
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRuthenium-catalyzed umpolung carboxylation of hydrazones with CO2†
Si-Shun
Yan
a,
Lei
Zhu
b,
Jian-Heng
Ye
a,
Zhen
Zhang
a,
He
Huang
a,
Huiying
Zeng
 c,
Chao-Jun
Li
*cd,
Yu
Lan
*b and
Da-Gang
Yu
*ae
c,
Chao-Jun
Li
*cd,
Yu
Lan
*b and
Da-Gang
Yu
*ae
aKey Laboratory of Green Chemistry & Technology of Ministry of Education, College of Chemistry, Sichuan University, 29 Wangjiang Road, Chengdu 610064, P. R. China. E-mail: dgyu@scu.edu.cn
bSchool of Chemistry and Chemical Engineering, Chongqing University, Chongqing 400030, P. R. China. E-mail: lanyu@cqu.edu.cn
cThe State Key Laboratory of Applied Organic Chemistry, Lanzhou University, Lanzhou 730000, P. R. China
dDepartment of Chemistry and FQRNT Centre for Green Chemistry and Catalysis, McGill University, 801 Sherbrooke St. W., Montreal, Quebec H3A 0B8, Canada. E-mail: cj.li@mcgill.ca
eState Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, P. R. China
First published on 30th April 2018
Abstract
The first ruthenium-catalyzed umpolung carboxylation of hydrazones with CO2 to generate important aryl acetic acids is reported. Besides aldehyde hydrazones, a variety of ketone hydrazones, which have not been successfully applied in previous umpolung reactions with other reactive electrophiles, also show high reactivity and selectivity under mild conditions. Moreover, this operationally simple protocol features good functional group tolerance, is readily scalable, and offers easy derivation of important structures, including bioactive felbinac and adiphenine. Computational studies reveal that this umpolung reaction proceeds through the generation of a Ru-nitrenoid followed by concerted [4 + 2] cycloaddition with CO2.
Introduction
Carbon dioxide (CO2) has attracted significant attention as an ideal C1 source due to its high abundance, low cost, low toxicity and renewability.1 In numerous chemical transformations involving CO2,2 the synthesis of carboxylic acids through C–C bond formation is highly promising.3–5 Given that aryl acetic acids are present in myriad natural products, agrochemicals and drugs (Fig. 1),6 great effort has been devoted to the synthesis of this motif following various strategies.7,8 Notably, the transition metal-catalyzed reductive carboxylation of benzyl (pseudo)halides with CO2 has been well documented by Martin and He (Scheme 1a).8 However, the Ru-catalyzed carboxylation with CO2 has rarely been investigated and very limited success has been achieved.9 Herein, we report a novel Ru-catalyzed umpolung carboxylation of hydrazones with CO2 under mild conditions (Scheme 1b). This reaction features good functional group tolerance, high selectivity, broad substrate scope and facile scalability.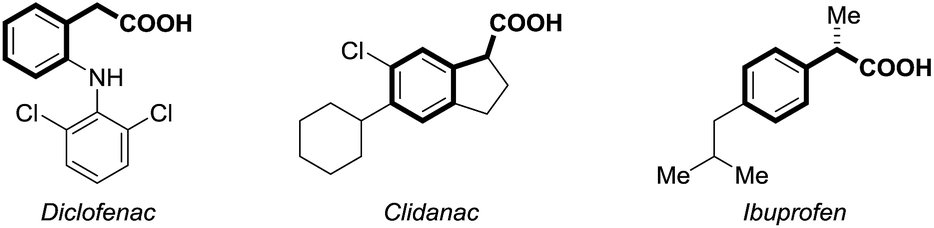 | ||
| Fig. 1 Aryl acetic acid-containing drugs. | ||
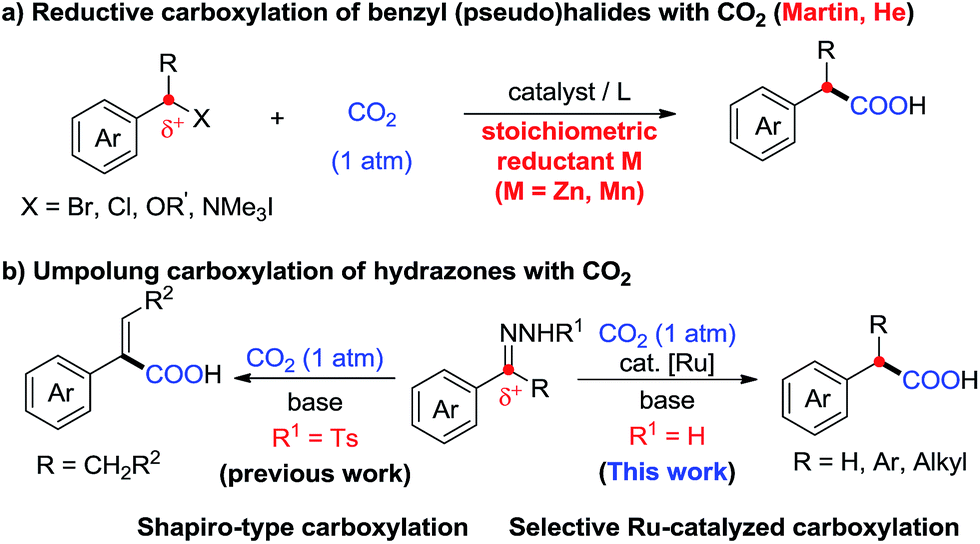 | ||
| Scheme 1 Synthesis of aryl acetic acids with CO2. | ||
An umpolung strategy creates new reactivity by reversing the inherent polarity of common functional groups and consequently allowing for new reactions with distinct bond formations.10 Recently, transition-metal-free umpolung carboxylations of imines and derivatives with CO2 have been well developed by Sato, Radosevich and Zhang to generate α-amino acids.11 In 2015, an elegant base-promoted Shapiro-type carboxylation of N-tosylhydrazones to generate acrylic acids was reported by Cheng (Scheme 1b).12 However, there is no report to generate important aryl acetic acids via the cleavage of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bonds with a catalytic system. As part of our continuing interest in advancing sustainable organic synthesis with CO2,13 we wondered whether transition metal-catalysis could resolve such a challenge with different reaction mechanisms. Recently, one of us has developed Ru-catalyzed umpolung reactions with carbonyls as carbanion equivalents.14 Considering that hydrazones are easily prepared from carbonyl compounds, which widely exist in nature and industry, this new strategy promises to be more sustainable and efficient for the synthesis of phenylacetic acids, especially for complex examples. At the outset of our investigations, however, it was unclear whether such a procedure could ever be implemented, since Ru-assisted Wolff–Kishner reduction15 takes place smoothly under mild reaction conditions. The azine byproduct could also be easily generated from the hydrazones. Moreover, a possible side reaction of base-promoted Shapiro-type carboxylation of ketone hydrazones to acrylic acids may occur to compete with our designed reactions.12
