
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dynamic self-assembled polymer: HCl responsive inversion of supramolecular polymer handedness†
Arpita
Paikar
and
Debasish
Haldar
 *
*
Department of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur, West Bengal 741252, India. E-mail: deba_h76@yahoo.com; deba_h76@iiserkol.ac.in; Fax: +91 3325873020; Tel: +91 3325873119
First published on 6th October 2017
Abstract
A series of discotic tricarboxyamides with varied amino acid side arm functionality and their HCl responsive diverse self-assembly behavior and formation of dynamic polymer is studied. Discotic trisamide 1 obtained from Boc protected L-Lys self-assembled into a long fibrillar aggregate-like supramolecular P helical polymer. However, on addition of HCl, the supramolecular helical handedness of the assemblies is completely inverted. Irrespective of the chiral centres configuration, supramolecular chiral bias is arising from molecular conformations. FE-SEM reveals the formation of right handed entangled polymeric fibers. However, left handed entangled fibers have appeared on addition of 0.12 M HCl. The trisamide 2 containing Boc protected L-Trp exhibits disk like morphology both in the presence and absence of 0.12 M HCl and does not show a change of supramolecular handedness. This demonstrates how remarkably distinct morphologies originate from stimuli responsive building blocks assembled in a subtly different manner.
Introduction
Supramolecular polymers are highly interesting for their dynamic character. Tuning the nature, directionality and spatial arrangement of stimuli responsive building blocks in self-assembled systems provides chiral supramolecular polymers with new features compared to the bulk materials.1 Understanding the chirality of supramolecular systems2–4 is highly important for biological sciences as well as material sciences mainly in the fields of chiral recognition and sensing,5 optoelectronic materials,6 chiroptical switches,7,8 asymmetric catalysts9,10 and separation processes,11 metal coordination,12 redox reactions,13 host–guest interactions,14 anions,15,16 solvent polarity,17 concentrations variations,18,19 light irradiation,20,21 high pressure22 and temperature23 on large discotic,24 dendritic,25,26 and gelator molecules.27 Feringa, van Esch and coworkers have reported the cyclohexane based C3 symmetric discotic hydrogelator containing three amino acid side groups.28–31 Meijer and coworkers have discussed analogous systems with 1,3,5-benzene tricarboxylic acid core.32–36 For these compounds, tuning experimental conditions promotes the selection of a predominant assembly.37 Changing the stereochemistry of the building blocks as well as experimental condition can control the supramolecular chiral amplification.38 However, the examples of designer supramolecular assemblies that can adopt diverse well-defined chiral nanostructures by simply changing the molecular conformation are relatively rare.39Herein, we report the conformational preferences in the trisamide system over the configuration of the chiral center to determine the helical handedness of supramolecular polymers. We have designed and synthesized a series of trisamides containing Boc protected L-lysine (Lys), L-tryptophan (Trp) and benzene-1,3,5-triamine and investigated their structures and stimuli responsive behavior. Interestingly the trisamide 1 containing (Boc)2-L-Lys formed one-dimensional supramolecular helical polymer. On protonation, the supramolecular helical hendedness of the polymeric assemblies has inverted. However, the analogue 2 containing (Boc)-L-Trp exhibits disk like morphology both in presence and absence of 0.12 M HCl and does not show change of supramolecular handedness.
Starting from commercially available 1,3,5-trinitrophenol (picric acid), 1,3,5-triaminobenzene was synthesized in single step in 86% yield, by refluxing with ammonium formate and Zn dust in methanol at 60 °C for 1 h (Scheme 1, ESI†). The trisamides 1 and 2 (Scheme 1) were synthesized by conventional solution phase DCC (dicyclohexylcarbodiimide) mediated coupling of 1,3,5-triaminobenzene with the (Boc)2-L-Lys and (Boc)-L-Trp following a high purity, as confirmed by 1H-NMR (nuclear magnetic resonance), 13C-NMR, FT-IR (Fourier-transform infrared) and mass spectrometry (MS) analysis (ESI†). For C3-symmetric trisamides 1 and 2 the design principle explored was to incorporate chiral amino acid side chains that are responsive to externational stimuli like protonation. The bulky Boc protective groups at side chains may force the amides groups out of the plane of the central aromatic core and facilitates the formation of three intermolecular hydrogen bonds for stack or dimer structure.
