
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLanthanide-based tools for the investigation of cellular environments
Emilie
Mathieu
,
Agnès
Sipos†
 ,
Ellen
Demeyere‡
,
Dulcie
Phipps§
,
Dimitra
Sakaveli¶
and
K. Eszter
Borbas
*
,
Ellen
Demeyere‡
,
Dulcie
Phipps§
,
Dimitra
Sakaveli¶
and
K. Eszter
Borbas
*
Department of Chemistry, Ångström Laboratory, Uppsala University, Box 523, 75120, Uppsala, Sweden. E-mail: eszter.borbas@kemi.uu.se
First published on 31st July 2018
Abstract
Biological probes constructed from lanthanides can provide a variety of readout signals, such as the luminescence of Eu(III), Tb(III), Yb(III), Sm(III) and Dy(III), and the proton relaxation enhancement of Gd(III) and Eu(II). For numerous applications the intracellular delivery of the lanthanide probe is essential. Here, we review the methods for the intracellular delivery of non-targeted complexes (i.e. where the overall complex structure enhances cellular uptake), as well as complexes attached to a targeting unit (i.e. to a peptide or a small molecule) that facilitates delivery. The cellular applications of lanthanide-based supramolecules (dendrimers, metal organic frameworks) are covered briefly. Throughout, we emphasize the techniques that can confirm the intracellular localization of the lanthanides and those that enable the determination of the fate of the probes once inside the cell. Finally, we highlight methods that have not yet been applied in the context of lanthanide-based probes, but have been successful in the intracellular delivery of other metal-based probes.
 Emilie Mathieu | Emilie Mathieu was born in Echirolles, France. She obtained her MSc in Chemistry in 2014 (Sorbonne University and the Ecole Normale Supérieure, Paris, France); she worked on the synthesis of gentiopicroside (Prof. Serge Thorimbert, Dr Luc Dechoux). She then worked on the role of copper in Alzheimer's disease (Prof. Tim Storr, Vancouver, Canada). During her PhD, which she obtained in September 2017, she studied manganese complex superoxide dismutase mimics and their cellular behavior under the supervision of Prof. Clotilde Policar and Dr Nicolas Delsuc. She is currently a postdoctoral fellow at Uppsala University, working on lanthanide coordination chemistry. |
 Agnès Sipos | Agnès Sipos was born in Meudon (France) in 1996. She obtained her BSc degree in 2017 from the Ecole Normale Supérieure in Paris after an internship in Christelle Hureau's group in Toulouse, where she did research on metal in a sperm peptide. She joined the Borbas group for an internship during her 1st year of master's. She is interested in inorganic chemistry and its applications to biology. She is preparing for the competitive teachers’ examination before finishing her master's degree. |
 Ellen Demeyere | Ellen Demeyere was born in 1997 in Ghent, Belgium. She earned her BSc in chemistry in 2018 from Ghent University after having completed an Erasmus exchange at Uppsala University, Sweden, where she worked in the research group of Eszter Borbas on the design and synthesis of luminescent lanthanide complexes. She will start her MSc studies in chemistry at Ghent University in the fall of 2018 with focus on organic chemistry. |
 Dulcie Phipps | Dulcie Phipps was born in Birmingham, England, but moved to Scotland when she was two years old and has lived there all her life. She is an undergraduate student at the University of Glasgow, with interest in nanoscience and in particular coordination chemistry. After graduating from her final year in 2019, Dulcie plans to find a PhD outside of the UK. |
 Dimitra Sakaveli | Dimitra Sakaveli was born in Heraklion of Crete, Greece. Dimitra completed her BSc in 2017 at the Department of Chemistry at the University of Crete, after completing an undergraduate thesis in the Laboratory of Bioinorganic Chemistry under the supervision of Professor A. Coutsolelos. In 2018 she worked in the Borbas lab at Uppsala University as part of a three month Erasmus+ internship. She is interested in materials chemistry and medical applications. Next year she will start an MSc in the Medical School at the University of Crete. |
 K. Eszter Borbas | Eszter Borbas grew up in Hungary and in Yemen. After completing her undergraduate studies at Eötvös University in Budapest with research work in József Rábai's group, she did her PhD at the Open University (with James Bruce), and she was a postdoc at North Carolina State University (with Jonathan Lindsey), and a Marie Curie fellow at Stockholm University in Sweden. She started her independent career in 2010 as a Swedish Research Council assistant professor. She is currently an associate professor at Uppsala University, where her research group works on lanthanide chemistry and photophysics, and on tetrapyrrole chemistry. |
Introduction
Probes based on lanthanide(III) ions are ubiquitous in the life sciences. The luminescence of Eu (red) and Tb (green), and, increasingly, Sm (orange), Dy (yellow), Yb and Nd (both near infrared; NIR) makes them excellent tools for spectroscopy or microscopy. Highly paramagnetic Gd(III) with its 7 unpaired electrons and isoelectronic Eu(II) are used as MRI contrast agents,1–6 although Eu(II) has interesting, and so far little explored photophysics.7–10Lanthanide luminescence is due to 4f–4f transitions. The f-electrons are buried and do not participate in bonding interactions with the ligand. Therefore, crystal field effects are small, and absorption and emission bands are sharp.11 The emission spectra of the lanthanide ions most often used in cellular imaging are shown in Fig. 1. The f–f transitions are forbidden by the Laporte selection rule, which can be relaxed by vibronic coupling or mixing with other orbitals. As a consequence, lanthanides have very weak molar extinction coefficients (∼1 L mol−1 cm−1),12 which implies that direct excitation of the Ln3+ ion will lead to very weak emission. This limitation is overcome by using a light-harvesting ‘antenna’, i.e. photosensitizer in the close vicinity of the metal ion, usually coupled to the ligand. Upon excitation, the antenna transfers its energy to the excited state of the Ln3+ ion, which can subsequently emit light (Fig. 1). The rate constant of the energy transfer between the antenna and the lanthanide ion is much higher compared to the luminescence emission lifetime of the lanthanide (kET ≫ klum).13 The efficiency of the energy transfer is dependent on the difference between the energy levels of the excited states of the ligand and the lanthanide. Quenching mechanisms include back energy transfer (BET, most often for Tb), photo-induced electron transfer (PeT, for the more reducible lanthanide(III) ions14,15), or coupling to X–H oscillators (X = O, N, C).16,17 Lanthanide emitter design has been covered in several recent reviews.18,19
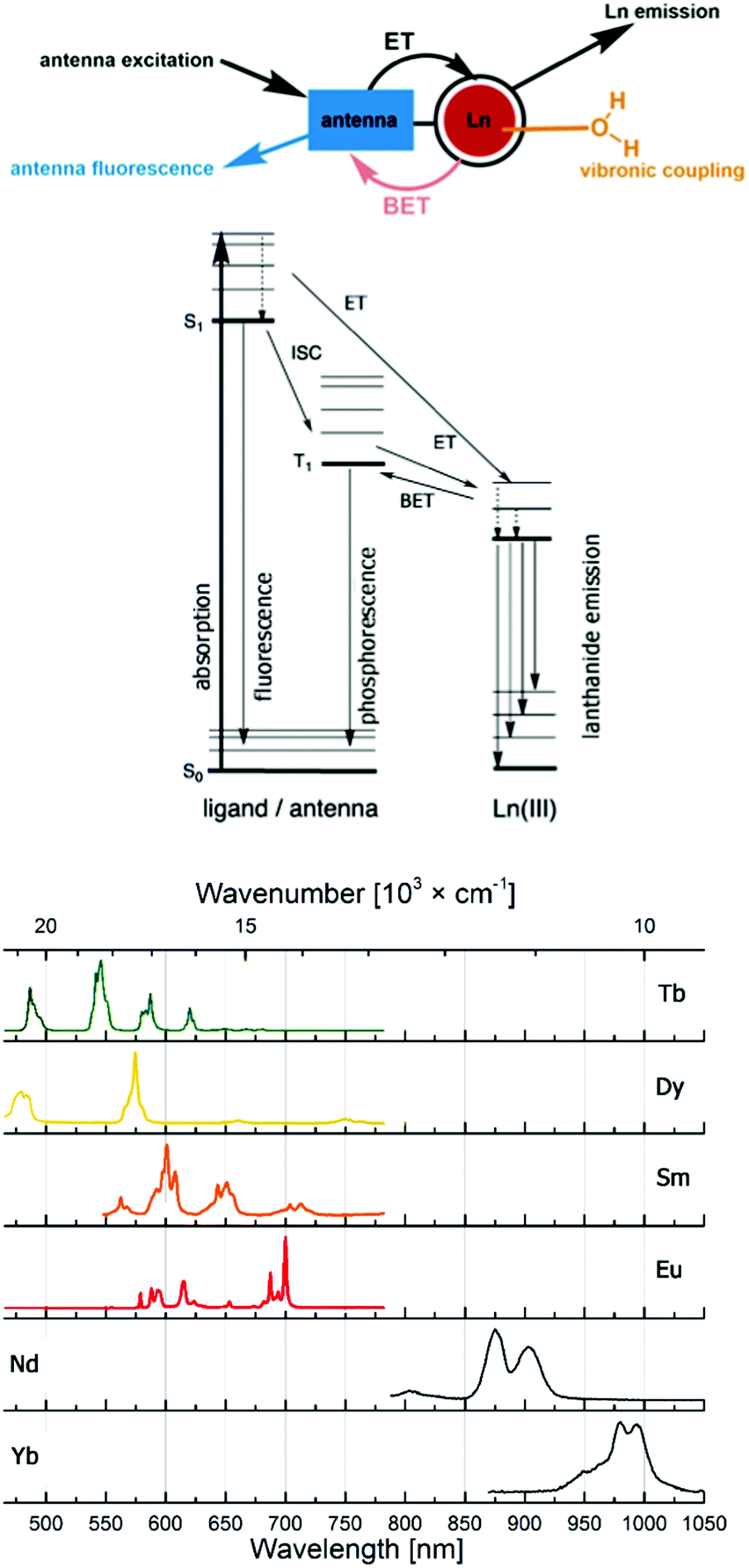 | ||
| Fig. 1 Antenna effect, with major deactivation pathways shown in colour, and productive pathways for lanthanide sensitization in black (top). Emission spectra of the most commonly used lanthanide(III) emitters (bottom). | ||
Lanthanide luminescence is characterized by long lifetimes (μs (Yb, Nd) to ms (Eu, Tb)) compared to that of organic fluorophores (ns).20 Large pseudo-Stokes shifts are observed, because the excitation wavelength of the antenna is usually in the 320–400 nm range, while that of the lanthanide emission is between 500 and 1500 nm. This makes lanthanide emitters less prone to auto-quenching. Furthermore, these complexes are more photostable than organic fluorophores.
Lanthanide luminescence is tailor-made for multiplex imaging. This includes not just the simultaneous detection of luminescent tags, but also the real-time monitoring of dynamic processes, and the measurements of analytes with changing concentrations, enzymes with changing activities, etc. One of the most challenging environments for doing this is in live cells, and not surprisingly, the dynamic multiplex detection of interacting analytes in living cells is not yet within reach. In order to achieve this goal, at least three things need to be optimized. (1) The design of the responsive probes needs to be optimized. These probes must of course be selective for their targets, have a large dynamic range, high water solubility and photostability, and low toxicity. Ideally, their design should be modular, and their synthesis short, convergent, and scalable.21 (2) The photophysical properties of the complexes need to be optimized. This comprises high quantum yields, which in turn necessitates the understanding of quenching pathways.14 (3) The probes must be able to enter target cells, and must be able to detect their target analytes.
Here, we will focus on the third aspect of probe design. We will present current strategies for delivering a lanthanide payload into cells, with an additional focus on the techniques that can confirm the internalization and the cellular localization of the metals. While we will focus on luminescent lanthanides, where appropriate, Gd(III) MRI contrast agents will be discussed. We will also briefly look at methods to target lanthanide complexes to the outside cell membranes. There are a number of relatively recent reviews on areas that partially overlap with the current topic: luminescent lanthanide labels in microscopy,22 lanthanide probes for detecting protein–protein interactions,23 responsive Eu-based probes,24 and the design of targeting extracellular receptors.25
Imaging techniques
The luminescent properties of lanthanide complexes are highly valuable to study their mechanism(s) of internalization in living cells. Cell penetration of these complexes can be followed in real time in living cells and give insight into uptake pathways and kinetics.26 Their long emission lifetimes limit signal contamination by endogenous chromophores (e.g. NAD(P)H, flavine, Trp, Tyr) that have nanosecond lifetimes.20Imaging techniques that take advantage of the photophysical properties of the lanthanides have been developed. Time-resolved fluorescence microscopy has been optimized to match the requirements of short and intense excitation pulses, tuneable delay times, and tuneable detection windows (Fig. 2).13,27–29 The introduction of a delay between an excitation pulse and the detection of the emission (10–200 μs) yields a luminescence signal free from the fluorescent background, which improves the signal-to-noise ratio.
