
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of amphiphilic copolymers based on acrylic acid, fluoroalkyl acrylates and n-butyl acrylate in organic, aqueous–organic, and aqueous media via RAFT polymerization†
N. S. Serkhacheva a,
O. I. Smirnova,
A. V. Tolkachevab,
N. I. Prokopova,
A. V. Plutalovac,
E. V. Chernikova*c,
E. Yu. Kozhunovad and
A. R. Khokhlovde
a,
O. I. Smirnova,
A. V. Tolkachevab,
N. I. Prokopova,
A. V. Plutalovac,
E. V. Chernikova*c,
E. Yu. Kozhunovad and
A. R. Khokhlovde
aMoscow Technological University, Institute of Fine Chemical Technologies, Vernadskogo pr., 86, 119571, Moscow, Russian Federation
bMax-Planck Institute of Colloids and Interfaces, Am Mühlenberg 1, 14476 Potsdam, Germany
cFaculty of Chemistry, Lomonosov Moscow State University, Lenin Hills, 1, bld.3, 119991, Moscow, Russian Federation. E-mail: chernikova_elena@mail.ru
dFaculty of Physics, Lomonosov Moscow State University, Lenin Hills, 1, bld.2, 119991, Moscow, Russian Federation
eInstitute of Advanced Energy Related Nanomaterials, University of Ulm, Albert Einstein Allee, 11, D-89081, Ulm, Germany
First published on 5th May 2017
Abstract
Amphiphilic narrow dispersed copolymers of acrylic acid and fluoroalkyl acrylates are synthesized via reversible addition–fragmentation chain transfer (RAFT) polymerization using dibenzyl trithiocarbonate in DMF solution. These copolymers and polyacrylic acid containing the trithiocarbonate group within the polymeric chain are used for the synthesis of triblock copolymers in organic, aqueous–organic or aqueous media by chain extension with n-butyl acrylate or its mixture with fluoroalkyl acrylates. Both dispersion polymerization and emulsifier-free emulsion polymerization lead to self-assembling of triblock copolymers in the core–shell particles; the former route provides the formation of microstructured polymer films with a percolated structure.
Introduction
Nowadays, progress in the reversible-deactivation radical polymerization (RDRP) allows the synthesis of nanoscale objects with controlled morphology directly during the course of synthesis.1–10 This route is possible for block copolymers, in particular for amphiphilic block copolymers, which experience phase separation in a solvent which is selective towards one of the blocks.7,8,11 After solvent removal, the resultant nanostructured objects are expected to find applications as high performance materials.When the synthesis of amphiphilic block copolymers is carried out in the solvent, which is non-selective for both blocks, the self-assembly can be induced by a slow addition of a non-solvent which is selective for one of the blocks. In this case, spherical micelles are usually formed.1,2 However this scenario seems to be more complicated as compared with the direct self-assembly in the course of the synthesis. The latter can be achieved when the grown living polymer is chain-extended with a monomer, which either is insoluble in a selected solvent or forms a polymer which is insoluble in this solvent. The resulting block copolymer is expected to self-assemble into nanosized self-stabilized particles of a given morphology. This behavior is mostly controlled by the ratio of the block lengths.7,8 The advantage of this technique, coined as polymerization-induced self-assembly (PISA), over the traditional emulsion and dispersion polymerization is concerned with the absence of an additional surfactant or a stabilizer, which are added to the polymerization system to provide stability of the formed particles.8
All known RDRP techniques have been already described when studying the synthesis of colloidally stable micro- to nanoobjects by means of PISA.4–10 Among them, the reversible addition–fragmentation chain transfer (RAFT) processes offer evident advantages, including the resistance to the functional groups of monomers, reaction medium, and temperature.12–14 These advantages provide an easy access to the emulsion, aqueous dispersion and organic dispersion block copolymerization of various hydrophilic and hydrophobic monomers.
In the literature on the block copolymer synthesis via RDRP and in particular via PISA technique,15–18 no information on the use of fluoropolymers as hydrophobic blocks is available. However, due to their unique properties such as high thermal and chemical stability, hydrophobicity, low refractive index, and surface energy, fluoropolymers seem to be attractive for many applications in optical, electrical, electronic, and medical devices.19–31 Among these polymers, special concern is focused on partially fluorinated polyacrylates and polymethacrylates. They can be easily synthesized via radical or anionic polymerization and they are soluble in certain organic solvents.
Recently, the interest reverted to the synthesis of various fluorinated poly(meth)acrylates due to the progress achieved in the RDRP. Let us mention several reports on homo- and copolymerization of fluoro(meth)acrylates like 2,2,3,4,4,4-hexafluorobutyl methacrylate, 2,2,2-trifluoroethyl methacrylate, 2-(perfluoro hexyl)ethyl methacrylate, dodecafluoroheptyl methacrylate, pentafluorophenyl methacrylate, 2,2,3,3,4,4,4-heptafluorobutyl- and 2,2,3,4,4,4,-hexafluorobutyl acrylates via the RAFT process.32–37 Moreover, using the atom transfer radical polymerization and the RAFT polymerization, new amphiphilic block copolymers based on fluoroalkyl methacrylates were synthesized by the solution polymerization and their self-assembly in bulk and solution was examined.38 Despite this obvious progress, only limited number of studies deals with the synthesis of amphiphilic block copolymers containing the fluorinated block.15,16 In these studies, poly(methacrylic acid) with the terminal dithiobenzoate group was used to produce the block copolymers with 2,2,2-trifluoroethyl methacrylate and 2,2,3,4,4,4-hexafluorobutyl acrylate (HFBA) or with dodecafluoroheptyl methacrylate in the emulsifier-free emulsion polymerization.
In the present research, we addressed the problems on the controlled synthesis of two- and three-component amphiphilic copolymers of various structures (random and block-random) based on fluoroalkyl acrylates. The aim of this research is to reveal the general and specific features of the RAFT copolymerization of fluoroalkyl acrylates in organic, aqueous–organic, and aqueous media and to reveal its advantages and limitations.
The choice of fluoroalkyl acrylate is primarily related to the presence of a long fluoroalkyl group, which provides the low glass transition temperature of the polymer and its high hydrophobicity which may be useful for the coating applications. In contrast to numerous studies, in which the monofunctional RAFT agent was used for the synthesis of amphiphilic block copolymers, we propose the use of bifunctional trithiocarbonates. This class of the RAFT agents provides the synthesis of the ABA triblock copolymers in two stages. Recently we have reported the synthesis of polyacrylic acid-block-poly(butyl acrylate)-block-polyacrylic acid in the solution, emulsifier-free emulsion polymerization and dispersion polymerization.39,40 From our point of view, this strategy may be successfully applied for the fluoroalkyl acrylates.
Results and discussion
RAFT copolymerization of fluoroalkyl acrylates with acrylic acid in N,N-dimethyl formamide
The specific feature of the radical copolymerization of polar and non-polar monomers is concerned with the effect of the solvent on the relative monomer reactivity and, hence, on the copolymerization kinetics.41–44 This effect can be explained by the formation of the complexes between the monomer and/or the propagating radical and the solvent, solvation of the transition state, and the preferential solvation of the reactant (the bootstrap effect). In the latter case,44–47 the local concentration of the monomer near the active center is supposed to change as compared with its average concentration in the medium due to the preferential monomer sorption. As a result, relative monomer reactivity and copolymerization kinetics are changed. The typical example is the styrene (S)–acrylic acid (AA) copolymerization: the higher the polarity of the solvent, the more pronounced is the difference between the monomer reactivities. For example, for the copolymerization in bulk rAA = 0.15 and rS = 0.25,48 in N,N-dimethyl formamide (DMF) rAA = 0.05–0.08, rS = 1.03–1.60,49,50 in 1,4-dioxane rAA = 0.13, rS = 0.25.51The RAFT copolymerizations of HFBA with acrylic acid as well as 2,2,3,3,4,4,5,5-octafluoropentyl acrylate (OFPA) with acrylic acid were conducted in the polar solvent DMF (50 vol%) using dibenzyl trithiocarbonate (BTC) as the RAFT agent and azo-bis-isobutyronitrile (AIBN) as an initiator at 80 °C. The final aim of these experiments was the synthesis of water-soluble narrow dispersed amphiphilic copolymers with the MW below 10 kDa for the further application in the emulsion and dispersion copolymerization of fluoroalkyl acrylates.
Fig. 1 shows the copolymerization kinetics in the HFBA–acrylic acid and OFPA–acrylic acid systems. Conventional radical copolymerization of HFBA and acrylic acid (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90 mol%) initiated by AIBN (1 × 10−3 mol L−1) in DMF proceeds with a high rate (Fig. 1a, curve 1). However, the increase in the HFBA content in the monomer feed mixture up to 50 mol% causes the inhibition of the polymerization and the polymerization rate slows down (Fig. 1a, curve 3). This result seems to be unexpected due to the higher activity of fluoroalkyl acrylates as compared with that of the conventional acrylic monomers.22 Hence, we may suppose that complexation between the monomers or between the monomers and the solvent takes place. In contrast to free monomers, these complexes are unable to propagate.52 This effect becomes more pronounced with increasing HFBA concentration.
:90 mol%) initiated by AIBN (1 × 10−3 mol L−1) in DMF proceeds with a high rate (Fig. 1a, curve 1). However, the increase in the HFBA content in the monomer feed mixture up to 50 mol% causes the inhibition of the polymerization and the polymerization rate slows down (Fig. 1a, curve 3). This result seems to be unexpected due to the higher activity of fluoroalkyl acrylates as compared with that of the conventional acrylic monomers.22 Hence, we may suppose that complexation between the monomers or between the monomers and the solvent takes place. In contrast to free monomers, these complexes are unable to propagate.52 This effect becomes more pronounced with increasing HFBA concentration.
