
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric synthesis of chromene skeletons via organocatalytic domino reactions of in situ generated ortho-quinone methide with malononitrile and β-functionalized ketone†
Lili
Zhang‡
a,
Xiao
Zhou‡
a,
Pengfei
Li
b,
Zhantao
Liu
a,
Yang
Liu
a,
Yong
Sun
a and
Wenjun
Li
 *a
*a
aDepartment of Medicinal Chemistry, School of Pharmacy, Qingdao University, Qingdao, 266021, China. E-mail: liwj@qdu.edu.cn
bDepartment of Chemistry, South University of Science and Technology of China, Shenzhen, 518055, China
First published on 10th August 2017
Abstract
Enantioselective organocatalytic domino reactions of in situ generated ortho-quinone methides with malononitrile and β-functionalized ketones have been developed. This strategy could generate various chiral chromenes in high yields (up to 99%) and stereoselectivities (up to >99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 e.r.) in the presence of 5 mol% of a bifunctional organocatalyst. Gram-scale and useful synthetic transformations of this process are also presented.
:1 e.r.) in the presence of 5 mol% of a bifunctional organocatalyst. Gram-scale and useful synthetic transformations of this process are also presented.
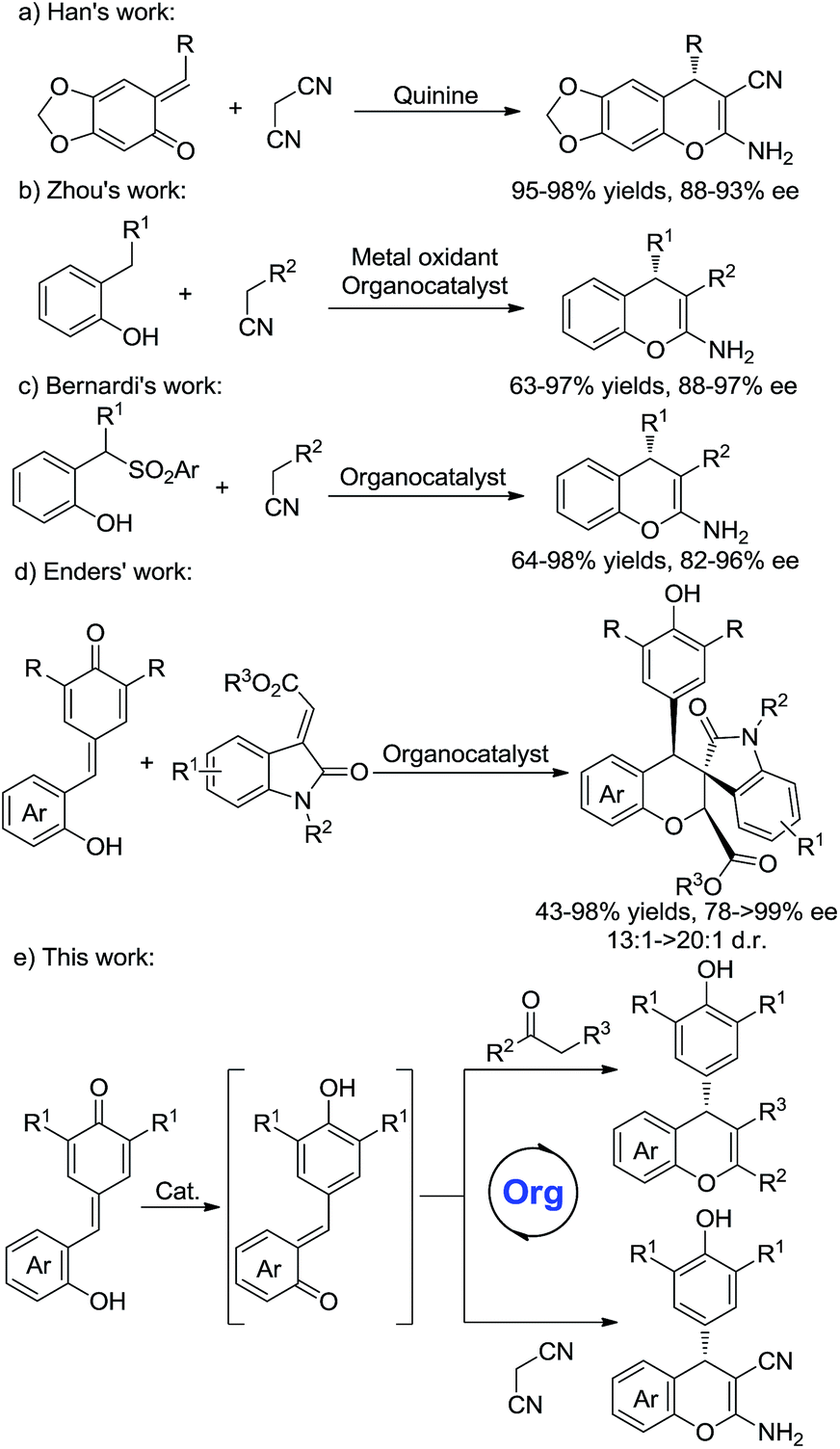
As an important structure in biologically active compounds, the chromene skeleton is a pervasive structural moiety in a plethora of pharmaceuticals.1 In particular, 2-amino-4H-chromenes are widely found in many biologically active molecules and exhibit various biological activities.2 Compared with racemic 2-amino-4H-chromenes, studies on the synthesis of chiral 2-amino-4H-chromenes are still rare.3,4 Wang et al. reported a chiral thiourea-catalyzed Mannich/cyclization sequence and conjugate additions of nitroalkanes to 2-iminochromenes.4a,b The Feng group developed metal complex catalyzed cascade reactions.4d Despite significant progress in this area, a few issues, such as unsatisfactory yields, poor stereoselectivities and limited substrate scope, have not yet been resolved. Hence, the development of an efficient protocol to access purely chiral chromenes remains in high demand.
