
Pd/NHC-catalyzed cross-coupling reactions of nitroarenes†
Myuto
Kashihara
a,
Rong-Lin
Zhong
b,
Kazuhiko
Semba
 a,
Shigeyoshi
Sakaki
b and
Yoshiaki
Nakao
*a
a,
Shigeyoshi
Sakaki
b and
Yoshiaki
Nakao
*a
aDepartment of Material Chemistry, Graduate School of Engineering, Kyoto University, Nishikyo-ku, Kyoto 615-8510, Japan. E-mail: nakao.yoshiaki.8n@kyoto-u.ac.jp
bFukui Institute for Fundamental Chemistry, Kyoto University, Sakyo-ku, Kyoto 606-8130, Japan
First published on 17th July 2019
Abstract
N-Heterocyclic carbene (NHC) ligands effective for the cross-coupling of nitroarenes were identified. A rational design of the NHC ligand structures enabled significant reduction of catalyst loadings compared with the previous system employing BrettPhos as a phosphine ligand. Experimental and theoretical studies to compare these ligands gave some insights into high activity of the newly developed NHC ligands.
Denitrative transformations of nitroarenes are advantageous in synthetic chemistry because they serve as an important class of chemical feedstocks readily available from simple nitration of aromatic compounds.1 In addition, well-established functionalisations of nitroarenes including SNAr/SEAr/VNS and/or C–H functionalisation2,3 to afford multi-substituted nitroarenes in a site-selective manner make denitrative transformations highly attractive to access a variety of substituted arenes. Conventionally, the replacement of the NO2 group with various functional groups could be achieved in 3 steps including reduction, diazotisation, and Sandmeyer/cross-coupling reactions. Direct transformations of nitro groups have been therefore of high demand to upgrade the synthetic utility of nitroarenes. Some examples of such single-step transformations of Ar–NO2 bonds have been reported but lacked generality in terms of scope of nitroarenes.4 The difficulty in the use of nitroarenes for cross-coupling reactions is partly derived from reduction of the NO2 group by low-valent metal catalysts.5 Nevertheless, we previously reported that the combination of palladium as a metal center and BrettPhos6a as a supporting ligand enabled the unprecedented oxidative addition of Ar–NO2 bonds to palladium(0) to enable the Suzuki–Miyaura coupling,7a Buchwald–Hartwig amination,7b and reductive denitration of nitroarenes.7c Although these coupling reactions opened a novel aspect in chemistry of nitroarenes, there still remained serious issues from a practical point of view such as high loadings of precious Pd (>5 mol%) and expensive Buchwald-type ligands6 (10–20 mol%). Phosphine ligands could also be deactivated through oxidation by the NO2 group.
To deviate from phosphine ligands, we turned our attention to the use of NHC ligands.8 In 2005, the groups of Lassaletta and Glorius independently reported the use of imidazo[1,5-a]pyridinylidenes,9a,b which appeared to be a hybrid form of the Buchwald-type ligands and NHC ligands (Scheme 1). Subsequently, some derivatives were investigated and published.9 Despite being structural mimics of the Buchwald-type ligands, they have rarely been applied to metal-catalysed reactions. We conceived the use of imidazo[1,5-a]pyridinylidene bearing an Ar group at the C5 position as a supporting ligand in the cross-coupling reactions of nitroarenes. NHC ligands generally possess higher electron-donicity and tolerance toward oxidation than phosphine ligands. We expected that the NHC ligands could facilitate the rate-determining oxidative addition of Ar–NO2 bonds and elongate the catalyst lifetime by preventing ligand oxidation.