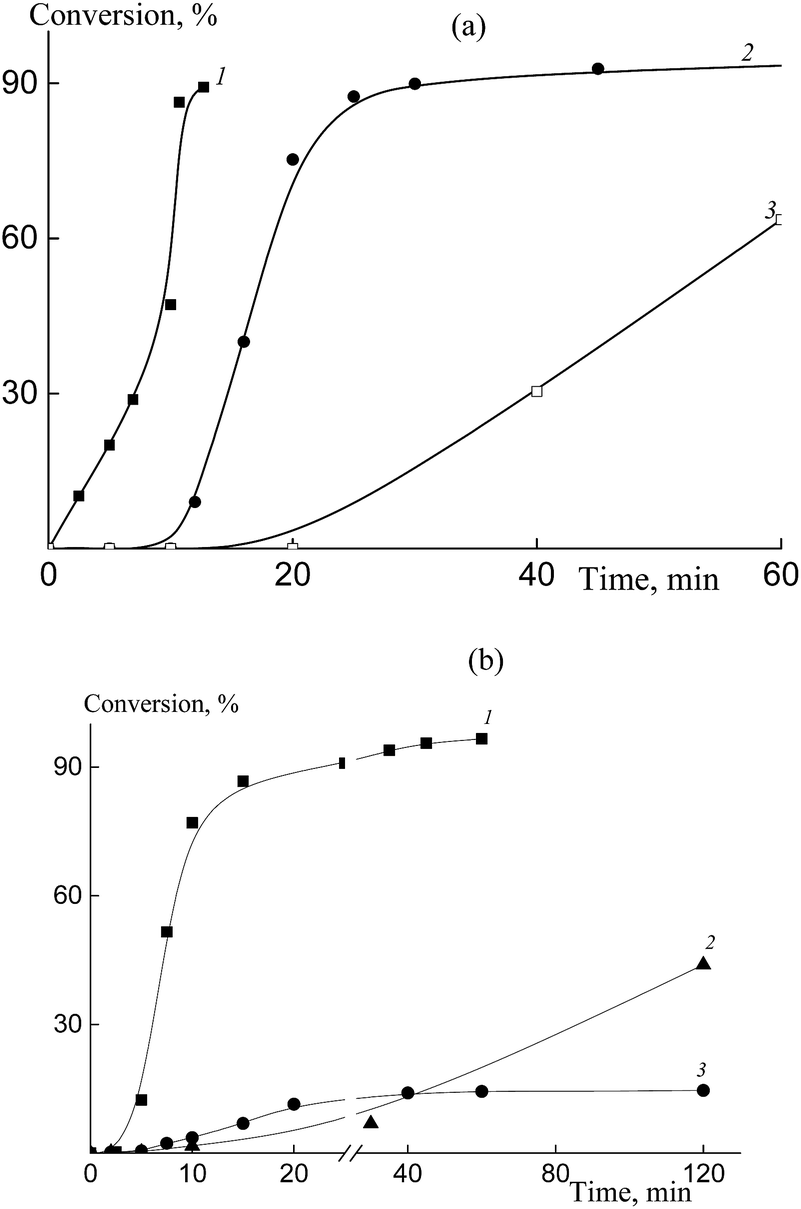 | ||
| Fig. 1 The time dependence of the conversion in the copolymerization of acrylic acid and HFBA (a), acrylic acid and OFPA (b) in DMF at 80 °C in the presence of BTC and AIBN (1 × 10−3 mol L−1). (a) [BTC] = 0 (1, 3) and 6 × 10−3 mol L−1 (2), HFPA in the feed: 10 (1, 2) and 50 mol% (3); (b) [BTC] = 6 × 10−3 mol L−1, OFPA in the feed: 10 (1), 20 (2) and 30 mol% (3). | ||
In the RAFT copolymerization in the presence of BTC (6 × 10−3 mol L−1), the retardation of the polymerization becomes more pronounced and comes into play even at 10 mol% of HFBA (Fig. 1a, curve 2). The copolymerization is completely inhibited at 20 mol% of HFBA. This tendency is preserved when HFBA is replaced by a more reactive monomer, OFPA (Fig. 1b). As the molar fraction of OFPA in the monomer feed increases, the polymerization slows down. The less pronounced effect of OFPA on the copolymerization kinetics may be explained by the higher reactivity of its propagating radical.52
The additional retardation in the RAFT copolymerization rate is provided by the specific features of the RAFT mechanism (Scheme 1). When the symmetrical RAFT agents, e.g. trithiocarbonates R–S–C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S)–S–R, are used, three specific reactions of the reversible chain transfer occur. Each of the above reactions results in the formation of the intermediate radicals (Int-1–Int-3), which are capable not only to experience fragmentation with the release of a new radical and the polymeric RAFT agent, but also they can participate in the termination reactions with the propagating radicals.53 In the latter case, the overall concentration of the propagating radicals decreases, thus leading to further retardation of the polymerization.53
S)–S–R, are used, three specific reactions of the reversible chain transfer occur. Each of the above reactions results in the formation of the intermediate radicals (Int-1–Int-3), which are capable not only to experience fragmentation with the release of a new radical and the polymeric RAFT agent, but also they can participate in the termination reactions with the propagating radicals.53 In the latter case, the overall concentration of the propagating radicals decreases, thus leading to further retardation of the polymerization.53
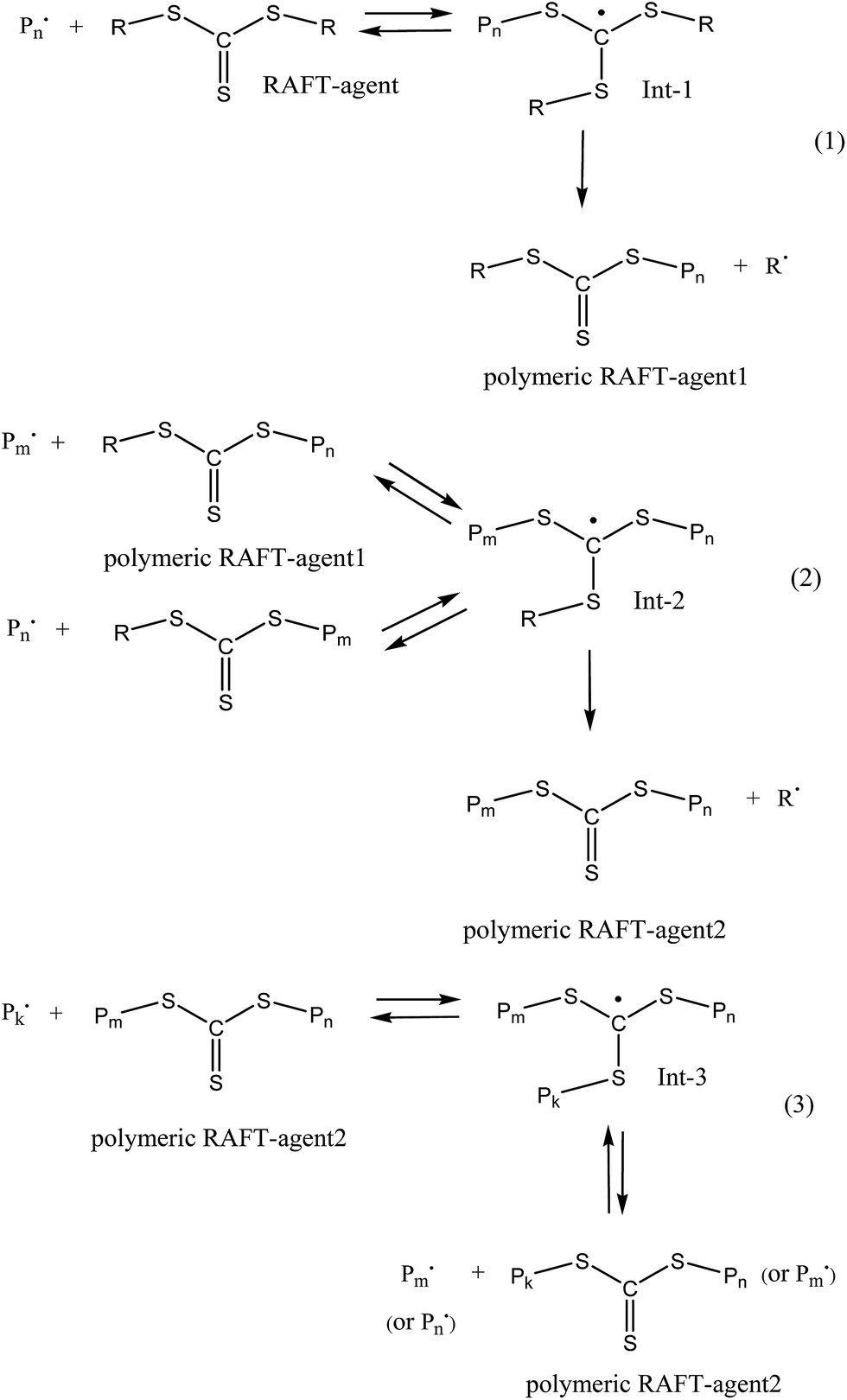 | ||
| Scheme 1 The general scenario of the RAFT mechanism in the presence of symmetrical trithiocarbonates –R–S(CS)–S–R. | ||
The copolymers formed in the absence of BTC are characterized by a broad MWD and by a relatively high MW. The number average molecular weight Mn of these copolymers is virtually independent of the monomer conversion. For example, for the copolymer prepared at a conversion of 47% from the monomer mixture of HFBA and acrylic acid (10:90 mol%) in the presence of AIBN (10−3 mol L−1), Mn = 110 kDa and Đ = 2.93, whereas, for the copolymer prepared from the equimolar mixture of HFBA and acrylic acid at a conversion of 63%, Mn = 96 kDa and Đ = 1.96.
In contrast, Mn and the MWD of the copolymers formed in both HFBA–acrylic acid and OFPA–acrylic acid systems in the presence of BTC depend on the monomer conversion (Fig. 2). The SEC curves are seen to be shifted to higher MW values with increasing monomer conversion (Fig. 2a and b). The Mn values calculated from the SEC analysis linearly increase with conversion (Fig. 2c, lines 1, 2) and this behavior agrees with the RAFT mechanism.12–14 At the same time, dispersity (Đ) slightly increases in the course of the polymerization, but its values are below 1.5 (Fig. 2c, 1′, 2′). The slight increase in the dispersity is typical of the RAFT polymerization of acrylate monomers in the solution, and this behavior is usually provided by the side chain transfer reactions.12
 | ||
| Fig. 2 The SEC curves normalized by the unit area for copolymers of (a) acrylic acid and HFBA (95:5 mol%) and (b) acrylic acid and OFPA (90:10 mol%) synthesized via the RAFT copolymerization in the presence of BTC in DMF at 80 °C. (a) [BTC] = 5 × 10−2 mol L−1 and [AIBN] = 1 × 10−3 mol L−1, conversion: 18.3 (1), 41.2 (2), 72.7 (3), and 96.6% (4); (b) [BTC] = 6 × 10−3 mol L−1 and [AIBN] = 1 × 10−3 mol L−1, conversion: 0.3 (1), 12.4 (2), 51.6 (3), 77.5 (4), 86.7 (5), and 93.9% (6). (c) Dependence of Mn (1, 2) and Đ (1′, 2′) of the synthesized copolymers (1, 1′ – a, 2, 2′ – b). | ||
When the chromatograph is not equipped with the MALLS detector but has only the refractive index detector, the SEC method allows estimation of relative molecular masses only rather than absolute ones. Hence, for copolymers, this technique may be correctly used to prove the living nature of the process only when the composition of the copolymers is preserved at the constant level or slightly changes. Due to the low amount of fluoroalkyl acrylate (5–10 mol%), the change in the copolymer composition in the course of copolymerization may be ignored. Hence, the linear increase in Mn with conversion in both systems is the strong argument in favor of the RAFT mechanism.
Reactivity ratios of the studied monomers are unknown. Nevertheless, some speculations on the monomer unit distribution in the chain can be advanced. Typically, the hydrophobic monomer (HFBA or OFPA in our case) is far more reactive (by 1–2 orders of magnitude) as compared with the hydrophilic monomer (acrylic acid) in the copolymerization in the polar solvent such as DMF.54,55 Even taking into account the low content of fluoroalkyl acrylate (below 10 mol%), it will be readily consumed at low monomer conversions. According to the simple calculations (Fig. S1, ESI†), the formation of the acrylic acid–fluoroalkyl acrylate dyads is more preferable than the formation of the fluoroalkyl acrylate–fluoroalkyl acrylate dyads. When the monomer conversion approaches 40–50%, fluoroalkyl acrylate is completely consumed. In the course of the polymerization, the molar fraction of acrylic acid–acrylic acid dyads is high due to the high content of this monomer in the feed mixture. Hence, as a result of the living mechanism and the use of the symmetrical trithiocarbonate (BTC) that provides the chain growth in two directions56 with respect to trithiocarbonate group, one can expect the formation of the random copolymers, in which fluoroalkyl acrylate units will be located closer to the both tails of the macromolecules.
One of the important features of the RDRP processes is concerned with the ability of the formed polymers to participate in the chain extension.1–3 This assumption was proved as follows, the copolymers of acrylic acid and HFBA (Cop-H5, Table 1), acrylic acid and OFPA (Cop-O2, Table 1) were synthesized by the RAFT copolymerization in the presence of BTC and added to the monomer mixtures containing AIBN and DMF. In the case of the RAFT mechanism, these copolymers should serve as the polymeric RAFT agents and provide the living process.12
| Abbreviation | Comonomer | Mn × 10−3 | Đ |
|---|---|---|---|
| Pol-A | — | 8.4 | 1.33 |
| Cop-H2 | HFBA, 2 mol% | 6.6 | 1.30 |
| Cop-H5 | HFBA, 5 mol% | 5.8 | 1.39 |
| Cop-O2 | OFPA, 2 mol% | 4.8 | 1.20 |
| Cop-O10 | OFPA, 10 mol% | 5.0 | 1.35 |
As follows from Fig. 3a and b, the copolymer with the higher MW is initially formed in both systems. This fact allows us to conclude that the polymeric RAFT agent is totally consumed at low monomer conversions. This behavior highlights the high efficiency of Cop-H5 and Cop-O2 as the RAFT agents.12 At the same time, a new high molecular weight shoulder is seen in the corresponding chromatograms and the MWDs become bimodal. Later, in the course of the experimental run, the position of the mode with the lower MW (mode 1) is preserved, but its intensity decreases.
