
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Review: a comprehensive summary of a decade development of the recombinase polymerase amplification
Jia
Li
 a,
Joanne
Macdonald
*bc and
Felix
von Stetten
*ad
a,
Joanne
Macdonald
*bc and
Felix
von Stetten
*ad
aLaboratory for MEMS Applications, IMTEK – Department of Microsystems Engineering, University of Freiburg, Georges-Köhler-Allee 103, 79110 Freiburg, Germany. E-mail: Felix.von.Stetten@Hahn-Schickard.de; Tel: +49 761 203-73243
bInflammation and Healing Research Cluster, Genecology Research Centre, School of Science and Engineering, University of the Sunshine Coast, Qld, Australia. E-mail: jmacdon1@usc.edu.au; Tel: +61 7 5456 5944
cDivision of Experimental Therapeutics, Columbia University, New York, NY, USA. E-mail: jm2236@columbia.edu
dHahn-Schickard, Georges-Köhler-Allee 103, 79110 Freiburg, Germany
First published on 14th November 2018
Abstract
Nucleic acid amplification has permeated every field in the life sciences since the introduction of the classic polymerase chain reaction (PCR) method in 1983. Yet, despite its fundamental reach, PCR has been constrained within the walls of a laboratory, due to its requirement for a sophisticated thermocycling machine, limiting external application in low-resource settings. New isothermal amplification strategies are seeking to break through traditional laboratory boundaries by providing nucleic acid replication at constant temperatures. Of these methods, recombinase polymerase amplification (RPA) is one of the fastest developing, experiencing rapid uptake and market, even though it was introduced comparatively late. Critically, RPA's technology potentiates highly accessible and sensitive nucleic acid amplification outside of laboratory, and even self-testing. Here we provide a comprehensive review of the equipment-free simplicity of RPA over its first decade of development. Our review includes key knowledge of RPA technology, such as its reaction components, mechanism, sensitivities and specificities, and distinctive detection methods. The review also provides know-how for developing RPA assays, and information about commercially available RPA reaction kits and accessories. We summarise critical RPA experimental tips and issues available through data mining the published literature, to assist researchers in mastering the RPA reaction. We also outline influential hotspots of RPA development, and conclude with outlooks for future development and implications for eclipsing PCR and further revolutionising the life sciences.
 Jia Li | Dr Jia Li did her undergraduate degree in the University of Sydney, working on the structure and activity relationship of carborane phosphonium salts for the Boron Neutron Capture Therapy (BNCT). She then went to England for a research project about onco-protein–protein interactions at the University of Leeds. She did her PhD under Dr Joanne Macdonald's supervision at the University of the Sunshine Coast, developing rapid and novel virus detection biosensors towards point-of-care. Afterwards, she has been working as a post-doc in the University of Freiburg under Alexander von Humboldt Fellowship, researching in assay development for micro-total diagnostic systems. |
 Joanne Macdonald | Dr Joanne Macdonald's research focuses on the molecular engineering of advanced devices, including a DNA automaton able to play tic-tac-toe against a human opponent, and rapid field-based biosensors for the detection of pathogens. She is an Associate Professor in Molecular Engineering at the University of the Sunshine Coast (QLD, Australia), holds a joint appointment in Clinical Medical Sciences at Columbia University (New York, USA), and is the Chief Technical Officer of the biosensing company BioCifer Pty Ltd. Dr Macdonald received her PhD in Microbiology in 2003 from the University of Queensland, Australia. |
 Felix von Stetten | Professor Felix von Stetten studied agricultural sciences and biotechnology. He completed his PhD in Microbiology in 1999 from the Technical University of Munich, Germany. Thereafter, he joined in the diagnostic industry, where he was involved in the development of methods for sample preparation, real-time PCR and DNA-arrays. Subsequently, he joined the Laboratory for MEMS Applications at IMTEK, University of Freiburg, where he was involved in lab-on-a-chip research. In 2008, he became head of the Hahn-Schickard Lab-on-a-Chip division. Now he is associate director of the Hahn-Schickard-Institut für Mikroanalysesysteme, and apl. professor at IMTEK, University of Freiburg. |
1. Introduction and overview
Nucleic acid amplification (NAA) in vitro, the artificial replication of genetic material, has infiltrated all areas of life sciences and biotechnology, such as pathogen detection, cancer research, cloning, sequencing, genetic engineering, synthetic biology, genotyping, mutagenesis, forensic identification of crimes, drug discovery, molecular archaeology, food testing, wellness and lifestyle testing etc. This explosive revolution began with the invention of the polymerase chain reaction (PCR) by Kary Mullis in 1983.1 Fundamentally, PCR is a cyclic process that performs exponential amplification from a single nucleic acid molecule to billions of copies in vitro, by providing successive temperatures favourable to nucleic acid replication processes (strand denaturation, primer annealing, and enzymatic extension). Increasing molecular quantities makes the handling and subsequent applications of nucleic acids easier, reducing the requirement for use of toxic radioactive probes to track molecular presence, and spawning immense creativity around applications for use. Yet, as valuable as PCR is, the requirement for a sophisticated thermocycler to provide the cyclic heating and cooling process, has largely bound PCR to implementation within the walls of a laboratory, impeding its application in low-resource settings.Recent advances in isothermal nucleic acid amplification have provided simplified incubation conditions for artificial nucleic acid replication, requiring only a constant temperature rather than thermocycling. The single temperature incubation reduces equipment requirements, opening new avenues to break through the boundaries of the laboratory and perform amplification in low-resource settings. The elimination of repeated heating and cooling steps also provides a second advantage for low-resource implementation, through reduced amplification times. Faster reactions occur not only because of a reduction in heating and cooling times, but also because multiple molecular reactions can proceed asynchronously rather than being forced to operate sequentially within an artificial heating and cooling cycle. Since the early 1990s, a plethora of isothermal nucleic acid amplification methods have adopted various reaction mechanisms. The most well-established methods are exemplified by nucleic acid sequence-based amplification (NASBA, also known as transcription mediated amplification, TMA), signal-mediated amplification of ribonucleic acid (RNA) technology (SMART), helicase-dependent amplification (HDA), recombinase polymerase amplification (RPA), rolling circle amplification (RCA), multiple displacement amplification (MDA), loop-mediated isothermal amplification (LAMP) and strand displacement amplification (SDA); readers can refer to details of these methods in a few reviews.2–5 One technology in particular, recombinase polymerase amplification (RPA), is experiencing rapid development and increasing market share (Fig. 1), despite its comparatively late introduction, due to its simplified equipment requirements and fast reaction times.
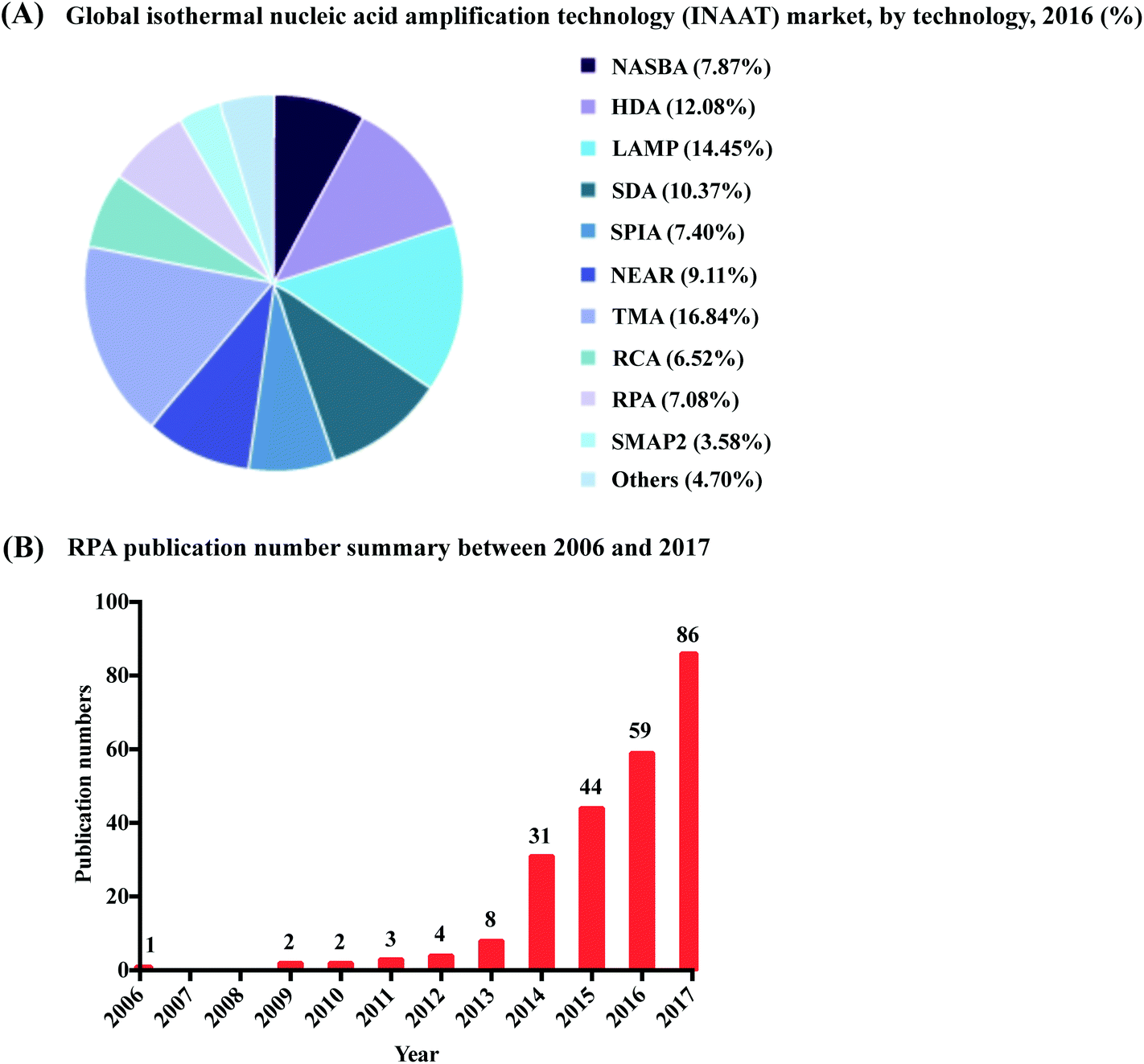 | ||
| Fig. 1 Summary of RPA market share and publication numbers from its first introduction. (A): RPA market share percentage in the isothermal nucleic acid amplification technology. Reprinted and reproduced with permission from ref. 7. Copyright 2018 Grand View Research, Inc. (B): RPA publication numbers from year 2006 to 2017 based on collected data from web of science. | ||
RPA was first introduced in 2006 by Niall Armes from ASM Scientific Ltd (Cambridge, United Kingdom, founded by the Wellcome Trust Sanger Institute).6 Although RPA has not yet occupied a large market share percentage in the isothermal nucleic acid amplification technology (according to the data from the Grand View Research report,7Fig. 1A), it is experiencing the most rapid uptake. More than 250 publications about RPA have been published so far, with a consistent increase in RPA publication numbers in the last six years; noticeably, the RPA publication number started growing exponentially from year 2014 (Fig. 1B). Among these publications, five RPA review papers were published in consecutive years from 2014 to 2018, respectively. The review of Zaghloul and El-shahat focuses on the application of RPA for hepatitis C virus diagnosis;8 the review of Moore and Jaykus emphasises RPA assays developed for the detection of enteric viruses;9 the reviews of James and Macdonald,10 Daher et al.11 and Lobato and O'Sullivan12 describe and summarise the characteristics and strengths for applications of RPA in point-of-care (POC) diagnostics. In comparison, here we provide a comprehensive review that focuses on the radical properties and development potential of RPA. Beginning with an introduction of the key aspects of RPA technology, namely reaction components and mechanisms. We subsequently provide know-how about developing RPA assays, including design and selection of oligonucleotides (primer, probe and template); the information about commercially available RPA reaction kits and accessories are also provided. For those interested in the technical implementation of RPA, we summarise critical RPA experimental tips and issues available through data mining the published literature to assist researchers better master RPA reaction. This is followed by elucidating the clinical/field performance of RPA via collated data such as sensitivity and specificity from RPA literature. We also describe some distinctive RPA detection methods for those who want to detect RPA assay signal using the methods other than the commonly used PCR detection methods. To understand the critical significance of this technology for eclipsing PCR and breaking out of the boundaries of the laboratory walls, we discuss the development hot-spots of RPA, including quantitative RPA, multiplex RPA reaction, mobile RPA diagnostic, integrated RPA assays on microfluidics and one-step RPA assays. Our review then concludes with outlooks of future development of RPA.
2. Recombinase polymerase amplification (RPA) reaction
The prominence of RPA as revolutionary method to eclipse PCR stems from its specific reaction components (Table 1) and mechanism. For a successful RPA assay, the nuances are hinged on the intrinsic factors, the design of the primers, probe and nucleic acid template; and are related to the extrinsic factors, such as reaction temperature and agitation, tolerability to mismatches, inhibitors and background DNA. In addition, the nucleic acid labelling during RPA, and RPA amplicon clean-up and post-amplification treatment are also important details for successful RPA detection. This section provides these practical information summarised from the RPA literature to serve as a guideline for RPA assay design. In addition, readers can also get information about commercially available RPA reaction kits and accessories (Tables 2 and 3). At the end, this section elucidates the clinical/field performance of RPA via data mining of RPA literature, which are also succinctly collated (Tables 4 and 5).| Reaction components | Typical concentration | Functions | Ref. |
|---|---|---|---|
| T4 UvsX protein | 120 ng μL−1 | Recombinase that possesses pairing and strand-transfer activity that is important in genetic recombination, DNA repair and replication (or E. coli RecA; recombinase is a central component in the related processes of recombinational DNA repair and homologous genetic recombination that is the ortholog of the UvsX protein). | 13 and 14 |
| T4 UvsY protein | 60 ng μL−1 | Recombinase loading factor that is classified as a recombination-mediator protein that stimulates the single-stranded DNA-dependent ATPase activity of T4 UvsX and lowers the critical concentration of T4 UvsX required for activity. | 15 |
| T4 gp32 | 600 ng μL−1 | Single-stranded binding (SSB) protein is involved in DNA replication, repair and recombination, and binds preferentially to single-stranded DNA. The T4 UvsX, T4 UvsY and T4 gp32 proteins work co-operatively to initiate the RPA reaction via unwinding, D-loop formation and stabilisation of the DNA template. | 16 and 17 |
| Bacillus subtilis DNA polymerase I (Bsu) or Staphylococcus aureus polymerase (Sau) | Bsu: 30 ng μL−1; Sau: 8.6 or 12.8 μg | DNA polymerase synthesises new DNA templates homologous to the target nucleic acid, by extending nucleotide building blocks from the bound primers, complementary to the original target nucleic acid sequence or “template”. | 18 and 19 |
| Deoxynucleotide triphosphate (dNTP, N = A, T, C, G) | 200 μM each | An equimolar solution of dATP, dCTP, dGTP and dTTP are building blocks used by the DNA polymerase to synthesise new templates. | — |
| Forward and reverse primers | Usually at 420 nM each, but can be varied in the concentration range of 150 nM to 600 nM | Primers are critical to directing the amplification event to the nucleic acid target of interest through homologous binding. After binding, the primers provide the essential 3′-OH for polymerase to perform strand extension. | 20 |
| DNA template | — | The oligonucleotide that the primers bind to for the synthesis of exact new oligonucleotides | — |
| Carbowax20M (a high molecular weight polyethylene glycol (PEG)) | PEG 35K (5%) | The crowding reagent is a good mimic of the real biomacromolecules condition in vivo and facilitates amplification, as the crowding agents can enhance the catalytic activity of the enzymes. | 6 and 21–24 |
| Dithiothreitol | 2 mM | Stablisation of the enzymes by baring free sulfhydryl groups. | 25 |
| Phosphocreatine | 50 mM | The three components form the energy-supply system for the activities of the recombinase and the DNA polymerase. | 26 |
| Creatine kinase | 100 ng μL−1 | ||
| Adenosine triphosphate (ATP) | 3 mM | ||
| Tris(hydroxymethyl)aminomethane (Tris) | 50 mM (pH 7.9) | The two components serve to stabilise and solubilise the DNA in solution. | 27 and 28 |
| Potassium acetate | 100 mM | ||
| Magnesium acetate | 14 mM | Acting as a cofactor for the performance of the enzymes. The RPA reaction initiates once the magnesium acetate is added. | 20 |
| Product name | Category | Nucleic acid detection | Compatible general detection method | Product information |
|---|---|---|---|---|
| TwistAmp® Basic | Lyophilised kit | DNA | Gel electrophoresis | The lyophilised kits contain pre-mixed enzymes and reagents necessary for the amplification, the user needs only supply primers and template (and dNTPs for the liquid kits). The RT kits afford one-step RNA amplification, which contain pre-mixed enzymes and reagents necessary for the amplification. The user need only supply primers, template and RNase inhibitor. |
| TwistAmp® Basic RT | Lyophilised kit | RNA | ||
| TwistAmp® Liquid Basic | Liquid kit | DNA | ||
| TwistAmp® Liquid Basic RT | Liquid kit | RNA | ||
| TwistAmp® exo | Lyophilised kit | DNA | Real-time fluorogenic probe-based | Recommended for users who want to combine TwistDx's RPA amplification technology with the use of TwistDx's proprietary fluorescent TwistAmp® exo probe in a homogenous format. The lyophilised kits contain pre-mixed enzymes and reagents necessary for the amplification, the user needs only supply primers, probe and template (and dNTPs for the liquid kits). The RT kits afford one-step RNA amplification, which contain pre-mixed enzymes and reagents necessary for the amplification. The user need only supply primers, probe, template and RNase inhibitor. |
| TwistAmp® exo RT | Lyophilised kit | RNA | ||
| TwistAmp® Liquid exo | Liquid kit | DNA | ||
| TwistAmp® Liquid exo RT | Liquid kit | RNA | ||
| TwistAmp® fpg | Lyophilised kit | DNA | Real-time and end-point fluorogenic probe-based | Tailored for users who want to combine TwistDx's amplification technology with an alternative TwistDx reporter probe system – fluorescent TwistAmp® fpg probe other than the TwistAmp® exo probe system in a homogenous format. The kit contains pre-mixed enzymes and reagents necessary for the amplification, the user needs only supply primers, probe, template and RNase inhibitor. |
| TwistAmp® nfo | Lyophilised kit | DNA | Lateral flow strip | Designed for users who want to detect the amplicons based on sandwich assays. The kit contains pre-mixed enzymes and reagents necessary for the amplification, the user need only supply primers, probe and template. |
| TwistAmp® exo +ListeriaM | Food safety lyophilised kit | DNA (Listeria monocytogenes hly gene) | Real-time fluorogenic probe-based | The kits contain pre-mixed enzymes, oligos and reagents necessary for detection of specific genes of Listeria monocytogenes and Campylobacter species respectively in less than 10 minutes. User must perform sample preparation. |
| TwistAmp® exo +Campylobacter | DNA (Campylobacter species including jejuni and coli) | |||
| TwistGlow® Salmonella | Food safety lyophilised kit | DNA (Salmonella enterica INVA gene) | Real-time and end-point fluorogenic probe-based | The kits contain pre-mixed enzymes, oligos and reagents necessary, users need only add DNA, with provided buffer and magnesium acetate to the reactions. The kits also feature internal control DNA and probes in the lyophilised pellets, and lysis buffer for a two-step lysis of up to 5 μL of sample. The turn-around time of the Glow kit and the Flow kit are less than 10 minutes and approximately 20 minutes, respectively. |
| TwistFlow® Salmonella | Lateral flow strip |
| Product name | Category | Compatible general detection method | Product information |
|---|---|---|---|
| Twirla™ Portable Mixing Incubator | Device for incubation | Gel electrophoresis and lateral flow strip | Small and portable, and incubates up to 6 RPA reactions at optimal temperature. It can be powered by battery or mains-power via micro USB. Magnetic mixing of RPA reactions is also possible when fitted with Micro Balls (0.2 mL; dispensed with Micro Ball Dispenser) in the reaction tube. |
| T8-ISO Instrument | Device for incubation and detection | Real-time | Incubates up to 8 RPA reactions with 2 channel fluorescence detection per tube; the testing temperature range is from 37 °C to 65 °C. It can be powered by mains-power via micro USB or PowerGorilla external battery. Magnetic mixing of RPA reactions is also possible when fitted with Micro Balls (0.2 mL; dispensed with Micro Ball Dispenser) in the reaction tube. It can also be adapted into a T8-ISO Carry Case when travelling to demanding environments. |
| T16-ISO Instrument | Device for incubation and detection | Real-time | An advanced version of the T8-ISO Instrument, which incubates up to 16 RPA reactions with 3 channel fluorescence detection per tube. |
| Milenia HybriDetect 1 | Device for lateral flow detection | Lateral flow strip | Single-plex detection designed to detect a biotin and FITC/FAM labelled amplicon. Detection is based on sandwich assay using gold nanoparticles as tracer. |
| Milenia HybriDetect 2 | Device for lateral flow detection | Lateral flow strip | Duplex detection designed to simultaneously detect two amplicons labelled with FITC (or FAM)/biotin and/or FITC (or FAM)/DIG. Detection is based on sandwich assay using gold nanoparticles as tracer. |
| PCRD Nucleic Acid Detection | Device for lateral flow detection | Lateral flow strip | Duplex detection designed to simultaneously detect two amplicons labelled with DIG/biotin and/or FITC (or FAM)/biotin. Detection is based on sandwich assay using carbon nanoparticles as tracer and is performed in an open cartridge. |
| U-Star disposable nucleic acid lateral flow detection units | Device for lateral flow detection | Lateral flow strip | Single-plex detection designed to detect a biotin and FITC (or FAM) labelled amplicon. Detection is based on sandwich assay using carbon nanoparticles as tracer and is performed in a sealed cartridge. |
| Analyte(s) | Detection method | Limit of detection | Ref. |
|---|---|---|---|
| a CFU: colony forming unit. | |||
| BlaCTX-M-15 antimicrobial resistance gene | Electrowetting-on-dielectric (EWOD)-based digital droplets end-point fluorescent detection | 5.6 fg (∼a single DNA copy) | 80 |
| RNA polymerase beta subunit (RPOB) gene of Mycobacterium tuberculosis | Electrochemical detection using gold nanoparticles on a solid phase | 1 CFUa | 103 |
| Early secretory antigenic target-6 (ESAT-6) gene of Mycobacterium tuberculosis | Electrochemical detection on a screen-printed carbon electrode (SPCE) | 1 CFUa | 102 |
| Serotype-specific Enteritidis sequence fragment sdfl of Salmonella Enterica | Real-time fluorescent detection | 1 CFUa | 119 |
| Genomic DNA of Plasmodium falciparum 3D7 | Real-time waveguide-based detection | <1 parasite per μL | 120 |
| Leishmania donovani kinetoplast minicircle DNA | Real-time fluorescent detection | 1 cell | 121 |
| β-Conglutin for Lup an 1 anaphylactic allergen | (Competitive) lateral flow strip detection | 0.17 attomol | 39 |
| CeuE gene of Campylobacter jejuni; hipO gene of Campylobacter coli | Real-time fluorescent detection | 1 CFUa ml−1 in pure culture and chicken broth without enrichment | 122 |
| Lentivviruses harboring genome fragment of Zika virus | Clustered regularly interspaced short palindromic repeats (CRISPR)-based end-point fluorescent detection | 2 attomol L−1 | 123 |
| B1 gene of Toxoplasma gondii | Lateral flow strip detection | 0.1 oocyst | 71 |
| Small subunit ribosomal RNA (18S RNA) gene of Plasmodium knowlesi | Real-time fluorescent detection | 1 plasmid | 124 |
| IS6110 gene of Mycobacterium tuberculosis H37Rv genomic DNA | Real-time silicon photonic microring-based detection | 3.2 genomic DNA copies (= single cell of H37Rv) | 113 |
| Analyte(s) | Detection method | Limit of detection | Clinical/field sample(s) | Clinical sensitivity | Clinical specificity | Benchmark method | Limit of detection of benchmark method | Clinical sensitivity of benchmark method compared to RPA | Clinical specificity of benchmark method compared to RPA | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a RT: reverse transcription. | ||||||||||
