
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal syntheses of schilancidilactones A and B, schilancitrilactone A, and 20-epi-schilancitrilactone A via late-stage nickel-catalyzed cross coupling†
Hengtao
Wang
a,
Xiunan
Zhang
a and
Pingping
Tang
 *ab
*ab
aState Key Laboratory and Institute of Elemento-Organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: ptang@nankai.edu.cn
bCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300071, China
First published on 30th August 2017
Abstract
The first total syntheses of schilancidilactones A and B, schilancitrilactone A, and 20-epi-schilancitrilactone A have been accomplished using a nickel-catalyzed cross coupling of alkyl bromide with vinyl stannane as the final step. The other key steps include late-stage C(sp3)–H bromination, the oxidative cleavage of a diol to provide the requisite ketone and ester for schilancidilactones A and B, and Dieckmann-type condensation to generate the A ring of schilancitrilactone A and 20-epi-schilancitrilactone A.
Schisandraceae triterpenoids have held great interest for synthetic organic chemists because of their molecular structures and diverse biological properties.1 In 2011, Yang and co-workers made a breakthrough in the total synthesis of schindilactone A.2 Since then, rubriflordilactone A has been synthesized by Li3 and Anderson,4 respectively. Our group has disclosed the total syntheses of schilancitrilactones B and C (4 and 5, Fig. 1).5 Recently, the syntheses of propindilactone G,6 rubriflordilactone B,7 19-dehydroxyl arisandilactone A,8 and lancifodilactone G acetate9 have been accomplished.
 | ||
| Fig. 1 Schilancidilactones A and B, schilancitrilactones A, B and C, and 20-epi-schilancitrilactone A. | ||
Schilancidilactones A and B and schilancitrilactone A (1–3, Fig. 1) were isolated by Sun and co-workers from the stems of Schisandra lancifolia.10 Preliminary biological assays indicated that schilancidilactone A (1) showed biological activities for inhibiting HIV-1, and schilancitrilactone A (3) exhibited antifeedant activity, while schilancidilactone B (2) was not tested for further bioactivities due to the limited amount isolated. Compared with schilancitrilactones B and C (4 and 5), schilancidilactones A and B (1 and 2) each possess a 7/5/5/5 tetracyclic core bearing eight stereocenters, and schilancitrilactone A (3) and its epimer (3′) each contain a 5/5/7/5/5/5 hexacyclic core bearing eleven stereocenters. The synthesis of these molecules is challenging. To the best of our knowledge, no syntheses of schilancidilactones A and B (1 and 2), schilancitrilactone A (3) and its epimer 3′ have been reported to date. In this communication, we present the first total syntheses of schilancidilactones A and B (1 and 2), schilancitrilactone A (3), and 20-epi-schilancitrilactone A (3′) using late-stage nickel-catalyzed intermolecular cross coupling for C–C bond formation as a key step.