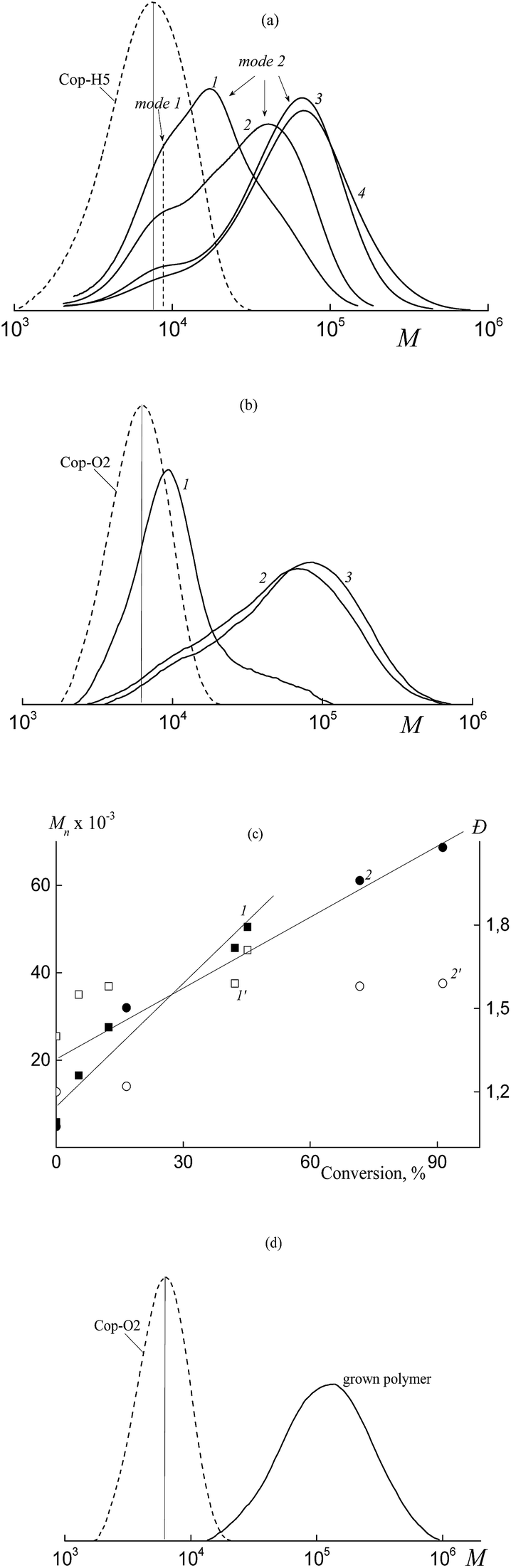 | ||
| Fig. 3 The SEC curves normalized by the unit area for the copolymers of (a) acrylic acid and HFBA (95:5 mol%), (b and d) acrylic acid and OFPA (90:10 mol%) synthesized via the RAFT copolymerization in the presence of the RAFT agents in DMF (a and b) and 1,4-dioxane (d). (a) [Cop-H5] = 3 × 10−3 mol L−1 and [AIBN] = 5 × 10−4 mol L−1, conversion: 5.3 (1), 12.4 (2), 42.2 (3), and 77.2% (4); (b) [Cop-O2] = 6 × 10−3 mol L−1 and [AIBN] = 1 × 10−3 mol L−1, conversion: 0.6 (1), 71.7 (2) and 91.3% (3). (c) Dependences of Mn (1, 2) and Đ (1′, 2′) of the synthesized copolymers (1, 1′ – a, 2, 2′ – b). | ||
As the monomer conversion increases, the position of mode 2 is shifted to higher MW.
Evaluation of the average MW and the dispersity of the second mode shows that Mn increases linearly, whereas the dispersity slightly increases with increasing monomer conversion (Fig. 3c). As the concentration of the polymeric RAFT agent increases, the growth rate of Mn decreases with conversion. This is the characteristic feature of the living process.13,14
The molecular weight characteristics of all synthesized copolymers are summarized in Table S1 (ESI†).
Therefore, the question concerning the nature of the unusual transformation of the MWDs with conversion in the experiments with the chain extension arises. The DLS was used for the study of the solutions of copolymer Cop-O2 in both DMF and the reaction mixture. In these experiments, the copolymer was taken in the same concentration as in the copolymerization. The aggregative potency of amphiphilic copolymers is known to be very sensitive to the thermodynamic quality of the solvent.57
DMF is a good solvent for polyacrylic acid (PAA) and a relatively good solvent for poly(OFPA); however, acrylic acid and OFPA are non-solvents for poly(acrylic acid).58 Hence, in DMF at a Cop-O2 concentration of 3 wt%, the particles with the number-average diameter Dn ∼ 7.5 nm are observed (Fig. S2a, ESI†). As a rough approximation, for the copolymer with Mn ∼ 5000, the value of Dn is assumed to be close to the size of the individual macromolecules (the chain length above 15 nm). Hence, as expected, Cop-O2 in DMF exists as unimers.
In the mixture of DMF–acrylic acid–OFPA under the same concentration of Cop-O2, aggregation takes place, and the particles with Dn ∼ 410 nm are observed (Fig. S2b, ESI†). This result allows us to conclude that the thermodynamic quality of this mixed solvent becomes worse and aggregation becomes preferable. In other words, when acrylic acid and fluoroalkyl acrylate are copolymerized in DMF in the presence of the polymeric RAFT agent when the aggregates are formed, the “living” mechanism is violated due to the lower access of the trithiocarbonate group to the propagating radical. This behavior provides a partial loss in the control over the molecular weight characteristics of the grown copolymer.
The same trend was observed for the solution of the Cop-H5 copolymer in DMF and in the reaction mixture. Hence, aggregation of the formed block copolymers may be considered as the main reason behind the violation of the living mechanism.
When DMF is replaced by 1,4-dioxane, no aggregation of Cop-O2 is observed both in 1,4-dioxane and its mixture with acrylic acid and OFPA (Dn ∼ 6.6 and 7.7 nm respectively). Hence, less polar 1,4-dioxane enhances the thermodynamic quality of the mixed solvent and prevents the aggregation. As a result, the copolymer formed at 99% conversion upon the copolymerization of acrylic acid and OFPA in the presence of Cop-O2 is characterized by the unimodal MWD (Fig. 3d). Hence, the suppressed aggregation of the macromolecules can improve the control over the MWD.
Therefore, the specific features of the synthesis of amphiphilic copolymers were revealed when polymerization was performed in the polar solvent DMF in the presence of the polymeric RAFT agent. The aggregation of macromolecules of the polymeric RAFT agent prevents control over polymerization. This undesirable phenomenon can be diminished by replacing DMF with 1,4-dioxane probably due to the improved thermodynamic quality of the solvent.
Structure and glass transition of copolymers were analyzed for the acrylic acid and HFBA copolymer prepared at a monomer conversion of 76% via the RAFT polymerization mediated by BTC. First, the copolymer was subjected to methylation and then it was analyzed by the NMR spectroscopy (Fig. S3, ESI†) and DSC. In the 1H NMR spectrum (Fig. S3a†), the resonance signals corresponding to the protons of the main chain (1.5–2.2 ppm), methyl group (CH3O, 3.52 ppm), the protons of methylene and methyne groups of fluoroalkyl substituent (4.55 and 5.93–6.02 ppm), aromatic protons of benzyl group (7.13–7.23 ppm) are observed. The signals of the protons of the leaving groups of the RAFT agent, which are present in the copolymer structure, can provide the independent evidence in favor of the RAFT mechanism.12–14 The 19F NMR spectrum (Fig. S3b†) shows the resonance signals corresponding to F atoms of fluoroalkyl substituent (−214.49, −(119.51–117.02) and −73.45 ppm). The copolymer composition was calculated from the values of the integral intensities of the protons of CH3O– group of methylated acrylic acid and –CH2O– group of HFBA. This copolymer contains 11.6 mol% of HFBA. Mn was calculated under the assumption that two benzyl groups exist in the structure of the copolymer, and the calculated value is equal to 6300. This value is lower as compared with the value obtained by the SEC measurements but it approaches the theoretical value of 6900. Hence, the BTC may be considered as a relatively efficient RAFT agent for the copolymerization of acrylic acid and fluoroalkyl acrylates.
According to the DCS data, the copolymer has one glass transition temperature which is equal to 41.1 °C.
Hence, for the first time, we demonstrated and proved the possibility of the direct synthesis of the narrow dispersed amphiphilic copolymers of acrylic acid with fluoroalkyl acrylates. The RAFT copolymerization of HFBA and OFPA with acrylic acid proceeds via the RAFT mechanism in the presence of BTC in DMF. The most serious limitation of this process is concerned with the retardation of the copolymerization, which is boosted with increasing the content of fluoroalkyl acrylate content in the monomer feed mixture.
Copolymerization of butyl acrylate and 2,2,3,4,4,4-hexafluorobutyl acrylate in DMF in the presence of trithiocarbonates
According to the literature data, heptafluorobutyl acrylate is capable of homo- and copolymerization with butyl acrylate in the solution of 1,4-dioxane in the presence of 2-cyano-2-propyl benzodithioate.36,37 Hence, we expected that the RAFT mechanism can be also achieved for the RAFT copolymerization of BA and HFBA.Fig. 4 shows the copolymerization kinetics of BA and HFBA initiated by AIBN in DMF in the presence of BTC (curve 1), poly(acrylic acid) trithiocarbonate (Pol-A, curves 2–4) and Cop-H5 (curve 5). In all cases, concentrations of AIBN, RAFT agent and monomers are kept constant, while the monomer ratio is varied. As is seen, polymerization rate decreases when BTC is replaced by Pol-A (curves 1 and 2). When the content of butyl acrylate in monomer feed decreases, provided all other conditions are the same, the polymerization rate increases (curves 2–4). Finally, the polymerization rate increases when Pol-A is replaced by Cop-H5 (curve 5).
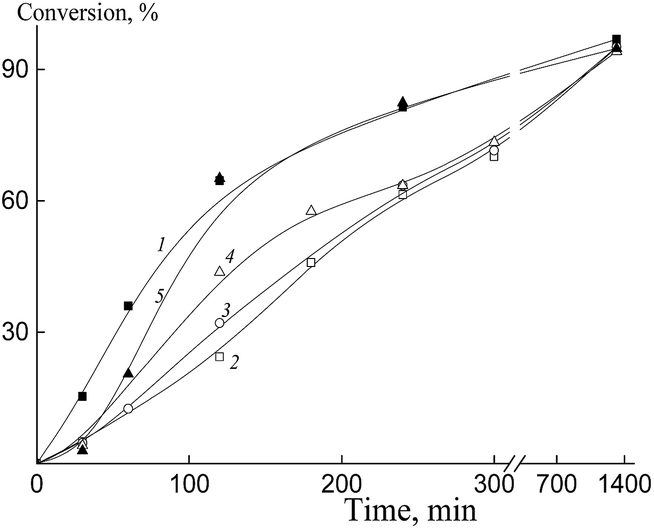 | ||
| Fig. 4 The time dependence of the conversion for the copolymerization of butyl acrylate and HFBA in DMF initiated by AIBN (5 × 10−4 mol L−1) in the presence of the RAFT agent (3 × 10−3 mol L−1). The RAFT agent: BTC (1), Pol-A (2–4) and Cop-H5 (5). Butyl acrylate:HFBA = 90:10 (1, 2), 85:15 (3), 80:20 (4, 5). Total monomer concentration is 2 mol L−1. | ||
Fluorinated (meth)acrylates are known to be more reactive as compared with their non-fluorinated analogs.22 Hence, when the molar fraction of HFBA in the monomer feed increases, the average propagation rate coefficient also increases and, as a result, the copolymerization rate becomes higher.
Recently, we have shown that, in the RAFT copolymerization of polar–non-polar monomer pair, relative monomer reactivities could be adjusted not only by varying the polarity of the solvent, but also by governing the hydrophilicity/hydrophobicity of the polymeric RAFT agent.59 The similar effect of the macroinitiator and the macromonomer on the relative monomer reactivities was also described for the case of the stable free radical polymerization60 and for the conventional radical polymerization.61 Hence, one can conclude that the similar effect of variations in the relative monomer reactivities is observed for the systems under study in the presence of Pol-A and Cop-H5.
When BTC is used as the RAFT agent, the formed copolymers are characterized by the relatively narrow MWD; Mn increases linearly with increasing monomer conversion, whereas the dispersity of the copolymers remains lower as compared with the corresponding values typical of the conventional radical polymerization (Fig. 5a and 6). In this case, a random copolymer is expected to be produced.