| Nucleocapsid (N) gene of bovine coronavirus | Real-time fluorescent detection | 10 to 100 RNA copies (19 RNA copies by probit analysis) | 16 fecal and 14 nasal swab specimens collected from cattle showing intestinal and/or respiratory manifestations | 100% | 100% | Real-time RTa-PCR | 1000 RNA copies | The same | The same | 125 |
| Chlamydia trachomatis CDS2 gene | Lateral flow strip detection | 5–12 pathogens per reaction | 70 self-collected first void morning urine samples from young adults (19 males and 51 females) | 83% | 100% | Roche Cobas Amplicor CT assay | — | Higher | The same | 92 |
| cAMP factor (cfb) gene of Group B Streptococci | Real-time fluorescent detection | 98 genome copies | 50 vaginal/anal samples collected from women | 96% | 100% | Real-time PCR | — | Higher | The same | 126 |
| DNA target sequence specific to Cryptosporidium spp. | Lateral flow strip detection | 100 oocysts per mL stool | A total of 10 human stool samples clinically verified to contain Cryptosporidium by a reference laboratory and 11 stool samples from healthy volunteers presumed to be uninfected | 100% | 100% | Real-time PCR | — | Lower | The same | 127 |
| 5′-Untranslated region of Yellow fever virus (YFV) | Real-time fluorescent detection on the tube scanner | 44 genomic copies per reaction in YFV RNA extracts; 21 genomic copies per reaction of YFV-spiked human plasma samples | 34 samples of monospecific pools of wild-caught mosquitoes collected from Kedougou, southern Senegal | 80% | 100% | Real-time RTa-PCR | 8 genomic copies per reaction in YFV RNA extracts | Higher | The same | 117 |
| Real-time fluorescent detection on the microfluidic platform | 27 RNA samples of mosquito pools | 71.4% | 100% | Higher | The same | |||||
| IS6110 gene of Mycobacterium tuberculoss (MTB) | Real-time fluorescent detection | 6.25 fg | 121 specimens including induced and expectorated sputum (n = 119) and respiratory washes (bronchial and tracheal, n = 2) collected from a total of 101 tuberculosis suspect cases (no more than 3 specimens/individual were tested) | 87.5% | 95.4% | Culture | — | Higher | Higher | 128 |
| IS1081 gene of Mycobacterium tuberculoss | 20 fg | 91.4% | 100% | Higher | The same | |||||
| Giardia beta giardin gene | Lateral flow strip detection | 103–103.5 cysts per mL of stool | 104 clinical stool samples | 73% | 96% | Real-time PCR | 102.5 cysts per mL of stool | Higher | Higher | 129 |
| IS6110 gene of Mycobacterium tuberculoss | Real-time photonic detection | 10−6-fold diluted MTB sample | 33 clinical samples including 13 smear and culture positive samples and 20 smear and culture negative samples | 100% | 100% | Conventional microscopy of smear and solid culture | — | The same | The same | 130 |
| A highly conserved 3′-untranslated region that cover DENV 1–4 | Real-time fluorescent detection | DENV serotype 1: 237 RNA copies; DENV serotype 2: 618 RNA copies; DENV serotype 3: 363 RNA copies; DENV serotype 4: 383 RNA copies | Inactivated DENV 1–4 spiked plasma and 31 DENV positive samples in Kedougou region in Senegal | 98% | — | Real-time RTa-PCR | — | Higher | — | 131 |
| RNA of 90 plasma samples extracted and tested between 2012–2013 by RT-PCR in Bangkok (Thailand) | 72% | — | Higher | — | ||||||
| 47 kDa gene sequence from the Karp strain of Orientia tsutsugamushi (47-RPA) and the 17 kDa gene sequence from the Wilmington strain of Rickettsia typhi | Lateral flow strip detection | 47 kDa gene: 53 DNA copies per reaction | 10 positive and 10 negative human samples | 80% | 100% | Real-time PCR | 47 kDa gene: 10 DNA copies per reaction | Higher | Higher | 95 |
| 17 kDa gene: 20 copies per reaction | — | — | — | 17 kDa gene: 6 DNA copies per reaction | — | — | ||||
| Ribosomal 18S DNA of Entamoeba histolytica | Lateral flow strip detection | 2.5 fg from serial dilutions of pure DNA extracted from parasites; 40 parasites from spiked stool sample | 32 samples of DNA extracted from clinical samples | 100% | 100% | Real-time PCR | 2.5 fg from serial dilutions of pure DNA extracted from parasites | The same | The same | 132 |
| A sequence designed based on ITS sequences of the Madurella mycetomatis type strain CBS 109801 | Gel electrophoresis detection | 0.23 ng of DNA | 12 patient biopsy specimens | 100% | 100% | Conventional PCR | — | The same | The same | 133 |
| Ebola virus (EBOV) nucleocapsid sequence | Real-time fluorescent detection | 5 genomic copies per reaction of a molecular RNA standard; 15 genomic copies per reaction in EBOV-spiked human plasma samples | 928 post-mortem swab samples | 100% | 100% | Real-time RTa-PCR | — | The same | The same | 134 |
| Orf virus (ORFV) DNA polymerase gene segments | Real-time fluorescent detection | 100 DNA copies | 22 samples collected from suspected cases of Orf, 8 nasal swabs collected from experimentally infected sheep and 5 samples obtained from healthy goats | 86% | 100% | Real-time PCR | — | Higher | The same | 135 |
| Leader peptidase A (LepA) gene of Streptococcus pneumoniae | Real-time fluorescent detection | 4.1 genome equivalents per reaction | 15 blood samples including 11 confirmed culture positive and 4 confirmed culture negative for Streptococcus pneumoniae | 100% | 100% | Real-time PCR | 5.1 genome equivalents per reaction | The same | The same | 97 |
| Orf virus (ORFV) DNA polymerase gene segments | Lateral flow strip detection | 80 copies per reaction of DNA plasmid | 24 ORFV-spiked tissues samples, 53 samples collected from goats with suspected ORFV infection, 8 nasal swabs samples and 5 tissues samples from healthy goats | 100% | 100% | Real-time PCR | — | The same | The same | 64 |
| Leishmania donovani (LD) kinetoplast minicircle DNA | Real-time fluorescent detection | 100 DNA copies applying the LD DNA linearised plasmid; 1 genomic DNA copy | 96 buffy coats and skin biopsies collected from visceral leishmaniasis, asymptomatic and post-kala-azar dermal leishmaniasis | 100% | 100% | Real-time PCR | — | The same | The same | 121 |
| Highly pathogenic porcine reproductive and respiratory syndrome virus (HP-PRRSV) NSP2 gene | Real-time fluorescent detection | 70 RNA copies per reaction | 68 tissue samples and 10 serum samples collected from suspected pigs of HP-PRRSV, 35 serum samples and 12 tissue samples collected from healthy pigs | 97.6% | 100% | Real-time RTa-PCR | — | Higher | The same | 136 |
| 100% conserved sequence of a major capsid protein gene of all cyprinid herpesvirus 3 strains | Gel electrophoresis detection | 10 copies of genomic DNA | 12 confirmed latently infected fish and 1 confirmed uninfected fish | 100% | 100% | Real-time PCR | — | Lower | The same | 66 |
| cAMP factor (cfb) gene of Group B Streptococci | Real-time fluorescent detection | 6.25–12.5 genome equivalents | 124 clinical samples | 100% | 100% | Real-time PCR | 3.1–6.25 genome equivalents | The same | The same | 137 |
| Non-structure protein 1 (nsP1) of Chikungunya virus (CHIKV) | Real-time fluorescent detection | 80 genome copies of extracted RNA from CHIKV isolate LR strain | 58 suspect Chikungunya fever cases | 100% | 100% | Real-time RTa-PCR | 80 genome copies of extracted RNA from CHIKV isolate LR strain | The same | The same | 87 |
| A sequence designed in NS2A region conserved among all Zika virus lineages | Real-time fluorescent detection | 21 RNA copies | 25 positive and 9 negative urine samples collected during the Zika virus epidemic in Tuparetama, Brazil | 92% | 100% | Real-time RTa-PCR | — | Higher | The same | 138 |
| G-protein-coupled chemokine receptor (GPCR) gene of lumpy skin disease virus (LSDV) | Real-time fluorescent detection | 100 DNA copies (179 DNA copies by probit analysis) | 12 negative skin samples and 22 skin nodules of suspected LSDV-infected cattle collected during the summer of 2012 in Dakahlia Governorate, Egypt | 100% | 100% | Real-time PCR | 37 DNA copies | The same | The same | 139 |
| IS900 gene of Mycobacterium avium subsp. paratuberculosis (MAP) | Real-time fluorescent detection | 16 plasmid copies per μL; 500 fg genomic DNA/reaction | Archived DNA of MAP positive blood (n = 14), sperm (n = 18), faecal (n = 12) and tissue (n = 4) samples and 20 MAP-negative faecal samples | 89.5% | — | Real-time PCR | 1 plasmid copies per μL; 50 fg genomic DNA/reaction | Higher | — | 140 |
| T1E4 gene of prostate cancer | Real-time fluorescent detection | 1000 RNA copies | 9 urine samples obtained from prostate cancer and 2 urine samples from healthy individuals | 90% | 100% | Real-time RTa-PCR | — | The same | The same | 141 |
| NS1 gene of porcine parvovirus (PPV) | Real-time fluorescent detection | 300 DNA copies | 101 clinical tissue samples (serum, liver, kidney, lymph node, spleen and duodenum) collected from pig farms with suspected cases of PPV in Gansu province, China, and 27 clinical samples (serum, kidney and duodenum) collected from healthy pigs | 94.4% | 100% | Real-time PCR | — | Higher | The same | 54 |
| Nucleocapsid gene of type 2 porcine reproductive and respiratory syndrome virua (PRRSV) | Real-time fluorescent detection | 100 RNA copies (690 RNA copies by probit analysis) | 60 clinical samples (lymph node, lung, spleen and liver) collected from diseased pigs suspected of having PRRS from 5 pigs farms in Hebei province, China from 2015–2016 | 100% | 100% | Real-time RTa-PCR | 100 RNA copies | The same | Lower | 142 |
| Cytochrome b gene of Theileria annulata | Lateral flow strip detection | 2 pg genomic DNA | 17 anticoagulated blood samples collected from tropical theileriosis endemic areas in Gansu province, China | 100% | 100% | Real-time PCR | — | The same | The same | 67 |
| pirA-like gene of Vibrio owensii | Real-time fluorescent detection | 2 plasmid copies (2.84 plasmid copies by probit analysis) | 138 clinical shrimp obtained from immersion bioassay, including 70 shrimp acute hepatopancreatic necrosis disease (AHPND) infected shrimp and 68 non-AHPND infected shrimp | 100% | 100% | Real-time PCR | — | Lower | Lower | 143 |
| rRNA gene of Fasciola hepatica | Gel electrophoresis detection | 1.6 pg μL−1 DNA copies | 102 human stool samples selected from banked specimens | 87.8% | 100% | Real-time PCR | 1.6 pg μL−1 DNA copies | Lower | The same | 144 |
| Lateral flow strip detection | 1.0 pg μL−1 DNA copies | 95.2% | 90.4% | Lower | Higher | |||||
| N gene of pest des petits ruminants virus (PPRV) | Real-time fluorescent detection | 100 plasmid copies | 32 clinical samples collected from suspected cases of PPRV in Gansu province, China and 5 samples obtained from healthy sheep | 90% | 100% | Real-time RTa-PCR | 150 plasmid copies | Higher | The same | 145 |
| Lateral flow strip detection | 10 plasmid copies | 90% | 100% | Higher | The same | |||||
| ITS2 gene of Phytophthora infestans | Real-time fluorescent detection | 50 fg μL−1 of genomic DNA | 24 potato leaf samples collected from fields with and without visible symptoms of late blight infections in New Brunswick and Quebec provinces, Canada, respectively | 60% | 100% | LAMP | 50 fg μL−1 of genomic DNA | Higher | Lower | 146 |
| ORF2 gene of porcine circovirus type 2 (PCV2) | Real-time fluorescent detection | 100 plasmid copies | 65 clinical samples (spleen, inguinal lymph node, tonsil, lung and serum) collected from suspected PCV2 infection pigs from 8 pig farms in Shandong province, China; 37 clinical samples (inguinal lymph node, tonsil, lung and serum) collected from Gansu Province, China, and 10 PCV1 positive samples conserved in the laboratory | 100% | 100% | Real-time PCR | 80 plasmid copies | The same | The same | 69 |
| Lateral flow strip detection | 100 plasmid copies | 100% | 100% | The same | The same | |||||
| gD gene of pseudorabies virus | Real-time fluorescent detection | 100 DNA copies | 76 clinical samples (tonsil, heart, spleen, lymph nodes, lung and serum) collected from pig farms in Shandong province, China, and 26 clinical samples (lymph nodes, tonsil and serum) collected from healthy pigs | 93.3% | 100% | Real-time PCR | — | Higher | The same | 70 |
| Lateral flow strip detection | 160 DNA copies | 93.3% | 100% | Higher | The same | |||||
| B1 gene of Toxoplasma gondii | Lateral flow strip detection | 0.1 oocysts per reaction | 35 soil samples and 15 water samples collected from parks, residential areas, schools and gutterways in Lanzhou city, Gansu rovince, China, during August 2016 | 100% | 100% | Nested PCR | 1 oocyst/reaction | The same | The same | 71 |
| RNA transcript of TMPRSS2:ERG (a fusion gene for prostate cancer) | RPA fluocculation assay | 105 RNA copies | Clinical urine specimens from 10 metastatic castration-resistant promising prostate cancer patients and 5 healthy control patients | 70% | 100% | Conventional RTa-PCR | — | The same | The same | 101 |
| VP2 gene of porcine parvovirus | Real-time fluorescent detection | 100 DNA copies (103 DNA copies by probit analysis) | 115 clinical samples (lymph node, lung, spleen, kidney and duodenum collected from pigs with reproductive disorders, diarrhea or respiratory disease in Hebei province, China from 2014 to 2016 | 100% | 100% | Real-time PCR | 100 DNA copies | The same | The same | 147 |
| G-protein-coupled chemokine receptor (GPCR) gene of Capripoxvirus | Real-time fluorescent detection | 300 plasmid copies | 107 clinical samples (liver, lung, kidney, spleen, skin and blood) collected from 14 suspected sheep and 6 suspected goats in Gansu province which were characterised by pyrexia, excessive salivation and generalised pock lesions in the skin during the period of October 2014 to August 2015 | 97% | 100% | Real-time PCR | — | Higher | The same | 148 |
| Lateral flow strip detection | 300 plasmid copies | 97% | 100% | Higher | The same | |||||
| Nucleocapsid protein gene of canine distemper virus | Real-time fluorescent detection | 9.4 RNA copies (31.8 RNA copies by probit analysis) | 32 nasal/oropharyngeal swabs collected from 20 dogs of both sexes (various breeds and ages) from the animal hospital of Agricultural University of Hebei and 12 raccoon dogs from the farms in Hebei Province, China from 2014 to 2016 | 100% | 100% | Real-time RTa-PCR | 94 RNA copies | The same | The same | 149 |
| imp gene of Candidatus Phytoplasma oryzae | Real-time fluorescent detection | 1–10 plasmid copies | 66 Napier grass samples from various geographical locations in western Kenya | 100% | 57.1% | Real-time PCR | — | Lower | The same | 79 |
| Lateral flow strip detection | 10–100 plasmid copies | — | — | |||||||
| imp gene of Candidatus Phytoplasma mali | Real-time fluorescent detection | 10 copies of cloned plasmid | 38 roots of field samples from apple (Malus domestica) trees collected in autumn 2014, in spring 2015 and in June 2016 in private orchards or in the experimental field of the Institute for Fruit Growing in Samochvalovichi, Belarus | 100% | 100% | Real-time PCR | — | The same | The same | 72 |
| Lateral flow strip detection | 10 copies of cloned plasmid | 100% | 100% | The same | The same | |||||
| N gene of rabies | Real-time fluorescent detection | 1000 RNA copies per μL of strains SAD B19, Bobcat USA and Kelev | A panel of RNA from 33 field samples | 97% | — | Real-time PCR | 1 RNA copies per μL of strains SAD B19, Bobcat USA and Kelev | Higher | — | 150 |
| KRAS oncogenic mutation gene G12D on Exon 12 | Real-time silicon photonic microring-based detection | 1% to 100% of the mutant cells | 70 frozen tissues samples from colorectal cancer patients in Bio-Resource Center of Asian Medical Center, including 24 samples with the G12D mutation (34.3%), 26 samples with G13D mutation (37.1%) and 20 samples with no mutation (28.6%) | 100% | 100% | Conventional PCR | 30% to 100% of the mutant cells | Lower | The same | 151 |
| KRAS oncogenic mutation genes G13D on Exon 13 | 100% | 100% | Lower | The same | ||||||
| A consensus region that covers all 7 S-segment clades of Crimean-Congo Hemorrhagic fever virus (CCHFV) | Real-time fluorescent detection | 500 RNA copies (251 RNA copies by probit analysis) | 21 extracted patient sera samples obtained in relation to outbreaks of CCHFV in 2013–2015 in Tajikistan | 88% | 100% | Real-time PCR | — | Higher | The same | 152 |
| Canine parvovirus 2 (CPV-2) nucleocapsid protein gene | Real-time fluorescent detection | 10 copies of recombinant plasmid | 91 fecal swab samples collected from the dogs from 2012 to 2016 | 100% | 100% | Real-time PCR | 10 copies of recombinant plasmid | The same | The same | 153 |
| G gene of bovine ephemeral fever virus (BEFV) | Lateral flow strip detection | 8 plasmid copies per reaction (corresponding to 24 RNA copies) | 104 clinical blood specimens and 24 tissue samples including 16 lung tissue specimens, 8 lymph gland specimens collected from suspected dairy cattle cases of BEFV infections in eastern China | 97.89% | 90.91% | Real-time PCR | — | Higher | Higher | 74 |
| IS900 gene of Mycobacterium avium subsp. paratuberculosis | Lateral flow strip detection | 8 plasmid copies per reaction | 320 individual fecal samples collected between September 2016 and September 2017 from 10 different dairy farms located in 10 distinct geographic regions of Shandong province, China | 100% | 97.63% | Real-time PCR | 8 plasmid copies per reaction | The same | Higher | 76 |
| Fno FSC771 hypothetical protein gene of Francisella noatunensis subsp. Orientalis | Real-time fluorescent detection | 10 plasmid copies (15 plasmid copies by probit analysis) | Samples of spleen (n = 78), head kidney (n = 78) and water (n = 5) | 100% | 84,89% | Real-time PCR | 10 plasmid copies (11 plasmid copies by probit analysis) | The same | Higher | 154 |
| VP1 gene of Enterovirus 71 subgenotype C4 (EV71-C4) | Real-time fluorescent detection | 3.767![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log10 genomic copies (LGC) log10 genomic copies (LGC) |
Stool samples (n = 44) collected in 2017 by Shenzhen Center for Disease Control and Prevention | 100% | 100% | Real-time PCR | 2.026log10 genomic copies (LGC) |
The same | The same | 155 |
| Stool samples (n = 134) collected from patients with suspected hand–foot–mouth disease at the pediatrics department of Zhujiang Hospital (Southern Medical University, Guangzhou, China) in 2009 | 89.5% | 100% | Lower | The same | ||||||
| 56 Kda gene of a Karp-like strain of Orientia tsutsugamushi | Lateral flow strip detection | 10 copies (recombinant plasmid); 12 copies of genomic DNA | 62 animal (including Apodemus agrarius, Rattus norvegicus, Microtus fortis and Neomys fodiens) organ samples including 5 infected animals trapped in the wild, 2 infected in the laboratory and 55 uninfected animals trapped in the wild | 100% | 100% | Real-time PCR | 12 copies of genomic DNA | The same | The same | 156 |
| 23S rRNA gene of Coxiella burnetii | Lateral flow strip detection | 10 copies (recombinant plasmid); 7 copies of genomic DNA | DNA of spleens from 9 5-week old C57BL/6 female Coxiella burnetii-infected mice and 9 control PBS-infected mice | 100% | 100% | Real-time PCR | 7 copies of genomic DNA | The same | The same | 157 |
2.1 Reaction components
The fundamental reaction mechanism of RPA relies on a synthetically engineered adaptation of a natural cellular process called homologous recombination, a key process in DNA metabolism. The standard RPA reaction reagents comprise three key proteins (recombinase, recombinase loading factor and single-stranded binding protein), which subsequently co-ordinate with ancillary components such as deoxyribonucleic acid (DNA) polymerase, crowding agent, energy/fuel components (e.g. adenosine triphosphate, ATP) and salt molecules to perform the RPA reaction mechanism (Fig. 2).6 The detailed reaction components, and their typical concentration and function are provided in Table 1: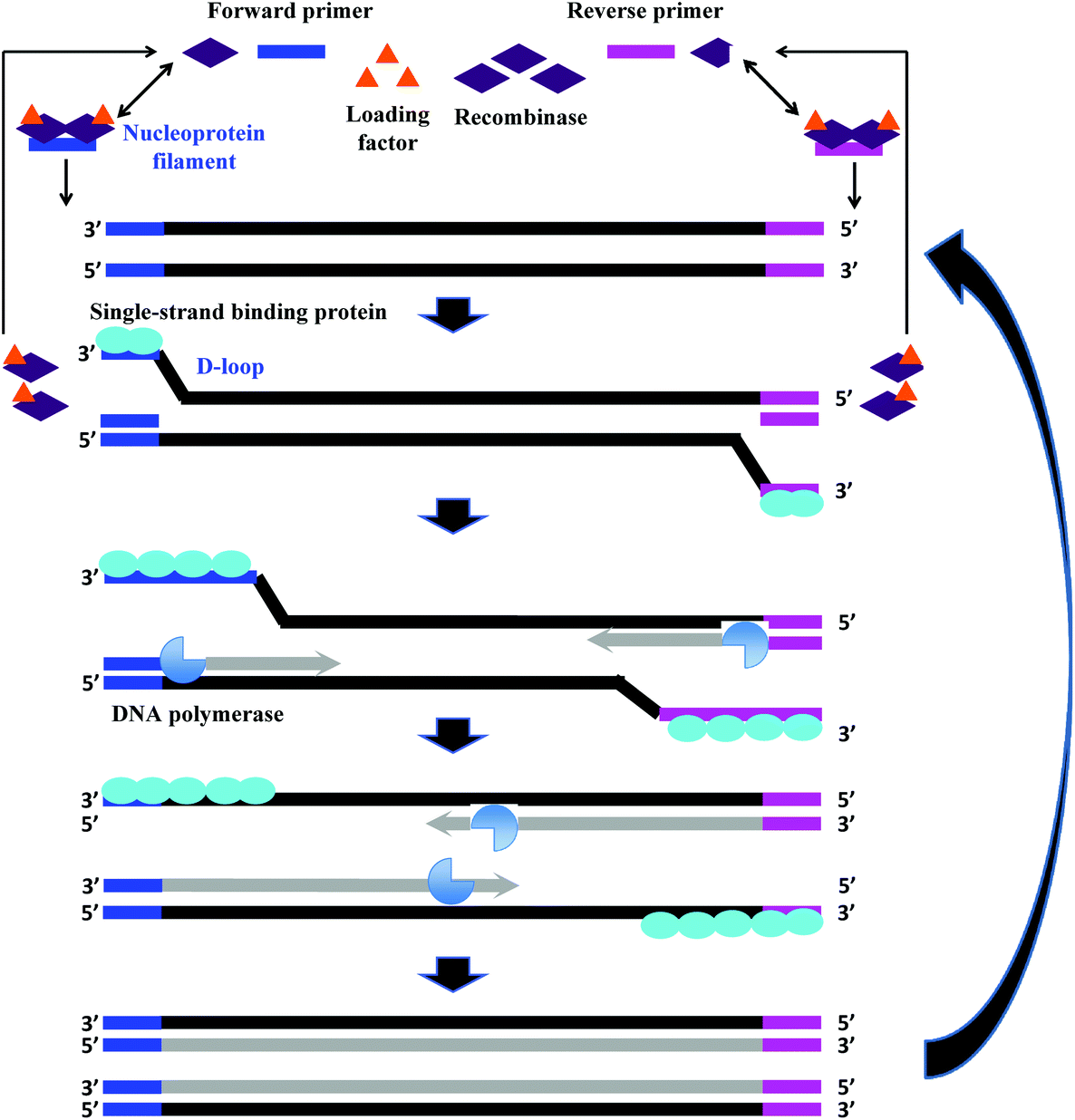 | ||
| Fig. 2 RPA reaction mechanism. The reaction starts from the binding of the recombinase (T4 UvsX) to the primers with the help of the loading factor (T4 UvsY). This forms a nucleoprotein filament that searches for the homologous sequence in the double-stranded DNA. Once the homology is located, the complex invades the duplex DNA, forming a D-loop structure to initiate a strand exchange reaction while the unwound strand is stabilised by the single-stranded binding proteins (T4 gp32). The recombinase (Bsu or Sau) disassembles from the nucleoprotein filament once the strand exchange is performed, and will be available for the next pair of primers. Next, the DNA polymerase extends from the 3′ end of primers. As the polymerisation continues, the two parental strands begin to separate and eventually form two duplexes, and then the whole process repeats.31 | ||
2.2 Mechanism
RPA starts with the binding of the T4 UvsX protein (recombinase), assisted by the T4 UvsY (loading factor), to the primers to form a nucleoprotein filament. The resulting complex searches for homologous sequences in duplex DNA (Fig. 2).6 Once the homology is located, the complex invades the double-stranded DNA, forming a D-loop structure. One side of the D-loop is double-stranded where the primer hybridises with the template strand, initiating a strand exchange reaction, whereas the other side of the D-loop remains single-stranded – the unwound complementary strand that is stabilised by the SSB proteins (T4 gp32).29,30 Subsequently, the recombinase disassembles from the nucleoprotein filament and becomes immediately available to initiate another strand displacement reaction with a new primer. Primer incorporation allows the DNA polymerase (Bsu or Sau) to initiate the synthesis from the free 3′-OH at the end of the primer. As the polymerisation continues, the two parental strands continue to separate. Incorporation of both forward and reverse primers enables strand synthesis to occur in both directions simultaneously, and ultimately results in the exponential accumulation of amplified duplex DNA, consisting of the sequence between the forward and reverse primers.During RPA, the formation of the recombinase-primer complex is the rate limiting for the D-loop formation.29 It was reported that the D-loop formation was most efficient at the stoichiometries at which the T4 UvsX protein fully coated the primers but did not bind substantially to the double-stranded DNA.30 The SSB proteins and the T4 UvsY (together with the ATP) have been shown to be essential for cooperating in strand exchange reaction along with the T4 UvsX protein.13,30,32 However, the presence of both of these proteins requires a higher concentration of T4 UvsX protein than what is required in the presence of only one of these proteins.29,33,34 The SSB proteins can stimulate the strand exchange reaction if T4 UvsX degrades.33 Importantly, the T4 UvsY protein neutralises the competition between the SSB proteins and the T4 UvsX for binding sites on the primers preventing the SSB from binding the primer from initiating the recombination event.35 When the primer concentration is low, the SSB proteins inhibits the strand exchange activity of the T4 UvsX protein.35 However, once the T4 UvsY protein is supplied, the T4 UvsY protein is able to invade the SSB proteins-covered primers to promote the binding of T4 UvsX protein to primers (from a site that is adjacent to the bound T4 UvsY protein), thereby displacing the SSB proteins from the primers.35