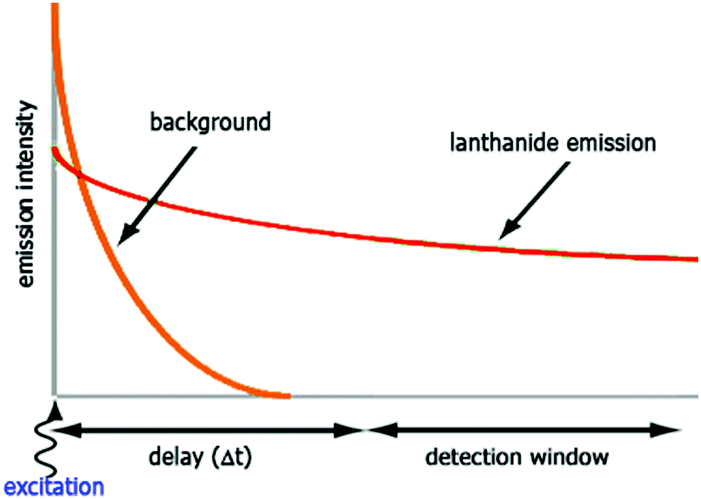 | ||
| Fig. 2 Time-resolved luminescence measurements. The sample is irradiated with a pulse at the excitation wavelength, and the emission intensity is recorded after a delay time (Δt) for a defined period (detection window). | ||
Multi-photon absorption microscopy is suitable for the study of lanthanide complexes. The wavelength used to excite the antenna (320–400 nm) can lead to photo-damage in cells. Two- and three-photon30 excitation spectroscopies use excitation wavelengths in the far visible/near-IR range (700–900 nm).31–34 Further developments have extended this method to NIR emitters (Yb, Nd).31–33 This technique can be used to follow the distribution of luminescent europium complexes in living T24 human cancer cells during mitosis as displayed in Fig. 3.34
 | ||
| Fig. 3 Intracellular distribution of [Na]3[EuL1G3] in live cells after mild saponin permeabilization. Adapted from ref. 34 with permission from the Royal Society of Chemistry. A mitotic cell 24 h after treatment is shown, and dye solution was added to the culture medium after saponin was rinsed out. Transmitted light DIC images of different stages of mitosis (A–C) and the corresponding [Na]3[EuL1G3] 2P luminescence images (D–F). | ||
Finally, lifetime imaging microscopy enables mapping of emission lifetimes in cells, which gives useful information on the environment of the complex. In short, the intensity decay in each pixel is fit by an exponential (or a multi-exponential) decay. The emission lifetime (τ) value for each pixel is extracted and a map showing the different τ values is constructed.13,20,32–34 Changes in emission lifetimes can give indications on O2 concentration in cells,35–37 colocalization with a FRET (Förster resonance energy transfer) acceptor,34 and changes in polarity, viscosity or lipophilicity.38
Mechanisms governing cell penetration
The subcellular localization of lanthanide complexes depends primarily on the mechanism(s) by which they are taken up by the cell. A combination of luminescence microscopy and the use of inhibitors or promoters of known uptake pathways has been used to better understand how the complexes are internalized in cells, and is presented in the following. Uptake pathways depend on the type of cell, and the size, charge and lipophilicity of the compounds. The cellular uptake mechanism(s) of non-functionalized lanthanide complexes is first described and followed by the modifications that have been made to lanthanide complexes to enhance or limit their cell penetration, as well as to direct them to subcellular compartments.Passive diffusion
The different uptake pathways are depicted in Fig. 4.26,39,40 The existence of a concentration gradient between the extracellular media and the cell can lead to a passive diffusion of complexes inside the cell.26 Measurements of cell penetration at low temperature (4–5 °C) help discriminate between passive and active internalization of compounds, but low temperature measurements imply the underestimation of the contribution of the translocation mechanism.41 Metabolic inhibitors can also give insights into the type of mechanism involved. The use of sodium azide,42 iodoacetate,42,43 or 2-deoxyglucose26 has been described in the literature. Indeed, active pathways are energy-dependent and are thus inhibited when working at low temperature.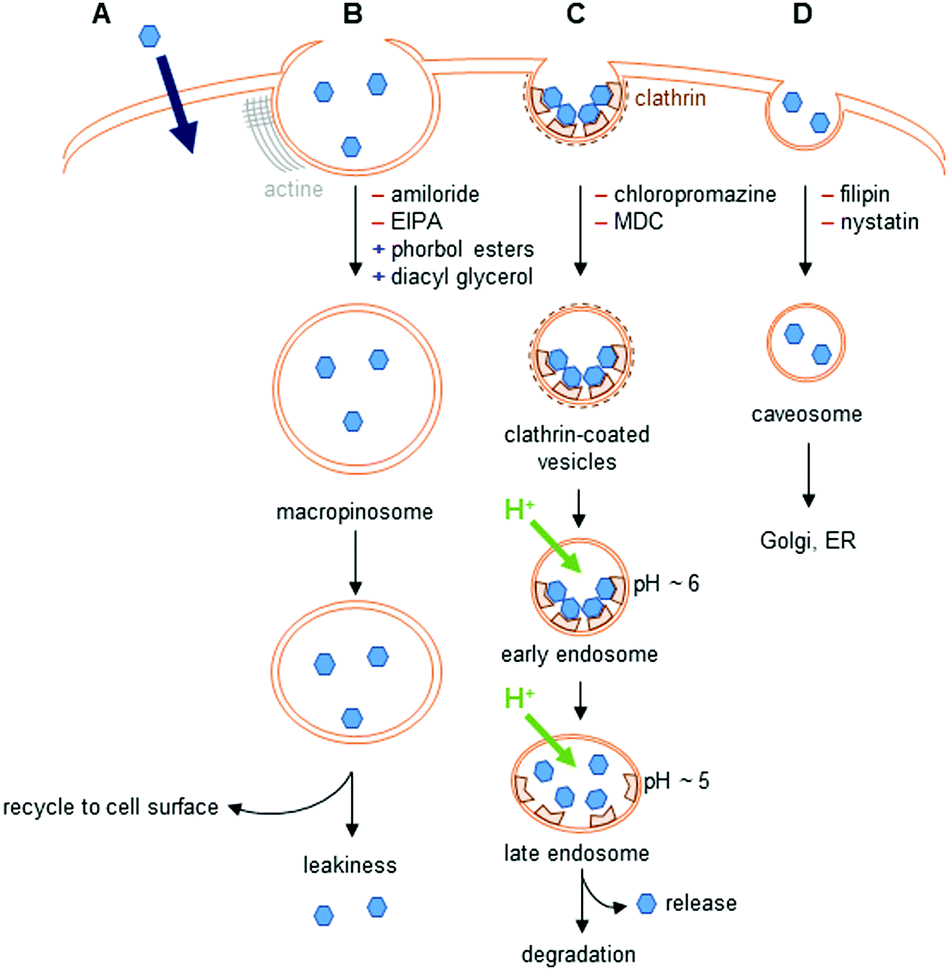 | ||
| Fig. 4 Uptake pathways in cells. (A) Passive diffusion. (B) Macropinocytosis. Amiloride and 5-(N-ethyl-N-isopropyl)amiloride (EIPA) are inhibitors of this pathway, whereas phorbol esters and diacyl glycerol promote it. (C) Clathrin-mediated endocytosis. Interaction with receptors at the cell surface leads to the formation of clathrin-coated vesicles. This pathway is inhibited by chlorpromazine, or monodansylcadaverine (MDC); however, these two inhibitors also disturb macropinocytosis. (D) Caveolin-dependent endocytosis. This pathway is inhibited by filipin and nystatin. | ||
Endocytosis
The endocytic pathways proceed through an invagination of the cell membrane that is energy-dependent, and lead to the internalization of macromolecules or solutes. There are two endocytic categories, phagocytosis, which occurs in macrophages, and pinocytosis, which is common to all cell types. Four pathways of pinocytosis are usually acknowledged: macropinocytosis, clathrin-mediated endocytosis, caveolin-mediated endocytosis, and clathrin/caveola-independent endocytosis.26,39,40Macropinocytosis corresponds to the actin-dependent formation of a cup-shaped ruffle at the cell surface, which closes to form a macropinosome.39 This vesicle is large, and complexes can leak into the cell. The macropinosome is then recycled to the cell surface. This pathway does not involve interaction with receptors.
In clathrin-mediated endocytosis, interaction of the complex with receptors at the surface of the cell leads to the formation of clathrin-coated vesicles.39 These vesicles mature into early, then late endosomes, before fusing with lysosomes, leading to the degradation of their content. During the maturation, the pH of the vesicles drops to ∼5. Endosomal release to lysosomes, the Golgi apparatus, or the nucleus has been observed. The content of endosomes can also be recycled to the cell surface.
Caveolin-dependent endocytosis is a small invagination of the cell membrane that leads to the formation of caveosomes in cells. These caveosomes are usually directed to the Golgi apparatus or to the endoplasmic reticulum (ER). This mechanism is involved in the uptake of, e.g., folic acid. Caveolin-dependent endocytosis was first described as an uptake mechanism that does not lead to lysosomal degradation, but this is now questioned as some evidence indicates that the vesicles formed fuse with lysosomes.40
Other uptake pathways that are mediated by interactions with receptors at the cell surface and are clathrin- and caveola-independent have also been described.
Different inhibitors and promoters can help investigate the mechanism(s) of cell penetration of lanthanide complexes, even though they are usually not specific inhibitors of one given pathway.26,39,40 Because these compounds interfere with cellular homeostasis, they can show non-specific toxicity and should be used with caution.26,40 Amiloride and its derivatives are known inhibitors of macropinocytosis that impact Na+/H+ exchange at the cell membrane and decrease the submembranous pH, whereas phorbol esters and diacyl glycerol promote macropinocytosis. Among the other inhibitors described in the literature, wortmannin is frequently used. However, this inhibitor is not specific, and can also inhibit clathrin- and caveolin-mediated endocytosis. Clathrin-mediated endocytosis is inhibited by chlorpromazine, monodansylcadaverine (MDC) and dynasore. However, these inhibitors can impact other endocytic pathways: the first two inhibiting macropinocytosis, while dynasore acts on caveolin-mediated endocytosis. K+ depletion or the use of a hypertonic medium enriched with sucrose also inhibits clathrin-mediated endocytosis. These methods are non-selective, and can perturb several endocytic pathways, as well as cellular physiology. Finally, inhibitors of caveolin-dependent endocytosis include filipin and nystatin, with pronounced toxicity.40
Colocalization with stains specific for endosomes or lysosomes can be used to obtain further information on the uptake mechanism(s).26,40 Co-incubation with fluorescently labelled low-density lipoproteins (LDLs) or transferrin enables clathrin-mediated endocytosis.
For cell-impermeable lanthanides, techniques, such as micro-injection,27,44 reversible plasma membrane permeabilization,45 and osmotic lysis of pinocytic vesicles,29,45 have been used. Co-incubation with compounds that increase cell permeability such as DMSO,30,46,47 saponin,34 or TMPyP48 has also been described.