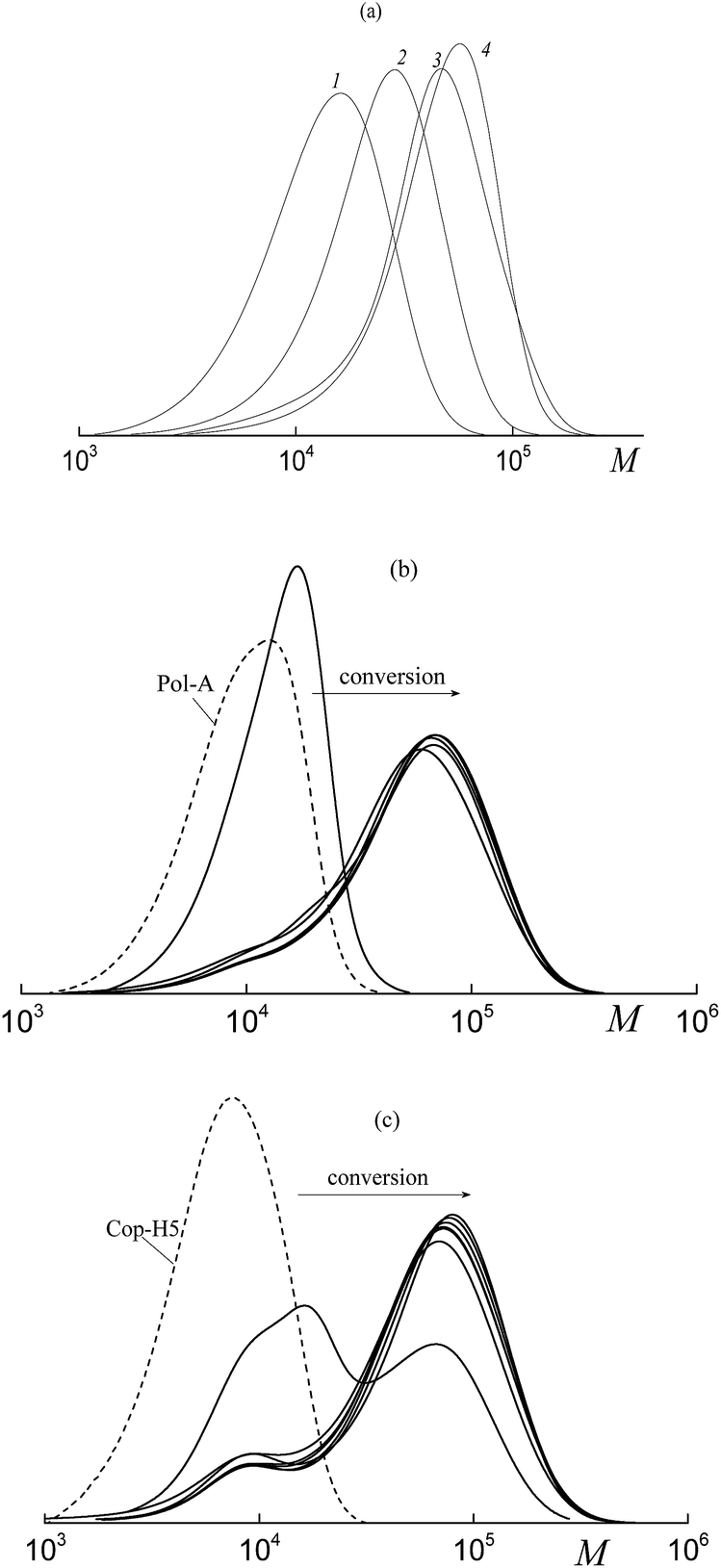 | ||
| Fig. 5 The SEC curves normalized by the unit area for the copolymers of butyl acrylate and HFBA synthesized in DMF in the presence of BTC (a), Pol-A (b) and Cop-H5 (c). [AIBN] = 5.0 × 10−4 mol L−1, [BTC] = [Pol-A] = [Cop-H5] = 3.0 × 10−3 mol L−1. (a) HFBA, 10 mol%, conversion 15.3 (1), 36.0 (2), 64.6 (3), and 96.9% (4); (b) and (c) HFBA, 20 mol%. | ||
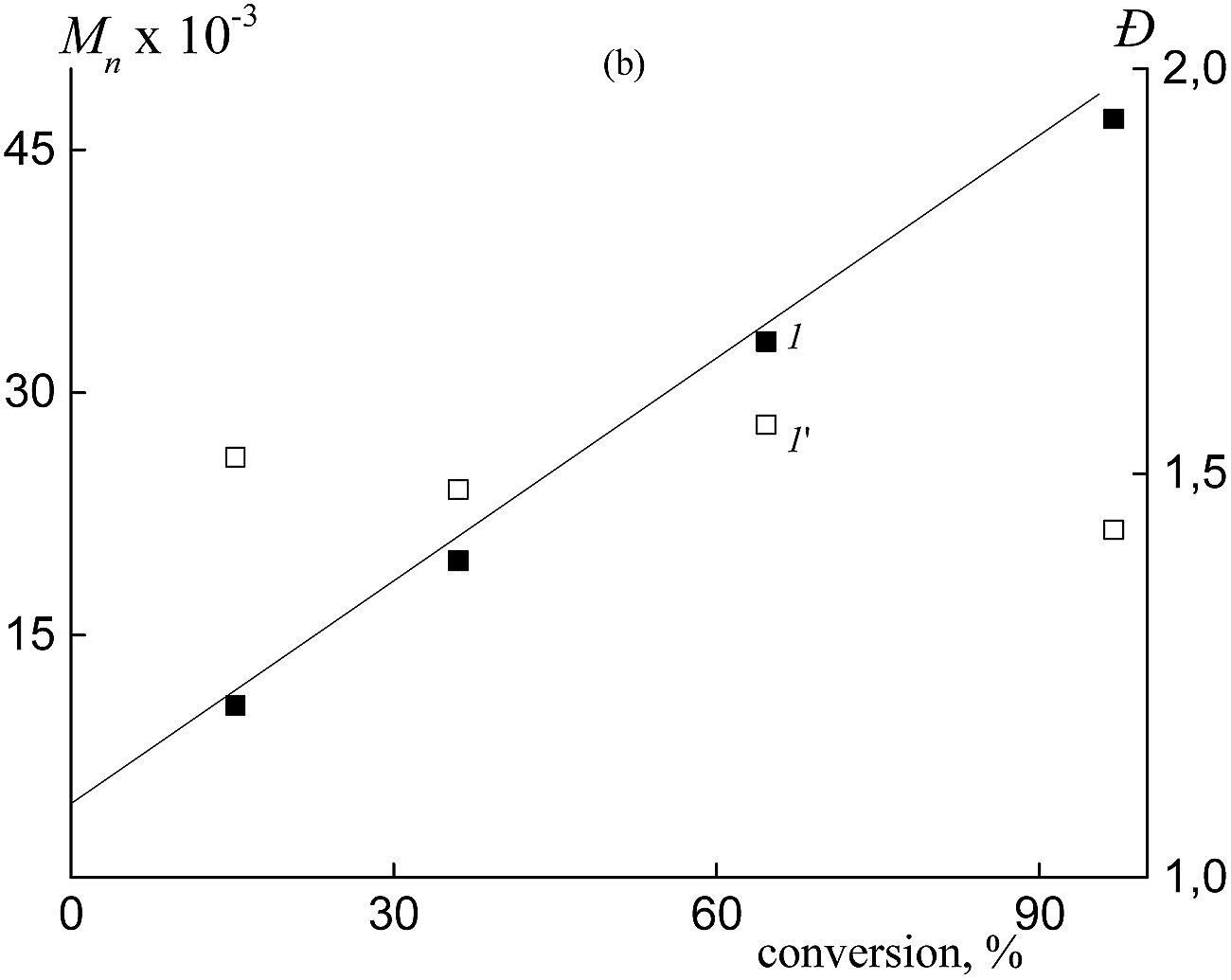 | ||
| Fig. 6 Dependences of Mn (1) and Đ (1′) on conversion for the synthesized copolymers in the presence of BTC. | ||
However, when Pol-A or Cop-H5 is used and when the living mechanism operates, the amphiphilic triblock copolymers of the following structure should be produced: poly(acrylic acid)-block-poly(BA-co-HFBA)-block-poly(acrylic acid) and poly(acrylic acid-co-HFBA)-block-poly(BA-co-HFBA)-block-poly(acrylic acid-co-HFBA). According to Scheme 1, both polymeric RAFT agents contain the trithiocarbonate group within the chain. Hence, the formation of triblock copolymers proceeds via the insertion of new monomers between the trithiocarbonate group and the terminal monomer unit of the attached polymeric substituent.13,14,62

Fig. 5b and c show the transformation of the MWDs with conversion for the products of the copolymerization of BA and HFBA (at a molar ratio of 80:20) in the presence of Pol-A (b) and Cop-H5 (c). When the molar ratio of BA and HFBA is equal to 90:10 and 85:15 in the presence of Pol-A, the corresponding SEC curves of the products are given in the ESI (Fig. S4†). The transformation of the MWD upon polymerization is seen to be similar to the above-described system composed of acrylic acid–fluoroalkyl acrylate–polymeric RAFT agent (Fig. 3). Evidently, at the early stages of monomer conversion, both polymeric RAFT agents, Pol-A and Cop-H5 are rapidly consumed to produce the triblock copolymer. Noteworthy is that, the higher the molar fraction of HFBA in the monomer feed, the lower the conversion, at which the second mode appears in the SEC curves. The same behavior is observed when, instead of Pol-A, Cop-H5 is used. Molecular weight characteristics of the synthesized copolymers are summarized in Table S2.†
Hence, both polymeric RAFT agents are efficient for the solution copolymerization of BA and HFBA which provides a rapid formation of block copolymers at early stages. However, as the monomer conversion increases, the control over the molar mass characteristics of the formed triblock copolymers is violated. Evidently, the final product contains two fractions. The first fraction is the block copolymer with Mn ∼ 12 kDa and Đ ∼ 1.3 and the second fraction is the block copolymer with Mn ∼ 47–60 kDa and Đ ∼ 1.4. According to the above reasoning, the possible reason behind this behavior is concerned with the aggregation of the block copolymers formed in the course of the synthesis.
Triblock copolymers synthesized in these systems at high conversions were subjected to methylation and analyzed by 1H and 19F NMR spectroscopy and DSC. Typical NMR spectra are presented in Fig. S5 (ESI†), and the copolymer composition and Tg values are listed in Table 2. As the content of HFBA in the monomer feed increases, its content in the copolymer also increases. The corresponding DSC curves show the two glass transition temperatures. The first glass transition temperature corresponds to the copolymer BA and HFBA; and its value increases with increasing HFBA content in the copolymer. The second Tg can be attributed to the methylated blocks of Pol-A and Cop-H5. These results together with the GPC data proved the formation of triblock copolymers.
| HFBA in monomer feed, mol% | Copolymer composition, mol% | Tg,1, °C | Tg,2, °C | ||
|---|---|---|---|---|---|
| MA (AA) | BA | HFBA | |||
| a Note: *Pol-A was used as the RAFT agent, **Cop-H5 was used as the RAFT agent, ***the sample was not analyzed by DSC. | |||||
| 10 | 11.5 | 79.0 | 9.5 | −37.8 | 6.0 |
| 15 | 11.3 | 74.9 | 13.8 | −34.5 | 6.0 |
| 20 | 11.7* | 70.0 | 18.3 | −31.0 | 6.0 |
| 9.1** | 71.3 | 19.6 | —*** | — | |
Hence, copolymerization of butyl acrylate with HFBA in the presence of BTC leads to the formation of the narrow dispersed copolymers. For the synthesis of amphiphilic block-random copolymers, the use of hydrophilic/amphiphilic polymeric RAFT agent in DMF appears to be nearly unsuitable.