2.3 Template, primers, probe and their designs
RPA was initially demonstrated to be a nucleic acid amplification method for DNA,6 later it was shown that RNA also could be the template by addition of reverse transcriptase (e.g. Murine Leukemia virus (MuLV) reverse transcriptase) in the same reaction tube.36 Regardless of nucleic acid template type, the recommended RPA amplicon length should be below 500 nucleotides for efficient amplification. Most published RPA papers have applied amplicon lengths between 100 and 250 nucleotides, which usually incur fast and efficient amplification. However, shorter amplicons (79 nucleotides;37 94 nucleotides38–40 and longer amplicon up to 1500 nucleotides6 and 1941 nucleotides41 have also been reported.Unlike PCR, the length of RPA primers is relatively long (a recommended minimum of 30 nucleotides, but typically between 32 and 35 nucleotides). Shorter PCR primers (typically between 18 and 25 nucleotides) can also be used in the RPA reaction but may decrease the reaction speed and sensitivity.42 Application of short PCR primers in RPA has been demonstrated by Mayboroda et al.,43 Martorell et al.,37 Wang et al.44 and Fuller et al.45 The latter two authors have shown that the PCR primers used in RPA resulted higher analytical sensitivity of detection compared to their usage in PCR: RPA detected 100 DNA copies of genetically modified GTS 40-3-2 soybean and 3.5 pg of genomic DNA of Agrobacterium spp. respectively, while the benchmark method PCR detected 1000 DNA copies and 350 pg of genomic DNA respectively.44,45
The company that sells the commercialised RPA reagents, TwistDx™ Ltd (see section 2.4 and Tables 2 and 3 for more details) provides additional probes that can be incorporated during the RPA reaction. The TwistAmp™ exo probe (typically between 46 and 52 nucleotides) and the TwistAmp® fpg probe (typically between 32 and 35 nucleotides) are used for fluorogenic real-time detection (Fig. 3A and B). These two probes are usually labelled with a fluorophore, a quencher (e.g. Black Hole Quencher) that is in close proximity to the fluorophore, to temporarily deter the fluorescent signal, and a blocker (e.g. C3-spacer, a phosphate, a biotin-TEG or an amine) at the 3′-end serving to prevent polymerase extension from the 3′-end (Fig. 3A and B). The real-time detection is based on cleavage of fluorogenic probes at an abasic site (also known as an apurinic/apyrimidinic site that is a location in DNA (less often in RNA), which has neither a purine nor a pyrimidine base) between the fluorophor and the quencher. The abasic site can either be tetrahydrofuran (THF) or a dSpacer (a derivative of the THF) or a dR group (the deoxyribose of the abasic site via a C–O–C linker). The E. coli exonuclease III cleaves the TwistAmp™ exo probe at a THF or a dSpacer site, while the glycosylase/lyase E. coli fpg cleaves the TwistAmp™ fpg probe at the dR position (Fig. 3A and B). After the enzymatic cleavage, the TwistAmp® exo probe can serve as a forward primer. However, the TwistAmp™ fpg probe cannot serve as a primer due to different catalytic mode (beta-elimination) of the glycosylase/lyase E. coli fpg protein, which does not generate an extendable 3′-OH group but a 3′-phosphate group.46
 | ||
| Fig. 3 RPA probes. A: TwistAmp™ exo probe. This probe is cleaved by the E. coli exonuclease III at the abasic site (e.g. tetrahydrofuran, THF) to depart the fluorophore from the quencher and generate an extensible 3′-OH group for polymerisation. B: TwistAmp™ fpg probe. This probe is cleaved by the glycosylase/lyase E. coli fpg at the dR position (the deoxyribose of the abasic site via a C–O–C linker) to depart the fluorophore from the quencher and generate a 3′-phosphate group which is non-extensible for polymerisation. C: TwistAmp™ LF probe. This probe is cleaved by the Nfo endonucleases IV at the abasic site (e.g. tetrahydrofuran, THF) to generate an extensible 3′-OH group for polymerisation. The DNA polymerases extend and displace from 3′-ends of the primers and cleaved probe to produce the minor amplicons (from the forward and reverse primers) and a displaced strand. The displaced strand combines with the labelled reverse primer, and leads to the production of a dual-labelled amplicon (the major amplicon) for the down-stream sandwich assay detection.31 | ||
A third probe, the TwistAmp™ LF (typically between 46 and 52 nucleotides), is used for lateral flow strip detection (Fig. 3C). This probe is labelled at the 5′-end (e.g. with fluorescein), has a blocker at the 3′-end, and an internal abasic site (THF or dSpacer). The Nfo endonucleases IV cleaves at this abasic site of the TwistAmp™ LF probe, and generates an extendable 3′-OH group for polymerisation. However, unlike the E. coli exonuclease III which degrades most of the amplicons during RPA reaction, the Nfo endonuclease IV generates a slower signal and incomplete cleavage to avoid amplicon degradation (also see section 2.8).46 Therefore, the TwistAmp™ LF probe can also be used for cases when gel electrophoresis (GE) is chosen as a detection method.
To select suitable RPA templates and to design primers and probes, users can refer to the criteria suggested in the TwistAmp™ reaction kit manual.20 In brief, (1) GC content of the DNA template should be between 40% and 60%, and should avoid long homo-polymer tracks, few direct/inverted repeats and palindromes; (2) GC content of the primers should be between 30% and 70%, and should avoid long tracks of guanines at the 5′ end but recommend cytidines; and (3) guanines and cytidines are recommended at 3′-end of the primer for improved performance. From the RPA literature, it is further recommended that users evaluate the melting temperature, hybridisation stability, secondary structures and dimer formations among these oligonucleotides.47 Specific softwares such as BioEdit version 7.0.5.3,48 Primer3,49 UNAFold,50 mFOLD,51 Oligoanalyzer 3.1 (IDT, Leuven, Belgium), PrimerQuest software (Integrated DNA Technologies, Coralville, IA) and Visual OMP (DNA software, MI, USA) have been used in the literature for analysing RPA oligonucleotide properties. An effective way to avoid primer dimer formation is to employ self-avoiding molecular recognition systems (SAMRS), by including SAMRS nucleotides 2-aminopurine-2′deoxyriboside (A*), 2′-deoxy-2-thiothymidine (T*), 2′-deoxyinosine (G*) and N4-thyl-2′-deoxycytidine (C*) in the primers.52 The inclusion of these SAMRS nucleotides strategically replaces the hydrogen-bonding units from natural A pairs with T (and G pairs with C) to SAMRS A* pairs with natural T, SAMRS T* pairs with natural A, SAMRS G* pairs with natural C and SAMRS C* pairs with natural G. However, the SAMRS A* and SAMRS G* nucleotides do not interact with the SAMRS T* and SAMRS C* nucleotides respectively no matter what their concentration, and in this way, the undesired products due to primer dimer can be avoided during nucleic acid amplification.52,53 Such SAMRS system has been demonstrated in RPA reaction by Sharma and co-workers, and was shown to successfully eliminate RPA artifacts.53 In addition, caution is needed to avoid overlap between the primer and probe which has shown to impede the desired amplification efficiency.54,55 Collectively speaking, it is sufficient to follow the described guidelines as a starting point for in silico optimal design of the RPA oligonucleotide candidates, however, the resulting candidates should be screened through RPA reactions to select the preferred final oligonucleotide set applicable for a specific RPA assay (e.g. TwistDx™ Ltd recommends designing five forward and reverse primers, and 3 probes).
2.4 Commercial kits and instrumentation by TwistDx™
All the RPA reagents are available for commercial purchase through TwistDx™, a subsidiary of Abbott.56 The company provides various kits for RPA reactions that can be customised towards specific applications by the end user. The company also sells RPA kits for the detection of specific food-borne pathogens (e.g. Listeria monocytogenes, Campylobacter and Salmonella enterica) (Table 2). The company not only provides RPA reagents in liquid format, but also in lyophilised pellet format which allows in-field application. These lyophilised pellets have shelf-lives up to 12 weeks at 25 °C or up to 3 weeks at 45 °C.57 In addition, TwistDx™ offers a custom freeze-drying service to create RPA reaction pellets containing primers, probes, and concentrations of protein components or other components (e.g. internal control DNA or RNA species), which can be encased in various holding vessels with different volumes.58Apart from various RPA reaction kits, TwistDx™ also develops tailor-made devices and accessories for RPA reactions; these devices and accessories enable incubation, dispensing, mixing, detection, power supply and portability (Table 3 and Fig. 4). The Twirla™ device is a hand-held sized battery-powered incubator, which allows up to six parallel RPA reactions and subsequent end-point detection (e.g. gel electrophoresis and lateral flow strip detection; Fig. 4A) (note that the Twirla™ is an upgraded version of an earlier Twista® device that does not support constant mixing during incubation, which has now been discontinued). Alternatively, the T8-ISO allows up to eight parallel incubations and two-channel real-time fluorescent detection per tube (Fig. 4B). The T16-ISO is an advanced version of the T8-ISO, which supports up to sixteen parallel reactions with three-channel fluorescent detection per tube (Fig. 4C). The T8-ISO and the T16-ISO can be powered by mains-power supply, micro USB, or PowerGorilla external battery (Fig. 4D). Moreover, all three incubators mentioned so far support magnetic mixing (programmed or constant) when fitted with Micro Ball(s) (0.2 mL; dispensed with Micro Ball Dispenser) in the reaction tube (Fig. 4E). For lateral flow strip detection, TwistDx™ provides four different lateral flow devices: Milenia HybriDetect 1, Milenia HybriDetect 2 (Fig. 4F), PCRD Nucleic Acid Detection (Fig. 4G) and U-Star Disposable Nucleic Acid Lateral Flow Detection Units (Fig. 4H). The Milenia HybriDetect 1 and the U-Star Disposable Nucleic Acid Lateral Flow Detection Units allow single-plex detection while the other two devices allow duplex detection. All the lateral flow devices except for the PCRD Nucleic Acid Detection device are based on sandwich assay using gold nanoparticles as tracer; the PCRD Nucleic Acid Detection device employs carbon nanoparticles, which can be more sensitive than the gold nanoparticles.59,60 The Milenia HybriDetect are provided as strips, whereas the PCRD Nucleic Acid Detection strips are encased in a semi-sealed cartridge, and the U-Star Disposable Nucleic Acid Lateral Flow Detection strips are embedded in a sealed cartridge designed to enable the RPA reaction to flow to the strips in a completely closed environment that prevents cross-contamination of amplified products.
 | ||
| Fig. 4 RPA devices and accessories. (A): Twirla™ Portable Mixing Incubator. (B): T8-ISO Instrument. (C): T16-ISO Instrument. (D): PowerGorilla external battery. (E): Micro Balls (0.2 mL, left) and Micro Ball Dispenser (right). (F): Milenia HybriDetect lateral flow strips. (G): PCRD Nucleic Acid Detection device. (H): U-Star Disposable Nucleic Acid Lateral Flow Detection Units device. Reprinted and reproduced with permission from TwistDx™ Limited. Copyright 2009–2018 TwistDx™ Limited. | ||
2.5 Influence of temperature and agitation
For RPA reactions to achieve optimal efficiency and analytical sensitivity, the choice of target sequence and the designs of corresponding primers and probe are the intrinsic determinants, however, the reaction temperature and agitation during RPA reaction are two of the most important contributing extrinsic factors.The recommended RPA reaction temperature is between 37 °C and 42 °C,42 and Crannell et al.61 and Wang et al.44 have also demonstrated that RPA reaction can be performed using body temperature, which can be used advantageously for in-field application. However, several research groups have studied RPA reaction temperatures that lie outside of the recommended range.38,44,45,60,62–78 The largest temperature range was tested between 15 °C and 50 °C;62,64,69,70,76 and results indicated the marginal reaction temperature to produce a positive result should be greater than 30 °C.62–64,66,67,69,71,74,76,77 However, Sun et al.65 and Poulton and Webster60 showed that temperature as low as 25 °C could still generate a positive signal after RPA amplification and subsequent lateral flow strip detection. Moreover, Lillis et al.63 showed that the ambient temperature also had an effect on RPA reaction: the RPA reaction was unstable if the ambient temperature was below 10 °C, however, extension of the reaction time could improve positive results attainability. Such reaction temperature range studies indicate that RPA reaction does not require precise temperature control.
While reaction temperature provides a suitable working environment for the RPA enzymes, agitation increases the interactions among the RPA components in a homogenous reaction solution. TwistDx™ recommends the user performing two times mixing steps for the RPA reaction, one is at the beginning of the process and the other is after 4 minutes of the reaction. The former is to mix all the RPA reagents to initiate the reaction, the latter is to prevent from local depletion of the reaction reagents, thereby increasing the reaction rate. Wambua et al.79 reported that threshold fluorescence values were reached in 5–8 minutes when agitation was performed after 4 minutes, whereas the time to reach detectable levels ranged between 8 and 14 minutes without this agitation. In addition, constant shaking throughout the RPA reaction has been shown to further accelerate the RPA reaction rate, achieve more stable positive results and improve sensitivity, especially when the template concentration is close to the limit of detection.57,62,80 Kersting et al.62 reported that constant shaking resulted in faster and more stable signals on the lateral flow strips than with the recommended two-shaking event. Kalsi et al.80 also reported that continuous mixing of microdroplets from a RPA exo assay led to faster time to result, increased fluorescence and improved sensitivity. In addition, Moody et al.81 built up a mathematical model and showed that mechanical stirring is better than manually shaking to eliminate inter-operator variations and obtain consistent quantitative experimental result; yet the ideal mixing frequency is assay dependent, and should be determined prior to the reaction.57,81 Nevertheless, if a shaking condition is not available, Lillis et al.57 demonstrated that a decrease of the reaction volume (e.g. from 50 μL to 5 μL) could compensate for the shaking effect, as smaller volume increased interactions between the reagents and oligonucleotides required for the amplification.
2.6 Tolerability to mismatches, inhibitors and background DNA
Apart from temperature and agitation, tolerability to mismatches, inhibitors and background DNA are other vital factors for efficient and sensitive RPA reaction. RPA has the ability to tolerate mismatches, and the highest mismatch tolerability reported so far is nine nucleotide base pairs across the primer and probe binding sites.82–89 Studies also showed that the mismatches at the 5′-end or centre of primers only mildly affect the RPA reaction, but mismatches located at the 3′-end of primers significantly affect the reaction.84,86 This is consistent with the RPA reaction mechanism (see section 2.2), since the polymerase extends the primers and probe (once cleaved) from the 3′-terminus. A useful application for such mismatch sensitivity at the 3′-end is to distinguish single nucleotide polymorphism (SNP). Yamanaka et al.90 applied this property to differentiate polymorphisms for the tobacco use disorder genes; the DNA polymerase extension was efficient when the 3′-terminal base of a primer matched its target, whereas the DNA polymerase extension was inefficient or non-existent when the 3′-terminal base was mismatched.90 However, of the general RPA mismatch tolerability (outside of the 3′-end of the primer) can be advantageous, as it enables some flexibility in primer design for highly polymorphic targets, where long conserved target regions are hard to locate. Conversely, the drawback of such mismatch tolerability is a tendency towards non-specific detection of closely-related species. Indeed, non-specific detections have been observed by Patel et al.,87 Moore et al.88 and Yang et al.69 when detecting chikungunya virus, epidemic human noroviruses and porcine circovirus Type 2, respectively.When testing clinical or field samples, numerous substances (e.g. inhibitors) are either present or could be introduced during sample preparation and processing steps, which can potentially interfere with nucleic acid amplification. RPA has been demonstrated to tolerate certain (PCR) inhibitors, including: (1) haemoglobin (20 g L−1), heparin (0.5 U) and urine (1.25%) showed no effect on RPA reaction;62,91 and (2) haemoglobin (50 g L−1), ethanol (4% v/v) and urine (up to 5%), which only slightly affected reactions.62,91,92 However, RPA reaction was totally inhibited in the presence of SDS (0.05% v/v) and urine (10%).62,91 It was also observed that RPA reactions were more susceptible to inhibitors when the DNA template concentration was close to the limit of detection.62,91 However, it is also important to carefully consider the choice of extraction buffer or incubation medium for the sample preparation and processing steps, as these working solutions may also contain potential inhibitors. For example, Valasevich and Schneider72 found that Cetyltrimethyl ammonium bromide (CTAB) DNA extraction buffer strongly inhibited RPA reaction. Similarly, Liu et al.93 found that selenite cystine broth (bacterial enrichment medium) significantly affected RPA reactions, resulting in a large number of primer dimers that led to false positive results on the lateral flow strip detection.
In addition to tolerating inhibitors, RPA is capable of amplifying target nucleic acids in the presence of background DNA.94–97 However, similar to the tolerability for inhibitors, the tolerability for background DNA is also concentration dependent. Clancy et al.97 observed that the RPA reaction was significantly inhibited when 400 ng of background human DNA was present, but was much less inhibited when 200 ng of background human DNA was present. Rohrman and Richards-Kortum94 showed that RPA was completely inhibited by 0.5 μg of sheared salmon sperm DNA when 50 copies of human immunodeficiency virus-1 (HIV-1) target DNA were present, while only inhibited by 2 or 5 μg of sheared salmon sperm DNA when 103 or 106 copies of the target DNA were present respectively. In addition, Rohrman and Richards-Kortum94 also pointed out that the primer, probe and target sequences used in the assay could influence the maximum background DNA concentration tolerability. Both HIV-1 and Plasmodium falciparum RPA assays were completely inhibited by 2 μg of sheared salmon sperm DNA respectively when 103 copies of HIV-1 and Plasmodium falciparum target DNAs were present.94 However, when the same amount of target DNA were present (103 copies), the Entamoeba histolytica and Giardia duodenalis assays were completely inhibited only by 1 and 0.5 μg of sheared salmon sperm DNA, respectively.94
2.7 Nucleic acid labelling during RPA
One vital process for diverse down-stream RPA applications (e.g. lateral flow strip detection and enzyme-linked immunosorbent assay) is to incorporate labels into nucleic acid template during RPA reaction, so that the incorporated labels allow capture, detection and/or assist the signal generation of RPA assays. Such nucleic acid labelling can be achieved terminally using 5′-labelled primers or internally via labelled nucleotides.39,40,98–104 The labels used for nucleic acid labelling can be fluorescent entity (e.g. fluorescein), ligand (e.g. biotin) or even a short segment of nucleotides (overhang). Most terminal nucleic acid labelling using RPA employs both 5′-labelled forward and backward primers, such that the amplicons possess dual-labels that can be captured and detected by corresponding recognition molecules in down-stream assays. However, RPA only tolerates to certain labels via 5′-labelling process. Crannell et al.105 reported a failure of RPA incorporation of five different 5′-labels (Cy5, Cy3, bromodeoxyuridine, tetrachlorofluorescein and hexachlorofluorescein) compared to successful incorporation with three 5′-labels, Alexa Fluor488, fluorescein and digoxigenin.For internal nucleic acid labelling during RPA, the reaction mixture can be supplemented with labelled nucleotides, mostly using digoxigenin-dUTPs, which randomly substitute dTTPs during polymerase extension to create labelled amplicons.101–104 In comparison to the terminal labelling, the internal labelling allows more labels to be incorporated into a single nucleic acid template, thus having more binding opportunities in down-stream assays. However, terminal labelling can be a better choice when the down-stream application is for a sandwich assay, as the two labels incorporated via terminal labelling are further apart (separated by the length of amplicon), which could prevent steric hindrance of binding if the labels were too close together.
