
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePredicting the DNP-SENS efficiency in reactive heterogeneous catalysts from hydrophilicity†
Eva
Pump
 a,
Anissa
Bendjeriou-Sedjerari
a,
Jasmine
Viger-Gravel
b,
David
Gajan
c,
Baptiste
Scotto
a,
Manoja K.
Samantaray
a,
Edy
Abou-Hamad
d,
Andrei
Gurinov
d,
Walid
Almaksoud
a,
Zhen
Cao
a,
Anne
Lesage
c,
Luigi
Cavallo
a,
Lyndon
Emsley
*b and
Jean-Marie
Basset
*a
a,
Anissa
Bendjeriou-Sedjerari
a,
Jasmine
Viger-Gravel
b,
David
Gajan
c,
Baptiste
Scotto
a,
Manoja K.
Samantaray
a,
Edy
Abou-Hamad
d,
Andrei
Gurinov
d,
Walid
Almaksoud
a,
Zhen
Cao
a,
Anne
Lesage
c,
Luigi
Cavallo
a,
Lyndon
Emsley
*b and
Jean-Marie
Basset
*a
aKing Abdullah University of Science and Technology (KAUST), KAUST Catalysis Center (KCC), Thuwal, 23955-6900, Saudi Arabia. E-mail: jeanmarie.basset@kaust.edu.sa
bInstitut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland. E-mail: lyndon.emsley@epfl.ch
cInstitut de Sciences Analytiques (CNRS/ENS-Lyon/UCB-Lyon 1), Université de Lyon, Centre de RMN à Très Hauts Champs, 69100 Villeurbanne, France
dKing Abdullah University of Science and Technology (KAUST), Core Labs, Thuwal, 23955-6900, Saudi Arabia
First published on 30th April 2018
Abstract
Identification of surfaces at the molecular level has benefited from progress in dynamic nuclear polarization surface enhanced NMR spectroscopy (DNP SENS). However, the technique is limited when using highly sensitive heterogeneous catalysts due to secondary reaction of surface organometallic fragments (SOMFs) with stable radical polarization agents. Here, we observe that in non-porous silica nanoparticles (NPs) (dparticle = 15 nm) some DNP enhanced NMR or SENS characterizations are possible, depending on the metal-loading of the SOMF and the type of SOMF substituents (methyl, isobutyl, neopentyl). This unexpected observation suggests that aggregation of the nanoparticles occurs in non-polar solvents (such as ortho-dichlorobenzene) leading to (partial) protection of the SOMF inside the interparticle space, thereby preventing reaction with bulky polarization agents. We discover that the DNP SENS efficiency is correlated with the hydrophilicity of the SOMF/support, which depends on the carbon and SOMF concentration. Nitrogen sorption measurements to determine the BET constant (CBET) were performed. This constant allows us to predict the aggregation of silica nanoparticles and consequently the efficiency of DNP SENS. Under optimal conditions, CBET > 60, we found signal enhancement factors of up to 30.
Introduction
Immobilization of organometallic compounds on supports represents a promising avenue in the design of ideal single site catalysts.1 Recent progress in this area has been made by entering the catalytic cycle directly with surface organometallic fragments (SOMFs).2–4 Generally, a SOMF consists of a metal center (group IV to group VIII), a ligand such as a hydride, an alkyl, a carbene-hydride, a carbene, or an imido used to target a given reaction (e.g. low temperature hydrogenolysis of polyolefins, ethylene polymerization, alkane, olefin or imine metathesis…)1,5,6 and an oxide or chemically modified inorganic support.7,8 Some of the most common supports used in surface organometallic chemistry (SOMC) are silica nanoparticles (NPs).1,9,10 Surface silanols generated by a high temperature dehydroxylation process under vacuum (700 °C, 10−5 mbar) act as anchor sites for early transition metals leading to the design of well-defined heterogeneous catalysts.11 This strategy, also known as “Catalysis by Design”, requires methods for unambiguous characterization of these species at the atomic level, usually through advanced spectroscopic techniques such as EXAFS and multi-dimensional solid-state NMR (SS NMR) spectroscopy.1,10,12,13 The low intrinsic sensitivity of NMR is a major drawback for atomic level characterization of SOMFs due to prohibitively low signal-to-noise ratios and long experimental times. This problem can be overcome by an emerging technique called dynamic nuclear polarization surface enhanced NMR spectroscopy (DNP SENS).14–16 DNP SENS can increase the signal intensity of conventional NMR spectra by two orders of magnitude, and has been applied to a range of surface systems.17–28 Recently, it has led to the first complete three-dimensional structure of a SOMF on a silica surface.29 The method requires the addition of stable radicals as sources of polarization, usually nitroxide biradicals such as TEKPol,14,16 which is usually added through incipient wetness impregnation (IWI)30 and acts as a source of unpaired electrons. In situ microwave (μwave) irradiation transfers electron polarization to the surrounding nuclei (usually protons), and is followed by spin diffusion and cross-polarization (CP) to the nuclei of interest.16,31 The presence of a biradical species means that DNP SENS has only rarely been applicable in SOMC, due to the reactivity of nitroxide radicals with the SOMF.32,33 Therefore this precludes the characterization of the most interesting catalytic species. There is thus a need for a non-destructive strategy for DNP SENS characterization of SOMFs. A range of different formulations for DNP SENS have recently been proposed.19,27,28,34–39,67 In particular it has been shown that reactive surface species can be studied either by protecting them inside mesoporous supports and using bulky radicals that are too large to enter the pores,27,39 or by using polarizing agents incorporated into dendrimers in order to avoid contact between the radical and the surface species.28 These approaches led to DNP signal enhancements for the surface species (εC, CP (SOMF)) of between 10 and 30. However, neither strategy is trivial to implement. Moreover, Perras et al.40 recently demonstrated that the radicals may physisorb on surfaces, and thus that the radical concentration should be optimized as a function of specific surface area in alumina, silica and mesoporous carbon samples. In the case of reactive surface species, the affinity between the radical and the surface will lead to the decomposition of active sites grafted on the support.Here, we introduce a strategy based on controlled aggregation for reactive samples which can be applied to silica nanoparticles. Aggregation leads to exclusion of bulky biradicals, thereby avoiding direct contact between the SOMF and the polarization agent. We show that the DNP SENS efficiency can be predicted by measuring the hydrophilicity of the surfaces. We find signal enhancement factors of up to 30, corresponding to a reduction in experimental acquisition time by a factor of 900 with respect to conventional SSNMR. In this way we investigate a library of highly sensitive supported organometallic complexes bearing alkyl-ligands of various coordinations and sizes: Ti(CH2tBu)4I, Zr(CH2tBu)4II, WMe6III, W(![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CtBu)(CH2tBu)3IV, ZnMe2V and Ga(iBu)3VI.