Synthesis of amphiphilic block copolymers by emulsifier-free emulsion polymerization
Prior to the use of the synthesized copolymers of acrylic acid and fluoroalkyl acrylates in the heterophase polymerization for the stabilization of the formed polymeric particles, their surface activity should be evaluated. In these experiments, we used the copolymers of acrylic acid and OFPA containing 2 and 10 mol% of OFPA (Cop-O2 and Cop-O10, Table 1) as well as the copolymer of acrylic acid and HFBA containing 2 mol% of HFBA (Cop-H2, Table 1). All copolymers were synthesized in DMF in the presence of BTC and contain trithiocarbonate group within the chain. As the reference, poly(acrylic acid) prepared under similar conditions was used (Pol-A, Table 1).Fig. 7a shows the surface tension isotherm for the aqueous solutions containing 0.1 M NaCl of polymer Pol-A and copolymers Cop-H2, Cop-O2, and Cop-O10, at pH 5.4 and T = 25 °C at the water–air interface. Oligomeric PAA containing trithiocarbonate group (Pol-A) is seen to exhibit low (poor) surface-active properties. This potency can be improved by the introduction of fluoroalkyl acrylate units.
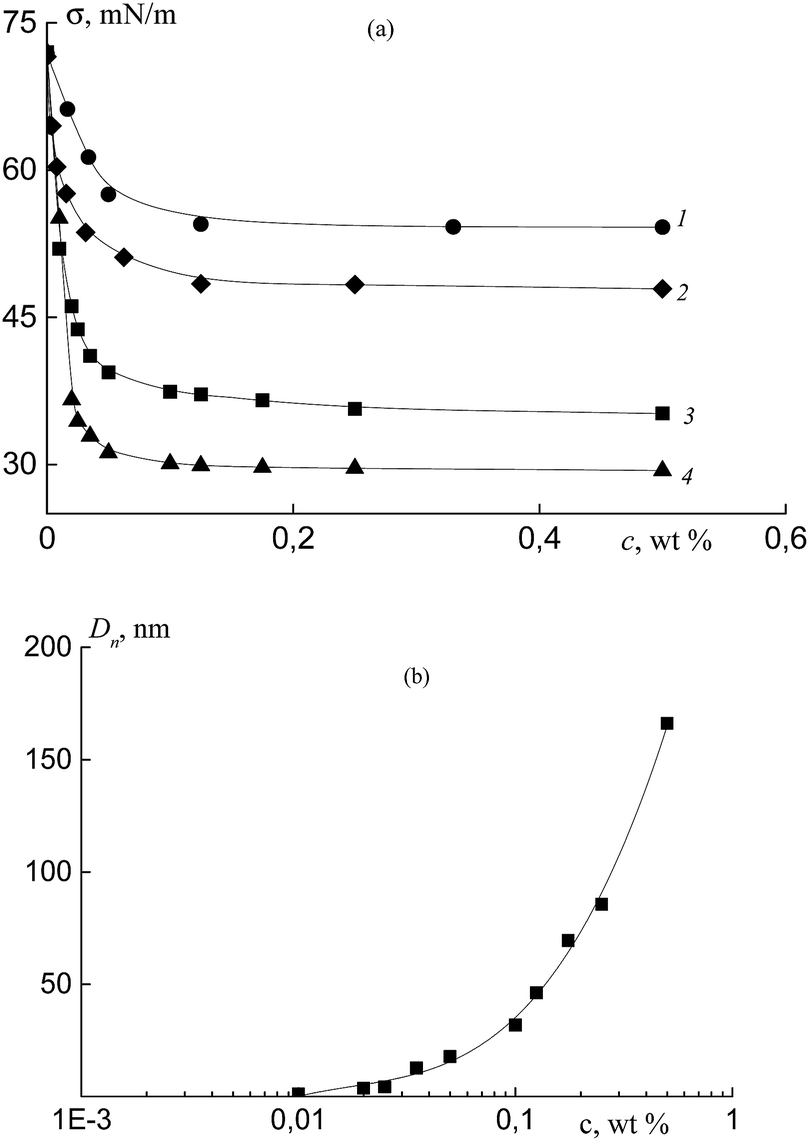 | ||
| Fig. 7 (a) Surface tension isotherms for aqueous solutions containing 0.1 M NaCl of polymer Pol-A (1) and copolymers Cop-H2 (2), Cop-O2 (3) and Cop-O10 (4) at pH 5.4 and T = 25 °C on the water–air interface. (b) Dependence of number-average diameters of the particles Dn in aqueous solution containing 0.1 M NaCl of copolymer Cop-O2 on its concentration at pH 5.4 and T = 25 °C. | ||
Evidently, due to the increase in the fluoroalkyl acrylate content in the copolymer as well as the increase in the content of the fluorine atoms in the alkyl substituent of the acrylate monomer, surface tension decreases. For Pol-A, Cop-H2, Cop-O2 and Cop-O10, critical micelle concentration (CMC) is equal to 0.06, 0.04, 0.03, and 0.02 wt%, respectively. These results agree with the DLS data. When the CMC is attained, the average diameter of the particles initially increases by several times, and this case corresponds to the formation of micelles, as the polymer concentration is further increased, the diameter exponentially increases, and this tendency is indicative of the formation of intermicellar aggregates (Fig. 7b).
In the case of the RAFT technique, the emulsifier-free emulsion polymerization is based on the concept that the hydrophilic polymeric RAFT agent which is easily soluble in the aqueous medium, is able to perform two functions upon the polymerization of the hydrophobic monomer which is poorly soluble in water.8 The first function is to serve as the polymeric RAFT agent and the second function is to stabilize the forming polymer–monomer particles. As was shown previously,39,40 upon the addition of polyacrylic acid with the group located within the chain during the emulsion polymerization of BA, the stable dispersion of triblock copolymers PAA–PBA–PAA can be formed.
Fig. 8 shows the kinetics of emulsifier-free emulsion copolymerization of butyl acrylate with HFBA (1, 2) and OFPA (3, 4) in the presence of Pol-A (1), Cop-H5 (2), Cop-O2 (3) and Cop-O10 (4). As the hydrophobicity of the polymeric RAFT agent increases, copolymerization rate slightly increases. This behavior allows us to conclude that the better steric stabilization provides the earlier completion of the formation of the monomer–polymer particles, where the polymerization takes place. Hence, the induction period corresponding to the formation of particles becomes shorter and the polymerization rate generally increases. The same tendency is observed with increasing monomer content in the reaction media.
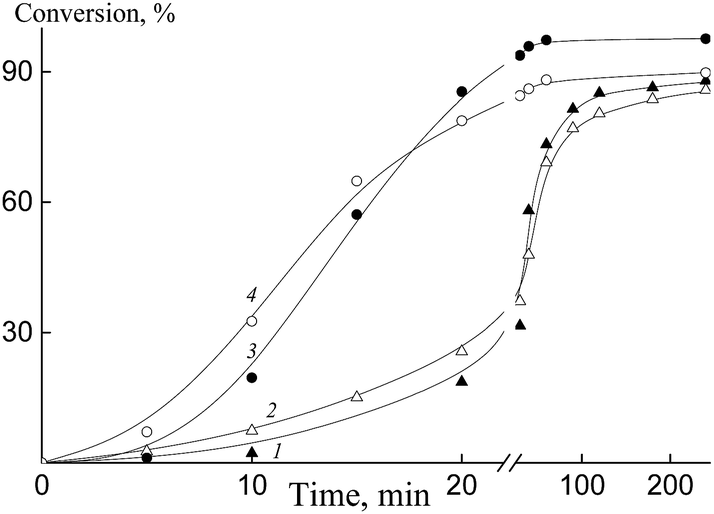 | ||
| Fig. 8 Dependences of conversion on the duration of polymerization for the emulsion copolymerization of BA and fluoroalkyl acrylate at 70 °C. HFBA (1, 2) and OFPA (3, 4). [Pol-A] (1) = [Cop-H5] (2) = 7.7 × 10−4 mol L−1, [PSK] = 2.6 × 10−4 mol L−1, [BA]/[HFBA] = 90/10 mol mol−1, monomers:water = 1:6 v/v; [Cop-O2] (3) = [Cop-O10] (4) = 5.0 × 10−3 mol L−1, [PSK] = 5.0 × 10−2 mol L−1, [BA]/[OFPA] = 80/20 mol mol−1, monomers:water = 1:4 v/v. | ||
The number-average hydrodynamic diameter of the particles Dn changes within the first 10–15 min of polymerization (up to 15–20% of conversion); then, this value remains invariable (Fig. 9). As the hydrophobicity of the polymeric RAFT agent increases, this constant value is achieved more quickly. The dispersions of the particles keep their stability for, at least, several weeks.
 | ||
| Fig. 9 Dependences of number-average diameter Dn of the particles on monomer conversion in the emulsion polymerization initiated by PSK at 70 °C. Monomers:water = 1:4 v/v, [BA]/[OFPA] = 80/20 mol mol−1, Cop-O2 (1), Cop-O10 (2). | ||
Fig. 10 shows the transformation of the MWDs with conversion for the products of the emulsion copolymerization of BA and HFBA (molar ratio 90:10) in the presence of Pol-A (a) and Cop-H5 (b) and of BA and OFPA (molar ratio 80:20) in the presence of Cop-O2 (c) and Cop-O10 (d). At low conversions, the initial polymeric RAFT agent is rapidly consumed, and the triblock copolymer is formed. Then, the MWD becomes bimodal and this behavior is similar to the above-described systems (Fig. 3a and b, 5b and c).
 | ||
| Fig. 10 The SEC curves normalized by the unit area for the products of the emulsion copolymerization of (a and b) butyl acrylate and HFBA (90:10 mol mol−1) and (c and d) butyl acrylate and OFPA (80:20 mol mol−1) at 70 °C initiated by PSK. (a) In the presence of Pol-A, conversion: 3.2 (1), 19.9 (2), 44.4 (3), 58.4 (4), and 81.4% (5); (b) in the presence of Cop-H5, conversion: 7.4 (1), 19.9 (2), 35.5 (3), 47.9 (4), and 85.8% (5); (c) in the presence of Cop-O2, conversion: 1.1 (1), 19.6 (2), 57.1 (3), 85.4 (4), 93.8 (5), and 95.9% (6); (d) in the presence of Cop-O10, conversion: 7.1 (1), 32.6 (2), 64.9 (3), 84.5 (4), and 86.1% (5). | ||
However, MW of the triblock copolymers formed upon the emulsion polymerization is higher than that of the block copolymers formed in the organic solvent (Fig. 10 and 5) and, in the first case, the rate of the polymerization is several times higher than that in the second case. For example, for the triblock copolymer formed via the emulsion polymerization at limited conversions (Fig. 10b), Mn = 1030 kDa and Đ = 1.58. As the concentration of the initiator increases (by ∼2 orders of magnitude) and the concentration of the polymeric RAFT agent increases by a factor of 10 (Fig. 10c), the block copolymer with high MW is formed: Mn = 90 kDa and Đ = 1.70. The molecular weight characteristics of the block copolymers are summarized in Table S3 (ESI†).
The triblock copolymers obtained upon the copolymerization of butyl acrylate and HFBA in the presence of Pol-A and Cop-H5 at limited conversions and subsequent methylation were characterized by the NMR and DSC methods (Fig. S6, ESI† and Table 3). The content of BA units was found to be similar in both block copolymers due to the same content of monomers in the monomer feed mixture and similar monomer conversion, whereas the content of acrylic acid and HFBA is different due to the different nature of the polymeric RAFT agent used in the synthesis, Pol-A or Cop-H5. As a result, Tg corresponding to the block of the BA/HFBA copolymer is the same for both blocks, whereas Tg corresponding to the block originated from the polymeric RAFT agent is different.
| RAFT agent | Copolymer composition, mol% | Tg,1, °C | Tg,2, °C | ||
|---|---|---|---|---|---|
| MA (AA) | BA | HFBA | |||
| Pol-A | 13.1 | 80.1 | 6.8 | −40.6 | 9.5 |
| Cop-H5 | 7.0 | 83.3 | 9.7 | −40.4 | −8.2 |
The morphology of the resulting aqueous dispersions after their drying was studied by the TEM method. Independently of the nature of both polymeric RAFT agent and fluoroalkyl acrylate and of their content in the reaction media, the whole pattern remains the same: individual spherical particles (200–400 nm) and their aggregates (∼1 μm) with the core–shell structure are formed. The hydrophobic block of the BA and HFBA or BA and OFPA copolymer produces the inner core, whereas the short blocks of PAA or its copolymers with HFBA or OFPA form the outer shell. The typical example of the TEM image is presented in Fig. 11. Similar results were reported in ref. 15 and 16 for the diblock copolymer of poly(methacrylic acid) and poly(dodecafluoroheptyl methacrylate) and for the diblock copolymer of poly(methacrylic acid) and poly(2,2,2-trifluoroethyl methacrylate-co-HFBA). In all our studies on the dispersion systems, all attempts to obtain the nanostructured films with the percolating structure failed. The possible reason is likely to be associated with an extremely high difference between the lengths of hydrophilic and hydrophobic blocks.
 | ||
| Fig. 11 The TEM images of the films after emulsion copolymerization of BA and OFPA in the presence of Cop-O2 and drying of the as-formed dispersions. | ||
Hence, the emulsifier-free emulsion copolymerization of butyl acrylate with HFBA in the presence of the hydrophilic/amphiphilic polymeric RAFT agents leads to the formation of high molecular weight triblock copolymers with the bimodal MWD. However, dispersions of the self-assembled triblock copolymers remain stable for, at least, one month after the synthesis.