In a retrosynthetic analysis (Scheme 1), we envisioned that schilancidilactones A and B (1 and 2), schilancitrilactone A (3), and 20-epi-schilancitrilactone A (3′) might be synthesized by the late-stage nickel-catalyzed intermolecular cross coupling of vinyl stannane 6 with alkyl bromides 7 and 10, respectively. Alkyl bromide 7 was expected to arise by the oxidative cleavage of a diol, followed by late-stage C(sp3)–H bromination at the C20 center of compound 8, which in turn could be constructed from compound 9 by a series of steps. Alkyl bromide 10 would arise from compound 11 through Dieckmann-type condensation to generate the A ring and late-stage C(sp3)–H bromination at the C20 center. Compound 11 in turn could be prepared from building blocks 12 and 13 using the chemistry developed in our total syntheses of 4 and 5. Building blocks 6, 9 and 13 were common intermediates in our total syntheses of 4 and 5.5
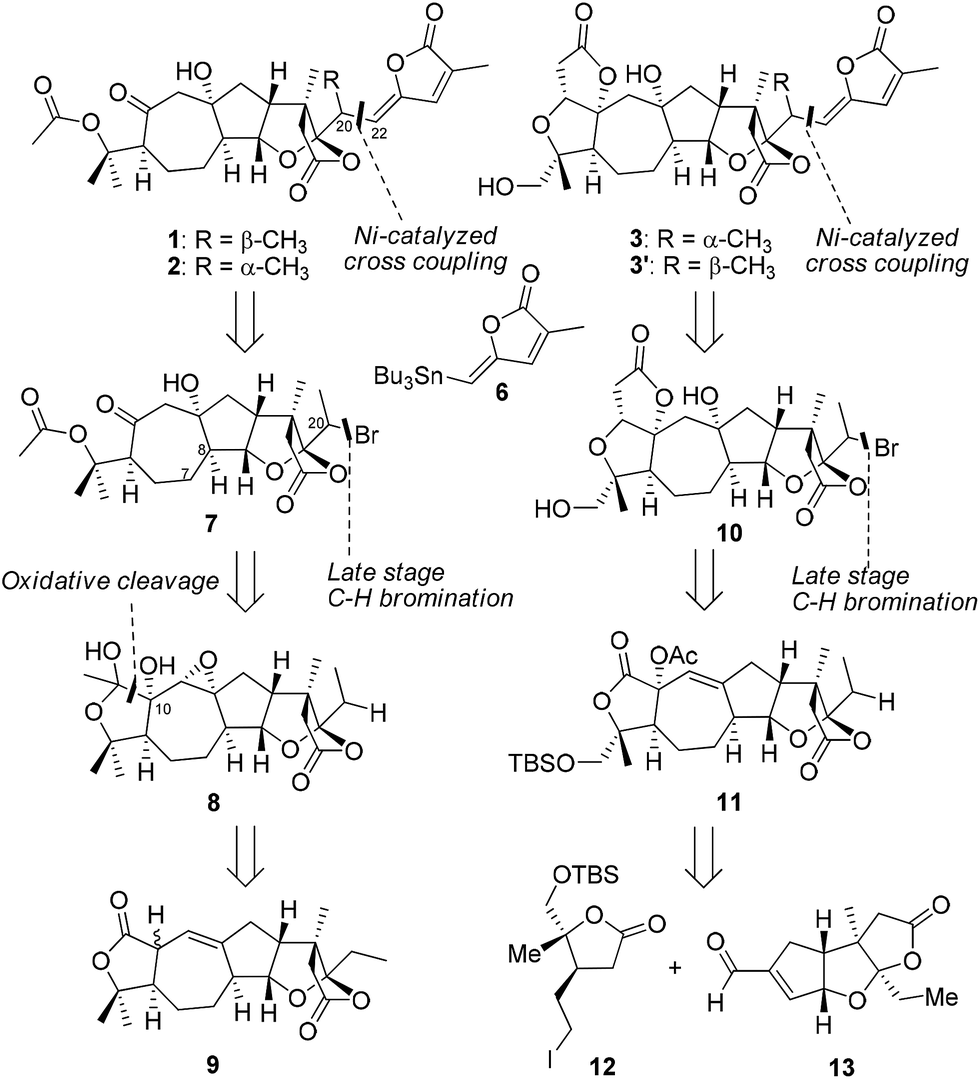 | ||
| Scheme 1 The retrosynthetic analysis of schilancidilactones A and B, schilancitrilactone A, and 20-epi-schilancitrilactone A. | ||
The synthesis commenced with the production of alkyl bromide 7 (Scheme 2). The treatment of compound 9 with LDA in THF and the subsequent reaction with O2 in the presence of P(OMe)3 gave the desired alcohol 14 in 77% yield.11 The epoxidation of 14 with VO(acac)2 and TBHP gave epoxide 15 in 97% yield as a single isomer.12 The configuration of epoxide 15 was determined by the X-ray crystallographic analysis. The addition of methyl magnesium bromide provided the corresponding diol 8 (d.r. = 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 at C1), followed by the oxidative cleavage of the diol using PDC to give ketone 16 in 48% overall yield (2 steps).13 Subsequently, intermediate 16 underwent reductive ring opening with SmI2 to give alcohol 17 in 60% yield,14 which was converted to the corresponding alkyl bromide 7 through late stage C(sp3)–H bromination at C20 with pyridinium tribromide (PyHBr3) in 81% yield (d.r. = 7:1 at C20).15 The selectivity of bromination at C20 rather than C19 might result from less steric hindrance at C20 in compound 17. The initial attempts to achieve late-stage C(sp3)–H iodination at C20 failed.
:1 at C1), followed by the oxidative cleavage of the diol using PDC to give ketone 16 in 48% overall yield (2 steps).13 Subsequently, intermediate 16 underwent reductive ring opening with SmI2 to give alcohol 17 in 60% yield,14 which was converted to the corresponding alkyl bromide 7 through late stage C(sp3)–H bromination at C20 with pyridinium tribromide (PyHBr3) in 81% yield (d.r. = 7:1 at C20).15 The selectivity of bromination at C20 rather than C19 might result from less steric hindrance at C20 in compound 17. The initial attempts to achieve late-stage C(sp3)–H iodination at C20 failed.