Quantification of uptake
Ln complexes are usually detected by luminescence methods, which, however, do not give quantitative information on the amount of complex internalized. Luminescence emission can be quenched by endogenous electron-rich molecules (urate, ascorbate, citrate, ATP, phosphate, lactate) through electron or charge transfer quenching.49,50–55 Interaction with proteins, DNA or RNA decreases the overall emission intensity.50,53,55,56 Other molecules such as promoters or inhibitors used to study lanthanide internalization can also quench the luminescence emission.57 Self-quenching due to aggregation has been reported.57 Thus, alternative methods providing quantitative information on the accumulation of Ln complexes should be used, such as inductively-coupled plasma mass spectrometry (ICP-MS), or atomic absorption spectroscopy (AAS).26,58Stability of Ln complexes in cells
A final factor to consider is the inertness and stability of lanthanide complexes in cells. Careful analysis of the excitation and emission spectra of the lanthanides (especially those of Eu species) in cells as well as their emission lifetimes can help determine the integrity of the emitter. In some cases, evidence of interactions with endogenous species, e.g. proteins, can be obtained.Helicates
Bünzli and coworkers studied emissive bimetallic lanthanide complexes with helical structures in cells.59 The uptake mechanism of [Eu2(LC2)3] in HeLa cells was studied in depth.60,61 Time-resolved microscopy was used to follow the internalization of the complex. Lanthanide luminescence was observed from inside the cells after 15 min incubation. The uptake of [Eu2(LC2)3] was decreased at 4 °C, demonstrating that the uptake proceeds through an active mechanism.61 The accumulation of the complex was determined in the presence of sodium azide (inhibitor of cellular respiration), sodium iodoacetate (inhibitor of anaerobic glycolysis), a hypertonic sucrose treatment (inhibitor of clathrin-dependent endocytosis), or potassium depletion (inhibitor of clathrin-dependent endocytosis).60 The first two treatments lead to a decrease in ATP production.42,43 A lower uptake of the complex following these two treatments was observed, which suggests that [Eu2(LC2)3] is taken up in cells through an active pathway. In addition, the uptake of [Eu2(LC2)3] decreased when cells were treated with sucrose, or when potassium is depleted from the cell culture medium. These observations additionally suggest that the complex enters the cells through an endocytic pathway. However, a change in cell morphology can be observed for K+ depletion treatment, and to a lesser extent for hypertonic treatment. An important message here is that cell pathway inhibitors and promoters should be used with caution to avoid the non-specific toxicity and perturbation of cell homeostasis.39 Interestingly, co-staining experiments with BIODIPY FL labeled low density lipoprotein (LDL) or transferrin were run.61 LDL and transferrin uptake followed a clathrin-mediated endocytosis. LDL interacts with the LDL receptor at the cell surface, which leads to the formation of clathrin-coated vesicles (CCV), which are released from late endosomes. Transferrin is an iron transporter that is recognized by the transferrin receptor on the cell membrane and internalized in CCV.26,39 Thus both markers are indicative of a clathrin-mediated endocytosis. Co-localization of the helicate [Eu2(LC2)3] and the probes was observed, which suggests a clathrin-mediated endocytosis. To note, [Ln2(LC2)3] (Ln = Sm, Tb) were also studied in HeLa cells and are taken up by the cell (Fig. 5).61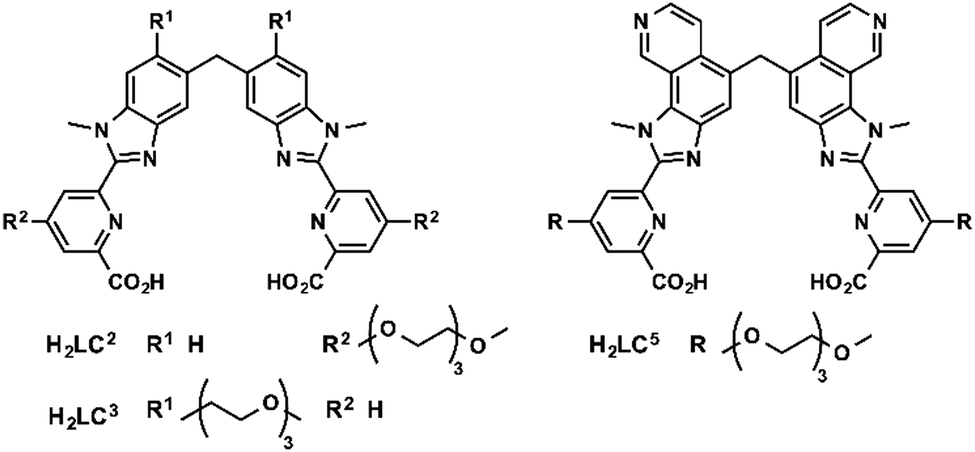 | ||
| Fig. 5 Structures of H2LCn ligands. | ||
One limitation of [Eu2(LC2)3] is its short excitation wavelength (330 nm). [Eu2(LC5)3] with a pyridine moiety on the benzimidazole is excitable at lower energies (365 nm). The uptake of [Eu2(LC5)3] in HeLa cells was followed by time-resolved luminescence microscopy. Similarly to [Eu2(LC2)3], [Eu2(LC5)3] was detected inside the cells after only 15 min, and showed a punctuated distribution that is consistent with an endocytic mechanism.62 In another study, the uptake of [Eu2(LC3)3] was investigated by measuring the brightness of the complex in cells at different temperatures (4 and 37 °C) and at different times of incubation.63 As for the other helicates, a relatively rapid uptake was observed after 15 min of incubation. Shifting the temperature to 4 °C decreased the accumulation of the complex in cells, which corresponds to an active uptake mechanism. The punctuated distribution of the complex within the cell of the complex suggests an endocytic mechanism. In summary, studies of lanthanide helicates in cells demonstrate that they enter through an active mechanism, which proceeds through an endocytic pathway; [Eu2(LC2)3] might enter cells via a clathrin-mediated endocytosis.60,61
Azamacrocycles
Parker and co-workers investigated in depth the uptake mechanisms of Eu(III)–cyclen complexes (12-N4) by systematically varying the charge, lipophilicity, antenna, and bulkiness of the complexes (Fig. 6).52,53,55,64–72 Most of the complexes reported enter the cell by macropinocytosis,64,67,68 as demonstrated by the use of inhibitors (wortmannin, amiloride, chlorpromazine, filipin, sucrose, temperature decrease to 4–5 °C), or promoters (phorbol ester, fatty acid glycerol) of different cellular pathways and of inhibitors of endosome maturation (chloroquine, monensin). The accumulation of the complexes in cells was determined by luminescence microscopy and ICP-MS. Changes in the antenna, the linker, or the lanthanide do not seem to impact the cell uptake pathways of these complexes. However, different subcellular localization profiles have been seen, with species accumulating in lysosomes,52,55,64–68,70,72 mitochondria,53,64,67,70,72 or the nucleolus.64,67,68,71 Some compounds showed first an accumulation in mitochondria before relocating into lysosomes.73–75 The antenna and the nature of the linker seem to play an important part in the subcellular localization of the complexes.64,67 In contrast, neither the overall charge of the complex, nor the lanthanide ion influences the subcellular distribution.64,67 Notably, the accumulation of complexes in the nucleoli of cells was shown to be linked with more permeable cell membranes,64 which is observed in cells under stress conditions. Thus, a nucleolus localization may indicate that the complex perturbs the cell and changes the permeability of the cytoplasmic membrane.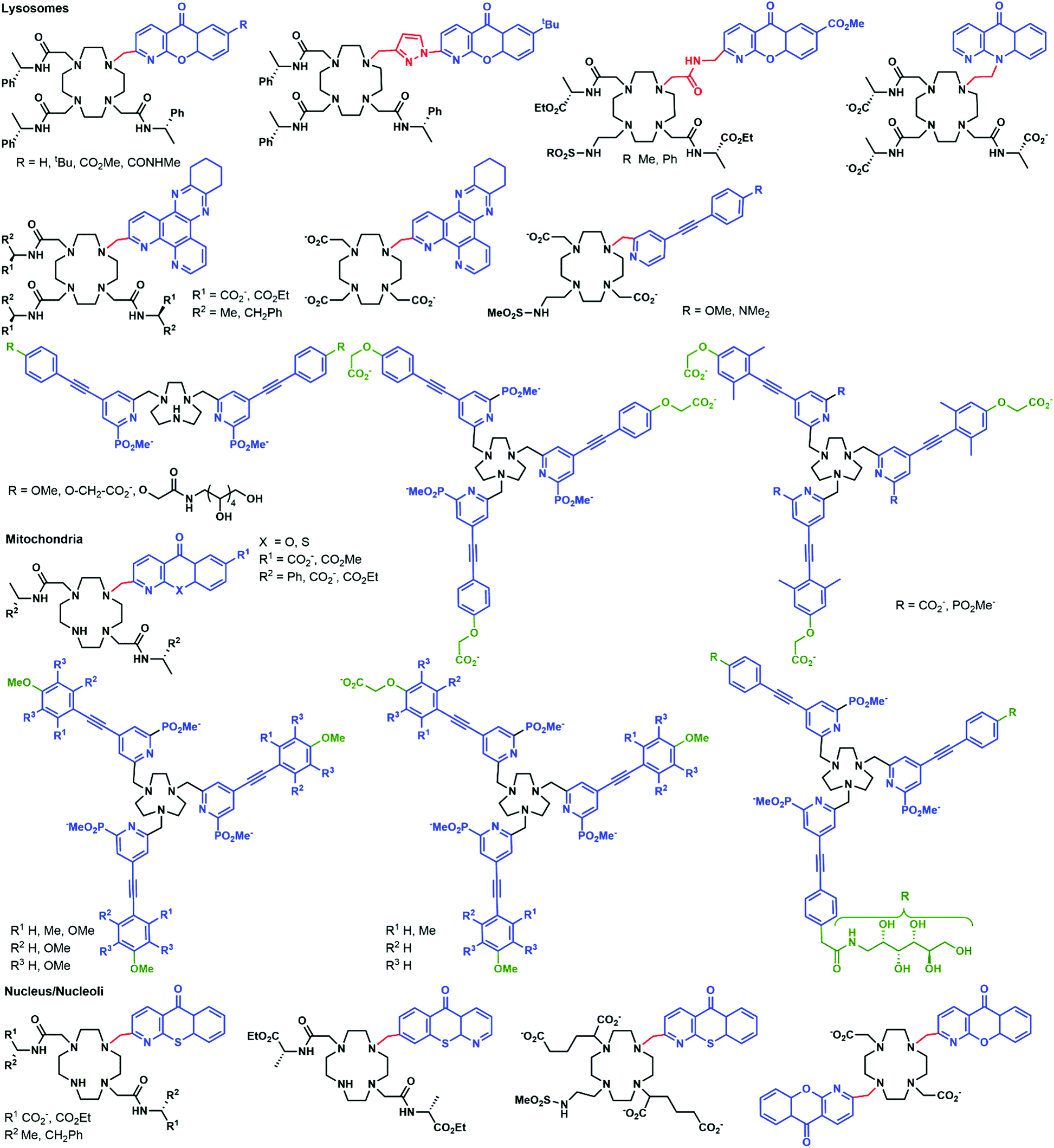 | ||
| Fig. 6 Structures of complexes studied by Parker and coworkers. | ||
Another prominent family of ligands is based on 9-N3 complexes with pyridylarylalkynyl antennae.57,74–78 The uptake of their lanthanide complexes occurs with macropinocytosis, in a similar manner to 12-N4 complexes. These compounds are localized in mitochondria,76,77 or lysosomes,74–76 with some showing a redistribution from mitochondria to lysosomes over time.57,75,76 Interestingly, other compounds were shown to first accumulate in mitochondria before relocating into the endoplasmic reticulum.72,76,78
In a recent paper, the impact of chirality on the cell uptake of Eu complexes was studied (Fig. 7).57 The authors showed that the racemic complex [EuL2] was taken up by the cells through macropinocytosis using various inhibitors and promoters of macropinocytosis (amiloride, wortmannin, phorbol ester, Di-Rac), clathrin-mediated endocytosis (chlorpromazine, sucrose), caveolin-dependent endocytosis (filipin), and inhibitors of the maturation of endosomes to lysosomes (chloroquine, monensin). The Λ- and Δ-enantiomers of [EuL2] and [EuL1] were internalized to different extents and had different subcellular localization profiles. The Λ-enantiomer of [EuL2] showed a greater accumulation in cells as demonstrated by luminescence and ICP-MS experiments, whereas the Δ-enantiomer was more abundant for [EuL1]. Macropinocytosis is not mediated by interaction with receptors at the cell surface; thus this behavior was not expected. As a potential explanation, the authors suggested that the enantiomers interact in a different manner with proteins possessing chiral binding pockets and are adsorbed to the cell surface. Thus, the internalization of one enantiomer is favored over the other. The subcellular distribution of the Λ- and Δ-enantiomers of [EuL2] and [EuL1] was studied using confocal microscopy and organelle-specific stains. The Λ-enantiomers accumulated preferentially in mitochondria, whereas the Δ-enantiomers were found in lysosomes. This difference in subcellular localization is explained by a faster relocalization of the Δ-enantiomers to the lysosomes.
