond fission processes. This Perspective provides a snapshot of the current state of knowledge as determined via gas phase (i.e. isolated molecule) studies of the primary photochemistry of families of hydrocarbon molecules (alkynes, alkenes, alkanes, aromatics and selected heteroatom containing analogues) and the corresponding radicals (including saturated and unsaturated hydrocarbon radicals). Different families show different and, in many cases, understandable propensities for dissociating from an excited electronic state or following non-adiabatic coupling (i.e. internal conversion) to high vibrational levels of the ground electronic state. The Perspective seeks to emphasise the potentially vast range of behaviours (dissociation timescales, product energy disposals, etc.) that can be expected to accompany internal conversion, reflecting the extent to which the tuning coordinate (i.e. the nuclear motions that tune the energy separation between the excited and ground state) projects onto the dissociation coordinate of interest (i.e. the breaking of the C–H bond).
1. Introduction
The recently concluded Cassini–Huygens mission has yielded a wealth of new data concerning the outer planets, their moons, and their atmospheres. The atmosphere of Saturn's moon Titan is now known to be composed mainly (∼98%) of nitrogen, with methane making up most of the remainder. Molecules in the upper atmosphere of Titan undergo photodissociation, by absorbing short wavelength (vacuum ultraviolet, VUV) solar radiation, yielding radical species. These, in turn, undergo chemical processing, forming a plethora of heavier hydrocarbons like ethane, propane, ethene, acetylene, methylacetylene, etc., and nitriles like hydrogen cyanide, acetonitrile, cyanoacetylene, etc.1–4 A quantitative knowledge of the primary photochemistry of such carbonaceous species is essential for any detailed analyses and modelling of the atmospheres of the outer planets and of moons like Titan.5Photoinduced C–H bond fissions are also of fundamental interest. The recent literature contains numerous articles, theoretical and experimental, that highlight the central role of excited states formed by electron promotion to an antibonding σ* orbital in facilitating bond fission.6–9 Such states, formed by exciting an electron from an occupied lone pair (n) or bonding (π or σ) orbital, are now recognised as pivotal in discussions of the UV photofragmentation dynamics in many broad families of molecules. Exemplars include: water, alcohols, phenols, ethers and their thio-analogues; ammonia, amines, azoles, etc.; unsaturated molecules like hydrogen cyanide, acetylene and their derivatives; and alkyl and aryl halides.7,10 nσ*/πσ* excited states have also been implicated in UV photoinduced ring-opening processes, the dynamics of which are also now attracting interest.11 Photoinduced C–H bond fissions in alkanes, as well as in alkenes, benzene and larger aromatic systems, are under-represented in most such discussions, however. The present work, in which we review and attempt to systematize prior studies of photoinduced C–H (and, on occasion, C–R, R = alkyl, aryl, etc.) bond fissions in both closed and open shell (i.e. radical) organic species, seeks to rectify this deficiency. Note, we have not attempted a comprehensive review of all hydrocarbon (and related) photochemistry. The foci of this article are the title photoinduced bond fissions in neutral precursors and under gas phase (collision-free) conditions, though we do note rival fragmentation channels when appropriate.
Fig. 1 shows potential energy curves (PECs) for the ground and a few of the lower lying excited electronic states of (a) H2, (b) methane and (c) acetylene that help set the scene for what follows. The highest (indeed the only) occupied molecular orbital (HOMO) in the ground (X1Σ+g) state of H2 is the 1σg orbital. Promoting one electron from this orbital to the antibonding (1σu*) orbital yields the a3Σ+u excited state. As Fig. 1(a) shows, the PEC for this state is repulsive. The a3Σ+u state is dissociative and is one of just two states (the other being the ground state) that correlate with the asymptotic products H(1s) + H(1s). The lowest lying bound excited state of H2 is the B1Σ+u state (attributable to a 2pσu←1σg orbital promotion), which correlates with the excited H(1s) + H(n = 2) products. [Note: all PECs reported here were calculated specifically for this article using the complete active space second order perturbation theory (CASPT2) method and MOLPRO2010.12 To avoid breaking the narrative, details of the methods, basis sets and active spaces employed in the various calculations are collected in the Appendix.]
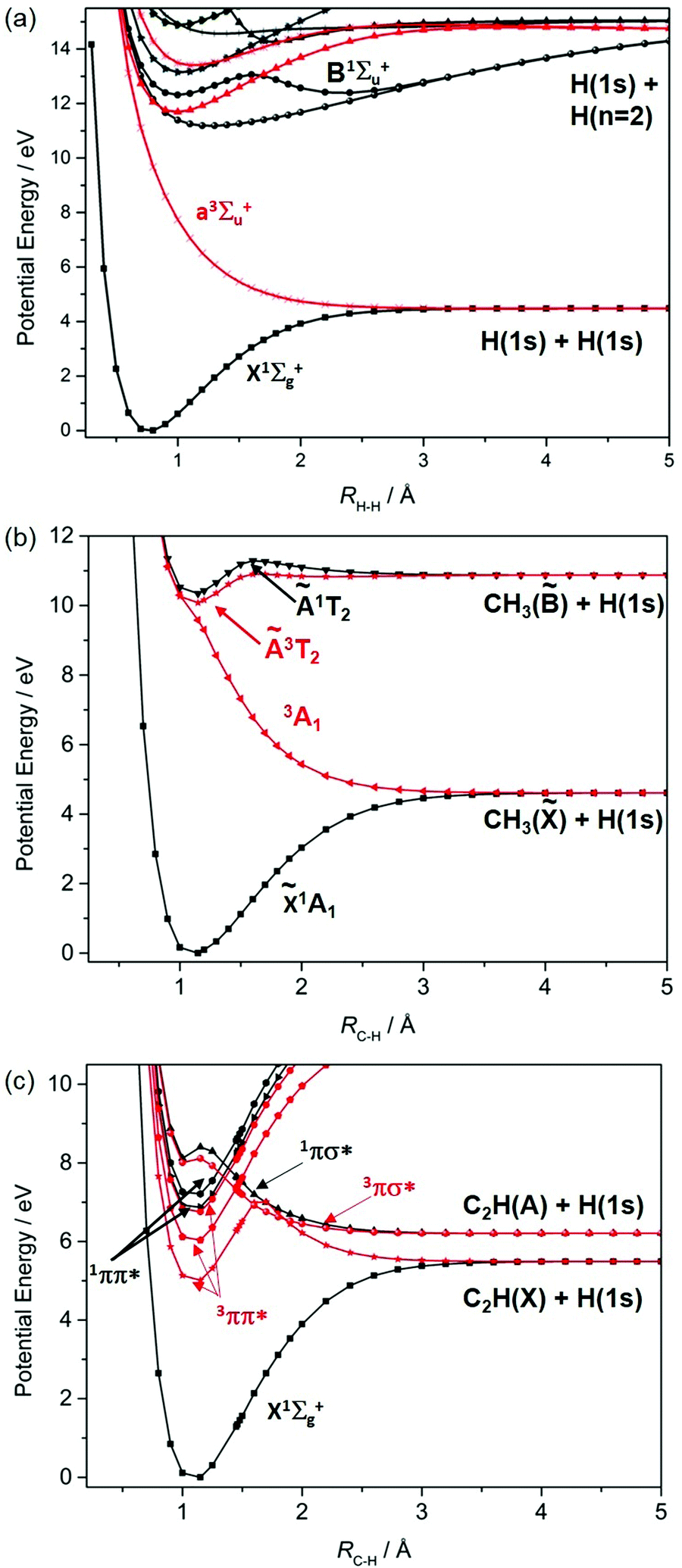 | ||
| Fig. 1 Singlet (in black) and triplet (in red) PECs for (a) H2 and, as a function of RC–H, for (b) CH4 and (c) HCCH. The latter two sets of curves are rigid body scans, calculated by progressively extending one C–H bond while holding the rest of the nuclear framework fixed at its ground state equilibrium geometry, with the constraint that the overall symmetry remains as C3v and C∞v, respectively. | ||
These PECs for H2 were established long ago,13 but are included to highlight synergies with a (limiting) cut through the multi-dimensional potential energy surfaces (PESs) for a saturated hydrocarbon like CH4. Fig. 1(b) shows the relevant cut, calculated by stepping one C–H bond length (RC–H), whilst holding the rest of the molecular framework at the ground state equilibrium geometry (i.e. retaining C3v symmetry in this case). As in H2, the lowest energy products (H(1s) + CH3(![[X with combining tilde]](https://www.rsc.org/images/entities/char_0058_0303.gif) )) each possess one unpaired electron but are otherwise non-degenerate. Their recombination yields one singlet state of CH4 (the ground state) and a dissociative triplet state with an electronic configuration at large RC–H that is dominated by a bond-localised σ*←σ excitation. As we will see, such a 3σσ* PES correlating to ground state products is a characteristic feature of all closed shell hydrocarbons. The first spin-allowed transition from the ground state of CH4 is to a singlet state formed by promoting an electron from the t2g HOMO to an orbital with substantial 3s Rydberg character in the Franck–Condon (FC) region. Fig. 1(b) shows this excited state correlating with an excited (Rydberg) state of CH3 upon extending RC–H. The much richer photofragmentation dynamics that prevail once the C3v symmetry constraint is lifted are described in Section 2.4, while the photochemistry exhibited by
)) each possess one unpaired electron but are otherwise non-degenerate. Their recombination yields one singlet state of CH4 (the ground state) and a dissociative triplet state with an electronic configuration at large RC–H that is dominated by a bond-localised σ*←σ excitation. As we will see, such a 3σσ* PES correlating to ground state products is a characteristic feature of all closed shell hydrocarbons. The first spin-allowed transition from the ground state of CH4 is to a singlet state formed by promoting an electron from the t2g HOMO to an orbital with substantial 3s Rydberg character in the Franck–Condon (FC) region. Fig. 1(b) shows this excited state correlating with an excited (Rydberg) state of CH3 upon extending RC–H. The much richer photofragmentation dynamics that prevail once the C3v symmetry constraint is lifted are described in Section 2.4, while the photochemistry exhibited by ![[B with combining tilde]](https://www.rsc.org/images/entities/char_0042_0303.gif) state CH3 radicals is detailed in Section 3.2.
state CH3 radicals is detailed in Section 3.2.
Fig. 1(c) shows corresponding scans for the ground and first few excited states of C2H2, calculated by extending RC–H with the remainder of the molecule maintaining its ground state (linear) minimum energy geometry. The additional state density (cf. CH4) arising as a result of the π (HOMOs) and the antibonding π* orbitals is obvious, as are the parallels with the corresponding PECs for the isoelectronic species HCN.7 Unlike the CH3 fragment formed upon C–H bond fission in CH4, the C2H radical formed by extending a C–H bond in C2H2 has two low-lying electronic states distinguished by whether the unpaired electron is in a pσ or pπ orbital. Fig. 1(c) shows that the former configuration constitutes the ground (X2Σ+) state of C2H, and that the H + C2H(X) products correlate with the ground state (and a dissociative 3σσ* state) of C2H2. The low-lying A2Π excited state of the radical also forms both singlet and triplet states upon recombining with an H atom. These 1Π and 3Π states of C2H2 (in the linear limit) derive from 3s/σ*←π(HOMO) excitations, i.e. excitations to states that have some 3s Rydberg character in the FC region but which acquire predominant σ* antibonding character upon extending RC–H. For compactness, we henceforth refer to such states simply as πσ* states. We also note that the first excited singlet (and triplet) states of C2H2 in the FC region arise from π*←π excitations. These valence excited states are bound in the RC–H coordinate, but have nonlinear minimum energy geometries and could predissociate by non-adiabatic coupling to one or more states that correlate with the lower energy asymptote(s).
We survey our current understanding of the UV photofragmentation dynamics of C2H2 and higher alkynes in Section 2.1, but a key distinction to note at this stage is the larger number of low-lying dissociation limits in a molecule like C2H2 (cf. the alkanes). Each dissociation limit in PE diagrams like those in Fig. 1 corresponds to a different electronic state of one or other dissociation product. In the specific cases featured here, one dissociation product is an H atom, the first excited state of which (with n = 2) lies 10.2 eV above the ground (n = 1) state. Thus, if we limit discussion to excitation energies below the ionisation limits of the molecules of interest (e.g. ∼12.6 eV in the case of CH4), the density of low lying product asymptotes is dictated by the electronic structure of the partner fragment. This is lower in a fragment like CH3 (for which, apart from the one unpaired valence electron, there are only σ bonds) than in, for example, C2H or HCO where the valence electrons also partition into less tightly bound π and, in the latter case, n orbitals. Indeed, much of the recent interest in molecular photofragmentations involving O–H (O–R), S–H (S–R) or N–H (N–R) bond fissions stems from conical intersections between PESs correlating to the various low-lying dissociation limits, and the non-adiabatic couplings enabled by these conical intersections.7
2. Hydrocarbon molecules
2.1 Acetylene, higher alkynes, alkyl analogues and nitriles
Acetylene photodissociation has also been investigated by monitoring the H atom products formed at several shorter excitation wavelengths: at λ = 193.3 nm (still within the π*←π band systems);24 at λ = 148.35 and 151.82 nm;25 at λ = 121.6 nm (the H Lyman-α wavelength)26 and at several wavelengths in the range 121 ≤ λ ≤ 133 nm27,28 chosen to match with peaks in the parent absorption spectrum. All reveal formation of C2H radicals in both their ground (X) and excited (A2Π) states, with the latter dominating at the higher excitation energies. These C2H(A) products are formed with extensive vibrational (predominantly in the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C stretch mode) but little rotational excitation, and the recoil velocities of the H atom partners are anisotropic (relative to the ε vector of the photolysis laser radiation). Emission attributable to C2H(A) photofragments has also been reported following excitation at many wavelengths λ ≤ 125 nm.29 Such energy disposal is consistent with dissociation following excitation to, or efficient predissociation of the photoprepared Rydberg states by, the lowest dissociative 1πσ* state7,25 depicted in Fig. 1(c). The H + C2H(X) products formed at these shorter excitation wavelengths, in contrast, show isotropic recoil velocity distributions that peak at a low total kinetic energy release (TKER) and are generally consistent with that expected on the basis of (slower) unimolecular decay after radiationless transfer to high levels of the ground (X, or S0) state.28
C stretch mode) but little rotational excitation, and the recoil velocities of the H atom partners are anisotropic (relative to the ε vector of the photolysis laser radiation). Emission attributable to C2H(A) photofragments has also been reported following excitation at many wavelengths λ ≤ 125 nm.29 Such energy disposal is consistent with dissociation following excitation to, or efficient predissociation of the photoprepared Rydberg states by, the lowest dissociative 1πσ* state7,25 depicted in Fig. 1(c). The H + C2H(X) products formed at these shorter excitation wavelengths, in contrast, show isotropic recoil velocity distributions that peak at a low total kinetic energy release (TKER) and are generally consistent with that expected on the basis of (slower) unimolecular decay after radiationless transfer to high levels of the ground (X, or S0) state.28
CCCH). No radical products have been reported following long wavelength (λ > 200 nm) excitation of this molecule. Reactions involving metastable C4H2* molecules formed by such photoexcitation were touted as a possible route to forming the larger polyynes and polycyclic aromatic hydrocarbons that contribute to the haze that cloaks Titan,30 but such a view was challenged following later measurements that showed that these C4H2* states have sub-μs lifetimes.31 The TKER spectra of the H + C4H products formed when exciting C4H2 at shorter wavelengths (in the range 127.5 ≤ λ ≤ 164.4 nm) chosen to match with resolved Rydberg features in the parent absorption spectrum show peaks sitting on a continuous background.32,33 Such structure reflects the formation of excited C4H(A2Π) state radicals, with specific vibrational excitation in the CCC bend and CC stretch modes. As with C2H2, these products arise via non-radiative transfer to the corresponding 1πσ* continuum, while the underlying (isotropic) signal – that peaks at low TKER – is logically attributed to unimolecular decay of highly internally excited C4H2(S0) molecules.
CH) photolysis at 193.3 nm were interpreted as showing acetylenic C–H bond fission as the dominant decay process,34–36 as has also been suggested in one recent trajectory surface-hopping theoretical study.37 However, H Rydberg atom photofragment translational spectroscopy (HRA-PTS) studies of propyne and its isomer allene (propadiene), at several wavelengths in the range 193.3 ≤ λ ≤ 213.3 nm, returned essentially identical TKER spectra with a form that matched well with that obtained by assuming an approximate statistical model predicated on population of all possible vibrational states of the propargyl (H2CCCH) product. Such behaviour was rationalised by assuming efficient coupling to high vibrational levels of the S0 state and isomerisation (including H atom migration) prior to fragmentation.38,39 This conclusion served to reinstate the original mechanistic proposal of Seki and Okabe,40 and is consistent with (i) observations that both H and D atom products are formed, with essentially identical translational energy distributions, in the vibrationally mediated photodissociation of CD3CCH using two photons with a total energy very similar to that of a single 193 nm photon,41 and (ii) translational spectroscopy studies of allene42 and propyne43 photolysis at 193 nm which both return a primary yield of H2 products that was about one tenth that of the H atoms.