N double bonds with a catalytic system. As part of our continuing interest in advancing sustainable organic synthesis with CO2,13 we wondered whether transition metal-catalysis could resolve such a challenge with different reaction mechanisms. Recently, one of us has developed Ru-catalyzed umpolung reactions with carbonyls as carbanion equivalents.14 Considering that hydrazones are easily prepared from carbonyl compounds, which widely exist in nature and industry, this new strategy promises to be more sustainable and efficient for the synthesis of phenylacetic acids, especially for complex examples. At the outset of our investigations, however, it was unclear whether such a procedure could ever be implemented, since Ru-assisted Wolff–Kishner reduction15 takes place smoothly under mild reaction conditions. The azine byproduct could also be easily generated from the hydrazones. Moreover, a possible side reaction of base-promoted Shapiro-type carboxylation of ketone hydrazones to acrylic acids may occur to compete with our designed reactions.12
Results and discussion
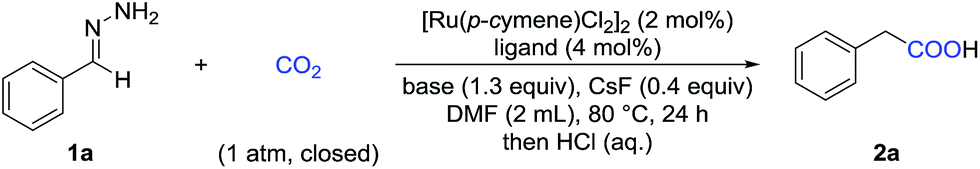
With these challenges in mind, we began our study by evaluating the reaction of benzaldehyde hydrazone 1a with CO2 (Table 1). To our delight, we found that many diphosphine ligands could promote this reaction (entries 1–3) and dppf gave the best result (entry 3), which might arise from higher nucleophilicity of the in situ generated Ru-complex. We also tested a variety of bases and found that inorganic bases were superior to organic bases in this reaction (entries 3–6). Increasing or decreasing the amount of base gave a lower yield (entries 7 and 8). CsF served as an efficient additive to enhance the yield (entry 11), which might arise from enhanced nucleophilicity with the fluoride anion as a strong hydrogen bond acceptor. Control experiments demonstrated that the Ru-catalyst was vital to this transformation (entry 12) and that both the ligand and the base were important (entries 9 and 10). No desired product was detected in the absence of CO2 (entry 13).|
|
|||
|---|---|---|---|
| Entry | Ligand | Base | Yieldb (%) |
| a Reaction conditions: 1a (0.4 mmol). b Yields were determined by crude 1H NMR using dibromomethane as an internal standard, and the isolated yields are given in parentheses. c Base (0.4 mmol). d Base (0.6 mmol). e No CsF. f No [Ru(p-cymene)Cl2]2. g N2 instead of CO2. DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene. N.D. = not detected. | |||
| 1 | dppp | Cs2CO3 | 44 |
| 2 | dppe | Cs2CO3 | 19 |
| 3 | dppf | Cs2CO3 | 85 (83) |
| 4 | dppf | K3PO4 | 51 |
| 5 | dppf | KOtBu | 80 |
| 6 | dppf | DBU | 27 |
| 7c | dppf | Cs2CO3 | 73 |
| 8d | dppf | Cs2CO3 | 72 |
| 9 | No | Cs2CO3 | 13 |
| 10 | dppf | No | 31 |
| 11e | dppf | Cs2CO3 | 78 |
| 12f | dppf | Cs2CO3 | N.D. |
| 13g | dppf | Cs2CO3 | N.D. |
With the optimized reaction conditions in hand, we then tested the scope of aldehyde hydrazones (Table 2), most of which were prepared from the corresponding aldehyde in one step and used without further purification, thus representing a significant advantage from a practical standpoint. Both electron-donating and electron-withdrawing groups on the phenyl ring were compatible. Different substituents at various positions on the arenes, including a bulky trifluoromethyl group at the ortho position (2r), did not hamper the reaction. Remarkably, a variety of functional groups, such as fluoro (2b), chloro (2c and 2l), bromo (2d, 2m and 2p), iodo (2e and 2q), ether (2g, 2j and 2k), ester (2i), nitro (2n), and cyano (2o), were all tolerated, providing a possibility for further transformations. Moreover, different kinds of fused rings (2s, 2t and 2u) and heterocycles, such as thiophene, benzothiophene and indole (2v, 2w and 2x), were compatible in this reaction. It is worth noting that a pharmaceutical agent felbinac (2f) and a known plant growth stimulant (2w) were both prepared readily and in a good yield with our strategy.