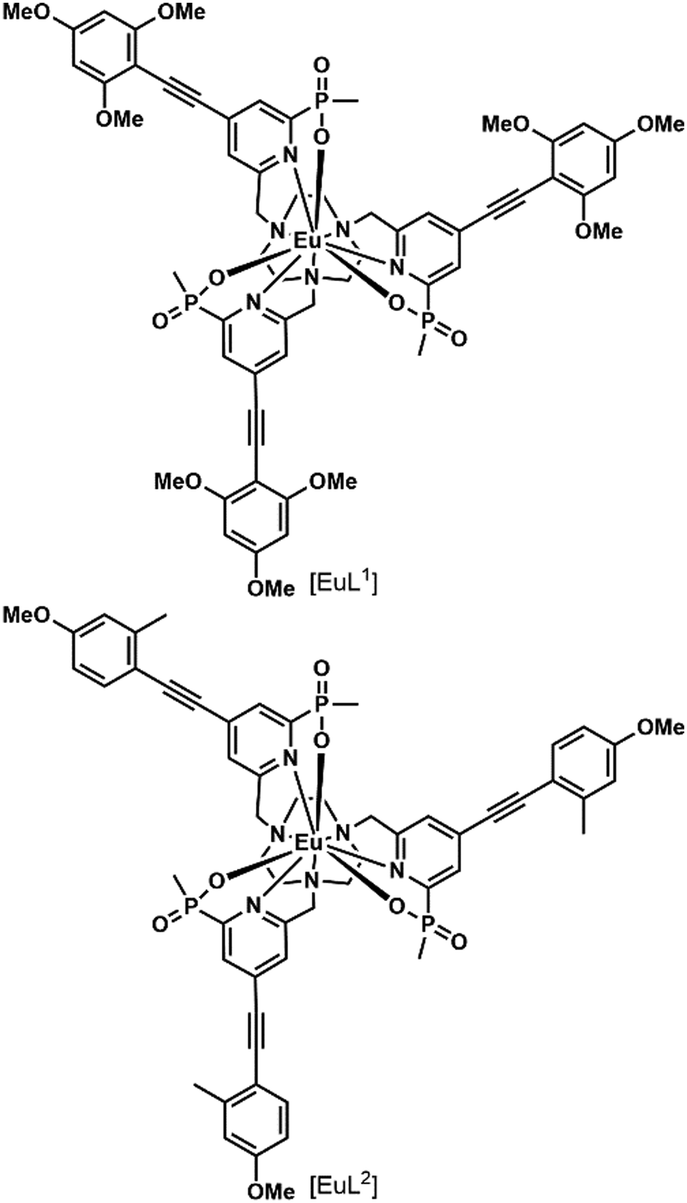 | ||
| Fig. 7 Structures of [EuL1–2] S-Δ enantiomers. | ||
Interestingly, [EuL2] luminescence was shown to be quenched by a promoter of macropinocytosis, phorbol ester. This stresses that measuring only the brightness of Ln complexes could be misleading as no increase in luminescence is observed in cells and this could lead to the wrong conclusion regarding the uptake mechanism. This illustrates the importance of reliably quantifying the accumulation of Eu complexes in cells by a luminescence-independent method, and the need for proper controls to ensure that the increase or decrease in luminescence corresponds to an increase or decrease in uptake.
[EuL1] was shown to self-aggregate in water, which quenched its luminescence. This may also occur in cells as the authors demonstrated that reduction of the concentration of incubation to 3 μM from 30 μM resulted in brighter images.
d–f heteronuclear complexes
There are numerous examples of d- and f-heteronuclear complexes with a d-block containing fragment as a light harvesting antenna and the f-element as the emitter, or that combine the luminescence of the lanthanide with the biological activity of the d-block element.51,52 Information on the uptake pathway of such complexes is scarce.53 Ln(III)–Pt(II) theranostics combine Eu or Tb luminescence with the DNA-binding of the Pt moiety.53,54 The uptake mechanism of the terbiplatin complex in HeLa cells was investigated using endocytic inhibitors and confocal microscopy. Chlorpromazine, cytochalasin-D, and rottlerin inhibited internalization, suggesting a macropinocytosis pathway. In HeLa and H460 cells europlatin and terbiplatin localized in the nucleoli. Based on previous work with azamacrocycles, this could be an indication that the complexes disturb the cells and change their permeability.34Prokaryotes
There are only a handful of examples of lanthanide complexes entering prokaryotic cells. We reported the internalization of a β-galactose-functionalized responsive Eu complex (Fig. 8).79 Cleavage of the galactose by a β-galactosidase turned on the red Eu emission by the in situ formation of the sensitizing antenna. In the case of galactose-caged probes, a response was only observed in LacZ cells. As the enzymes were localized inside the cells, the probes had to enter the bacteria to get turned on. The probes were activated very rapidly, as when cells were incubated with 10 μM Eu complex, the resultant Eu intensity was essentially identical at 10, 20 and 30 minutes. This suggests an uptake that is much faster than that observed for non-galactosylated complexes, which had to be incubated overnight.15 However, the uptake mechanisms were not investigated for either set of complexes, and differences between the bacterial strains may contribute to some of the observed differences.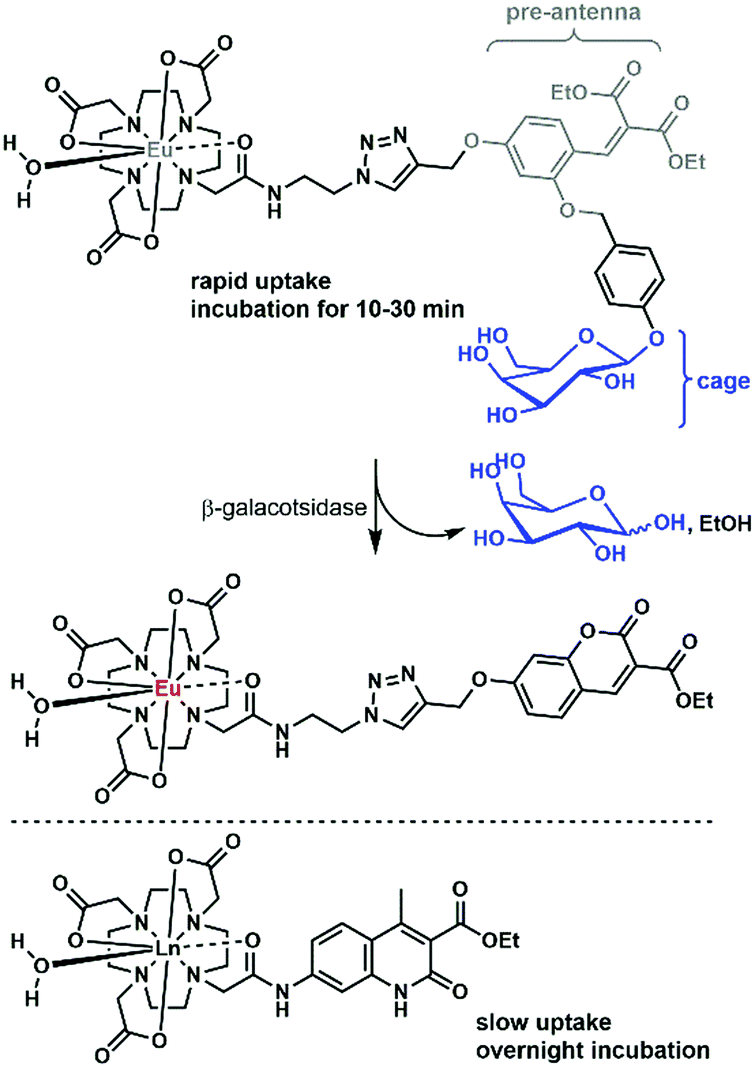 | ||
| Fig. 8 Selected complexes taken up by bacteria. | ||
An Eu probe utilizing a similar in situ antenna formation strategy as the complex in Fig. 8 could be used to detect DNA and RNA. The complex was grafted onto a DNA strand, while a triscarboxyethylphosphine (TCEP) was placed on a second strand. The two strands could be brought together by a complementary DNA or RNA template (the analyte), which triggered the Staudinger reduction of the azide, and the cyclization of the pre-antenna. The end result was the turn-on of Eu emission. This turn-on probe could be used for RNA detection in bacteria.80
Overview
A wide variety of lanthanide complexes seems to enter cells through macropinocytosis. Helicates might enter cells through a different pathway, but this hypothesis needs further investigation. The diversity of the complexes taken up by this pathway (charges, lipophilicity, bulkiness) should point further research toward a better understanding of the parameters that drive the uptake by macropinocytosis. The cell culture medium abounds with proteins, of which albumin represents the main part. Lanthanide complexes have shown affinity for albumin.49,81 Their internalization in cells could thus be driven by their interactions with proteins. The study of the speciation of the lanthanide complexes in the cell culture medium and in cells would provide valuable information that could help decipher the pathways by which they internalized. In this regard, the tools developed to study the mode of action of metallodrugs, e.g. mass spectrometry-based techniques, can be used to study Ln complexes.82Methods of lanthanide delivery
Complexes targeted to the exterior of the cell
There are numerous reports detailing the inability of ‘free’ lanthanide(III) ions to enter intact cells. Similarities between spectroscopically silent Ca2+ and versatile lanthanides have enabled the investigation of calcium-binding sites in cell membranes and calcium channels.83,84 Significant differences are often seen within the lanthanide series due to the decrease in the ionic radius from 106 ppm to 86 ppm upon going from La3+ to Yb3+.84,85 In the absence of the ionophore A23187, at concentrations up to 400 μM, Yb3+, Tb3+, Eu3+, La3+, Sm3+ and Gd3+ were unable to mimic Ca2+ in triggering K+-release. In the presence of A23187 even concentrations <10 μM were sufficient, in accord with the ionophore being necessary for the lanthanides to enter the cell.86 In a study on the effect of lanthanide(III) ions on insulin release in β-cells the localization of Sm3+ and Tm3+ was confirmed to be restricted to the plasma membrane by electron microscopy.85 EuCl3 (pH 7.4 solution) and the complex of Eu with albumin had different distributions when injected intravenously into rats. The salt was taken up by the liver, while the albumin complex was excreted rapidly into the urine, suggestive of a weak interaction between Eu3+ and albumin in serum.87 Intriguingly, La2(CO3)2 is a potential non-calcium phosphate binding drug. Its surprisingly low systemic toxicity is ascribed to its low solubility in physiological fluids. The resulting [La3+] <3 pm L−1 is low enough to make the lanthanide's calcium-like behaviour negligible.88Despite free lanthanides in general being cell-impermeable, Cheng et al. found by confocal scanning microscopy that erythrocytes are in fact permeable to these ions.89 Intracellular fluorescein fluorescence (introduced by labelling with fluorescein isothiocyanate) was quenched by uncomplexed lanthanides in a size- and total orbital angular momentum-dependent fashion. Overall anionic species, such as the −3 charged dicitrate complexes, were even more effective, as they could enter cells via anion channels.89
In prokaryotes systematic studies are lacking. However, electrogenic Ca2+ uptake in Azotobacter vinelandii was inhibited by LaCl3, TbCl3 and PrCl3 (20 μM) due to the similarity of the lanthanide(III) ions to the natural substrate. However, the presence of Ln, Tb or Pr was not investigated in the cells.90
Parker and collaborators from Cisbio Bioassays synthesized para-substituted aryl-alkynyltriazacyclononane (TACN) Eu(III) complexes.91 These highly water-soluble carboxylated or sulfonated complexes had overall negative charges, which prevent nonspecific binding to the cell membrane. Binding to the cholecystokinin 2 receptor (CCK2) expressed in the membrane of the HEK293 cells was possible with SNAP-tag technology. Successful binding was confirmed by time resolved FRET microscopy with the Eu(III) as the donor and a red fluorescent agonist of CCK2 as the acceptor.91
In another study, eight Gd–DOTA derivatives were prepared that targeted the metabotropic glutamate receptor subtype 5 (mGluR5).92 mGluR5 is part of the excitatory glutamate neurotransmitter system, involving emotions, the cognitive and motivation actions of the brain. Gd–DOTA complexes with a glutarate arm (Gd–DOTA–GA) were linked to specific allosteric antagonists of mGluR5. The complexes were tested in rat astrocytes. Two of the eight complexes bound selectively to the receptor and were taken up by the cells.92
Covalent membrane labelling of HEK293 cells was also possible with upconverting nanocrystals (UCNs) doped with Nd3+.93 The crystals can absorb 808 nm light and emit at 480 nm, have a minimized absorption of water, and are inert to thermal effects, unlike other UCNs that absorb 980 nm light. Membrane labelling was achieved by azide–alkyne cycloaddition. Cell-surface azides were introduced metabolically from N-azidoacetylmanosamine, and reacted with dibenzylcyclooctyne-functionalized UCNs. The membrane localization was confirmed by confocal microscopy and flow cytometry (FMS) analysis. UCNs bound to the membrane could act as NIR-activatable photoswitches. The emission wavelength of the UCNs matches the absorption of channelrhodopsin-2 (ChR2, 480 nm), which enabled remote control in zebrafish larvae of calcium channels expressing light-gated ChR2.93 Interestingly, PEG–phospholipid-coated UCNs not covalently anchored to the membrane are taken up most likely by nonspecific endocytosis.94
MRI contrast agents that label the cell membrane are interesting alternatives to tags targeting intracellular structures. Gd complexes attached to hydrophobic 4-, 10-, or 12-carbon alkyl chains were prepared.95 Low micromolar concentrations of these complexes with 10- or 12-carbon chains labelled the cell membranes of a range of adhering and non-adhering cells within 40 min. DFRET confirmed that the complexes bound to the membrane with the metal site facing the extracellular place, while the hydrophobic parts were arranged in the membrane. The labelled cells had normal doubling times and their morphologies were normal compared to the control cells.95
Small molecule targeting
Numerous receptors are known to be overexpressed in malignant cells. Lanthanide probes bearing specific targeting moieties can help with the detection of these cells. One such overexpressed receptor is the folate receptor (FR), which has a high affinity for folic acid (FA). When lanthanide complexes were attached to a folic acid targeting moiety, compounds with shorter FA–chelate distances were more efficiently internalized.96 It was also found that synthetically more accessible pteroic acid (PTE) functionalized complexes are suboptimal substitutes for folic acid, since lower cellular uptake could be observed for these species.96 When Eu chelates were bound to three different positions of FA, no noticeable difference between the cellular uptakes of the compounds could be seen.97 The substitution of FA with methotrexate (MTX), an anti-folate, resulted in higher cytotoxicity for all the MTX complexes compared to their corresponding FA ones.98In all cases, where incubation was performed on both FR(−) and FR(+) cells,97,98 selective internalization in FR(+) cells could be observed. This, and a reduced amount of cellular uptake when competing experiments with free FA were performed, indicates that the FA complexes are internalized through a FR-mediated mechanism.99 In the latter, the molecules are taken up by caveolae, and after binding with the FR and sealing the compartment, the ligand gets dissociated from the FR using a proton pump. This induces ligand transport via the reduced folate carrier (RFC), an anion exchange channel. This mechanism is somewhat controversial, since in certain cell lines, coupling of FR and RFC has been observed;100,101 in others it has been concluded that they work independently.102,103 High cellular uptake for MTX-functionalized complexes was seen in a FR(−) cell. This indicates that an alternative mechanism must be active,98 and indeed, the RFC has been recognized as the major route for anti-folate complexes.104 Kalber et al. synthesized a Gd–DOTA–folate complex that could enter both FR(−) and FR(+) cells, but in higher amounts for the latter cell type.105 Again, the conclusion must be that, in addition to the FR mediated mechanism, a second, unknown mechanism takes place. Similar findings were reported for the uptake of the non-lanthanide-bound folic acid derivative, 5-methyltetrahydrofolate.106
GLUTs are membrane-bound glucose transporters, and like the FR, they are overexpressed in several malignant cells.107 However, only a handful of metal complexes (e.g. glucose- and galactose-appended Ir(III) bipyridines108), and no lanthanide labels have been targeted to these receptors. The asialoglycoprotein receptor (ASGP-R) has a high affinity for galactose. Studies on cellular uptake via this receptor have been performed with radioactive analogues (111In and 153Sm) of MRI contrast agents.109,110 High cellular uptake in ASGP-R(+) cells compared to almost no uptake in ASGP-R(−) cells indicated that the compounds are internalized via a receptor-mediated mechanism. However, it seems that yet again a second mechanism also operates, as it was found that cellular uptake was reduced less than expected in competition experiments with D-galactose and asialofetuin.