PTS studies of propyne and allene following excitation at shorter wavelengths (157 nm44 and 121.6 nm39), in contrast, return isomer specific H atom velocity distributions. The photolysis of both molecules, at both wavelengths, yields H atoms with velocity distributions that peak at low KEs attributable to decay of highly internally excited S0 molecules, but propyne also yields more H atoms with higher KE attributable to excited state acetylenic C–H bond fission – reminiscent of that seen in the short wavelength photolysis of both C2H2 and C4H2. Analysis of the photofragment fluorescence excitation spectra obtained following tuneable VUV excitation of propyne and allene also reveals isomer specific photodissociation dynamics at short excitation wavelengths.45 Though outside the scope of a review focussed on photoinduced C–H bond fissions, we note that both CH3 and CH2 products have also been reported in the 157 nm photolysis of propyne, but the dynamics of the C–C bond fission process(es) leading to these products remains unclear.44
H + C3N fragments are the major dissociation products when cyanoacetylene is excited at 193.3 nm, but the low quantum yield estimated for this channel (0.3 ± 0.05) implies substantial population of metastable excited states.47 The TKER distribution of the H atom products derived from ion imaging studies is intriguing.46 The energy provided by a 193 nm photon is ∼0.6 eV above the calculated C–H bond dissociation energy, D0(H–CCCN). Necessarily, therefore, the H atoms are slow, but their velocity distribution is sharply peaked and implies a substantial partitioning of the available energy (i.e. the photon energy less the bond dissociation energy) into product translation. Such energy disposal is characteristic of dissociation on a repulsive excited state PES, encouraging the suggestion that the photofragmentation of cyanoacetylene following excitation at 193.3 nm proceeds via coupling to the 1πσ* continuum. Analogy with C2H2, C4H2 (see above) and HCN (see below) has further encouraged the suggestion that the C3N partner fragments are formed in the first excited A2Π state,46 but the energy resolution of the data reported thus far is insufficient to allow substantiation of this prediction.
2.2 Ethene, higher alkenes, polyenes and carbonyl containing analogues
Much recent effort has been devoted to unravelling details of the nuclear motions and couplings that drive IC from the 1ππ* state to the S0 state. Theory62–65 has identified roles for two general classes of conical intersections, one in regions of configuration space associated with twisted and pyramidal geometries, the other near an ethylidene (CH3CH) configuration that involves an H atom migrating across what (prior to photoexcitation) was the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond. The PECs shown in Fig. 2(a) have been calculated for a sequence of geometries along a linearly interpolated internal coordinate (LIIC) connecting the optimised ground state geometry to that of the minimum energy conical intersection (MECI) linking the 1ππ* and ground state PESs, and are included primarily to illustrate the essentially barrierless nature of this excited state decay route. Two molecular dynamics (MD) studies64,66 also proposed a rival deactivation pathway via the 1π3s Rydberg state. Ultrafast pump–probe ion yield studies (involving both C2H4 and C2D4 excitation at 162 nm67) and a more recent ultrafast pump–probe photoelectron spectroscopy study (at 156 nm68) both serve to validate this suggestion, though another (at 159 nm69) found no evidence for the participation of any Rydberg state in the non-radiative decay of the 1ππ* state. Excited state C–H bond fission involving nuclear motion on a 1πσ* PES analogous to that shown in Fig. 1(c), leading to electronically excited C2H3 radicals, should be possible at sufficiently high excitation energies – as shown in Fig. 2(b), and as noted long ago by Evleth and Sevin70 – but we are not aware of any experimental demonstrations of such an excited state channel competing successfully with the ultrafast non-radiative decay to the S0 state.
C double bond. The PECs shown in Fig. 2(a) have been calculated for a sequence of geometries along a linearly interpolated internal coordinate (LIIC) connecting the optimised ground state geometry to that of the minimum energy conical intersection (MECI) linking the 1ππ* and ground state PESs, and are included primarily to illustrate the essentially barrierless nature of this excited state decay route. Two molecular dynamics (MD) studies64,66 also proposed a rival deactivation pathway via the 1π3s Rydberg state. Ultrafast pump–probe ion yield studies (involving both C2H4 and C2D4 excitation at 162 nm67) and a more recent ultrafast pump–probe photoelectron spectroscopy study (at 156 nm68) both serve to validate this suggestion, though another (at 159 nm69) found no evidence for the participation of any Rydberg state in the non-radiative decay of the 1ππ* state. Excited state C–H bond fission involving nuclear motion on a 1πσ* PES analogous to that shown in Fig. 1(c), leading to electronically excited C2H3 radicals, should be possible at sufficiently high excitation energies – as shown in Fig. 2(b), and as noted long ago by Evleth and Sevin70 – but we are not aware of any experimental demonstrations of such an excited state channel competing successfully with the ultrafast non-radiative decay to the S0 state.
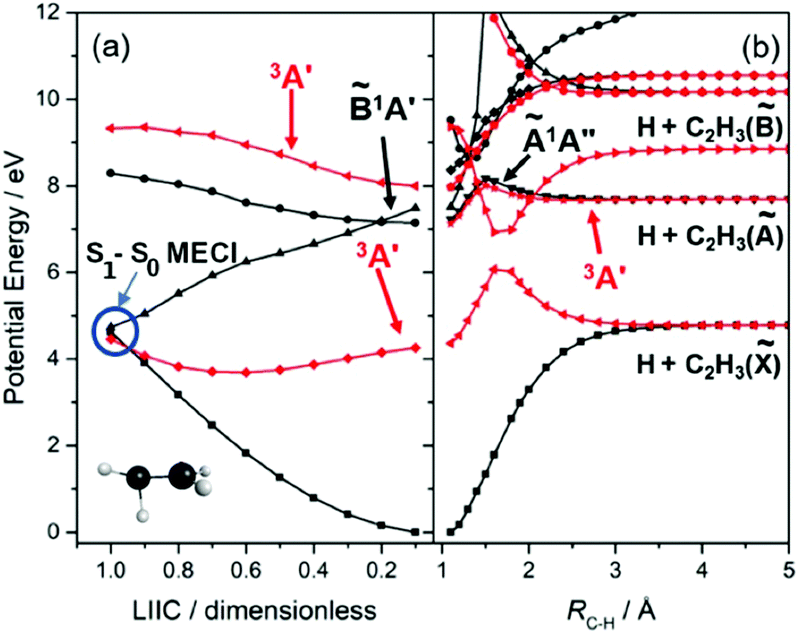 | ||
| Fig. 2 Singlet (in black) and triplet (in red) PECs for C2H4. Panel (a) is for a sequence of geometries along a LIIC connecting the optimised ground state geometry to that of the MECI linking the 1ππ* and S0 PESs, the structure of which is shown. The curves shown in panel (b) are from a rigid body scan wherein RC–H is extended at planar geometries, with the rest of the nuclear framework held at its ground state equilibrium geometry. | ||
PTS studies of propene (and selectively deuterated isotopomers of propene) following photoexcitation at λ = 157 nm identified no fewer than eight dissociation channels, of which the triple fragmentation to C2H2 + CH3 + H products is dominant. The small kinetic energy releases and minimal recoil anisotropies of all products again imply that dissociation proceeds on the electronic ground state PES following non-radiative transfer from the photoexcited state.73–75 Chin and Lee76 reported relative probabilities (derived via RRKM calculations) for various two- and three-body decay channels of 1-butene molecules on the S0 PES (calculated by electronic structure methods) at total energies appropriate for photoexcitation at 193 nm and 157 nm and predicted CH2CHCH2 + CH3 radicals (i.e. products arising from a C–C bond fission after an initial H atom migration) as the dominant fragments at both excitation wavelengths.
UV excitation of cyclic dienes induces broadly similar photophysics. Theory (ab initio MD simulations)86 and experiment (time-resolved photoelectron spectroscopy)86,87 imply that the ultrafast non-radiative decay following π*←π excitation in 1,3-cyclopentadiene, for example, is driven by initial (in-plane) motion along the bond-alternation coordinate followed by out-of-plane torsional motion about the CC double bonds – similar to the nuclear motions that follow π*←π excitation of C2H488 – to access a conical intersection with the S0 PES. Photoinduced ring-opening of 1,3-cyclohexadiene following π*←π excitation has been studied more extensively.89–95 Again, the 1ππ* excited state decays on an ultrafast timescale by non-adiabatic coupling (probably via an optically dark excited state) to the S0 PES. Again, the topography of the PESs encourages C–C bond extension and torsion around the CC double bonds, thereby priming the molecule to ring-open fully (to 1,3,5-hexatriene) or to revert to the ring-closed structure on the S0 PES – albeit with sufficient internal excitation to fragment further. In both cases, the identity of final decomposition products remains an open question.
Nonetheless, the available data implies that the rates of non-radiative decay following π*←π excitation of these CC bond containing molecules (including allene38,39,44 and fulvenallene96,97) are all so fast that rival excited state fragmentation pathways are unable to compete. Internal conversion to high vibrational levels of the S0 state is the norm and C–H bond fission is a major decay pathway. But, increases in molecular size and in excitation energy also translate into increased complexity: the number of energetically accessible product channels increases, as does the likelihood that some of these products are formed in multiple isomeric forms and may be susceptible to further (and possibly unintentional) photo-processing.
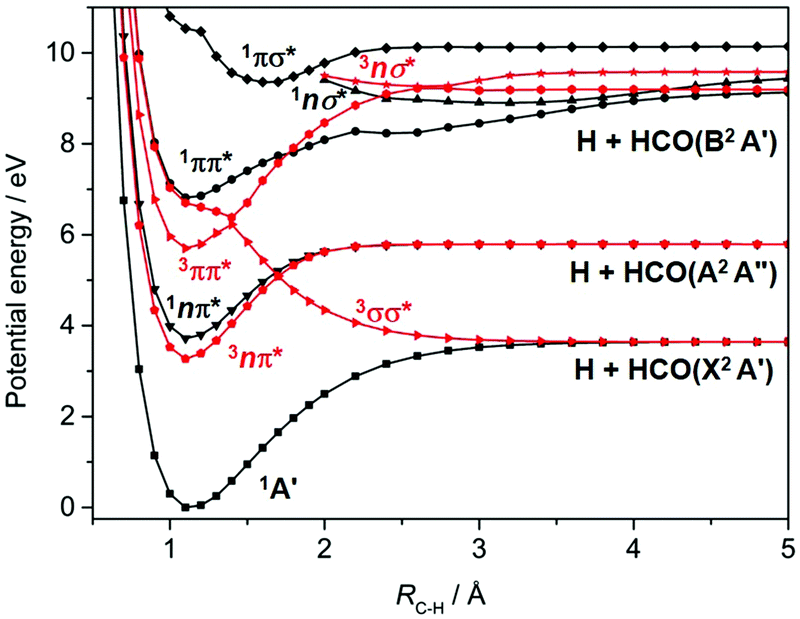 | ||
| Fig. 3 Rigid body singlet (in black) and triplet (in red) PECs for HCHO plotted as a function of RC–H for planar geometries, with the rest of the nuclear framework maintained at its ground state equilibrium geometry. | ||
Most recent interest in HCHO photochemistry has focussed on the rival H2 + CO molecular product channel and, particularly, unravelling signatures of the ‘roaming’ contribution to this product yield. Roaming in this case is now understood in terms of a frustrated C–H bond fission, wherein the H atom all but escapes from the long range attractive part of the S0 PES, returns, re-encounters and reacts with the HCO partner to yield the molecular products.103,104 The recent ion imaging studies104 have also identified a three body fragmentation channel yielding H + H + CO products, with an energetic threshold of 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 410 cm−1, which constitutes ∼5% of the total product yield when exciting at λ = 266 nm. Dynamical studies of C–H bond fission following excitation of HCHO at shorter wavelengths are scarce, but ab initio theory – Fig. 3 in the context of motion along RC–H, and prior coupled multi-surface photodynamics studies focussing on the CO stretch and symmetric HCH bend coordinates105 – indicate a wealth of non-adiabatic couplings and possible non-radiative decay pathways following excitation to the 1ππ* and/or low-lying Rydberg states at wavelengths λ < 180 nm.
410 cm−1, which constitutes ∼5% of the total product yield when exciting at λ = 266 nm. Dynamical studies of C–H bond fission following excitation of HCHO at shorter wavelengths are scarce, but ab initio theory – Fig. 3 in the context of motion along RC–H, and prior coupled multi-surface photodynamics studies focussing on the CO stretch and symmetric HCH bend coordinates105 – indicate a wealth of non-adiabatic couplings and possible non-radiative decay pathways following excitation to the 1ππ* and/or low-lying Rydberg states at wavelengths λ < 180 nm.
HCO radicals have been reported following long wavelength (λ ∼ 300 nm) excitation of propenal (acrolein, CH2CHCHO) via its S1–S0 (π*←n) transition, and explained in terms of dissociation after intersystem crossing to the lowest triplet (T1) PES.115 PTS studies following excitation at shorter wavelengths (193 nm (π*←π excitation)116,117 and at 157 nm118) identify H + CH2CHCO and C2H3 + HCO as the dominant primary fragmentation pathways. Analysis of the product translational and angular distributions suggests roles for fragmentation following radiationless transfer to both the S0 and T1 PESs, but interpretation is complicated by the wealth of possible isomerisation and secondary fragmentation processes available at these high excitation energies.
H atom PHOFEX studies119 and PTS studies following excitation of FCHO in the range 218 ≤ λ ≤ 248 nm120 and at λ = 193 nm121 all provide unequivocal evidence of aldehydic C–H bond fission. The operation of the rival F + HCO bond fission process was first deduced from analysis of the times-of-flight (TOFs) of H atoms formed by unintended UV laser photolysis of the partner HCO fragments122 and then confirmed (at λ = 193 nm) by direct observation of the F atom and HCO radicals using universal ionization methods.121 Theory123,124 serves to confirm suggestions, based on the deduced product energy disposal, that the observed C–H bond fission occurs following ISC to the T1 PES (the analogue of the T1 (3σσ*) PES shown for HCHO in Fig. 3).
In the case of alkenes, our summary identifies IC to high vibrational levels of the S0 state and subsequent dissociation as the typical outcome following UV photoexcitation. Relative to the alkenes, the corresponding aldehydes have a lower lying 1nπ* excited state. This state can be populated at energies close to the C–H dissociation limit, and typically shows orders of magnitude slower IC rates. This allows an opportunity for decay via (traditionally much slower) ISC to the T1 PES, and the formation of ground state radical products with obviously non-statistical internal and/or translational energy distributions. The extent to which this photochemical difference persists when the simple aldehydes are excited at shorter (VUV) excitation wavelengths remains unclear.
![[b with combining tilde]](https://www.rsc.org/images/entities/char_0062_0303.gif) 1B1 state.131 Such observations would suggest that these products arise from CC bond cleavage on a dissociative excited state PES.
1B1 state.131 Such observations would suggest that these products arise from CC bond cleavage on a dissociative excited state PES.
2.3 Benzene and related aromatics
The UV photodissociation of many small aromatic hydrocarbons has been explored using a combination of multimass ion imaging techniques and complementary electronic structure theory.132 Benzene (C6H6) is the exemplar. Long wavelength excitation populates the S1 (Ã1B2u, 1ππ*) state via an electric dipole forbidden, but vibronically allowed, transition. The S1 fluorescence quantum yield drops rapidly at wavelengths λ < 244.5 nm, due to the opening of a new population loss process historically termed ‘channel three’.133 The mechanism of this decay has been a source of longstanding controversy, but recent ultrafast pump–probe photoelectron data have been interpreted as showing contributions from both IC to S0 (via a conical intersection at prefulvenoid geometries) and ISC to low lying triplet states.134 The PECs shown in Fig. 4(a), for a range of geometries along a LIIC connecting the optimised S0 state geometry to that of the prefulvenic MECI between the S1 and S0 state PESs, support previous findings regarding the comparative ‘flatness’ of the S1 PES (and a partner triplet PES) in this coordinate. Excitation at shorter wavelengths populates the S2 (1B1u, 1ππ*) state, and early PTS experiments identified C–H bond fission following IC to high vibrational levels of the S0 state as the dominant (probably the exclusive) fragmentation process when exciting at λ = 193 nm.135,136 H2-elimination leading to formation of o-C6H4 is a less endoergic process, but the energy of the transition state en route to these products on the S0 PES is too high for this rival decay channel to compete when exciting at λ = 193 nm. The prospects for excited state C–H bond fission in benzene appear somewhat like those for C2H4. As Fig. 4(b) shows, a rigid body scan in which one C–H bond, RC–H, is extended while imposing planarity and holding the rest of the nuclear framework at the ground state equilibrium geometry shows the expected triplet (3σσ*) repulsive PES correlating to the lowest dissociation limit, and identifies repulsive 1πσ* and (not shown) 3πσ* potentials correlating to the H + C6H5(Ã2A1) limit.