 | ||
| Scheme 2 The reagents and conditions: (a) LDA, THF, and −78 °C, then P(OMe)3 and O2, 77% yield; (b) VO(acac)2 (30 mol%), TBHP, DCM, rt, 97% yield; (c) MeMgBr, THF, and 0 °C, then PDC, DCM, rt, 48% yield for the 2 steps; (d) SmI2, THF, −78 °C, 60% yield; (e) PyHBr3, THF, rt, 81%, d.r. (at C20) = 7:1. LDA = lithium N,N-diisopropylamide, PDC = pyridinium dichromate, and PyHBr3 = pyridinium tribromide. | ||
With alkyl bromide 7 in hand, we attempted to finish the total syntheses of schilancidilactones A and B. In our initial synthetic design, we planned to take advantage of the intermolecular radical addition reaction to form the C20–C22 bond based on the chemistry developed in our total syntheses of 4 and 5. The traditional radical conditions (AIBN and Bu3SnH) led to the hydrodebromination product and no desired product was observed. Photoredox catalysis16 was also evaluated and no desired product was found. Inspired by recent advances in the nickel catalyzed cross coupling of alkyl halides for the formation of C–C bonds,17 we postulated our total syntheses of 1 and 2 to involve a late-stage cross coupling reaction with nickel to form the C20–C22 linkage. So we investigated the nickel catalyzed cross coupling reaction of alkyl bromide 18 with vinyl stannane 6 as the model study. Firstly, the conditions developed by the Fu group18 were tested, but no desired product was found (Table 1, entry 1). The hydrodebromination product was a major side product under this condition. With Ni(cod)2 as the catalyst, the various ligands were evaluated and bis(diphenylphosphino)methane (dppm) was found to give a 28% yield of 19 and 28% yield of 19′ (Table 1, entries 2–6). No desired product was observed with NiCl2 or Ni(acac)2 as the catalysts (Table 1, entries 7 and 8). The amounts of Ni(cod)2 and dppm were crucial for the reaction to proceed efficiently. When 40 mol% Ni(cod)2 and 60 mol% dppm were used, a total 80% yield was observed (Table 1, entry 10). After thorough optimization of the reaction conditions (see more details in the ESI†), reactions with 40 mol% Ni(cod)2 and 60 mol% dppm in 1,4-dioxane at 60 °C under N2 were found to give high yields of the desired product. The Z- and E-products might refer to the radical being involved in this cross coupling reaction. Perhaps unsurprisingly, no reaction occurred when a radical inhibitor, such as TEMPO, was added (Table 1, entry 11).
|
|
||
|---|---|---|
| Entry | Conditions | Yield (%) (19/19′)a |