Progesterone receptors (PR) can be found inside the cells. Steroid conjugated contrast agents are used for magnetic resonance imaging. The conjugation with these target moieties increases the ability of Gd chelates to associate with cells.111,112 Although the cellular uptake mechanism is still unclear, all experiments111,113–115 show moderately selective uptake for PR(+) cells, indicating that the presence of those receptors has an influence on the internalization. More hydrophilic complexes had lower cytotoxicity, but also lower membrane permeability.111,113–115 An overall negative charge on the complex decreased the internalization (Fig. 9).115
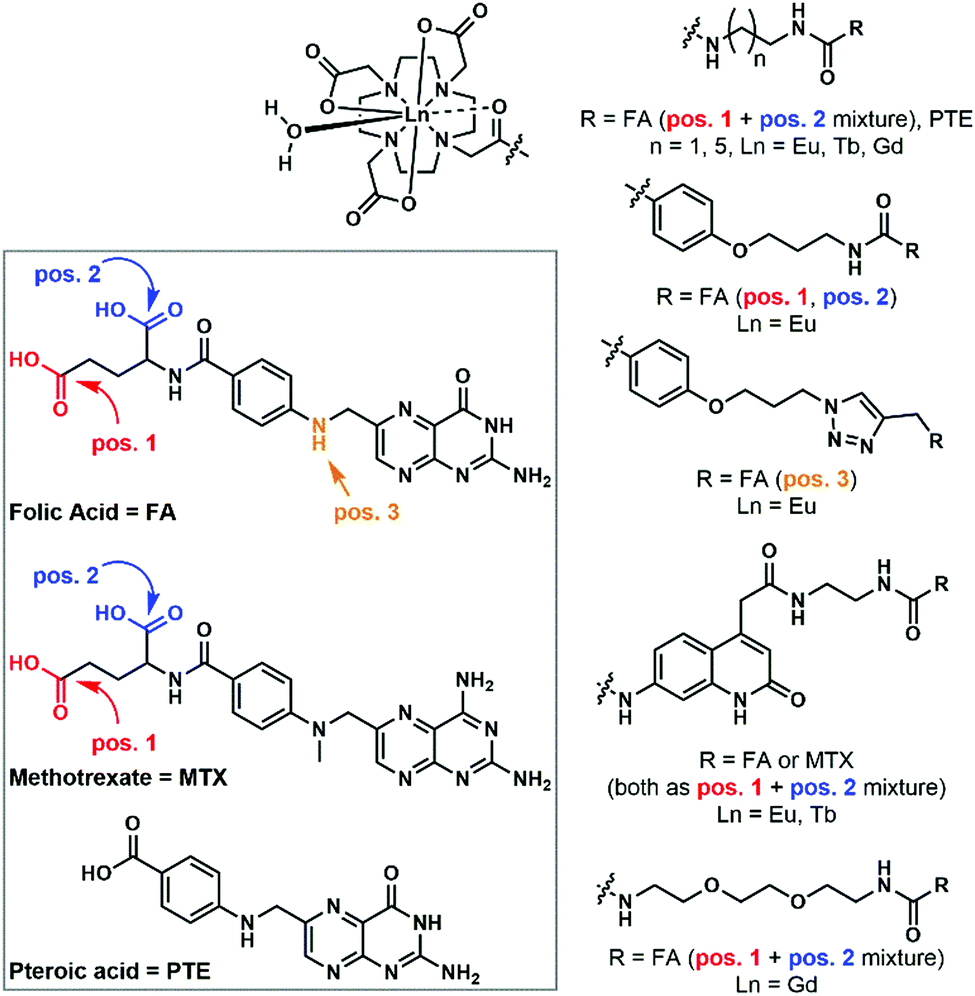 | ||
| Fig. 9 Folic acid and folic acid-analogue appended lanthanide complexes. | ||
Targeting with peptides
Cell penetrating peptides (CPPs) are <30 residue peptides that can cross the cell membrane or carry cell-impermeable cargos through the cell membrane.116 The majority have high positive charges and interact electrostatically with membrane proteins. A range of CPPs are known: some are derived from proteins, some are the combination of different protein parts, and some are synthetic (Table 1).41,117 So far no chimeric peptides have been used for metal uptake.| Name | Sequence | Target organelle | Metal cargo | Ref. |
|---|---|---|---|---|
| Peptides derived from proteins | ||||
| TPU | RQVKIWFQNRRMKQKK | Lanthanide | 121 | |
| Tat 49–57 | RKKRRQRRR | Nucleus | Ln, Zn, Tc, Rh | 132–135 |
| pVEC | LLIILRRRIRKQAHAHSK | — | — | — |
| NLS | ||||
| ALL1 | RKRKRK | Lanthanide | 120 | |
| SV40T | KKKRKVK | Co, Tc | 136–138 | |
| Chimeric peptides | ||||
| Transportan | GWTLNSAGYLLGKINLKALAALAKKIL | — | — | — |
| Pep-1 | KETWWETWWTEWSQPKKKRKV | — | — | — |
| MPG | GALFLGFLGAAGSTMGAWSQPKKKRKV | — | — | — |
| Synthetic peptides | ||||
| PolyArg | RRR⋯RRR | Lanthanide, Ru | 51, 73, 112, 127 and 139–141 | |
| MAP | KLALKLALKALKAALKLA | — | — | — |
| R6W3 | — | — | — | |
| MPP | FxrFxKFxrFxK | Mitochondria | Ru | 123 |
| Guanidinium | Perinuclear | Lanthanide | 51, 124 | |
Studies of the uptake mechanisms of CPPs have shown that they are taken up by cells via different pathways as evidenced by the parameters that influence the uptake mechanism.116,118,119 In particular, the uptake mechanism does not depend on the type of peptide used, but rather on the combination of the cargo and the peptide, interactions with the cell membrane components (lipids, proteins, polysaccharides), and the local concentration of the peptide.116,118,119 At low concentrations (below 2 μM) direct translocation seems to be favoured,118 whereas at higher concentrations (10 μM and above) CPPs are taken up by endocytosis. This criterion is not exclusive however, and both uptake mechanisms can be observed at the same time.116,118,119 At higher concentrations, the transduction mechanism, the direct uptake of CPPs dependent of intracellular processes, has also been described.118
Peptides can help metal complexes cross the membrane and can precisely target one subcellular localisation (Table 1). The nucleus can be targeted with Nuclear Localisation Sequences (NLSs) such as SV4073,120,121 appended to the CPPs. When this sequence is used in combination with lanthanides, a second sequence performs the role of membrane transport. The HIV Trans Activator of the Transcription (Tat) peptide shows also a predominant localisation in the nucleus. The synthetic mitochondria targeted peptide (Mitochondria Penetrating Peptide (MPP))122 sequence is composed of an alternating sequence of a synthetic cyclohexylalanine residue and a D-arginine or a lysine. The lipophilicity of MPP facilitates subcellular targeting, as a high log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P is beneficial for mitochondrial localization, whereas low logP peptides stay in the cytoplasm and in the nuclei. The threshold value depends on the complex charge. These peptides had carried a Ru complex cargo.123 The guanidinium protein linked to a complex co-localizes with MitoTracker Orange in mitochondria.124 Poly-lysine with human transferrin and poly-lysine with Gd–DTPA were added onto a luciferase plasmid.125 The concentration of the Gd complex on poly-lysine had to be chosen carefully so as not to interfere with the interaction between the DNA polyanion and the cationic lysine polymers. The plasmid was taken up by the cells via the transferrin receptors. The co-transport of DNA and the MRI contrast agent was efficient, as with each plasmid more than a thousand metal complexes are taken up.
P is beneficial for mitochondrial localization, whereas low logP peptides stay in the cytoplasm and in the nuclei. The threshold value depends on the complex charge. These peptides had carried a Ru complex cargo.123 The guanidinium protein linked to a complex co-localizes with MitoTracker Orange in mitochondria.124 Poly-lysine with human transferrin and poly-lysine with Gd–DTPA were added onto a luciferase plasmid.125 The concentration of the Gd complex on poly-lysine had to be chosen carefully so as not to interfere with the interaction between the DNA polyanion and the cationic lysine polymers. The plasmid was taken up by the cells via the transferrin receptors. The co-transport of DNA and the MRI contrast agent was efficient, as with each plasmid more than a thousand metal complexes are taken up.
Mohandessi et al. reported the concentration-dependent passive cytoplasmic delivery of a Tb(III) complex functionalized with cell-penetrating peptides in MDCKII cells.126 At high incubation concentrations (20–60 μM instead of 5 μM), a diffuse distribution throughout the cytoplasm was observed by time-resolved fluorescence microscopy. Decreasing the temperature to 4 °C did not influence the concentration of the complex in cells, suggesting that a passive energy-independent mechanism takes place, which could be attributed to a transduction mechanism.118
Complex diffusion inside the cells is improved upon liberation of the cargo from the peptide. For this, disulfide bonds between the complex and the peptide sequence can be cleaved by intracellular thiols.120,127 Molecules with disulfide bonds stay longer in cells than those without, which could be interesting for imaging.
Peptides can be cytotoxic. This can be an advantage if targeting cancer cells; however, it can be a problem for imaging. Natural CPPs have a low toxicity, while the Tat sequence is known to induce apoptosis of hippocampal neurons128,129 and poly-Arg can induce microvascular macromolecule leakage.130 With this in mind, a lanthanide complex was equipped with two NLSs: ALL1 to improve the cellular uptake and SV40 to target the nucleus.120 To avoid an immune response, a human homologue (87% similar) to the Penetratin, a CPP from the Antennapedia homeodomain, was used to transport a contrast agent.121
The peptide can also be toxic for organelles. Mitochondria are sensitive to the hydrophobicity of MPP: highly hydrophobic compounds induce membrane disruption and apoptosis, whereas high concentrations of less hydrophobic ones can modify the membrane potential.131 These factors need to be taken into account when choosing the CPP.