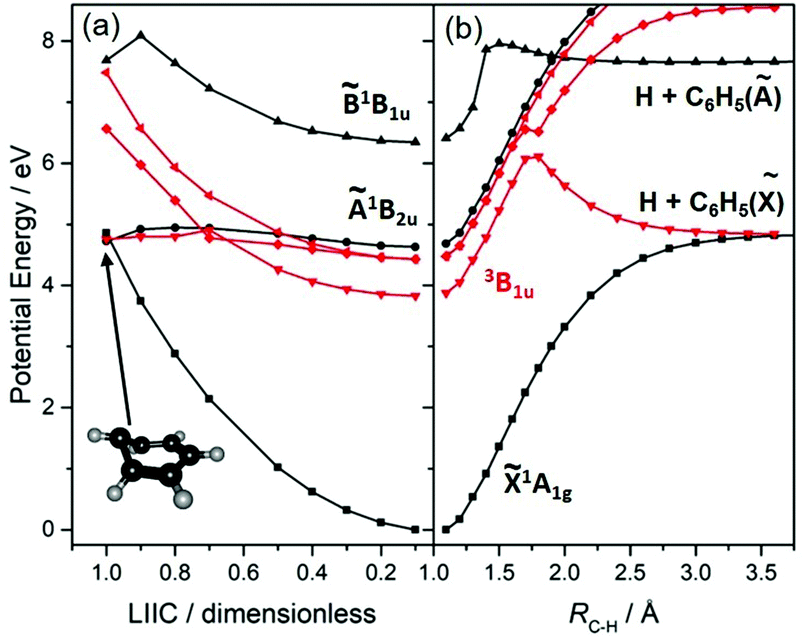 | ||
| Fig. 4 Singlet (in black) and triplet (in red) PECs for C6H6. Panel (a) is for a sequence of geometries along a LIIC connecting the optimised ground state geometry to that of the prefulvenic MECI linking the 1ππ* and S0 PESs, the structure of which is included as an inset. The PECs shown in panel (b) are from a rigid body scan wherein RC–H is extended at planar geometries with the rest of the nuclear framework fixed at its ground state equilibrium geometry. | ||
Molecules like toluene137 and m-xylene138 show similar fragmentation behaviour following excitation to their respective S2 states at λ = 193 nm, but experiments involving selectively deuterated precursors also reveal clear evidence for some isomerisation on the S0 PES prior to eventual unimolecular decay by both C–C and C–H bond fission, yielding C6H5 + CH3 and C6H5CH2 + H products (in the case of toluene). Dissociation following IC to high levels of the S0 state has been similarly advanced to account for the product energy disposals measured following 193 nm excitation of, for example, ethyl-, n-propyl-, isopropyl- and butylbenzene.132 Notably different dissociation dynamics have been reported following excitation to the S1 states of ethyl- and n-propylbenzene at λ = 248 nm.139,140 C6H5CH2 + CH3 and C6H5CH2 + C2H5 products are still observed with appearance rates and kinetic energy distributions consistent with that expected for the dissociation of vibrationally ‘hot’ S0 molecules. However, these are dwarfed by an additional yield of faster products that have been attributed to dissociation following ISC to the T1 PES – reminiscent of the deduced involvement of both singlet and triplet decay pathways following UV excitation of benzene in the channel three region.
2.4 Alkanes
1B1) products have been detected via their → ã fluorescence following excitation at λ < 133 nm.149 H2 products formed by two photon excitation in the range 210 ≤ λ ≤ 230 nm have been investigated also.142 Photoexcitation, at least at the lower energies within this range, promotes an electron from the 1t2 HOMO to the 3s/σ* orbital. As Fig. 1(b) showed, the resulting S1 state correlates with electronically excited CH3 radicals upon extending RC–H,150 but both ab initio electronic structure151 and trajectory surface hopping dynamics152 calculations confirm efficient non-adiabatic coupling to the S0 PES.
Fig. 5 shows the geometries of two of the MECIs between the S1 and S0 PESs of CH4 calculated using the global reaction route mapping (GRRM) method.153–156 The lowest energy structures (of which CI1 is a representative) are sensibly consistent with dissociations evolving towards CH2(ã1A1) + H2 products, the latter of which have been shown (experimentally) to be formed both rotationally and vibrationally excited.142 The CI2 structure shown in Fig. 5 is reminiscent of that reported previously.151 The experimental finding that the CH3() fragments formed following excitation of CH4 at, for example, λ = 132.748 nm (Fig. 6) carry high levels of a-axis rotational angular momentum146 can be understood as the carry-over of the nuclear momenta developed en route from the FC region accessed by S1←S0 excitation to geometries like CI2. Experiment also reveals an increasing tendency for three-body fragmentation processes on tuning to shorter excitation wavelengths.146,148
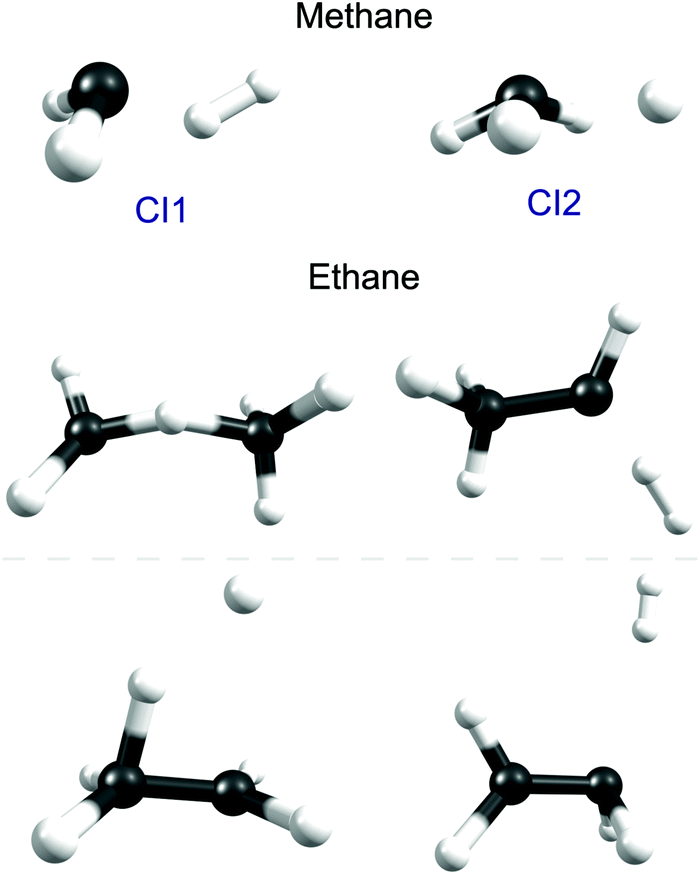 | ||
| Fig. 5 Geometries of selected low lying conical intersections between the S1 and S0 PESs of CH4 (upper) and C2H6 (below). | ||
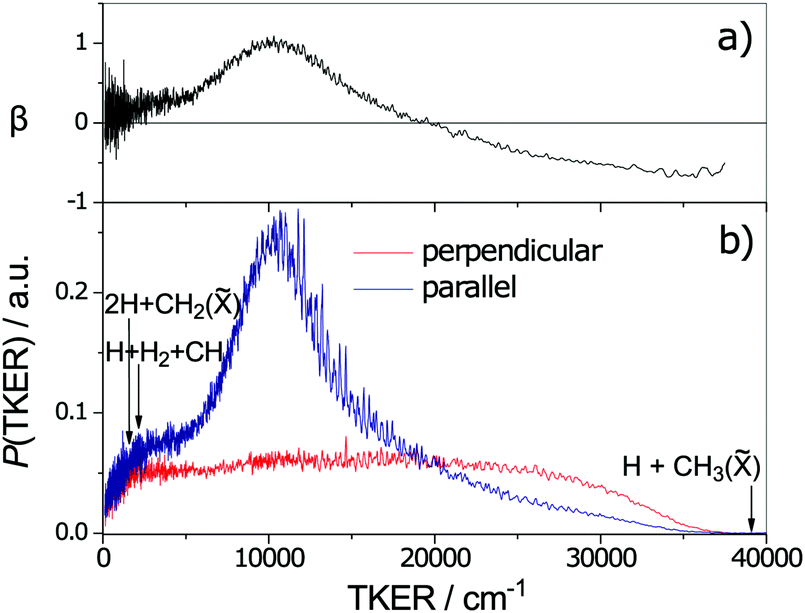 | ||
| Fig. 6 TKER distributions derived from TOF measurements of H atoms formed in the 132.748 nm photodissociation of jet-cooled CH4 molecules with the photolysis laser radiation polarised parallel (blue) and perpendicular (red) to the detection axis. The maximum possible TKERs of products arising via various three-body fragmentation processes are shown at the far left of the figure. Analysis of the fine structure evident in the TKER spectrum reveals that the CH3() fragments are formed with high rotational angular momentum, preferentially about the a-inertial axis. (Adapted from ref. 146, with the author's permission.) | ||
As the upper panel of Fig. 6 shows, the H + CH3() fragments display anisotropic, and TKER dependent, recoil velocity distributions.143,146 Such an observation is not without precedent. Similar behaviour has, for example, also been reported for the H + NH2() fragments from UV photodissociation of NH3157 and the O + NO(X) fragments from photolysis of NO2,158 and is a natural consequence of angular momentum conservation when, as here, the early time nuclear motions involve substantial motion transverse to the dissociation coordinate. Other striking aspects of the data shown in Fig. 6 include recognition that the decay of highly internally excited molecules formed by non-adiabatic coupling to the S0 PES need not be ‘slow’, nor that the product branching ratios be anything like ‘statistical’, nor that the product recoil distributions be isotropic. In many of the cases considered in Sections 2.2 and 2.3, like ethene or benzene, the nuclear motions that facilitate IC to the S0 state (e.g. torsion about the CC bond, or ring puckering) are essentially orthogonal to the bond fission coordinates of interest. Thus the parent vibrational motions activated by coupling from the photoexcited state to the S0 state need to evolve (by IVR) before sufficient energy accumulates in any bond destined to break. This takes time, during which the molecule can sample much of the S0 PES – as required for a ‘statistical’ fragmentation process. CH4 illustrates the alternative behavioural extreme; non-adiabatic coupling via CI2, for example, involves passage through geometries that are on a clear path from the initial FC region on the S1 PES to the H + CH3() asymptote, and the fragmentation after IC can be as ‘direct’ and the product energy disposal as ‘dynamically-determined’ as any direct dissociation on a repulsive excited state PES.
An early photolysis study using the Xe resonance lines (λ = 147.0 and 129.5 nm) deduced the participation of (at least) three fragmentation pathways, two involving elimination of H2 (with H3CCH and H2CCH2 as the co-fragments) and another yielding CH4 + CH2 products.161 As Fig. 5 shows, GRRM calculations identify suitable low-lying conical intersections between the S1 and S0 PESs of ethane to facilitate formation of each of these sets of products, plus another conical intersection consistent with direct C–H bond fission. H atom products have been reported following λ = 121.6 nm photolysis of both ethane and propane.162 The H atom velocity distributions determined in both cases appear very similar – isotropic, peaking at low TKER and with a weak tail extending to higher kinetic energies. These observations have been rationalised by invoking initial C–H bond fission, yielding a fast H atom along with an electronically excited R* (i.e. C2H5* or C3H7*) fragment (analogous to the excited CH3* product in Fig. 1(b)), followed by loss of a second (slow) H atom from the unimolecular decay of the primary R* fragment.162 Validation of this suggested mechanism by high level theory is still awaited.
The electronic absorption spectra of propane and of the larger alkanes all stretch to longer wavelengths; unlike ethane, there is no symmetry reason why excitation from the HOMO to the 3s Rydberg orbital should be forbidden in these higher alkanes. PTS studies following 157 nm photolysis of propane have identified H, H2 and CH3 loss channels, each with significant branching fractions and each with different associated kinetic energy releases.163 Experiments with selectively deuterated isotopomers revealed striking site specificities. Most of the H2 photoproducts are eliminated from the central C atom, and recent trajectory surface hopping dynamics calculations suggest that the (much smaller) H2 fractions attributed to 1,2- and 1,3-eliminations arise via ‘roaming’ mechanisms.164 The PTS experiments also suggest that most of the H atoms originate from the terminal CH3 groups, though Wu et al.163 note that both three-body fragmentation channels and unintended secondary photolysis of the primary radical photofragments can complicate interpretation of the observed H atom signals. No CH4 + C2H4 products were identified, despite this being the lowest energy product asymptote. Similarly detailed PTS studies of the H atom and H2 products arising in the 157 nm photolysis of several larger straight-chain, branched-chain and cyclic alkanes have also been reported.165 PTS studies of the 157 nm photolysis of cyclopropane identify C2H4 + CH2 as the dominant dissociation products, but also measure a significant (∼14%) yield of H atoms that are thought to arise via synchronous loss of two H atoms.166 As with the n-alkanes, the H2 yields from 157 nm photolysis of the branched chain and cyclic alkanes were found to gain in relative importance (cf. H atom loss) with increasing molecular size, hinting that the photodissociation dynamics of the larger alkanes is correlated with their flexibility and, in the cycloalkanes, with the ring strain. In all cases, neither the branching fractions nor the deduced translational energy disposals appear ‘statistical’ but, as the authors note, more theoretical investigations will be needed if we are to gain a detailed understanding of the fragmentation dynamics of these larger molecules.
3. Hydrocarbon radicals
3.1 Methylidyne (CH) and methylene (CH2)
The CH radical is included here mainly for completeness; ref. 172 lists many of the prior experimental and theoretical studies of this radical. As Fig. 7(a) shows, repulsive PECs attributable to σ*←σ and/or σ*←n excitations correlate to the H + C(3P) and H + C(1D) limits; wavelength dependent photodissociation cross-sections have been calculated for incorporation in astrophysical chemistry modelling.173 | ||
| Fig. 7 Doublet (in black) and quartet (in red) PECs along RC–H for the ground and first few excited states of (a) CH and (b) CH3 radicals. The latter PECs were calculated by extending RC–H at planar geometries, holding the rest of the framework at its ground state equilibrium geometry. | ||
The photodissociation of CH2 radicals has also been investigated theoretically.174–178 The ground state of CH2 has 3Σ− symmetry when linear, which reduces to 3B1 at its bent equilibrium geometry. As Fig. 8 shows, in the linear limit, the ground state of CH2 correlates with excited products (H + CH(4Σ−)) upon extending RC–H, while the first excited triplet state (the 3Π state formed by a 3s/σ*←n electron promotion) correlates with ground state (H + CH(X2Π)) products. The degeneracy of the 3A′ and 3A′′ components of the 3Π state is lifted upon HCH angle bending. The 3A′ PEC is relatively insensitive to ∠HCH, but the crossing between the ground 3Σ− and 3Π(A′′) PECs develops into a conical intersection at extended RC–H. The singlet PECs are more sensitive to changes in ∠HCH. Bending causes the first excited 1Δ state to split into its A′ (ã1A1 at C2v) and A′′ (1B1) components. Both interact strongly with the corresponding A′ and A′′ components of the repulsive 1Π (3s/σ*←n) state, with the result that both correlate adiabatically with ground state H + CH(X) products.
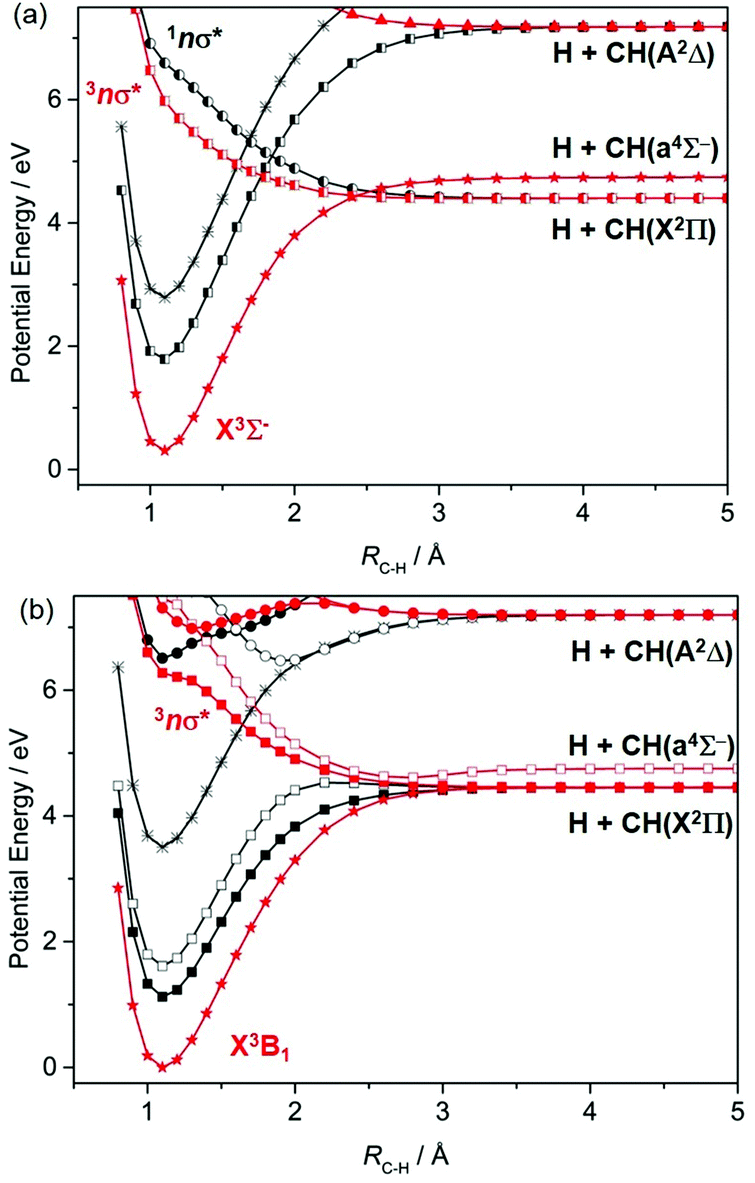 | ||
| Fig. 8 Singlet (in black) and triplet (in red) PECs along RC–H for the ground and first few excited states of the CH2 radical for (a) ∠HCH = 180° and (b) ∠HCH = 133° (the equilibrium bond angle in the 3B1 ground state). States of ′ and ′′ symmetry in (b) are distinguished by filled and open symbols, respectively. | ||
In terms of parent → product correlations, the lowest triplet and singlet states of the CH2 radical show parallels with those for the analogous (singlet) states of H2O or H2S.7 Long wavelength excitation to the 3A′ state of CH2 can be expected to yield CH(X) radicals with modest internal excitation – as observed for the case of 193 nm photolysis of the CH2(X) products arising in the near UV photolysis of H2CCO.179 Relative to H2O or H2S, 3CH2() has an equilibrium bond angle much closer to 180°, so one can predict that molecules excited to the 3Π(A′′) state will also be likely to funnel through the conical intersection at linear geometries and dissociate to H + CH(X) products. CH2 radicals photoexcited from the lowest singlet (ã) state will likely yield these products also, but analogy with H2O or H2S suggests that the resulting CH(X) fragments will be highly rotationally excited – given the large change in bond angle from the initial state (∠HCH = 102°) required to reach the conical intersection at linear geometries and extended RC–H.