Synthesis of amphiphilic block copolymers by dispersion polymerization
Dispersion polymerization was used as an alternative approach to the synthesis of the self-assembled dispersions of block copolymers. The reaction medium used for the dispersion polymerization is known to be compatible with a monomer and serves as a non-solvent for the polymer.10,11 Therefore, polymerization starts in the homogeneous medium, but when the propagating species reach their critical length, which depends on the chemical nature of the monomer and thermodynamic quality of the solvent, they precipitate into an individual phase and form the particles in which further polymerization proceeds. Aggregation of the particles is usually suppressed by the addition of a stabilizer to the system.Previously, we showed that Pol-A can provide the RAFT mechanism in the dispersion polymerization of BA, which takes place in the methanol–water mixture.40 In this case, upon polymerization, the linear increase in both Mn was observed.
These encouraging results allow us to expect the occurrence of the RAFT mechanism in the BA and HFBA dispersion copolymerization. However, as the first step, the dispersion copolymerization of BA and HFBA in the methanol–water mixture (70:30 v/v) at 65 °C was conducted; the volume ratio of the monomer–medium phases was 1:7. BTC was used as the RAFT agent and no additional stabilizes were used.
Even in this case, the “living” mechanism is observed. The SEC curves are seen to be shifted to higher MW values with increasing monomer conversion (Fig. 12a).
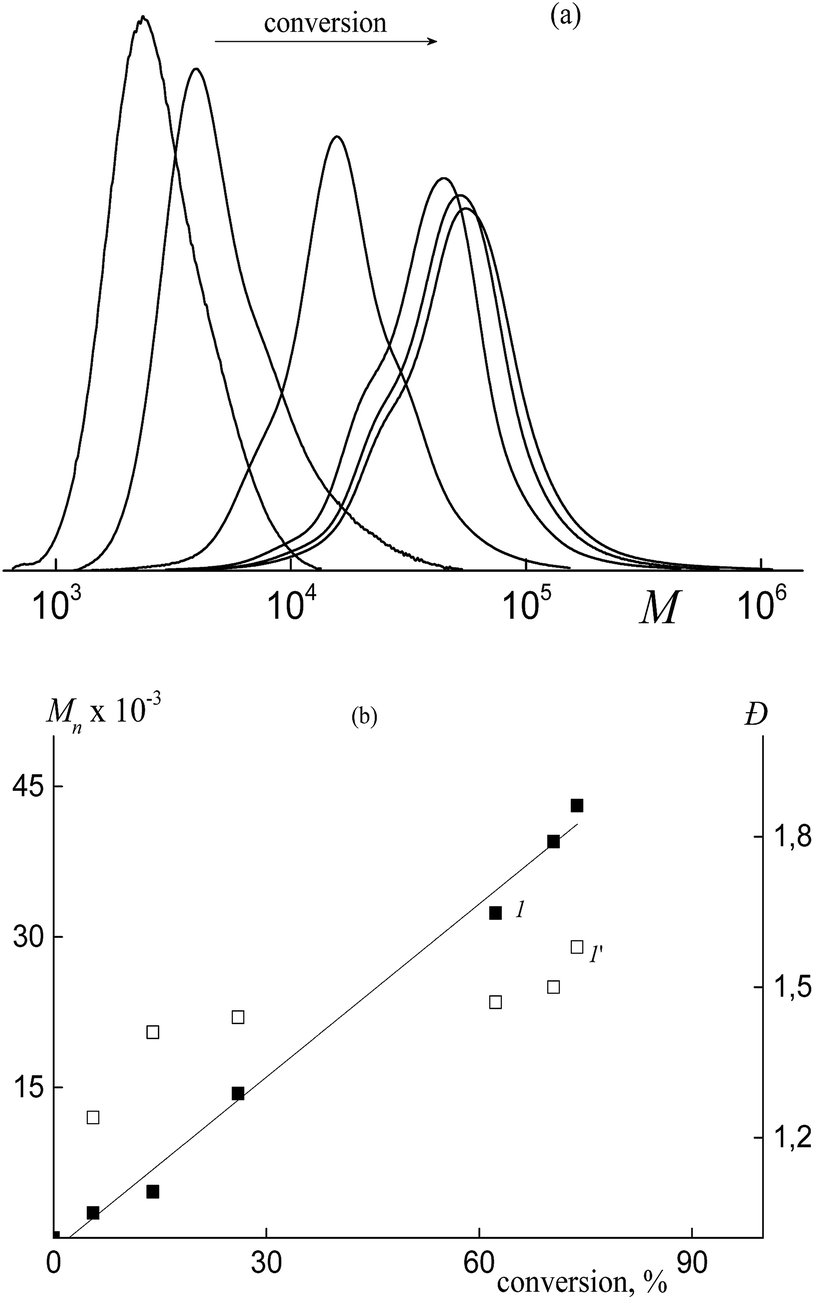 | ||
| Fig. 12 (a) The SEC curves normalized by the unit area for the copolymers of butyl acrylate and HFBA synthesized in the presence of BTC by the dispersion polymerization. HFBA, 10 mol% in monomer feed, methanol:water = 70:30 vol%, monomers:media = 1:7 v/v, [BTC] = 2.2 × 10−3 mol L−1, [AIBN] = 1.1 × 10−3 mol L−1. (b) Dependences of Mn (1) and Đ (1′) on conversion for the synthesized copolymers. | ||
Moreover, Mn increases linearly with increasing conversion, whereas the dispersity remains lower as compared with that of the conventional radical mechanism (Fig. 12b). However, the MWD broadens in the course of the polymerization as evidenced by the shoulders both in low and high MW regions. This behavior may be provided by the discontinuous growth of macromolecules within the particles of different sizes and composition.
In our subsequent experiments, BTC was replaced by Pol-A, the methanol–water ratio was also varied. As the content of water in the system (at a constant value of monomers to media ratio) increases, the thermodynamic quality of the solvent towards the polymer becomes worse, and the polymerization rate increases (Fig. 13). Dependences of the conversion on the reaction time are seen to be S-shaped, and this profile is typical of the dispersion polymerization. When polymerization is conducted in the presence of Pol-A, its rate is higher as compared with that of the system containing BTC.
 | ||
| Fig. 13 Dependence of monomer conversion on the reaction time for the dispersion copolymerization of BA and HFBA in the methanol–water mixture at 65 °C in the presence of BTC (1) and Pol-A (2–4), [BTC] = [Pol-A] = 2.2 × 10−3 mol L−1, [AIBN] = 1.1 × 10−3 mol L−1, 10 mol% of HFBA. Volume ratio (%) of methanol:water = 90:10 (2), 80:20 (3), and 70:30 (1, 4). | ||
The particle size distribution of the dispersions after polymerization in the presence of Pol-A is unimodal. For example, the number-average hydrodynamic diameter Dn of the particles is equal to 60 nm immediately after the completion of the polymerization in the methanol–water mixture (methanol:water = 80:20 vol%). After the dialysis, rinsing with water, and centrifugation, this value increases up to 320 nm. Diameter Dn, estimated after the dialysis increases to 490 nm when the water content in the reaction medium decreases (methanol:water = 90:10 vol%). Then, when the water content increases (methanol:water = 70:30 vol%) this value shrinks down to 305 nm. When Pol-A is replaced by BTC, provided all other conditions are the same, Dn markedly increases from 305 to 420 nm.
Triblock copolymers produced via the copolymerization in the presence of Pol-A are characterized by the unimodal MWD (Fig. 14). The corresponding SEC curves are shifted to higher MW with increasing monomer conversion and they are seen to be slightly broadened due to the shoulder in the lower MW region. This pattern can be explained by the fact that a certain fraction of the formed triblock copolymer (the lower MW shoulder) participates only in the stabilization of the polymer particles, whereas another fraction is also involved in the RAFT mechanism of the polymerization. With increasing water content in the initial reaction mixture, the control over the molecular weight characteristics of the polymerization products is improved (Fig. 14c).
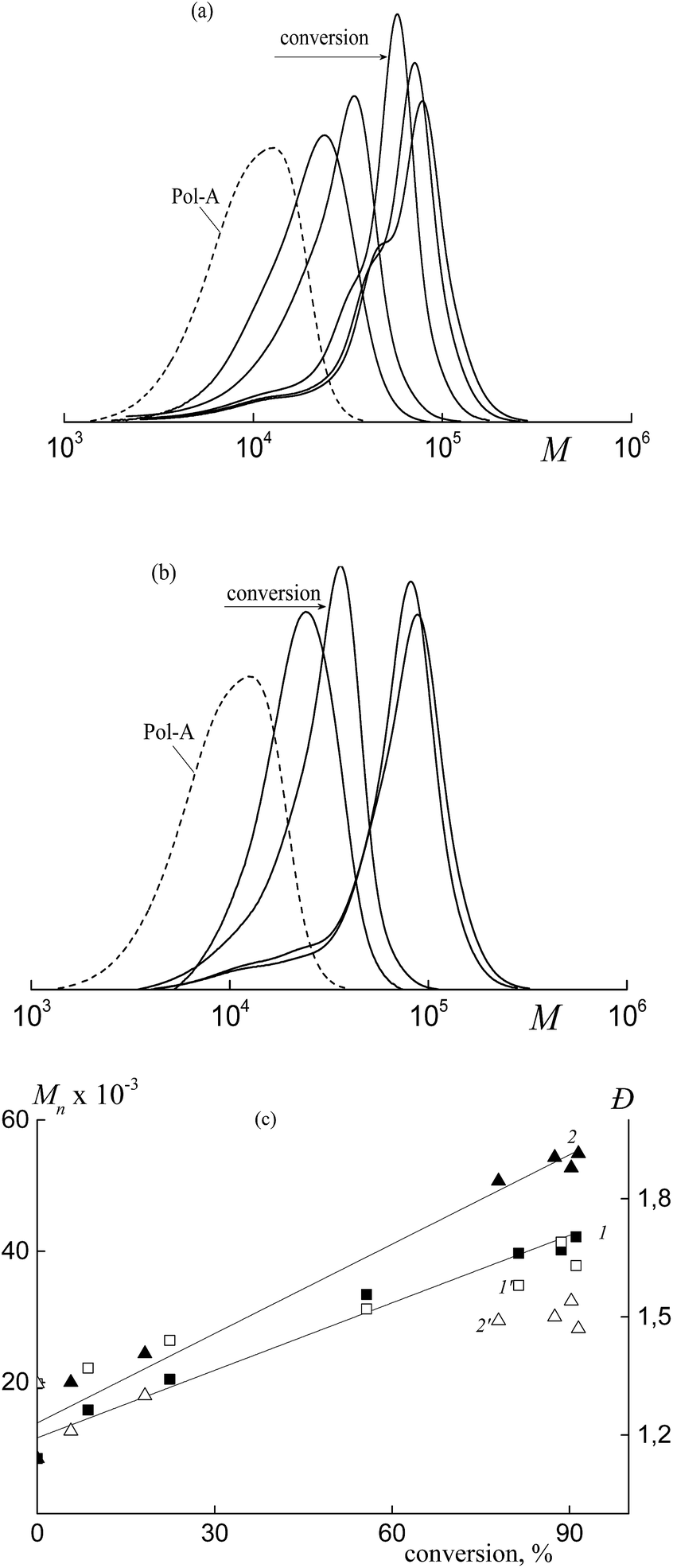 | ||
| Fig. 14 (a) The SEC curves normalized by the unit area for the block copolymers of butyl acrylate and HFBA synthesized in the presence of Pol-A by dispersion polymerization. HFBA, 10 mol% in monomer feed, [Pol-A] = 2.2 × 10−3 mol L−1, [AIBN] = 1.1 × 10−3 mol L−1, monomers:media = 1:7 v/v, methanol:water = 80:20 (a) and 70:30 vol% (b). (c) Dependences of Mn (1, 2) and Đ (1′, 2′) on conversion for the synthesized block copolymers; water content in the reaction mixtures 20 (1, 1′) and 30 vol% (2, 2′). | ||
The triblock copolymers produced via the dispersion polymerization are characterized by the lower MW and a narrower MWD as compared with the products of the emulsion polymerization (Fig. 10).