| a Reaction conditions: 1 (0.4 mmol), 1 atm of CO2, [Ru(p-cymene)Cl2]2 (0.008 mol), dppf (0.016 mmol), Cs2CO3 (0.52 mmol), CsF (0.16 mmol), DMF (2 mL), 80 °C, and 24 h; isolated yields. |
|---|

|
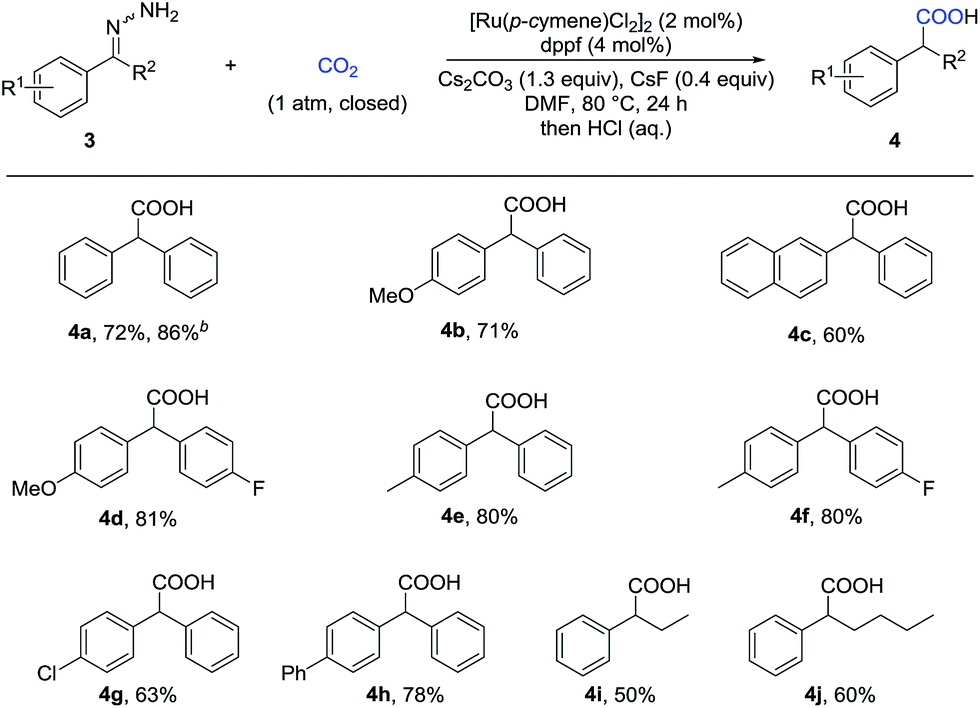
Given that α-substituted phenylacetic acids are present in a large collection of bioactive molecules, we wondered whether our procedure could be extended to ketone hydrazones, which have not been successfully applied in previous umpolung reactions with other reactive electrophiles.14 Although Wolff–Kishner reduction was anticipated, we were delighted to discover that benzophenone hydrazone reacted smoothly to give the diphenylacetic acid 4a in 72% or 86% yield with minor modification of the reaction conditions (Table 3). Furthermore, the substrates with different substituents, including fluoro, chloro, and methoxyl, afforded the desired products in moderate to good yields (4b–4h). Besides the benzophenone hydrazones, readily-available ketone hydrazones 3i and 3j furnished the corresponding α-alkylphenylacetic acid products in synthetically useful yields, showing the further utility of this process.
To further demonstrate the utility of this umpolung carboxylation, we conducted a gram-scale synthesis of 3-bromophenylacetic acid 2m in 83% yield (Scheme 2). An antihyperlipidemic drug, fenofibrate 3k′, could be subjected to this method to produce the acid 4k in 60% isolated yield. Moreover, the product 4a was readily converted in one step to adiphenine (5), an inhibitor of the nicotinic receptor.16a The antiproliferative agent 6 and the known histone deacetylase inhibitor 7 were efficiently accessed by respective amidations in good yields (see the ESI† for details).16b–d
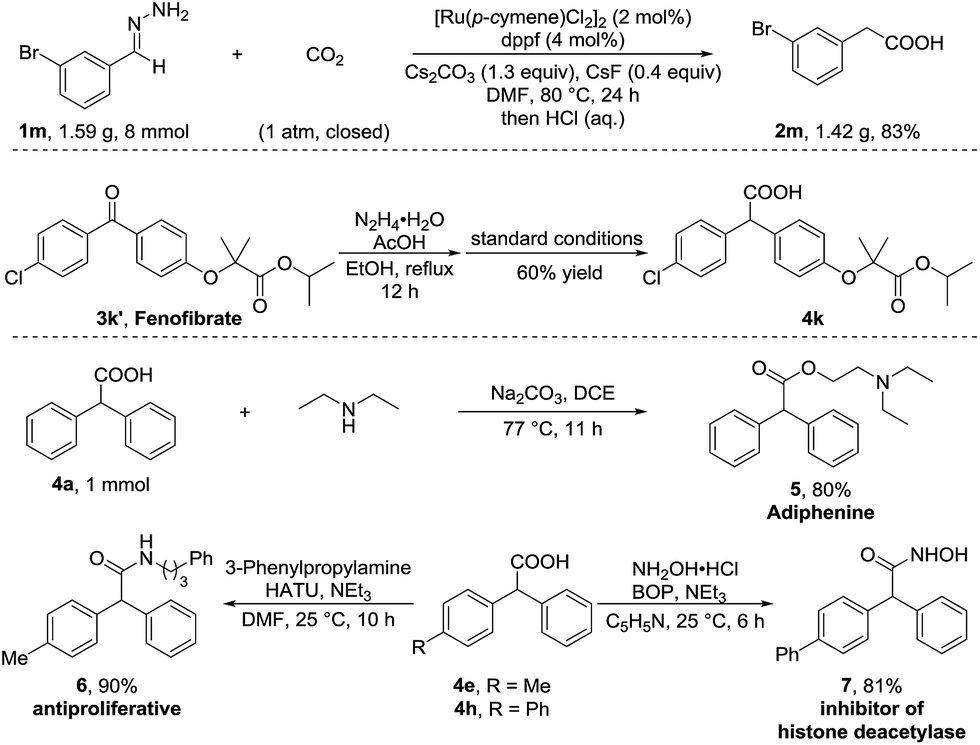 | ||
| Scheme 2 Gram-scale synthesis and transformations. | ||
On the basis of preliminary mechanistic studies and previous reports,14 a plausible mechanism is proposed to account for this transformation. As shown in Fig. 2, catalyst A could be generated (see the ESI† for details) which undergoes ligand exchange with hydrazone and Cs2CO3 to give complex B, which then undergoes two step deprotonation to generate a Ru-nitrenoid complex D.17 After a concerted hetero-[4 + 2] cycloaddition with CO2, a six-membered Ru-cycle E can be formed. Then the release of N2, a driving force for this reaction, and the following protonation-ligand exchange provide the desired product and regenerate the active catalyst B. Alternatively, the intermediate F could also be generated from Dvia sequential isomerization, carbenation, and CO2 insertion (Path-B).