ortho-Quinone methides (o-QMs) are highly reactive intermediates and are easily obtained from various precursors.5 Due to their distinctive electrophilic properties, o-QMs have been widely explored in organic chemistry. In recent years, much attention has been made in their use in catalytic asymmetric reactions.6 For instances, Sigman and co-workers reported a palladium-catalyzed asymmetric dialkoxylation of 2-propenyl phenols through a palladium ortho-quinone methide intermediate.6a Lectka et al. developed an organocatalyzed [4 + 2] cycloaddition reaction of o-QMs with silylketene acetals to afford coumarin derivatives in good enantioselectivities.6b More recently, o-QMs have been utilized for the synthesis of chiral chromenes.7 Schneider and Rueping independently developed Brønsted acid catalyzed conjugate addition/cyclodehydration reactions of β-diketones with in situ generated o-QMs for the synthesis of 4H-chromenes.7a,b Moreover, the Han group reported quinine-catalyzed annulations of o-QMs with malononitrile to furnish chiral 2-amino-4H-chromenes in high stereoselectivities (Scheme 1a).7c Later, Zhou et al. developed a novel method for the asymmetric synthesis of 2-amino-4H-chromenes from in situ generated o-QMs and active methylene compounds (Scheme 1b).7d Bernardi's group reported a bifunctional squaramide-catalyzed reaction of o-QMs generated in situ from 2-(1-arylsulfonyl-alkyl)phenols with active methylene compounds (Scheme 1c).4e
 | ||
| Scheme 1 Synthesis of chiral chromenes via o-quinone methides. | ||
Although some progress have been made in the synthesis of chiral chromenes, the substrate scope is still limited. Very recently, Enders's group have developed organocatalytic domino oxa-Michael/1,6-addition reactions to synthesize functionalized chromenes (Scheme 1d).8 Inspired by this work, we envisioned that an organocatalytic 1,6-conjugate addition and subsequent cycloaddition reactions of in situ generated o-QMs with malononitrile and β-functionalized ketones might provide a novel approach for the construction of various types of chiral chromenes (Scheme 1e).
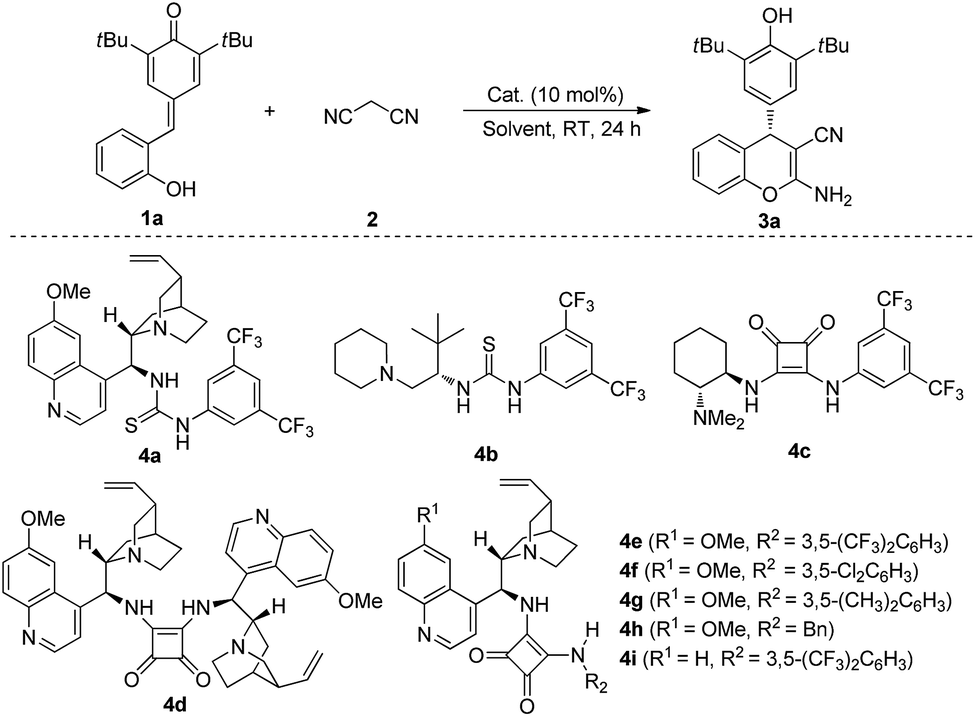
We started our preliminary investigation with the reaction between quinone methide 1a and malononitrile 2 in the presence of catalyst 4a. To our delight, 84% yield and 85:15 e.r. were obtained (Table 1, entry 1). With the initial experimental results in hand, we then switched our attention to other organocatalysts. The assessment of catalysts indicated that bifunctional catalyst 4e, which was pioneered by Rawal and co-workers,9 was the most efficient catalyst to furnish the desired product 3a in 86% yield and 93:7 e.r. (Table 1, entry 5). After identifying catalyst 4e as the best catalyst, we investigated the role of solvent in this process. Further investigations revealed that solvents also played a key role in this transformation. For instances, moderate e.r. were obtained when toluene, PhCF3, anisole, xylenes and dichloroethane were utilized (Table 1, entries 10–14). Switching the solvent to chloroform afforded the product 3a in 89% yield and 94:6 e.r. (Table 1, entry 15). To further optimize the reaction conditions, we changed the concentration and reaction temperature. The results showed that e.r. would improve to 97.5:2.5 with 90% yield when 1.0 mL of CHCl3 was used (Table 1, entry 16). Further increasing the volume of CHCl3 would not improve e.r. (Table 1, entry 17). 90% yield and 95:5 e.r. were observed when we reduced the reaction temperature to 0 °C for 48 h with 1.0 mL of CHCl3 (Table 1, entry 18). Gratifyingly, if we decreased the catalyst loading to 5 mol%, the desired product 3a could be still obtained in 97% yield with 97.5:2.5 e.r. (Table 1, entry 19).