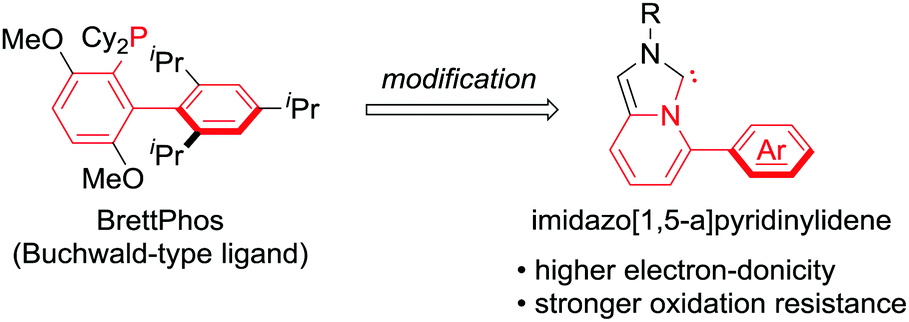 | ||
| Scheme 1 Design of imidazo[1,5-a]pyridinylidene ligands for the cross-coupling of nitroarenes. | ||
We examined the Suzuki–Miyaura coupling of 4-nitroanisole (1a) and phenylboronic acid (2a) in the presence of 1.0 mol% Pd(acac)2 and 2.0 mol% L19i (eqn (1)). In contrast to the use of BrettPhos, which resulted in only 6% of the desired product 3a, the use of L1 drastically improved the yield of 3a to 60%.
Motivated by the preliminary result, we screened various imidazo[1,5-a]pyridinylidenes as ligands in the reaction of 1a with 2a using 1.0 mol% Pd (Scheme 2). The HCl adduct of L1 could be used directly without any loss of the yield.10 Regarding the substituent on nitrogen, electron-withdrawing 3,5-bistrifluoromethyphenyl in L2 and even the phenyl group in L3 were not suitable at all, while sterically hindered 2,6-diisopropylphenyl in L4 and 2,6-dimethoxyphenyl in L5 deteriorated the catalytic activity as well, though they were electron-donating. Cycloalkyl substituents seemed good for this system, except for the cyclopropyl group in L6, which could react with Pd(0).11L9 showed the best performance among these, producing 3a in 61% yield. The