 | ||
| Scheme 1 The trisamides 1 and 2. | ||
For trisamide, the hydrogen-bonded self-assembly promotes mainly dimers or stacks, which can be controlled by the nature of the substituent or experimental conditions (concentration) or external stimuli such as temperature, magnetic field, metal ions.39 Depending on the ratio of the number of pairs of hydrogen bond dipole vectors which are parallel or antiparallel to each other, the hydrogen-bonded stack of trisamide can exists in two conformations, 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 and 2:1.39 In 3:0 stacking, all the amide oxygens attached to central core are oriented in the same direction with respect to the central benzene plane (Fig. S1, ESI†). However, in 2:1 stacking, two amide oxygens attached to central core are on one side and the third one is on other side of the central benzene plane (Fig. S2, ESI†).39 From our previous reports, by X-ray crystallography it has proved that 3:0 conformation stack in right handed helical manner (P helix) and the 2:1 conformation stack in left handed helical manner (M helix) (Fig. 1). This is irrespective on the chirality of the side chains. A trisamide containing achiral β-alanine adopts 2:1 conformations and formed M helix (Fig. 1a).40 However, trisamide containing achiral γ-aminobutyric acid (Fig. 1b) or chiral methionine (Fig. 1c) adopts 3:0 conformations and formed P helix.41
:0 and 2:1.39 In 3:0 stacking, all the amide oxygens attached to central core are oriented in the same direction with respect to the central benzene plane (Fig. S1, ESI†). However, in 2:1 stacking, two amide oxygens attached to central core are on one side and the third one is on other side of the central benzene plane (Fig. S2, ESI†).39 From our previous reports, by X-ray crystallography it has proved that 3:0 conformation stack in right handed helical manner (P helix) and the 2:1 conformation stack in left handed helical manner (M helix) (Fig. 1). This is irrespective on the chirality of the side chains. A trisamide containing achiral β-alanine adopts 2:1 conformations and formed M helix (Fig. 1a).40 However, trisamide containing achiral γ-aminobutyric acid (Fig. 1b) or chiral methionine (Fig. 1c) adopts 3:0 conformations and formed P helix.41
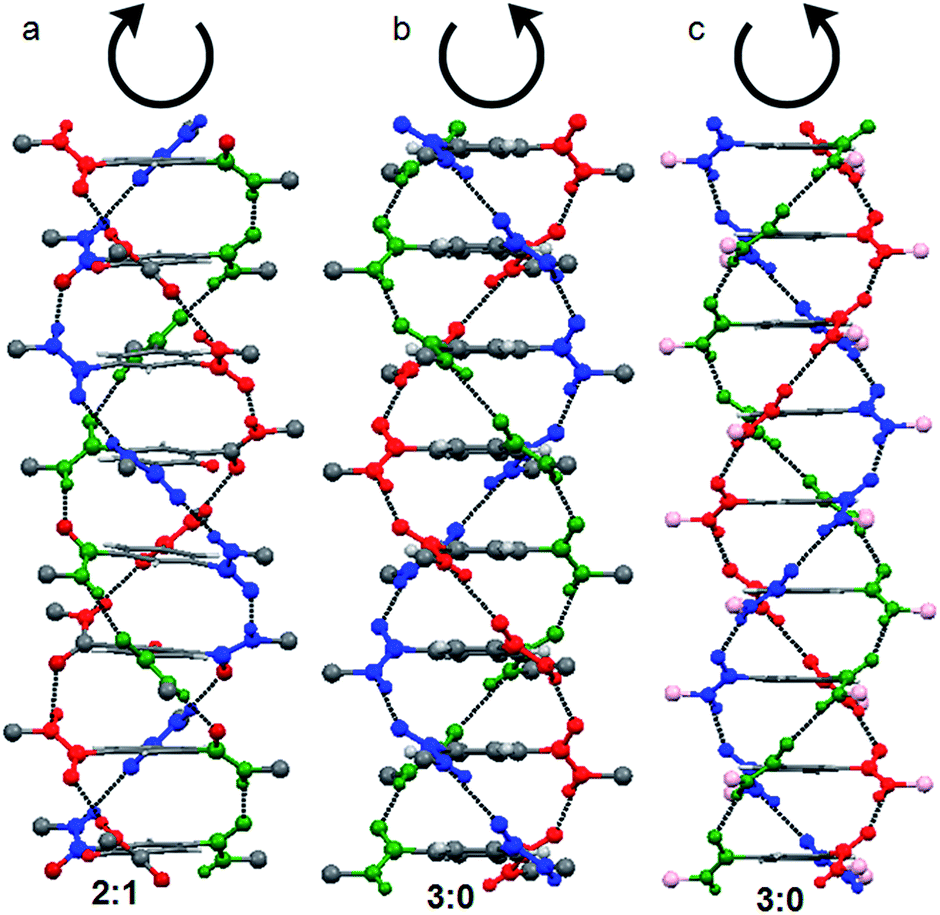 | ||
| Fig. 1 (a) M helix from a trisamide with 2:1 conformations containing achiral β-alanine. (b) P helix by a trisamide with 3:0 conformations containing achiral γ-aminobutyric acid. (c) The trisamide containing chiral methionine (pink balls) adopts 3:0 conformations and formed P helix. Amino acids side chains are omitted for clarity. | ||
Results and discussion
The supramolecular propensities of the reported trisamides 1 and 2 containing chiral (Boc)2-L-Lys and (Boc)-L-Trp have been studied using a wide variety of different spectroscopic techniques in methanol solution. The typical UV-Vis absorption spectra of trisamide 1 in methanol (0.13232 mM) show an absorption band at 208 nm, responsible for π to π* transition (Fig. S3, ESI†). However, the 10 nm bathochromic shift of the absorption band with increasing concentration indicates J-type stacking between the trisamide 1 molecules.2 There is also another band at 360 nm. Compairing with studies, we conclude that trisamide 1 formed one-dimensional supramolecular columnar stacks. The absorption spectra of trisamide 2 in methanol (0.06788 mM) also show an absorption bands at 208 nm and 280 nm but only intensity increase with increasing concentration (Fig. S4, ESI†). Hence the trisamides 1 and 2 have different aggregation propensities.The methanol solution of trisamide 1 is light green in colour. With increasing trisamide 1 concentration in methanol solution the intensity of the colour increases. But we observed that on addition of 0.12 M HCl the solution become colourless (inset of Fig. 2a). There is a possibility of Boc-NH groups deprotection in presence of acid. But in the solution phase synthesis procedure we have work up the trisamides with 2 M HCl and there is no Boc-NH groups de protection as it was confirmed by 1H-NMR, 13C-NMR, FT-IR and mass spectrometry analysis. So, the self-assembly of the trisamide 1 is responsive towards HCl. We have further explore this phenomenon using a wide variety of different spectroscopic techniques. The UV-Vis titration of trisamide 1 methanol solution (1.984 × 10−4 M) with gradual addition of 0.12 M HCl shows the gradual increase of the absorption band intensity at 208 nm (Fig. 2a). However, the absorption band at 360 nm disappeared (Fig. 2a). On the other hand, very light green colour of trisamide 2 in methanol solution become intense green colour by addition of 0.12 M HCl (inset of Fig. 2b). The UV-Vis titration of trisamide 2 methanol solution (3.451 × 10−5 M) with gradual addition of HCl shows the gradual increase of the absorption bands intensities at 208 nm and 280 nm (Fig. 2b). So, protonation can tuned self-assembly of the trisamides.
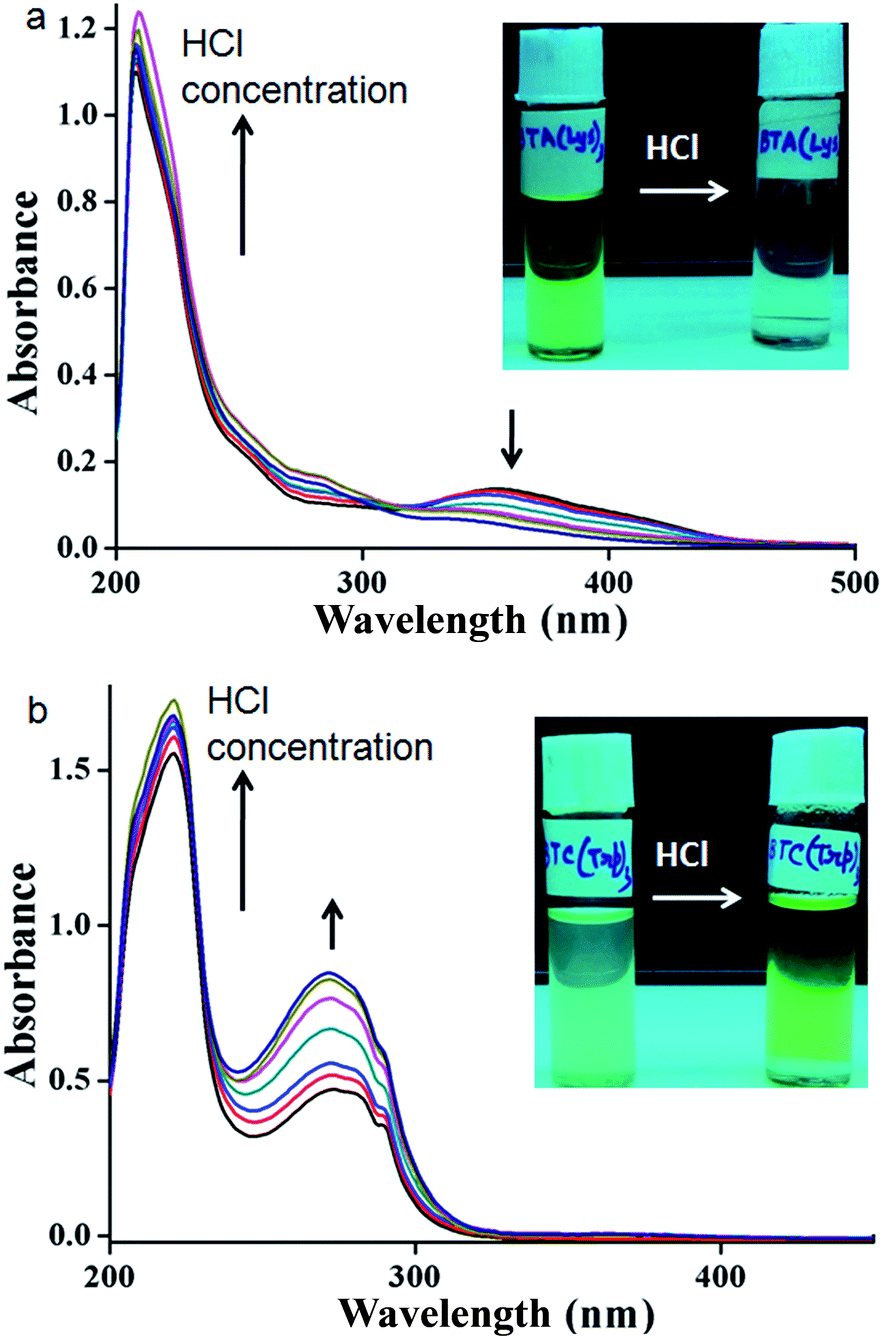 | ||
| Fig. 2 The UV-Vis titration of (a) trisamide 1 and (b) trisamide 2 methanol solution (1.984 × 10−4 M) and (3.451 × 10−5 M) respectively with gradual addition of 0.12 M HCl. Inset: colour change by addition of HCl. | ||
FT-IR spectroscopy is an excellent technique to investigate the HCl responsive self-assembly of trisamide 1. The region 3500–3200 cm−1 is important for the N–H stretching vibrations and 1800–1500 cm−1 corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration. The intense band at 3382 cm−1 is for six Boc-NH groups which are hydrogen bonded (Fig. 3a). The band at 3201 cm−1 is responsible for three core amide NH groups which are hydrogen bonded to form the columnar stack (Fig. 3a). The trisamide 1 shows the amide I band at 1682 cm−1 and amide II band at 1523 cm−1. On addition of HCl the N–H stretching vibrations appears at 3406 cm−1 (Fig. 3a). The 3201 cm−1 brand is now very broad. The amide I bands are now split in 1:2 ratio and appear at 1689 and 1640 cm−1 (Fig. 3a). The intensity of amide II band at 1523 cm−1 is very low (Fig. 3a). Fig. S5, ESI† shows the FT-IR spectra of 2.
O stretching vibration. The intense band at 3382 cm−1 is for six Boc-NH groups which are hydrogen bonded (Fig. 3a). The band at 3201 cm−1 is responsible for three core amide NH groups which are hydrogen bonded to form the columnar stack (Fig. 3a). The trisamide 1 shows the amide I band at 1682 cm−1 and amide II band at 1523 cm−1. On addition of HCl the N–H stretching vibrations appears at 3406 cm−1 (Fig. 3a). The 3201 cm−1 brand is now very broad. The amide I bands are now split in 1:2 ratio and appear at 1689 and 1640 cm−1 (Fig. 3a). The intensity of amide II band at 1523 cm−1 is very low (Fig. 3a). Fig. S5, ESI† shows the FT-IR spectra of 2.
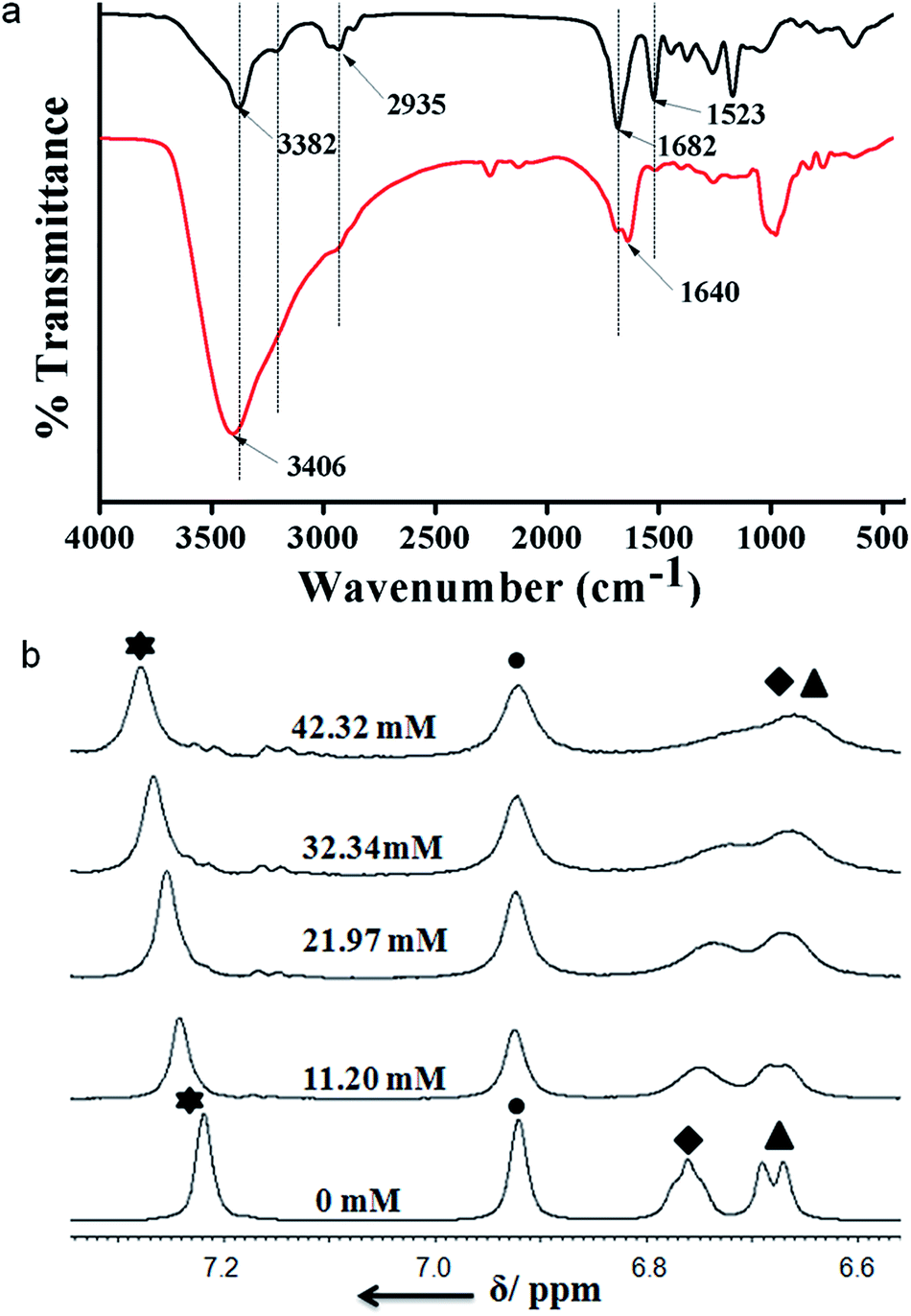 | ||
| Fig. 3 (a) FT-IR spectra of trisamide 1 in absence of HCl (black) and in presence of HCl (red). (b) The acid dependence of NH chemical shifts of trisamide 1 at varying concentrations of HCl. Star: core CONH, circle: core aromatic Hs, square: Lys side chain NHs, triangle: Lys NHs. | ||
Moreover, to investigate the effect of HCl on trisamide 1, the NMR titration experiments were performed. To a solution of compound 1 1.082 × 10−2 (M) in CD3OD the 0.57 (M) HCl solution was gradually added by an amount of 10 μL at room temperature and the NMR spectra were recorded. The results of the NMR titrations of trisamide 1 have been shown in the Fig. 3b. The NMR titrations data show that the NH protons get exposed to the acid and there are significant shift of NH protons for the trisamide 1 with gradually increase in HCl concentration. From the stack plot, the core amide NH exhibits upfield shift suggest the strong hydrogen bond formation. Whereas the protons of central aromatic core show no shift i.e. the stacking structure is not changing. The Boc protected NHs of Lys exhibits downfield shift as well as broadening suggest that these NHs are open to acid. Fig. S6, ESI† shows the acid dependence NH chemical shifts of 2.