2.8 Amplicon clean-up and post-amplification treatment
The above-mentioned issues have considered the conditions both before and during RPA reactions. In addition, post-reaction procedures are critical for successful RPA signal detections, and should be determined according to the intended use of RPA amplicons. The generation of RPA amplicons are RPA reaction kit dependent. Usage of the TwistAmp® Basic kit (also the Basic RT kits) produces a single amplicon from the forward and reverse primers. Conversely, usage of the TwistAmp® exo kit (also the exo RT kits) and the TwistAmp® fpg kits do not produce a single amplicon, the former is due to the exonuclease present in the reaction mixture digesting most of the amplicons during RPA reaction,42 and the latter is due to the glycosylase/lyase E. coli fpg cleavage generating a non-extensible 3′-phosphate group (also see section 2.3).46 For the TwistAmp® nfo kit, however, two types of amplicons are generated, due to the DNA polymerase displacement activity to the probe-primed template: a dual-labelled amplicon emerges as a short product from the probe and one of the primers, whereas a singlely-labelled amplicon emerges as a longer product from the forward and reverse primers (Fig. 3C) (note that only the dual-labelled product will generate a positive signal in the test zone of a lateral flow strip detection based on a sandwich assay).105–108Nevertheless, the RPA amplicons are initially associated with proteins and crowding agents, and the resulting DNA–protein-crowding agent complexes prevent direct use of DNA molecules for gel electrophoresis detection.109,110 This is because these complexes affect the proper migration of the amplicons in gel electrophoresis, leading to a lump of smears on the gel pattern. Several methods have been reported in the literature to process RPA amplicons before gel electrophoresis detection, these include protein denaturation by heating (at 65 °C or 95 °C for 10 minutes) and detergent treatment (e.g. sodium dodecyl sulfate, SDS), enzymatic digestion (e.g. proteinase K), protein sedimentation via high-speed centrifugation and purification using commercial DNA clean-up kit.37,109–113 Among these methods, the heating method worked equivalently to the methods by proteinase K digestion or SDS treatment.109,111 However, heating at 65 °C for 10 minutes showed better result than that of heating at higher temperature (95 °C).111 SDS treatment (20% in loading buffer) generated brighter and thicker gel bands than the proteinase K digestion method (0.2 mg mL−1 or 20 mg mL−1);109 heating at 65 °C for 10 minutes generated equivalent brightness gel bands to the ones generated by SDS treatment method (5% or 10% in the loading buffer) in Londono and co-workers’ results, however, Kapoor and co-workers showed that the SDS treatment method (5% in the loading buffer) resulted in brighter gel bands than the heating method (65 °C for 10 minutes) but also resulted in a smear-like pattern above the target band.111,112 In comparison to the heating, proteinase K digestion and SDS treatment methods, usage of the commercial DNA clean-up kit produced only the target band but in a much lower band intensity.37,109,111 In addition, as an alternative method, centrifugation (3 minutes) to pellet RPA proteins showed equivalent performance to the heating method (65 °C for 10 minutes).110
As with lateral flow strip detection, direct usage of RPA amplicons is possible, but it is recommended to dilute the amplicons with the running buffer (e.g. 1/100 dilution) before running on the strip to (1) improve its wicking performance114 and (2) avoid “ghost band” effects.45,54,115–117 Notably, Powell and co-workers pointed out that the viscous wicking problem on the lateral flow strip can be mitigated by replacing the high molecular weight PEG (5.5% 35 kDa; see also section 2.1) with the low molecular weight PEG (6.5% 3 kDa) in the RPA reagent formulae.114 Moreover, Powell and co-workers also developed methodology to alleviate the dilution step for lateral flow strip detection. They found that sometimes the RPA amplicons are being rendered unavailable in the “RPA globule” (the core of nucleic acid amplification which contains localised RPA reagents; Fig. 5A), the formation of which is highly associated with the PEG, for binding to the test line of lateral flow strip. However, applying a dual-labelled probe (two labels are connected via short length linkers) enabled escape from the “RPA globule” after the enzymatic cleavage, permitting amplicon ready for down-stream sandwich assay detection (Fig. 5B).114
 | ||
| Fig. 5 Influence of TwistAmp® nfo formulae and reaction mechanism on lateral flow strip detection. (A) The TwistAmp® nfo amplicons tend to be trapped in a RPA globule (a core of nucleic acid amplification that contains localised RPA reagents), and are consequently impended for binding to the test line of lateral flow strip. (B) The introduction of a dual-labelled probe (two labels are connected via short length linkers) allows escape from the RPA globule trap after enzymatic cleavage. The escaped cleaved dual-labelled probes are readily available for the down-stream sandwich assay detection. Reprinted and reproduced with permission from ref. 114. Copyright 2017 Powell et al. Published by Elsevier Inc. | ||
2.9 Sensitivities and specificities
The sensitivities and specificities of RPA can be evaluated in two categories, namely analytical and clinical (or field). The analytical sensitivity indicates the lowest amount of analyte that an assay can detect (also known as limit of detection); the analytical specificity is the ability of an assay to measure one particular analyte rather than others in a sample.118 In comparison, the clinical sensitivity is the percentage of correct detection of positive clinical samples, while the clinical specificity is the percentage of correct detection of negative clinical samples.From the analytical sensitivity and specificity perspective, RPA is very sensitive and can detect as little as a few molecules (copies) of the analyte, which approaches the analytical sensitivity of PCR (Table 5). Furthermore, ultra-sensitive detection down to even a single copy of the analyte can also be achieved in RPA (Table 4). In most cases, RPA is very specific for distinguishing one species from other non-closely related species, however, the natural function of these enzymes for performing homology directed repair becomes a disadvantage of RPA to discriminate towards closely-related species, especially when these species share high sequence similarity.69,87,88 Conversely, this indicates that RPA can tolerate to a certain degree of primers or probe mismatches to the target sequence (more details see section 2.6).
Apart from measuring the analytical sensitivity and analytical specificity, many researchers have applied RPA for testing clinical or field samples, and compared the results with a standard method (mostly PCR). From the summary Table 5, the clinical sensitivity of RPA is only half as sensitive as the benchmark method, whereas the clinical specificity of RPA is most of time as specific as the benchmark method. These results indicate that RPA may (in some cases only), mis-detect a positive sample (false negative), but is unlikely to show a false positive. In short, RPA is still at the beginning of undergoing clinical/field test evaluation, but not yet matured to be a routine test in the clinical/field settings.
3. Distinctive RPA detection methods
In the proceeding sections, we have discussed how RPA adapts to commonly used PCR detection methods, such as real-time fluorescent detection, gel electrophoresis and lateral flow strip detection. However, a myriad of different detection methods have been coupled with RPA, including flocculation assay, electrochemical, chemiluminescent, silicon microring resonator (SMR)-based photonic and surface-enhanced Raman scattering (SERS) detections. This section describes these distinctive RPA detection methods and discusses their advantages, which have enabled RPA assays to be more efficient and sensitive, yet sometimes simpler and faster.3.1 Flocculation assay detection
Flocculation assay detection is based on a bridging flocculation phenomenon in colloid chemistry described by Ruehrwein and Ward (in 1952),158 and later explained by La Mer and Healy (in 1960s).159–161 The basic principle of bridging flocculation involves the use of long polymers to cross-link multiple particles and thus flocculate out of solution at a specific buffer condition (e.g. pH and salt concentration; Fig. 6). A RPA reaction in combination with a flocculation assay detection was first demonstrated by Wee and co-workers, where RPA amplicons greater than 100 bp (from a plant pathogen) that resembled long polymers were precipitated onto a magnetic bead surface (at low buffer pH).162 The resulted conjugated particles underwent flocculation due to cross-linking of magnetic beads via RPA amplicons. The flocculation could only be triggered with amplified nucleic acids of lengths above 100 nucleotides, which is much longer than standard RPA primers; RPA reaction with no or non-target template did not produce such long “DNA polymer segments” and thus incurred no flocculation.162 Following from this first application, such RPA-flocculation assays were further applied to detect gene-specific DNA methylation,163Mycobacterium tuberculosis96 and prostate cancer biomarkers (TMPRSS2:ERG).164 Analytical sensitivity of these detections were 10% methylation (from 5 ng of starting material),163 10 bacteria colony forming unit (CFU)96 and 105 copies of TMPRSS2:ERG RNA (equivalent to a single cell),164 respectively. In addition, the detection of TMPRSS2:ERG biomarker underwent clinical sample test, and achieved 70% clinical sensitivity and 100% clinical specificity in comparison to the standard method reverse transcription-PCR.164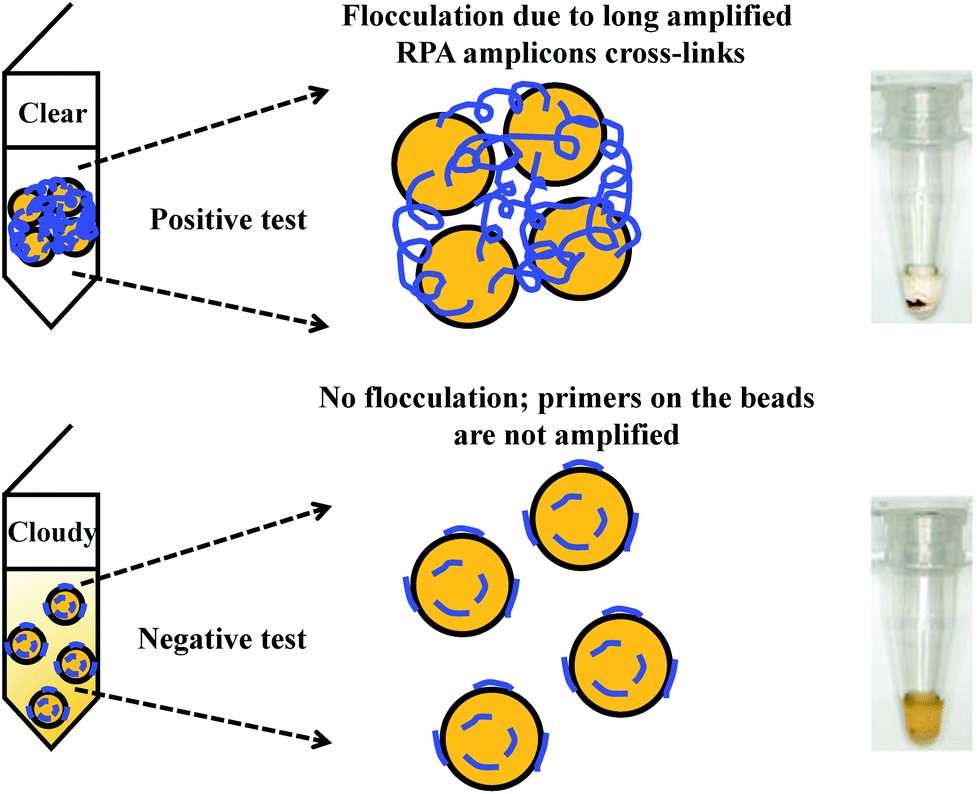 | ||
| Fig. 6 RPA in combination with flocculation assay detection. The RPA amplicons are incubated with magnetic beads at low pH buffer condition. Consequently, the precipitated RPA amplicons on the magnetic bead surfaces cross-link multiple other RPA-magnetic bead conjugates, and thus flocculate out of solution, causing a sharp transition between solution phase and flocculate. RPA reactions with no or non-target template do not produce long “DNA polymer segments”, hence no flocculation takes place. Reprinted and reproduced with permission from ref. 163. Copyright 2015 Springer Nature. | ||
The flocculation assay detection is a good alternative to the commonly used end-point detection methods (e.g. lateral flow strip detection) for quick qualitative detection of RPA amplicons. The total detection time is within ten minutes and only requires a minimum amount of RPA amplicons (10% of the reaction volume). Such detection method does not involve any DNA labelling or modifications during RPA reaction, and it also does not require equipment to confirm the RPA amplicons. The detection is a sharp transition between solution phase and flocculate, and is therefore better as naked-eye visualisation in comparison to other colourimetric visualisation detections.
3.2 Electrochemical detection
Another alternative detection strategy for RPA is electrochemical detection, which employs electrochemically active compounds to produce a signal in relation to the amplified nucleic acids. Most electrochemical approaches for RPA detection measure amperometric signals from RPA-enzyme-linked immunosorbent (ELISA) assay or RPA-enzymatic assay (more details please refer to literature37,38,43,104) due to the electrochemically active property of 3,3′,5,5′-tetramethylbenzidine (TMB) (Fig. 7).68,101,102,165–167 Results suggest that electrochemical detection could be up to 105-fold more sensitive than optical detection (by ELISA).166 Alternatively, Ng et al.103 and Lau et al.100 have applied gold nanoparticles as signal transducers for RPA amplicons detection. The gold nanoparticles can selectively bind to the RPA amplicons via specific conjugation, and transfers concentration of RPA amplicons into a measurable electrochemical signal. Again, such electrochemical detection exhibited high analytical sensitivity; Ng et al. was able to detect as low as 1 CFU of Mycobacterium tuberculosis bacilli DNA, while Lau et al. could detect down to 214 pM of Pseudomonas syringae (pathogenic bacteria for crops) DNA, which is 100 times more sensitive than RPA-GE detection. Recently, Tsaloglou et al.168 applied [Ru(NH3)6]3+ as an electroactive mediator for RPA-electrochemical detection. The [Ru(NH3)6]3+ can bind to the double-stranded DNA analogous to the intercalating fluorescent dye SYBR Green I, consequently causing a drop in the diffusion-controlled current as more double-stranded DNAs are synthesised during RPA. This ruthenium compound-based electrochemical detection achieved 11 CFU mL−1 of Mycobacterium tuberculosis analytical sensitivity, which is even more sensitive than the GeneXpert MTB/RIF (Cepheid Inc.) detection (a World Health Organisation recommended tuberculosis diagnostic system that employs PCR real-time fluorescent detection; 131 CFU mL−1).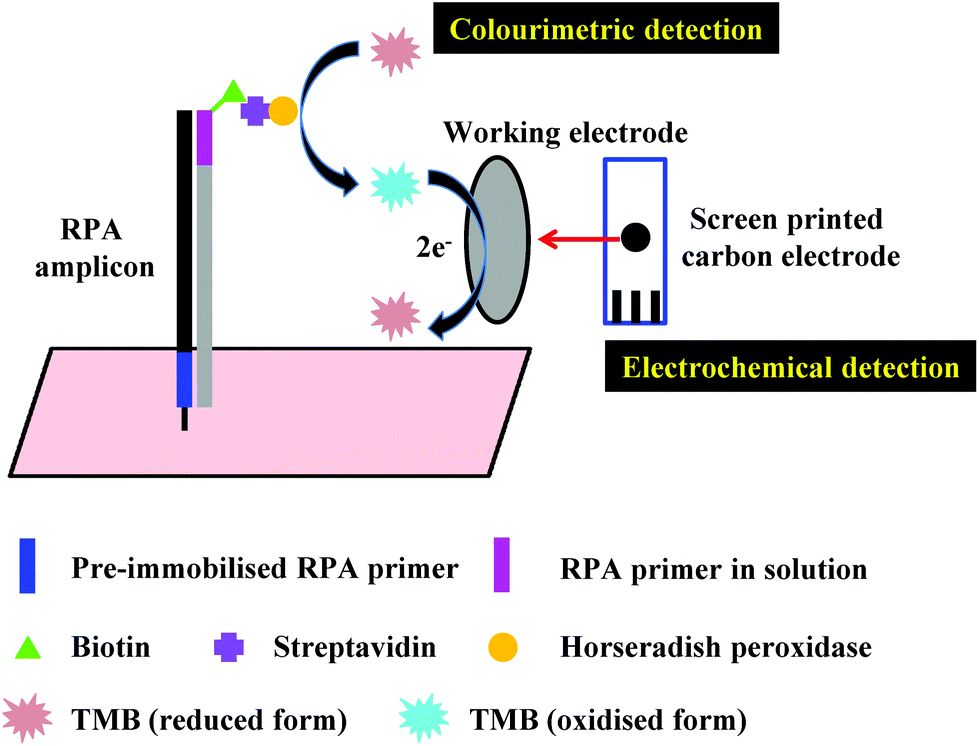 | ||
| Fig. 7 RPA in combination with electrochemical detection. The example demonstrates electrochemical detection from a RPA-enzymatic assay. The RPA-enzymatic assay is performed in an asymmetric manner, where one of the RPA primers is pre-immobilised on the solid surface, and another RPA primer, together with the DNA template and RPA reagents are free in the solution. After the RPA reaction, streptavidin-conjugated horseradish peroxidase is added, and followed by addition of electrochemically active 3,3′,5,5′-tetramethylbenzidine (TMB). The generated electrochemical signal can be detected on a screen printed carbon electrode.103 | ||
It is obvious that the RPA-electrochemical detection is advantageous for high analytical sensitivity. In addition, RPA-electrochemical detection is fast, low cost and field amenable, as it can be implemented easily with small components manufactured by inexpensive materials (e.g. screen printed carbon electrodes) with signal measured using a portable device (e.g. μSTAT 400 bipotentiostat/galvanostat).101
3.3 Chemiluminescent detection
As an alternative to the fluorescent probe-based detection that requires a light source, RPA reactions can also be detected via chemiluminescence. The chemiluminescent detection converts chemical energy into the emission of visible light (luminescence) as the result of an oxidation or hydrolysis reaction.169 Seidel's research group has been applying RPA coupled with chemiluminescent detection of water-borne microbes on flow-based microarrarys.170,171 Their chemiluminescent detection employs conversion of energy from oxidation between luminol and peroxide catalysed by horseradish peroxidase to give off luminescent signals detected by a charge-coupled device (CCD) camera (Fig. 8).170,171 Seidel and co-workers first demonstrated a triplex detection of Human adenovirus 41(HAdV 41), Phi X 174 (a virus) and the bacterium Enterococcus faecalis on the chemiluminescent flow-based microarray, and achieved the limit of detections of 35 genomic units (GU) μL−1, 1 GU μL−1 and 5 × 103 GU μL−1, respectively.170 The limit of detection of HAdV 41 was in the same range as the TaqMan-qPCR result reported by Heim et al. (15 GU μL−1);172 the limit of detection of Enterococcus faecalis was one order of magnitude lower than the reported RPA-electrochemical detection (of a Francisella tularensis, 2 × 104 GU μL−1) by Del Rio et al.166 Seidel and co-workers’ second demonstration was a duplex detection of Legionella spp. and Legionella pneumophila, and they achieved the limit of detection of 87 GU μL−1 and 26 GU μL−1, respectively.171 The Legionella spp. could be quantified over four log-steps while the Legionella pneumophila could be quantified over five log-steps.171 In short, the chemiluminescent detection is highly sensitive, having long and stable dynamic range, and is a good candidate for coupling with portable detection device (e.g. a smart phone) in comparison to the fluorescent probe-based detection.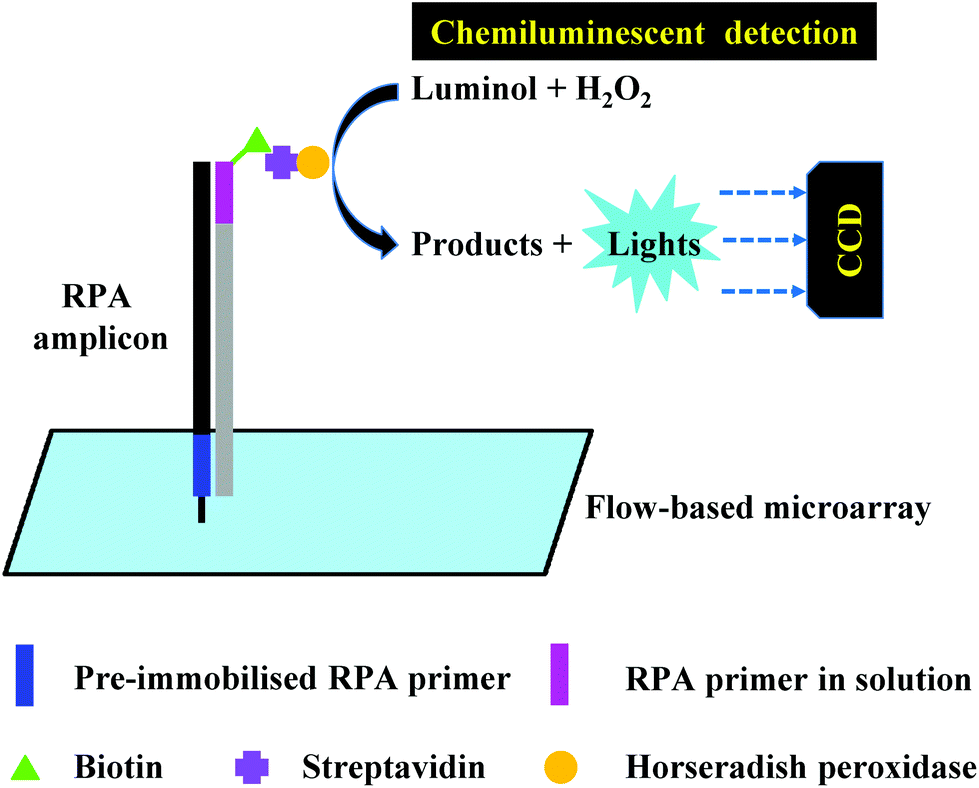 | ||