The synthesized triblock copolymers were analyzed by the NMR spectroscopy (Fig. S7, ESI† and Table 4). Composition of hydrophobic block synthesized up to similar conversions is seen to be independent of the chemical nature of the RAFT agent (BTC or Pol-A) and the composition of the dispersion medium. In the system with the minimum water content, composition of the hydrophobic block appears to be different from other systems due to the low limited conversion (15%). This difference allows us to conclude that the reactivity of monomers in the polymerization is different.
| Methanol:water, v/v |
Copolymer composition, mol% | Tg,1, °C | Tg,2, °C | ||
|---|---|---|---|---|---|
| MA (AA) | BA | HFBA | |||
| a Note: *synthesis was conducted in the presence of BTC. | |||||
| 90:10 |
76.6 | 19.8 | 3.6 | −47.1 | 9.2 |
| 80:20 |
25.8 | 66.7 | 7.5 | −39.3 | 9.0 |
| 70:30 |
25.1 | 67.2 | 7.7 | −40.9 | 10.2 |
| —* | 90.2 | 9.8 | −42.6 | — | |
The DSC curves of the methylated samples of block copolymers show two glass transition as in the case of the block copolymers synthesized in DMF and via the emulsion polymerization. The first Tg corresponds to the block of the BA and HFBA copolymer; the second Tg corresponds to the methylated blocks of PAA (Table 4). Changes in the composition of the methanol–water mixture has no effect on Tg at the same monomer conversion due to the constant monomer feed composition upon the synthesis. For the hydrophobic block in the block copolymer obtained in the presence of Pol-A and copolymer obtained in the presence of BTC, a slight difference in Tg can be provided by the effect of short hydrophilic PAA blocks. For the hydrophobic block in the block copolymer prepared at a monomer conversion of 15% from the mixture containing 10 vol% of water, a marked difference in Tg may be explained by the low MW of this block as compared with other blocks. By analyzing these data in comparison with the results listed in Table 2, one can conclude that, when the hydrophobic blocks have similar composition but lower length (i.e. at a higher molar fraction of the hydrophilic block), both values of Tg are different. The reasons behind this phenomenon are not clear and require further investigation.
Fig. 15 shows the TEM micrographs of the thin polymer films prepared from the dispersions and subjected to the dialysis. The film is seen to contain spherical particles with the hexagonal packing; their size depends on the polymerization conditions and agrees with the results of the light scattering measurements for the solution of the dispersions.
 | ||
| Fig. 15 The TEM images of the thin films prepared from the dispersions produced by the dispersion copolymerization of BA and HFBA in the presence of Pol-A in the methanol–water mixture with the following composition: 80:20 (a) and 70:30 vol% (b), and after the dialysis. | ||
Hence, dispersion copolymerization of butyl acrylate and HFBA mediated by the hydrophilic polymeric RAFT agent allows the controlled synthesis of the amphiphilic triblock copolymers. The formed dispersions are able to produce microstructured films with the percolating structure of the hydrophilic channels. Hence, this approach seems to be most promising as compared with all other approaches studied in this research.
Experimental
Materials and polymer synthesis
Prior to use, all monomers, acrylic acid (AA), 2,2,3,3,4,4,5,5-octafluoropentyl acrylate (OFPA), 2,2,3,4,4,4-hexafluorobutyl acrylate (HFBA) and n-butyl acrylate (BA), and the solvent, N,N-dimethyl formamide (DMF), were distilled under the reduced pressure. Before its use, azobis-isobutyronitrile (AIBN) was recrystallized from anhydrous methanol, potassium persulfate (PSK) was used without purification. The RAFT agent – dibenzyl trithiocarbonate (BTC) was synthesized and characterized as described elsewhere.63All experiments on the polymer synthesis were conducted in a glass reactor equipped with a water jacket, a stirrer and a condenser in an inert atmosphere.
The RAFT copolymerization of acrylic acid with fluoroalkyl acrylate (HFBA or OFPA) was conducted in DMF at 80 °C. The reaction mixtures for copolymerization were prepared by the dissolution of the required amounts of BTC (5 × 10−2 or 6 × 10−3 mol L−1) in the mixture of DMF and acrylic acid, followed by the dropwise addition of fluoroalkyl acrylate to the as-prepared solution. The monomer–solvent volume ratio was equal to 1. The prepared solutions were poured into the reactor and purged with an inert gas for 30 min to remove oxygen. Then, the reactor was pre-heated to 80 °C and the required amount of AIBN dissolved in DMF was added to the reaction mixture until the initiator concentration in the medium became equal to 1 × 10−3 mol L−1. The reactor was purged with an inert gas for 10 min and the polymerization was carried out upon stirring (80 rpm) at 80 °C. Aliquot samples were taken from the reaction mixture within regular time intervals and the polymers were dried under vacuum.
Synthesis of the block copolymers in DMF was conducted according to the same protocol. The polymeric RAFT agent (see Table 1), polyacrylic acid or copolymer of acrylic acid with fluoroalkyl acrylate synthesized via the RAFT polymerization with BTC was dissolved in DMF. The BA and HFBA mixture (BA:HFBA = 90:10, 85:15 and 80:20, molar ratio) was slowly added to the as-prepared solution. The initiator dissolved in DMF was added after the removal of oxygen and pre-heating to 75 °C. Block copolymerization was carried out at 75 °C. Aliquot samples were taken from the reaction mixture within regular time intervals and the polymers were dried under vacuum.
Polymeric RAFT agents were synthesized according to the following general procedure. BTC (1 × 10−1 mol L−1) and AIBN (1 × 10−3 mol L−1) were dissolved in DMF, the monomers (acrylic acid and fluoroalkyl acrylate) were added to this solution. The monomer–solvent volume ratio was equal to 1. Polymerization was carried out in an inert atmosphere at 80 °C for 24 h. Then, the reaction mixture was diluted with DMF and subjected to the dialysis against water. The polymers were lyophilized from the water solutions. The synthesized polymeric RAFT agents and their characteristics are given in Table 1.
Dispersion copolymerization of the BA and HFBA mixture (BA:HFBA = 90:10, molar ratio) was conducted in an inert atmosphere in the methanol/water medium (CH3OH:H2O = 90:10, 80:20 and 70:30, volume ratio) at the monomer–medium volume ratio was equal to 1:7. BTC and polyacrylic acid trithiocarbonate (Pol-A) were used as the RAFT agents. The RAFT agent and AIBN (RAFT agent:AIBN = 2:1, molar ratio) were dissolved in the methanol–water mixture; then, BA and HFBA were added. Oxygen was removed according to the above-described protocol. Polymerization was carried upon stirring (80 rpm) at 65 °C.
Emulsifier-free emulsion copolymerization of BA and HFBA as well as BA and OFPA was conducted in an inert atmosphere upon stirring (300 rpm) at 70 °C. Molar ratio of the monomers was equal to 90:10 for BA and HFBA and 80:20 for BA and OFPA. The monomer–aqueous phase volume ratio was equal to 1:6 for the copolymerization of BA and HFBA and 1:4 for the copolymerization of BA and OFPA.
In the general procedure, the polymeric RAFT agent (Pol-A, Cop-H5, Cop-O2, Cop-O10) was dissolved in the bidistilled water and then, pH was adjusted to 5.4. Monomers were added to the aqueous solution. The reactor with the reactor mixture was purged with nitrogen. Then, the required amount of PSK aqueous solution was added to the reactor and repeatedly purged with nitrogen. [Pol-A] = [Cop-H5] = 7.7 × 10−4 mol L−1, [PSK] = 2.6 × 10−4 mol L−1; [Cop-O2] = [Cop-O10] = 5.0 × 10−3 mol L−1, [PSK] = 5.0 × 10−2 mol L−1.
Instrumentation
For all samples, conversion was estimated by gravimetry. Before analysis, all copolymers containing the units of acrylic acid were subjected to methylation by diazomethane.64The average molecular weights and dispersity (Đ) were determined by the size exclusion chromatography (SEC). The SEC measurements were performed in DMF containing 0.1 wt% of LiBr at 50 °C with a flow rate of 1.0 mL min−1 using a GPC-120 “PolymerLabs” chromatograph equipped with refractive index and with two columns PLgel 5 μm mixed B for MW range of 5 × 102 to 1 × 107. The SEC system was calibrated using the narrow dispersed linear poly(methyl methacrylate) ranging from 800 to 2 × 106 g mol−1.
For polymeric RAFT agents, surface tension isotherms were collected at room temperature and pH 5.4 at the water–air interface in the aqueous solution of 0.1 M NaCl using a KRUSS K100MK2 processor tensiometer (Germany).
The average diameter and ζ-potential of the particles of polymeric dispersions were measured by a Zetasizer Nano-ZS “Malvern” photon analyzer (Great Britain) equipped with a He–Ne laser as a light source (λ = 633 nm and a power of light source of 5 mW); the particle size was measured within the interval from 0.6 to 6000 nm. The initial dispersions were diluted with bidistilled water and dedusted by filtration. The measurements were conducted at room temperature and at a scattering angle of 173° in the automatic mode according to the standard procedure.
The thermal effects upon heating of the samples were studied by the differential scanning calorimetry using a Netzsch DSC 204 “Phoenix” (Germany) under dry nitrogen atmosphere at the flow rate 100 mL min−1 within the temperature range from −80 to +180 °C with a heating rate of 5 K min−1. The weight of the samples for the DSC measurements was 4–6 mg; the samples were placed into the aluminum pans with the punctured cap. The results were analyzed using a Netzsch Proteus software. The glass transition temperature (Tg) was calculated from the inflection point of the second heating curve.
1H and 19F NMR spectra of the copolymers were recorded on a VARIAN MR-400 spectrometer (USA) at 400 MHz using CDCl3 or dimethyl sulfoxide-d6 as a solvent. The signals of the protons of the deuterated solvents were used as internal standards. The spectra were processed using the MestReNova software.
For the TEM studies, few microliters of the dispersion were cast onto a collodion-coated copper grid and dried at ambient temperature. Then, the particles were contrasted with a Pt–Ir–C alloy. The TEM images were collected on a TESLA BS 500 transmission electron microscope (Czech Republic) at an acceleration voltage of 60 kV.
The microphotographs of the emulsion particles were obtained using a Leo 912 AB Omega (Karl Zeiss) transmission electron microscope (TEM) operating at an accelerating voltage of 100 kV; 1 μL droplets of the dilute emulsion solutions were deposited onto the carbon-coated copper TEM grids and dried at room temperature.
Conclusions
The present research addresses the problems of the controlled synthesis of the amphiphilic copolymers of various structures based on acrylic acid, fluoroalkyl acrylates and n-butyl acrylate. To accomplish this goal, the process proceeds via the RAFT route (1) in the organic solvent, and via the emulsion polymerization (2) and dispersion polymerization (3). This complex approach allows the comparison of the specific features of the above processes and evaluation of their potential for the block copolymer synthesis. Our special interest is focused on the self-assembly of triblock copolymers within the core–shell particles which preserve their nanoscale structure in the polymeric films.According to the experimental results, in the case of both fluoroalkyl acrylates, HFBA an OFPA, copolymerization with acrylic acid in DMF or 1,4-dioxane markedly slows down. This effect is strongly enhanced as the molar fraction of fluoroalkyl acrylate molar in the monomer feed slightly increases. This result seems to be rather unexpected because the RAFT homopolymerization rate for both monomers is high and, upon the conventional radical copolymerization, no retardation is observed. Nevertheless, the RAFT mechanism is preserved when copolymerization proceeds in the presence of BTC.