bulky adamantyl groups in L12 and L13, and 3,5-di-tert-butyl-4-methoxyphenyl in L14 retarded the reaction. L15 and L16, which were expected to be more electron-donating than L1, unfortunately failed to improve the catalytic activity. Similarly, introducing an electron-donating methyl substituent on the backbone in L17 did not bring any positive effects. By analogy with the Buchwald-type phosphines, the properties of the C5-aryl group were found to be important. L18 and L19 were less active than L1 in line with the competition of the Buchwald phosphines (SPhos and RuPhos respectively vs. XPhos or BrettPhos) in our previous report.7a To our surprise, NHC bearing a hydroxymethyl group L20 marked higher yield of 3a than L19.12
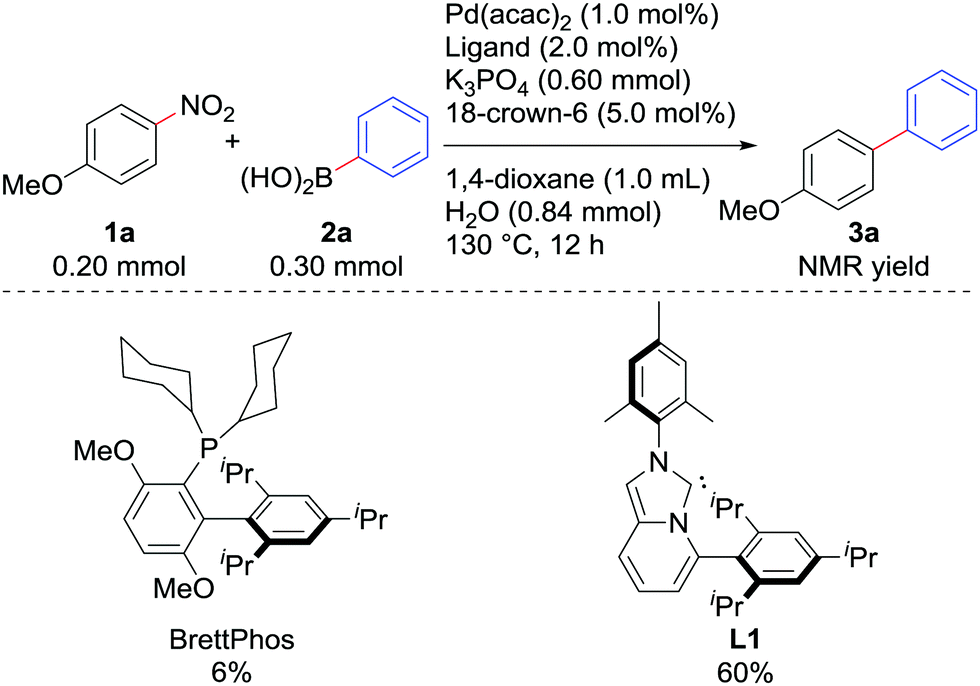 | (1) |
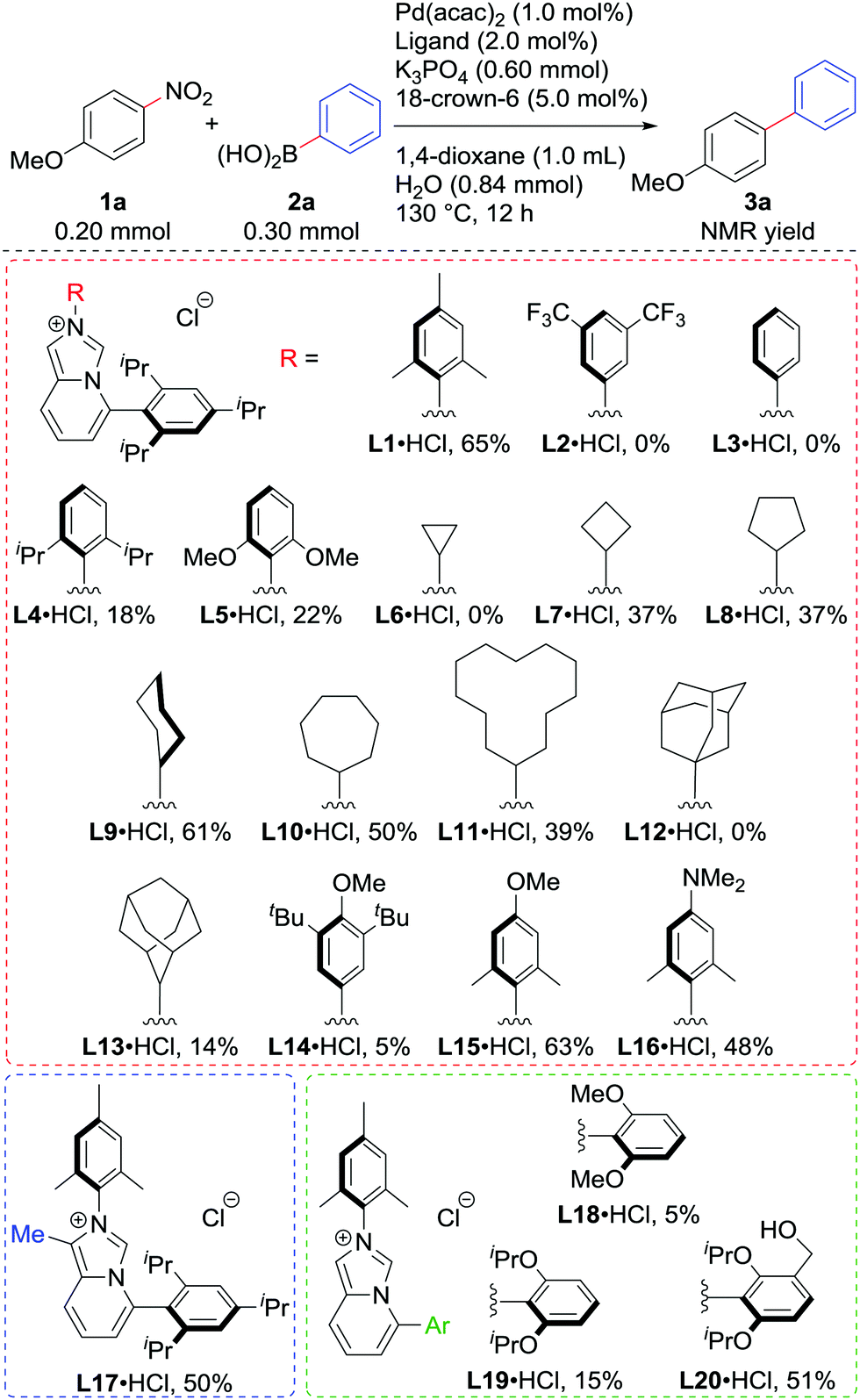 | ||
| Scheme 2 Optimisation of ligand structures. | ||
To make this system more efficient, we made an attempt to use (L1)Pd complexes as catalyst precursors (Table 1). (L1)Pd(acac)Cl was prepared and examined first, but the yield was similar to the case where Pd(acac)2 and L1·HCl were independently used. Another complex (L1)Pd(allyl)Cl proved to be effective to afford 3a in 76%.