| Fig. 8 RPA in combination with chemiluminescent detection. The example demonstrates chemiluminescent detection using luminol and peroxide catalysed by horseradish peroxidase on flow-based microarray. The luminescent signal is detected by the CCD camera.170 | ||
3.4 Silicon microring resonator (SMR)-based photonic detection
Detection of RPA reactions can also be performed using silicon microring resonator (SMR)-based photonic detection, which involves performing nucleic acid amplification in an asymmetric manner (pre-immobilised on one of the primers on the SMR, and all the other oligonucleotides and reagents are free in the solution, see also section 4.2) in the evanescent field of a resonator waveguide.173–175 The binding of nucleic acids to pre-immobilised primers induces changes in the refractive index proximal to the waveguide surface. As the nucleic acid amplification progresses, the wavelength shift due to binding can be monitored in real-time on the SMR (Fig. 9). The SMR-based photonic detection is an alternative detection method for fluorophore-based real-time detection, yet is label-free and much more sensitive. Shin et al.130 achieved the limit of detection for a 10−4-fold diluted sample for Mycobacterium tuberculosis detection using real-time PCR, while they could detect down to a 10−6-fold diluted sample when using RPA-SMR detection. Sabaté del Río et al.176 demonstrated that RPA-SMR (2 fg μL−1) was 1000 times more sensitive than the real-time PCR (5 pg μL−1) for the detection of Francisella tularensis. Jin et al.151 showed that real-time PCR could only detect greater than 30% of the mutant allele in wild-type KRAS (a mutant gene in colorectal cancer) populations, while the RPA-SMR detection could detect 1% to 100% of the mutant allele. Since several silicon microrings can be accommodated on the resonator surface, a multiplexed RPA assay is achievable simultaneously on multiple microrings in parallel (also see section 4.2). Liu et al.113 demonstrated a duplex detection of IS6110 and IS1081 insertion sequences of Mycobacterium bovis Bulgarian BCG using RPA-SMR assay, and achieved 3.2 and 12 genomic DNA copies per reaction analytical sensitivity respectively. Dao et al.177 also demonstrated a duplex RPA-SMR assay for the detection of Salmonella Typhimurium and Brucella ovis, and they could identify 50 CFU (in 10 mL urine) and 100 CFU (in 10 mL urine) respectively. In comparison to the multiplex fluorophore-based real-time detection, the multiplex SMR-based photonic detection mitigates potential signal interferences due to fluorophore spectrum overlap, as the wavelength shifts are measured individually on each silicon microring.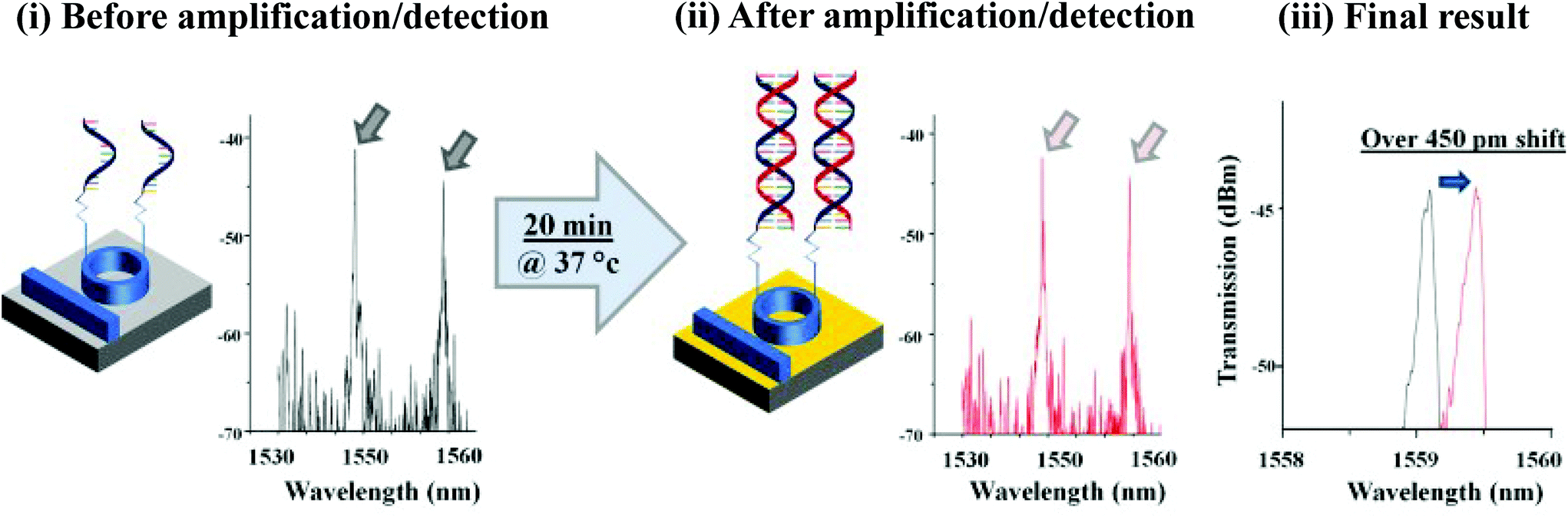 | ||
| Fig. 9 RPA in combination with silicon microring resonator (SMR)-based photonic detection. An asymmetric RPA reaction is performed on the SMR, where one of the RPA primers is pre-immobilised on the SMR, and another RPA primer, DNA template together with RPA reagents are free in the solution. The binding of DNAs to the pre-immobilised primers induces changes in the refractive index proximal to the waveguide surface, and can be monitored in real-time. The signal is detected as a wavelength shift. Reprinted and reproduced with permission from ref. 130. Copyright 2015 Elsevier B.V. All rights reserved. | ||
3.5 Surface-enhanced Raman scattering (SERS) detection
The final method to be highlighted for RPA detection is surface-enhanced Raman scattering (SERS). This is the phenomenon of when a laser excites nanoscale roughened metal surfaces (e.g. gold or silver), which resonantly drives surface charges, creating a highly localised (plasmonic) light field. When a molecule is absorbed, or lies close to, the enhanced field at the surface, a large enhancement in the Raman signal can be observed.178 SERS is a highly sensitive spectroscopic detection technique, which shows narrow and distinct spectral peaks of the detection molecules, and is particularly prominent for multiple target molecules detection (Fig. 10). Wang et al.179 demonstrated a duplex RPA-SERS assay for the detection of two prostate cancer biomarkers T1E4 and RN7SL1. The peaks assigned to the phosphate backbone (913 cm−1), purines (560, 742 and 1334 cm−1) and pyrimidines (1632 cm−1) could readily be observed in the SERS spectral profiles of both T1E4 and RN7SL1 target amplicons. Lau et al.98 demonstrated a triplex RPA-SERS detection of plant pathogens (Botrytis cinerea, Pseudomonas syringae and Fusarium oxysporum); distinct peaks corresponding to the three pathogens (due to the labelled Raman reporters) could be clearly displayed on the spectrum. Their results also showed that the RPA-SERS detection (2.3 DNA copies) was 100 and 10000 times more sensitive than RPA-Gel electrophoresis (GE) (2.32 × 102 DNA copies) and PCR-GE (2.3 × 104 DNA copies) detections, respectively. Koo et al.,99 however, has demonstrated the up-to-date highest multiplexity of RPA-SERS assay, a penta-plex detection of prostate cancer biomarkers (T1E4, T1E5, PCA3, ARV7 and RN7SL1). Distinct peaks corresponding to each prostate cancer biomarker was well-separated and clearly observed at 1075 cm−1 (T1E4), 1175 cm−1 (T1E5), 1285 cm−1 (PCA3), 1338 cm−1 (ARV7) and 1380 cm−1 (RN7SL1). In comparison to the SMR-based photonic detection, the SERS detection is also a better suited alternative to the fluorescent-based detection, especially for multiplexed detection. The multiplex ability of SMR-based photonic detection is limited by the number of silicon microrings that can be accommodated on the resonator surface, while the multiplex ability of SERS-based detection is limited by the number of Raman reporters (typically labelled to the target molecules) that produce sharp and unique peaks in the SERS detection.
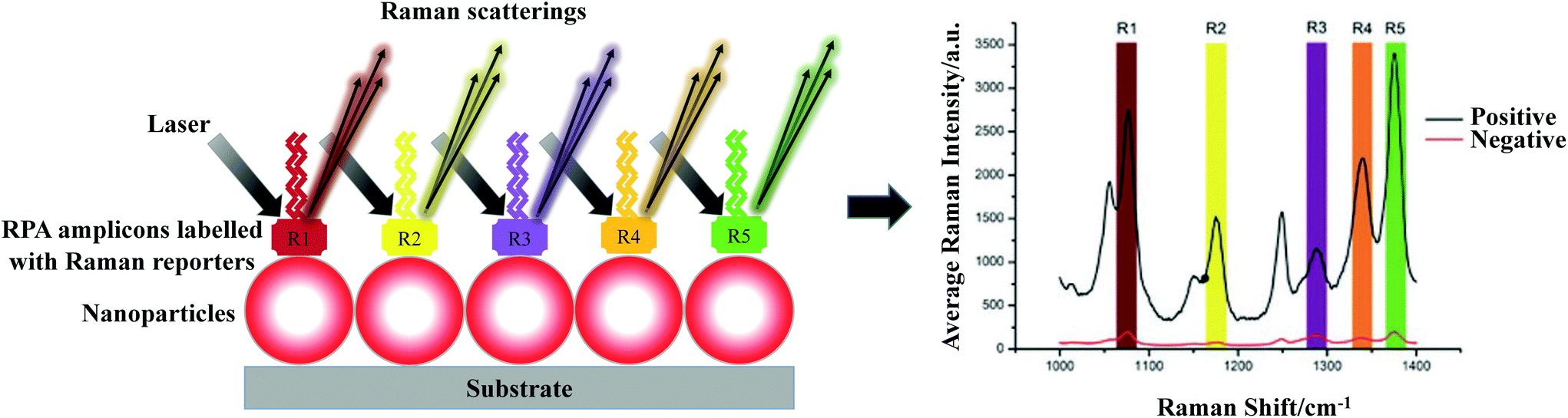 | ||
| Fig. 10 RPA in combination with surface-enhanced Raman scattering (SERS) detection. The example demonstrates a penta-plex RPA-SERS assay. The RPA amplicons labelled with Raman reporters are absorbed onto metal nanoparticles. When the laser excites these nanoparticles, a large enhancement in the Raman signals can be observed due to surface charge induced plasmonic light field surrounding the nanoparticles. The signal is displayed as narrow and distinct spectral peaks of the Raman reporters, correlating to different target templates. Reprinted and reproduced with permission from ref. 99. Copyright 2016 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
4. Hot spots for RPA development
As demonstrated in the proceeding sections, RPA is a fast developing nucleic acid amplification technique with a growing number of applications and detection systems. In this section, we consider some of the upcoming “hot spots” for RPA development, and discuss their progress and limitations. In particular, we discuss critical advancements in moving RPA into an accurate quantitative technique through the use of digital RPA, as well as increasing the detection capacity through multiplexing. We also outline progress in realising the ability of RPA to break through the bounds of the laboratory for true field implementation, either through mobile laboratory setups (such as the “RPA in a suitcase”), microfluidic integration, or simple single-step RPA procedures.4.1 Quantitative RPA – digital RPA
One of the most important developments of RPA is to quantify nucleic acid amplification, as quantitative nucleic acid amplification can be informative of gene expression levels, enabling better understanding of biological mechanisms, such as gene regulation during replication or pharmacological treatments, or distinguishing symptomatic from asymptomatic infection. However, unlike the classical real-time PCR (or quantitative PCR), real-time RPA is not considered to be robust for nucleic acid quantification. This is because the RPA reaction employs a chemical start (by adding the magnesium acetate) rather than a thermal start (by increasing reaction temperature to 95 °C), thus the reaction starting point cannot be precisely controlled. In addition, PCR is synchronised during each thermal cycle, as the annealing occurs only at low temperature, while there is no such synchronisation in RPA, as the annealing takes places all the time at the optimal reaction temperature range. Because of these reasons, RPA generates non-linear calibration curves for quantification.Nevertheless, the quantification of nucleic acids can be achieved using digital RPA that alleviates calibration curves by the ability of absolute quantification. Digital RPA involves fractionating a nucleic acid sample into thousands to hundred thousands of micro- to pico-litre volume compartments. Nucleic acid amplification subsequently occurs in each individual compartment. The amplification is usually detected via fluorescent probe, and compartments that encase nucleic acid from the sample will give rise to fluorescent signals. At the end of amplification, counting the positive and negative droplets gives precise, absolute quantification of the initial amount of nucleic acid template based on Poisson statistics (the targets end up in partitions independently and with a fixed rate). The published digital RPA reactions can be based on pre-fabricated partition and droplet partition of nucleic acid sample (Fig. 11).
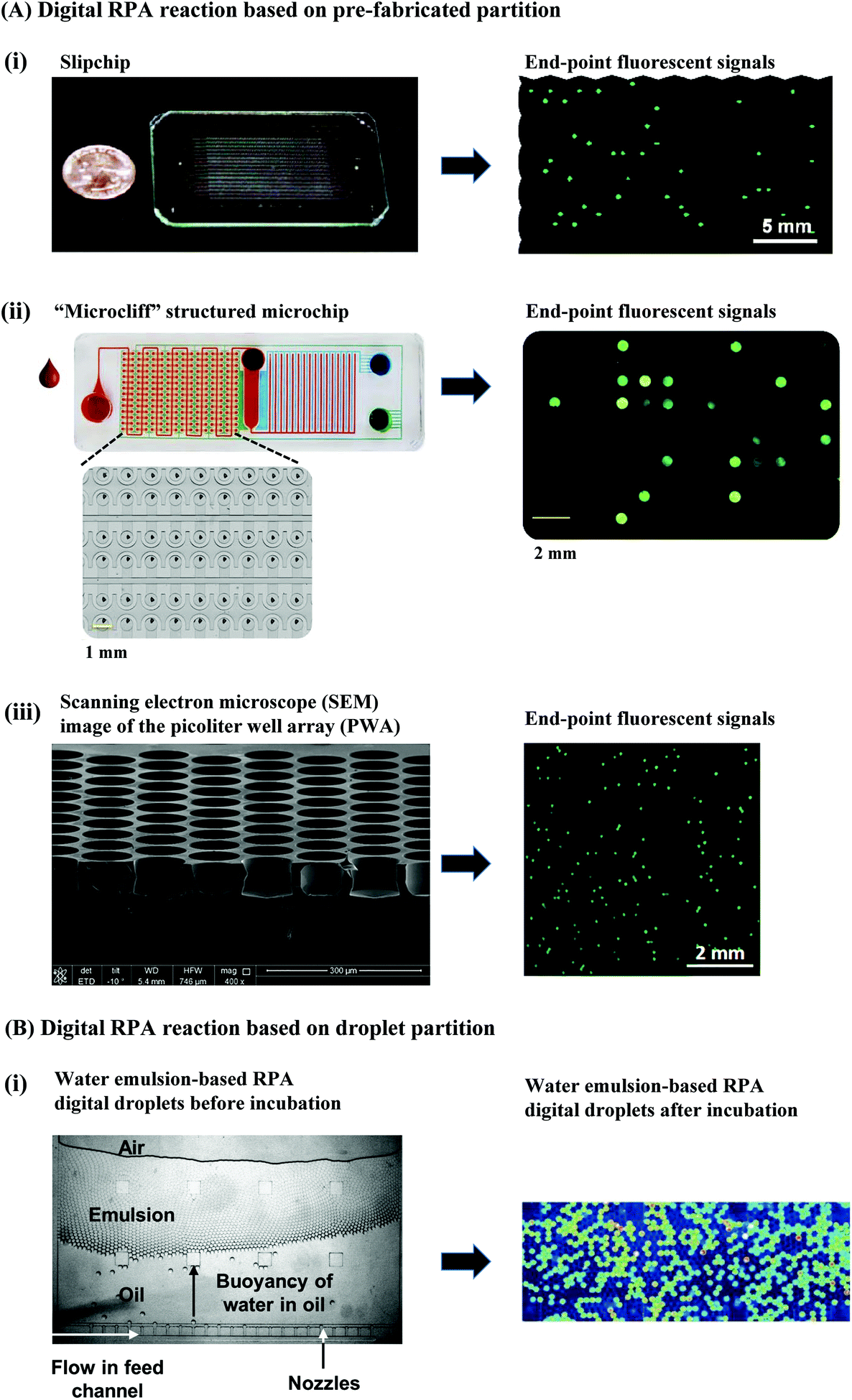 | ||
| Fig. 11 Digital RPA. (A): Digital RPA reaction based on pre-fabricated compartments. (i): Digital RPA on a slipchip. The slipchip contains top and bottom plates that contain wells and ducts which are pre-loaded with RPA reaction reagents respectively. The RPA reaction is initiated only when the top and bottom plate are brought in contact to each other and slipped to a specific configuration. The wells that encase the target DNA will give rise to an end-point fluorescent signal. Reprinted with permission from ref. 180. Copyright 2011 American Chemical Society. (ii): Pre-fabricated partition-based digital RPA on a “microcliff” structured microchip. RPA reagents and extracted DNA are delivered into the microchip via passive polydimethylsiloxane (PDMS) degas pumping. The wells that trap target DNA will give rise to an end-point fluorescent signal. Reprinted and reproduced with permission from ref. 183. Copyright 2017 Yeh et al. (iii) Pre-fabricated partition-based digital RPA on a pico-litre well array (PWA) chip. The mixture of RPA reagents and Listeria monocytogenes genomic DNA is evenly distributed into the wells (314 pL per well) using a scraping blade. The pico-litre wells that trap target DNA will give rise to an end-point fluorescent signal. Reprinted and reproduced with permission from ref. 184. Copyright 2016 Li et al. (B): Digital RPA reaction based on droplet partition. (i) Water-in-oil emulsion-based digital droplet RPA reaction produced by multiple nozzles. Mixture of RPA reagents and Listeria monocytogenes DNA is dispersed into continuous oil phase. The droplets that encapsulate the Listeria monocytogenes DNA will give rise to an end-point fluorescent signal. Reprinted and reproduced with permission from ref. 185. Copyright 2015 The Royal Society of Chemistry. | ||
The pre-fabricated partition employs a large amount of “mini-cavities” – wells that can hold a defined volume, which allows massively parallel nucleic acid amplifications. One demonstration of such pre-fabricated partition-based digital RPA is the slipchip.180,181 The slipchip contains two plates (top and bottom) containing wells and ducts which are pre-loaded with RPA reaction reagents respectively. The RPA reaction is initiated only when the top and bottom plates are aligned in a specific configuration by slipping. Tsaloglou et al.181 demonstrated a slipchip which allowed 8 parallel RPA reactions (each of the eight reactions comprises 500 nL of volume) for the detection of Clostridium difficile, and achieved a limit of detection of 1000 DNA copies. Shen et al.180 also demonstrated a slipchip that allowed 1550 parallel RPA reactions (each with 9 nL of volume) for methicillin-resistant Staphylococcus aureus (MRSA) detection, and achieved a limit of detection of 300 copies per mL of genomic DNA (Fig. 11A(i)). The other demonstration is on a “microcliff” structured microchip (Fig. 11A(ii)).182,183 Unlike the slipchip, the sample (nucleic acid and RPA reagents) delivery in this microchip is via passive polydimethylsiloxane (PDMS) degas pumping that alleviates external pumps and power source. RPA incubation starts once the sample is delivered into the wells. The “microcliff” structured microchip demonstrated by Yeh et al.182,183 encased 200 to 1500 wells (30–100 nL per well), which allowed detection of 10–105 copies per μL of MRSA DNA. Another demonstration of pre-fabricated partition-based digital RPA is on a pico-liter well array (PWA) chip (27000 wells, with each well of 314 pico-litres, in a 6 cm2 area; Fig. 11A(iii)).184 In this chip, the sample was evenly distributed into the wells using a scraping blade. The chip was sealed with oil to prevent evaporation before RPA incubation. The PWA chip could quantify serial dilutions of Listeria monocytogenes genomic DNA from 9 × 10−1 to 4 × 10−3 copies per well with an average error of less than 11% (n = 15).184
The digital RPA by droplet partitions is based on water-in-oil emulsion and is generated by dispersing liquid within an oil phase.185,186 In this case, each generated droplet acts as an individual micro-reactor, which contains all the reagents for a miniaturised RPA reaction (in the volume range from nano-litre to pico-litre; Fig. 11B(i)). Schuler et al.185 is the first to demonstrate the RPA reaction in water emulsion-based droplets (also called digital droplet RPA, ddRPA), and they successfully quantified Listeria monocytogenes DNA (100, 215, 464 and 1000 copies) that was concordant to the number of copies measured with digital droplet PCR.
In summary, the digital RPA partitionates a RPA reaction into nano- or pico-litre sized cavities to allow absolute quantification of the starting nucleic acid. This provides an opportunity to reduce the quantitative variability associated with real-time fluorescent probe-based quantification, as it does not rely on calibration curves. In addition, the digital RPA quantification is of significant value when testing samples that only contain negligible quantity (prospectively circulating tumour cells) in the original sample; the nucleic acid amplification from such samples can be overwhelmed in the tube (bulk) reaction, whereas, they become conspicuous in the digital RPA reaction.