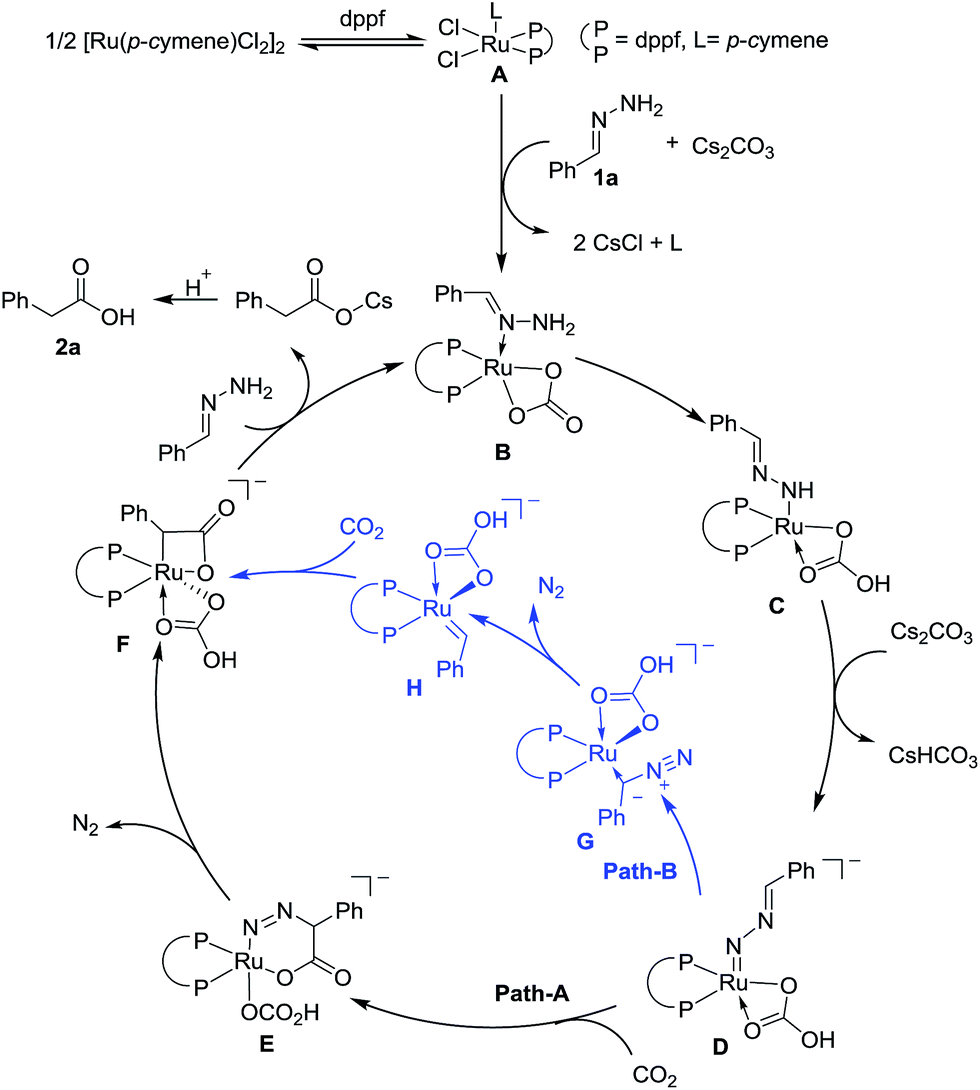 | ||
| Fig. 2 Proposed mechanism. | ||
To further identify the proposed mechanism, a density functional theory (DFT) method M06-L was employed to investigate this reaction.18 The reactant 1a coordinated Ru carbonate species B was considered as a starting complex (see the Fig. S1† for details). The first N–H bond cleavage proceeds via a transition state 8-ts (Fig. S2†), with a free energy barrier of 23.9 kcal mol−1, to generate the amino-Ru intermediate C. Subsequently, the second step deprotonation occurs rapidly with the assistance of Cs2CO3, leading to the formation of the Ru-nitrenoid intermediate D.
After the formation of Ru-nitrenoid D, in Path-A (Fig. 3), the six-membered Ru-cycle complex E was obtained through an intermolecular hetero-[4 + 2] cycloaddition with CO2, with a free energy barrier of 22.4 kcal mol−1.19 The subsequent C–N bond cleavage leads to the generation of the benzylruthenium intermediate F, which is exergonic by 34.1 kcal mol−1 as a result of N2 release. The phenylacetate 14 was then released after the following protonation-ligand exchange, and the active catalyst B was also concomitantly regenerated. Meanwhile, the calculation of activation free energy of Path-B has also been conducted. The isomerization of the Ru-nitrenoid intermediate D could afford the diazoalkane-coordinated Ru complex G, which is endergonic by 13.0 kcal mol−1. The subsequent carbenation could proceed rapidly to afford the Ru-carbenoid H with an energy barrier of 1.8 kcal mol−1. CO2 insertion via a transition state 17-ts then generates the common benzylruthenium intermediate F. The overall activation free energy of Path-B is determined to be 26.6 kcal mol−1, which is 4.2 kcal mol−1 higher than that of the [4 + 2] cycloaddition step in Path-A. Therefore, the carbenation pathway (Path-B) turns out to be unfavorable in comparison with Path-A.