| Pd/L1 | Yield (%) |
|---|---|
| Pd(acac)2 (1.0 mol%) + L1·HCl (2.0 mol%) | 65 |
| (L1)Pd(acac)Cl (1.0 mol%) | 61 |
| (L1)Pd(allyl)Cl (1.0 mol%) | 76 |
We then carried out some analyses to verify the properties of L1. Fig. 1 shows the time-course of the Suzuki–Miyaura coupling of 1a with 2a catalysed by 5.0 mol%Pd/BrettPhos and 1.0 mol%Pd/L1. The former system turned out to be deactivated within 3 h,13 whereas the coupling proceeded with the latter system much faster and the yield of 3a kept increasing even after 4 h. These reaction profiles obviously revealed two significant effects associated with L1: rate-acceleration and longer catalyst lifetime. The higher reaction rate was also supported by DFT calculations. The activation barrier for the rate-limiting oxidative addition of 1a to (L1)Pd0 was calculated to be 27.2 kcal mol−1, which was smaller than that of (BrettPhos)Pd0 (30.1 kcal mol−1).14 This difference was likely to derive from their HOMO energies. The higher HOMO level of (L1)Pd0 could enable the faster oxidative addition of the Ar–NO2 bond (see the ESI† for details). Experimentally, a large difference of %VBur15 between (BrettPhos)AuCl and (L1)AuCl was noted (59.5% vs. 51.9%,9i respectively), illustrating that L1 occupied less space around the Pd center, possibly allowing easier access of the substrate to Pd than with BrettPhos. Furthermore, the rigid skeleton of L1 could inhibit its flip in the coordination sphere, unlike BrettPhos which could show two different coordination modes. This rigidity could partly contribute to the robustness of the (L1)Pd system in collaboration with reluctance of oxidation by nitroarenes.
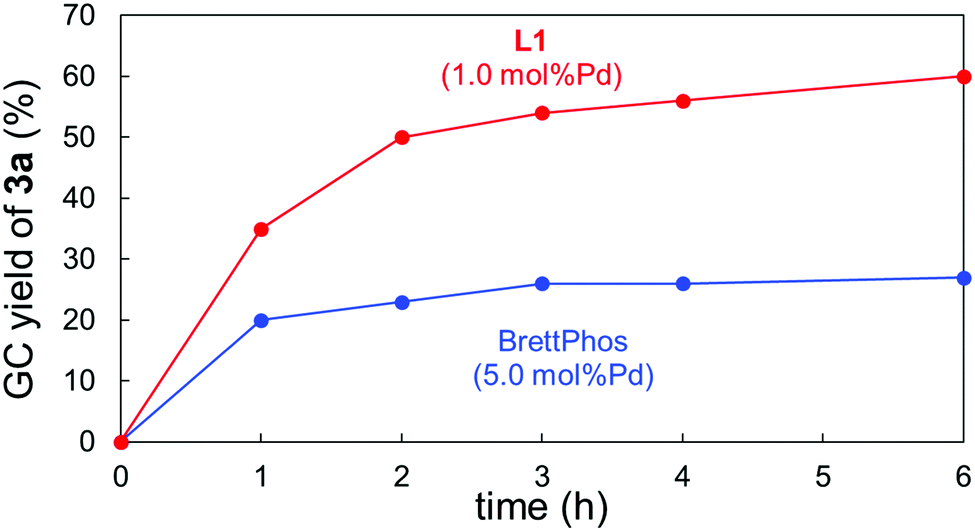 | ||
| Fig. 1 Time-courses of the coupling of 4-nitroanisole (1a) and phenylboronic acid (2a). | ||
We also checked the reactivity of the new catalyst to several other substrate sets (Scheme 3). The use of boronic acid neopentylglycol ester in combination with a catalyst derived from Pd(acac)2 and L1·HCl slightly improved the yield of 3a. Couplings of nitronaphthalene and F-containing arylboronic acids proceeded very smoothly to give 3b and 3c. A nitroarene bearing an electron-withdrawing trifluoromethyl group could be reacted, though the yield of biaryl 3d was relatively low as observed in our original report.7a In all the cases, the new catalytic system performed much better than 1.0 mol% Pd/BrettPhos. Moreover, 2,6-dimethylnitrobenzene, which was too sterically demanding to cross-couple under the previous conditions, afforded biaryl 3e by the Pd/L1 catalyst, possibly due to the reduced %VBur of L1 compared with BrettPhos.