CtBu)(CH2tBu)3IV, ZnMe2V and Ga(iBu)3VI.
Attraction between silica NPs is mainly governed by hydrogen bonding41,42 and van der Waals interactions43 and leads to aggregation. Hydrophilic silica NPs (e.g. Aerosil-200 dehydroxylated at 700 °C) aggregate to a much higher extent in solvents with low polarity compared to NPs that have hydrophobic surfaces.44–48 Surface modification of silica by SOMFs has three main effects: (i) when there is lower concentration of remaining [SiOH] on the NP surface, agglomeration due to the formation of inter-particle hydrogen bonds between two nanoparticles is weakened, (ii) the presence of bulky SOMFs on the surface reduces aggregation of NPs due to steric hindrance48 and (iii) the presence of hydrophobic alkyl groups in the SOMFs in non-polar solvent also reduces the propensity for aggregation.49 We expect all of this to have an effect on DNP efficiency. For example, the presence of surface methyl groups has been shown in the past to strongly affect DNP enhancements.50
With these observations in mind, here we suggest three parameters that influence the aggregation of NPs and can determine the applicability of DNP SENS for characterization of reactive species: ligand/solvent polarity, ligand size, and SOMF surface loading. A controlled aggregation of silica NPs allows for the protection of the SOMF from TEKPol while maintaining a reasonable transfer of polarization through the solvent inside the interparticle space of the aggregates. Ideal conditions are found based on the loading of the SOMF, which can be measured by elemental analysis, and the hydrophobicity of the material, which is obtained from nitrogen sorption measurements (CBET, vide infra).51–53
Results and discussion
Reactions of organometallic complexes I–VI were performed with various SOMF loadings (of 0.2 (low), 0.5 (med) and 1.0 eq. [SOMF]/g [SiOH] (high)). The reactions were either conducted at room temperature (I, II, IV, V and VI) or −40 °C (III) with partially dehydroxylated silica at 700 °C SiO2, 700 (0.30 ± 0.05 mmol of [SiOH]/g of SiO2, 700) in n-pentane leading to SOMFs 1–6 (Table 1) according to literature protocols.54–59 More detailed information regarding grafting conditions and characterization can be found in the ESI.† FT-IR spectra (Fig. S1, ESI†) generally illustrate the decrease of the [SiOH]-vibrational band intensity at 3740 cm−1 and the increase of the intensity of ν(C–H) (3100–2700 cm−1) and δ(CH) bands (1569 cm−1 and 1361 cm−1) with increasing SOMF concentration. The SOMF concentration and the carbon content were determined by elemental analysis (ESI†), and the results are summarized in Table 1.
| Entry | Sample | Structure | c SOMF (mmol gsilica−1) | c carbon (mmol gsilica−1) | C BET |
|---|---|---|---|---|---|
| a Late transition metals are prone to open siloxane bridges leading to a higher than expected carbon/metal ratio. b The detection limit of the instrument is 0.2 wt% carbon. | |||||
| 1 | 0 |
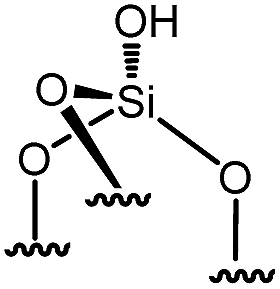
|
0 | 0 | 81 |
| 2 | 1-med |