|
|
||||
|---|---|---|---|---|
| Entry | Cat. | Solvent | Yieldb (%) | e.r.c |
| a Reaction conditions: a mixture of 1a (0.05 mmol), 2 (0.06 mmol) and catalyst (10 mol%) in the solvent (0.3 mL) was stirred at room temperature for 24 h. b Isolated yield. c Determined by HPLC analysis. d 1.0 mL of CHCl3 was used. e 1.5 mL of CHCl3 was used. f The reaction was conducted at 0 °C for 48 h. g 5 mol% of 4e was used for 48 h. | ||||
| 1 | 4a | CH2Cl2 | 84 | 85:15 |
| 2 | 4b | CH2Cl2 | 70 | 82:18 |
| 3 | 4c | CH2Cl2 | 98 | 13:87 |
| 4 | 4d | CH2Cl2 | 65 | 74:26 |
| 5 | 4e | CH2Cl2 | 86 | 93:7 |
| 6 | 4f | CH2Cl2 | 87 | 91:9 |
| 7 | 4g | CH2Cl2 | 85 | 87:13 |
| 8 | 4h | CH2Cl2 | 98 | 79:21 |
| 9 | 4i | CH2Cl2 | 87 | 91:9 |
| 10 | 4e | Toluene | 58 | 89:11 |
| 11 | 4e | PhCF3 | 82 | 81:19 |
| 12 | 4e | Anisole | 34 | 90:10 |
| 13 | 4e | Xylenes | 63 | 87:13 |
| 14 | 4e | DCE | 86 | 88:12 |
| 15 | 4e | CHCl3 | 89 | 94:6 |
| 16d | 4e | CHCl3 | 90 | 97.5:2.5 |
| 17e | 4e | CHCl3 | 94 | 95:5 |
| 18d,f | 4e | CHCl3 | 90 | 95:5 |
| 19d,g | 4e | CHCl3 | 97 | 97.5:2.5 |
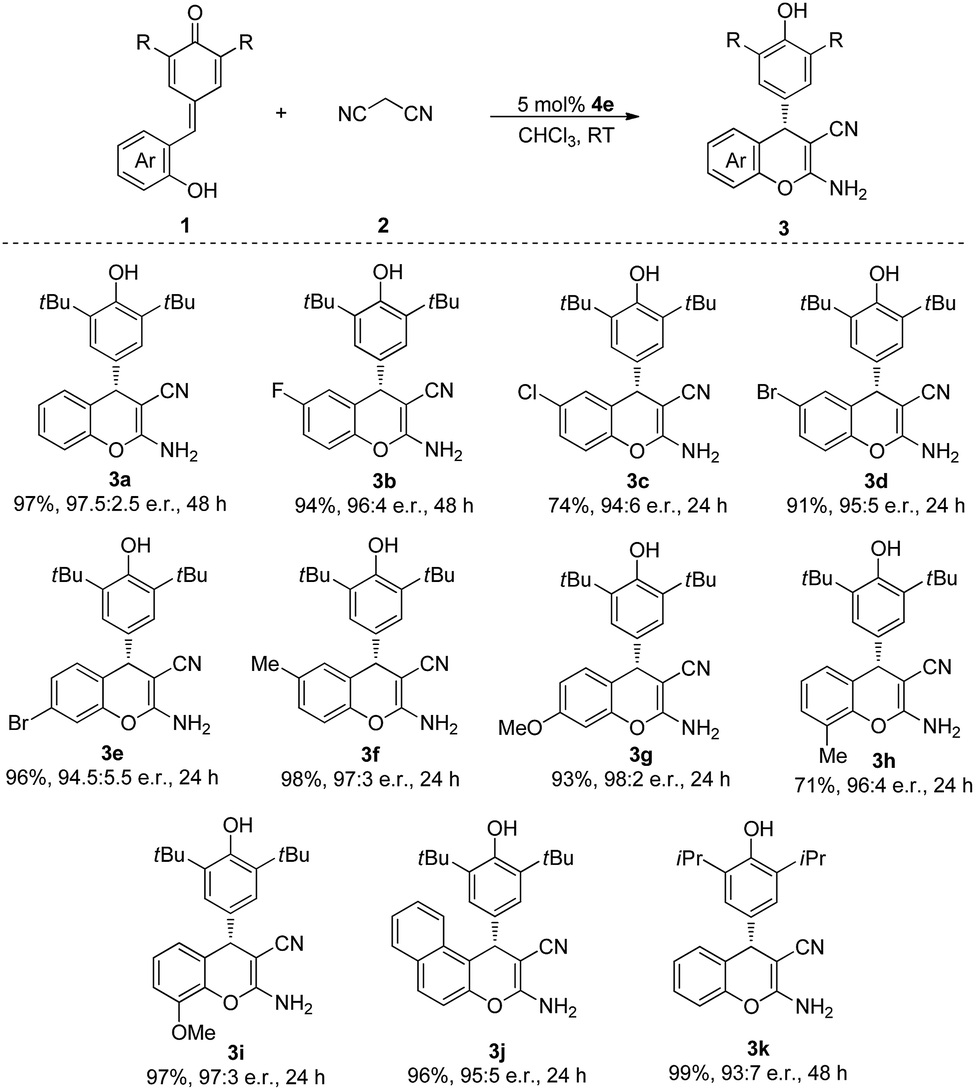
With the optimized conditions in hand, we then tested the substrate scope of this cascade process. As indicated in Table 2, QMs 1, which contained various functional groups were surveyed. The substrates bearing both electron-withdrawing groups (F, Cl, Br) and electron-donating groups (Me, OMe) in para, meta and ortho positions of the phenyl ring were all tolerated in this reaction to afford the corresponding products in excellent yields and e.r. (3a–i). Furthermore, the substrate 1j, which contained naphthyl moiety, also participated in this process and gave the desired product 3j in 96% yield and 95:5 e.r. after 24 h at room temperature. When we replaced the tert-butyl group of the QMs by isopropyl group, the desired product 3k was obtained in 99% yield and 93:7 e.r.