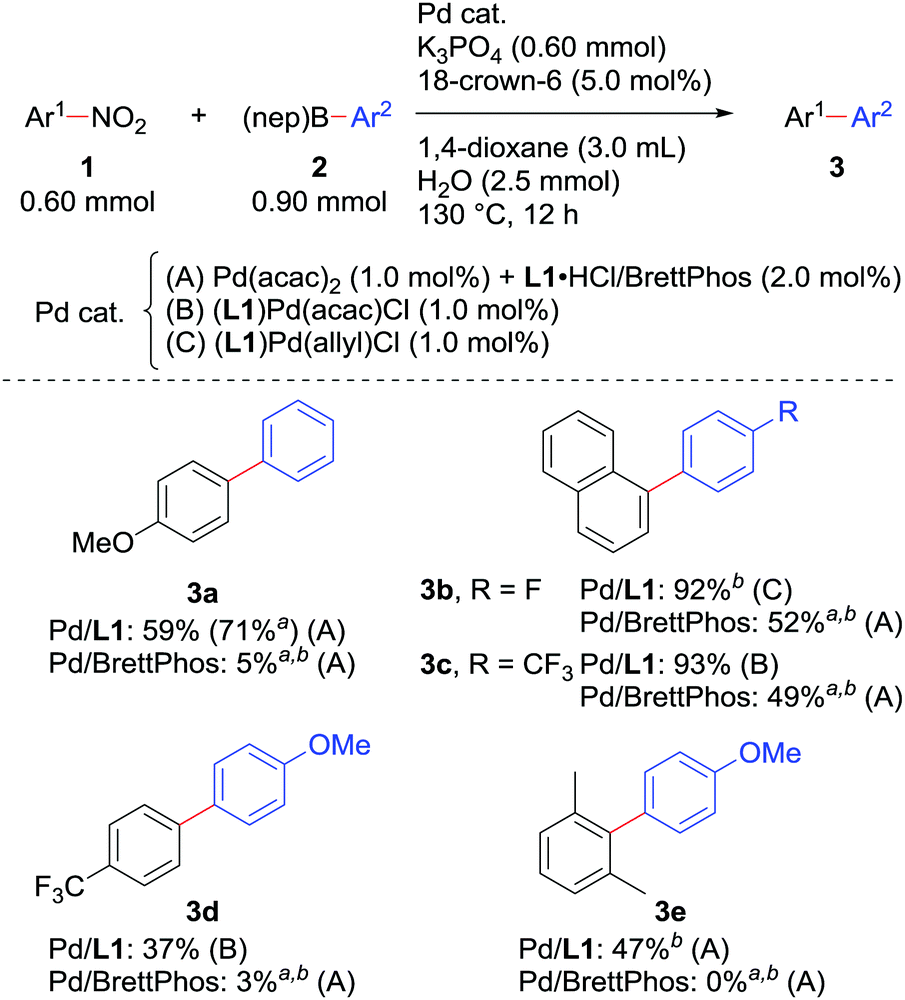 | ||
Scheme 3 Scope of the Suzuki–Miyaura coupling of nitroarenes. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) NMR yields determined using 1,3,5-trimethoxybenzene as an internal standard. bAr2–B(OH)2 was used instead of Ar2–B(nep). NMR yields determined using 1,3,5-trimethoxybenzene as an internal standard. bAr2–B(OH)2 was used instead of Ar2–B(nep). | ||
The Pd/NHC system developed herein catalysed not only the Suzuki–Miyaura coupling, but also the Buchwald–Hartwig amination and reductive denitration of nitroarenes (Scheme 4). Aniline (4) could be coupled with 1a to afford diarylamine 5 by using 1.0 mol% (L1)Pd(acac)Cl as a catalyst precursor. Denitration of 1a proceeded well with (L9)Pd(acac)Cl, delivering anisole (7) in 62% yield. Both reactions again afforded the products in yields much higher than those catalysed by 1 mol% Pd/BrettPhos.
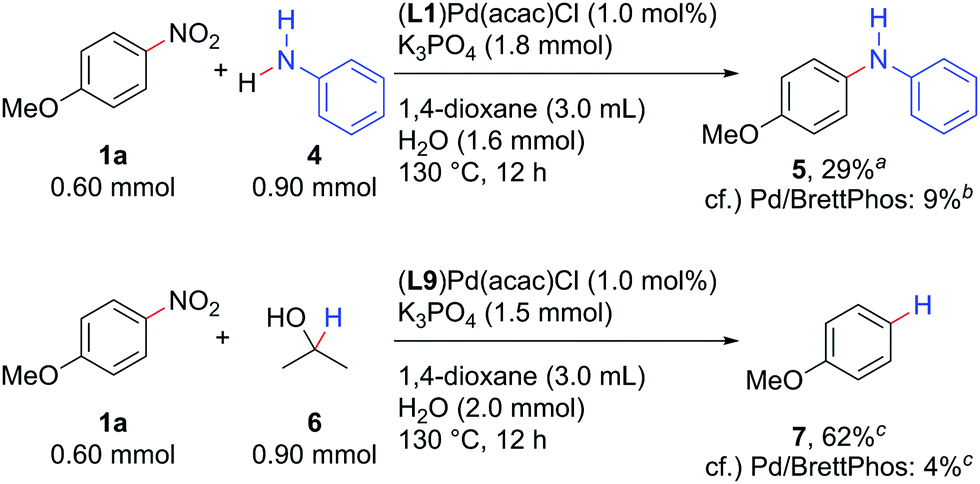 | ||
| Scheme 4 The Buchwald–Hartwig amination and reductive denitration of nitroarenes. aIsolated yield. bNMR yield determined using 1,3,5-trimethoxybenzene as an internal standard. cGC yields determined using n-C13H28 as an internal standard. | ||
In conclusion, we have developed new reaction conditions employing imidazo[1,5-a]pyridinylidene as NHC ligands for the cross-coupling reactions of nitroarenes. The Pd/NHC catalysts showed much higher activity than the Pd/BrettPhos system. Some insights into the reasons for the improved performance by the Pd/NHC catalyst are shown in terms of experimental and theoretical studies. Further applications of the Pd/NHC catalyst to other reactions are in progress.
This work was supported by the “JST CREST program Grant Number JPMJCR14L3 in Establishment of Molecular Technology towards the Creation of New Functions”, the “JSPS KAKENHI Grant Number JP15H05799 in Precisely Designed Catalysts with Customized Scaffolding”, and TOSOH corporation.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) N. Ono, The Nitro Group in Organic Synthesis, Wiley-VCH, New York, 2001 Search PubMed; (b) G. Booth, Nitro Compounds, Aromatic, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, New York, 2012 Search PubMed.
- M. Mąkosza and J. Winiarski, Acc. Chem. Res., 1987, 20, 282 Search PubMed.