In order to gain more information on the structural changes of these trisamides in presence of acid, we performed circular dichroism (CD) measurements. The positive cotton effect observed (185 nm) in the CD spectra of trisamide 1 in methanol (1.474 × 10−5 M) is suggesting a supramolecular right handed helix (Fig. 4a) which is consistent with the handedness of the helices formed by the tricarboxyamide containing L-methionine methyl ester with 3:0 conformation in the crystalline state.41 On addition of HCl, (4 × 10−5 M) the helical handedness of the supramolecular polymer reverses to negative cotton effect (Fig. 4a). The CD spectra is suggesting a left handed supramolecular polymer which is consistent with the handedness of the helices formed by the tricarboxyamide containing β-alanine methyl ester with 2:1 conformation in crystal.40 This suggest that only 2.713 equivalent HCl is enough for the chirality change of tricarboxamide 1. However the trisamide 2 in methanol (1.474 × 10−5 M) has a positive band at 185 nm which does not change after addition of HCL (4 × 10−5 M) (Fig. 4b). There also a negative band at 200 nm and 217 nm which does not shift in presence of acid (Fig. 4b). The CD experiments also performed with trisamide containing achiral β-alanine and γ-aminobutyric acid in absence and presence of HCl. The γ-aminobutyric acid containing trisamide shows a positive band at 210 nm which does not shift in presence of acid (Fig. S7, ESI†). Another trisamide containing β-alanine shows a negative band at 200 nm and after addition of HCl it remain in the same conformation (Fig. S8, ESI†). To examine the supramolecular polymer formation, we have performed the DOSY NMR experiment with trisamide 1. From DOSY experiment, trisamide 1 have viscosity coefficient 3.047 × 10−14 m2 s−1, however after addition of HCl it shows viscosity coefficient 2.416 × 10−14 m2 s−1. This is due to the supramolecular polymer formation (Fig. S9, ESI†).
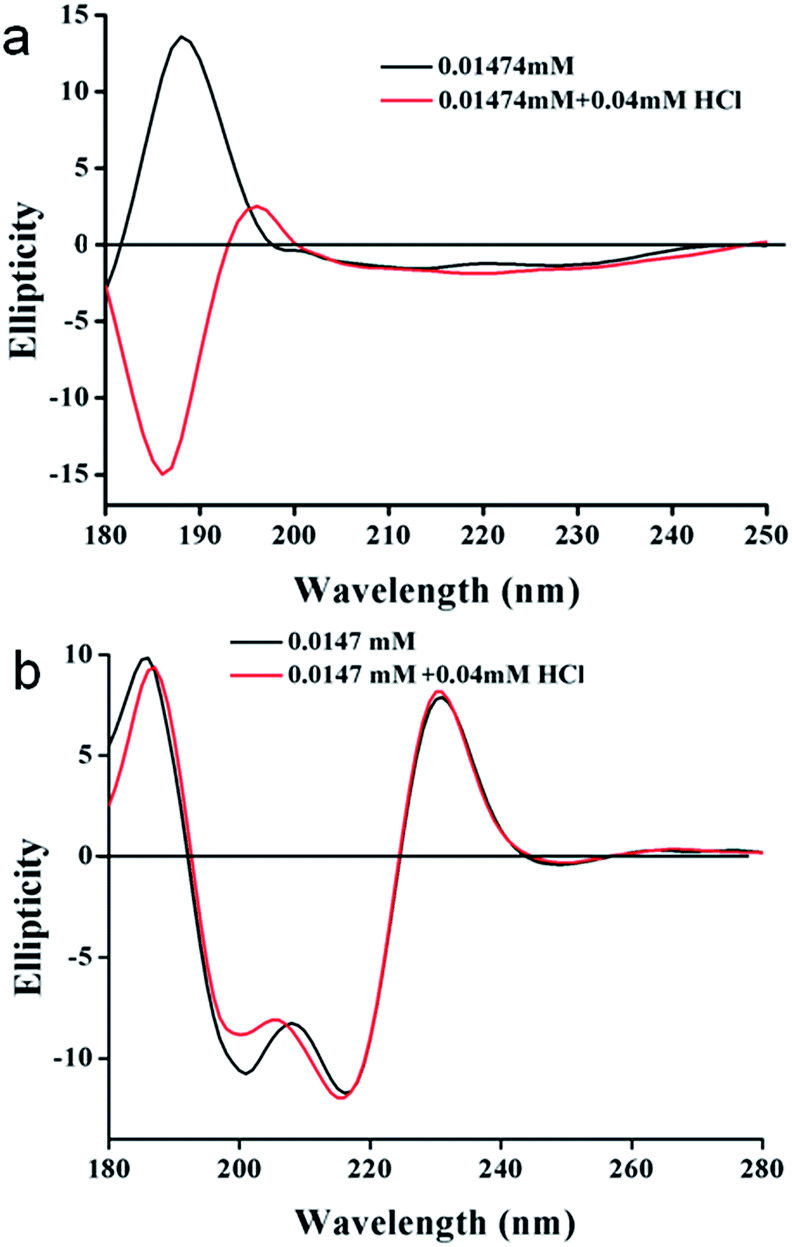 | ||
| Fig. 4 (a) CD spectra of trisamide 1 in methanol without HCl (black) and in presence of HCl (red). (b) CD spectra of trisamide 2 in methanol without HCl (black) and in presence of HCl (red). | ||
The morphology of trisamide 1 was observed by field emission scanning electron microscopy (FE-SEM). The analysis of the morphology of trisamide 1 by FE-SEM reveals the formation of right handed entangled polymeric fibers (Fig. 5b). After addition of 0.12 M HCl, left handed entangled fibers have appeared. The trisamide 2 exhibits disk like morphology (Fig. 5c) which is consistent with the morphology of the tricarboxyamides that formed dimer in crystal.42 On addition of 0.12 M HCl, the trisamide 2 does not exhibit significant change, only the disks are talking to each other (Fig. 5d).