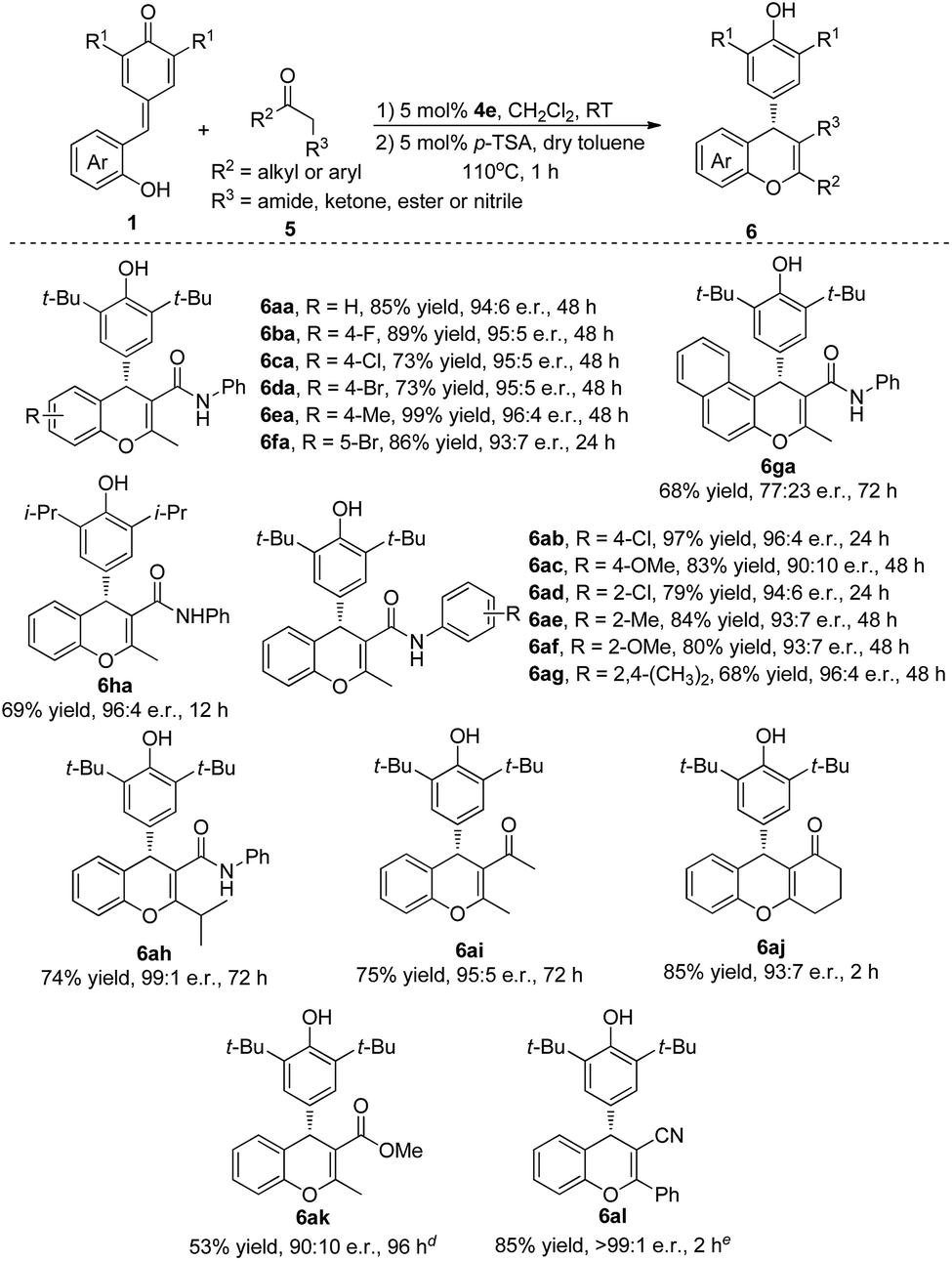
Inspired by the success, we shifted our focus to β-functionalized ketones to access chiral chromenes with more functional groups. As shown in Table 3, we started our investigation with reaction between quinone methide 1a and β-keto amide 5a. To our delight, the desired product 6aa was obtained in 85% yield and 94:6 e.r. through a cascade reaction in the presence of bifunctional catalyst 4e and a subsequent dehydration catalyzed by p-toluenesulfonic acid (for optimal conditions, see ESI†). The substrates QMs 1, which bearing electron-withdrawing or electron-donating groups in the different position of phenyl ring, underwent this transformation to give the desired products 6aa–fa in excellent yields (73–99%) and e.r. (93:7–96:4). A naphthyl- and hydroxy-substituted QM also took part in this process and gave the desired product 6ga in 68% yield and 77:23 e.r. Replacing the tert-butyl group of the QMs by isopropyl group also furnished the desired product 6ha in 69% yield and 96:4 e.r. Encouraged by the above results, the generality of β-functionalized ketones 5 was also evaluated. A wide range of β-keto amides (5b–h) was tolerated under the reaction conditions to provide the corresponding products 6ab–ah in 68–97% yields and 90:10–99:1 e.r. Gratifyingly, 1,3-diketones 5i and 5j, α-ester ketone 5k and β-ketonitrile 5l also participated in this transformation successfully to give the desired products 6ai–al in 53–85% yields and 90:10 to >99:1 e.r.