- (a) L. Caron, L.-C. Campeau and K. Fagnou, Org. Lett., 2008, 10, 4533 CAS; (b) P. Guo, J. M. Joo, S. Rakshit and D. Sames, J. Am. Chem. Soc., 2011, 133, 16338 CAS; (c) V. O. Iaroshenko, A. Gevorgyan, O. Davydova, A. Villinger and P. Langer, J. Org. Chem., 2014, 79, 2906 CAS; (d) V. O. Iaroshenko, A. Gevorgyan, S. Mkrtchyan, T. Grigoryan, E. Movsisyan, A. Villinger and P. Langer, ChemCatChem, 2015, 7, 316 CAS; (e) Z. Yi, Y. Aschenaki, R. Daley, S. Davick, A. Schnaith, R. Wander and D. Kalyani, J. Org. Chem., 2017, 82, 6946 CAS.
- (a) J. R. Beck, Tetrahedron, 1978, 34, 2057 CAS; (b) J. Holt, F. Tjosås, J. M. Bakke and A. Fiksdahl, J. Heterocycl. Chem., 2004, 41, 987 CAS; (c) S. D. Kuduk, R. M. DiPardo and M. G. Bock, Org. Lett., 2005, 7, 577 CAS; (d) F. Tjosaas and A. Fiksdahl, Molecules, 2006, 11, 130 CAS; (e) L. Arias, H. Salgado-Zamora, H. Cervantes, E. Campos, A. Reyes and E. C. Taylor, J. Heterocycl. Chem., 2006, 43, 565 CAS; (f) P. Beier, T. Pastýříková, N. Vida and G. Iakobson, Org. Lett., 2011, 13, 1466 CAS; (g) M. Mondal, S. K. Bharadwaj and U. Bora, New J. Chem., 2015, 39, 31 CAS; (h) T. Begum, M. Mondal, M. P. Borpuzari, R. Kar, P. K. Gogoi and U. Bora, Eur. J. Org. Chem., 2017, 3244 CAS; (i) S. S. Bahekar, A. P. Sarkate, V. M. Wadhai, P. S. Wakte and D. B. Shinde, Catal. Commun., 2013, 41, 123 CAS; (j) H. Tian, A. Cao, L. Qiao, A. Yu, J. Chang and Y. Wu, Tetrahedron, 2014, 70, 9107 CAS; (k) T. B. Nguyen and P. Retailleau, Org. Lett., 2017, 19, 4858 CAS; (l) D. W. Lamson, P. Ulrich and R. O. Hutchins, J. Org. Chem., 1973, 38, 2928 CAS; (m) R. Fielden, O. Meth-Cohn and H. Suschitzky, J. Chem. Soc., Perkin Trans. 1, 1973, 696 CAS; (n) A. G. Giumanini and G. Verardo, Can. J. Chem., 1997, 75, 469 CAS; (o) C. W. Rees and S. C. Tsoi, Chem. Commun., 2000, 415 CAS; (p) R. El-Berjawi and P. Hudhomme, Dyes Pigm., 2018, 159, 551 CAS.
- (a) R. S. Berman and J. K. Kochi, Inorg. Chem., 1980, 19, 248 CAS; (b) K. Osakada, R. Sato and T. Yamamoto, Organometallics, 1994, 13, 4645 CAS.
- (a) B. P. Fors, D. A. Watson, M. R. Biscoe and S. L. Buchwald, J. Am. Chem. Soc., 2008, 130, 13552 CAS; (b) R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461 CAS; (c) E. J. Cho, T. D. Senecal, T. Kinzel, Y. Zhang, D. A. Watson and S. L. Buchwald, Science, 2010, 328, 1679 CAS; (d) A. Aranyos, D. W. Old, A. Kiyomori, J. P. Wolfe, J. P. Sadighi and S. L. Buchwald, J. Am. Chem. Soc., 1999, 121, 4369 CAS; (e) A. V. Vorogushin, X. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 8146 CAS; (f) X. Wu, B. P. Fors and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 9943 CAS; (g) D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338 CAS; (h) B. P. Fors and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 12898 CAS; (i) A. C. Sather and S. L. Buchwald, Acc. Chem. Res., 2016, 49, 2146 CAS.