3.2 Alkyl radicals
state) is populated by promoting an electron from the singly occupied molecular orbital, the non-bonding carbon 2pz orbital (n), to an orbital that is best viewed as 3s Rydberg in the FC region but gains increasing σ* antibonding valence character upon extending one C–H bond. The CH3(–) absorption shows poorly resolved rovibronic structure, which is sharper in CD3.180 Photoexcitation within this band results in bond fission, yielding H(D) atoms together with CH2(CD2) fragments. The atomic fragments show an anisotropic (perpendicular) recoil velocity distribution181 – consistent with the – parent transition assignment (i.e. the H(D) atoms recoil in the plane perpendicular to the parent transition moment, which is aligned along the C3 axis) and the short excited state lifetime (∼60 fs in the case of CH3182). As Fig. 9(a) shows, excitation of the CH3(–) origin transition (at λ = 216 nm) yields CH2 fragments in their first excited ã1A1 state (the lowest energy singlet state) with little rovibrational excitation.181,183–185 Such an outcome matches theoretical expectations.186,187 As Fig. 7(b) showed, the ground state of the CH3 radical correlates with H + CH2() products. The other parent state correlating to this lowest dissociation asymptote is a repulsive quartet state. The state of CH3 correlates with H + CH2(ã1A1) products (consistent with the experimental observation) and the very specific product energy disposal implies negligible non-adiabatic coupling between the and state PESs in the regions of configuration space sampled during the dissociation process. But, tuning to somewhat higher excitation energies, the H atoms formed following (two photon) excitation to the 3pz Rydberg state of the CH3 radical are reported to show a broad, isotropic velocity distribution, peaking at low TKER values but extending to (and even beyond) the upper limit allowed by energy conservation, even when assuming ground state CH2() as the co-fragment.185 This very different energy disposal has been rationalised in terms of a sequence of non-adiabatic couplings that allow access to, and eventual dissociation on, the ground state PES.185,187
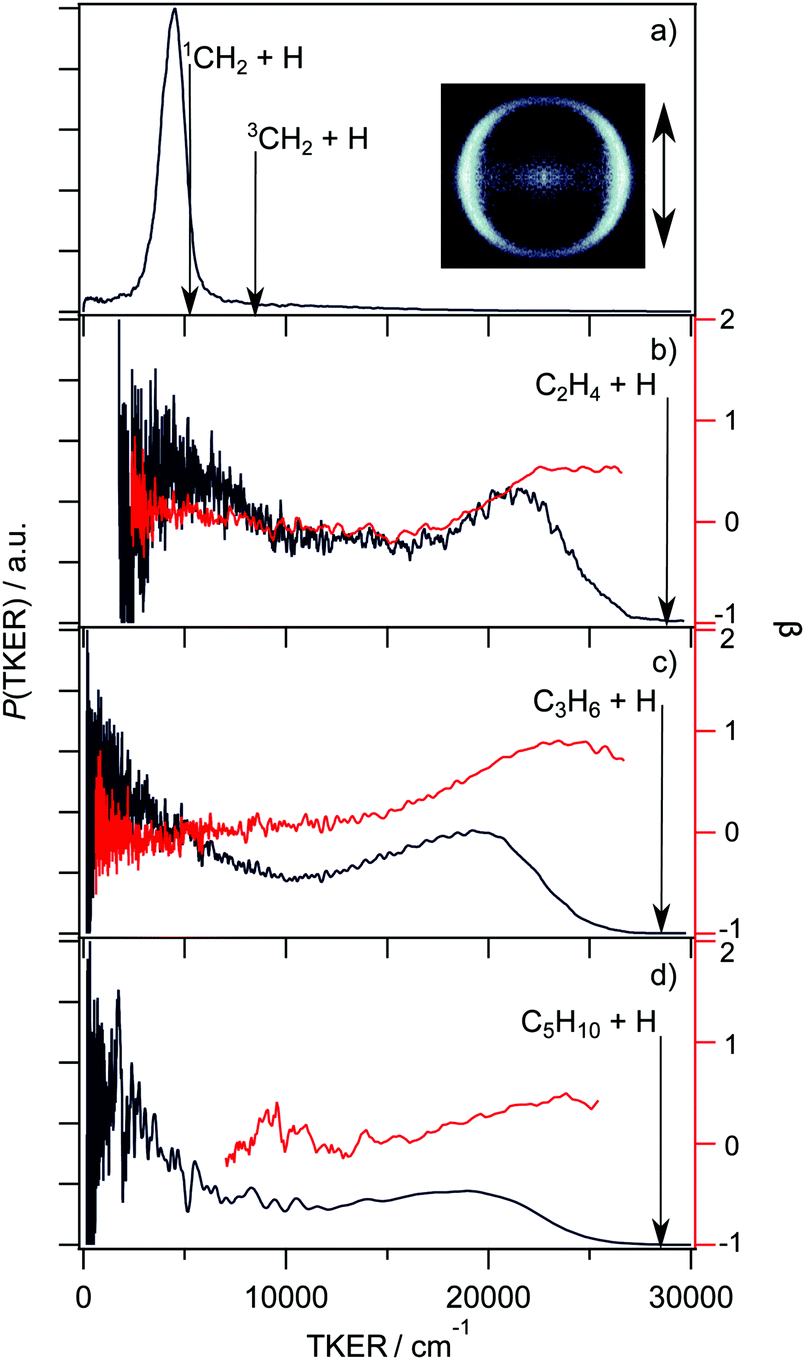 | ||
| Fig. 9 TKER distributions (in black, left hand axis) and TKER dependent recoil anisotropy parameters (β, in red, right hand axis) derived from velocity or TOF measurements of H atoms formed by UV photoinduced C–H bond fission of the following alkyl radicals: (a) CH3 at λ = 216 nm (with the H+ ion image obtained with vertically polarised photolysis laser radiation shown alongside), (b) C2H5 at λ = 245 nm, (c) n-C3H7 at λ = 245 nm and (d) n-C5H11 at λ = 240 nm. (Adapted from ref. 185, 188, 195 and 197, respectively.) | ||
state of CH3) could occur both directly – as in CH3, but to relatively much more stable (H + C2H4) products in this case – and indirectly, following non-adiabatic coupling to the ground state via one or more of the conical intersections linking the parent à and state PESs at distorted geometries.186,190 Questions remain, however. Analogy with methyl (Fig. 7(b)) suggests that the PES associated with 3s/σ*←n electron promotion should correlate to H + 1CH3CH (i.e. methyl carbene (ethylidene)) products, the latter of which can isomerise to the minimum energy ground state structure (ethene), but theory190,191 shows that the problem needs to be treated in higher dimensionality and that H atom ejection is preceded by H atom migration to a bridged excited state structure. Neither the H/D branching ratios measured when photolyzing partially deuterated ethyl radicals at λ = 250 nm nor the deduced production rate of the low TKER H/D atom products accord with expectations based on ‘statistical’ unimolecular decay from highly vibrationally excited ground state radicals.189 Direct dynamics calculations have variously suggested roles for a minor channel leading to electronically excited triplet C2H4(ã3B1u) + H products,192 and for a roaming channel leading to H2 + C2H3 products. These C2H3 products are then proposed to release an H atom, which could offer a possible rationale for the reported slow build-up rate of the atomic products,193 though it is difficult to reconcile the observed H atom velocity distribution with such a mechanism.194 Finally, we note that C–C bond fission is also thermodynamically allowed when exciting in this wavelength range, but we are not aware of any reports of such product formation.
The UV absorption spectra of the n- and iso-propyl radicals mimic that of the ethyl radical, as do the TKER spectra returned by HRA-PTS studies of these radicals at various wavelengths in the range 230 ≤ λ ≤ 260 nm.195 The TKER spectra are bimodal, as shown for the case of the n-propyl radical in Fig. 9(c), with fast (anisotropic, particularly in the case of n-propyl) and slow (isotropic) components. Again, the product TKER distributions stretch to much higher values than in the case of methyl radical photodissociation, reflecting the much greater relative stability of the H + alkene (cf. H + carbene) product pair.195 Experiments involving partially deuterated propyl radicals show site-specific loss of the β H atom, confirming conclusions reached in earlier studies of the relative H and D atom yields following 248 nm photolysis of (selectively deuterated) n-propyl radicals formed by 222 nm photolysis of the corresponding bromopropane.196 Again, the measured velocity distributions imply two dissociation pathways leading to the H + alkene (propene, in this case) products: one, wherein the photoexcited n(3s/σ*) state radicals evolve directly to ground state fragments, the other, indirect, involving unimolecular decay after non-adiabatic coupling to high vibrational levels of the ground state. The thermodynamic threshold for C–C bond fission in n-propyl (to CH3 + C2H4 products) lies below that for C–H bond fission but there do not yet appear to be any studies reporting formation of such C–C bond fission products. Very similar PTS data has been reported for the H + C5H10 products from photolysis of the n-pentyl radical at wavelengths in the range 236 ≤ λ ≤ 254 nm,197 as illustrated in Fig. 9(d).
C–C bond fission is a significant contributor following UV photoexcitation of t-butyl radicals, however. The relative stabilisation of this radical by the pendant methyl groups is reflected in the lowering of its ionisation potential (6.87 eV,198cf. 9.84 eV for CH3199) and of the energies of the associated excitations to Rydberg states. The UV absorption spectrum of the t-butyl radical shows well separated bands centred at λ ∼ 333, 253 and 233 nm that have been assigned to, respectively, 3s/σ*←n, 3p←n and 3d←n excitations.200 Time resolved photoelectron spectroscopy data obtained following excitation at λ = 330 and 266 nm were interpreted as implying an important role for excited state C–C bond extension,201 but PTS measurements were required to confirm the importance of both C–C and C–H bond fission pathways (yielding CH3 + CH3CCH3 and H + (CH3)2CCH2 products, respectively) following excitation at λ = 248 nm. Neither product TKER distribution nor the deduced product branching fraction approximate that expected on the basis of ‘statistical’ dissociation on the ground state PES202 and later ion imaging studies194 confirmed that the H atom photofragments from photolysis at wavelengths within all three of these UV absorption bands display bimodal velocity distributions reminiscent of those from photolysis of the smaller alkyl radicals. All of these data imply a substantial role for excited state C–H bond fission processes following UV photoexcitation of alkyl radicals.
O stretch mode) and translationally excited, with an anisotropic recoil velocity distribution (β ∼ −0.7), but a smaller fraction of the products shows much less translational and higher (undefined) internal excitation.206
All these products arise via non-adiabatic coupling at a conical intersection between the excited 22A (2n(3s/σ*)) and ground (12A) PESs at extended RO–H bond lengths. Molecules that pass through this conical intersection upon increasing RO–H are proposed to dissociate directly, yielding the translationally excited products. Conversely, dissociating molecules that initially follow the adiabatic path at the conical intersection have insufficient energy to access the H + HCHO(ã3A2) limit, resample the region of conical intersection, couple to the ground state and eventually dissociate to yield the more internally excited, less translationally excited products.205 Fragmentations yielding both trans- and cis-HCOH products from photoinduced C–H bond fissions are identified with increasing (but still small) relative yields once above their respective formation threshold energies.206,207 The measured energy disposals suggest that these HCOH products also arise from the decay of internally excited ground state CH2OH radicals formed via non-adiabatic coupling at the 22A/12A conical intersection in the RO–H stretch coordinate.