Intracellular imaging with macromolecules
Biological material has low autofluorescence in the NIR region, which means that signals from lanthanides can be easily differentiated from signals from the cells; there is a good signal-to-noise ratio and good detection sensitivity. Another advantage is that NIR photons have minimal interaction with biological media so will not perturb the live cells, and they scatter less than photons from the visible range. The NIR emitting lanthanides have low intrinsic quantum yields and are extremely sensitive to quenching with X–H oscillators (X = O, N, C). One way to overcome the often low brightness of these NIR emitters is to incorporate a large number of absorbing and emitting units into supramolecular edifices. Three types of supramolecular materials will be discussed: metal–organic frameworks, metallacrowns and dendrimers.One of the key requirements of a MOF is that it should enter the cells and release its cargo efficiently. Nanoparticles enter cells via endocytosis.144 Endocytosis can occur on a macro- or microscale; as far as MOFs are concerned the process is on a microscale. Microscale endocytosis can be divided into groups depending on the protein coating on the vesicles that form upon cell penetration.144
The effect of functionalizing and extending the organic 1,4-benzenedicarboxylate (BDC) linker of a Zr-based MOF, UiO-66, on cellular uptake was investigated.145 The fluorescent and hydrophilic molecule calcein was used in conjunction with flow cytometry analysis and confocal microscopy to determine the efficiency of MOF internalization. The Zr-MOFs were functionalized with –NO2, –Br and –NH2, or their linkers were extended with naphthalene-2,6-dicarboxylic acid or 4,4′-biphenyldicarboxylic acid and incubated with HeLa cells. MOFs with functionalized ligands were internalized more efficiently than MOFs with extended linkers. However, they were more susceptible to lysosomal degradation, to which extended linker MOFs were resistant. Increasing uptake efficiency with decreasing particle size was found. Increased granularity was seen by flow cytometry for larger MOFs compared to smaller ones.145 These results on surface chemistry modifications should be applicable to lanthanide-based MOFs.
The first sensitized NIR emission came from Yb3+-containing MOFs. The MOF structure could be varied while keeping the chromophore linker the same, which tuned the photophysical properties of the MOFs by modifying the structures.146 The ligand used was 4,4′-[(2,5-dimethoxy-1,4-phenylene)di-2,1-ethenediyl]bis-benzoic acid (H2PVDC), due to its ability to absorb strongly in the visible range and to sensitize the Yb3+. X-ray diffraction showed that Yb-PVDC-1 crystallized in the high symmetry Fddd space group, and the chains are made up of alternating octa- and hexa-coordinated Yb3+ ions, with the octa-coordinated ions having two water molecules coordinated. The arrangements of the ligands could allow for weak interactions between one another, possibly decreasing the excitation energy. A second MOF, Yb-PVDC-2, was also synthesized, which still contained octa- and hexa-coordinated Yb3+ ions, but the ligands were then parallel. The lowest energy excitation of Yb-PVDC-2 red-shifted to 500 nm, compared to 470 nm in Yb-PVDC-1, possibly due to the close π–π interactions. The quantum yield of Yb-PVDC-2 was five times higher than that of Yb-PVDC-1, which is most likely due to the coordinated water molecules, which are absent in Yb-PVDC-2.146
Yb-PVDC-3 was prepared via reverse microemulsion synthesis, which yielded a nanoscale version of the MOF that could be used for imaging in living cells.143 Lifetime measurements show that the Yb3+ are in two types of environments. Those with longer lifetimes are in the inside of the MOF structure, and those with shorter lifetimes are on the edges/faces of the MOF. The external Yb3+ ions are more susceptible to quenching from their surroundings; this explains their shorter lifetime values.
Nano-Yb-PVDC-3 exhibited both biological stability and photostability, and had an IC50 of 100 μg L−1, which is suitable for live cell imaging. Nano-Yb-PVDC-3 was incubated with HeLa and NIH3T3 cells. Confocal microscopy and inductively coupled plasma measurements show that nano-Yb-PVDC-3 is indeed internalized into the cell. Further analysis is required to examine the mechanism of cell penetration. The quantum yields for the nanoMOF are reported to be relatively low: 1.0 (±0.3) × 10−4 in water and 5.2 (±0.8) × 10−5 in aqueous 0.1 M Hepes buffer. This is where the advantage of using a MOF comes into play, as it maximizes the number of cations and chromophores per unit volume, which increases the number of emitted photons to the level that Yb luminescence is readily detected even in such challenging milieux.
Yb-PVDC-3 was photostable. Stability in cells was tested by monitoring the cells with SEM. As the lanthanide emission can only be detected if it is sensitized by the antenna, the fact that emission could be detected throughout the test meant that the MOF remained intact in the cell and is therefore biologically stable.
So far the use of metallacrowns in cells has been limited to necrotic cells, where they are used as alternative cell markers. Detection of cell necrosis is important so as to determine cell viability. Currently used is propidium iodide,151 an organic fluorophore that is susceptible to rapid photobleaching, to which lanthanide-based metallacrowns are resistant.
Recently, a series of lanthanide metallacrowns with the Ln3+[Zn(II)MCquinHA] structure (where Ln3+ = Nd, Er, Yb) were synthesized. The quin ligand enables excitation with low energy light, while the rigid structure that keeps solvent molecules away from the lanthanide afforded high quantum yields.150 While these complexes were poorly water soluble, a new generation of metallacrowns could overcome this problem.152 By replacing the chromophoric linker with the one based on pyrazinehydroxamic acid (H2pyzHA), highly water-soluble compounds (Ln3+[Zn(II)MCpyzHA], Ln3+ = Yb, Nd) were obtained that maintained the impressive NIR luminescence of their predecessors. HeLa cells were incubated with Yb3+[Zn(II)MCpyzHA] to examine necrotic cell accumulation, cell viability and photostability. It was found the metallacrowns were both photophysically and biologically stable.152 This metallacrown was also found, in a separate study, to be suitable for in vivo cell fixation and counterstaining upon exposure to UVA light.153
Examples for the incorporation of NIR-emitting lanthanide ions into dendrimers are scarce. Two dendrimers based on cyclam cores with PAMAM appendages and incorporating Nd3+ were designed. Dansyl4 and a larger Dansyl8 were synthesised; the former was able to coordinate Nd3+ but unable to sensitize it, whereas by increasing the number of chromophoric dendrons from four to eight, sensitisation was seen.156 No investigations into the biological applications of these molecules have been reported, most likely due to the unfavourable cationic nature of the compounds.
Substituting parts of PAMAM with a total of 32 2,3-naphthalimide chromophores gave rise to a dendrimer which was capable of accommodating eight lanthanide ions.157 Photophysical studies showed that the triplet states of the ligands were at a particularly favourable energy position for Sm3+ sensitization. Sm(III)-G3P-2,3-Nap exhibited sharp visible and NIR emission bands. The emission was shown to be sensitized by the chromophores of the dendrimer. Furthermore, an Alamar Blue assay of HeLa and NIH3T3 cells showed that cell viability was higher than 95% at a concentration of 1 μM and the dendrimer was non-toxic up to 24 h, indicating that the compound can be used for live cell imaging. Confocal microscopy showed that the probe was taken up by cells and most likely distributed amongst lysosomes.
Conclusions
The transition from a test tube to living cells is a challenging one even for a selective, sensitive, highly water-soluble chemical probe. The requirements placed on lanthanide complexes by the delicate homeostasis of the cells are stringent. Not only do they have to perform to a comparable extent in the biological medium as they do in buffered water, but they have to be non-toxic, they have to be resistant to degradation for the period of the experiment, and they should not produce degradation products that interfere with the system under investigation.A special challenge is due to the fact that many of the currently available lanthanide probes are coordination complexes with the possibility of dissociating in dilute solutions. Therefore, thermodynamic data on complexes with new ligand systems are highly valuable. Of note, several recent reports have detailed the uptake of moderately stable complexes of simple bi- and tridentate ligands into living cells.158,159 Given the synthetic accessibility of these systems, and the efficiency with which they are internalized, it would be of great importance to understand the role of these attractively simple ligands in cellular uptake, and to investigate whether their benefits could be transferred to traditional tri- and tetraazamacrocyclic ligands.
There are still large unexplored areas in this field, from the factors governing complex uptake and egress in bacteria to the fates of the probes in both eukaryotic and prokaryotic cells. Recently emerging lanthanide(II) based smart contrast agents open up new avenues of investigation due to their larger, softer metal ions and the classes of ligands that accommodate them.9,160 These structural features, as well as their accessible redox chemistry, will likely impact their biological behaviour.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Swedish Research Council (project grant 2017-04077 to K. E. B.) and the Carl Tryggers Foundation (postdoctoral fellowship for E. M.). E. D., D. P. and D. S. are Erasmus students. The Ecole Normale Supérieure (Paris, http://www.chimie.ens.fr/?q=en) is gratefully acknowledged for financial support through an Erasmus fellowship to A. S. We thank Daniel Kovacs for collecting the spectra in Fig. 1.Notes and references
- L. Burai, R. Scopelliti and E. Toth, Chem. Commun., 2002, 2366–2367 RSC.
- E. Toth, L. Burai and A. E. Merbach, Coord. Chem. Rev., 2001, 216–217, 363–382 CrossRef.
- L. Burai, E. Toth, S. Seibig, R. Scopelliti and A. E. Merbach, Chem. – Eur. J., 2000, 6, 3761–3770 CrossRef.
- S. Seibig, E. Toth and A. E. Merbach, J. Am. Chem. Soc., 2000, 122, 5822–5830 CrossRef.
- ed. A. Merbach, L. Helm and E. Toth, The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging, John Wiley & Sons Ltd, 2013 Search PubMed.
- L. A. Ekanger, L. A. Polin, Y. Shen, E. M. Haacke, P. D. Martin and M. J. Allen, Angew. Chem., Int. Ed., 2015, 54, 14398–14401 CrossRef PubMed.
- L. A. Ekanger, L. A. Basal and M. J. Allen, Chem. – Eur. J., 2017, 5, 1145–1150 CrossRef PubMed.
- L. A. Ekanger, D. R. Mills, M. M. Ali, L. A. Polin, Y. Shen, E. M. Haacke and M. J. Allen, Inorg. Chem., 2016, 55, 9981–9988 CrossRef PubMed.
- M. J. Allen, Synlett, 2016, 1310–1317 CrossRef PubMed.
- A. N. W. Kuda-Wedagedara, C. Wang, P. D. Martin and M. J. Allen, J. Am. Chem. Soc., 2015, 137, 4960–4963 CrossRef PubMed.
- A. de Bettencourt-Dias, Luminescence of Lanthanide Ions in Coordination Compounds and Nanomaterials, John Wiley & Sons Ltd, 2014, ch. 01, pp. 1–48 Search PubMed.
- K. Binnemans, Coord. Chem. Rev., 2015, 295, 1–45 CrossRef.
- A. Beeby, S. W. Botchway, I. M. Clarkson, S. Faulkner, A. W. Parker, D. Parker and J. A. G. Williams, J. Photochem. Photobiol., B, 2000, 57, 83–89 CrossRef.
- D. Kovacs and K. E. Borbas, Coord. Chem. Rev., 2018, 364, 1–9 CrossRef.
- D. Kovacs, X. Lu, L. S. Mészáros, M. Ott, J. Andres and K. E. Borbas, J. Am. Chem. Soc., 2017, 139, 5756–5767 CrossRef PubMed.
- R. M. Supkowski and W. D. Horrocks, Jr., Inorg. Chim. Acta, 2002, 340, 44–48 CrossRef.
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, S. A. S. de, J. A. G. Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493–504 RSC.
- J.-C. G. Bunzli, Coord. Chem. Rev., 2015, 293–294, 19–47 CrossRef.
- J.-C. G. Bünzli and S. V. Eliseeva, in Lanthanide Luminescence: Photophysical, Analytical and Biological Aspects, ed. P. Hänninen and H. Härmä, Springer, Berlin, Heidelberg, 2011, pp. 1–45 Search PubMed.
- K. Y. Zhang, Q. Yu, H. Wei, S. Liu, Q. Zhao and W. Huang, Chem. Rev., 2018, 118, 1770–1839 CrossRef PubMed.
- E. Pershagen and K. E. Borbas, Coord. Chem. Rev., 2014, 273-274, 30–46 CrossRef.
- M. Sy, A. Nonat, N. Hildebrandt and L. J. Charbonniere, Chem. Commun., 2016, 52, 5080–5095 RSC.