| a Reaction conditions: a mixture of 1a–h (0.05 mmol), 5a–l (0.06 mmol) and cat. 4e (5 mol%) in CH2Cl2 (1.0 mL) was stirred at room temperature for 2–96 h. After column chromatography, the intermediate product was treated with 5 mol% of p-TSA in dry toluene (0.5 mL) at 110 °C for 1 h. b Isolated yield. c Determined by HPLC analysis. d 20 mol% catalyst was used and the reaction was conducted at 110 °C for 3 h. e The reaction was conducted at 110 °C for 24 h. |
|---|
|
To test the synthetic utility of our method, 3a was prepared on a gram scale. As shown in Scheme 2, the desired product 3a was obtained in 92% yield with 97.5:2.5 e.r. under optimal reaction conditions. As indicated in Scheme 3, some useful synthetic transformations of this process were also presented. Treatment of 3a with another equivalent of malononitrile in the presence of triethylamine in EtOH at reflux afforded benzopyranopyridine 7 in 53% yield and 90:10 e.r. Furthermore, the AlCl3-mediated de-tert-butylation of 5aa was also attempted and the desired product 8 was obtained in 92% yield and 93:7 e.r. (Scheme 3).
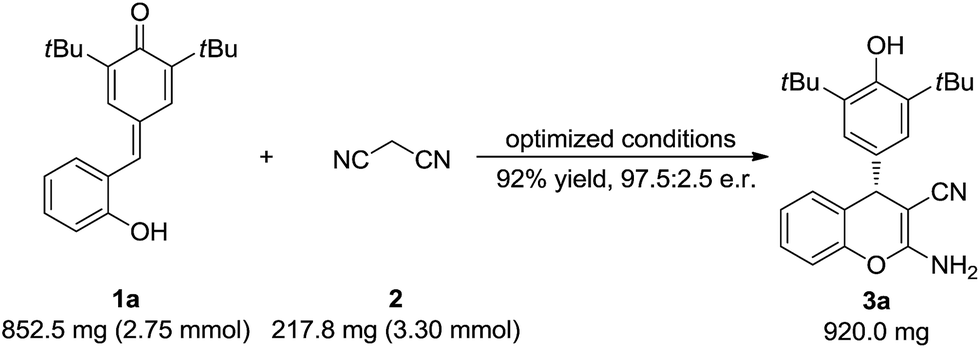 | ||
| Scheme 2 Gram-scale synthesis of 3a. | ||
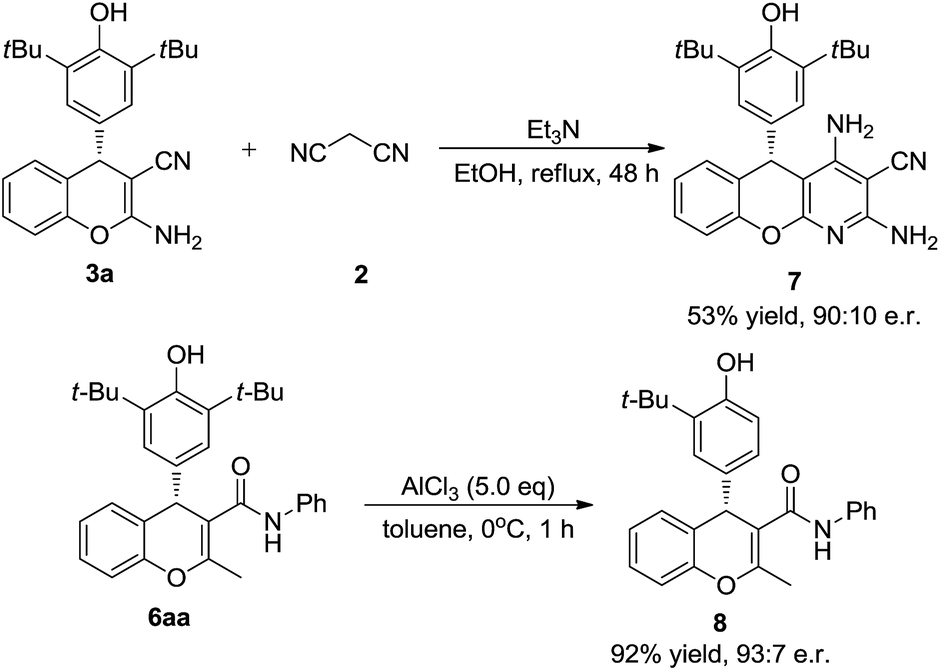 | ||
| Scheme 3 Synthetic transformations. | ||
To verify the mechanism of this process, a control experiment using TBS-protected substrate 9 was performed in the presence of catalyst 4e. The experiment showed that product 10 could be obtained in 87% yield after 72 h at room temperature (Scheme 4). Meanwhile, 50:50 e.r. was observed, which indicated that the hydroxy group is important in the enantioselectivity-determining step and this process proceeded through o-QM intermediate. As shown in Scheme 5, a plausible mechanism is proposed to explain the reaction process. First of all, substrate 1d would be transformed into o-QM 1d′ in the presence of bifunctional catalyst 4e and the 1,6-conjugated addition of o-QM 1d′ with