4.2 Multiplex RPA
While quantitative RPA is critical to measure the amount of a single genetic marker in a system, multiplex testing is critical for assessing a series of different genetic markers, which can be equally important in gaining an improved understanding of a biological system, or for disease monitoring or detection. Multiplexing is highly preferred from a time and precision of understanding, as it greatly increases the result output per sample input in comparison to the single-plex detection within defined analytical turn-around time. Multiplex RPA reaction has been demonstrated either in a single tube (homogenous) or in a parallel fashion (sometimes refers to heterogeneous). The single-tube multiplex RPA can be performed using single or multiple nucleic acid templates. For the former, Kim et al.122 performed a duplex detection of Campylobacter species: Campylobacter coli and Campylobacter jejuni (detection limits of each: 1 CFU mL−1 in pure culture and in chicken broth samples without enrichment; 3log CFU g−1 in non-enriched egg and chicken meat samples; 1 CFU g−1 in enriched egg and chicken meat samples), and Wang et al.187 conducted a duplex assay to differentiate wild-type and gE-deleted vaccine pseudorabies (limit of detection of 100 viral DNA copies each). However, the highest level of multiplexing is a triplex assay demonstrated by Piepenburg and co-workers for the detection of MRSA I, II and III genotypes (detection limits are 10 genomic copies each).6 For single-tube multiplex RPA using multiple different nucleic acid templates, a few duplex and triplex assays have been demonstrated for the detection of bacteria,126,188,189 parasites,105 fungus,98 genetic modified soybeans21 and prostate cancer biomarkers.179 In particular, Song and co-workers demonstrated the highest multiplexity via a 16-plex assay for pathogen detection by combing RPA reactions with LAMP on a microchip, which was termed a two-stage isothermal amplification (Fig. 12A).190 In Song's demonstration, a first-stage RPA reaction was applied to simultaneously amplify sixteen different DNA targets in the RPA reactor, while a second-stage LAMP reaction was performed in sixteen branching chambers that stemmed from the RPA reactor to amplify and detect the resultant RPA amplicons in parallel. Each of the branching chambers contained a specific set of primers and probe for amplifying a specific pathogen among a pool of sixteen RPA amplicons from the first stage reaction.190 The RPA-LAMP assay could detect down to 1 plaque-forming units (PFU) using Zika virus-American strain (mex 2-81, Mexico) as a demonstration (amplifying the DNA template with the present of 16 primer pairs), with a linear correlation between 5 PFU and 500 PFU.190
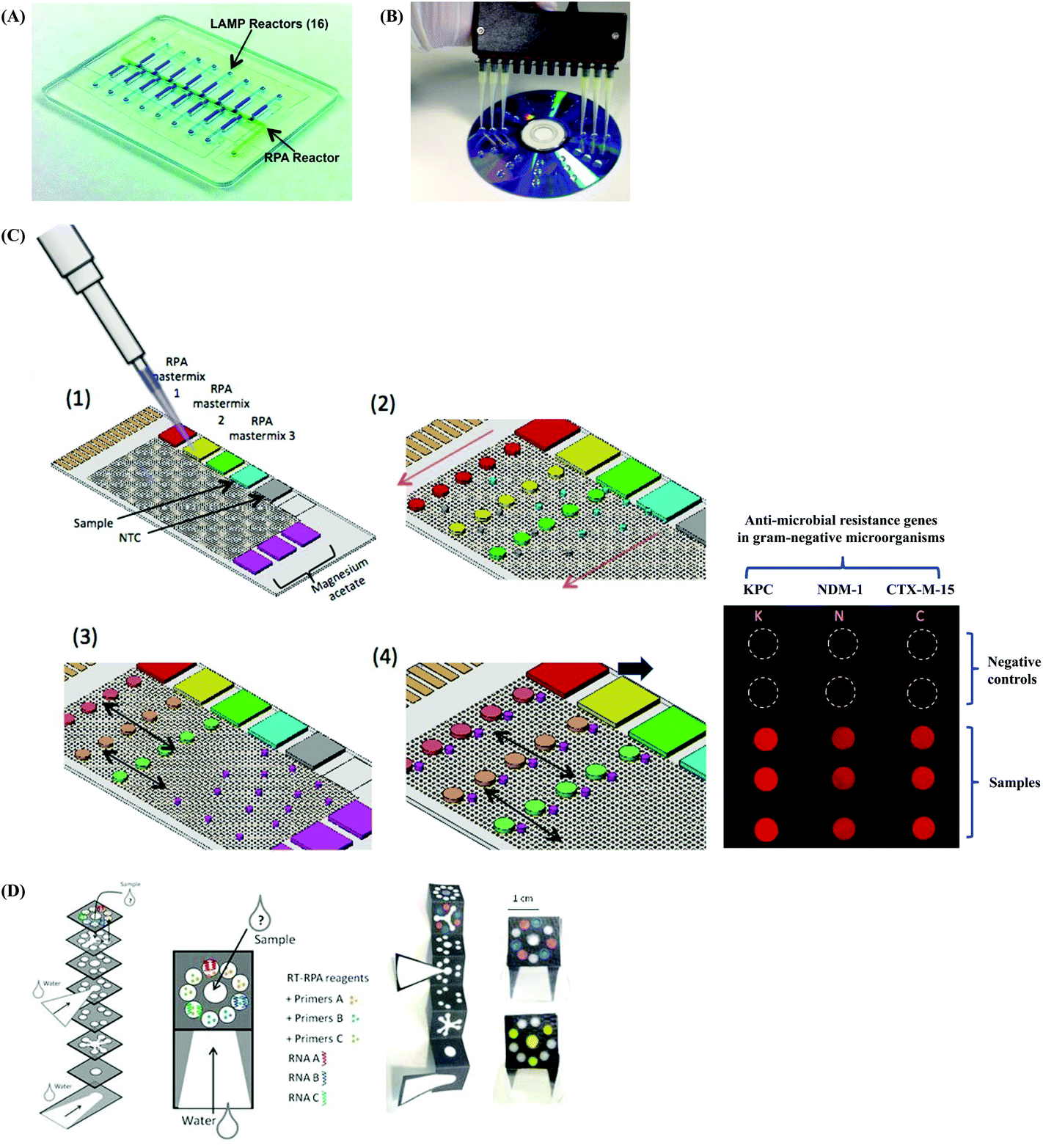 | ||
| Fig. 12 Multiplex RPA. (A): Example of single-tube multiplex RPA via a two-stage isothermal amplification (RPA in combination with LAMP). Sixteen different DNA targets are amplified using RPA in the RPA reactor at the first stage, followed by metering the resulting mixture of RPA amplicons into sixteen branching LAMP reactors that have been pre-stored with set of LAMP primers and probe for a specific pathogen detection respectively. If the DNA targets are present in the sample and are successfully amplified by RPA, the branching LAMP reactors will produce fluorescent signals. Reprinted with permission from ref. 190. Copyright AMERICAN ASSOCIATION FOR CLINICAL CHEMISTRY, INC. (B): Example of in-parallel multiplex RPA that adopts homogeneous assay format on a DVD. RPA reaction mixtures are pipetted onto a DVD as a microarray. Reprinted with permission from ref. 191. Copyright 2014 Elsevier Ltd. All rights reserved. (C) Example of in-parallel multiplex RPA that adopts homogeneous assay format on electrowetting-on-dielectric (EWOD) surface. The example demonstrates a triplex RPA reaction (each sample with 3 replicates and duplicates negative control) for the detection of anti-microbial resistance genes in Gram-negative microorganisms. The mixing of different components of RPA reagents can be performed at desired time-points and in a sequential manner, as the RPA reaction droplets can be individually manipulated (dispensing, splitting or mixing) via the phenomenon of EWOD. The RPA reaction droplets that contain the DNA target will give rise to an end-point fluorescent signal. Reprinted and reproduced with permission from ref. 192. Copyright 2017 Kalsi et al. (D) Example of in parallel multiplex RPA that adopts homogeneous assay format on a 3D origami paper-based device. Schematic view of the 3D origami paper-based device showing the location of freeze-dried biological components (real-time RPA reagents with three different primers and associated RNA templates) on wax patterned areas (left). Application of three different coloured dyes to display the multiplex RPA detection (right). After supplying the sample, dipping into the water and being folded, the input sample will flow along the defined patterns via capillary force between adjacent paper layers, and finally reach to the desired areas for RPA amplification. Reprinted with permission from ref. 193. Copyright 2017 Springer Nature. | ||
Collectively speaking, the single-tube multiplex RPA reaction saves both reaction space and volume. However, further increasing multiplexing levels is very challenging. In the case of using a single nucleic acid template, the multiplex capacity is mainly restricted by the capacity to accommodate multiple genes within a certain length of consensus sequence. However, the main restriction becomes the non-specific interactions among multiple oligonucleotides (primers, probes and nucleic acid templates) when using multiple nucleic acid templates. Although Song and co-workers’ methodology alleviates such restriction (as long as there were few correct RPA amplicons generated in the first-stage reaction, despite concurrently produced non-specific amplicons, these correct RPA amplicons would be specifically further amplified via a second-stage LAMP reaction), their methodology is still limited by the capacity of primer concentrations in the first-stage RPA reaction.
In contrast to single-tube multiplex RPA, performing multiplex RPA in a parallel fashion omits the non-specific interactions among multiple oligonucleotides. This is because (1) each single-plex RPA reaction occurs next to each other but is independent of each other (performed as homogeneous assays) or (2) the RPA reaction is performed in an asymmetrical manner (heterogenous assay), where one of the RPA primer is pre-immobilised on a matrix while the other RPA primer remains in the solution together with the DNA template (and probe), in this way, only the RPA amplicons will be captured on the matrix for detection.
The in-parallel multiplex RPA that adopts homogeneous assay format can be performed either in solution or on a solid phase. For the former, multiple amplicons from collateral single-plex RPA reactions (in tubes) are combined together and submitted to a subsequent multiplex detection method, exemplified by quantum dot beads-coated microwell detection (duplex),194 quantum dot-based flow cytometry detection (penta-plex)195 and SERS detection (penta-plex).99 For the latter, one demonstration is on the digital video disk (DVD) by Maquieira research group, and the resulting signals can be detected by a DVD player (Fig. 12B).188,191,194 The other demonstration is on an electrowetting-on-dielectric (EWOD) surface, where the EWOD-based RPA reaction droplets are controlled using electric potential by varying electric energy across the thin dielectric film between the liquid and conducting substrate.80,192 Kalsi et al.192 demonstrated a triplex EWOD-based RPA reaction in a parallel format for the detection of three antibiotic resistant genes (CTX-M-15, KPC and NDM-1). Each single-plex droplet RPA reaction was independent of other single-plex droplet RPA reactions, and could perform a set of programmed operations including move, merge, split, dispense and mix in an automatic manner (Fig. 12C). The EWOD-based RPA reaction achieved the limit of detections of 1000 DNA copies of each antibiotic resistant gene, which is 100 times less sensitive than the bench-top RPA reactions.192 Another demonstration is on paper by Magro and co-workers, which they constructed a wax patterned five-layer paper device in three-dimensional (3D) origami configuration (Fig. 12D).193 Freeze-dried RPA reaction reagents were pre-stored on multiple individual wax patterned areas. Once the liquid sample mixture was pipetted onto the sample inlet, the input liquid sample mixture could be distributed via capillary force between adjacent paper layers by folding, and ultimately reached specific areas that contained pre-stored reagents, to initiate RPA reactions.
Unlike the in-parallel multiplex RPA that adopts homogeneous assay format, the demonstrations of the in-parallel multiplex RPA that adopts heterogeneous assay takes place only on solid surfaces. The majority of such assay performs multiplex RPA in an asymmetric manner on a microarray. Kersting et al.196 performed triplex RPA in a 4 × 6 microarray for the detection of Salmonella enterica, Neisseria gonorrhoeae and MRSA on the epoxy-silanised glass slides, and achieved detection limits of 10 CFU, 100 CFU and 10 CFU respectively. Kunze et al.170 conducted triplex RPA in a 5 × 5 microarray for the detection of Enterococcus faecalis, Human adenovirus 41 (HADV 41) and Phi X 174 virus on the poly(propylene glycol) diamine (DAPPG) coated glass slides (also see section 3.3), and achieved detection limits of 35 GU μL−1, 1 GU μL−1 and 5 × 103 GU μL−1 respectively. Another demonstration of such an assay is the performance of multiple single-plex RPA reactions on multiple silicon microrings fitted on a resonator surface, demonstrated by Liu et al.113 and Dao et al.177 (more details refer to section 3.4).
Taken together, the multiplex RPA in a parallel fashion has a much higher assay throughput, circumvents cross-reaction (among oligonucleotides) issues, and possesses higher multiplex capacity compared to the single-tube multiplex RPA. However, its degree of multiplexing is limited by (1) the number of available unique labels conjugated to multiple nucleic acid templates which permit distinctive detection; (2) the size of the reaction surface; and (3) EWOD-based RPA reaction is also limited by the minimum volume of the droplet that permits an effective reaction.
4.3 RPA in a suitcase for mobile laboratory
The ultimate purpose of isothermal amplification such as RPA is to take the nucleic acid testing away from the centralised lab to the field or resource-limited settings. Abd El Wahed and co-workers turned this thought into reality by miniaturising the RPA diagnostic into a standard suitcase (56 cm × 45.5 cm × 26.5 cm by size), dubbed diagnostics-in-a-suitcase (Dias; costs around 5000 Euro; Fig. 13A).197 The Dias contained all the reagents and equipment necessary for the real-time RPA assay to detect the emerging avian influenza A (H7N9) virus at the site of an outbreak. The reagents and the equipment were fixed in the foam at the base of the suitcase which acted as a shock absorber during transport; a solar panel and a power pack provided the power support; and the storage box was refillable whenever needed.197 The RPA-Dias detection of influenza A (H7N9) was successful, and achieved detection limits of 10 and 100 RNA copies for the detection of H gene and N gene, respectively. | ||
| Fig. 13 Mobile RPA assay. (A): RPA in-a-suitcase. The standard suitcase (56 cm × 45.5 cm × 26.5 cm by size) contains all equipment and reagents for performing the real-time RPA assay. The equipment and reagents are imbedded into the foam or fixed for stable portability. Reprinted with permission from ref. 197. Copyright 2015 Abd El Wahed et al. Published by Elsevier B.V. (B): A mobile laboratory set up from the “RPA in-a-suitcase”. The mobile lab is arranged into 4 sites in close proximity, including the RNA extraction, master mix, sample mix and detection. The RNA extraction area encompasses magnetic separator stand, vortex, rotator, 1.5–2 mL Eppendorf tube rack, automatic 100–1000 μL micopipette, micropipette tips, digital timer, 1.5 mL disposable plastic Eppendorf tubes and a waste container with autoclavable plastic bags. Both the master mix and sample mix areas contain vortex, minicentrifuge, automatic 1–10 and 10–100 μL micopipettes, micropipette tips, scissor and 0.2 mL tubes rack. The detection was done using the tubescanner (Twista device, TwistDx, Cambridge, UK). Reprinted with permission from ref. 131. Copyright 2015 Abd El Wahed et al. | ||
This highly promising demonstration of the first RPA-Dias inspired Abd El Wahed and co-workers to perform further testing. They carried the Dias (a total weight of 23 kilograms including the aluminium case) to the field of both of Kedougou (Senegal) and Bangkok (Thailand), and set up a mobile laboratory for the real-time RPA detection of dengue virus (1–4 serotypes).131 The mobile lab was organised into 4 sites, including the RNA extraction, master mix, sample mix and detection (using a tubescanner, TwistDx, Cambridge, UK) in close proximity (Fig. 13B).131 All the reagents were cold-chain independent; the power was supplied either from a motor vehicle battery (via inverter) or from the solar panel. The real-time RPA assay performed successfully in such an open-air environment and observed no influence of dust on the assay quality. The clinical sensitivities and specificities tested in Kedougou (Senegal) and Bangkok (Thailand) were 98% and 100% and 72% and 100%, respectively.131 Later, Abd El Wahed and co-workers also carried the Dias to the local hospitals in Matoto (Guinea)134 and Mymensingh (Bangladesh)121 for the on-site detection of Ebola virus and Leishmania donovani, respectively. The real-time RPA Ebola assay was able to correctly include positives (91% of clinical sensitivity) and exclude negatives (100% of clinical specificity);134 while the real-time RPA Leishmania donovani assay achieved 100% clinical sensitivity and 100% clinical specificity.121 The demonstration of RPA-Dias brings the rapid nucleic acid testing to the site of need, in particular benefiting the places with poor laboratory infrastructure. With its continuous development, there is no doubt that the RPA-Dias will become pervasive within five years.
4.4 Microfluidic integration of RPA assays
Apart from multiplex RPA reactions and mobile RPA reactions that provide increasing diagnostic efficiency and portability respectively, a growing attention has been focused on developing RPA assays that encompasses both of the two merits. The solution is to integrate RPA amplification with other chemical processes on a miniaturised device using microfluidics. The major chemical processes to combine with RPA amplification for an intact diagnosis are sample preparation, amplification and signal detection. There have been a few examples of integrating RPA amplification with signal detection, however, only few demonstrations have been performed integrating all of the three processes. Branavan et al.198 developed a microfluidics that integrated RPA amplification and fluorescent signal detection for the detection of sexually transmitted infections; Renner et al.47 developed a degas-actuated microfluidics that integrated multiplex RPA amplification and fluorescent signal detection for the detection of ESKAPE (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacter spp.) bacterial pathogens; and Hu et al.199 developed thin film transistor-based microfluidics that integrated RPA amplification and electronic signal detection for the detection of anti-microbial resistance genes. We note that all these three examples require the input of manually pre-mixed RPA reaction reagents. For fully integrated assays, one demonstration is the self-powered integrated microfluidic by Yeh et al.183 The self-powered integrated microfluidic integrated sample preparation (plasma separation from blood cells), digital RPA amplification (pre-partition-based) and detection (fluorescent-probe based) in an automatic manner via a built-in vacuum battery system (that frees the chip from external pumps sources for pumping).183 The self-powered integrated microfluidic was able to detect 2 × 105 copies per μL of HIV-1 RNA spiked in human blood within 18 minutes, and was able to quantitatively detect 10 to 105 copies per μL of MRSA DNA in water or directly from spiked human whole blood.183 Another demonstration of fully integrated assays is the application of centrifugal microfluidics that perform all the required procedure for RPA reaction in an automatic manner governing by centrifugal forces. Zengerle's research group was the first one to demonstrate such performance. They developed a foil disc (Fig. 14A) that performs metering of the RPA rehydration buffer to re-suspend RPA reaction pellet, and subsequently aliquot the resulting mixture into five parallel reaction chambers that were pre-stored with oligonucleotides (primers, probe and DNA template).200 The foil disc was fitted onto a modified Rotor-Gene 2000, which executed centrifugation to manipulate the liquid movement between different chambers on the foil disc and real-time fluorescent signal detection. Followed up this demonstration, they developed another foil disc that integrated sample preparation (DNA extraction from blood plasma), nucleic acid amplification and fluorescent detection of Bacillus anthracis and Francisella tularensis.201 And this time, they tailor-made a portable LabDisk Player (approximately 18 × 28×15 cm3 and 2 kilograms by weight) which could process the integrated chemical processes on the foil disc in an automatic manner. The fully integrated microfluidic RPA assay detected 6 × 104 genome equivalents of Bacillus anthracis and 6 × 106 genome equivalents of Francisella tularensis in less than 45 minutes.201 Their centrifugal microfluidics is the first true example of fully integrated RPA assay device, with the entire procedure from DNA extraction through to detection, in a fully automated fashion. Later, Kim et al.202 also developed a centrifugal microfluidics that integrated sample lysis, RPA amplification, metering, dilution and lateral flow strip detection of food-borne pathogen (Salmonella enteritidis; Fig. 14B). The centrifugal microfluidics was fitted onto a computer-controlled unit that included a spinning motor, a laser for sample lysis and a local heating apparatus to support RPA assay procedure. Their demonstration achieved the limit of detections of 10 CFU mL−1 and 100 CFU mL−1 in phosphate-buffered saline (PBS) and milk respectively with a total analysis time of 30 minutes.202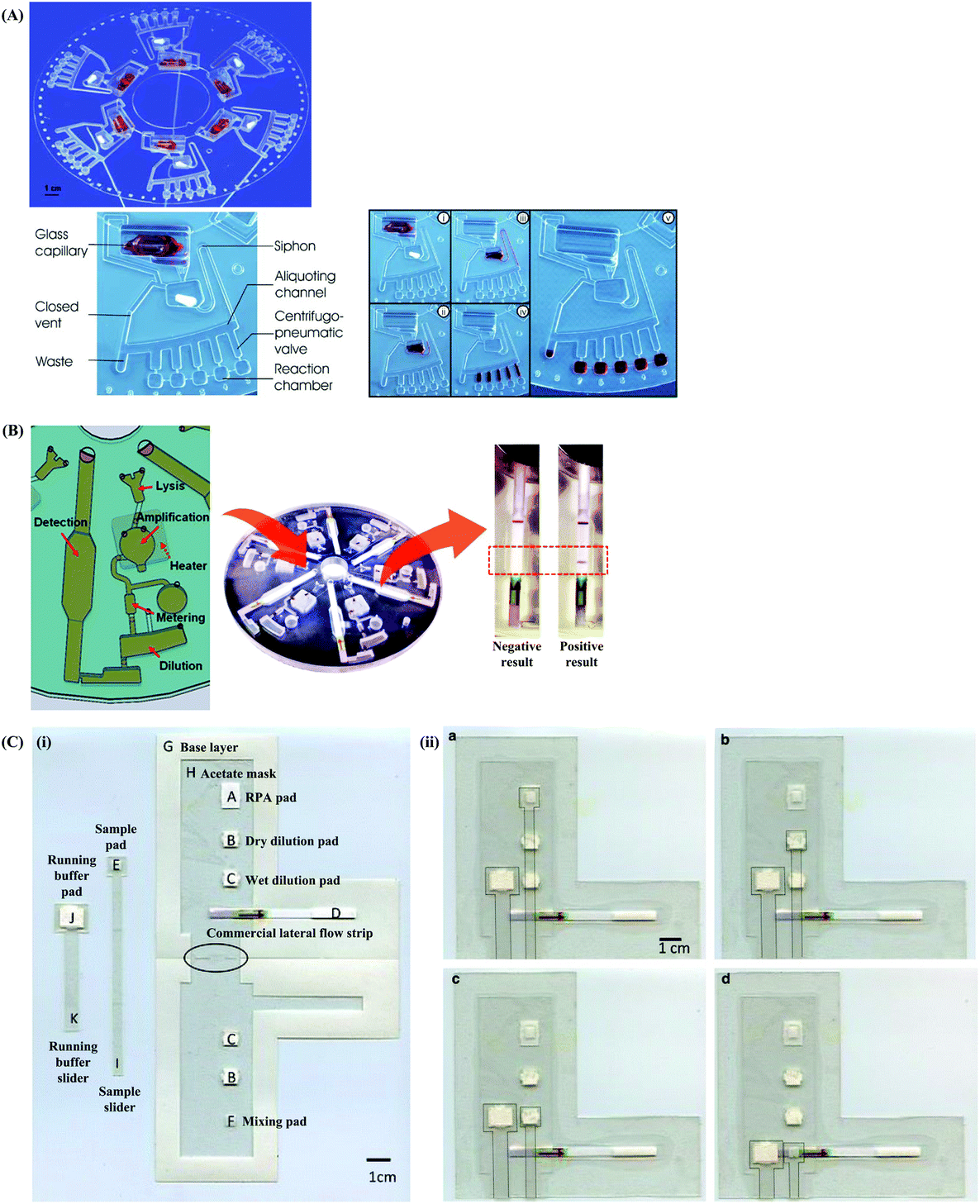 | ||
| Fig. 14 Integrated RPA assays on microfluidics. (A): Centrifugal microfluidics that integrates RPA reagents metering, mixing and aliquoting, amplification and fluorescent signal detection. Photograph of a foil disc assembled with RPA reaction reagents (liquid and lyophilisate reagents) featuring 6 fluidic structures (top), each capable of processing 5 geometrically multiplexed assays. Detailed fluidic structure for one unit is denoted; the buffer is replaced by red ink for demonstration purposes (bottom left). Elucidation of automated RPA assay on a centrifugal microfluidic (bottom right); fluids movement between chamber to chamber are manipulated via centrifugal force. (i) Inside the glass capillary contains the RPA liquid reagents, including DNA template, primers, probe, rehydration buffer, magnesium acetate, while the lyophilisate chamber contains the lyophilised pellet; (ii) the RPA liquid reagents is spun into the lyophilisate chamber after the glass capillary is crushed; (iii) the siphon allows valving between the lyophilisate chamber and the aliquoting structure; (iv) the 50 μL RPA liquid reagents volume is split into 5 × 10 μL aliquots; and (v) the fluid fills the reaction chambers via a centrifugal pneumatic valve. Reprinted with permission from ref. 200. Copyright 2010 Royal Society of Chemistry. (B): Centrifugal microfluidics that integrated sample lysis, RPA amplification, metering, dilution and lateral flow strip detection. Reprinted with permission from ref. 202. Copyright 2014 American Chemical Society. (C): Paper-based microfluidics that integrates RPA amplification, dilution and lateral flow strip detection. (i) Layout and components of the device; (ii) operations of the device: (a) sample slider at position 1, mixing RPA reagents and target to initiate the amplification (30 minutes of incubation); (b) after amplification, the sample slider pulled down to position 2, in contact with dry dilution pads to absorb RPA buffer (10 minutes of duration); (c) the sample slider pulled down to position 3, in contact with wet dilution pads, for dilution with TBST buffer (10 minutes of duration); and (d) the sample slider pulled down to position 4 into contact with the lateral flow strip, and the running buffer slider is pulled down to activate the lateral flow detection (5 minutes of duration). Reprinted with permission from ref. 106. Copyright 2015 Cordray and Richards-Kortum. | ||
The foil-based microfluidics described above are a compact and affordable platform for performing integrated RPA assays, but they require complicated and expensive design and fabrication. Nonetheless, paper-based microfluidics can be an alternative for performing integrated RPA assays, as they can also conduct many of the functions of the foil-based microfluidics. Cordray and Richards-Kortum106 developed a paper-based microfluidics that integrated RPA amplification, dilution and lateral flow strip detection. All the reagents for this three processes were pre-stored in different paper pads, and were mixed with each other sequentially by pulling the sliders (Fig. 14C).106 Their paper-based integrated RPA assay could detect as few as 50 synthetic Plasmodium DNA copies, which was equivalent to the analytical sensitivity of the bench-top RPA-GE assay. In comparison, Magro et al.193 developed a 3D origami paper-based device that integrated multiplex RPA amplification and fluorescent signal detection (refer to section 4.2). The pre-stored reagents and oligonucleotides in different wax-patterned areas were rehydrated once the device is folded and dipped into water. Although the integration of RPA amplification and signal detection showed successful paper-based microfluidics, it required human intervention to complete the integrated RPA assay, as opposed to a fully integrated RPA assay on foil-based microfluidics. Moreover, it is very challenging to integrate sample preparation with RPA amplification and signal detection on paper-based microfluidics, not to mention in an automated execution. The reason is that it is very difficult to manipulate liquid movement on paper merely via capillary force, which is a weak force, and thus results in a slow moving process (despite of the liquid has a high wettability and the matrix is very hydrophilic and porous).