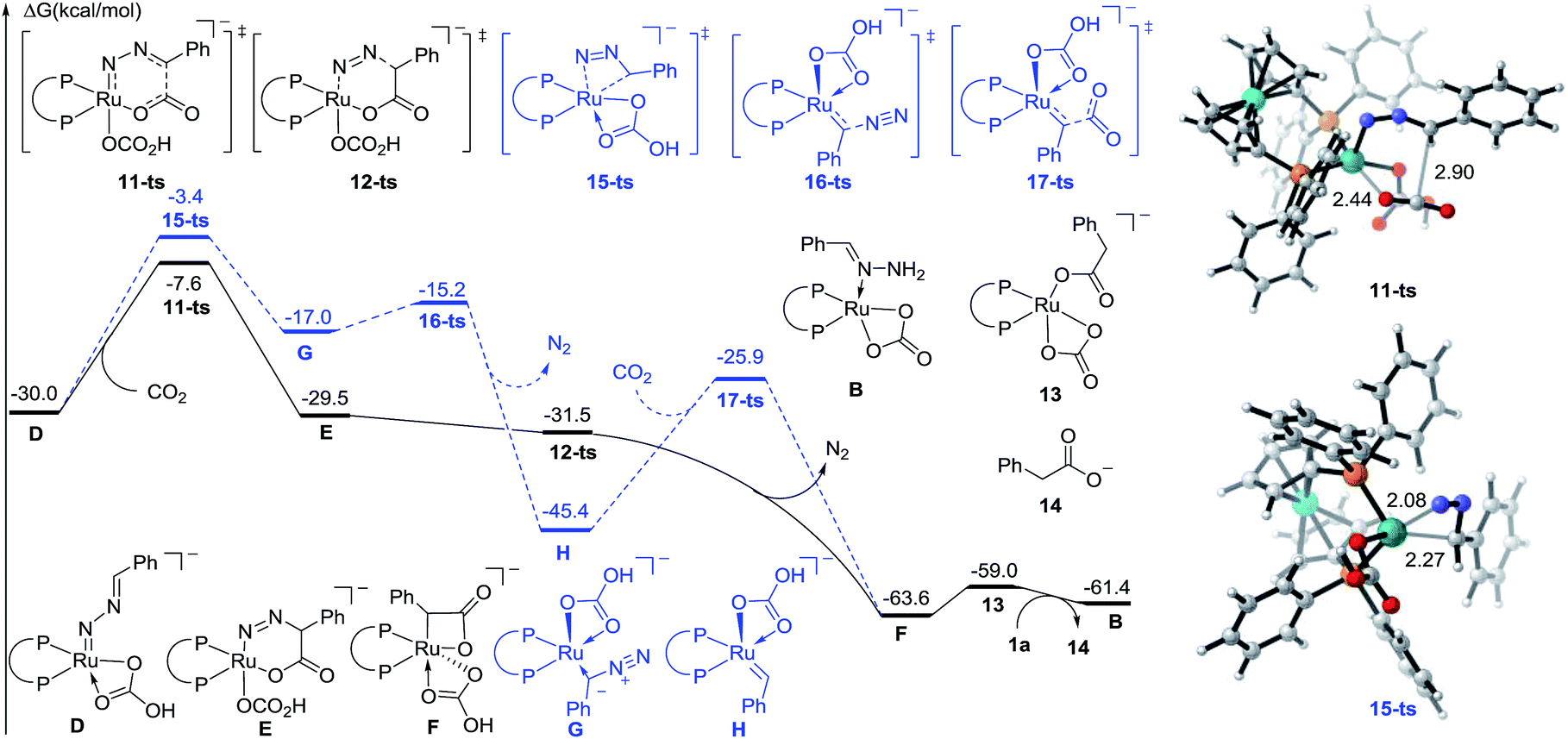 | ||
| Fig. 3 Free energy profiles of Path-A (plain) and Path-B (dashed) for ruthenium-catalyzed carboxylation. The distances are represented in angstroms. | ||
Moreover, Fig. 4 shows the variation of the natural bond orbital (NBO)20 atomic charges on benzylic carbon during the catalytic cycle. It demonstrates that the increasing of the electron density on benzylic carbon is accompanied by the two step N–H cleavage, which clearly indicates the umpolung of the reactants. Thus, the Ru-nitrenoid D exhibits enhanced reactivity with CO2.
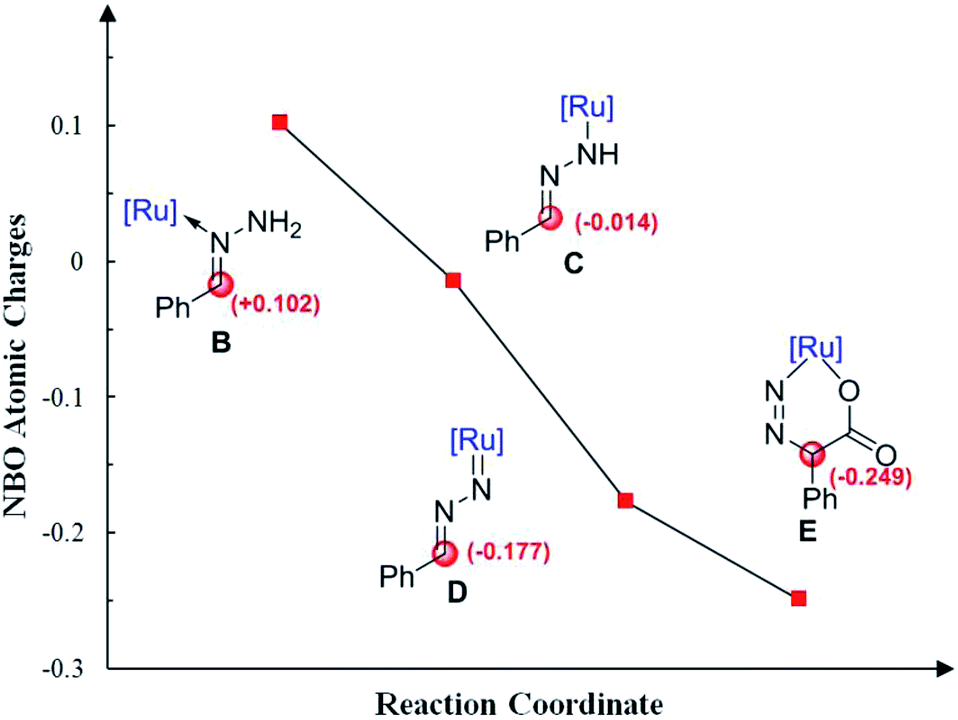 | ||
| Fig. 4 Calculated NBO atomic charges on benzylic carbon during the catalytic cycle. | ||
Conclusions
In summary, we have realized the first Ru-catalyzed umpolung carboxylation of hydrazones with CO2 for synthesizing important aryl acetic acids under mild conditions. The only byproduct in this reaction is N2 while overall a high atom efficiency is realized displaying significant advantages in carboxylations. Besides aldehyde hydrazones, a variety of ketone hydrazones, which have not been successfully applied in previous umpolung reactions with other reactive electrophiles, also show high reactivity and selectivity in this reaction. This methodology displays a broad substrate scope, functional group tolerance, scalability and easy derivation of important structures. With a deeper understanding of this reaction, DFT calculations reveal the fundamental of this transformation with the generation of a Ru-nitrenoid and intermolecular [4 + 2] cycloaddition with CO2.Conflicts of interest
Acknowledgements
We thank Prof. Xiaofeng Wu (Leibniz-Institut für Katalyse) and Prof. Chao Liu (Lanzhou Institute of Chemical Physics) for reproducing the results. We thank Prof. Jason J. Chruma (Sichuan University) and Prof. Bi-Jie Li (Tsinghua University) for helpful manuscript revisions. We thank the National Natural Science Foundation of China (21502124, 21772020, and 21372266), the “973” Project from the MOST of China (2015CB856600), the Recruitment Program of Global Experts (Short-Term B for C. -J. Li and Young Talents for D. -G. Yu) and the Fundamental Research Funds for the Central Universities for financial support.Notes and references
- (a) M. Aresta, Carbon Dioxide as Chemical Feedstock, Wiley-VCH, Weinheim, 2010 Search PubMed; (b) L. Ackermann, Angew. Chem., Int. Ed., 2011, 50, 3842 CrossRef CAS PubMed.