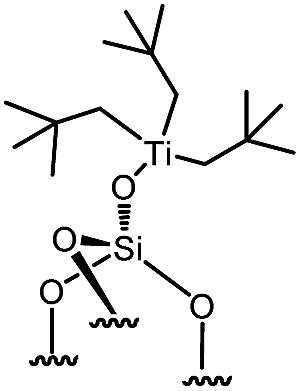
|
0.179 | 2.70 | 43 |
| 3 | 1-low | 0.099 | 1.59 | 57 | |
| 4 | 2-med |
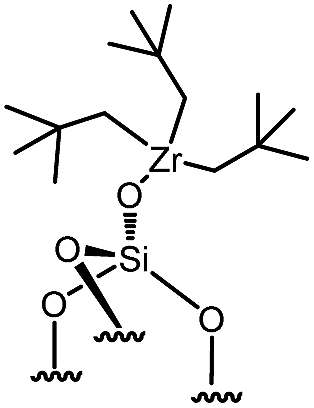
|
0.153 | 1.93 | 54 |
| 5 | 2-low | 0.044 | 0.76 | 58 | |
| 6 | 3-med |
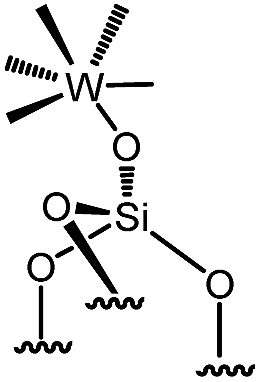
|
0.125 | 0.65 | 65 |
| 7 | 3-low | 0.070 | 0.34 | 75 | |
| 8 | 4-high |
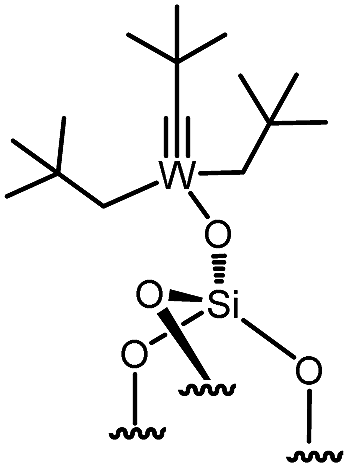
|
0.234 | 3.59 | 32 |
| 9 | 4-med | 0.114 | 1.80 | 50 | |
| 10 | 4-low | 0.065 | 0.89 | 65 | |
| 11 | 5-med |
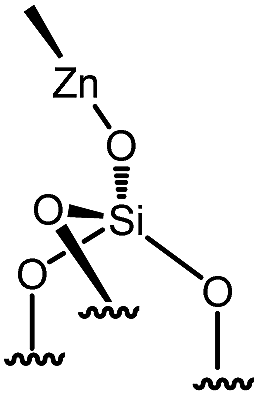
|
0.103 | 0.19a | 71 |
| 12 | 5-low | 0.049 | 0.02b | 78 | |
| 13 | 6-high |
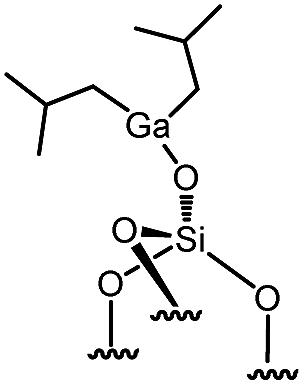
|
0.200 | 2.41a | 42 |
| 14 | 6-med | 0.110 | 1.39a | 54 | |
Additionally, nitrogen sorption measurements were performed to determine the value of CBET, which is used to assess the surface polarity of the silica.51–53 More precisely, CBET expresses the strength of the adsorbate–adsorbent interaction and is related to the excess enthalpy of condensation of nitrogen molecules adsorbed on the first monolayer. The CBET value is determined from the BET analysis of the portion of the isotherm between relative pressures (p/p0) of 0.05 and 0.20 according to the following eqn (1):
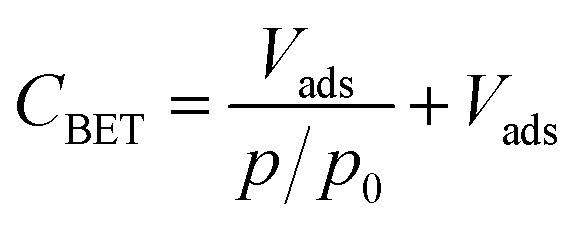 | (1) |
The aggregation hypothesis is also supported by free energy calculations (details are provided in the ESI†),61–63 which reflect the effective interaction between particles in solution. Calculations were performed on molecular T8-silsesquioxane models (Fig. S4, ESI†). Non-functionalized [SiO1.5(OH)]8 (M-0, black in Fig. 1) shows a strong tendency towards aggregation between these particles due to the hydrogen-bonding interactions between their OH groups. After functionalization of four [SiOH] with –tBu(CMe3) (M-Me, blue in Fig. 1) or –C(CH2tBu)3, (M-CH2tBu, red in Fig. 1) the attraction between two silsesquioxane molecules decreases with increasing bulkiness of the ligand.
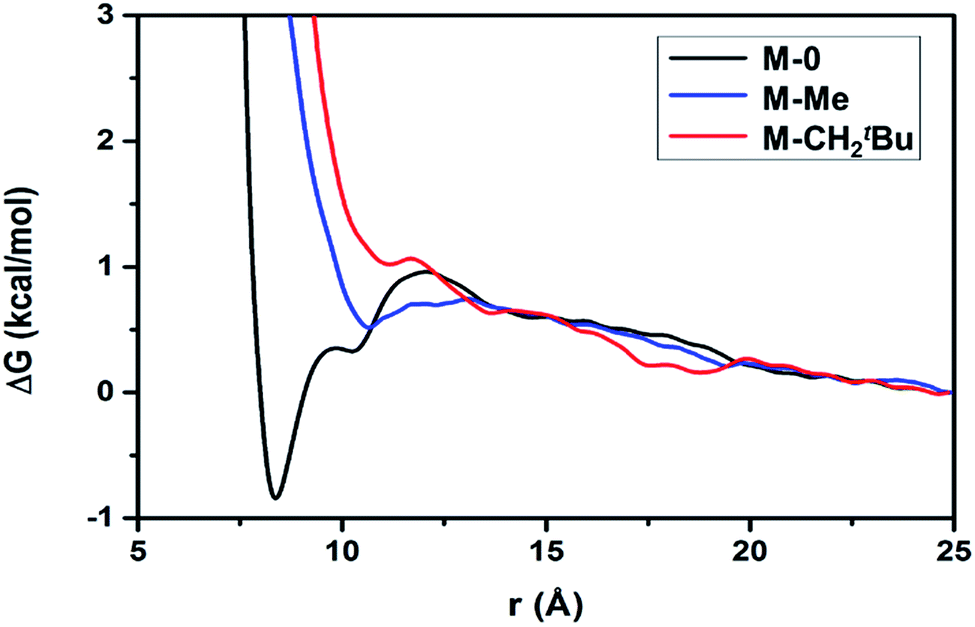 | ||
| Fig. 1 Free energy as a function of distance (r) between centers of masses of two prototypical silica particles M-0, M-Me or M-CH2tBu illustrates the trend of aggregation between these two particles in o-DCB solution. M-0, M-Me, and M-CH2tBu correspond to [SiO1.5(OH)]8, [SiO1.5(OtBu)0.5(OH)0.5]8, and [SiO1.5(OC(CH2tBu)3)0.5(OH)0.5]8, respectively. Details of the free energy calculation are summarized in the ESI.† | ||
The materials remain more hydrophilic (higher CBET) with lower carbon content which holds true for (i) changing the ligand from –CH2tBu to –iBu to –Me, (ii) changing the number of ligands and (iii) decreasing the SOMF loading from high (cSOMF > 0.18 mmol gsilica−1) to med (0.1 < cSOMF < 0.18 mmol gsilica−1) to low (cSOMF < 0.1 mmol gsilica−1). The CBET value is around 75 for lower loadings of SOMFs having lower carbon contents (e.g.5-low, 5-med, and 3-low) and decreases to around 30 for higher loadings of SOMFs with higher carbon content (e.g.1-med, 4-high, and 6-high). The results are illustrated in Fig. 2a.