 | ||
| Fig. 5 FE-SEM images of trisamide 1 (a) without acid, (b) after addition of HCl. (c) and (d) FE-SEM images of trisamide 2 without HCl and with 0.12 M HCl respectively. | ||
To investigate the topology of the supramolecular polymer, atomic force microscopic (AFM) studies were performed. The trisamide 1 solutions in methanol were drop-casted on a microscopic glass cover slip and investigated by AFM. The trisamide 1 shows right handed helical supramolecular polymer fibers (Fig. 6a). However, after addition of 0.12 M HCl, it shows left handed helical fibers (Fig. 6b).
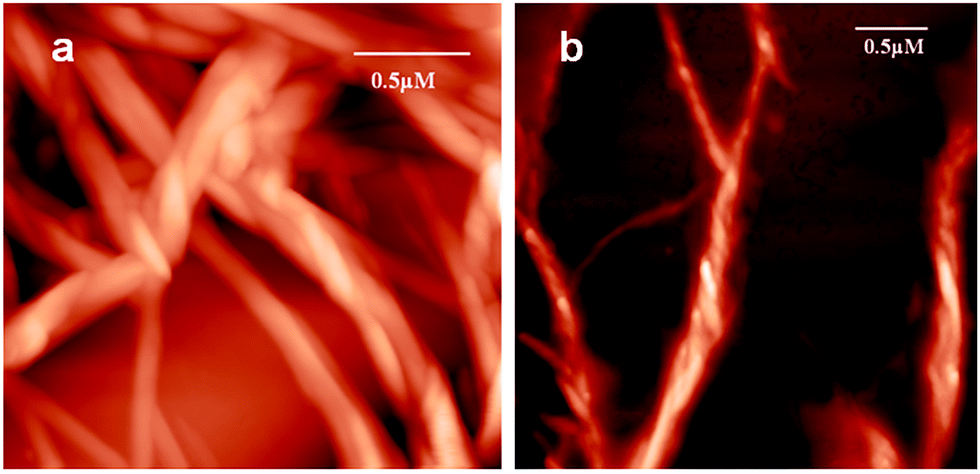 | ||
| Fig. 6 AFM images of helical supramolecular polymer of trisamide 1 (a) without acid, (b) after addition of 0.12 M HCl. | ||
Experimental
General methods and materials
All L-amino acids (L-lysine, L-tryptophan, and trinitrophenol) were purchased from Sigma chemicals. HOBt (1-hydroxybenzotriazole) and DCC (dicyclohexylcarbodiimide) were purchased from SRL. Synthesis. The peptides were synthesized by conventional solution-phase methodology by using a racemization free fragment condensation strategy. The Boc group was used for N-terminal protection and. Couplings were mediated by dicyclohexylcarbodiimide/1-hydroxybenzotriazole (DCC/HOBt). The products were purified by column chromatography using silica gel (100–200 mesh) as the stationary phase and n-hexane–ethyl acetate mixture as eluent. All the intermediates were characterized by 500 MHz and 400 MHz 1H NMR and mass spectrometry. The final compound was fully characterized by 500 MHz and 400 MHz 1H NMR spectroscopy, 13C NMR spectroscopy (125 MHz, 100 MHz), mass spectrometry, and IR spectroscopy.:1) as eluent. Yield: 1.562 g (1.41 mmol, 53.4%). 1H NMR (CDCl3, 500 MHz, δ in ppm): 6.568 (s, 3H, triamine NH protons), 6.0317 (s, 3H, TAB), 5.4141 (s, 3H, NH), 4.1120 (s, 3H, NH Boc), 3.0823–3.0747 (d, 3H, J = 1.92 CαH Lys), 1.8325–1.7632 and 1.6800–1.6012 (2m, 18H, Cβ+γ+ωH of Lys), 1.3781 (s, 54H, Boc). 13C NMR (CDCl3, 125 MHz, δ in ppm): 175.5228, 156.6967, 156.1335, 80.3308, 79.6014, 54.4205, 40.3461, 40.3461, 34.0851, 32.3809, 30.0484, 22.9715.