- M. Rajendran, E. Yapici and L. W. Miller, Inorg. Chem., 2014, 53, 1839–1853 CrossRef PubMed.
- S. Shuvaev, M. Starck and D. Parker, Chem. – Eur. J., 2017, 23, 9974–9989 CrossRef PubMed.
- N. Sim and D. Parker, Chem. Soc. Rev., 2015, 44, 2122–2134 RSC.
- C. A. Puckett, R. J. Ernst and J. K. Barton, Dalton Trans., 2010, 39, 1159–1170 RSC.
- K. Hanaoka, K. Kikuchi, S. Kobayashi and T. Nagano, J. Am. Chem. Soc., 2007, 129, 13502–13509 CrossRef PubMed.
- M. Rajendran and L. W. Miller, Biophys. J., 2015, 109, 240–248 CrossRef PubMed.
- G. Nivriti and L. W. Miller, Cytometry, Part A, 2010, 77, 1113–1125 Search PubMed.
- G.-L. Law, K.-L. Wong, C. W.-Y. Man, W.-T. Wong, S.-W. Tsao, M. H.-W. Lam and P. K.-S. Lam, J. Am. Chem. Soc., 2008, 130, 3714–3715 CrossRef PubMed.
- L.-O. Palsson, R. Pal, B. S. Murray, D. Parker and A. Beeby, Dalton Trans., 2007, 5726–5734 RSC.
- A. D’Aléo, A. Picot, A. Beeby, J. A. Gareth Williams, B. Le Guennic, C. Andraud and O. Maury, Inorg. Chem., 2008, 47, 10258–10268 CrossRef PubMed.
- A. D'Aleo, A. Bourdolle, S. Brustlein, T. Fauquier, A. Grichine, A. Duperray, P. L. Baldeck, C. Andraud, S. Brasselet and O. Maury, Angew. Chem., Int. Ed., 2012, 51, 6622–6625 CrossRef PubMed.
- A. Grichine, A. Haefele, S. Pascal, A. Duperray, R. Michel, C. Andraud and O. Maury, Chem. Sci., 2014, 5, 3475–3485 RSC.
- A. Jana, E. Baggaley, A. Amoroso and M. D. Ward, Chem. Commun., 2015, 51, 8833–8836 RSC.
- A. T. Bui, A. Grichine, A. Duperray, P. Lidon, F. Riobe, C. Andraud and O. Maury, J. Am. Chem. Soc., 2017, 139, 7693–7696 CrossRef PubMed.
- E. R. H. Walter, J. A. G. Williams and D. Parker, Chem. Commun., 2017, 53, 13344–13347 RSC.
- Y. Ning, J. Tang, Y.-W. Liu, J. Jing, Y. Sun and J.-L. Zhang, Chem. Sci., 2018, 9, 3742–3753 RSC.
- I. A. Khalil, K. Kogure, H. Akita and H. Harashima, Pharmacol. Rev., 2006, 58, 32–45 CrossRef PubMed.
- T.-G. Iversen, T. Skotland and K. Sandvig, Nano Today, 2011, 6, 176–185 CrossRef.
- C. Bechara and S. Sagan, FEBS Lett., 2013, 587, 1693–1702 CrossRef PubMed.
- V. P. Torchilin, R. Rammohan, V. Weissig and T. S. Levchenko, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8786–8791 CrossRef PubMed.
- D. L. Foxall, K. M. Brindle, I. D. Campbell and R. J. Simpson, Biochim. Biophys. Acta, 1984, 804, 209–215 CrossRef.
- A. Mohamadi and L. W. Miller, Bioconjugate Chem., 2016, 27, 2540–2548 CrossRef PubMed.
- H. E. Rajapakse, N. Gahlaut, S. Mohandessi, D. Yu, J. R. Turner and L. W. Miller, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13582–13587 CrossRef PubMed.
- V. Placide, A. T. Bui, A. Grichine, A. Duperray, D. Pitrat, C. Andraud and O. Maury, Dalton Trans., 2015, 44, 4918–4924 RSC.
- T. Zhang, X. Zhu, C. C. W. Cheng, W.-M. Kwok, H.-L. Tam, J. Hao, D. W. J. Kwong, W.-K. Wong and K.-L. Wong, J. Am. Chem. Soc., 2011, 133, 20120–20122 CrossRef PubMed.
- B. Song, G. Wang, M. Tan and J. Yuan, J. Am. Chem. Soc., 2006, 128, 13442–13450 CrossRef PubMed.
- R. A. Poole, C. P. Montgomery, E. J. New, A. Congreve, D. Parker and M. Botta, Org. Biomol. Chem., 2007, 5, 2055–2062 RSC.
- R. A. Poole, G. Bobba, M. J. Cann, J.-C. Frias, D. Parker and R. D. Peacock, Org. Biomol. Chem., 2005, 3, 1013–1024 RSC.
- F. Kielar, A. Congreve, G.-L. Law, E. J. New, D. Parker, K.-L. Wong, P. Castreňo and J. D. Mendoza, Chem. Commun., 2008, 2435–2437 RSC.
- F. Kielar, G.-L. Law, E. J. New and D. Parker, Org. Biomol. Chem., 2008, 6, 2256–2258 RSC.
- D. G. Smith, G.-l. Law, B. S. Murray, R. Pal, D. Parker and K.-L. Wong, Chem. Commun., 2011, 47, 7347–7349 RSC.
- G.-L. Law, D. Parker, S. L. Richardson and K.-L. Wong, Dalton Trans., 2009, 8481–8484 RSC.
- E. J. New, D. Parker and R. D. Peacock, Dalton Trans., 2009, 672–679 RSC.
- J. Yu, D. Parker, R. Pal, R. A. Poole and M. J. Cann, J. Am. Chem. Soc., 2006, 128, 2294–2299 CrossRef PubMed.
- A. T. Frawley, H. V. Linford, M. Starck, R. Pal and D. Parker, Chem. Sci., 2018, 9, 1042–1049 RSC.
- M. Groessl and C. G. Hartinger, Anal. Bioanal. Chem., 2013, 405, 1791–1808 CrossRef PubMed.
- J.-C. G. Bünzli, Interface Focus, 2013, 3, 20130032 CrossRef PubMed.
- B. Song, C. D. B. Vandevyver, A.-S. Chauvin and J.-C. G. Buenzli, Org. Biomol. Chem., 2008, 6, 4125–4133 RSC.
- A.-S. Chauvin, S. Comby, B. Song, C. D. B. Vandevyver and J.-C. G. Bunzli, Chem. – Eur. J., 2008, 14, 1726–1739 CrossRef PubMed.
- E. Deiters, B. Song, A.-S. Chauvin, C. D. B. Vandevyver, F. Gumy and J.-C. G. Bünzli, Chem. – Eur. J., 2009, 15, 885–900 CrossRef PubMed.
- A.-S. Chauvin, S. Comby, B. Song, C. D. B. Vandevyver, F. Thomas and J.-C. G. Bünzli, Chem. – Eur. J., 2007, 13, 9515–9526 CrossRef PubMed.
- E. J. New, D. Parker, D. G. Smith and J. W. Walton, Curr. Opin. Chem. Biol., 2010, 14, 238–246 CrossRef PubMed.
- S. Shuvaev, R. Pal and D. Parker, Chem. Commun., 2017, 53, 6724–6727 RSC.
- D. G. Smith, B. K. McMahon, R. Pal and D. Parker, Chem. Commun., 2012, 48, 8520–8522 RSC.
- E. J. New, A. Congreve and D. Parker, Chem. Sci., 2010, 1, 111–118 RSC.
- E. J. New and D. Parker, Org. Biomol. Chem., 2009, 7, 851–855 RSC.
- C. P. Montgomery, E. J. New, L. O. Palsson, D. Parker, A. S. Batsanov and L. Lamarque, Helv. Chim. Acta, 2009, 92, 2186–2213 CrossRef.
- C. P. Montgomery, B. S. Murray, E. J. New, R. Pal and D. Parker, Acc. Chem. Res., 2009, 42, 925–937 CrossRef PubMed.
- R. Pal and D. Parker, Org. Biomol. Chem., 2008, 6, 1020–1033 RSC.
- B. S. Murray, E. J. New, R. Pal and D. Parker, Org. Biomol. Chem., 2008, 6, 2085–2094 RSC.
- S. J. Butler, M. Delbianco, L. Lamarque, B. K. McMahon, E. R. Neil, R. Pal, D. Parker, J. W. Walton and J. M. Zwier, Dalton Trans., 2015, 44, 4791–4803 RSC.
- M. Starck, R. Pal and D. Parker, Chem. – Eur. J., 2016, 22, 570–580 CrossRef PubMed.
- S. J. Butler, B. K. McMahon, R. Pal, D. Parker and J. W. Walton, Chem. – Eur. J., 2013, 19, 9511–9517 CrossRef PubMed.
- S. J. Butler, L. Lamarque, R. Pal and D. Parker, Chem. Sci., 2014, 5, 1750–1756 RSC.
- J. W. Walton, A. Bourdolle, S. J. Butler, M. Soulie, M. Delbianco, B. K. McMahon, R. Pal, H. Puschmann, J. M. Zwier, L. Lamarque, O. Maury, C. Andraud and D. Parker, Chem. Commun., 2013, 49, 1600–1602 RSC.
- B. K. McMahon, R. Pal and D. Parker, Chem. Commun., 2013, 49, 5363–5365 RSC.
- E. Pershagen, J. Nordholm and K. E. Borbas, J. Am. Chem. Soc., 2012, 134, 9832–9835 CrossRef PubMed.
- H. Saneyoshi, Y. Ito and H. Abe, J. Am. Chem. Soc., 2013, 135, 13632–13635 CrossRef PubMed.
- F. Kielar, C. P. Montgomery, E. J. New, D. Parker, R. A. Poole, S. L. Richardson and P. A. Stenson, Org. Biomol. Chem., 2007, 5, 2975–2982 RSC.
- C. G. Hartinger, M. Groessl, S. M. Meier, A. Casini and P. J. Dyson, Chem. Soc. Rev., 2013, 42, 6186–6199 RSC.
- H. Simpkins, M. Figliomeni and M. Rosen, Biochim. Biophys. Acta, 1988, 972, 25–32 CrossRef.
- K. C. Fernando and G. J. Barritt, Biochim. Biophys. Acta, 1995, 1268, 97–106 CrossRef.
- P. R. Flatt, P. O. Berggren, E. Gylfe and B. Hellman, Endocrinology, 1980, 107, 1007–1013 CrossRef PubMed.
- J. Sneddon, Biochem. Pharmacol., 1987, 36, 3723–3730 CrossRef PubMed.
- D. Bingham and M. Dobrota, Biometals, 1994, 7, 142–148 CrossRef PubMed.
- S. J. P. Damment and M. Pennick, Clin. Pharmacokinet., 2008, 47, 553–563 CrossRef PubMed.
- Y. Cheng, Q. Huo, J. Lu, R. Li and K. Wang, J. Biol. Inorg. Chem., 1999, 4, 447–456 CrossRef PubMed.
- P. Zimniak and E. M. Barnes, Jr., J. Biol. Chem., 1980, 255, 10140–10143 Search PubMed.
- M. Delbianco, V. Sadovnikova, E. Bourrier, G. Mathis, L. Lamarque, J. M. Zwier and D. Parker, Angew. Chem., Int. Ed., 2014, 53, 10718–10722 CrossRef PubMed.
- S. Gottschalk, J. Engelmann, G. A. Rolla, M. Botta, D. Parker and A. Mishra, Org. Biomol. Chem., 2013, 11, 6131 RSC.
- X. Ai, L. Lyu, Y. Zhang, Y. Tang, J. Mu, F. Liu, Y. Zhou, Z. Zuo, G. Liu and B. Xing, Angew. Chem., Int. Ed., 2017, 56, 3031–3035 CrossRef PubMed.
- N. S. Hwan, B. Y. Mi, P. Y. Il, K. J. Hyun, K. H. Min, C. J. Sig, L. K. Taek, H. Taeghwan and S. Y. Doug, Angew. Chem., Int. Ed., 2011, 50, 6093–6097 CrossRef PubMed.
- Q. Zheng, H. Dai, M. E. Merritt, C. Malloy, C. Y. Pan and W.-H. Li, J. Am. Chem. Soc., 2005, 127, 16178–16188 CrossRef PubMed.
- Z. Du, G. N. Borlace, R. D. Brooks, R. N. Butler, D. A. Brooks and S. E. Plush, Inorg. Chim. Acta, 2014, 410, 11–19 CrossRef.
- S. Quici, A. Casoni, F. Foschi, L. Armelao, G. Bottaro, R. Seraglia, C. Bolzati, N. Salvarese, D. Carpanese and A. Rosato, J. Med. Chem., 2015, 58, 2003–2014 CrossRef PubMed.