The fragmentation dynamics of the CH2OH radical change markedly when exciting at energies in the region of the 3px and 3pz Rydberg states. Now the excitation energies exceed the H + HCHO(ã3A2) dissociation limit, and an additional slow H atom yield is identified consistent with fragmentations that follow this adiabatic route past the 22A/12A conical intersection at extended RO–H. The HCOH/HCHO product yield ratio increases when exciting on 3p←n resonances, the (higher energy) cis-isomer is the favoured HCOH product, and the deduced energy disposal in the HCOH products is isomer-dependent and clearly non-statistical.207 These findings have been rationalised by assuming initial excitation to both the 32A (3p) and 22A (3s) Rydberg states, the former of which can decay by passage through successive conical intersections in the RC–H stretch coordinate en route to both cis- and trans-HCOH products in their respective ground states.208
The corresponding 3s←n absorption of the 1-hydroxyethyl (CH3CHOH) radical extends to λ > 500 nm. As with the CH2OH radical, studies involving selectively deuterated precursors and long excitation wavelengths identify O–H bond fission (leading to fast H atoms with an anisotropic recoil velocity distribution along with CH3CHO co-products) as the exclusive fragmentation channel, and theory209 has again identified a conical intersection between the 22A (2n(3s/σ*)) and 12A (ground state) PESs in the RO–H stretch coordinate that successfully explains this observation. A second, slow component apparent in the H atom velocity distributions measured following excitation at λ ≤ 320 nm has been attributed to the onset of rival C–H bond fission yielding vinyl alcohol (enol) partner fragments.210
The methoxy (CH3O) radical is an isomer of hydroxymethyl. C–O bond fission is the dominant outcome following photoexcitation of CH3O radicals in the wavelength range 282 ≥ λ ≥ 267 nm211 and we have not found any reports of C–H bond fission following UV photoexcitation of CH3O. The photodissociation of ethoxy (C2H5O) radicals and their deuterated analogues has been investigated, however, by PTS methods at three wavelengths in the range 208 ≤ λ ≤ 240 nm, revealing the operation of three dissociation channels – yielding H + CH3CHO, CH3 + HCHO and OH + C2H4 products – with roughly equal probabilities.212 The product branching ratios, their respective TKER distributions, and the anisotropic recoil velocity distribution of, particularly, the OH + C2H4 products all suggest a non-statistical fragmentation process and that the necessary isomerisation to CH2CH2OH prior to OH loss probably occurs on an excited state PES.212 Photodetachment of the ethoxide anion at λ = 388 nm yields ethoxy radicals, in both the 2A′′ and first excited Ã2A′ states. A fraction of these radicals are formed with sufficient internal energy to fragment, yielding CH3 + HCHO products only. C–H bond fission would be possible on energetic grounds, but is not observed.213
Turning to the sulfur analogues, C–S bond fission has been observed following excitation of the thiomethoxy (CH3S) radical in the wavelength range 360 ≥ λ ≥ 340 nm and at λ = 219 nm.214 C–H bond fission has also been recognised following excitation of CH3S radicals in the ranges 213 ≤ λ ≤ 220 nm215 and 344 ≤ λ ≤ 362 nm.216 The co-fragment in both cases is H2CS, and dissociation in the latter case is assumed to occur after IC to the ground electronic state (which, in this scenario, must occur in competition with the excited state predissociation to CH3 + S fragments).214
3.3 Unsaturated aliphatic radicals (and related species)
C and C–H bond lengths.217–219 Laser induced fluorescence measurements of the B–X system and of the B state fluorescence lifetimes return an upper limit value for D0(CC–H) ≤ 39388 ± 7 cm−1,220 which is lower than that of the analogous C–H bond in acetylene (D0(HCC–H) = 46074 ± 8 cm−1 (ref. 15)). The fastest H atoms evident in early PTS studies of C2H2 at λ = 193 nm can be attributed to secondary photolysis of the nascent C2H radicals,24 and ab initio calculations of the PESs for the ground and first few excited states of C2H along RC–H217–219 provide some rationale for the spread of electronic states in which the resulting C2 products are observed.221
Photofragmentation studies involving the various C3H2 isomers are still rare. Propargylene (HCCCH) has a triplet ground state. Anion photodetachment spectroscopy228 has revealed two low-lying singlet excited states, and confirmed the lower of two triplet excited states identified in earlier matrix isolation spectroscopy studies (with absorption maxima at λ ∼ 300 nm and ∼245 nm229). Ion imaging studies of the H atoms formed following λ = 250 nm photolysis of propargylene biradicals return an isotropic velocity distribution peaking at low kinetic energies reminiscent of that found in many other systems where dissociation occurs after IC to the ground state PES. In this case, however, complementary non-adiabatic surface hopping calculations suggest that dissociations from excited triplet states also contribute to the measured dissociation yield.230
The two carbene isomers, propadienylidene (H2CCC) and cyclopropenylidene (c-C3H2), both have singlet ground states. Features attributable to vibronic levels of the second excited singlet (S2 or 1B1) state of c-C3H2 have been identified by resonance enhanced multiphoton ionization (REMPI) studies at λ ∼ 270 nm. Ultrafast pump–probe ion yield studies and ab initio dynamics simulations both imply fast (<1 ps) non-adiabatic coupling to the S1 state and thence to the S0 state, yielding H atoms with a kinetic energy distribution consistent with that expected from the statistical decay of vibrationally excited ground state C3H2 radicals.231 The calculations suggest that the majority co-fragment is c-C3H, but also reveal some propensity for ring-opening. Time resolved ion yield studies reveal similarly fast excited state decay when exciting the H2CCC isomer at λ ∼ 250 nm (to its S3 or ![[C with combining tilde]](https://www.rsc.org/images/entities/char_0043_0303.gif) 1A1 state),232 and the measured Doppler profiles of the H atom fragments are again consistent with IC and subsequent unimolecular decay of vibrationally ‘hot’ S0 species.233
1A1 state),232 and the measured Doppler profiles of the H atom fragments are again consistent with IC and subsequent unimolecular decay of vibrationally ‘hot’ S0 species.233
band spanning the region 360–500 nm,236 attributed to an n←π promotion, and a more intense, unstructured – (π*←π) absorption centred at λ ∼ 230 nm.237 The lowest energy dissociation limit (yielding H + C2H2 products) lies at ∼12000 cm−1. Only the ground state of C2H3 correlates diabatically with H + C2H2(X) products. However, the measured lifetimes of the lowest vibrational levels of the Ã2A′′ state are just a few ps (and these lifetimes decrease further with increasing energy), pointing to the presence of efficient non-adiabatic coupling routes to the ground state PES.236 Ion imaging studies of the H atom products formed following excitation to several low lying vibrational levels of the C2H3(Ã) state confirmed C2H2 production238 and HRA-PTS experiments at λ = 366.2 nm and λ = 327.4 nm showed that the C2H2 products are formed with an inverted vibrational population distribution (in the CC stretch mode (ν2), with or without one quantum of the bending mode (ν4 or ν5)). The H atom co-fragments associated with these two progressions of peaks show, respectively, preferential parallel and perpendicular recoil anisotropies.235Ab initio electronic structure plus dynamics calculations identify various non-adiabatic pathways by which photoexcited à state radicals can return to the ground state, the lowest energy of which is promoted by nuclear motions consistent with the vibrational activity observed in the products.239
Vinyl radical photodissociation has also been investigated at λ = 243.2 nm, by imaging the H atom products. Excitation at this wavelength populates the 2A′′ excited state, at energies above the thresholds for forming an H atom with the vinylidene (H2CC) radical or with C2H2 in its lowest triplet excited state. Image analysis suggests that the former (or, possibly, C2H2 molecules with internal energies sufficient that interconversion between the H2CC and HCCH geometries is facile) is the major molecular photoproduct.234
The C3H5 radical exists as four isomers: 1-propenyl (CH3CHCH), 2-propenyl (CH2CCH3), cyclo-propyl (c-C3H5) and allyl (CH2CHCH2). The two propenyl isomers are simply β- and α-CH3 substituted vinyl radicals. Theory predicts that both will show a similar pattern of excited valence states to that of the allyl radical.240 PTS studies following excitation of 1-propenyl radicals in the range 224 ≤ λ ≤ 248 nm reveal the formation of H atom products with recoil velocity distributions that are consistent with unimolecular decay to H + C3H4 products following IC to the ground state.241 Accompanying quasi-classical trajectory calculations suggest propyne as the dominant C3H4 product, but also suggest that C–C bond fission (yielding CH3 + C2H2 products) is the dominant decay path for the vibrationally ‘hot’ ground state radicals – in accord with conclusions reached in earlier PTS studies of the λ = 193 nm photolysis of the 1-propenyl radical products from photodissociation of cis-1-bromopropene.242
Allyl is the simplest hydrocarbon radical with a conjugated π-electron system and, as such, has long been viewed as a benchmark system – for experiment and theory. The allyl radical exhibits diffuse banded absorption in the 410–370 nm region and another UV absorption centred at λ ∼ 225 nm, and excitations attributed to 3s and 3p Rydberg states have been identified by REMPI spectroscopy.243 PTS studies following excitation at λ = 351 nm and at λ = 248 nm identified C–H bond fission as the dominant decay channel at both wavelengths,244 with a minor (∼5%) contribution from C–C bond fission at λ = 248 nm.245 Trajectory calculations, run on an ab initio calculated ground state PES at a total energy appropriate for 248 nm photon absorption, reproduce the observed dominance of C–H bond fission and predict allene as the major C3H4 product246 – in accord with the conclusions reached by measuring relative H/D atom yields following photoexcitation of strategically deuterated allyl radicals at λ = 248.2 nm.247 Three different mechanisms have been proposed as contributors to the observed (minor) yield of C–C bond fission products following excitation at λ = 248 nm – with both C2H2 and H2CC (vinylidene) identified as partners to the CH3 co-fragment.246 HRA-PTS studies of the H atom products formed following excitation of allyl to the 2A1(3s), 2B2(3py) and Ẽ2B1(px) excited states (at various wavelengths in the range 216 ≤ λ ≤ 248 nm) serve to reinforce the conclusion that dissociation occurs after IC to the ground state PES.243 Femtosecond time resolved photoelectron imaging studies of 2-methylallyl radicals following photoexcitation at various wavelengths in the range 236 ≤ λ ≤ 241 nm similarly conclude that the excited state population undergoes rapid IC to the ground state PES.248
Studies of the 248 nm photolysis of cyclopentadienyl radicals have identified two major fragmentation channels yielding, respectively, C3H3 + C2H2 and H + C5H4 products with the latter product identified as the ethynylallene isomer.249 Once again, the deduced product branching ratio and the respective product energy disposals are broadly consistent with fragmentation occurring after IC to the ground state PES.
and à states of the HCO radical are a Renner–Teller pair,250 the H–CO bond is weak (D0(H–CO) = 5083 ± 8 cm−1) and the photodissociation dynamics following excitation at visible wavelengths have been explored by measuring linewidths of the predissociating parent resonances,251 the energy disposal in the resulting CO products,252 the recoil anisotropies of the H and CO photofragments,253 and by velocity map imaging254 and HRA-PTS studies255 of the H atom products. The last of these studies provides a particularly clear illustration of quantum interference effects in the decay of vibronically state selected HCO(A) radicals to H + CO(X,v,J) products.255 Studies of the energy disposal in the CO products from predissociation of vibrationally state selected HCO() radicals (formed by stimulated emission pumping via the state) have also been reported.256 The photochemistry of the acetyl radical has received much less attention, but it too shows a broad visible absorption.257 Theory confirms the low barrier to H3C–CO bond fission,258 but the Doppler broadened lineshapes of the H/D atom photofragments measured following (multiphoton) 243 nm excitation of acetone, acetaldehyde and acetic acid have been taken as evidence of (one photon induced) C–H bond fission of the primary acetyl radical photoproducts.259
H + CH2CO (and CH3 + CO) products have been found following preparation of state vinoxy (CH2CHO) radicals by photodetachment of the vinoxide anion at λ = 347 nm.260 These same product pairs were also identified from the unimolecular decay of highly internally excited vinoxy radicals formed in the photolysis of chloroacetaldehyde at λ = 193 nm261 and λ = 157 nm.262 Again, IC to the ground state PES was assumed to precede fragmentation, and theory has identified conical intersections that could facilitate non-adiabatic transfer between the , Ã and state PESs.263 A similar fragmentation mechanism has been invoked to account for the (dominant) CH3 + CH2CO and (minor) C2H5 + CO product yields and TKER distributions observed when exciting 1- (or i-)methylvinoxy (CH3COCH2) radicals at λ = 225, 248, and 308 nm.264
The methylene amidogen (H2CN) radical is also isoelectronic with the vinyl radical. TOF measurements of the H atoms formed following photoexcitation of H2CN in the range 274 ≤ λ ≤ 288 nm reveal a near isotropic recoil velocity distribution and that most of the energy above that required for C–H bond fission is partitioned into internal energy of the HCN co-fragment.265Ab initio theory confirms that the photoprepared 2A1 state of H2CN correlates with an excited state of HCN and that the observed fragmentation proceeds via non-adiabatic coupling to the ground state PES.266
3.4 Phenyl, benzyl and larger aromatic radicals
As noted in Section 2.3, deep-UV photolysis of benzene yields vibrationally excited phenyl (C6H5) radicals that can spontaneously decay to yield another H atom and a C6H4 (o-benzyne) fragment. HRA-PTS studies following photolysis of jet-cooled C6H5 radicals in the range 215 ≤ λ ≤ 268 nm – within the2A1–2A1 (π*←π) absorption system267 – return isotropic velocity distributions and H atom production rates compatible with C–H bond fission and formation of o-C6H4 products after IC to the ground state PES.268 Further support for such a fragmentation mechanism is provided by traditional PTS studies at 193 nm, which identify a (minor) channel yielding C2H2 (along with n-C4H3) products in addition to one or more channels yielding H atom products.269 Theory suggests that the molecular fragments, and part of the H atom yield formed at these short excitation wavelengths, arise from the unimolecular decay after ring-opening on the ground state PES.270
Such behaviour, i.e. non-adiabatic coupling to, and subsequent fragmentation on the ground state PES to yield products with branching ratios and energy disposals that are broadly consistent with statistical models of unimolecular decay, appears to be the ‘usual’ fate of aryl radicals following UV excitation. For example, PTS studies of photodissociation of benzyl (C6H5CH2) radicals following excitation in the range 228 ≤ λ ≤ 270 nm and at λ = 248 nm identify C–H bond fission as the dominant decay path (with fulvenallene as the predominant co-fragment), with a minor contribution from the rival CH3 + benzyne product channel.271,272 Ion imaging studies of the H atoms formed by photolysis of o- and p-xylyl radicals at wavelengths λ ∼ 310 nm and ∼250 nm are similarly consistent with C–H bond fission after IC to the ground state PES.273 These data all serve to illustrate the extent of isomerisation (ring opening and H atom transfer) that is required after accessing the ground state PES in order to sample the lowest energy fragmentation pathways. But these studies also return H atom formation rates that, whilst still slow, are considerably (one or more orders of magnitude) faster than predicted by RRKM calculations for the fully thermalized ground state radical at the relevant excitation energy.271,273 Might this be a hint that, even in these larger radicals and over these longer timescales, the decay of the ‘hot’ ground state species is influenced by the dynamical process(es) by which they are formed? In terms of energy disposal at least, this photophysical behaviour appears to extend to heteroaryl radicals also. The o-pyridyl radical is isoelectronic with phenyl, and the recoil velocity distributions of the H atoms formed following excitation of o-pyridyl radicals in the range 224 ≤ λ ≤ 246 nm are consistent with C–H bond fission (yielding cyanovinylacetylene co-fragments) after IC and isomerisation initiated by ring-opening at the C–N bond.274 Similar data has been reported (and similar conclusions reached) for the photodissociation of m-pyridyl radicals following excitation at similar UV wavelengths.275
4. Conclusions
Experiment and theory are now revealing many details of the rich photochemistry displayed by hydrocarbon molecules. The UV absorption spectra of the smaller alkynes show resolvable fine structure at long wavelengths, reflecting the fact that the lowest levels of the first 1ππ* states of these molecules lie below the threshold energy for C–H bond fission. Molecules excited to such levels fluoresce. C–H bond fission sets in upon tuning to higher energies, however, initially by coupling to one or more of the 3ππ* PESs that correlate to the lowest dissociation products. At yet shorter excitation wavelengths, this ISC channel is overtaken by a 1πσ*-state mediated C–H bond fission channel, reminiscent of that identified in numerous molecules containing X–H (X = heteroatom) bonds.7The studies of alkenes reported to date reveal very different photofragmentation behaviour. The first excited states again arise via π*←π transitions, but the electron promotion drives torsional motion about what (hitherto) was the CC bond and facilitates efficient non-adiabatic coupling to the S0 state. This accounts for the near ubiquitous finding that the photofragmentation of alkenes can be rationalised in terms of the unimolecular decay of highly internally excited S0 molecules. The singlet state formed by 3s/σ*←π excitation in ethene correlates diabatically with electronically excited C2H3 fragments, so any potential rival 1πσ*-state mediated C–H bond fission channel in alkenes would likely only reveal itself at shorter excitation wavelengths than investigated thus far. Similar considerations apply in the case of the aromatic molecules studied to date, though ISC provides a (relatively) more important non-radiative decay path when exciting at UV wavelengths.
Turning now to the alkanes, the recent studies of methane photophysics conclude that dissociation also occurs after efficient non-adiabatic coupling to the S0 state. However, at least in the case of CH4, the nuclear motions that promote the non-adiabatic coupling are closely aligned to the eventual fragmentation coordinate and an IC-driven dissociation thus has many of the dynamical hallmarks traditionally associated with a direct dissociation occurring on a repulsive excited state PES.
Extending such photodynamical studies to larger hydrocarbons will be challenging. These molecules contain just C and H atoms, and multiple C–C and C–H bonds. The parent molecules, and the radical products from C–H bond fission, can often exist in several isomeric forms. Products from the decay of hot S0 molecules are often formed with sufficient internal energy that they can undergo further unimolecular decay (i.e. a triple fragmentation process viewed from the perspective of the photoexcited parent molecule). The primary radical products are also prone to unintended secondary photolysis. Any such secondary dissociation products add to, and potentially confuse the interpretation of, the measured velocity distributions and the yields of the lighter fragments (e.g. H atoms, H2 molecules, etc.) that are typically most accessible to experimental study. Such challenges are also present when investigating hydrocarbon radicals, and are compounded by the need to produce a sufficient dense, pure and internally cold source of the radical.
We note some generic thermochemical and spectroscopic differences between hydrocarbon radicals and molecules. The C–H bonds in a hydrocarbon radical (R) are generally ‘weaker’ than those in the corresponding closed shell hydrocarbon precursor (RH), but the extent of the weakening can be very system dependent. For example, as shown in Section 3.3, D0(CC–H) is ∼85% that of D0(HCC–H), whereas the β C–H bond dissociation energy in the vinyl radical is only ∼30% that of a C–H bond in ethene. The experimental bond dissociation energies reflect not just the intrinsic ‘strength’ of the C–H bond of interest, but also the additional stabilization or destabilization of the dissociation products. The big difference in the latter case can be traced to the facts that (i) C–H bond fission in the vinyl radical generates an additional π bond upon forming the product (C2H2) and (ii) the C–H bonds in C2H2 are stronger due to the sp hybridization. The ground state hydrocarbon radicals have a partially filled HOMO. Electron promotions from (and to) this orbital typically support more valence excited states than in the corresponding closed shell RH molecule. Further, this odd electron will generally have a lower binding energy than that of the electrons in the HOMO of the RH molecule. Thus, the first ionization potential of R will be below that of RH and, more significantly from the viewpoint of the UV spectroscopy, so too will be the energies of the Rydberg states converging to this limit.
This offers one crumb of comfort to those exploring the photofragmentation of hydrocarbon radicals. In many cases, the products from photoinduced C–H bond fission in a hydrocarbon radical will be a closed shell molecule – and thus immune to unintended photochemistry at the UV wavelengths under investigation. Based on the limited available data, the alkyl radicals stand out from most of the other hydrocarbon radicals considered in this study – by yielding some C–H bond fission products with high translational energies and anisotropic recoil velocity distributions consistent with excited state dissociation mediated by the 2n(3s/σ*) PES. For all but the very simplest unsaturated hydrocarbon radicals, in contrast, the dissociation products formed upon photoexcitation, their relative yields, and their translational energy distributions, all appear – at least on a first glance – to be broadly consistent with expectations based on the ‘statistical’ decomposition of highly vibrationally excited ground state species. But, as noted at several points in this Perspective, there are a sufficient number of niggling inconsistencies with regard to estimated product yields, or product production rate constants, to encourage caution. The fragmentation of highly internally excited ground state radicals (and molecules) should only be expected to display truly ‘statistical’ characteristics if the internal energy in the ‘hot’ species has had time to become fully randomised. The internal energy distribution in a species immediately after radiationless transfer to its ground state PES must reflect specific nuclear motions that promote the non-adiabatic coupling, and is thus most unlikely to involve statistical population of all of the energetically accessible vibrational levels. Intramolecular vibrational redistribution, towards this statistical limit, will occur in competition with unimolecular decay, and the relatively probabilities of these processes will be sensitively dependent upon the nuclear motions imprinted during the non-adiabatic coupling and the topography of the ground state PES that the species sample thereafter.