malononitrile 2 formed the intermediated A in the presence of catalyst 4e. Subsequently, the intramolecular oxa-nucleophilic addition took place to afford the intermediated B, which would be transformed into the final product 3d through tautomerization process. The absolute configuration of the adduct 3d and 6da were unambiguously determined by X-ray crystallography.10
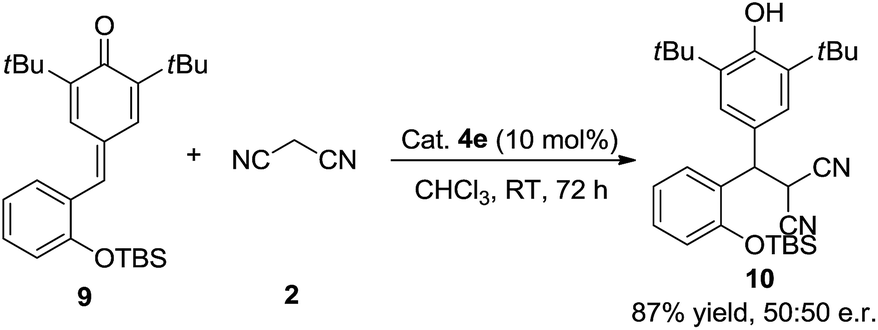 | ||
| Scheme 4 The mechanistic study. | ||
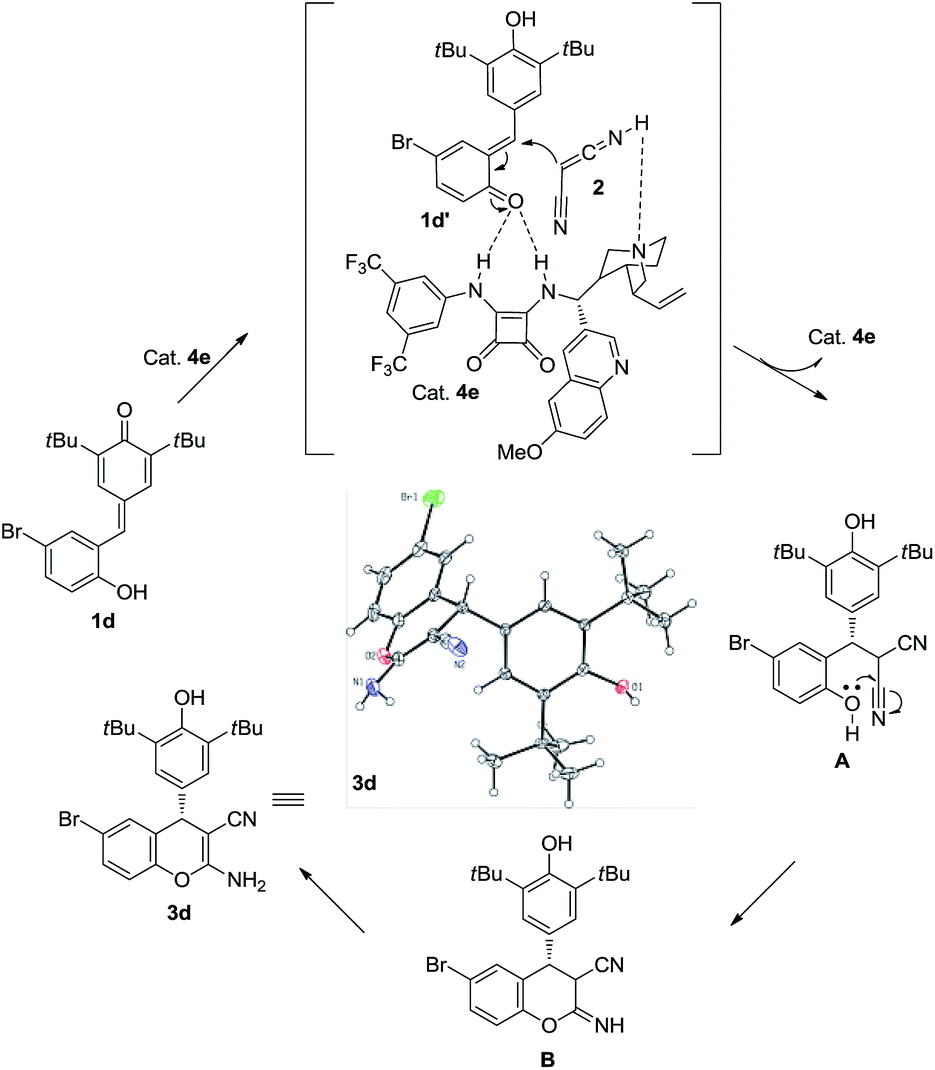 | ||
| Scheme 5 Plausible mechanism. | ||
In summary, we have developed an asymmetric organocatalytic domino reactions of in situ generated ortho-quinone methides with malononitrile and β-functionalized ketones for the synthesis of chiral chromenes in high yields and enantioselectivities. This strategy provides an efficient and convenient pathway to synthesize chiral chromene skeletons. Further investigation regarding the utilization of this organocatalytic procedure in the preparation of natural products is underway.
Acknowledgements
The authors acknowledge the financial support from the start-up grant from Qingdao University and National Natural Science Foundation of China (No. 21502043).Notes and references
- For selected examples of natural molecules containing the chromene scaffold, see: (a) The Flavanoids: Advances in Research, ed. J. B. Harborne, Chapman and Hall, London, 1994 Search PubMed; (b) V. S. Parmar, S. C. Jain, K. S. Bisht, R. Jain, P. Taneja, A. Jha, O. D. Tyagi, A. K. Prasad, J. Wengel, C. E. Olsen and P. M. Boll, Phytochemistry, 1997, 46, 597 CrossRef CAS; (c) B. A. Bohm, J. B. Choy and A. Y.-M. Lee, Phytochemistry, 1989, 28, 501 CrossRef CAS; (d) G. A. Iacobucci and J. G. Sweeny, Tetrahedron, 1983, 39, 3005 CrossRef CAS.