- (a) T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS PubMed; (b) K. Huang, C.-L. Sun and Z.-J. Shi, Chem. Soc. Rev., 2011, 40, 2435 RSC; (c) M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510 CrossRef CAS PubMed; (d) Y. Tsuji and T. Fujihara, Chem. Commun., 2012, 48, 9956 RSC; (e) M. He, Y. Sun and B. Han, Angew. Chem., Int. Ed., 2013, 52, 9620 CrossRef CAS PubMed; (f) L. Zhang and Z. Hou, Chem. Sci., 2013, 4, 3395 RSC; (g) M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709 CrossRef CAS PubMed; (h) C. Maeda, Y. Miyazaki and T. Ema, Catal. Sci. Technol., 2014, 4, 1482 RSC; (i) C. S. Yeung and V. M. Dong, Top. Catal., 2014, 57, 1342 CrossRef CAS; (j) Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed; (k) Z. Zhang, T. Ju, J.-H. Ye and D.-G. Yu, Synlett, 2017, 28, 741 CrossRef CAS.
- For reviews, see: (a) M. Börjesson, T. Moragas, D. Gallego and R. Martin, ACS Catal., 2016, 6, 6739 CrossRef PubMed; (b) S. Wang, G. Du and C. Xi, Org. Biomol. Chem., 2016, 14, 3666 RSC; (c) Q. Zhu, L. Wang, C. Xia and C. Liu, Chin. J. Org. Chem., 2016, 36, 2813 CrossRef CAS; (d) Q.-W. Song, Z.-H. Zhou and L.-N. He, Green Chem., 2017, 19, 3707 RSC; (e) W. Zhang, N. Zhang, C. Guo and X. Lu, Chin. J. Org. Chem., 2017, 37, 1309 CrossRef CAS; (f) X.-F. Wu and F. Zheng, Top. Curr. Chem., 2017, 375, 4 CrossRef PubMed; (g) J. Luo and I. Larrosa, ChemSusChem, 2017, 10, 3317 CrossRef CAS PubMed; (h) Y.-Y. Gui, W.-J. Zhou, J.-H. Ye and D.-G. Yu, ChemSusChem, 2017, 10, 1337 CrossRef CAS PubMed; (i) A. Tortajada, F. Juliá-Hernández, M. Börjesson, T. Moragas and R. Martin, Angew. Chem., Int. Ed., 2018 DOI:10.1002/anie.201803186.
- For selected examples of carboxylation of electrophiles with CO2, see: (a) A. Correa and R. Martin, J. Am. Chem. Soc., 2009, 131, 15974 CrossRef CAS PubMed; (b) T. Fujihara, K. Nogi, T. Xu, J. Terao and Y. Tsuji, J. Am. Chem. Soc., 2012, 134, 9106 CrossRef CAS PubMed; (c) H. Tran-Vu and O. Daugulis, ACS Catal., 2013, 3, 2417 CrossRef CAS PubMed; (d) T. Moragas, J. Cornella and R. Martin, J. Am. Chem. Soc., 2014, 136, 17702 CrossRef CAS PubMed; (e) Y. Liu, J. Cornella and R. Martin, J. Am. Chem. Soc., 2014, 136, 11212 CrossRef CAS PubMed; (f) K. Nogi, T. Fujihara, J. Terao and Y. Tsuji, Chem. Commun., 2014, 50, 13052 RSC; (g) X. Wang, Y. Liu and R. Martin, J. Am. Chem. Soc., 2015, 137, 6476 CrossRef CAS PubMed; (h) K. Nogi, T. Fujihara, J. Terao and Y. Tsuji, J. Org. Chem., 2015, 80, 11618 CrossRef CAS PubMed; (i) T. Mita, Y. Higuchi and Y. Sato, Chem.–Eur. J., 2015, 21, 16391 CrossRef CAS PubMed; (j) M. Börjesson, T. Moragas and R. Martin, J. Am. Chem. Soc., 2016, 138, 7504 CrossRef PubMed; (k) F. Juliá-Hernández, T. Moragas, J. Cornella and R. Martin, Nature, 2017, 545, 84 CrossRef PubMed; (l) K. Shimomaki, K. Murata, R. Martin and N. Iwasawa, J. Am. Chem. Soc., 2017, 139, 9467 CrossRef CAS PubMed; (m) Q.-Y. Meng, S. Wang and B. König, Angew. Chem., Int. Ed., 2017, 56, 13426 CrossRef CAS PubMed.