| a The yields were determined by 1H NMR spectroscopy with benzyl chloride as the internal standard. 19/19′ were each formed as a mixture of the diastereomers at the C20 position (see more details in the ESI). | ||
| 1 | NiCl2 (10 mol%), 2,2-bipyridine, KOtBu, t-BuOH/i-BuOH | 0 |
| 2 | Ni(cod)2 (10 mol%), 2,2-bipyridine, dioxane | 0 |
| 3 | Ni(cod)2 (10 mol%), dppf, dioxane | Trace |
| 4 | Ni(cod)2 (10 mol%), dppm, dioxane | 28/28 |
| 5 | Ni(cod)2 (10 mol%), PPh3, dioxane | Trace |
| 6 | Ni(cod)2 (10 mol%), dppp, dioxane | 0 |
| 7 | NiCl2 (10 mol%), dppm, dioxane | 0 |
| 8 | Ni(acac)2 (10 mol%), dppm, dioxane | 0 |
| 9 | Ni(cod)2 (25 mol%), dppm, dioxane | 30/30 |
| 10 | Ni(cod)2 (40 mol%), dppm, dioxane | 40/40 |
| 11 | Ni(cod)2 (40 mol%), dppm, TEMPO, dioxane | 0 |
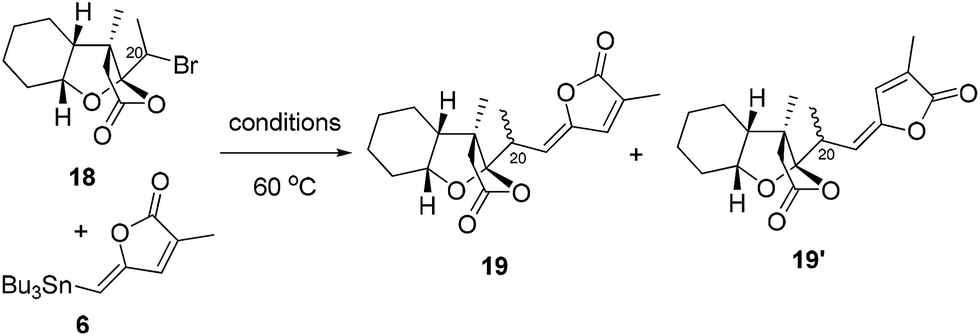
With the optimum reaction conditions, schilancidilactones A (1, 36%) and B (2, 7%) were synthesized from alkyl bromide 7 and vinyl stannane 6 in 43% total yield (Scheme 3). The characterization data obtained for synthetic 1 and 2 were identical to the data reported for the natural products.10a
 | ||
| Scheme 3 The reagents and conditions: (a) 6, Ni(cod)2 (40 mol%), dppm (60 mol%), 1,4-dioxane, 60 °C, 36% yield for 1, 7% yield for 2. cod = 1,5-cyclooctadiene and dppm = bis(diphenylphosphino)methane. | ||
Next, we directed our attention to the syntheses of schilancitrilactone A (3) and 20-epi-schilancitrilactone A (3′). Scheme 4 illustrates the preparation of building block alkyl iodide 12. Alcohol 205 (d.r. at C4 = 2:1) was converted to alkyl iodide 21 with I2, in the presence of Ph3P and imidazole, in 62% yield, together with 21′ in 31% yield.19 The structure of isomer 21′ was confirmed by X-ray crystallographic analysis. The methanolysis of iodolactone 21 with NaOMe provided epoxide 22,20 followed by selective epoxide opening and efficient cyclization to deliver lactone 23. The protection of the primary hydroxyl group in 23 as a TBS ether furnished the desired alkyl iodide 12 in 70% yield for the two steps.
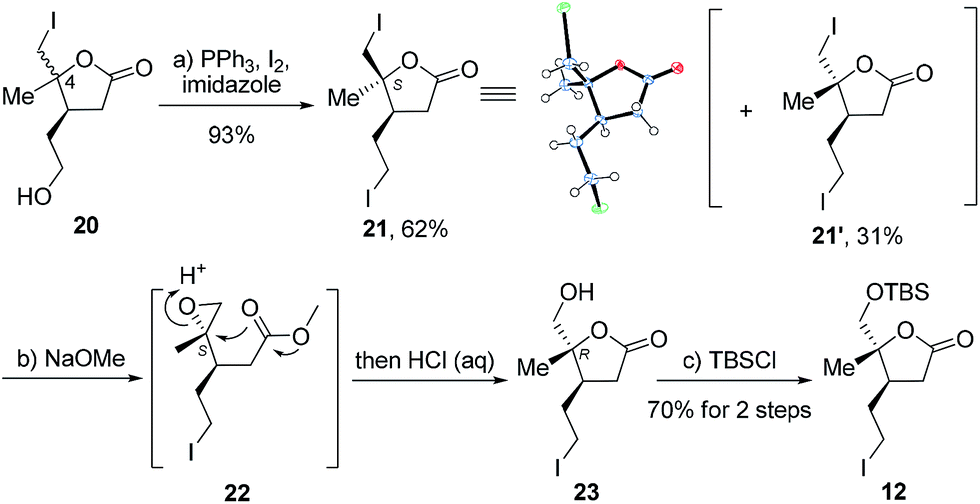 | ||
| Scheme 4 The reagents and conditions: (a) 20 (d.r. at C4 = 2:1), PPh3, I2, imidazole, 0 °C to rt, THF, 93% yield; (b) NaOMe and MeOH/THF, then HCl, rt; (c) TBSCl, imidazole, DCM, rt, 70% yield for the 2 steps. | ||