NMR experiments
All NMR studies were carried out on a Bruker AVANCE 500 MHz and Jeol 400 MHz spectrometer at 278 K. Compound concentrations were in the range 1–10 mM in CDCl3 and DMSO-d6.FT-IR spectroscopy
All reported solution FT-IR spectra were obtained with a Perkin Elmer Spectrum RX1 spectrophotometer.UV-Vis spectroscopy
UV-Vis absorption spectra were recorded on a UV-Vis spectrophotometer (Hitachi).Mass spectra
Mass spectra were recorded on a Q-Tof Micro YA263 high resolution (Waters Corporation) mass spectrometer by positive-mode electrospray ionization.CD spectroscopy
Circular dichroism absorption spectra were recorded Jasco J-815 CD spectrophotometer.Field emission scanning electron microscopy
Morphologies of all reported compounds were investigated using field emission-scanning electron microscopy (FE-SEM). A small amount of solution of the corresponding compound was placed on a clean silicon wafer and then dried by slow evaporation. The material was then allowed to dry under vacuum at 30 °C for two days. The materials were gold-coated, and the micrographs were taken in an FE-SEM apparatus (Jeol Scanning Microscope-JSM-6700F).Atomic force microscopy
The morphology of the reported compound was investigated by atomic force microscopy (AFM). A drop of the sample solution in methanol were placed on a clean microscope cover glass and then dried by slow evaporation. The material was then allowed to dry under vacuum at 30 °C for two days. Images were taken with an NTMDT instrument, model no. AP-0100 by semicontact-mode.Conclusions
In summary, we have shown the addition of HCl reverses the helical handedness of the supramolecular polymer obtained from the self-assembly of chiral amino acid modified C3-symmetric trisamide. The intermolecular three fold hydrogen bonds break and amide functional groups re-organized in the opposite side of central aromatic core by virtue of interaction with HCl. Irrespective of chiral centres configuration, supramolecular chiral bias is arising from molecular conformations. FE-SEM reveals the formation of right handed entangled polymeric fibers. However, left handed entangled fibers have appeared on addition of 0.12 M HCl. The trisamide 2 containing Boc protected L-Trp exhibits disk like morphology both in presence and absence of 0.12 M HCl and does not show change of supramolecular handedness. The acid responsive supramolecular chirality inversion strategy has potential for other building blocks that form multiple competing chiral supramolecular structures.Conflicts of interest
Authors do not have any conflict of interest.Acknowledgements
A. Paikar acknowledges the UGC, India for research fellowship. We acknowledge Dr Rangeet Bhattacharyya, Department of Physical Sciences, and Ipshita Chakraborty, Department of Chemical Sciences, Indian Institute of Science education and Research Kolkata for their help in DOSY experiments.Notes and references
- T. Aida, E. W. Meijer and S. Stupp, Science, 2012, 335, 813–817 CrossRef CAS.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 166, 13752–13990 CrossRef.
- M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS.
- M. A. Mateos-Timoneda, M. Crego-Calama and D. N. Reinhoudt, Chem. Soc. Rev., 2004, 33, 363–372 RSC.
- L. You, D. Zha and E. V. Anslyn, Chem. Rev., 2015, 115, 7840–7892 CrossRef CAS.
- B. Narayan, K. K. Bejagam, S. Balasubramanian and S. J. George, Angew. Chem., Int. Ed., 2015, 54, 13053–13057 CrossRef CAS.
- J. J. D. de Jong, L. N. Lucas, R. M. Kellogg, J. van Esch and B. L. Feringa, Science, 2004, 304, 278–281 CrossRef CAS.
- J. Kim, J. Lee, W. Y. Kim, H. Kim, S. Lee, H. C. Lee, Y. S. Lee, M. Seo and S. Y. Kim, Nat. Commun., 2015, 6, 6959 CrossRef CAS.
- T. Hasegawa, Y. Furusho, H. Katagiri and E. Yashima, Angew. Chem., Int. Ed., 2007, 46, 5885–5888 CrossRef CAS.
- M. Raynal, F. Portier, P. W. N. M. van Leeuwen and L. Bouteiller, J. Am. Chem. Soc., 2013, 135, 17687–17690 CrossRef CAS.
- K. Shimomura, T. Ikai, S. Kanoh, E. Yashima and K. Maeda, Nat. Chem., 2014, 6, 429 CrossRef CAS.
- F. Freire, J. M. Seco, E. Quinoa and R. Riguera, Angew. Chem., Int. Ed., 2011, 50, 11692–11696 CrossRef CAS.
- E. Ohta, H. Sato, S. Ando, A. Kosaka, T. Fukushima, D. Hashizume, M. Yamasaki, K. Hasegawa, A. Muraoka, H. Ushiyama, K. Yamashita and T. Aida, Nat. Chem., 2011, 3, 68–73 CrossRef CAS.
- R. Katoono, S. Kawai, K. Fujiwara and T. Suzuki, Chem. Sci., 2015, 6, 6592–6600 RSC.
- H. Miyake, H. Kamon, I. Miyahara, H. Sugimoto and H. Tsukube, J. Am. Chem. Soc., 2008, 130, 792–793 CrossRef CAS.