Clearly, there remains a pressing need for further studies – particularly at shorter excitation wavelengths – to test the extent to which the characteristics of fragmentations of ‘hot’ ground state molecules/radicals formed via non-adiabatic coupling from a higher excited state deviate for expectations based on statistics. Fortunately, we can anticipate considerable progress in the near future, given the relentless advances in experimental capability (e.g. the increasing availability of intense, short pulse duration, tuneable (V)UV sources for time-resolved pump (photolysis)–probe (e.g. universal photoionization) product imaging experiments) and in the accuracy and efficiency of electronic structure methods and in the treatment of non-adiabatic excited state dynamics. Determining and explaining the dynamics of a specific photofragmentation channel is a fascinating and rewarding intellectual challenge, but we should not forget that, in many cases, there is still a need to establish reliable branching ratios (quantum yields) for competing fragmentation channels and how these vary with photolysis wavelength. Such information is a key yet, in many cases still poorly determined, part of the input to models of the atmospheres of the outer planets in our solar system and beyond.276
Conflicts of interest
There are no conflicts to declare.Appendix
The ground state minimum energy geometries were optimised using Møller–Plesset second order perturbation theory (MP2)277 coupled to Dunning's cc-pVDZ basis set.278 Unrelaxed, rigid-body potential energy profiles were constructed using complete-active space second order perturbation theory (CASPT2),279,280 coupled to Dunning's aug-cc-pVDZ basis set. The CASPT2 calculations were based on a state-averaged complete-active space self-consistent field (SA-CASSCF) reference wavefunction. An imaginary level shift of 0.5 EH was used to aid convergence and to mitigate intruder state effects.The active space used was species specific. C2v symmetry and a full-valence active space was used for the CH, CH2 and CH3 radicals. For CH4 and HCHO, Cs symmetry was used. The CH4 calculations used an (8,8) active space (i.e. 8 electrons in 8 orbitals, six of A′ and two of A′′ symmetry) while those for HCHO used a (6,5) active space (3 A′ and 2 A′′ orbitals). The C2H2 calculations used C2v symmetry and a (10,8) active space comprising 4 A1, 2 B1 and 2 B2 orbitals, while those for C2H4 assumed Cs symmetry and used a (4,4) active space (2 A′ and 2A′′ orbitals). For benzene, Cs symmetry and an active space of 8 electrons in 8 orbitals (6 A′′ and 2 A′) was used. The optimisations of the conical intersections for C2H2 and benzene used, respectively, (2,2) and (6,6) active spaces. These calculations made use of the 6-31G(d) basis set. All optimisations were carried out in Gaussian 09281 whilst the potential energy scans were performed in MOLPRO 2010.1.12
The MECIs shown in Fig. 5 were obtained by performing the seam model function (SMF)/single-component artificial force induced reaction (SC-AFIR) method, with spin-flip time-dependent density functional theory (SF-TDDFT), as implemented in a developmental version of the global reaction route mapping program (GRRM).153–156 The searches were started from ground state equilibrium structures, using the BHHLYP functional and 6-31G(d) basis set.
Additional outputs that underpin the ab initio calculations reported in this paper have been placed in the University of Bristol's research data repository and can be accessed using the following DOI: http://10.5523/bris.3tm9tsqgl5w3n2bdykoltp78q9.
Acknowledgements
MNRA is grateful to the Engineering and Physical Sciences Research Council for funding through a Programme Grant (EP/L005913). RAI is grateful to Prof. Satoshi Maeda and Dr Yu. Harabuchi for access to computational resources and a development version of the GRRM code and helpful discussions. JSZ acknowledges the support from the US National Science Foundation (NSF CHE-1566636).References
- J. Cui, R. V. Yelle, V. Vuitton, J. H. Waite, W. T. Kasprzak, D. A. Gell., H. B. Niemann, I. C. F. Muller-Wodarg, N. Borggren, G. G. Fletcher, E. L. Patrick, E. Raaen and B. A. Magee, Icarus, 2009, 200, 581–615 CrossRef CAS.
- V. G. Kunde, A. C. Aikin, R. A. Hanel, D. E. Jennings, W. C. Maguire and R. E. Samuelson, Nature, 1981, 292, 686–688 CrossRef CAS.
- V. A. Krasnopolsky, Icarus, 2009, 201, 226–256 CrossRef CAS.
- J. C. Loison, E. Hebrard, M. Dobrijevic, K. M. Hickson, F. Caralp, V. Hue, G. Gronoff, O. Venot and Y. Benilan, Icarus, 2015, 247, 218–247 CrossRef CAS.
- Y. L. Yung and W. B. DeMore, Photochemistry of Planetary Atmospheres, Oxford University Press, 1999 Search PubMed.
- A. L. Sobolewski, W. Domcke, C. Dedonder-Lardeux and C. Jouvet, Phys. Chem. Chem. Phys., 2002, 4, 1093–1100 RSC.
- M. N. R. Ashfold, G. A. King, D. Murdock, M. G. D. Nix, T. A. A. Oliver and A. G. Sage, Phys. Chem. Chem. Phys., 2010, 12, 1218–1238 RSC.
- G. M. Roberts and V. G. Stavros, Chem. Sci., 2014, 5, 1698–1722 RSC.
- M. N. R. Ashfold, D. Murdock and T. A. A. Oliver, Annu. Rev. Phys. Chem., 2017, 68, 63–82 CrossRef CAS PubMed.
- A. G. Sage, T. A. A. Oliver, D. Murdock, M. B. Crow, G. A. D. Ritchie, J. N. Harvey and M. N. R. Ashfold, Phys. Chem. Chem. Phys., 2011, 13, 8075–8093 RSC.
- M. N. R. Ashfold, M. Bain, C. S. Hansen, R. A. Ingle, T. N. V. Karsili, B. Marchetti and D. Murdock, J. Phys. Chem. Lett., 2017, 8, 3440–3451 CrossRef CAS PubMed.
- H. J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, W. Győrffy, D. Kats, T. Korona and R. Lindh, et al., MOLPRO, version 2015.1, A Package of ab initio Programs, see http://www.molpro.net Search PubMed.
- See, for example, T. E. Sharp, At. Data Nucl. Data Tables, 1971, 2, 119–169 CrossRef.
- T. Suzuki and N. Hashimoto, J. Chem. Phys., 1999, 110, 2042–2050 CrossRef CAS.
- D. H. Mordaunt and M. N. R. Ashfold, J. Chem. Phys., 1994, 101, 2630–2631 CrossRef CAS.
- N. Yamakita, S. Iwamoto and S. Tsuchiya, J. Phys. Chem. A, 2003, 107, 2597–2605 CrossRef CAS.
- P. B. Changala, J. H. Baraban, A. J. Merer and R. W. Field, J. Chem. Phys., 2015, 143, 084310 CrossRef PubMed.
- D. H. Mordaunt, M. N. R. Ashfold, R. N. Dixon, P. Löffler, L. Schnieder and K. H. Welge, J. Chem. Phys., 1998, 108, 519–526 CrossRef CAS.
- J. Jiang, C. A. Saladrigas, T. J. Erickson, C. L. Keenan and R. W. Field, J. Chem. Phys., 2018, 149, 174309 CrossRef PubMed.
- Q. Cui and K. Morokuma, Chem. Phys. Lett., 1997, 272, 319–327 CrossRef CAS.
- R. P. Schmid, T. Arusi-Parpar, R.-J. Li, I. Bar and S. Rosenwaks, J. Chem. Phys., 1997, 107, 385–391 CrossRef CAS.
- I. Bar and S. Rosenwaks, Mol. Phys., 2012, 110, 2673–2686 CrossRef CAS.
- J. Zhang, C. W. Riehn, M. Dulligan and C. Wittig, J. Chem. Phys., 1995, 103, 6815–6818 CrossRef CAS.
- B. A. Balko, J. Zhang and Y. T. Lee, J. Chem. Phys., 1991, 94, 7958–7966 CrossRef CAS.
- Y. Zhang, K. Yuan, S. Yu, D. H. Parker and X. Yang, J. Chem. Phys., 2010, 133, 014307 CrossRef PubMed.
- J.-H. Wang, Y.-T. Hsu and K. Liu, J. Phys. Chem. A, 1997, 101, 6593–6602 CrossRef CAS.
- P. Löffler, D. Lacombe, A. Ross, E. Wrede, L. Schnieder and K. H. Welge, Chem. Phys. Lett., 1996, 252, 304–310 CrossRef.
- P. Löffler, E. Wrede, L. Schnieder, J. B. Halpern, W. M. Jackson and K. H. Welge, J. Chem. Phys., 1998, 109, 5231–5246 CrossRef.
- S. Boyé, A. Campos, S. Douin, C. Fellows, D. Gauyacq, N. Shafizadeh, Ph. Halvick and M. Boggio-Pasqua, J. Chem. Phys., 2002, 116, 8843–8855 CrossRef.
- R. E. Bandy, C. Lakshminarayan, R. K. Frost and T. S. Zwier, Science, 1992, 258, 1630–1633 CrossRef CAS PubMed.
- R. Silva, W. K. Gichuhi, C. Huang, M. B. Doyle, V. V. Kislov, A. M. Mebel and A. G. Suits, PNAS, 2008, 105, 12713–12718 CrossRef CAS PubMed.
- S. R. Yu, S. Su, Y. W. Zhang, D. X. Dai, K. J. Yuan and X. M. Yang, J. Chem. Phys., 2013, 139, 124307 CrossRef PubMed.
- H. Z. Wang, S. R. Yu, S. Su, D. X. Dai, K. J. Yuan and X. M. Yang, J. Phys. Chem. A, 2015, 119, 11313–11319 CrossRef CAS PubMed.
- S. Satyapal and R. Bersohn, J. Phys. Chem., 1991, 95, 8004–8006 CrossRef CAS.
- C.-K. Ni, J. D. Huang, Y. T. Chen, A. H. Kung and W. M. Jackson, J. Chem. Phys., 1998, 110, 3320–3325 CrossRef.
- W. Sun, K. Yokoyama, J. C. Robinson, A. G. Suits and D. M. Neumark, J. Chem. Phys., 1999, 110, 4363–4368 CrossRef CAS.
- S. Ghosh, A. K. Rauta and B. Maiti, Phys. Chem. Chem. Phys., 2016, 18, 8219–8227 RSC.
- R. H. Qadiri, E. J. Feltham, E. E. H. Cottrill, N. Taniguchi and M. N. R. Ashfold, J. Chem. Phys., 2002, 116, 906–912 CrossRef CAS.
- R. H. Qadiri, E. J. Feltham, N. H. Nahler, R. P. Garcia and M. N. R. Ashfold, J. Chem. Phys., 2003, 119, 12842–12851 CrossRef CAS.
- K. Seki and H. Okabe, J. Phys. Chem., 1992, 96, 3345–3349 CrossRef CAS.
- X. Chen, Y. Ganot, I. Bar and S. Rosenwaks, J. Chem. Phys., 2000, 113, 5134–5137 CrossRef CAS.
- W. M. Jackson, D. S. Anex, R. E. Continetti, B. A. Balko and Y. T. Lee, J. Chem. Phys., 1991, 95, 7327–7336 CrossRef CAS.
- J. C. Robinson, N. E. Sveum, S. J. Goncher and D. M. Neumark, Mol. Phys., 2005, 103, 1765–1783 CrossRef CAS.
- S. Harich, J. J. Lin, Y. T. Lee and X. M. Yang, J. Chem. Phys., 2000, 112, 6656–6665 CrossRef CAS.
- K. Alnama, S. Boyé-Péronne, S. Douin, F. Innocenti, J. O’Reilly, A.-L. Roche, N. Shafizadeh, L. Zuin and D. Gauyacq, J. Chem. Phys., 2007, 126, 044304 CrossRef PubMed.
- R. Silva, W. K. Gichuhi, V. V. Kislov, A. Landera, A. M. Mebel and A. G. Suits, J. Phys. Chem. A, 2009, 113, 11182–11186 CrossRef CAS PubMed.
- K. Seki, M. Q. He, R. Z. Liu and H. Okabe, J. Phys. Chem., 1996, 100, 5349–5353 CrossRef CAS.
- R. Eng, T. Carrington, C. H. Dugan, S. V. Filseth and C. M. Sadowski, Chem. Phys., 1987, 113, 119–130 CrossRef CAS.
- J. Z. Guo, R. Eng, T. Carrington and S. V. Filseth, J. Chem. Phys., 2000, 112, 8904–8909 CrossRef CAS.
- G. P. Morley, I. R. Lambert, M. N. R. Ashfold, K. N. Rosser and C. M. Western, J. Chem. Phys., 1992, 97, 3157–3165 CrossRef CAS.
- P. A. Cook, S. R. Langford, M. N. R. Ashfold and R. N. Dixon, J. Chem. Phys., 2000, 113, 994–1004 CrossRef CAS.
- C. R. Bucher and K. K. Lehmann, Chem. Phys. Lett., 1998, 294, 173–180 CrossRef CAS.
- G. A. West and M. J. Berry, J. Chem. Phys., 1974, 61, 4700–4716 CrossRef CAS.
- A. J. Merer and R. S. Mulliken, Chem. Rev., 1969, 69, 639–656 CrossRef CAS.
- K. Alnama, S. Boyé-Péronne, A.-L. Roche and D. Gauyacq, Mol. Phys., 2007, 105, 1743–1756 CrossRef CAS.
- B. A. Balko, J. Zhang and Y. T. Lee, J. Chem. Phys., 1992, 97, 935–942 CrossRef CAS.
- J. J. Lin, D. W. Hwang, Y. T. Lee and X.-M. Yang, J. Chem. Phys., 1998, 109, 2979–2982 CrossRef CAS.
- J. J. Lin, C. C. Wang, Y. T. Lee and X. Yang, J. Chem. Phys., 2000, 113, 9668–9677 CrossRef CAS.
- S.-H. Lee, Y. T. Lee and X. Yang, J. Chem. Phys., 2004, 120, 10983–10991 CrossRef CAS PubMed.
- S.-H. Lee, Y.-C. Lee and Y. T. Lee, J. Phys. Chem. A, 2006, 110, 2337–2344 CrossRef CAS PubMed.
- K. Alnama, S. Boyé, S. Douin, F. Innocenti, J. O’Reilly, A.-L. Roche, N. Shafizadeh, L. Zuin and D. Gauyacq, Phys. Chem. Chem. Phys., 2004, 6, 2093–2100 RSC.
- A. Viel, R. P. Krawczyk, U. Manthe and W. Domcke, J. Chem. Phys., 2004, 120, 11000–11010 CrossRef CAS PubMed.
- M. Barbatti, J. Paier and H. Lischka, J. Chem. Phys., 2004, 121, 11614–11624 CrossRef CAS PubMed.
- T. Mori, W. J. Glover, M. S. Schuurman and T. J. Martinez, J. Phys. Chem. A, 2012, 116, 2808–2818 CrossRef CAS PubMed.
- T. K. Allison, H. Tao, W. J. Glover, T. W. Wright, A. M. Stooke, C. Khurmi, J. van Tilborg, Y. Liu, R. W. Falcone, T. J. Martinez and A. Belkacem, J. Chem. Phys., 2012, 136, 124317 CrossRef CAS PubMed.
- B. Sellner, M. Barbatti, T. Müller, W. Domcke and H. Lischka, Mol. Phys., 2013, 111, 2439–2450 CrossRef CAS.
- K. Kosma, S. A. Trushin, W. Fuss and W. E. Schmid, J. Phys. Chem. A, 2008, 112, 7514–7529 CrossRef CAS PubMed.
- E. G. Champenois, N. H. Shivaram, T. W. Wright, C.-S. Yang, A. Belkacem and J. P. Cryan, J. Chem. Phys., 2016, 144, 014303 CrossRef PubMed.
- T. Kobayashi, T. Horio and T. Suzuki, J. Phys. Chem. A, 2015, 119, 9518–9523 CrossRef CAS PubMed.
- E. M. Evleth and A. Sevin, J. Am. Chem. Soc., 1981, 103, 7414–7422 CrossRef CAS.
- G. Wu, A. E. Boguslavskiy, O. Schalk, M. S. Schuurman and A. Stolow, J. Chem. Phys., 2011, 135, 164309 CrossRef PubMed.
- G. M. P. Just, B. Negru, D. Park and D. M. Neumark, Phys. Chem. Chem. Phys., 2012, 14, 675–680 RSC.
- S.-H. Lee, Y.-Y. Lee, Y. T. Lee and X. M. Yang, J. Chem. Phys., 2003, 119, 827–838 CrossRef CAS.
- S.-H. Lee, Y. T. Lee and X. M. Yang, J. Chem. Phys., 2004, 120, 10992–10999 CrossRef CAS PubMed.
- S.-H. Lee and Y. T. Lee, Chem. Phys. Lett., 2004, 395, 311–315 CrossRef CAS.
- C.-H. Chin and S.-H. Lee, J. Chem. Phys., 2012, 136, 024308 CrossRef PubMed.
- D. R. Cyr and C. C. Hayden, J. Chem. Phys., 1996, 104, 771–774 CrossRef CAS.
- V. Blanchet, M. Z. Zgierski, T. Seidemann and A. Stolow, Nature, 1999, 401, 52–54 CrossRef CAS.
- F. Assenmacher, M. Gutmann, G. Höhlneicher, V. Stert and W. Radloff, Phys. Chem. Chem. Phys., 2001, 3, 2981–2982 RSC.
- W. Fuss, W. E. Schmid and S. A. Trushin, Chem. Phys. Lett., 2001, 342, 91–98 CrossRef CAS.
- A. Makida, H. Igarashi, T. Fujiwara, T. Sekikawa, Y. Harabuchi and T. Taketsugu, J. Phys. Chem. Lett., 2014, 5, 1760–1765 CrossRef CAS PubMed.