We now moved to the stage for the completion of the total syntheses of schilancitrilactone A (3) and its epimer (3′). Based on the chemistry developed in our total syntheses of 4 and 5 and the precedent research by Yang’s group (Scheme 5),2,5,6 lactone 12 was treated with LDA in THF and the resulting enolate was reacted with aldehyde 13 to give compound 24 in 88% yield (d.r. = 9:1 at C19), which then underwent dehydration to obtain a 3:1 mixture of diene 25 in 93% yield.21 Under the Luche conditions [CuI, Zn],22 product 26 was prepared in 56% yield (d.r. = 7:1 at C10) through intramolecular radical cyclization. The oxidation of compound 26 by reaction with LDA in the presence of O2 and P(OMe)3 gave alcohol 27, which could be converted into acetate 11 in 93% yield. The treatment of acetate 11 with LDA formed lactone 28 through intramolecular Dieckmann-type condensation in 82% yield,2,6 followed by dehydration with Martin’s sulfurane to give the unsaturated lactone 29 in 75% yield.23 The selective reduction of 29 with L-selectride generated lactone 30, which underwent hydration under Mukaiyama conditions [Co(acac)2, PhSiH3, O2] to install a tertiary alcohol and give compound 31.24 Finally, we used late-stage C(sp3)–H bromination followed by nickel catalyzed cross coupling to finish the total synthesis of schilancitrilacetone A (3) along with its C20-epimer (3′). The spectra and physical properties of schilancitrilacetone A (3) are identical to those reported for the natural product.10b
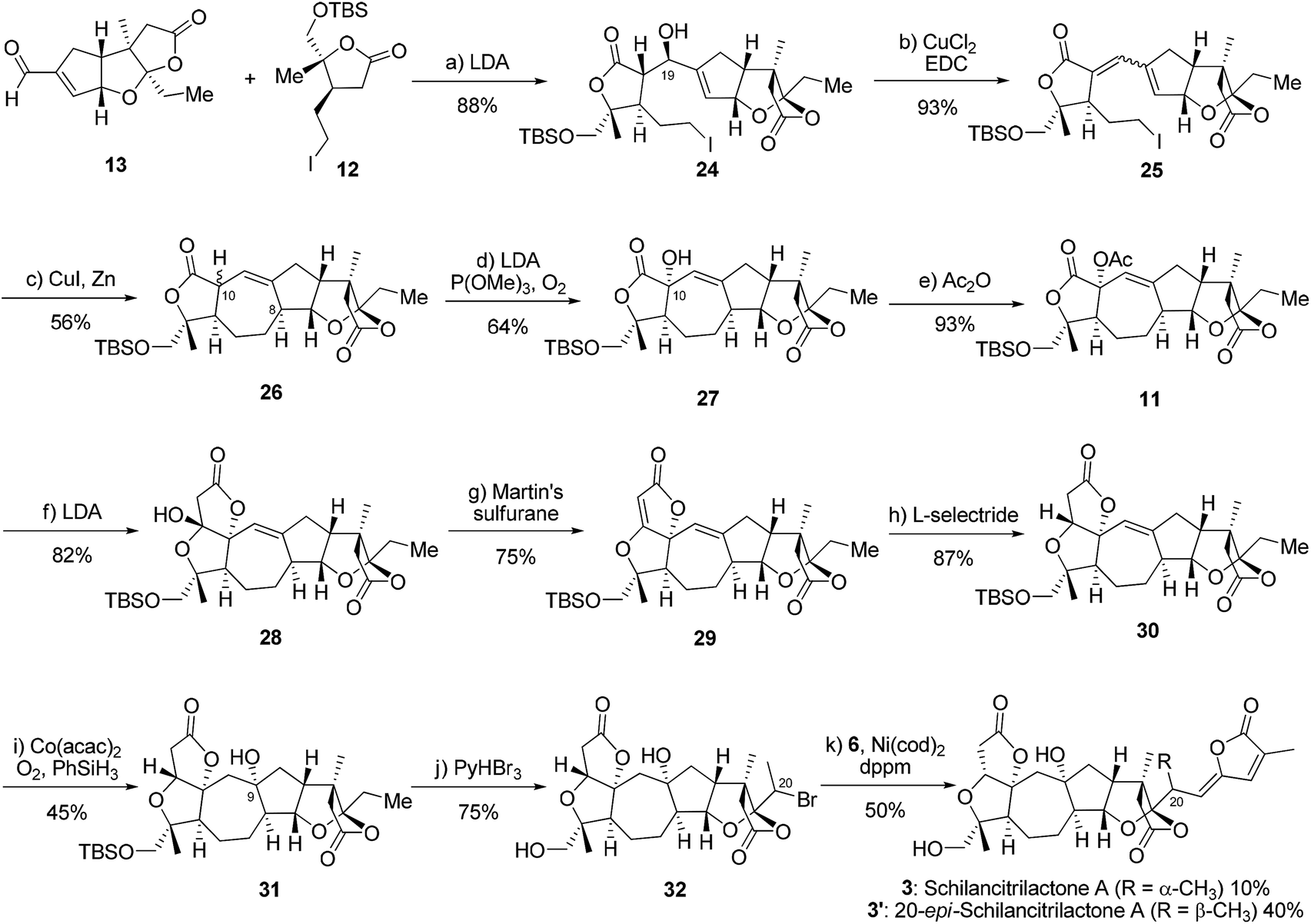 | ||
| Scheme 5 The reagents and conditions: (a) LDA, THF, −78 °C, then 13, 88% yield, d.r. (at C19) = 9:1; (b) CuCl2 (50 mol%), EDC, toluene, 80 °C, 93% yield; (c) CuI, Zn, pyridine/H2O, ultrasound, rt, 56% yield, d.r. (at C10) = 7:1; (d) LDA, THF, and −78 °C, then P(OMe)3, O2, 64% yield for 27, d.r. (at C10) = 2.7:1; (e) Ac2O, Et3N, DCM, rt, 93% yield; (f) LDA, THF, −78 °C, 82% yield; (g) Martin’s sulfurane, DCM, rt, 75% yield; (h) L-selectride, THF, −78 °C, 87% yield; (i) Co(acac)2 (20 mol%), PhSiH3, O2, 1,4-dioxane, rt, 45% yield for 31, d.r. (at C9) = 1.3:1; (j) PyHBr3, THF, rt, 75% yield, d.r. (at C20) = 2:1; (k) 6, Ni(cod)2 (40 mol%), dppm (60 mol%), 1,4-dioxane, 60 °C, 10% yield for 3, 40% yield for 20-epimer 3′. EDC = 1-(3-N,N-dimethylaminopropyl)-3-ethylcarbodiimide, Martin’s sulfurane = bis-[α,α-bis(trifluoromethyl)benzenemethanolato]-diphenysulfur, and L-selectride = lithium tri-sec-butylborohydride. | ||
Conclusions
In summary, we accomplished the first total syntheses of schilancidilactones A and B, schilancitrilactone A, and 20-epi-schilancitrilactone A. A nickel-catalyzed intermolecular cross coupling of alkyl bromide with vinyl stannane was developed to form the C–C bond in the late stage as a key step. In this way, the right hand moieties present in this family of natural products were prepared in the final step of each total synthesis. This strategy shows promise for entry into other derivatives and analogues by way of a common intermediate, which may facilitate the biological studies of Schisandraceae titerpenoids.