Overall, developing a microfluidically integrated RPA assay is a transition towards the realisation of fully automated sample-in-answer-out RPA assay to increase the diagnostic efficiency and portability. Nevertheless, the sample preparation step is the bottleneck to achieve this ultimate goal. In particular, some samples (e.g. blood) require multiple processing steps before the extracted nucleic acids are ready for the subsequent amplification and detection. Zengerle's research group and Kim and colleagues’ work by employing multiple sample preparation chambers together with the amplification chamber (and detection chamber) on a single foil-based microfluidics, demonstrates one feasible way, albeit with challenging and costly microfluidics design and fabrication.
4.5 One-step RPA assays
A simple and feasible way to facilitate full automation of the RPA assay without introducing multiple sample preparation chambers is to perform a one-step RPA assay, or in other words, to perform RPA amplification directly from the crude sample (in the same tube). RPA has been able to amplify nucleic acids directly from crude samples, such as plant tissue extract,111,203 crude sap,79,112 soil and water samples,71 food samples (eggs and chicken meat)119 and vaginal swab lysate.137Nevertheless, Choi and co-workers were the first to demonstrate a fully integrated RPA assay on foil-based microfluidics that was facilitated by the one-step assay approach. Choi et al. employed direct PCR buffer in RPA reaction for the detection of food poisoning bacteria (Salmonella enterica, Escherichia coli O157:H7 or Vibrio parahaemolyticus).204,205 The direct PCR buffer was able to lyse the bacterial cells, inactivate amplification inhibitors from the lysed sample and was compatible to the RPA reaction system.204 The entire assay completed within 30 minutes, which was much shorter than the time required for a non-sample preparation integrated microfluidic or a sample preparation integrated microfluidic system (e.g. at least 60 minutes). Although the direct RPA assay is a shortcut to assist full automation of foil-based microfluidics, there is no rule of thumb for the direct RPA assay buffer. This is because each sample has different lysis difficulty levels, and the effective lysis buffer may not be compatible with the RPA system and may also have potential impact on fluid movement within the microfluidics. Therefore, it is a trail-and-error process to identify such effective direct RPA buffers.
5. Conclusions and future perspectives
The PCR is a revolutionary watershed in nucleic acid test, however, isothermal nucleic acid amplification, as an alternative to PCR, has a lower requirement in heating conditions and performs nucleic acid replication more rapidly. RPA was a comparatively late introduced isothermal amplification method, however, it is one of the fastest developing methods. This is due to its true isothermal properties, simple reaction scheme, fast reaction time and excellent reports of sensitivity and specificity. In this review, we provide comprehensive knowledge of RPA technology: from its reaction components and mechanism, to the design of RPA assay and detection methods. In particular, we summarise many experimental tips from practical implementation, dispose clinical/field performance data and point out focused RPA development, to help researchers make better use of RPA and make contributions to boost its development.RPA has just passed its first decade of development, and is now stepping into a next stage of evolution. We have considered some critical issues for future development. Firstly, more attention is needed for developing field-amenable sample preparation methods including concentration, extraction and purification, as this would largely facilitate a complete RPA assay for on-site or field application. Secondly, developing portable and fully automated RPA diagnostic devices is of paramount importance not only to further increase the diagnostic efficiency, but more importantly to bring RPA diagnostic closer to the layperson and people who live in places with poor healthcare infrastructure. Thirdly, RPA can be applied for developing wearable sensors by virtue of its close-to-body reaction temperature. Imagine if people can simply take some samples from their own body (e.g. body fluid) and perform a fast RPA assay using their body temperature to detect potential pathogens, this could revolutionise RPA diagnostics to be a self-testing. Fourthly, unlike real-time PCR which is controlled by heat cycle, RPA employs continuous amplification after reaction initiation; it would be beneficial to develop a method or mechanism to precisely control the starting point of each RPA amplification cycle, so as to assist obtaining reliable quantitative RPA results (other than the absolute quantification by digital RPA). Fifthly, the current market share of RPA (7.08%) is far less than that of PCR, and is less in comparison to other isothermal nucleic acid amplification methods (e.g. SDA and LAMP). To boost its market share, more studies in comparative clinical validations between RPA and existing PCR assays can be performed. In addition, practical websites or software should be developed to ease and streamline the RPA primers and probe design, and screen for optimal oligonucleotide pairs for the reaction. Rationally speaking, RPA may not replace the status of PCR in years to come, but it can be a versatile complement of PCR. At this time point, RPA technology is maturing for application in the clinic, however, it is still at a transition period towards on-site or field application. With its continuous fast development, we foresee that RPA technology will ultimately become robust mobile and point-of-need applications in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by Alexander von Humboldt Fellowship. We also acknowledge the financial support from the Baden-Württemberg Ministry for Economy, Labour and Housing (project “IDAK” AZ 7-4332.62-HSG/69) and the Queensland Government, Department of Science, Information Technology, Innovation and the Arts (DSITIA, Australia).References
- K. Mullis, F. Faloona, S. Scharf, R. Saiki, G. Horn and H. Erlich, Cold Spring Harbor Symp. Quant. Biol., 1986, 51(Part 1), 263–273 CrossRef CAS PubMed
.
- J. Li and J. Macdonald, Biosens. Bioelectron., 2014, 64C, 196–211 Search PubMed
- P. Craw and W. Balachandran, Lab Chip, 2012, 12, 2469–2486 RSC
- P. Gill and A. Ghaemi, Nucleosides, Nucleotides Nucleic Acids, 2008, 27, 224–243 CrossRef CAS PubMed
- Y. Zhao, F. Chen, Q. Li, L. Wang and C. Fan, Chem. Rev., 2015, 115, 12491–12545 CrossRef CAS PubMed
- O. Piepenburg, C. H. Williams, D. L. Stemple and N. A. Armes, PLoS Biol., 2006, 4, e204 CrossRef PubMed
- Isothermal nucleic acid amplification technology (INAAT) market analysis by product, by technology (NASBA, HDA, LAMP, SDA, SPIA, NEAR, TMA, RCA, RPA, SMAP2), and segment forecasts, 2018–2025, Report GVR-1-68038-588-5, Grand View Research, 2017.
- H. Zaghloul and M. El-Shahat, World J. Hepatol., 2014, 6, 916–922 CrossRef PubMed
- M. D. Moore and L.-A. Jaykus, Future Virol., 2017, 12, 421–429 CrossRef CAS
- A. James and J. Macdonald, Expert Rev. Mol. Diagn., 2015, 15, 1475–1489 CrossRef CAS PubMed
- R. K. Daher, G. Stewart, M. Boissinot and M. G. Bergeron, Clin. Chem., 2016, 62, 947–958 CrossRef CAS PubMed
- I. M. Lobato and C. K. O'Sullivan, TrAC, Trends Anal. Chem., 2018, 98, 19–35 CrossRef CAS
- J. Griffith and T. Formosa, J. Biol. Chem., 1985, 260, 4484–4491 CAS
- M. Bianchi, B. Riboli and G. Magni, EMBO J., 1985, 4, 3025–3030 CrossRef CAS PubMed
- J. M. Boyle and N. Symonds, Mutat. Res., 1969, 8, 431–439 CrossRef CAS PubMed
- Y. Shamoo, A. M. Friedman, M. R. Parsons, W. H. Konigsberg and T. A. Steitz, Nature, 1995, 376, 362–366 CrossRef CAS PubMed
- B. M. Alberts and L. Frey, Nature, 1970, 227, 1313–1318 CrossRef CAS PubMed
- T. Okazaki and A. Kornberg, J. Biol. Chem., 1964, 239, 259–268 CAS
- M. Euler, Y. Wang, P. Otto, H. Tomaso, R. Escudero, P. Anda, F. T. Hufert and M. Weidmann, J. Clin. Microbiol., 2012, 50, 2234–2238 CrossRef CAS PubMed
- RPA Assay Design, https://www.twistdx.co.uk/en/support/rpa-assay-design-2, (accessed Oct 4th, 2018).
- D. Chandu, S. Paul, M. Parker, Y. Dudin, J. King-Sitzes, T. Perez, D. W. Mittanck, M. Shah, K. C. Glenn and O. Piepenburg, BioMed. Res. Int., 2016, 2016, 3145921 Search PubMed
- S. B. Zimmerman and B. Harrison, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 1871–1875 CrossRef CAS
- R. J. Ellis, Trends Biochem. Sci., 2001, 26, 597–604 CrossRef CAS PubMed
- Y. Sasaki, D. Miyoshi and N. Sugimoto, Biotechnol. J., 2006, 1, 440–446 CrossRef CAS PubMed
- S. Fjelstrup, M. B. Andersen, J. Thomsen, J. Wang, M. Stougaard, F. S. Pedersen, Y.-P. Ho, M. S. Hede and B. R. Knudsen, Sensors, 2017, 17, 1201 CrossRef PubMed
- Creatine kinase, https://en.wikipedia.org/wiki/Creatine_kinase, (accessed Oct 4, 2018).
- Tris, https://en.wikipedia.org/wiki/Tris, (accessed Oct 4th, 2018).
- cambio, Lab Reagents: Smart Buffers and Reagents, https://www.cambio.co.uk/24/1283/77/products/potassium-acetate/#tab-2, (accessed July 27th, 2018).
- T. Yonesaki and T. Minagawa, EMBO J., 1985, 4, 3321–3327 CrossRef CAS PubMed
- L. D. Harris and J. Griffith, J. Biol. Chem., 1987, 262, 9285–9292 CAS
- TwistDxLtd., TwistAmp® combined instruction manual, https://www.twistdx.co.uk/docs/default-source/RPA-assay-design/ta01cmanual-combined-manual_revo_v1–3a.pdf?sfvrsn=6, (accessed Oct 4th, 2018).
- M. M. Cox and I. R. Lehman, J. Biol. Chem., 1982, 257, 8523–8532 CAS
- L. D. Harris and J. D. Griffith, J. Mol. Biol., 1989, 206, 19–27 CrossRef CAS PubMed
- T. Yonesaki and T. Minagawa, J. Biol. Chem., 1989, 264, 7814–7820 CAS
- K. Hashimoto and T. Yonesaki, J. Biol. Chem., 1991, 266, 4883–4888 CAS
- M. Euler, Y. Wang, O. Nentwich, O. Piepenburg, F. T. Hufert and M. Weidmann, J. Clin. Virol., 2012, 54, 308–312 CrossRef CAS PubMed
- S. Martorell, S. Palanca, A. Maquieira and L. A. Tortajada-Genaro, Anal. Biochem., 2017, 544, 49–56 CrossRef PubMed
- M. Jauset-Rubio, J. Sabate Del Rio, T. Mairal, M. Svobodova, M. S. El-Shahawi, A. S. Bashammakh, A. O. Alyoubi and C. K. O'Sullivan, Anal. Bioanal. Chem., 2017, 409, 143–149 CrossRef CAS PubMed
- M. Jauset-Rubio, M. Svobodova, T. Mairal, C. McNeil, N. Keegan, M. S. El-Shahawi, A. S. Bashammakh, A. O. Alyoubi and C. K. O'Sullivan, Anal. Chem., 2016, 88, 10701–10709 CrossRef CAS PubMed
- M. Jauset-Rubio, M. Svobodova, T. Mairal, C. McNeil, N. Keegan, A. Saeed, M. N. Abbas, M. S. El-Shahawi, A. S. Bashammakh, A. O. Alyoubi and O. S. CK, Sci. Rep., 2016, 6, 37732 CrossRef CAS PubMed
- J. Wang, J. Wang, Y. Geng and W. Yuan, Can. J. Vet. Res., 2017, 81, 308–312 Search PubMed
- TwistAmp® manuals, https://www.twistdx.co.uk/en/support/manuals/twistamp-manuals, (accessed Oct 4th, 2018).
- O. Mayboroda, A. Gonzalez Benito, J. Sabate del Rio, M. Svobodova, S. Julich, H. Tomaso, C. K. O'Sullivan and I. Katakis, Anal. Bioanal. Chem., 2016, 408, 671–676 CrossRef CAS PubMed
- R. Wang, F. Zhang, L. Wang, W. Qian, C. Qian, J. Wu and Y. Ying, Anal. Chem., 2017, 89, 4413–4418 CrossRef CAS PubMed
- S. L. Fuller, E. A. Savory, A. J. Weisberg, J. Z. Buser, M. I. Gordon, M. L. Putnam and J. H. Chang, Phytopathology, 2017, 107, 1062–1068 CrossRef PubMed
-
O. Piepenburg and N. A. Armes, DNA glycosylase/lyase and AP endonuclease substrates, Alere San Diego, Inc., US20110053153 A1, 2011
- L. D. Renner, J. Zan, L. I. Hu, M. Martinez, P. J. Resto, A. C. Siegel, C. Torres, S. B. Hall, T. R. Slezak, T. H. Nguyen and D. B. Weibel, Appl. Environ. Microbiol., 2017, 83, e02449–e02416 CrossRef PubMed
-
T. A. Hall, BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT, Nucleic acids symposium series, 1999, vol. 41, pp. 95–98 Search PubMed
- T. Koressaar and M. Remm, Bioinformatics, 2007, 23, 1289–1291 CrossRef CAS PubMed
- N. R. Markham and M. Zuker, Methods Mol. Biol., 2008, 453, 3–31 CrossRef CAS PubMed
- M. Zuker, Nucleic Acids Res., 2003, 31, 3406–3415 CrossRef CAS PubMed
- S. Hoshika, F. Chen, N. A. Leal and S. A. Benner, Nucleic Acids Symp. Ser., 2008, 129–130 CrossRef CAS PubMed
- N. Sharma, S. Hoshika, D. Hutter, K. M. Bradley and S. A. Benner, ChemBioChem, 2014, 15, 2268–2274 CrossRef CAS PubMed
- Y. Yang, X. Qin, W. Zhang, Y. Li and Z. Zhang, Mol. Cell. Probes, 2016, 30, 300–305 CrossRef CAS PubMed
- X. Xia, Y. Yu, L. Hu, M. Weidmann, Y. Pan, S. Yan and Y. Wang, Arch. Virol., 2015, 160, 987–994 CrossRef CAS PubMed
- TwistDxLtd., TwistDx, https://www.twistdx.co.uk, (accessed Oct 4th, 2018).
- L. Lillis, J. Siverson, A. Lee, J. Cantera, M. Parker, O. Piepenburg, D. A. Lehman and D. S. Boyle, Mol. Cell. Probes, 2016, 30, 74–78 CrossRef CAS PubMed
- TwistDxLtd., Custom freeze-drying, https://www.twistdx.co.uk/en/products/custom-freeze-drying, (accessed May 3rd, 2018).
- G. Posthuma-Trumpie, J. Wichers, M. Koets, L. J. M. Berendsen and A. Amerongen, Anal. Bioanal. Chem., 2012, 402, 593–600 CrossRef CAS PubMed
- K. Poulton and B. Webster, Anal. Biochem., 2018, 546, 65–71 CrossRef CAS PubMed
- Z. A. Crannell, B. Rohrman and R. Richards-Kortum, PLoS One, 2014, 9, e112146 CrossRef PubMed
- S. Kersting, V. Rausch, F. F. Bier and M. von Nickisch-Rosenegk, Malar. J., 2014, 13, 99 CrossRef PubMed
- L. Lillis, D. Lehman, M. C. Singhal, J. Cantera, J. Singleton, P. Labarre, A. Toyama, O. Piepenburg, M. Parker, R. Wood, J. Overbaugh and D. S. Boyle, PLoS One, 2014, 9, e108189 CrossRef PubMed
- Y. Yang, X. Qin, G. Wang, J. Jin, Y. Shang and Z. Zhang, Virol. J., 2016, 13, 46 CrossRef PubMed
- K. Sun, W. Xing, X. Yu, W. Fu, Y. Wang, M. Zou, Z. Luo and D. Xu, Parasites Vectors, 2016, 9, 476 CrossRef PubMed
- M. A. Prescott, A. N. Reed, L. Jin and M. K. Pastey, J. Aquat. Anim. Health, 2016, 28, 173–180 CrossRef CAS PubMed
- F. Yin, J. Liu, A. Liu, Y. Li, J. Luo, G. Guan and H. Yin, Vet. Parasitol., 2017, 237, 125–129 CrossRef CAS PubMed
- J. S. Del Rio, I. M. Lobato, O. Mayboroda, I. Katakis and C. K. O'Sullivan, Anal. Bioanal. Chem., 2017, 409, 3261–3269 CrossRef CAS PubMed
- Y. Yang, X. Qin, Y. Sun, G. Cong, Y. Li and Z. Zhang, BioMed. Res. Int., 2017, 2017, 8403642 Search PubMed
- Y. Yang, X. Qin, W. Zhang, Z. Li, S. Zhang, Y. Li and Z. Zhang, Mol. Cell. Probes, 2017, 33, 32–35 CrossRef CAS PubMed
- Y. D. Wu, M. J. Xu, Q. Q. Wang, C. X. Zhou, M. Wang, X. Q. Zhu and D. H. Zhou, Vet. Parasitol., 2017, 243, 199–203 CrossRef CAS PubMed
- N. Valasevich and B. Schneider, J. Phytopathol., 2017, 165, 762–770 CrossRef CAS
- E. Tian, Q. Liu, H. Ye, F. Li and Z. Chao, Molecules, 2017, 22, 2261–2270 CrossRef PubMed
- P. Hou, G. Zhao, H. Wang, C. He, Y. Huan and H. He, Mol. Cell. Probes, 2018, 38, 31–37 CrossRef CAS PubMed
- M. Y. Lai, C. H. Ooi and Y. L. Lau, Am. J. Trop. Med. Hyg., 2018, 98, 700–703 CrossRef CAS PubMed
- Z. Guimin, W. Hongmei, H. Peili, H. Chengqiang and H. Hongbin, J. Vet. Sci., 2017, 19, 242–250 Search PubMed
- G. Zhao, H. Wang, P. Hou, C. He and H. He, J. Vet. Sci., 2018, 19, 242–250 CrossRef PubMed
- H. Soliman and M. El-Matbouli, J. Fish Dis., 2018, 41, 761–772 CrossRef CAS PubMed
- L. Wambua, B. Schneider, A. Okwaro, J. O. Wanga, O. Imali, P. N. Wambua, L. Agutu, C. Olds, C. S. Jones, D. Masiga, C. Midega, Z. Khan, J. Jores and A. Fischer, Mol. Cell. Probes, 2017, 35, 44–56 CrossRef CAS PubMed
- S. Kalsi, M. Valiadi, M. N. Tsaloglou, L. Parry-Jones, A. Jacobs, R. Watson, C. Turner, R. Amos, B. Hadwen, J. Buse, C. Brown, M. Sutton and H. Morgan, Lab Chip, 2015, 15, 3065–3075 RSC
- C. Moody, H. Newell and H. Viljoen, Biochem. Eng. J., 2016, 112, 193–201 CrossRef CAS
- D. S. Boyle, D. A. Lehman, L. Lillis, D. Peterson, M. Singhal, N. Armes, M. Parker, O. Piepenburg and J. Overbaugh, mBio, 2013, 4, e00135-13 CrossRef PubMed
- A. Abd El Wahed, A. El-Deeb, M. El-Tholoth, H. Abd El Kader, A. Ahmed, S. Hassan, B. Hoffmann, B. Haas, M. A. Shalaby, F. T. Hufert and M. Weidmann, PLoS One, 2013, 8, e71642 CrossRef CAS PubMed
- R. K. Daher, G. Stewart, M. Boissinot, D. K. Boudreau and M. G. Bergeron, Mol. Cell. Probes, 2015, 29, 116–121 CrossRef CAS PubMed
- N. Yehia, A. S. Arafa, A. Abd El Wahed, A. A. El-Sanousi, M. Weidmann and M. A. Shalaby, J. Virol. Methods, 2015, 223, 45–49 CrossRef CAS PubMed