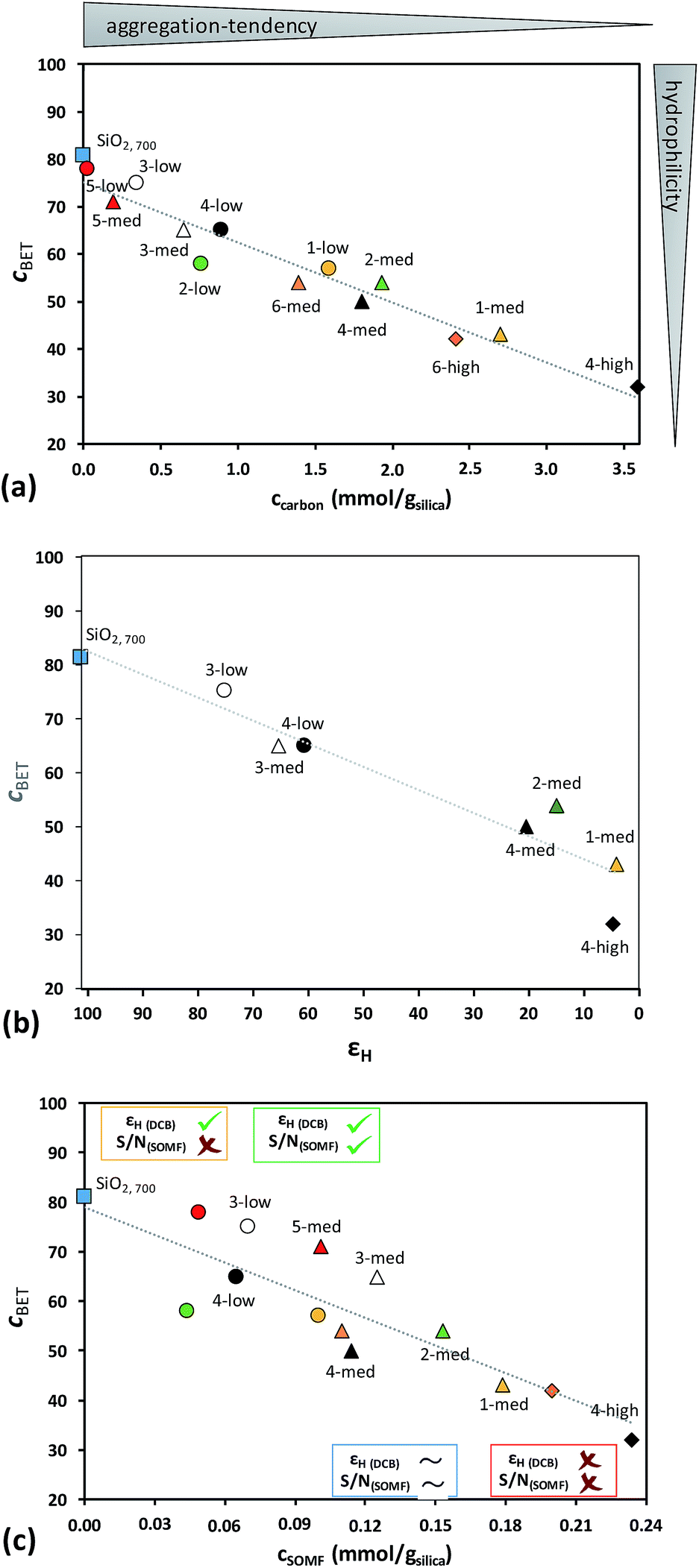 | ||
| Fig. 2 (a) Correlation of CBET and ccarbon of SOMFs 1–6 at various concentrations; (b) correlation of CBET with 1H enhancement factors εH (o-DCB) and (c) correlation of CBET and cSOMF of SOMFs 1–6 at different concentrations. The various regions (yellow, blue, red and green) indicate where DNP SENS measurements are possible and where they are not. S/N(SOMF) corresponds to the signal to noise ratio of the SOMF. | ||
We have taken seven silica NPs loaded with various SOMFs at different concentrations along the trend line in Fig. 2b and investigated them in more detail by DNP SENS: 1-med (CBET = 43), 2-med (CBET = 54), 3-low (CBET = 75), 3-med (CBET = 65), 4-low (CBET = 65), 4-med (CBET = 50) and 4-high (CBET = 32). DNP SENS experiments of 3 and 4 are particularly interesting, as the results can be compared to previous experiments where the SOMF was protected inside mesopores of MCM-41.27
For DNP-SENS experiments, ortho-dichlorobenzene (o-DCB)64 was used as the solvent because its carbon resonances (δ = 115–140 ppm) do not overlap with the expected 13C chemical shifts of SOMFs for most investigations27 (though it has recently been shown that the solvent resonances can be suppressed if needed).65 The maximum solvent 1H enhancement, εH, for pure SiO2, 7000 impregnated with a 16 mM solution of TEKPol in o-DCB obtained here was εH = 101 (the TEKPol is inert on silica) and serves in the following as an indicator for estimating the destruction of TEKPol by reaction with the SOMF. Typically, TEKPol (biradical) is added in default (0.6 μmol) with respect to the SOMF (1.6–4.7 μmol). Depending on the SOMF loading, a SOMF/radical ratio of at least ≥1 is used for DNP SENS experiments. This ratio suggests that the radical should be completely destroyed in all cases if we assume full accessibility of the SOMFs (Table S1, ESI†). Consequently, no enhancement should be detected. For example, when SOMFs 3 and 4 were immobilized on SBA15 (dpore = 6.0 nm),27 we were not able to detect signal enhancements by DNP SENS after the samples were impregnated with TEKPol. We found by quantitative EPR that the radical concentration for 4-high was 94% lower as compared to non-functionalized silica nanoparticles 0 (1 h after impregnation), and decreased further with time (99% lower after 4.5 h) (Table S5 and Fig. S9, ESI†).
Comparing the measured CBET value with the experimental solvent 1H enhancement factors obtained from DNP SENS experiments in o-DCB (Fig. 2b), we observe a correlation between increasing εH and increasing hydrophilicity of silica nanoparticles.
Low levels of functionalization of the silica NPs (such as 1-low, 2-low, and 3-med having a CBET > 60) lead to aggregation of the silica NPs, which partially prevents reaction between TEKPol and the SOMF and hence enables reasonable solvent enhancements (εH > 60). A value in the range of 50 < CBET < 60 is observed for silica NPs loaded with SOMFs bearing –CH2tBu ligands at a medium concentration (2-med and 4-med). The concomitant 1H enhancements are 10 < εH < 20. A higher degree of functionalization (1-med and 4-high; CBET < 50) leads to a higher dispersion of silica NPs and hence SOMFs are more exposed to react with TEKPol, leading to low 1H enhancements (εH < 5).