- (a) W. Kemnitzer, S. Kasibhatla, S. Jiang, H. Zhang, J. Zhao, S. Jia, L. Xu, C. Crogan-Grundy, R. Denis, N. Barriault, L. Vaillancourt, S. Charron, J. Dodd, G. Attardo, D. Labrecque, S. Lamothe, H. Gourdeau, B. Tseng, J. Drewe and S. X. Cai, Bioorg. Med. Chem. Lett., 2005, 15, 4745 CrossRef CAS; (b) W. Kemnitzer, J. Drewe, S. Jiang, H. Zhang, Y. Wang, J. Zhao, S. Jia, J. Herich, D. Labreque, R. Storer, K. Meerovitch, D. Bouffard, R. Rej, R. Denis, C. Blais, S. Lamothe, G. Attardo, H. Gourdeau, B. Tseng, S. Kasibhatla and S. X. Cai, J. Med. Chem., 2004, 47, 6299 CrossRef CAS.
- (a) D. R. Anderson, S. Hegde, E. Reinhard, L. Gomez, W. F. Vernier, L. Lee, S. Liu, A. Sambandam, P. A. Snider and L. Masih, Bioorg. Med. Chem. Lett., 2005, 15, 1587 CrossRef CAS PubMed; (b) J. L. Wang, D. Liu, Z. Zhang, S. Shan, X. Han, S. M. Srinvasula, C. M. Croce, E. S. Alnemeri and Z. Huang, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 7124 CrossRef CAS PubMed; (c) P. Vachal and E. N. Jacobsen, J. Am. Chem. Soc., 2002, 124, 10012 CrossRef CAS; (d) T. Okino, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2003, 125, 12672 CrossRef CAS; (e) T. Okino, Y. Hoashi, T. Furukawa and Y. T. X. Xu, J. Am. Chem. Soc., 2005, 127, 119 CrossRef CAS.
- For the synthesis of chiral 2-amino-4H-chromenes, see: (a) Q. Ren, W. Y. Siau, Z. Y. Du, K. Zhang and J. Wang, Chem.–Eur. J., 2011, 17, 7781 CrossRef CAS; (b) W. Li, H. Liu, X. Jiang and J. Wang, ACS Catal., 2012, 2, 1535 CrossRef CAS; (c) Z. Dong, X. Liu, J. Feng, M. Wang, L. Lin and X. Feng, Eur. J. Org. Chem., 2011, 137 CrossRef CAS; (d) W. Chen, Y. Cai, X. Fu, X. Liu, L. Lin and X. Feng, Org. Lett., 2011, 13, 4910 CrossRef CAS; (e) L. Caruana, M. Mondatori, V. Corti, S. Morales, A. Mazzanti, M. Fochi and L. Bernardi, Chem.–Eur. J., 2015, 21, 6037 CrossRef CAS.