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the State Key Laboratory of Elemento-Organic Chemistry for the generous start-up financial support. This work was supported by the National Key Research and Development Program of China (2016YFA0602900), NSFC (21421062, 21522205, 21672110).Notes and references
- Selected reviews on Schisandraceae triterpenoids: (a) W.-L. Xiao, R.-T. Li, S.-X. Huang, J.-X. Pu and H.-D. Sun, Nat. Prod. Rep., 2008, 25, 871 RSC; (b) Y.-M. Shi, W.-L. Xiao, J.-X. Pu and H.-D. Sun, Nat. Prod. Rep., 2015, 32, 367 RSC; (c) X. Li, P. H. Cheong and R. G. Carter, Angew. Chem., Int. Ed., 2017, 56, 1704 CrossRef CAS PubMed Selected examples of the synthetic efforts for Schisandraceae triterpenoids: (d) Y. Tang, L. Deng, Y. Zhang, G. Dong, J. Chen and Z. Yang, Org. Lett., 2005, 7, 593 CrossRef CAS PubMed; (e) Y. Tang, Y. Zhang, M. Dai, T. Luo, L. Deng, J. Chen and Z. Yang, Org. Lett., 2005, 7, 885 CrossRef CAS PubMed; (f) Y. Zhang, Y. Tang, T. Luo, J. Shen, J. Chen and Z. Yang, Org. Lett., 2006, 8, 107 CrossRef CAS PubMed; (g) D. Fischer and E. A. Theodorakis, Eur. J. Org. Chem., 2007, 4193 CrossRef CAS; (h) Y. Zhang, W. Ren, Y. Lan, Q. Xiao, K. Wang, J. Xu, J. Chen and Z. Yang, Org. Lett., 2008, 10, 665 CrossRef CAS PubMed; (i) Q. Wang and C. Chen, Org. Lett., 2008, 10, 1223 CrossRef CAS PubMed; (j) L. A. Paquette and K. W. Lai, Org. Lett., 2008, 10, 2111 CrossRef CAS PubMed; (k) K. W. Lai and L. A. Paquette, Org. Lett., 2008, 10, 2115 CrossRef CAS PubMed; (l) L. A. Paquette and K. W. Lai, Org. Lett., 2008, 10, 3781 CrossRef CAS PubMed; (m) K. Matcha, S. Maity, C. K. Malik and S. Ghosh, Tetrahedron Lett., 2010, 51, 2754 CrossRef CAS; (n) M. F. Hossain, K. Matcha and S. Ghosh, Tetrahedron Lett., 2011, 52, 6473 CrossRef CAS; (o) A. Bartoli, G. Chouraqui and J.-L. Parrain, Org. Lett., 2012, 14, 122 CrossRef CAS PubMed; (p) V. A. Ignatenko, Y. Han and G. P. Tochtrop, J. Org. Chem., 2013, 78, 12229 CrossRef CAS PubMed; (q) B. Gockel, S. S. Goh, E. J. Puttock, H. Baars, G. Chaubet and E. A. Anderson, Org. Lett., 2014, 16, 4480 CrossRef CAS PubMed; (r) Y. Wang, Z. Li, L. Lv and Z. Xie, Org. Lett., 2016, 18, 792 CrossRef CAS PubMed; (s) N. Grimblat, T. S. Kaufman and A. M. Sarotti, Org. Lett., 2016, 18, 6420 CrossRef CAS PubMed; (t) Y. Wang, Y. Zhang, Z. Li, Z. Yang and Z. Xie, Org. Chem. Front., 2017, 4, 47 RSC.