- J. C. Robinson, S. A. Harris, W. Z. Sun, N. E. Sveum and D. M. Neumark, J. Am. Chem. Soc., 2002, 124, 10211–10224 CrossRef CAS.
- M. Olivucci, J. N. Ragazos, F. Bernardi and M. A. Robb, J. Am. Chem. Soc., 1993, 115, 3710–3721 CrossRef CAS.
- A. Komainda, B. Ostojic and H. Köppel, J. Phys. Chem. A, 2013, 117, 8782–8793 CrossRef CAS PubMed.
- B. G. Levine and T. J. Martinez, J. Phys. Chem. A, 2009, 113, 12815–12824 CrossRef CAS PubMed.
- T. S. Kuhlman, W. J. Glover, T. Mori, T. Møller and T. J. Martinez, Faraday Discuss., 2012, 157, 193–212 RSC.
- O. Schalk, A. E. Boguslavskiy and A. Stolow, J. Phys. Chem. A, 2010, 114, 4058–4064 CrossRef CAS PubMed.
- H. L. Tao, B. G. Levine and T. J. Martinez, J. Phys. Chem. A, 2009, 113, 13656–13662 CrossRef CAS PubMed.
- M. Garavelli, C. S. Page, P. Celani, M. Olivucci, W. E. Schmid, S. A. Trushin and W. Fuss, J. Phys. Chem. A, 2001, 105, 4458–4469 CrossRef CAS.
- K. Kosma, S. A. Trushin, W. Fuss and W. E. Schmid, Phys. Chem. Chem. Phys., 2009, 11, 172–181 RSC.
- S. Adachi, M. Sato and T. Suzuki, J. Phys. Chem. Lett., 2015, 6, 343–346 CrossRef CAS PubMed.
- C. C. Pemberton, Y. Zhang, K. Saita, A. Kirrander and P. M. Weber, J. Phys. Chem. A, 2015, 119, 8832–8845 CrossRef CAS PubMed.
- M. P. Minitti, J. M. Budarz, A. Kirrander, J. S. Robinson, D. Ratner, T. J. Lane, D. Zhu, J. M. Glownia, M. Kozina, H. T. Lemke, M. Sikorski, Y. Feng, S. Nelson, K. Saita, B. Stankus, T. Northey, J. B. Hastings and P. M. Weber, Phys. Rev. Lett., 2015, 114, 255501 CrossRef CAS PubMed.
- O. Schalk, T. Geng, T. Thompson, N. Baluyot, R. D. Thomas, E. Tapavicza and T. Hansson, J. Phys. Chem. A, 2016, 120, 2320–2329 CrossRef CAS PubMed.
- A. R. Attar, A. Bhattacherjee, C. D. Pemmaraju, K. Schnorr, K. D. Closser, D. Prendergast and S. R. Leone, Science, 2017, 356, 54–58 CrossRef CAS PubMed.
- J. Giegerich and I. Fischer, Phys. Chem. Chem. Phys., 2013, 15, 13162–13168 RSC.
- I. A. Ramphal, M. Shapero, C. Halbach-Morris and D. M. Neumark, Phys. Chem. Chem. Phys., 2017, 19, 29305–29314 RSC.
- N. Hobday, M. S. Quinn, K. Nauta, D. U. Andrews, M. J. T. Jordan and S. H. Kable, J. Phys. Chem. A, 2013, 117, 12091–12103 CrossRef CAS PubMed.
- W. S. Hopkins, H.-P. Loock, B. Cronin, M. G. D. Nix, A. L. Devine, R. N. Dixon and M. N. R. Ashfold, J. Chem. Phys., 2007, 127, 064301 CrossRef PubMed.
- M. J. Dulligan, M. F. Tuchler, J. Zhang, A. Kolessov and C. Wittig, Chem. Phys. Lett., 1997, 276, 84–91 CrossRef CAS.
- W. S. Hopkins, H.-P. Loock, B. Cronin, M. G. D. Nix, A. L. Devine, R. N. Dixon, M. N. R. Ashfold, H.-M. Yin, S. J. Rowling, A. Büll and S. H. Kable, J. Phys. Chem. A, 2008, 112, 9283–9289 CrossRef CAS PubMed.
- P. Zhang, S. Maeda, K. Morokuma and B. J. Braams, J. Chem. Phys., 2009, 130, 114304 CrossRef PubMed.
- D. Townsend, S. A. Lahankar, S. K. Lee, S. D. Chambreau, A. G. Suits, X. Zhang, J. Rheinecker, L. B. Harding and J. M. Bowman, Science, 2004, 306, 1158–1161 CrossRef CAS PubMed.
- M. S. Quinn, D. U. Andrews, K. Nauta, M. J. T. Jordan and S. H. Kable, J. Chem. Phys., 2017, 147, 013935 CrossRef PubMed.
- S. Gomez-Carrasco, T. Muller and H. Köppel, J. Phys. Chem. A, 2010, 114, 11436–11449 CrossRef CAS PubMed.
- K. L. K. Lee, M. S. Quinn, A. T. Maccarone, K. Nauta, P. L. Houston, S. A. Reid, M. J. T. Jordan and S. H. Kable, Chem. Sci., 2014, 5, 4633–4638 RSC.
- H. K. Li, P. Y. Tsai, K. C. Hung, T. Kasai and K. C. Lin, J. Chem. Phys., 2015, 142, 041101 CrossRef PubMed.
- B. W. Toulson, K. M. Kapnas, D. A. Fishman and C. Murray, Phys. Chem. Chem. Phys., 2017, 19, 14276–14288 RSC.
- P. Morajkar, A. Bossolasco, C. Schoemaecker and C. Fittschen, J. Chem. Phys., 2014, 140, 214308 CrossRef PubMed.
- Y. C. Han, P. Y. Tsai, J. M. Bowman and K. C. Lin, Phys. Chem. Chem. Phys., 2017, 19, 18628–18634 RSC.
- L. Rubio-Lago, G. A. Amaral, A. Arregui, J. G. Izquierdo, F. Wang, D. Zaouris, T. N. Kitsopoulos and L. Bañares, Phys. Chem. Chem. Phys., 2007, 9, 6123–6127 RSC.
- T. Y. Kang, S. W. Kang and H. L. Kim, Chem. Phys. Lett., 2007, 434, 6–10 CrossRef CAS.
- S.-H. Lee, J. Chem. Phys., 2009, 131, 174312 CrossRef PubMed.
- P. Y. Tsai, H. K. Li, T. Kasai and K. C. Lin, Phys. Chem. Chem. Phys., 2015, 17, 23112–23120 RSC.
- S.-H. Jen and I.-C. Chen, J. Chem. Phys., 1999, 111, 8448–8453 CrossRef CAS.
- B. M. Haas, T. K. Minton, P. Felder and J. R. Huber, J. Phys. Chem., 1991, 95, 5149–5159 CrossRef CAS.
- C. Chaudhuri and S. H. Lee, Phys. Chem. Chem. Phys., 2011, 13, 7312–7321 RSC.
- C.-H. Chin, C. Chaudhuri and S.-H. Lee, J. Chem. Phys., 2011, 135, 044301 CrossRef PubMed.
- T. W. R. Hancock and R. N. Dixon, J. Chem. Soc., Faraday Trans., 1997, 93, 2707–2719 RSC.
- C. L. Reed, M. Kono, S. R. Langford, R. N. Dixon and M. N. R. Ashfold, J. Chem. Soc., Faraday Trans., 1997, 93, 2721–2729 RSC.
- S. H. Lee, C.-Y. Wu, S.-K. Yang and Y.-P. Lee, J. Chem. Phys., 2005, 123, 074326 CrossRef PubMed.
- C. Maul, C. Dietrich, T. Haas, K.-H. Gericke, H. Tachikawa, S. R. Langford, M. Kono, C. L. Reed, R. N. Dixon and M. N. R. Ashfold, Phys. Chem. Chem. Phys., 1999, 1, 767–772 RSC.
- H. Tachikawa, Phys. Chem. Chem. Phys., 1999, 1, 2675–2679 RSC.
- W.-H. Fang and R.-Z. Liu, J. Chem. Phys., 2001, 115, 5411–5417 CrossRef CAS.
- E. A. Wade, H. Clauberg, S. K. Kim, A. Mellinger and C. B. Moore, J. Phys. Chem. A, 1997, 101, 732–739 CrossRef CAS.
- J. Liu, F. Y. Wang, H. Wang, B. Jiang and X. M. Yang, J. Chem. Phys., 2005, 122, 104309 CrossRef PubMed.
- G. T. Fujimoto, M. E. Umstead and M. C. Lin, Chem. Phys., 1982, 65, 197–203 CrossRef CAS.
- E. J. Feltham, R. H. Qadiri, E. E. H. Cottrill, P. A. Cook, J. P. Cole, G. G. Balint-Kurti and M. N. R. Ashfold, J. Chem. Phys., 2003, 119, 6017–6031 CrossRef CAS.
- Y. Liu, J. K. Yu, X. R. Huang and C. C. Sun, J. Chem. Phys., 2006, 125, 044311 CrossRef PubMed.
- H. Y. Xiao, S. Maeda and K. Morokuma, J. Phys. Chem. A, 2013, 117, 7001–7008 CrossRef CAS PubMed.
- I. C. Lu, S. H. Lee, Y. T. Lee and X. M. Yang, J. Chem. Phys., 2006, 124, 024324 CrossRef PubMed.
- C.-K. Ni and Y. T. Lee, Int. Rev. Phys. Chem., 2004, 23, 187–218 Search PubMed.
- R. J. Longfellow, D. B. Moss and C. S. Parmenter, J. Phys. Chem., 1998, 92, 5438–5449 Search PubMed.
- T. J. Penfold, R. Spesyvtsev, O. M. Kirkby, R. S. Minns, D. S. N. Parker, H. H. Fielding and G. A. Worth, J. Chem. Phys., 2012, 137, 204310 CrossRef CAS PubMed.
- A. Yokoyama, X. Zhao, E. J. Hintsa, R. E. Continetti and Y. T. Lee, J. Chem. Phys., 1990, 92, 4222–4233 CrossRef CAS.
- A. M. Mebel, S. H. Lin, X. M. Yang and Y. T. Lee, J. Phys. Chem. A, 1997, 101, 6781–6789 CrossRef CAS.
- C. K. Lin, C. L. Huang, J. C. Jiang, A. H. H. Chang, Y. T. Lee, S. H. Lin and C. K. Ni, J. Am. Chem. Soc., 2002, 124, 4068–4075 CrossRef CAS.
- C. L. Huang, J. C. Jiang, A. H. H. Chang, Y. T. Lee and C. K. Ni, J. Phys. Chem. A, 2003, 107, 4019–4024 CrossRef CAS.
- C. L. Huang, J. C. Jiang, S. H. Lin, Y. T. Lee and C. K. Ni, J. Chem. Phys., 2002, 116, 7779–7782 CrossRef CAS.
- C. L. Huang, J. C. Jiang, Y. T. Lee and C. K. Ni, J. Chem. Phys., 2002, 117, 7034–7040 CrossRef CAS.
- D. H. Mordaunt, I. R. Lambert, G. P. Morley, M. N. R. Ashfold, R. N. Dixon, L. Schnieder and K. H. Welge, J. Chem. Phys., 1993, 98, 2054–2065 CrossRef CAS.
- A. J. R. Heck, R. N. Zare and D. W. Chandler, J. Chem. Phys., 1996, 104, 4019–4030 CrossRef CAS.
- J.-H. Wang and K. Liu, J. Chem. Phys., 1998, 109, 7105–7112 CrossRef CAS.
- J.-H. Wang, K. Liu, Z. Y. Min, H. M. Su, R. Bersohn, J. Preses and J. Z. Larese, J. Chem. Phys., 2000, 113, 4146–4152 CrossRef CAS.
- P. A. Cook, M. N. R. Ashfold, Y.-J. Lee, K.-H. Jung, S. Harich and X. Yang, Phys. Chem. Chem. Phys., 2001, 3, 1848–1860 RSC.
- Y. W. Zhang, K. J. Yuan, S. R. Yu and X. M. Yang, J. Phys. Chem. Lett., 2010, 1, 475–479 CrossRef CAS.
- F. Z. Chen and C. Y. R. Wu, J. Quant. Spectrosc. Radiat. Transfer, 2004, 85, 195–209 CrossRef CAS.
- B. Gans, S. Boyé-Péronne, M. Broquier, M. Delsaut, S. Douin, C. E. Fellows, P. Halvick, J.-C. Loison, R. R. Lucchese and D. Gauyacq, Phys. Chem. Chem. Phys., 2011, 13, 8140–8152 RSC.
- L. C. Lee and C. C. Chiang, J. Chem. Phys., 1983, 78, 688–691 CrossRef CAS.
- A. M. Mebel, S.-H. Lin and C.-H. Chang, J. Chem. Phys., 1997, 106, 2612–2620 CrossRef CAS.
- R. van Harrevelt, J. Chem. Phys., 2006, 125, 124302 CrossRef PubMed.
- M. D. Lodriguito, G. Lendvay and G. C. Schatz, J. Chem. Phys., 2009, 131, 224320 CrossRef PubMed.
- S. Maeda, K. Ohno and K. Morokuma, Phys. Chem. Chem. Phys., 2013, 15, 3683–3701 RSC.
- S. Maeda, T. Taketsugu and K. Morokuma, J. Comput. Chem., 2014, 35, 166–173 CrossRef CAS PubMed.
- S. Maeda, T. Taketsugu, K. Ohno and K. Morokuma, J. Am. Chem. Soc., 2015, 137, 3433–3445 CrossRef CAS PubMed.
- S. Maeda, Y. Osada, Y. Harabuchi, T. Taketsugu, K. Morokuma and K. Ohno, GRRM, a developmental version, Hokkaido University, Sapporo, 2015 Search PubMed.
- D. H. Mordaunt, M. N. R. Ashfold and R. N. Dixon, J. Chem. Phys., 1996, 104, 6460–6471 CrossRef CAS.
- A. V. Demyanenko, V. Dribinski, H. Reisler, H. Meyer and C. X. W. Qian, J. Chem. Phys., 1999, 111, 7383–7396 CrossRef CAS.
- R. J. Buenker and S. D. Peyerimhoff, Chem. Phys., 1975, 8, 56–67 CrossRef CAS.
- U. Jacovella, C. J. Stein, M. Grütter, L. Freitag, C. Lauzin, M. Reiher and F. Merkt, Phys. Chem. Chem. Phys., 2018, 20, 1072–1081 RSC.
- H. Okabe and J. R. McNesby, J. Chem. Phys., 1961, 34, 668–669 CrossRef CAS.
- W. M. Jackson, R. J. Price II, D. D. Xu, J. D. Wrobel, M. Ahmed, D. S. Peterka and A. G. Suits, J. Chem. Phys., 1998, 109, 4703–4706 CrossRef CAS.
- S. M. Wu, J. J. Lin, Y. T. Lee and X. Yang, J. Chem. Phys., 2000, 112, 8027–8037 CrossRef CAS.
- A. K. Rauta and B. Maiti, J. Chem. Phys., 2018, 149, 044308 CrossRef PubMed.
- S. M. Wu, J. J. Lin, Y. T. Lee and X. Yang, J. Phys. Chem. A, 2000, 104, 7189–7199 CrossRef CAS.
- C. C. Wang, Y. T. Lee, J. J. Lin, J. Shu., Y.-Y. Lee and X. Yang, J. Chem. Phys., 2002, 117, 153–160 CrossRef CAS.
- K. Tonokura, Y. Matsumi and M. Kawasaki, J. Chem. Phys., 1991, 95, 5065–5071 CrossRef CAS.
- K. Tonokura, Y. Mo, Y. Matsumi and M. Kawasaki, J. Phys. Chem., 1992, 96, 6688–6693 CrossRef CAS.
- R. A. Brownsword, M. Hillenkamp, T. Laurent, R. K. Vatsa, H. R. Volpp and J. Wolfrum, J. Chem. Phys., 1997, 106, 1359–1366 CrossRef CAS.
- G. Amaral, K. S. Xu and J. S. Zhang, J. Phys. Chem. A, 2001, 105, 1115–1120 CrossRef CAS.
- M. Lucas, Y. L. Liu, R. Bryant, J. Minor and J. S. Zhang, Chem. Phys. Lett., 2015, 619, 18–22 CrossRef CAS.
- A. Kalemos, A. Mavridis and A. Metropoulos, J. Chem. Phys., 1999, 111, 9536–9548 CrossRef CAS.
- E. F. van Dishoeck, J. Chem. Phys., 1987, 86, 196–214 CrossRef CAS.
- R. A. Beärda, M. C. van Hemert and E. F. van Dishoeck, J. Chem. Phys., 1992, 97, 8240–8249 CrossRef.
- G. J. Kroes, E. F. van Dishoeck, R. A. Beärda and M. C. van Hemert, J. Chem. Phys., 1993, 99, 228–236 CrossRef CAS.
- R. A. Beärda, G. J. Kroes, M. C. van Hemert, B. Heumann, R. Schinke and E. F. van Dishoeck, J. Chem. Phys., 1994, 100, 1113–1127 CrossRef.
- R. A. Beärda, M. C. van Hemert and E. F. van Dishoeck, J. Chem. Phys., 1995, 102, 8930–8941 CrossRef.