- For reviews on the chemistry of o-QMs, see: (a) N. J. Willis and C. D. Bray, Chem.–Eur. J., 2012, 18, 9160 CrossRef CAS; (b) Quinone Methides, ed. S. E. Rokita, Wiley, New York, 2009, p. 412 Search PubMed; (c) R. W. Van DeWater and T. R. R. Pettus, Tetrahedron, 2002, 58, 5367 CrossRef CAS; (d) T. P. Pathak and M. S. Sigman, J. Org. Chem., 2011, 76, 9210 CrossRef CAS; (e) C. M. Beaudry, J. P. Malerich and D. Trauner, Chem. Rev., 2005, 105, 4757 CrossRef CAS.
- For selected examples of asymmetric reactions with o-QMs, see: (a) Y. Zhang and M. S. Sigman, J. Am. Chem. Soc., 2007, 129, 3076 CrossRef CAS; (b) E. Alden-Danforth, M. T. Scerba and T. Lectka, Org. Lett., 2008, 10, 4951 CrossRef CAS; (c) H. Lv, L. You and S. Ye, Adv. Synth. Catal., 2009, 351, 2822 CrossRef CAS; (d) T. P. Pathak, K. M. Gligorich, B. E. Welm and M. S. Sigman, J. Am. Chem. Soc., 2010, 132, 7870 CrossRef CAS; (e) M. Rueping, U. Uria, M. Y. Lin and I. Atodiresei, J. Am. Chem. Soc., 2011, 133, 3732 CrossRef CAS; (f) D. Wilcke, E. Herdtweck and T. Bach, Synlett, 2011, 1235 CAS; (g) Y. Luan and S. E. Schaus, J. Am. Chem. Soc., 2012, 134, 19965 CrossRef CAS; (h) H. Lv, W. Q. Jia, L. H. Sun and S. Ye, Angew. Chem., Int. Ed., 2013, 52, 8607 CrossRef CAS; (i) J. Izquierdo, A. Orue and K. A. Scheidt, J. Am. Chem. Soc., 2013, 135, 10634 CrossRef CAS; (j) Z. B. Wang, F. J. Ai, Z. Wang, W. X. Zhao, G. Y. Zhu, Z. Y. Lin and J. W. Sun, J. Am. Chem. Soc., 2015, 137, 383 CrossRef CAS; (k) W. Zhao, Z. Wang, B. Chu and J. Sun, Angew. Chem., Int. Ed., 2015, 54, 1910 CrossRef CAS; (l) S. Saha, S. K. Alamsetti and C. Schneider, Chem. Commun., 2015, 51, 1461 RSC; (m) S. Saha and C. Schneider, Chem.–Eur. J., 2015, 21, 2348 CrossRef CAS; (n) S. Saha and C. Schneider, Org. Lett., 2015, 17, 648 CrossRef CAS; (o) J.-J. Zhao, S.-B. Sun, S.-H. He, Q. Wu and F. Shi, Angew. Chem., Int. Ed., 2015, 54, 5460 CrossRef CAS; (p) B. Wu, Z. Yu, X. Gao, Y. Lan and Y. G. Zhou, Angew. Chem., Int. Ed., 2017, 56, 4006 CrossRef CAS; (q) Y. Xie and B. List, Angew. Chem., Int. Ed., 2017, 56, 4936 CrossRef CAS.
- (a) C. Hsiao, H. Liao and M. Rueping, Angew. Chem., Int. Ed., 2014, 53, 13258 CrossRef CAS; (b) O. El-Sepelgy, S. Haseloff, S. Alamsetti and C. Schneider, Angew. Chem., Int. Ed., 2014, 53, 7923 CrossRef CAS; (c) A. Adili, Z.-L. Tao, D.-F. Chen and Z.-Y. Han, Org. Biomol. Chem., 2015, 13, 2247 RSC; (d) B. Wu, X. Gao, Z. Yan, M. Chen and Y. G. Zhou, Org. Lett., 2015, 17, 6134 CrossRef CAS PubMed; (e) B. Wu, X. Gao, Z. Yan, W.-X. Huang and Y.-G. Zhou, Tetrahedron Lett., 2015, 56, 4334 CrossRef CAS.
- K. Zhao, Y. Zhi, T. Shu, A. Valkonen, K. Rissanen and D. Enders, Angew. Chem., Int. Ed., 2016, 55, 12104 CrossRef CAS PubMed.
- (a) J. P. Malerich, K. Hagihara and V. R. Rawal, J. Am. Chem. Soc., 2008, 130, 14416 CrossRef CAS; (b) Y. Zhu, J. P. Malerich and V. H. Rawal, Angew. Chem., Int. Ed., 2010, 49, 153 CrossRef CAS.
- CCDC 1526191 (3d) and 1545246 (6da) contain the supplementary crystallographic data for this paper.†.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1526191 and 1545246. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra08157j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |