
Probing ultrafast dynamics during and after passing through conical intersections†

Abstract
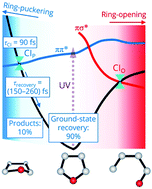
Time-resolved photoelectron spectroscopy using vacuum-UV probe pulses enables observing ultrafast dynamics during and after passing through conical intersections (CIs). The ring-puckering CI plays a prominent role following the ππ* photoexcitation of furan. More than 90% of the excited molecules safely return to the original ground state, while the remaining 10% transforms into isomers after passing through the puckering CI.
- This article is part of the themed collections: Photodissociation and reaction dynamics and 2018 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