- J. Suk, V. Ramesh Naidu, X. Liu, M. Soo Lah and K.-S. Jeong, J. Am. Chem. Soc., 2011, 133, 13938–13941 CrossRef CAS.
- M. Hutin and J. Nitschke, Chem. Commun., 2006, 1724–1726 RSC.
- S. G. Telfer and R. Kuroda, Chem.–Eur. J., 2005, 11, 57–68 CrossRef.
- S. Maity, P. Das and M. Reches, Sci. Rep., 2015, 5, 16365 CrossRef CAS.
- H. Krishna Bisoyi and Q. Li, Angew. Chem., Int. Ed., 2016, 55, 2994–3010 CrossRef.
- D. Pijper, M. G. M. Jongejan, A. Meetsma and B. L. Feringa, J. Am. Chem. Soc., 2008, 130, 4541–4552 CrossRef CAS.
- Y. Nagata, R. Takeda and M. Suginome, Chem. Commun., 2015, 51, 11182–11185 RSC.
- Z. Huang, S.-K. Kang, M. Banno, T. Yamaguchi, D. Lee, C. Seok, E. Yashima and M. Lee, Science, 2012, 337, 1521–1526 CrossRef CAS.
- A. R. A. Palmans, J. A. J. M. Vekemans, E. E. Havinga and E. W. Meijer, Angew. Chem., Int. Ed., 1997, 36, 2648–2651 CrossRef CAS.
- M. Peterca, M. R. Imam, C.-H. Ahn, V. S. K. Balagurusamy, D. A. Wilson, B. M. Rosen and V. Percec, J. Am. Chem. Soc., 2011, 133, 2311–2328 CrossRef CAS.
- Y. Li, M. Wang, T. J. White, T. J. Bunning and Q. Li, Angew. Chem., Int. Ed., 2013, 52, 8925–8929 CrossRef CAS.
- G.-F. Liu, L.-Y. Zhu, W. Ji, C.-L. Feng and Z.-X. Wei, Angew. Chem., Int. Ed., 2016, 55, 2411–2415 CrossRef CAS.
- A. Heeres, C. Van Der Pol, M. Stuart, A. Friggeri, B. L. Feringa and J. van Esch, J. Am. Chem. Soc., 2003, 125, 14252–14253 CrossRef CAS.
- K. J. C. Van Bommel, C. Van Der Pol, I. Muizebelt, A. Friggeri, A. Heeres, A. Meetsma, B. L. Feringa and J. Van Esch, Angew. Chem., Int. Ed., 2004, 43, 1663–1667 CrossRef CAS PubMed.
- A. Friggeri, C. Van Der Pol, K. J. C. Van Bommel, A. Heeres, M. C. A. Stuart, B. L. Feringa and J. Van Esch, Chem.–Eur. J., 2005, 11, 5353–5361 CrossRef CAS.
- A. Brizard, M. Stuart, K. Van Bommel, A. Friggeri, M. de Jong and J. Van Esch, Angew. Chem., Int. Ed., 2008, 47, 2063–2066 CrossRef CAS.
- J. A. Berrocal, F. D. Meo, M. García-Iglesias, R. P. J. Gosens, E. W. Meijer, M. Linares and A. R. A. Palmans, Chem. Commun., 2016, 52, 10870–10873 RSC.
- M. M. J. Smulders, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2008, 130, 606–611 CrossRef CAS PubMed.
- K. P. Van Den Hout, R. Martín-Rapún, J. A. J. M. Vekemans and E. W. Meijer, Chem.–Eur. J., 2007, 13, 8111–8123 CrossRef CAS PubMed.
- P. J. M. Stals, M. M. J. Smulders, R. Martín-Rapún, A. R. A. Palmans and E. W. Meijer, Chem.–Eur. J., 2009, 15, 2071–2080 CrossRef CAS.
- P. Besenius, K. P. Van Den Hout, H. M. H. G. Albers, T. F. A. De Greef, L. L. C. Olijve, T. M. Hermans, B. F. M. De Waal, P. H. H. Bomans, N. A. J. M. Sommerdijk, G. Portale, A. R. A. Palmans, M. H. P. Van Genderen, J. A. J. M. Vekemans and E. W. Meijer, Chem.–Eur. J., 2011, 17, 5193–5203 CrossRef CAS.
- T. R. Canrinus, F. J. R. Cerpentier, B. L. Feringa and W. R. Browne, Chem. Commun., 2017, 53, 1719–1722 RSC.
- X. r. Caumes, A. Baldi, G. Gontard, P. Brocorens, R. Lazzaroni, N. Vanthuyne, C. Troufflard, M. Raynal and L. Bouteiller, Chem. Commun., 2016, 52, 13369–13372 RSC.
- K. K. Bejagam, C. Kulkarni, S. J. George and S. Balasubramanian, Chem. Commun., 2015, 51, 16049–16052 RSC.
- A. Paikar, A. Pramanik and D. Haldar, RSC Adv., 2015, 5, 31845–31851 RSC.
- P. Jana, A. Paikar, S. Bera, S. K. Maity and D. Haldar, Org. Lett., 2014, 16, 3841 CrossRef.
- S. Bera, S. K. Maity and D. Haldar, CrystEngComm, 2014, 16, 4834–4841 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of compounds, 1H NMR, 13C NMR, solid state FTIR spectra, Fig. S1–S17. See DOI: 10.1039/c7ra08035b |
| This journal is © The Royal Society of Chemistry 2017 |