Thus, we see that for successful characterization of the SOMFs by DNP SENS the aggregation tendency of the silica NPs (deduced from the CBET value) and the concentration of SOMFs are significant, as illustrated in Fig. 2c. Note that in this study all data were recorded at a constant radical concentration. The surface area of the silica samples here is around 200 m2 g−1, and the 16 mM radical concentration used is the optimal TEKPol concentration determined by Perras et al. for silica with 186 m2 g−1 surface area,40 as was determined previously.64
Analysis of the data in Fig. 2c leads to the following conclusions:
(I) For CBET < 60 and cSOMF > 0.17 mmol g−1 (1-med and 4-high), the (almost) fully passivated SOMF-covered silica NPs are more hydrophobic compared to SiO2, 700 leading to better dispersed particles in non-polar solvents. The SOMF is not protected by the interparticle space and the radical/surface interaction leads to (almost) complete decomposition of the polarizing agent. Hence, (almost) no enhancement εH and surface signal are obtained (Fig. 2 and S7, ESI†).
(II) For CBET < 60 and 0.095 < cSOMF < 0.17 mmol g−1 (2-med and 4-med), the polarization agent is partially destroyed, as observed by the low solvent εH enhancements (10–20), corresponding to a long build-up time (TB,ON = 18.7 s). The 13C CP MAS NMR spectra of 2-med (S/N = 20) as well as 4-med (S/N = 28) showed a low signal-to-noise ratio for C(![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) H3)3 after 10
H3)3 after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 16000 scans, respectively (Fig. S7, ESI†). The long build-up time indicates that the biradical is significantly decomposed (a lower radical content in solution will increase TB);66 however the observed solvent enhancement shows that the residual biradical does succeed in providing a modest enhancement in the surface species.
000 and 16000 scans, respectively (Fig. S7, ESI†). The long build-up time indicates that the biradical is significantly decomposed (a lower radical content in solution will increase TB);66 however the observed solvent enhancement shows that the residual biradical does succeed in providing a modest enhancement in the surface species.
(III) For CBET > 60 and 0.075 < cSOMF < 0.13 mmol g−1 (3-med), we obtain the most beneficial properties to enable characterization of the SOMF by DNP SENS. For SOMF 3-med, 1H MAS, 13C (S/N = 13 for Me1 after 288 scans), and 29Si CP MAS DNP SENS spectra (S/N = 27 after 1024 scans) with reasonable S/N ratios are acquired within 1.5 hours (Fig. 3) with εH (o-DCB) = 65.4(0.6), εC, CP (3-med) = 27(9) and εSi, CP (3-med) = 36(3) and with 8, 288 and 1024 scans, respectively. A 1H–13C HETCOR DNP SENS spectrum was recorded in 10 hours (128 scans, 96 increments, t1 = 3 s; Fig. S6, ESI†), which would take 375 days without using DNP SENS.
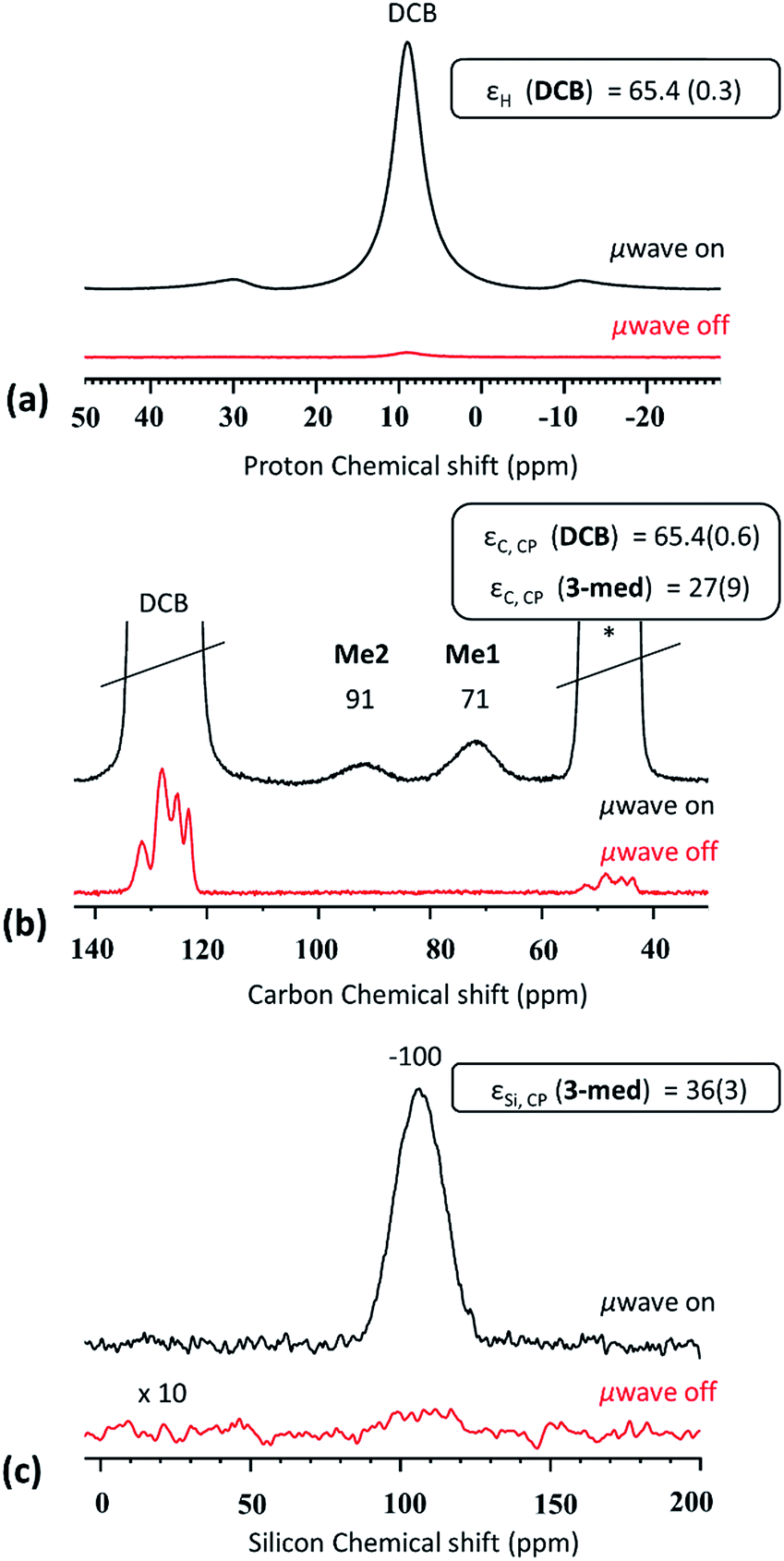 | ||
| Fig. 3 (a) DNP SENS 1H MAS, (b) 13C CP MAS and (c) 29Si CP MAS (100 K, 400 MHz/263 GHz gyrotron) of 3-med in 16 mM TEKPol o-DCB solution obtained at 8 kHz MAS. The red line represents the spectra without microwave irradiation (μwave off), and the black line represents experiments with microwave irradiation (μwave on). The stars indicate the spinning side band. | ||
The success of the experiments is attributed to the aggregation of the SOMF-covered silica NPs in non-polar solvent preventing the interaction between the radical and the reactive surface species inside the aggregates. Note that a small amount of reactive surface species is located on the outside of the aggregates, leading to slight decomposition of the bi-radicals, as indicated by a fairly short build-up time (TB,ON = 6.1 s) but the latter is longer than in a bulk 16 mM TEKPol solution (TB,ON = 4 s). Hence, the destruction of the polarization agent is mainly avoided. The 1H surface enhancement εC, CP ∼ 30 is lower than the bulk solvent enhancement which is a signature of polarization relay from outside the aggregates.
(IV) For CBET > 60 and cSOMF < 0.075 mmol g−1 (3-low and 4-low), a high solvent enhancement is detected (εH (o-DCB) = 80), but the intensities of SOMF signals in 13C CP MAS DNP SENS experiments remain weak (Fig. S5 and S6, ESI†). The short build-up time (TB,ON = 4.8 s), close to the value expected for bulk 16 mM TEKPol, indicates that the polarizing agent does not interact with the reactive surface species. Indeed, the high CBET leads to a good protection of the SOMF inside the aggregates, and the low SOMF concentration leads to a negligible quantity on the aggregate surface.
Conclusions
In conclusion, the propensity of silica NPs for aggregation can be controlled and yield high DNP SENS efficiency for reactive surface species. Differences in the DNP SENS results are attributed to the modification of the spatial properties of silica aggregates upon grafting of surface organometallic fragments. In the regime where hydrophilic nanoparticles form aggregates that allow the solvent (here o-DCB) to enter the interparticle space, but do not allow entry of the bulky TEKPol radicals (obtained when CBET > 60 and the SOMF concentration is modest (∼0.13 mmol gsilica−1)), enhancement factors of up to 30 are obtain for the reactive surface species. High SOMF concentrations (cSOMF > 0.2 mmol gsilica−1) lead to hydrophobic silica nanoparticles (CBET < 35) which are highly dispersed in the non-polar solvent (o-DCB), and hence the SOMF is accessible for reaction with TEKPol, and DNP SENS cannot be applied. Similarly silica nanoparticles having a low SOMF concentration (<0.10 mmol gsilica−1) remain hydrophilic (CBET > 60) and create aggregates where neither the TEKPol nor the solvent can enter the interparticle space, and DNP SENS is also inefficient. Contact of the solvent o-DCB with both SOMF and TEKPol is important to guarantee the transfer of polarization.Finally, we note that the quality of the DNP SENS spectrum of SOMF 3 on silica nanoparticles (3-med) and encapsulated inside MCM41 (3-MCM41)27 is similar. However, the two approaches are complementary. The MCM41-based approach is tunable, but requires a specific synthetic approach, whereas the approach here can be applied to native systems if they fulfill the criteria for agglomeration, without inducing any change in catalytic activity.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work received support from the King Abdullah University of Science and Technology (KAUST) and the European Research Council (ERC Advanced Grant No. 320860). We thank Dr Olivier Ouari and Paul Tordo of the University of Aix-Marseille for providing the TEKPol radical.Notes and references
- J. D. A. Pelletier and J.-M. Basset, Acc. Chem. Res., 2016, 49, 664–677 CrossRef CAS PubMed
.
-
C. Li and Y. Liu, Bridging Heterogeneous and Homogeneous Catalysis: Concepts, Strategies, and Applications, Wiley, 2014 Search PubMed
- M. P. Conley, A. J. Rossini, A. Comas-Vives, M. Valla, G. Casano, O. Ouari, P. Tordo, A. Lesage, L. Emsley and C. Coperet, Phys. Chem. Chem. Phys., 2014, 16, 17822–17827 RSC
-
B. Trewyn, Heterogeneous Catalysis for Today's Challenges: Synthesis, Characterization and Applications, Royal Society of Chemistry, 2015 Search PubMed
- C. Coperet, M. Chabanas, R. P. Saint-Arroman and J. M. Basset, Angew. Chem., Int. Ed., 2003, 42, 156–181 CrossRef CAS PubMed
- C. Copéret, D. P. Estes, K. Larmier and K. Searles, Chem. Rev., 2016, 116, 8463–8505 CrossRef PubMed
- A. Bendjeriou-Sedjerari, J. M. Azzi, E. Abou-Hamad, D. H. Anjum, F. A. Pasha, K. W. Huang, L. Emsley and J. M. Basset, J. Am. Chem. Soc., 2013, 135, 17943–17951 CrossRef CAS PubMed
- A. Bendjeriou-Sedjerari, J. Sofack-Kreutzer, Y. Minenkov, E. Abou-Hamad, B. Hamzaoui, B. Werghi, D. H. Anjum, L. Cavallo, K.-W. Huang and J.-M. Basset, Angew. Chem., Int. Ed., 2016, 55, 11162–11166 CrossRef CAS PubMed
- R. Anwander, Chem. Mater., 2001, 13, 4419–4438 CrossRef CAS
- C. Copéret, A. Comas-Vives, M. P. Conley, D. P. Estes, A. Fedorov, V. Mougel, H. Nagae, F. Núñez-Zarur and P. A. Zhizhko, Chem. Rev., 2016, 116, 323–421 CrossRef PubMed
- L. T. Zhuravlev, Colloids Surf., A, 2000, 173, 1–38 CrossRef CAS
-
J.-M. Basset, E. Callens and N. Riache, in Handbook of Metathesis, Wiley-VCH Verlag GmbH & Co. KGaA, 2015, pp. 33–70 Search PubMed