- For selected examples of carboxylation of nucleophiles with CO2, see: (a) M. Shi and K. M. Nicholas, J. Am. Chem. Soc., 1997, 119, 5057 CrossRef CAS; (b) K. Ukai, M. Aoki, J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2006, 128, 8706 CrossRef CAS PubMed; (c) T. Ohishi, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2008, 47, 5792 CrossRef CAS PubMed; (d) H. Ochiai, M. Jang, K. Hirano, H. Yorimitsu and K. Oshima, Org. Lett., 2008, 10, 2681 CrossRef CAS PubMed; (e) C. S. Yeung and V. M. Dong, J. Am. Chem. Soc., 2008, 130, 7826 CrossRef CAS PubMed; (f) L. Zhang, J. Cheng, T. Ohishi and Z. Hou, Angew. Chem., Int. Ed., 2010, 49, 8670 CrossRef CAS PubMed; (g) I. I. F. Boogaerts and S. P. Nolan, J. Am. Chem. Soc., 2010, 132, 8858 CrossRef CAS PubMed; (h) O. Vechorkin, N. Hirt and X. Hu, Org. Lett., 2010, 12, 3567 CrossRef CAS PubMed; (i) K. Nemoto, H. Yoshida, N. Egusa, N. Morohashi and T. Hattori, J. Org. Chem., 2010, 75, 7855 CrossRef CAS PubMed; (j) H. Mizuno, J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2011, 133, 1251 CrossRef CAS PubMed; (k) Z. Xin, C. Lescot, S. D. Friis, K. Daasbjerg and T. Skrydstrup, Angew. Chem., Int. Ed., 2015, 54, 6862 CrossRef CAS PubMed; (l) W.-Y. Gao, H. Wu, K. Leng, Y. Sun and S. Ma, Angew. Chem., Int. Ed., 2016, 55, 5472 CrossRef CAS PubMed; (m) A. Banerjee, G. R. Dick, T. Yoshino and M. W. Kanan, Nature, 2016, 531, 215 CrossRef CAS PubMed; (n) S. Fenner and L. Ackermann, Green Chem., 2016, 18, 3804 RSC; (o) J. Luo, S. Preciado, P. Xie and I. Larrosa, Chem.–Eur. J., 2016, 22, 6798 CrossRef CAS PubMed; (p) K. Michigami, T. Mita and Y. Sato, J. Am. Chem. Soc., 2017, 139, 6094 CrossRef CAS PubMed.
- (a) A. L. Blobaum and L. J. Marnett, J. Biol. Chem., 2007, 282, 16379 CrossRef CAS PubMed; (b) J. E. Milne, T. Storz, J. T. Colyer, O. R. Thiel, M. Dilmeghani Seran, R. D. Larsen and J. A. Murry, J. Org. Chem., 2011, 76, 9519 CrossRef CAS PubMed; (c) R. D. Grigg, J. W. Rigoli, R. Van Hoveln, S. Neale and J. M. Schomaker, Chem.–Eur. J., 2012, 18, 9391 CrossRef CAS PubMed.
- For selected examples, see: (a) H. L. Finkbeiner and G. D. Cooper, J. Org. Chem., 1962, 27, 3395 CrossRef CAS; (b) C. M. Williams, J. B. Johnson and T. Rovis, J. Am. Chem. Soc., 2008, 130, 14936 CrossRef CAS PubMed; (c) M. D. Greenhalgh and S. P. Thomas, J. Am. Chem. Soc., 2012, 134, 11900 CrossRef CAS PubMed; (d) P. Shao, S. Wang, C. Chen and C. Xi, Org. Lett., 2016, 18, 2050 CrossRef CAS PubMed; (e) S. Kawashima, K. Aikawa and K. Mikami, Eur. J. Org. Chem., 2016, 2016, 3166 CrossRef CAS; (f) K. Murata, N. Numasawa, K. Shimomaki, J. Takaya and N. Iwasawa, Chem. Commun., 2017, 53, 3098 RSC; (g) V. R. Yatham, Y. Shen and R. Martin, Angew. Chem., Int. Ed., 2017, 56, 10915 CrossRef CAS PubMed; (h) M. Gaydou, T. Moragas, F. Juliá-Hernández and R. Martin, J. Am. Chem. Soc., 2017, 139, 12161 CrossRef CAS PubMed.
- (a) T. León, A. Correa and R. Martin, J. Am. Chem. Soc., 2013, 135, 1221 CrossRef PubMed; (b) A. Correa, T. León and R. Martin, J. Am. Chem. Soc., 2014, 136, 1062 CrossRef CAS PubMed; (c) S. Zhang, W.-Q. Chen, A. Yu and L.-N. He, ChemCatChem, 2015, 7, 3972 CrossRef CAS; (d) T. Moragas, M. Gaydou and R. Martin, Angew. Chem., Int. Ed., 2016, 55, 5053 CrossRef CAS PubMed.
- L. Wu, Q. Liu, I. Fleischer, R. Jackstell and M. Beller, Nat. Commun., 2014, 5, 3091 Search PubMed.
- (a) D. Seebach, Angew. Chem., Int. Ed., 1979, 18, 239 CrossRef; (b) N. Marion, S. Díez-González and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2988 CrossRef CAS PubMed; (c) X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511 RSC; (d) D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307 CrossRef CAS PubMed.
- (a) T. Mita, J. Chen, M. Sugawara and Y. Sato, Angew. Chem., Int. Ed., 2011, 50, 1393 CrossRef CAS PubMed; (b) T. Mita, J. Chen, M. Sugawara and Y. Sato, Org. Lett., 2012, 14, 6202 CrossRef CAS PubMed; (c) T. Mita, M. Sugawara, H. Hasegawa and Y. Sato, J. Org. Chem., 2012, 77, 2159 CrossRef CAS PubMed; (d) T. Mita, Y. Higuchi and Y. Sato, Chem.–Eur. J., 2013, 19, 1123 CrossRef CAS PubMed; (e) A. A. Sathe, D. R. Hartline and A. T. Radosevich, Chem. Commun., 2013, 49, 5040 RSC; (f) T. Mita, Y. Higuchi and Y. Sato, Org. Lett., 2014, 16, 14 CrossRef CAS PubMed; (g) T. Mita, J. Chen and Y. Sato, Org. Lett., 2014, 16, 2200 CrossRef CAS PubMed; (h) T. Mita, M. Sugawara, K. Saito and Y. Sato, Org. Lett., 2014, 16, 3028 CrossRef CAS PubMed; (i) C.-X. Guo, W.-Z. Zhang, H. Zhou, N. Zhang and X.-B. Lu, Chem.–Eur. J., 2016, 22, 17156 CrossRef CAS PubMed.