- (a) M. R. Yadav, M. Nagaoka, M. Kashihara, R.-L. Zhong, T. Miyazaki, S. Sakaki and Y. Nakao, J. Am. Chem. Soc., 2017, 139, 9423 CAS; (b) F. Inoue, M. Kashihara, M. R. Yadav and Y. Nakao, Angew. Chem., Int. Ed., 2017, 56, 13307 CAS; (c) M. Kashihara, M. R. Yadav and Y. Nakao, Org. Lett., 2018, 20, 1655 CAS; (d) K. K. Asahara, T. Okita, A. N. Saito, K. Muto, Y. Nakao and J. Yamaguchi, Org. Lett., 2019, 21, 4721 CAS.
- For reviews, see: (a) E. A. B. Kantchev, C. J. O’Brien and M. G. Organ, Angew. Chem., Int. Ed., 2007, 46, 2768 CAS; (b) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612 Search PubMed; (c) J. C. Y. Lin, R. T. W. Huang, C. S. Lee, A. Bhattacharyya, W. S. Hwang and I. J. B. Lins, Chem. Rev., 2009, 109, 3561 CAS; (d) M. Poyatos, J. A. Mata and E. Peris, Chem. Rev., 2009, 109, 3677 CAS; (e) G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151 CAS; (f) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CAS.
- (a) M. Alcarazo, S. J. Roseblade, A. R. Cowley, R. Fernández, J. M. Brown and J. M. Lassaletta, J. Am. Chem. Soc., 2005, 127, 3290 CAS; (b) C. Burstein, C. W. Lehmann and F. Glorius, Tetrahedron, 2005, 61, 6207 CAS; (c) M. Nonnenmacher, D. Kunz, F. Rominger and T. Oeser, Chem. Commun., 2006, 1378 CAS; (d) A. Fürstner, M. Alcarazo, H. Krause and C. W. Lehmann, J. Am. Chem. Soc., 2007, 129, 12676 Search PubMed; (e) J. T. Hutt and Z. D. Aron, Org. Lett., 2011, 13, 5256 CAS; (f) E. Y. Tsui and T. Agapie, Polyhedron, 2014, 84, 103 CAS; (g) M. Espina, I. Rivilla, A. Conde, M. M. Díaz-Requejo, P. J. Pérez, E. Álvarez, R. Fernández and J. M. Lassaletta, Organometallics, 2015, 34, 1328 CAS; (h) C. T. Check, K. P. Jang, C. B. Schwamb, A. S. Wong, M. H. Wang and K. A. Scheidt, Angew. Chem., Int. Ed., 2015, 54, 4264 CAS; (i) Y. Kim, Y. Kim, M. Y. Hur and E. Lee, J. Organomet. Chem., 2016, 820, 1 CAS; (j) Y. Koto, F. Shibahara and T. Murai, Chem. Lett., 2016, 45, 1327 CAS; (k) Y. Koto, F. Shibahara and T. Murai, Org. Biomol. Chem., 2017, 15, 1810 CAS.
- Unreacted 1a (24%) and p-anisidine (5%) were also observed.
- (a) S. Ma, L. Lu and J. Zhang, J. Am. Chem. Soc., 2004, 126, 9645 CAS; (b) G. T. Jong and F. M. Bickelhaupt, ChemPhysChem, 2007, 8, 1170 Search PubMed.
- Although we confirmed that the hydroxymethyl group of L20 was lost by 1H NMR, its fate and the reason for the higher activity compared with L19 were unclear.
- BrettPhos was completely consumed to give BrettPhos oxide (∼80%) and a certain amount of biaryl via C(sp2)–P bond cleavage.
- The energy value for (BrettPhos)Pd is different from that in ref. 7a because we applied a different functional (see ESI† for details).
- (a) H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841 CAS; (b) L. Falivene, R. Credendino, A. Poater, A. Petta, L. Serra, R. Oliva, V. Scarano and L. Cavallo, Organometallics, 2016, 35, 2286 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc05055h |
| This journal is © The Royal Society of Chemistry 2019 |