- (a) Q. Xiao, W.-W. Ren, Z.-X. Chen, T.-W. Sun, Y. Li, Q.-D. Ye, J.-X. Gong, F.-K. Meng, L. You, Y.-F. Liu, M.-Z. Zhao, L.-M. Xu, Z.-H. Shan, Y. Shi, Y.-F. Tang, J.-H. Chen and Z. Yang, Angew. Chem., Int. Ed., 2011, 50, 7373 CrossRef CAS PubMed; (b) T.-W. Sun, W.-W. Ren, Q. Xiao, Y.-F. Tang, Y.-D. Zhang, Y. Li, F.-K. Meng, Y.-F. Liu, M.-Z. Zhao, L.-M. Xu, J.-H. Chen and Z. Yang, Chem.–Asian J., 2012, 7, 2321 CrossRef CAS PubMed; (c) Y. Li, Z.-X. Chen, Q. Xiao, Q.-D. Ye, T.-W. Sun, F.-K. Meng, W.-W. Ren, L. You, L.-M. Xu, Y.-F. Wang, J.-H. Chen and Z. Yang, Chem.–Asian J., 2012, 7, 2334 CrossRef CAS PubMed; (d) W.-W. Ren, Z.-X. Chen, D. Xiao, Y. Li, T.-W. Sun, Z.-Y. Zhang, Q.-D. Ye, F.-K. Meng, L. You, M.-Z. Zhao, L.-M. Xu, Y.-F. Tang, J.-H. Chen and Z. Yang, Chem.–Asian J., 2012, 7, 2341 CrossRef CAS PubMed.
- J. Li, P. Yang, M. Yao, J. Deng and A. Li, J. Am. Chem. Soc., 2014, 136, 16477 CrossRef CAS PubMed.
- S. S. Goh, G. Chaubet, B. Gockel, M. A. Cordonnier, H. Baars, A. W. Phillips and E. A. Anderson, Angew. Chem., Int. Ed., 2015, 54, 12618 CrossRef CAS PubMed.
- L. Wang, H. Wang, Y. Li and P. Tang, Angew. Chem., Int. Ed., 2015, 54, 5732 CrossRef CAS PubMed.
- (a) L. You, X.-T. Liang, L.-M. Xu, Y.-F. Wang, J.-J. Zhang, Q. Su, Y.-H. Li, B. Zhang, S.-L. Yang, J.-H. Chen and Z. Yang, J. Am. Chem. Soc., 2015, 137, 10120 CrossRef CAS PubMed; (b) L.-M. Xu, L. You, Z.-H. Shan, R.-C. Yu, B. Zhang, Y.-H. Li, Y. Shi, J.-H. Chen and Z. Yang, Chem.–Asian J., 2016, 11, 1406 CrossRef CAS PubMed; (c) J.-J. Zhang, L. You, Y.-F. Wang, Y.-H. Li, X.-T. Liang, B. Zhang, S.-L. Yang, Q. Su, J.-H. Chen and Z. Yang, Chem.–Asian J., 2016, 11, 1414 CrossRef CAS PubMed; (d) X.-T. Liang, L. You, Y.-H. Li, H.-X. Yu, J.-H. Chen and Z. Yang, Chem.–Asian J., 2016, 11, 1425 CrossRef CAS PubMed.
- P. Yang, M. Yao, J. Li, Y. Li and A. Li, Angew. Chem., Int. Ed., 2016, 55, 6964 CrossRef CAS PubMed.
- Y.-X. Han, Y.-L. Jiang, Y. Li, H.-X. Yu, B.-Q. Tong, Z. Niu, S.-J. Zhou, S. Liu, Y. Lan, J.-H. Chen and Z. Yang, Nat. Commun., 2017, 8, 14233, DOI:10.1038/ncomms14233.