- Z. Du, J. Sun, C. A. Bader, D. A. Brooks, M. Li, X. Li and S. E. Plush, J. Inorg. Biochem., 2018, 178, 32–42 CrossRef PubMed.
- R. G. Anderson, B. A. Kamen, K. G. Rothberg and S. W. Lacey, Science, 1992, 255, 410–411 CrossRef PubMed.
- P. D. Prasad, V. B. Mahesh, F. H. Leibach and V. Ganapathy, Biochim. Biophys. Acta, 1994, 1222, 309–314 CrossRef.
- B. A. Kamen, A. K. Smith and R. G. Anderson, J. Clin. Invest., 1991, 87, 1442–1449 CrossRef PubMed.
- K. H. Dixon, T. Mulligan, K. N. Chung, P. C. Elwood and K. H. Cowan, J. Biol. Chem., 1992, 267, 24140–24147 Search PubMed.
- G. R. Westerhof, G. Jansen, N. V. Emmerik, I. Kathmann, G. Rijksen, A. L. Jackman and J. H. Schornagel, Cancer Res., 1991, 51, 5507–5513 Search PubMed.
- G. R. Westerhof, J. H. Schornagel, I. Kathmann, A. L. Jackman, A. Rosowsky, R. A. Forsch, J. B. Hynes, F. T. Boyle, G. J. Peters and H. M. Pinedo, Mol. Pharmacol., 1995, 48, 459–471 Search PubMed.
- T. L. Kalber, N. Kamaly, P.-W. So, J. A. Pugh, J. Bunch, C. W. McLeod, M. R. Jorgensen, A. D. Miller and J. D. Bell, Mol. Imaging Biol., 2011, 13, 653–662 CrossRef PubMed.
- S. Miotti, M. Bagnoli, F. Ottone, A. Tomassetti, M. I. Colnaghi and S. Canevari, J. Cell. Biochem., 1997, 65, 479–491 CrossRef PubMed.
- R. A. Medina and G. I. Owen, Biol. Res., 2002, 35, 9–26 Search PubMed.
- W. H.-T. Law, L. C.-C. Lee, M.-W. Louie, H.-W. Liu, T. W.-H. Ang and K. K.-W. Lo, Inorg. Chem., 2013, 52, 13029–13041 CrossRef PubMed.
- M. M. Alauddin, A. Y. Louie, A. Shahinian, T. J. Meade and P. S. Conti, Nucl. Med. Biol., 2003, 30, 261–265 CrossRef PubMed.
- M. I. M. Prata, A. C. Santos, S. Torres, J. P. André, J. A. Martins, M. Neves, M. L. García-Martín, T. B. Rodrigues, P. López-Larrubia, S. Cerdán and C. F. G. C. Geraldes, Contrast Media Mol. Imaging, 2006, 1, 246–258 CrossRef PubMed.
- T. R. Townsend, G. Moyle-Heyrman, P. A. Sukerkar, K. W. MacRenaris, J. E. Burdette and T. J. Meade, Bioconjugate Chem., 2014, 25, 1428–1437 CrossRef PubMed.
- M. J. Allen, K. W. MacRenaris, P. N. Venkatasubramanian and T. J. Meade, Chem. Biol., 2004, 11, 301–307 CrossRef PubMed.
- J. Lee, M. J. Zylka, D. J. Anderson, J. E. Burdette, T. K. Woodruff and T. J. Meade, J. Am. Chem. Soc., 2005, 127, 13164–13166 CrossRef PubMed.
- P. A. Sukerkar, K. W. MacRenaris, T. R. Townsend, R. A. Ahmed, J. E. Burdette and T. J. Meade, Bioconjugate Chem., 2011, 22, 2304–2316 CrossRef PubMed.
- J. Lee, J. E. Burdette, K. W. MacRenaris, D. Mustafi, T. K. Woodruff and T. J. Meade, Chem. Biol., 2007, 14, 824–834 CrossRef PubMed.
- W. B. Kauffman, T. Fuselier, J. He and W. C. Wimley, Trends Biochem. Sci., 2015, 40, 749–764 CrossRef PubMed.
- S. Mohandessi, M. Rajendran, D. Magda and L. W. Miller, Chem. – Eur. J., 2012, 18, 10825–10829 CrossRef PubMed.
- R. Brock, Bioconjugate Chem., 2014, 25, 863–868 CrossRef PubMed.
- A. Walrant, S. Cardon, F. Burlina and S. Sagan, Acc. Chem. Res., 2017, 50, 2968–2975 CrossRef PubMed.
- S. Heckl and U. Vogel, J. Pharmacol. Exp. Ther., 2006, 319, 657–662 CrossRef PubMed.
- S. Heckl, J. Debus, J. Jenne, R. Pipkorn, W. Waldeck, H. Spring, R. Rastert, C. W. V. D. Lieth and K. Braun, Cancer Res., 2002, 62, 7018–7024 Search PubMed.
- K. L. Horton, K. M. Stewart, S. B. Fonseca, Q. Guo and S. O. Kelley, Chem. Biol., 2008, 15, 375–382 CrossRef PubMed.
- A. Martin, A. Byrne, C. S. Burke, R. J. Forster and T. E. Keyes, J. Am. Chem. Soc., 2014, 136, 15300–15309 CrossRef PubMed.
- J. Fernández-Carneado, M. Van Gool, V. Martos, S. Castel, P. Prados, J. de Mendoza and E. Giralt, J. Am. Chem. Soc., 2005, 127, 869–874 CrossRef PubMed.
- J. Faiz Kayyem, R. M. Kumar, S. E. Fraser and T. J. Meade, Chem. Biol., 1995, 2, 615–620 CrossRef.
- M. Shabnam, R. Megha, M. Darren and L. W. Miller, Chem. – Eur. J., 2012, 18, 10825–10829 CrossRef PubMed.
- P. J. Endres, K. W. MacRenaris, S. Vogt and T. J. Meade, Bioconjugate Chem., 2008, 19, 2049–2059 CrossRef PubMed.
- A. M. Prantner, V. Sharma, J. R. Garbow and D. Piwnica-Worms, Mol. Imaging, 2003, 2, 15353500200303106 Search PubMed.
- I. I. Kruman, A. Nath and M. P. Mattson, Exp. Neurol., 1998, 154, 276–288 CrossRef PubMed.
- M. Minnicozzi, M. A. M. Ramírez, R. W. Egan, G. J. Gleich, I. Kobayashi, D. Kim and W. N. Durán, Microvasc. Res., 1995, 50, 56–70 CrossRef PubMed.
- K. L. Horton, M. P. Pereira, K. M. Stewart, S. B. Fonseca and S. O. Kelley, ChemBioChem, 2012, 13, 476–485 CrossRef PubMed.
- R. Bhorade, R. Weissleder, T. Nakakoshi, A. Moore and C.-H. Tung, Bioconjugate Chem., 2000, 11, 301–305 CrossRef PubMed.
- R. Mishra, W. Su, R. Pohmann, J. Pfeuffer, M. G. Sauer, K. Ugurbil and J. Engelmann, Bioconjugate Chem., 2009, 20, 1860–1868 CrossRef PubMed.
- M. Sibrian-Vazquez, J. Ortiz, I. V. Nesterova, F. Fernández-Lázaro, A. Sastre-Santos, S. A. Soper and M. G. H. Vicente, Bioconjugate Chem., 2007, 18, 410–420 CrossRef PubMed.
- V. Polyakov, V. Sharma, J. L. Dahlheimer, C. M. Pica, G. D. Luker and D. Piwnica-Worms, Bioconjugate Chem., 2000, 11, 762–771 CrossRef PubMed.
- S. I. Kirin, I. Ott, R. Gust, W. Mier, T. Weyhermüller and N. Metzler-Nolte, Angew. Chem., Int. Ed., 2008, 47, 955–959 CrossRef PubMed.
- P. Haefliger, N. Agorastos, A. Renard, G. Giambonini-Brugnoli, C. Marty and R. Alberto, Bioconjugate Chem., 2005, 16, 582–587 CrossRef PubMed.
- F. Noor, A. Wüstholz, R. Kinscherf and N. Metzler-Nolte, Angew. Chem., Int. Ed., 2005, 44, 2429–2432 CrossRef PubMed.
- X. Zou, M. Rajendran, D. Magda and L. W. Miller, Bioconjugate Chem., 2015, 26, 460–465 CrossRef PubMed.
- M. J. Allen and T. J. Meade, J. Biol. Inorg. Chem., 2003, 8, 746–750 CrossRef PubMed.
- C. A. Puckett and J. K. Barton, J. Am. Chem. Soc., 2009, 131, 8738–8739 CrossRef PubMed.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef PubMed.
- A. Foucault-Collet, K. A. Gogick, K. A. White, S. Villette, A. Pallier, G. Collet, C. Kieda, T. Li, S. J. Geib, N. L. Rosi and S. Petoud, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17199–17204 CrossRef PubMed.
- C. Orellana-Tavra, S. A. Mercado and D. Fairen-Jimenez, Adv. Healthcare Mater., 2016, 5, 2261–2270 CrossRef PubMed.
- C. Orellana-Tavra, S. Haddad, R. J. Marshall, I. Abánades Lázaro, G. Boix, I. Imaz, D. Maspoch, R. S. Forgan and D. Fairen-Jimenez, ACS Appl. Mater. Interfaces, 2017, 9, 35516–35525 CrossRef PubMed.
- K. A. White, D. A. Chengelis, M. Zeller, S. J. Geib, J. Szakos, S. Petoud and N. L. Rosi, Chem. Commun., 2009, 4506–4508 RSC.
- G. Mezei, C. M. Zaleski and V. L. Pecoraro, Chem. Rev., 2007, 107, 4933–5003 CrossRef PubMed.
- T. N. Nguyen, C. Y. Chow, S. V. Eliseeva, E. R. Trivedi, J. W. Kampf, I. Martinic, S. Petoud and V. L. Pecoraro, Chem. – Eur. J., 2018, 24, 1031–1035 CrossRef PubMed.
- J. C. Lutter, C. M. Zaleski and V. L. Pecoraro, in Advances in Inorganic Chemistry, ed. R. van Eldik and R. Puchta, Academic Press, 2018, vol. 71, pp. 177–246 Search PubMed.
- E. R. Trivedi, S. V. Eliseeva, J. Jankolovits, M. M. Olmstead, S. Petoud and V. L. Pecoraro, J. Am. Chem. Soc., 2014, 136, 1526–1534 CrossRef PubMed.
- A. Krishan, J. Cell Biol., 1975, 66, 188–193 CrossRef PubMed.
- I. Martinić, S. V. Eliseeva and S. Petoud, J. Lumin., 2017, 189, 19–43 CrossRef.
- I. Martinic, S. V. Eliseeva, T. N. Nguyen, F. Foucher, D. Gosset, F. Westall, V. L. Pecoraro and S. Petoud, Chem. Sci., 2017, 8, 6042–6050 RSC.
- B. Klajnert and M. Bryszewska, Acta Biochim. Pol., 2001, 48, 9 Search PubMed.
- D. Astruc, E. Boisselier and C. Ornelas, Chem. Rev., 2010, 110, 1857–1959 CrossRef PubMed.
- Z. S. Pillai, P. Ceroni, M. Kubeil, J.-M. Heldt, H. Stephan and G. Bergamini, Chem. – Asian J., 2013, 8, 771–777 CrossRef PubMed.
- A. Foucault-Collet, C. M. Shade, I. Nazarenko, S. Petoud and S. V. Eliseeva, Angew. Chem., Int. Ed., 2014, 53, 2927–2930 CrossRef PubMed.
- J. H. S. K. Monteiro, D. Machado, L. M. de Hollanda, M. Lancellotti, F. A. Sigoli and A. de Bettencourt-Dias, Chem. Commun., 2017, 53, 11818–11821 RSC.
- V. V. Utochnikova, D. S. Koshelev, A. V. Medvedko, A. S. Kalyakina, I. S. Bushmarinov, A. Y. Grishko, U. Schepers, S. Bräse and S. Z. Vatsadze, Opt. Mater., 2017, 74, 191–196 CrossRef.
- N.-D. H. Gamage, Y. Mei, J. Garcia and M. J. Allen, Angew. Chem., Int. Ed., 2010, 49, 8923–8925 CrossRef PubMed.
Footnotes |
| † Exchange MSc student from Département de chimie, École normale supérieure, PSL University, 75005 Paris, France |
| ‡ Exchange BSc student from the Ghent University, Belgium |
| § Erasmus student from the University of Glasgow, Scotland |
| ¶ Erasmus student from the University of Crete, Greece |
| This journal is © The Royal Society of Chemistry 2018 |