- S. Sun, J. T. Yu, Y. Jiang and J. Cheng, J. Org. Chem., 2015, 80, 2855 CrossRef CAS PubMed.
- (a) W.-J. Yoo and C.-J. Li, Adv. Synth. Catal., 2008, 350, 1503 CrossRef CAS; (b) Z. Zhang, L.-L. Liao, S.-S. Yan, L. Wang, Y.-Q. He, J.-H. Ye, J. Li, Y.-G. Zhi and D.-G. Yu, Angew. Chem., Int. Ed., 2016, 55, 7068 CrossRef CAS PubMed; (c) J.-H. Ye, L. Song, W.-J. Zhou, T. Ju, Z.-B. Yin, S.-S. Yan, Z. Zhang, J. Li and D.-G. Yu, Angew. Chem., Int. Ed., 2016, 55, 10022 CrossRef CAS PubMed; (d) Z. Zhang, T. Ju, M. Miao, J.-L. Han, Y.-H. Zhang, X.-Y. Zhu, J.-H. Ye, D.-G. Yu and Y.-G. Zhi, Org. Lett., 2017, 19, 396 CrossRef CAS PubMed; (e) Y.-Y. Gui, N. Hu, X.-W. Chen, L.-L. Liao, T. Ju, J.-H. Ye, Z. Zhang, J. Li and D.-G. Yu, J. Am. Chem. Soc., 2017, 139, 17011 CrossRef CAS PubMed; (f) J.-H. Ye, M. Miao, H. Huang, S.-S. Yan, Z.-B. Yin, W.-J. Zhou and D.-G. Yu, Angew. Chem., Int. Ed., 2017, 56, 15416 CrossRef CAS PubMed; (g) J.-H. Ye, L. Zhu, S.-S. Yan, M. Miao, X.-C. Zhang, W.-J. Zhou, J. Li, Y. Lan and D.-G. Yu, ACS Catal., 2017, 7, 8324 CrossRef CAS; (h) Z.-B. Yin, J.-H. Ye, W.-J. Zhou, Y.-H. Zhang, L. Ding, Y.-Y. Gui, S.-S. Yan, J. Li and D.-G. Yu, Org. Lett., 2018, 20, 190 CrossRef CAS PubMed; (i) L. Zhu, J.-H. Ye, M. Duan, X. Qi, D.-G. Yu, R. Bai and Y. Lan, Org. Chem. Front., 2018, 5, 633 RSC; (j) Z. Zhang, C.-J. Zhu, M. Miao, J.-L. Han, T. Ju, L. Song, J.-H. Ye, J. Li and D.-G. Yu, Chin. J. Chem., 2018, 36, 430 CrossRef CAS; (k) L. Sun, J.-H. Ye, W.-J. Zhou, X. Zeng and D.-G. Yu, Org. Lett., 2018 DOI:10.1021/acs.orglett.8b01079; (l) G. Shen, W.-J. Zhou, X.-B. Zhang, G.-M. Cao, Z. Zhang, J.-H. Ye, L.-L. Liao, J. Li and D.-G. Yu, Chem. Commun., 2018 10.1039/C8CC03039A.
- (a) H. Wang, X.-J. Dai and C.-J. Li, Nat. Chem., 2016, 9, 374 CrossRef PubMed; (b) N. Chen, X.-J. Dai, H. Wang and C.-J. Li, Angew. Chem., Int. Ed., 2017, 56, 6260 CrossRef CAS PubMed; (c) X.-J. Dai, H. Wang and C.-J. Li, Angew. Chem., Int. Ed., 2017, 56, 6302 CrossRef CAS PubMed; (d) W. Wei, X.-J. Dai, H. Wang, C. Li, X. Yang and C.-J. Li, Chem. Sci., 2017, 8, 8193 RSC.
- M. E. Furrow and A. G. Myers, J. Am. Chem. Soc., 2004, 126, 5436 CrossRef CAS PubMed and references therein.
- (a) G. Spitzmaul, F. Gumilar, J. P. Dilger and C. Bouzat, Br. J. Pharmacol., 2009, 157, 804 CrossRef CAS PubMed; (b) P. Urbani, M. G. Cascio, A. Ramunno, T. Bisogno, C. Saturnino and V. D. Marzo, Bioorg. Med. Chem., 2008, 16, 7510 CrossRef CAS PubMed; (c) G. Autore, A. Caruso, S. Marzocco, B. Nicolaus, C. Palladino, A. Pinto, A. Popolo, M. S. Sinicropi, G. Tommonaro and C. Saturnino, Molecules, 2010, 15, 2028 CrossRef CAS PubMed; (d) B. Ma, Z. Chu, B. Huang, Z. Liu, L. Liu and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 2749 CrossRef CAS PubMed.
- (a) J. L. Jat, M. P. Paudyal, H. Gao, Q.-L. Xu, M. Yousufuddin, D. Devarajan, D. H. Ess, L. Kurti and J. R. Falck, Science, 2014, 343, 61 CrossRef CAS PubMed; (b) V. Bizet, L. Buglioni and C. Bolm, Angew. Chem., Int. Ed., 2014, 53, 5639 CrossRef CAS PubMed; (c) Y. Lin, L. Zhu, Y. Lan and Y. Rao, Chem.–Eur. J., 2015, 21, 14937 CrossRef CAS PubMed.
- The activation free energy of cycloaddition between amino-ruthenium C and CO2 is up to 38.1 kcal mol−1 (Fig. S3†), which suggests that the second N–H bond cleavage would be preferentially occur from amino-ruthenium C.
- For more computational details, please see in ESI.†.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization data. See DOI: 10.1039/c8sc01299g |
| This journal is © The Royal Society of Chemistry 2018 |