- D. Liu, T. Sun, K. Wang, Y. Lu, S. Zhang, Y. Li, Y. Jiang, J. Chen and Z. Yang, J. Am. Chem. Soc., 2017, 139, 5732 CrossRef CAS PubMed.
- (a) X. Luo, Y. Chang, X.-J. Zhang, J.-X. Pu, X.-M. Gao, Y.-L. Wu, R.-R. Wang, W.-L. Xiao, Y.-T. Zheng, Y. Lu, G.-Q. Chen, Q.-T. Zheng and H.-D. Sun, Tetrahedron Lett., 2009, 50, 5962 CrossRef CAS; (b) X. Luo, Y.-M. Shi, R.-H. Luo, S.-H. Luo, X.-N. Li, R.-R. Wang, S.-H. Li, Y.-T. Zheng, X. Du, W.-L. Xiao, J.-X. Pu and H.-D. Sun, Org. Lett., 2012, 14, 1286 CrossRef CAS PubMed.
- C. H. Byeon, C. Y. Chen, D. A. Ellis, D. J. Hart and J. Li, Synlett, 1998, 596 CrossRef CAS.
- K. B. Sharpless and R. C. Michaelson, J. Am. Chem. Soc., 1973, 95, 6136 CrossRef CAS.
- X.-M. Zhang, H. Shao, Y.-Q. Tu, F.-M. Zhang and S.-H. Wang, J. Org. Chem., 2012, 77, 8174 CrossRef CAS PubMed.
- G. A. Molander and G. Hahn, J. Org. Chem., 1986, 51, 2596 CrossRef CAS.
- Selected examples of application of PyHBr3 as brominated reagent: (a) C. Djerassi and C. R. Scholz, J. Am. Chem. Soc., 1948, 70, 417 CrossRef CAS PubMed; (b) D. C. Dong and J. T. Edward, J. Chem. Eng. Data, 1982, 27, 211 CrossRef; (c) P. G. Baraldi, A. Barco, S. Benetti, G. P. Pollini, E. Polo and D. Simoni, J. Org. Chem., 1985, 50, 23 CrossRef CAS; (d) N. Harada, J. Kohori, H. Uda, K. Nakanishi and R. Takeda, J. Am. Chem. Soc., 1985, 107, 423 CrossRef CAS; (e) S. Moon, L. M. Stuhmiller, R. K. Chadha and T. C. McMorris, Tetrahedron, 1990, 46, 2287 CrossRef CAS; (f) K. Hagiya, A. Yamasaki, T. Okuyama and T. Sugimura, Tetrahedron: Asymmetry, 2004, 15, 1409 CrossRef CAS; (g) E. Mernyak, E. Kozma, A. Hetenyi, L. Mark, G. Schneider and J. Woelfling, Steroids, 2009, 74, 520 CrossRef CAS PubMed.
- For selected reviews, see: (a) K. Zeitler, Angew. Chem., Int. Ed., 2009, 48, 9785 CrossRef CAS PubMed; (b) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527 CrossRef CAS PubMed; (c) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC; (d) J. Xuan and W. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828 CrossRef CAS PubMed; (e) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed.
- For selected reviews, see: (a) M. R. Netherton and G. C. Fu, Adv. Synth. Catal., 2004, 346, 1525 CrossRef CAS; (b) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417 CrossRef CAS PubMed; (c) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299 CrossRef CAS PubMed.
- D. A. Powell, T. Maki and G. C. Fu, J. Am. Chem. Soc., 2005, 127, 510 CrossRef CAS PubMed.
- E. J. Corey, S. G. Pyne and W.-G. Su, Tetrahedron Lett., 1983, 24, 4883 CrossRef CAS.
- P. A. Bartlett and J. Myerson, J. Am. Chem. Soc., 1978, 100, 3950 CrossRef CAS.
- (a) H. Sai and H. Ohmizu, Tetrahedron Lett., 1999, 40, 5019 CrossRef CAS; (b) H. Sai, T. Ogiku and H. Ohmizu, Tetrahedron, 2007, 63, 10345 CrossRef CAS.
- J. L. Luche and C. Allavena, Tetrahedron Lett., 1988, 29, 5369 CrossRef CAS.
- (a) R. J. Arhart and J. C. Martin, J. Am. Chem. Soc., 1972, 94, 5003 CrossRef CAS; (b) F. Yokokawa and T. Shioiri, Tetrahedron Lett., 2002, 43, 8679 CrossRef CAS.
- S. Isayama and T. Mukaiyama, Chem. Lett., 1989, 1071 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Data for new compounds and experimental procedures. CCDC 1535911 and 1535913. For the ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc03293e |
| This journal is © The Royal Society of Chemistry 2017 |