- L. Lillis, D. A. Lehman, J. B. Siverson, J. Weis, J. Cantera, M. Parker, O. Piepenburg, J. Overbaugh and D. S. Boyle, J. Virol. Methods, 2016, 230, 28–35 CrossRef CAS PubMed
- P. Patel, A. Abd El Wahed, O. Faye, P. Pruger, M. Kaiser, S. Thaloengsok, S. Ubol, A. Sakuntabhai, I. Leparc-Goffart, F. T. Hufert, A. A. Sall, M. Weidmann and M. Niedrig, PLoS Neglected Trop. Dis., 2016, 10, e0004953 CrossRef PubMed
- M. D. Moore and L. A. Jaykus, Sci. Rep., 2017, 7, 40244 CrossRef CAS PubMed
- J. Kissenkötter, S. Hansen, S. Böhlken-Fascher, O. G. Ademowo, O. E. Oyinloye, A. S. Bakarey, G. Dobler, D. Tappe, P. Patel, C.-P. Czerny and A. Abd El Wahed, Anal. Biochem., 2017, 544, 29–33 CrossRef PubMed
- E. S. Yamanaka, L. A. Tortajada-Genaro and Á. Maquieira, Microchim. Acta, 2017, 184, 1453–1462 CrossRef CAS
- A. Rosser, D. Rollinson, M. Forrest and B. L. Webster, Parasites Vectors, 2015, 8, 446 CrossRef CAS PubMed
- K. Krolov, J. Frolova, O. Tudoran, J. Suhorutsenko, T. Lehto, H. Sibul, I. Mager, M. Laanpere, I. Tulp and U. Langel, J. Mol. Diagn., 2014, 16, 127–135 CrossRef CAS PubMed
- H. B. Liu, Y. X. Zang, X. J. Du, P. Li and S. Wang, J. Dairy Sci., 2017, 100, 7016–7025 CrossRef CAS PubMed
- B. Rohrman and R. Richards-Kortum, Anal. Chem., 2015, 87, 1963–1967 CrossRef CAS PubMed
- C. C. Chao, T. Belinskaya, Z. Zhang and W. M. Ching, PLoS Neglected Trop. Dis., 2015, 9, e0003884 CrossRef PubMed
- B. Y. C. Ng, E. J. H. Wee, N. P. West and M. Trau, Sci. Rep., 2015, 5, 15027 CrossRef CAS
- E. Clancy, O. Higgins, M. S. Forrest, T. W. Boo, M. Cormican, T. Barry, O. Piepenburg and T. J. Smith, BMC Infect. Dis., 2015, 15, 481 CrossRef PubMed
- H. Y. Lau, Y. Wang, E. J. Wee, J. R. Botella and M. Trau, Anal. Chem., 2016, 88, 8074–8081 CrossRef CAS PubMed
- K. M. Koo, E. J. Wee, P. N. Mainwaring, Y. Wang and M. Trau, Small, 2016, 12, 6233–6242 CrossRef CAS PubMed
- H. Y. Lau, H. Wu, E. J. Wee, M. Trau, Y. Wang and J. R. Botella, Sci. Rep., 2017, 7, 38896 CrossRef CAS PubMed
- K. M. Koo, E. J. Wee and M. Trau, Theranostics, 2016, 6, 1415–1424 CrossRef CAS PubMed
- B. Y. C. Ng, E. J. H. Wee, N. P. West and M. Trau, ACS Sens., 2016, 1, 173–178 CrossRef CAS
- B. Y. Ng, W. Xiao, N. P. West, E. J. Wee, Y. Wang and M. Trau, Anal. Chem., 2015, 87, 10613–10618 CrossRef CAS PubMed
- S. Santiago-Felipe, L. A. Tortajada-Genaro, R. Puchades and A. Maquieira, Anal. Chim. Acta, 2014, 811, 81–87 CrossRef CAS PubMed
- Z. Crannell, A. Castellanos-Gonzalez, G. Nair, R. Mejia, A. C. White and R. Richards-Kortum, Anal. Chem., 2016, 88, 1610–1616 CrossRef CAS PubMed
- M. S. Cordray and R. R. Richards-Kortum, Malar. J., 2015, 14, 472 CrossRef PubMed
- J. Wang, L. Liu, R. Li, J. Wang, Q. Fu and W. Yuan, Arch. Virol., 2016, 161, 1015–1018 CrossRef CAS PubMed
- P. A. Tu, J. S. Shiu, S. H. Lee, V. F. Pang, D. C. Wang and P. H. Wang, J. Virol. Methods, 2017, 243, 98–104 CrossRef CAS PubMed
- L. Glais and E. Jacquot, Methods Mol. Biol., 2015, 1302, 207–225 CrossRef PubMed
- B. Babu, B. K. Washburn, S. H. Miller, K. Poduch, T. Sarigul, G. W. Knox, F. M. Ochoa-Corona and M. L. Paret, J. Virol. Methods, 2017, 240, 78–84 CrossRef CAS PubMed
- M. A. Londono, C. L. Harmon and J. E. Polston, Virol. J., 2016, 13, 48 CrossRef PubMed
- R. Kapoor, N. Srivastava, S. Kumar, R. K. Saritha, S. K. Sharma, R. K. Jain and V. K. Baranwal, Arch. Virol., 2017, 162, 2791–2796 CrossRef CAS PubMed
- Q. Liu, B. K. L. Lim, S. Y. Lim, W. Y. Tang, Z. Gu, J. Chung, M. K. Park and T. Barkham, Sens. Actuators, B, 2018, 255, 1595–1603 CrossRef CAS
- M. L. Powell, F. R. Bowler, A. J. Martinez, C. J. Greenwood, N. Armes and O. Piepenburg, Anal. Biochem., 2017, 543, 108–115 CrossRef PubMed
- O. A. Saldarriaga, A. Castellanos-Gonzalez, R. Porrozzi, G. C. Baldeviano, A. G. Lescano, M. B. de Los Santos, O. L. Fernandez, N. G. Saravia, E. Costa, P. C. Melby and B. L. Travi, PLoS Neglected Trop. Dis., 2016, 10, e0004638 CrossRef PubMed
- Y. D. Wu, D. H. Zhou, L. X. Zhang, W. B. Zheng, J. G. Ma, M. Wang, X. Q. Zhu and M. J. Xu, Parasitol. Res., 2016, 115, 3551–3555 CrossRef PubMed
- C. Escadafal, O. Faye, A. A. Sall, O. Faye, M. Weidmann, O. Strohmeier, F. von Stetten, J. Drexler, M. Eberhard, M. Niedrig and P. Patel, PLoS Neglected Trop. Dis., 2014, 8, e2730 CrossRef PubMed
- A. J. Saah and D. R. Hoover, Ann. Intern. Med., 1997, 126, 91–94 CrossRef CAS PubMed
- J. Y. Kim and J.-L. Lee, J. Food Saf., 2016, 36, 402–411 CrossRef CAS
- Q. Liu, J. Nam, S. Kim, C. T. Lim, M. K. Park and Y. Shin, Biosens. Bioelectron., 2016, 82, 1–8 CrossRef PubMed
- D. Mondal, P. Ghosh, M. A. Khan, F. Hossain, S. Bohlken-Fascher, G. Matlashewski, A. Kroeger, P. Olliaro and A. Abd El Wahed, Parasites Vectors, 2016, 9, 281 CrossRef PubMed
- J. Y. Kim and J.-L. Lee, Food Control, 2017, 73(Part B), 1247–1255 CrossRef CAS
- J. S. Gootenberg, O. O. Abudayyeh, J. W. Lee, P. Essletzbichler, A. J. Dy, J. Joung, V. Verdine, N. Donghia, N. M. Daringer, C. A. Freije, C. Myhrvold, R. P. Bhattacharyya, J. Livny, A. Regev, E. V. Koonin, D. T. Hung, P. C. Sabeti, J. J. Collins and F. Zhang, Science, 2017, 356, 438–442 CrossRef CAS PubMed
- M. Y. Lai, C. H. Ooi and Y. L. Lau, Am. J. Trop. Med. Hyg., 2017, 97, 1597–1599 CrossRef CAS PubMed
- H. M. Amer, A. Abd El Wahed, M. A. Shalaby, F. N. Almajhdi, F. T. Hufert and M. Weidmann, J. Virol. Methods, 2013, 193, 337–340 CrossRef CAS PubMed
- R. K. Daher, G. Stewart, M. Boissinot and M. G. Bergeron, Clin. Chem., 2014, 60, 660–666 CrossRef CAS PubMed
- Z. A. Crannell, A. Castellanos-Gonzalez, A. Irani, B. Rohrman, A. C. White and R. Richards-Kortum, Anal. Chem., 2014, 86, 2565–2571 CrossRef CAS PubMed
- D. S. Boyle, R. McNerney, H. Teng Low, B. T. Leader, A. C. Perez-Osorio, J. C. Meyer, D. M. O'Sullivan, D. G. Brooks, O. Piepenburg and M. S. Forrest, PLoS One, 2014, 9, e103091 CrossRef PubMed
- Z. A. Crannell, M. M. Cabada, A. Castellanos-Gonzalez, A. Irani, A. C. White and R. Richards-Kortum, Am. J. Trop. Med. Hyg., 2015, 92, 583–587 CrossRef CAS PubMed
- Y. Shin, A. P. Perera, W. Y. Tang, D. L. Fu, Q. Liu, J. K. Sheng, Z. Gu, T. Y. Lee, T. Barkham and M. Kyoung Park, Biosens. Bioelectron., 2015, 68C, 390–396 CrossRef PubMed
- A. Abd El Wahed, P. Patel, O. Faye, S. Thaloengsok, D. Heidenreich, P. Matangkasombut, K. Manopwisedjaroen, A. Sakuntabhai, A. A. Sall, F. T. Hufert and M. Weidmann, PLoS One, 2015, 10, e0129682 CrossRef PubMed
- G. Nair, M. Rebolledo, A. C. White Jr., Z. Crannell, R. R. Richards-Kortum, A. E. Pinilla, J. D. Ramirez, M. C. Lopez and A. Castellanos-Gonzalez, Am. J. Trop. Med. Hyg., 2015, 93, 591–595 CrossRef CAS PubMed
- S. A. Ahmed, W. W. van de Sande, M. Desnos-Ollivier, A. H. Fahal, N. A. Mhmoud and G. S. de Hoog, J. Clin. Microbiol., 2015, 53, 3280–3285 CrossRef CAS PubMed
- O. Faye, O. Faye, B. Soropogui, P. Patel, A. A. El Wahed, C. Loucoubar, G. Fall, D. Kiory, N. Magassouba, S. Keita, M. K. Konde, A. A. Diallo, L. Koivogui, H. Karlberg, A. Mirazimi, O. Nentwich, O. Piepenburg, M. Niedrig, M. Weidmann and A. A. Sall, Euro. Surveill., 2015, 20, 10–18 Search PubMed
- Y. Yang, X. Qin, G. Wang, Y. Zhang, Y. Shang and Z. Zhang, Virol. J., 2015, 12, 206 CrossRef PubMed
- Y. Yang, X. Qin, Y. Sun, T. Chen and Z. Zhang, Virus Genes, 2016, 52, 883–886 CrossRef CAS PubMed
- C. Clarke, L. O'Connor, H. Carre-Skinner, O. Piepenburg and T. J. Smith, BMC Microbiol., 2016, 16, 221–226 CrossRef PubMed
- A. Abd El Wahed, S. S. Sanabani, O. Faye, R. Pessoa, J. V. Patriota, R. R. Giorgi, P. Patel, S. Bohlken-Fascher, O. Landt, M. Niedrig, P. M. Zanotto, C. P. Czerny, A. A. Sall and M. Weidmann, PLoS Curr., 2017, 9, 1–10 Search PubMed
- M. A. Shalaby, A. El-Deeb, M. El-Tholoth, D. Hoffmann, C. P. Czerny, F. T. Hufert, M. Weidmann and A. Abd El Wahed, BMC Vet. Res., 2016, 12, 244 CrossRef PubMed
- S. Hansen, J. Schafer, K. Fechner, C. P. Czerny and A. Abd El Wahed, PLoS One, 2016, 11, e0168733 CrossRef PubMed
- K. M. Koo, E. J. H. Wee and M. Trau, Biosens. Bioelectron., 2017, 89, 715–720 CrossRef CAS PubMed
- J. C. Wang, W. Z. Yuan, Q. A. Han, J. F. Wang and L. B. Liu, J. Virol. Methods, 2017, 243, 55–60 CrossRef CAS PubMed
- L. Liu, L. Jiang, Y. Yu, X. Xia, Y. Pan, S. Yan and Y. Wang, Mol. Cell. Probes, 2017, 33, 4–7 CrossRef CAS PubMed
- M. M. Cabada, J. L. Malaga, A. Castellanos-Gonzalez, K. A. Bagwell, P. A. Naeger, H. K. Rogers, S. Maharsi, M. Mbaka and A. C. White Jr., Am. J. Trop. Med. Hyg., 2016, 96, 341–346 CrossRef PubMed
- Y. Yang, X. Qin, Y. Song, W. Zhang, G. Hu, Y. Dou, Y. Li and Z. Zhang, Virol. J., 2017, 14, 24 CrossRef PubMed
- M. Si Ammour, G. J. Bilodeau, D. M. Tremblay, H. Van der Heyden, T. Yaseen, L. Varvaro and O. Carisse, Plant Dis., 2017, 101, 1269–1277 CrossRef
- J. C. Wang, L. B. Liu, Q. A. Han, J. F. Wang and W. Z. Yuan, J. Virol. Methods, 2017, 248, 145–147 CrossRef CAS PubMed
- Y. Yang, X. Qin, X. Zhang, Z. Zhao, W. Zhang, X. Zhu, G. Cong, Y. Li and Z. Zhang, Virol. J., 2017, 14, 131 CrossRef PubMed
- J. Wang, J. Wang, R. Li, L. Liu and W. Yuan, BMC Vet. Res., 2017, 13, 241 CrossRef PubMed
- K. Schlottau, C. M. Freuling, T. Muller, M. Beer and B. Hoffmann, Virol. J., 2017, 14, 184 CrossRef PubMed
- C. E. Jin, S. S. Yeom, B. Koo, T. Y. Lee, J. H. Lee, Y. Shin and S. B. Lim, Oncotarget, 2017, 8, 83860–83871 Search PubMed
- L. C. Bonney, R. J. Watson, B. Afrough, M. Mullojonova, V. Dzhuraeva, F. Tishkova and R. Hewson, PLoS Neglected Trop. Dis., 2017, 11, e0006013 CrossRef PubMed
- Y. Geng, J. Wang, L. Liu, Y. Lu, K. Tan and Y.-Z. Chang, BMC Vet. Res., 2017, 13, 311 CrossRef PubMed
- K. Shahin, J. Gustavo Ramirez-Paredes, G. Harold, B. Lopez-Jimena, A. Adams and M. Weidmann, PLoS One, 2018, 13, e0192979 CrossRef PubMed
- D. Yin, Y. Zhu, K. Wang, J. Wang, X. Zhang, M. Han, Y. He, Q. Chen and G. Hu, Arch. Virol., 2018, 163, 2459–2463 CrossRef CAS PubMed
- Y. Qi, Q. Yin, Y. Shao, M. Cao, S. Li, H. Chen, W. Shen, J. Rao, J. Li, X. Li, Y. Sun, Y. Lin, Y. Deng, W. Zeng, S. Zheng, S. Liu and Y. Li, Int. J. Infect. Dis., 2018, 70, 42–50 CrossRef CAS PubMed
- Y. Qi, Q. Yin, Y. Shao, S. Li, H. Chen, W. Shen, J. Rao, J. Li, X. Li, Y. Sun, Y. Lin, Y. Deng, W. Zeng, S. Zheng, S. Liu and Y. Li, BioMed Res. Int., 2018, 2018, 1–10 CrossRef PubMed
- R. A. Ruehrwein and D. W. Ward, Soil Sci., 1952, 73, 485–492 CrossRef CAS
- V. K. La Mer, Discuss. Faraday Soc., 1966, 42, 248–254 RSC
- T. W. Healy and V. K. La Mer, J. Colloid Sci., 1964, 19, 323–332 CrossRef CAS
- R. H. Smellie and V. K. La Mer, J. Colloid Sci., 1958, 13, 589–599 CrossRef CAS
- E. J. Wee, H. Y. Lau, J. R. Botella and M. Trau, Chem. Commun., 2015, 51, 5828–5831 RSC
- E. J. Wee, T. Ha Ngo and M. Trau, Sci. Rep., 2015, 5, 15028 CrossRef CAS PubMed
- K. M. Koo, E. J. Wee, P. N. Mainwaring and M. Trau, Sci. Rep., 2016, 6, 30722 CrossRef CAS PubMed
- E. J. H. Wee, T. H. Ngo and M. Trau, Clin. Epigenet., 2015, 7, 1–9 CrossRef PubMed
- J. S. Del Rio, N. Yehia Adly, J. L. Acero-Sanchez, O. Y. Henry and C. K. O'Sullivan, Biosens. Bioelectron., 2014, 54, 674–678 CrossRef CAS PubMed
- J. S. Del Rio, M. Svobodova, P. Bustos, P. Conejeros and C. K. O'Sullivan, Anal. Bioanal. Chem., 2016, 408, 8611–8620 CrossRef CAS PubMed
- M. N. Tsaloglou, A. Nemiroski, G. Camci-Unal, D. C. Christodouleas, L. P. Murray, J. T. Connelly and G. M. Whitesides, Anal. Biochem., 2017, 543, 116–121 CrossRef PubMed
-
R. E. Farrell, in RNA Methodologies, ed. R. E. Farrell, Academic Press, San Diego, 4th edn, 2010, ch. 14, pp. 301–320 Search PubMed
- A. Kunze, M. Dilcher, A. Abd El Wahed, F. Hufert, R. Niessner and M. Seidel, Anal. Chem., 2016, 88, 898–905 CrossRef CAS PubMed
- C. Kober, R. Niessner and M. Seidel, Biosens. Bioelectron., 2018, 100, 49–55 CrossRef CAS PubMed
- A. Heim, C. Ebnet, G. Harste and P. Pring-Akerblom, J. Med. Virol., 2003, 70, 228–239 CrossRef CAS PubMed
- W. Bogaerts, P. De Heyn, T. Van Vaerenbergh, K. De Vos, S. K. Selvaraja, T. Claes, P. Dumon, P. Bienstman, D. Van Thourhout and R. Baets, Laser Photonics Rev., 2012, 6, 47–73 CrossRef CAS
- B. Koo, C. E. Jin, S. Y. Park, T. Y. Lee, J. Nam, Y. R. Jang, S. M. Kim, J. Y. Kim, S. H. Kim and Y. Shin, J. Biophotonics, 2017, 11, e201700167 CrossRef PubMed
- Y. Shin, A. P. Perera, K. W. Kim and M. K. Park, Lab Chip, 2013, 13, 2106–2114 RSC
- J. Sabaté del Río, T. Steylaerts, O. Y. Henry, P. Bienstman, T. Stakenborg, W. Van Roy and C. K. O'Sullivan, Biosens. Bioelectron., 2015, 73, 130–137 CrossRef PubMed
- T. N. T. Dao, E. Y. Lee, B. Koo, C. E. Jin, T. Y. Lee and Y. Shin, Anal. Biochem., 2017, 544, 87–92 CrossRef PubMed
- S. Schlucker, Angew. Chem., Int. Ed., 2014, 53, 4756–4795 CrossRef PubMed
- J. Wang, K. M. Koo, E. J. Wee, Y. Wang and M. Trau, Nanoscale, 2017, 9, 3496–3503 RSC
- F. Shen, E. K. Davydova, W. Du, J. E. Kreutz, O. Piepenburg and R. F. Ismagilov, Anal. Chem., 2011, 83, 3533–3540 CrossRef CAS PubMed
- M. N. Tsaloglou, R. J. Watson, C. M. Rushworth, Y. Zhao, X. Niu, J. M. Sutton and H. Morgan, Analyst, 2015, 140, 258–264 RSC
-
E.-C. Yeh and L. P. Lee, Presented in part at the 17th International Conference on Miniaturised Systems for Chemistry and Life Sciences, Freiburg, Germany, 27–31 October, 2013
- E.-C. Yeh, C.-C. Fu, L. Hu, R. Thakur, J. Feng and L. P. Lee, Sci. Adv., 2017, 3, 1–11 Search PubMed
- Z. Li, Y. Liu, Q. Wei, Y. Liu, W. Liu, X. Zhang and Y. Yu, PLoS One, 2016, 11, e0153359 CrossRef PubMed
- F. Schuler, F. Schwemmer, M. Trotter, S. Wadle, R. Zengerle, F. von Stetten and N. Paust, Lab Chip, 2015, 15, 2759–2766 RSC
- C. J. DeJournette, J. Kim, H. Medlen, X. Li, L. J. Vincent and C. J. Easley, Anal. Chem., 2013, 85, 10556–10564 CrossRef CAS PubMed
- J. Wang, L. Liu, J. Wang, X. Pang and W. Yuan, Anal. Biochem., 2017, 543, 122–127 CrossRef PubMed
- S. Santiago-Felipe, L. A. Tortajada-Genaro, S. Morais, R. Puchades and A. Maquieira, Food Chem., 2015, 174, 509–515 CrossRef CAS PubMed
- H. B. Liu, X. J. Du, Y. X. Zang, P. Li and S. Wang, J. Agric. Food Chem., 2017, 65, 10290–10299 CrossRef CAS PubMed
- J. Song, C. Liu, M. G. Mauk, S. C. Rankin, J. B. Lok, R. M. Greenberg and H. H. Bau, Clin. Chem., 2017, 63, 714–722 CrossRef CAS PubMed
- S. Santiago-Felipe, L. A. Tortajada-Genaro, S. Morais, R. Puchades and Á. Maquieira, Sens. Actuators, B, 2014, 204, 273–281 CrossRef CAS
- S. Kalsi, S. L. Sellars, C. Turner, J. M. Sutton and H. Morgan, Micromachines, 2017, 8, 1–12 CrossRef
- L. Magro, B. Jacquelin, C. Escadafal, P. Garneret, A. Kwasiborski, J. C. Manuguerra, F. Monti, A. Sakuntabhai, J. Vanhomwegen, P. Lafaye and P. Tabeling, Sci. Rep., 2017, 7, 1347 CrossRef PubMed
- K. Ming, J. Kim, M. J. Biondi, A. Syed, K. Chen, A. Lam, M. Ostrowski, A. Rebbapragada, J. J. Feld and W. C. Chan, ACS Nano, 2015, 9, 3060–3074 CrossRef CAS PubMed
- J. Kim, M. J. Biondi, J. J. Feld and W. C. W. Chan, ACS Nano, 2016, 10, 4742–4753 CrossRef CAS PubMed
- S. Kersting, V. Rausch, F. F. Bier and M. von Nickisch-Rosenegk, Mikrochim. Acta, 2014, 181, 1715–1723 CrossRef CAS PubMed
- A. Abd El Wahed, M. Weidmann and F. T. Hufert, J. Clin. Virol., 2015, 69, 16–21 CrossRef CAS PubMed
- M. Branavan, R. E. Mackay, P. Craw, A. Naveenathayalan, J. C. Ahern, T. Sivanesan, C. Hudson, T. Stead, J. Kremer, N. Garg, M. Baker, S. T. Sadiq and W. Balachandran, Med. Eng. Phys., 2016, 38, 741–748 CrossRef PubMed
- C. Hu, S. Kalsi, I. Zeimpekis, K. Sun, P. Ashburn, C. Turner, J. M. Sutton and H. Morgan, Biosens. Bioelectron., 2017, 96, 281–287 CrossRef CAS PubMed
- S. Lutz, P. Weber, M. Focke, B. Faltin, J. Hoffmann, C. Muller, D. Mark, G. Roth, P. Munday, N. Armes, O. Piepenburg, R. Zengerle and F. von Stetten, Lab Chip, 2010, 10, 887–893 RSC
- O. Strohmeier, B. Kanat, D. Bär, P. Patel, J. Drexler, M. Weidmann, T. v. Oordt, G. Roth, D. Mark, R. Zengerle and F. v. Stetten, Presented in part at the MicroTAS 2012, Okinawa, Japan, Oct. 28th to Nov. 1st 2012, 2012.
- T. H. Kim, J. Park, C. J. Kim and Y. K. Cho, Anal. Chem., 2014, 86, 3841–3848 CrossRef CAS PubMed
- G. Silva, J. Oyekanmi, C. K. Nkere, M. Bomer, P. L. Kumar and S. E. Seal, Anal. Biochem., 2018, 546, 17–22 CrossRef CAS PubMed
- G. Choi, J. H. Jung, B. H. Park, S. J. Oh, J. H. Seo, J. S. Choi, H. Kim do and T. S. Seo, Lab Chip, 2016, 16, 2309–2316 RSC
- G. Choi, J. H. Jung, B. H. Park, S. J. Oh and T. S. Seo, Presented in part at the 19th International Conference on Miniaturized Systems for Chemistry and Life Sciences, Gyeongju, Korea, October 25–29, 2015.
| This journal is © The Royal Society of Chemistry 2019 |