This conclusion is also confirmed by the following result. Both polymeric trithiocarbonates based on polyacrylic acid or amphiphilic copolymer of acrylic acid with fluoroalkyl acrylate are consumed rather rapidly and produce the triblock copolymers upon the polymerization of BA or its copolymerization with fluoroalkyl acrylate whatever the scenario of the synthesis.
However, the reaction route can be varied. The dispersion polymerization offers better control over the molecular weight characteristics and the morphology of the synthesized triblock copolymers at a certain methanol–water volume ratio. As a result, the nanostructured film with the percolating structure of the hydrophilic phase is produced.
To the contrary, in the case of the emulsion polymerization and polymerization in DMF, the living nature of the process is violated due to the partial participation of the newly formed block copolymer in the further chain extension. In both cases, the polymerization products are characterized by the bimodal MWD. However, in the former system, Mn of the formed product is about 1000 kDa whereas, in the latter case, Mn is about 50 kDa.
Hence, the dispersion copolymerization of BA and fluoroalkyl acrylates in the presence of hydrophilic (or amphiphilic) polymeric trithiocarbonates offers a powerful tool for the synthesis of self-assembling amphiphilic triblock copolymers with fluoroalkyl acrylates.
Acknowledgements
This research was financially supported by the Russian Science Foundation (project no. 15-03-30007, sections “Synthesis of amphiphilic block copolymers by emulsifier-free emulsion polymerization” and “Synthesis of amphiphilic block copolymers by dispersion polymerization”) and Russian Foundation for Basic Research (projects no. 17-03-00131 and 16-53-76007 (part of the Join project ERA.Net RUS Plus: SAS-MEM 254)). We are grateful to S. S. Abramchuk for his assistance in the TEM analysis. A. V. Tolkachev is grateful to the German Academic Exchange Service (Research Grants for Doctoral Candidates and Young Academics and Scientists, 2015/16, No. 57130104); AVT also is grateful to the colleagues from Max-Planck Institute of Colloids and Interfaces for their assistance in the polymer characterization.References
- Controlled and Living Polymerizations: From Mechanisms to Applications, ed. A. H. E. Müller and K. Matyjaszewski, Wiley-Interscience, New York, 2009 Search PubMed.
- Progress in Controlled Radical Polymerization: Materials and Applications, ed. K. Matyjaszewski, B. S. Sumerlin and N. V. Tsarevsky, Am. Chem. Soc., New York, 2013 Search PubMed.
- Controlled Radical Polymerization: Mechanisms, ed. K. Matyjaszewski, B. S. Sumerlin, N. V. Tsarevsky and J. Chiefari, Am. Chem. Soc., 2015, ACS Symposium Series, vol. 1187 Search PubMed.
- G. Delaittre, J. Nicolas, C. Lefay, M. Save and B. Charleux, Chem. Commun., 2005, 1, 614–616 RSC.
- S. Brusseau, F. D'Agosto, S. Magnet, L. Couvreur, C. Chamignon and B. Charleux, Macromolecules, 2011, 44, 5590–5598 CrossRef CAS.
- S. Sugihara, K. Sugihara, S. P. Armes, H. Ahmad and A. L. Lewis, Macromolecules, 2010, 43, 6321–6329 CrossRef CAS.
- J. Rieger, Macromol. Rapid Commun., 2015, 36, 1458–1471 CrossRef CAS PubMed.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- M. J. Derry, L. A. Fielding and S. P. Armes, Prog. Polym. Sci., 2016, 52, 1–18 CrossRef CAS.
- J.-T. Sun, C.-Y. Hong and C.-Y. Pan, Polym. Chem., 2013, 4, 873–881 RSC.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- Handbook of RAFT polymerization, ed. C. Barner-Kowollik, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 985–1076 CrossRef CAS.
- Y. Jiang, L. Li, J. Liu, R. Wang, H. Wang, Q. Tian and X. Li, J. Fluorine Chem., 2016, 183, 82–91 CrossRef CAS.
- S. Xu and W. Liu, J. Fluorine Chem., 2008, 129, 125–130 CrossRef CAS.
- Y. Chen, W. Luo, Y. Wang, C. Sun, M. Han and C. Zhang, J. Colloid Interface Sci., 2012, 369, 46–51 CrossRef CAS PubMed.
- H. Chen and Y. Luo, Macromol. Chem. Phys., 2011, 212, 737–743 CrossRef CAS.
- J. J. Reisinger and M. A. Hillmyer, Prog. Polym. Sci., 2002, 27, 971–1005 CrossRef CAS.
- K. S. Lee and J. S. Lee, Chem. Mater., 2006, 18, 4519–4525 CrossRef CAS.
- C. M. Timperley, R. E. Arbon and M. Bird, J. Fluorine Chem., 2003, 121, 23–31 CrossRef CAS.
- W. Yao, Y. Li and X. Huang, Polymer, 2014, 55, 6197–6211 CrossRef CAS.
- M. Eberhardt, R. Mruk, R. Zentel and P. Théato, Eur. Polym. J., 2005, 41, 1569–1575 CrossRef CAS.
- M. Eberhardt and P. Théato, Macromol. Rapid Commun., 2005, 26, 1488–1493 CrossRef CAS.
- M. Allmeroth, D. Moderegger, B. Biesalski, K. Koynov, F. Rösch, O. Thews and R. Zentel, Biomacromolecules, 2011, 12, 2841–2849 CrossRef CAS PubMed.
- M. I. Gibson, M. Danial and H.-A. Klok, ACS Comb. Sci., 2011, 13, 286–297 CrossRef CAS PubMed.
- G. B. H. Chua, P. J. Roth, H. T. T. Duong, T. P. Davis and A. B. Lowe, Macromolecules, 2012, 45, 1362–1374 CrossRef CAS.
- L. Nuhn, M. Hirsch, B. Krieg, K. Koynov, K. Fischer, M. Schnidt, M. Helm and R. Zentel, ACS Nano, 2012, 6(3), 2198–2214 CrossRef CAS PubMed.
- M. I. Gibson, E. Fröhlich and H.-A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4332–4345 CrossRef CAS.
- M. M. Herth, M. Barz, D. Moderegger, M. Allmeroth, M. Jahn, O. Thews, R. Zentel and F. Rösch, Biomacromolecules, 2009, 10, 1697–1703 CrossRef CAS PubMed.
- M. Barz, R. Luxenhofer, R. Zentel and A. V. Kabanov, Biomaterials, 2009, 30, 5682–5690 CrossRef CAS PubMed.
- T. Guo, D. Tang, M. Song and B. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5067–5075 CrossRef CAS.
- Q. Zhang, Q. Wang, Z. Luo, X. Zhan and F. Chen, Polym. Eng. Sci., 2009, 49, 1818–1824 CAS.
- N. M. L. Hansen, K. Jankova and S. Hvilsted, Eur. Polym. J., 2007, 43, 255–293 CrossRef CAS.
- Z. Qinghua, Z. Xiaoli, C. Fengqiu, S. Ying and W. Qiongyan, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1585–1594 CrossRef.
- B. R. Koiry, H.-A. Klok and N. K. Singha, J. Fluorine Chem., 2014, 165, 109–115 CrossRef CAS.
- B. R. Koiry, M. Moukwa and N. K. Singha, J. Fluorine Chem., 2013, 153, 137–142 CrossRef CAS.
- B. Jiang, L. Zhang, J. Shi, S. Zhou, B. Liao, H. Liu, J. Zhen and H. Pang, J. Fluorine Chem., 2013, 153, 74–81 CrossRef CAS.
- E. V. Chernikova, N. S. Serkhacheva, O. I. Smirnov, N. I. Prokopov, A. V. Plutalova, E. A. Lysenko and E. Y. Kozhunova, Polym. Sci., Ser. B, 2016, 58(6), 417–428 Search PubMed.
- E. V. Chernikova, A. V. Plutalova, K. O. Mineeva, I. R. Nasimova, E. Y. Kozhunova, A. V. Bol'shakova, A. V. Tolkachev, N. S. Serkhacheva, S. D. Zaitsev, N. I. Prokopov and A. B. Zezin, Polym. Sci., Ser. B, 2015, 57(6), 547–559 CrossRef CAS.
- R. Kerber and H. Glamann, Makromol. Chem., 1971, 144, 1–16 CrossRef CAS.
- A. Chapiro, Eur. Polym. J., 1973, 9(5), 417–427 CrossRef CAS.
- K. Plochocka and H. J. Harwood, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1978, 19, 240 CAS.
- K. J. Plochocka, Journal of Macromolecular Science, Part C, 1981, 20(1), 67–148 CrossRef.
- H. J. Harwood, Makromol. Chem., Macromol. Symp., 1987, 10/11(1), 331–334 CrossRef.
- Y. D. Semchikov, Makromol. Chem., Macromol. Symp., 1996, 111(1), 317–328 CrossRef CAS.
- Y. D. Semchikov, L. A. Smirnova, T. Y. Knyazeva, S. A. Bulgakova and V. I. Sherstyanykh, Eur. Polym. J., 1990, 26(8), 883–887 CrossRef CAS.
- E. C. Chapin, G. E. Ham and C. L. Mills, J. Polym. Sci., 1949, 4(5), 597–604 CrossRef CAS.
- A. V. Ryabov, Y. D. Semchikov and N. N. Slavnitskaya, Vysokomol. Soedin., Ser. A, 1970, 12(3), 553–560 CAS.
- S. Wang and G. W. Poehlein, J. Appl. Polym. Sci., 1993, 49(6), 991–1001 CrossRef CAS.
- R. Kerber, Makromol. Chem., 1966, 96, 30–40 CrossRef CAS.
- Handbook of Radical Polymerization, ed. K. Matyjaszewski and T. P. Davis, Wiley-Interscience, Hoboken, 2002 Search PubMed.
- Y. Kwak, A. Goto and T. Fukuda, Macromolecules, 2004, 37(4), 1219–1225 CrossRef CAS.
- O. V. Borisova, M. Yu. Zaremski, O. V. Borisov and L. Billon, Polym. Sci., Ser. B, 2013, 55(11–12), 573–576 CrossRef CAS.
- C. Lefay, B. Charleux, M. Save, C. Chassenieux, O. Guerret and S. Magnet, Polymer, 2006, 47, 1935–1945 CrossRef CAS.
- E. V. Chernikova, A. V. Plutalova, E. S. Garina and D. V. Vishnevetskii, Polym. Chem., 2016, 7, 3622–3632 RSC.
- G. Reiss, Prog. Polym. Sci., 2003, 28(7), 1107–1170 CrossRef.
- Polymer Handbook, ed. J. Brandrup, E. H. Immergut and E. A. Grulke, Wiley, New York, 1999 Search PubMed.
- D. V. Vishnevetskii, A. V. Plutalova, V. V. Yulusov, O. S. Zotova, E. V. Chernikova and S. D. Zaitsev, Polym. Sci., Ser. B, 2015, 57(3), 197–206 CrossRef CAS.
- O. V. Borisova, M. Y. Zaremski, O. V. Borisov and L. Billon, Polym. Sci., Ser. B, 2013, 55(11–12), 573–576 CrossRef CAS.
- Y. Tsukahara, N. Hayashi, X.-L. Jiang and Y. Yamashita, Polym. J., 1989, 21(5), 377–391 CrossRef CAS.
- E. V. Chernikova, D. V. Vishnevetskii, E. S. Garina, A. V. Plutalova, E. A. Litmanovich, B. A. Korolev, A. V. Shlyakhtin, Y. V. Kostina and G. N. Bondarenko, Polym. Sci., Ser. B, 2012, 54(3–4), 127–141 CrossRef CAS.
- E. V. Chernikova, P. S. Terpugova, E. S. Garina and V. B. Golubev, Polym. Sci., Ser. A, 2007, 49, 108–119 CrossRef.
- E. V. Vasilyeva, N. A. Kopylova, S. D. Zaitsev and Y. D. Semchikov, Polym. Sci., Ser. B, 2011, 53(9–10), 491–496 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, SEC data, theoretical dependences of copolymer composition and monomer unit distribution vs. conversion. See DOI: 10.1039/c7ra03203j |
| This journal is © The Royal Society of Chemistry 2017 |