- G.-J. Kroes, M. C. van Hemert, G. D. Billing and D. Neuhauser, J. Chem. Phys., 1997, 107, 5757–5770 CrossRef CAS.
- B. Bohn and F. Stuhl, J. Chem. Phys., 1995, 102, 8842–8845 CrossRef CAS.
- G. Herzberg, Proc. R. Soc. London, 1961, 262, 291–317 Search PubMed.
- G. R. Wu, B. Jiang, Q. Ran, J. H. Zhang, S. A. Harich and X. M. Yang, J. Chem. Phys., 2004, 120, 2193–2198 CrossRef CAS PubMed.
- S. G. Westre, P. B. Kelly, Y. P. Zhang and L. D. Ziegler, J. Chem. Phys., 1991, 94, 270–276 CrossRef CAS.
- S. H. S. Wilson, J. D. Howe, K. N. Rosser, M. N. R. Ashfold and R. N. Dixon, Chem. Phys. Lett., 1994, 227, 456–460 CrossRef CAS.
- S. W. North, D. A. Blank, P. M. Chu and Y. T. Lee, J. Chem. Phys., 1995, 102, 792–798 CrossRef CAS.
- S. M. Poullian, D. V. Chicharro, A. Zanchet, M. G. Gonzalez, L. Rubio-Lago, M. L. Senent, A. Garcia-Vela and L. Bañares, Phys. Chem. Chem. Phys., 2016, 18, 17054–17061 RSC.
- H. T. Yu, A. Sevin, E. Kassab and E. M. Evleth, J. Chem. Phys., 1984, 80, 2049–2059 CrossRef CAS.
- A. Zanchet, L. Bañares, M. L. Senent and A. Garcia-Vela, Phys. Chem. Chem. Phys., 2016, 18, 33195–33203 RSC.
- G. Amaral, K. S. Xu and J. S. Zhang, J. Chem. Phys., 2001, 114, 5164–5169 CrossRef CAS.
- M. Steinbauer, J. Giegerich, K. H. Fischer and I. Fischer, J. Chem. Phys., 2012, 137, 014303 CrossRef PubMed.
- A. S. Zyubin, A. M. Mebel and S. H. Lin, Chem. Phys. Lett., 2000, 323, 441–447 CrossRef CAS.
- A. Sevin, H. T. Yu and E. M. Evleth, THEOCHEM, 1983, 13, 163–178 CrossRef CAS.
- J. M. Hostettler, A. Bach and P. Chen, J. Chem. Phys., 2009, 130, 034303 CrossRef PubMed.
- A. Matsugi, J. Phys. Chem. Lett., 2013, 4, 4237–4240 CrossRef CAS PubMed.
- J. Giegerich and I. Fischer, J. Chem. Phys., 2015, 142, 044304 CrossRef PubMed.
- Y. Song, X. F. Zheng, W. D. Zhou, M. Lucas and J. S. Zhang, J. Chem. Phys., 2015, 142, 224306 CrossRef PubMed.
- Z. G. Wang, M. G. Matthews and B. Koplitz, J. Phys. Chem., 1995, 99, 6913–6916 CrossRef CAS.
- G. Sun, Y. Song and J. S. Zhang, Chin. J. Chem. Phys., 2018, 31, 439–445 CrossRef CAS.
- W. R. Stevens, S. H. Walker, N. S. Shuman and T. Baer, J. Phys. Chem. A, 2010, 114, 804–810 CrossRef CAS PubMed.
- J. A. Blush and P. Chen, J. Chem. Phys., 1993, 98, 3557–3559 CrossRef CAS.
- J. Pacansky, W. Koch and M. D. Miller, J. Am. Chem. Soc., 1991, 113, 317–328 CrossRef CAS.
- B. Noller, R. Maksimenka, I. Fischer, M. Armone, B. Engels, C. Alcaraz, L. Poisson and J.-M. Mestdagh, J. Phys. Chem. A, 2007, 111, 1771–1779 CrossRef CAS PubMed.
- B. Negru, G. M. P. Just, D. Park and D. M. Neumark, Phys. Chem. Chem. Phys., 2011, 13, 8180–8185 RSC.
- B. C. Hoffman and D. R. Yarkony, J. Chem. Phys., 2002, 116, 8300–8306 CrossRef CAS.
- D. R. Yarkony, J. Chem. Phys., 2005, 122, 084316 CrossRef PubMed.
- C. J. Xie, C. Malbon, D. R. Yarkony and H. Guo, J. Chem. Phys., 2017, 146, 224306 CrossRef PubMed.
- C. P. Rodrigo, C. C. Zhou and H. Reisler, J. Phys. Chem. A, 2013, 117, 12049–12059 CrossRef CAS PubMed.
- C. P. Rodrigo, S. Sutradhar and H. Reisler, J. Phys. Chem. A, 2014, 118, 11916–11925 CrossRef CAS PubMed.
- C. L. Malbon and D. R. Yarkony, J. Chem. Phys., 2017, 146, 134302 CrossRef PubMed.
- K. Samanta and D. R. Yarkony, Chem. Phys., 2010, 378, 110–117 CrossRef CAS.
- B. Karpichev, L. W. Edwards, J. Wei and H. Reisler, J. Phys. Chem. A, 2008, 112, 412–418 CrossRef CAS PubMed.
- D. L. Osborn, D. J. Leahy and D. M. Neumark, J. Phys. Chem. A, 1997, 101, 6583–6592 CrossRef CAS.
- A. E. Faulhaber, D. E. Szpunar, K. E. Kautzman and D. M. Neumark, J. Phys. Chem. A, 2005, 109, 10239–10248 CrossRef CAS PubMed.
- B. L. J. Poad, A. W. Ray and R. E. Continetti, J. Phys. Chem. A, 2013, 117, 12035–12041 CrossRef CAS PubMed.
- R. T. Bise, H. Choi, H. B. Pedersen, D. H. Mordaunt and D. M. Neumark, J. Chem. Phys., 1999, 110, 805–816 CrossRef CAS.
- S. H. S. Wilson, M. N. R. Ashfold and R. N. Dixon, J. Chem. Phys., 1994, 101, 7538–7547 CrossRef CAS.
- X. F. Zheng, Y. Song, J. Z. Wu and J. S. Zhang, Chem. Phys. Lett., 2008, 467, 46–51 CrossRef CAS.
- S.-K. Shih, S. D. Peyerimhoff and R. J. Buenker, J. Mol. Spectrosc., 1979, 74, 124–135 CrossRef CAS.
- D. Duflot, J.-M. Robbe and J.-P. Flament, J. Chem. Phys., 1994, 100, 1236–1246 CrossRef CAS.
- Q. Cui and K. Morokuma, J. Chem. Phys., 1998, 108, 626–636 CrossRef CAS.
- W. Y. Chiang and Y. C. Hsu, J. Chem. Phys., 2000, 112, 7394–7399 CrossRef CAS.
- A. M. Mebel, M. Hayashi, W. M. Jackson, J. Wrobel., M. Green, D. D. Xu and S. H. Lin, J. Chem. Phys., 2001, 114, 9821–9831 CrossRef CAS.
- A. Fahr, P. Hassanzadeh, B. Laszlo and R. E. Huie, Chem. Phys., 1997, 215, 59–66 CrossRef CAS.
- H.-J. Deyerl, I. Fischer and P. Chen, J. Chem. Phys., 1999, 111, 3441–3448 CrossRef CAS.
- S. J. Goncher, D. T. Moore, N. E. Sveum and D. M. Neumark, J. Chem. Phys., 2008, 128, 114303 CrossRef PubMed.
- X. F. Zheng, Y. Song and J. S. Zhang, J. Phys. Chem. A, 2009, 113, 4604–4612 CrossRef CAS PubMed.
- T. L. Nguyen, A. M. Mebel, S. H. Lin and R. I. Kaiser, J. Phys. Chem. A, 2001, 105, 11549–11559 CrossRef CAS.
- B. M. Broderick, N. Suas-David, N. Dias and A. G. Suits, Phys. Chem. Chem. Phys., 2018, 20, 5517–5529 RSC.
- D. L. Osborn, K. M. Vogelhuber, S. W. Wren, E. M. Miller, Y.-J. Lu, A. S. Case, L. Sheps, R. J. McMahon, J. F. Stanton, L. B. Harding, B. Ruscic and W. C. Lineberger, J. Am. Chem. Soc., 2014, 136, 10361–10372 CrossRef CAS PubMed.
- R. A. Seburg, E. V. Patterson and R. J. McMahon, J. Am. Chem. Soc., 2009, 131, 9442–9455 CrossRef CAS PubMed.
- J. Giegerich, J. Petersen, R. Mitrić and I. Fischer, Phys. Chem. Chem. Phys., 2014, 16, 6294–6302 RSC.
- M. S. Schuurman, J. Giegerich, K. Pachner, D. Lang, B. Kiendl, R. J. MacDonell, A. Krueger and I. Fischer, Chem. – Eur. J., 2015, 21, 14486–14495 CrossRef CAS PubMed.
- B. Noller, M. Margraf, C. Schröter, T. Schultz and I. Fischer, Phys. Chem. Chem. Phys., 2009, 11, 5353–5357 RSC.
- C. Groß, B. Noller and I. Fischer, Phys. Chem. Chem. Phys., 2008, 10, 5196–5201 RSC.
- M. Ahmed, D. S. Peterka and A. G. Suits, J. Chem. Phys., 1999, 110, 4248–4253 CrossRef CAS.
- K. S. Xu and J. S. Zhang, J. Chem. Phys., 1999, 111, 3783–3786 CrossRef CAS.
- M. Shahu, C. H. Yang, C. D. Pibel, A. McIlroy, C. A. Taatjes and J. B. Halpern, J. Chem. Phys., 2002, 116, 8343–8352 CrossRef CAS.
- A. Fahr, P. Hassanzadeh and D. B. Atkinson, Chem. Phys., 1998, 236, 43–51 CrossRef CAS.
- A. M. Mann, X. Chen, V. A. Lozovsky and C. B. Moore, J. Chem. Phys., 2003, 118, 4452–4455 CrossRef CAS.
- P. Zhang, S. Irle, K. Morokuma and G. S. Tschumper, J. Chem. Phys., 2003, 119, 6524–6538 CrossRef CAS.
- L. Koziol, S. V. Levchenko and A. I. Krylov, J. Phys. Chem. A, 2006, 110, 2746–2758 CrossRef CAS.
- M. Lucas, Y. Song, J. S. Zhang, C. Brazier, P. L. Houston and J. M. Bowman, J. Phys. Chem. A, 2016, 120, 5248–5256 CrossRef CAS PubMed.
- M. L. Morton, J. L. Miller, L. J. Butler and F. Qi, J. Phys. Chem. A, 2002, 106, 10831–10842 CrossRef CAS.
- Y. Song, M. Lucas, M. Alcaraz and J. S. Zhang, J. Phys. Chem. A, 2015, 119, 12318–12328 CrossRef CAS PubMed.
- D. Stranges, M. Stemmler, X. M. Yang, J. D. Chesko, A. G. Suits and Y. T. Lee, J. Chem. Phys., 1998, 109, 5372–5382 CrossRef CAS.
- D. Stranges, P. O’Keeffe, G. Scotti, R. Di Santo and P. L. Houston, J. Chem. Phys., 2008, 128, 151101 CrossRef CAS PubMed.
- C. Chen, B. Braams, D. Y. Lee, J. M. Bowman, P. L. Houston and D. Stranges, J. Phys. Chem. A, 2011, 115, 6797–6804 CrossRef CAS PubMed.
- H.-J. Deyerl, I. Fischer and P. Chen, J. Chem. Phys., 1999, 110, 1450–1462 CrossRef CAS.
- A. Röder, K. Issler, L. Poisson, A. Humeniuk, M. Wohlgemuth, M. Comte, F. Lepetit, I. Fischer, R. Mitric and J. Petersen, J. Chem. Phys., 2017, 147, 013902 CrossRef PubMed.
- M. Shapero, I. A. Ramphal and D. M. Neumark, J. Phys. Chem. A, 2018, 122, 4265–4272 CrossRef CAS PubMed.
- S. A. Ndenqué, R. Dawes and H. Guo, J. Chem. Phys., 2016, 144, 244301 CrossRef PubMed.
- J. C. Loison, S. H. Kable, P. L. Houston and I. Burak, J. Chem. Phys., 1991, 94, 1796–1802 CrossRef CAS.
- D. W. Neyer, S. H. Kable, J. C. Loison, P. L. Houston, I. Burak and E. M. Goldfield, J. Chem. Phys., 1992, 97, 9036–9045 CrossRef CAS.
- S. H. Kable, J. C. Loison, D. W. Neyer, P. L. Houston, I. Burak and R. N. Dixon, J. Phys. Chem., 1991, 95, 8013–8018 CrossRef CAS.
- J. Riedel, S. Dziarzhytski, A. Kuczmann, F. Renth and F. Temps, Chem. Phys. Lett., 2005, 414, 473–478 CrossRef CAS.
- S. Y. Han, X. F. Zheng, S. Ndengue, Y. Song, R. Dawes, D. Q. Xie, J. S. Zhang and H. Guo, Sci. Adv., 2019, 5, eaau0582 CrossRef PubMed.
- D. W. Neyer, X. Luo, I. Burak and P. L. Houston, J. Chem. Phys., 1995, 102, 1645–1657 CrossRef CAS.
- B. Rajakumar, J. E. Flad, T. Gierczak, A. R. Ravishankara and J. B. Burkholder, J. Phys. Chem. A, 2007, 111, 8950–8958 CrossRef CAS PubMed.
- W. T. Mao, Q. Li, F. Kong and M. B. Huang, Chem. Phys. Lett., 1998, 283, 114–118 CrossRef CAS.
- S. K. Shin, S. K. Kim, H. L. Kim and C. R. Park, J. Photochem. Photobiol., A, 2001, 143, 11–16 CrossRef CAS.
- D. L. Osborn, H. Choi, D. H. Mordaunt, R. T. Bise, D. M. Neumark and C. M. Rohlfing, J. Chem. Phys., 1997, 106, 3049–3066 CrossRef CAS.
- J. L. Miller, L. R. McCunn, M. J. Krisch, L. J. Butler and J. Shu, J. Chem. Phys., 2004, 121, 1830–1838 CrossRef CAS PubMed.
- C.-S. Lam, J. D. Adams and L. J. Butler, J. Phys. Chem. A, 2016, 120, 2521–2536 CrossRef CAS PubMed.
- S. Matsika and D. R. Yarkony, J. Chem. Phys., 2002, 117, 7198–7206 CrossRef CAS.
- B. Nichols, E. N. Sullivan, M. Ryazanov and D. M. Neumark, J. Phys. Chem. A, 2017, 121, 579–586 CrossRef CAS PubMed.
- E. J. Bernard, B. R. Strazisar and H. F. Davis, Chem. Phys. Lett., 1999, 313, 461–466 CrossRef CAS.
- A. Teslja, P. J. Dagdigian, M. Banck and W. Eisfeld, J. Phys. Chem. A, 2006, 110, 7826–7834 CrossRef CAS PubMed.
- J. G. Radziszewski, Chem. Phys. Lett., 1999, 301, 565–570 CrossRef CAS.
- Y. Song, M. Lucas, M. Alcaraz, J. S. Zhang and C. Brazier, J. Chem. Phys., 2012, 136, 044308 CrossRef PubMed.
- N. C. Cole-Filipiak, M. Shapero, B. Negru and D. M. Neumark, J. Chem. Phys., 2014, 141, 104307 CrossRef PubMed.
- A. M. Mebel and A. Landera, J. Chem. Phys., 2012, 136, 234305 CrossRef PubMed.
- Y. Song, X. F. Zheng, M. Lucas and J. S. Zhang, Phys. Chem. Chem. Phys., 2011, 13, 8296–8305 RSC.
- M. Shapero, N. C. Cole-Filipiak, C. Haibach-Morris and D. M. Neumark, J. Phys. Chem. A, 2015, 119, 12349–12356 CrossRef CAS PubMed.
- K. Pachner, M. Steglich, P. Hemberger and I. Fischer, J. Chem. Phys., 2017, 147, 084303 CrossRef PubMed.
- M. Lucas, J. Minor and J. S. Zhang, J. Phys. Chem. A, 2013, 117, 12138–12145 CrossRef CAS PubMed.
- M. Lucas, J. Minor, J. S. Zhang and C. Brazier, Chin. J. Chem. Phys., 2014, 27, 621–627 CrossRef CAS.
- E. Hébrard, M. Dobrijevic, Y. Bénilan and F. Raulin, J. Photochem. Photobiol., C, 2006, 7, 211–230 CrossRef.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618–622 CrossRef.
- T. H. Dunning, Jr., J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- K. Andersson, P. A. Malmqvist, B. O. Roos, A. J. Sadlej and K. Wolinski, J. Phys. Chem., 1990, 94, 5483–5488 CrossRef CAS.
- B. O. Roos, P. Linse, P. E. M. Siegbahn and M. R. A. Blomberg, Chem. Phys., 1982, 66, 197–207 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revis. B.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
Footnote |
| † Current address: EPFL SB ISIC LSU, CH H1 575 (Bâtiment CH), Station 6, CH-1015 Lausanne, Switzerland. |
| This journal is © the Owner Societies 2019 |