- F. Blanc, C. Coperet, A. Lesage and L. Emsley, Chem. Soc. Rev., 2008, 37, 518–526 RSC
- A. Lesage, M. Lelli, D. Gajan, M. A. Caporini, V. Vitzthum, P. Miéville, J. Alauzun, A. Roussey, C. Thieuleux, A. Mehdi, G. Bodenhausen, C. Copéret and L. Emsley, J. Am. Chem. Soc., 2010, 132, 15459–15461 CrossRef CAS PubMed
- M. Lelli, D. Gajan, A. Lesage, M. A. Caporini, V. Vitzthum, P. Mieville, F. Heroguel, F. Rascon, A. Roussey, C. Thieuleux, M. Boualleg, L. Veyre, G. Bodenhausen, C. Coperet and L. Emsley, J. Am. Chem. Soc., 2011, 133, 2104–2107 CrossRef CAS PubMed
- A. J. Rossini, A. Zagdoun, M. Lelli, A. Lesage, C. Copéret and L. Emsley, Acc. Chem. Res., 2013, 46, 1942–1951 CrossRef CAS PubMed
- M. P. Conley, R. M. Drost, M. Baffert, D. Gajan, C. Elsevier, W. T. Franks, H. Oschkinat, L. Veyre, A. Zagdoun, A. Rossini, M. Lelli, A. Lesage, G. Casano, O. Ouari, P. Tordo, L. Emsley, C. Copéret and C. Thieuleux, Chem.–Eur. J., 2013, 19, 12234–12238 CrossRef CAS PubMed
- W. R. Gruning, A. J. Rossini, A. Zagdoun, D. Gajan, A. Lesage, L. Emsley and C. Coperet, Phys. Chem. Chem. Phys., 2013, 15, 13270–13274 RSC
- O. Lafon, A. S. L. Thankamony, T. Kobayashi, D. Carnevale, V. Vitzthum, I. I. Slowing, K. Kandel, H. Vezin, J. P. Amoureux, G. Bodenhausen and M. Pruski, J. Phys. Chem. C, 2013, 117, 1375–1382 CAS
- M. P. Conley, C. Coperet and C. Thieuleux, ACS Catal., 2014, 4, 1458–1469 CrossRef CAS
- N. Eedugurala, Z. Wang, U. Chaudhary, N. Nelson, K. Kandel, T. Kobayashi, I. I. Slowing, M. Pruski and A. D. Sadow, ACS Catal., 2015, 5, 7399–7414 CrossRef CAS
- T. Gutmann, J. Liu, N. Rothermel, Y. Xu, E. Jaumann, M. Werner, H. Breitzke, S. T. Sigurdsson and G. Buntkowsky, Chem.–Eur. J., 2015, 21, 3798–3805 CrossRef CAS PubMed
- I. Romanenko, D. Gajan, R. Sayah, D. Crozet, E. Jeanneau, C. Lucas, L. Leroux, L. Veyre, A. Lesage, L. Emsley, E. Lacote and C. Thieuleux, Angew. Chem., Int. Ed., 2015, 54, 12937–12941 CrossRef CAS PubMed
- M. Werner, A. Heil, N. Rothermel, H. Breitzke, P. B. Groszewicz, A. S. Thankamony, T. Gutmann and G. Buntkowsky, Solid State Nucl. Magn. Reson., 2015, 72, 73–78 CrossRef CAS PubMed
- B. Hamzaoui, A. Bendjeriou-Sedjerari, E. Pump, E. Abou-Hamad, R. Sougrat, A. Gurinov, K. W. Huang, D. Gajan, A. Lesage, L. Emsley and J. M. Basset, Chem. Sci., 2016, 7, 6099–6105 RSC
- T. C. Ong, W. C. Liao, V. Mougel, D. Gajan, A. Lesage, L. Emsley and C. Coperet, Angew. Chem., Int. Ed., 2016, 55, 4743–4747 CrossRef CAS PubMed
- E. Pump, J. Viger-Gravel, E. Abou-Hamad, M. K. Samantaray, B. Hamzaoui, A. Gurinov, D. H. Anjum, D. Gajan, A. Lesage, A. Bendjeriou-Sedjerari, L. Emsley and J.-M. Basset, Chem. Sci., 2017, 8, 284–290 RSC
- W.-C. Liao, T.-C. Ong, D. Gajan, F. Bernada, C. Sauvee, M. Yulikov, M. Pucino, R. Schowner, M. Schwarzwalder, M. R. Buchmeiser, G. Jeschke, P. Tordo, O. Ouari, A. Lesage, L. Emsley and C. Coperet, Chem. Sci., 2017, 8, 416–422 RSC
- P. Berruyer, M. Lelli, M. P. Conley, D. L. Silverio, C. M. Widdifield, G. Siddiqi, D. Gajan, A. Lesage, C. Coperet and L. Emsley, J. Am. Chem. Soc., 2017, 139, 849–855 CrossRef CAS PubMed
-
K. P. de Jong, Synthesis of solid catalysts, John Wiley & Sons, 2009 Search PubMed
- G. J. Gerfen, L. R. Becerra, D. A. Hall, R. G. Griffin, R. J. Temkin and D. J. Singel, J. Chem. Phys., 1995, 102, 9494–9497 CrossRef CAS
- K. W. Huang and R. M. Waymouth, J. Am. Chem. Soc., 2002, 124, 8200–8201 CrossRef CAS PubMed
- T. Iwamoto, H. Masuda, S. Ishida, C. Kabuto and M. Kira, J. Am. Chem. Soc., 2003, 125, 9300–9301 CrossRef CAS PubMed
- H. Takahashi, S. Hediger and G. De Paepe, Chem. Commun., 2013, 49, 9479–9481 RSC
- C. Fernández-de-Alba, H. Takahashi, A. Richard, Y. Chenavier, L. Dubois, V. Maurel, D. Lee, S. Hediger and G. De Paëpe, Chem.–Eur. J., 2015, 21, 4512–4517 CrossRef PubMed
- L. Piveteau, T. C. Ong, A. J. Rossini, L. Emsley, C. Coperet and M. V. Kovalenko, J. Am. Chem. Soc., 2015, 137, 13964–13971 CrossRef CAS PubMed
- K. G. Valentine, G. Mathies, S. Bédard, N. V. Nucci, I. Dodevski, M. A. Stetz, T. V. Can, R. G. Griffin and A. J. Wand, J. Am. Chem. Soc., 2014, 136, 2800–2807 CrossRef CAS PubMed
- J. Viger-Gravel, P. Berruyer, D. Gajan, J.-M. Basset, A. Lesage, P. Tordo, O. Ouari and L. Emsley, Angew. Chem., Int. Ed., 2017, 56, 8726–8730 CrossRef CAS PubMed
- W. R. Gunther, V. K. Michaelis, M. A. Caporini, R. G. Griffin and Y. Roman-Leshkov, J. Am. Chem. Soc., 2014, 136, 6219–6222 CrossRef CAS PubMed
- F. A. Perras, L.-L. Wang, J. S. Manzano, U. Chaudhary, N. N. Opembe, D. D. Johnson, I. I. Slowing and M. Pruski, Curr. Opin. Colloid Interface Sci., 2018, 33, 9–18 CrossRef CAS
- J. Beckmann, D. Dakternieks, A. Duthie, M. L. Larchin and E. R. T. Tiekink, Appl. Organomet. Chem., 2003, 17, 52–62 CrossRef CAS
- I. S. Chuang and G. E. Maciel, J. Phys. Chem. B, 1997, 101, 3052–3064 CrossRef CAS
- L. Bergstrom, Adv. Colloid Interface Sci., 1997, 70, 125–169 CrossRef CAS
- B. Vincent, Z. Király, S. Emmett and A. Beaver, Colloids Surf., 1990, 49, 121–132 CrossRef CAS
- D. H. Lee, J. Jeong, S. W. Han and D. P. Kang, J. Mater. Chem. A, 2014, 2, 17165–17173 CAS
- S. D. Bhagat, Y.-H. Kim, K.-H. Suh, Y.-S. Ahn, J.-G. Yeo and J.-H. Han, Microporous Mesoporous Mater., 2008, 112, 504–509 CrossRef CAS
- N. Rakhshan and M. Pakizeh, Korean J. Chem. Eng., 2015, 32, 2524–2533 CrossRef CAS
- R. P. Bagwe, L. R. Hilliard and W. Tan, Langmuir, 2006, 22, 4357–4362 CrossRef CAS PubMed
- A. J. Worthen, V. Tran, K. A. Cornell, T. M. Truskett and K. P. Johnston, Soft Matter, 2016, 12, 2025–2039 RSC
- A. Zagdoun, A. J. Rossini, M. P. Conley, W. R. Gruning, M. Schwarzwalder, M. Lelli, W. T. Franks, H. Oschkinat, C. Coperet, L. Emsley and A. Lesage, Angew.
Chem., Int. Ed., 2013, 52, 1222–1225 CrossRef CAS PubMed
- L. Jelinek and E. S. Kovats, Langmuir, 1994, 10, 4225–4231 CrossRef CAS
- D. Brunel, A. Cauvel, F. Di Renzo, F. Fajula, B. Fubini, B. Onida and E. Garrone, New J. Chem., 2000, 24, 807–813 RSC
-
J. A. Osaheni and S. T. Buddle, US Pat., 6193412 B1, 2001
- M. K. Samantaray, E. Callens, E. Abou-Hamad, A. J. Rossini, C. M. Widdifield, R. Dey, L. Emsley and J. M. Basset, J. Am. Chem. Soc., 2014, 136, 1054–1061 CrossRef CAS PubMed
- E. Le Roux, M. Taoufik, M. Chabanas, D. Alcor, A. Baudouin, C. Coperet, J. Thivolle-Cazat, J. M. Basset, A. Lesage, S. Hediger and L. Emsley, Organometallics, 2005, 24, 4274–4279 CrossRef CAS
- M. Adachi, C. Nédez, X. X. Wang, F. Bayard, V. Dufaud, F. Lefebvre and J.-M. Basset, J. Mol. Catal. A: Chem., 2003, 204, 443–455 CrossRef
- F. Bini, C. Rosier, R. P. Saint-Arroman, E. Neumann, C. Dablemont, A. de Mallmann, F. Lefebvre, G. P. Niccolai, J. M. Basset, M. Crocker and J. K. Buijink, Organometallics, 2006, 25, 3743–3760 CrossRef CAS
- K. C. Szeto, A. Gallo, S. Hernández-Morejudo, U. Olsbye, A. De Mallmann, F. Lefebvre, R. M. Gauvin, L. Delevoye, S. L. Scott and M. Taoufik, J. Phys. Chem. C, 2015, 119, 26611–26619 CAS
- A. Hamieh, R. Dey, M. K. Samantaray, S. Abdel-Azeim, E. Abou-Hamad, Y. Chen, J. D. A. Pelletier, L. Cavallo and J.-M. Basset, Organometallics, 2016, 35, 2524–2531 CrossRef CAS
- J. J. Yuan, S. X. Zhou, G. X. Gu and L. M. Wu, J. Mater. Sci., 2005, 40, 3927–3932 CrossRef CAS
- Z. Cao, Y. Peng, S. Li, L. Liu and T. Yan, J. Phys. Chem. C, 2009, 113, 3096–3104 CAS
- N. Choudhury and B. M. Pettitt, J. Am. Chem. Soc., 2005, 127, 3556–3567 CrossRef CAS PubMed
- R. Zangi, M. Hagen and B. J. Berne, J. Am. Chem. Soc., 2007, 129, 4678–4686 CrossRef CAS PubMed
- A. Zagdoun, A. J. Rossini, D. Gajan, A. Bourdolle, O. Ouari, M. Rosay, W. E. Maas, P. Tordo, M. Lelli, L. Emsley, A. Lesage and C. Coperet, Chem. Commun., 2012, 48, 654–656 RSC
- J. R. Yarava, S. R. Chaudhari, A. J. Rossini, A. Lesage and L. Emsley, J. Magn. Reson., 2017, 277, 149–153 CrossRef CAS PubMed
- A. C. Pinon, J. Schlagnitweit, P. Berruyer, A. J. Rossini, M. Lelli, E. Socie, M. X. Tang, T. Pham, A. Lesage, S. Schantz and L. Emsley, J. Phys. Chem. C, 2017, 121, 15993–16005 CAS
- D. L. Silverio, H. A. van Kalkeren, T.-C. Ong, M. Baudin, M. Yulikov, L. Veyre, P. Berruyer, S. Chaudhari, D. Gajan, D. Baudouin, M. Cavaillés, B. Vuichoud, A. Bornet, G. Jeschke, G. Bodenhausen, A. Lesage, L. Emsley, S. Jannin, C. Thieuleux and C. Copéret, Helv. Chim. Acta, 2017, 100, e1700101 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: All experimental details such as detailed synthetic procedures, elemental analysis, dynamic nuclear polarization enhanced NMR spectroscopy, nitrogen sorption, transmission-electron microscopy, and computational results, as well as NMR data for all the spectra. See DOI: 10.1039/c8sc00532j |
| This journal is © The Royal Society of Chemistry 2018 |