
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of contact-killing non-leaching antimicrobial guanidyl-functionalized polymers via click chemistry
Kaimei Peng a,
Tao Zoua,
Wei Dinga,
Ruonan Wanga,
Jinshan Guob,
John Jay Roundb,
Weiping Tua,
Chao Liub and
Jianqing Hu*a
a,
Tao Zoua,
Wei Dinga,
Ruonan Wanga,
Jinshan Guob,
John Jay Roundb,
Weiping Tua,
Chao Liub and
Jianqing Hu*a
aSchool of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou 510640, China. E-mail: glennhu@scut.edu.cn
bAleo BME, Inc.200 Innovation Blvd, Suite 210A, State College, PA 16803, USA
First published on 10th May 2017
Abstract
An expedient and efficient method applied to synthesize click-suitable penta-substituted guanidines at room temperature is hereby described. The guanidine containing polyurethanes (PU-TMGs) were prepared via click reaction (copper catalyzed alkyne-azide [3 + 2] cycloaddition, CuAAC), which was performed either before or after polymerization to incorporate the guanidine. The guanidine-functionalized polymers exhibited strong contact-killing antibacterial activity against both Gram-negative and Gram-positive bacteria. There is around 99.9% killing of S. aureus and 98.0% killing of E. coli when TMG weight percentage content in PU-TMG is around 1%. Covalent conjugation of the guanidine groups to the polymers prevents leaching. The conjugation also reduces cytotoxicity of the materials and preserves long-term antimicrobial activity.
1 Introduction
Microbes are ubiquitous in nature. Although some of them are beneficial to human beings, most of them are harmful, and some are even fatal. According to the list of major concerns about health from World Health Organization (WHO), various health problems are caused by micro-organisms such as influenza, cholera and sanitation. Microbes may contaminate water and sanitation services, and infect thousands of people. Therefore, some processes such as cleaning, disinfection and sterilization of wastewater and air are certainly essential. Although natural products and small molecules are used to prevent the spread and growth of microbes, the unpleasant side effects of releasing biocides into the environment, and the emerging antibiotic-resistant bacteria have become major concerns. Just in the European Union alone,1 more than 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people each year are contracted by antibiotic-resistant microbe, such as methicillin-resistant staphylococcus aureus (MRSA), one of several “superbugs” that are a global threat.2,3
000 people each year are contracted by antibiotic-resistant microbe, such as methicillin-resistant staphylococcus aureus (MRSA), one of several “superbugs” that are a global threat.2,3
The increasing prevalence of antibiotic resistance is a serious emerging challenge and has been a driving force for the development of new anti-microbial materials. In recent years, the development of cationic antimicrobial polymers has become a promising strategy. Different with other antimicrobial agents, the cationic polymers and the negatively charged microbial cell membranes undergo non-specific electrostatic interactions instead of specific receptor–protein interactions. Those types of interactions present minimal risk for the creation of mechanisms of bacterial resistance.4 As a result of the critical attribute, quaternary ammonium polymers have widely used as cationic antimicrobial materials. However, the weak interactions between the quaternary ammonium and the phosphate groups of phospholipids within the cell membranes and the poor alkaline stability of the material at elevated temperatures are challenges that still need to be addressed.5
In recent years, guanidine-based polymers have attracted significant attention as powerful, and selective anti-microbial materials because the selective interactions occur at between guanidine groups and negatively charged bacterial cell-membrane instead of electrically neutral mammalian cell membranes, and nontoxic antimicrobial materials. Guanidine groups not only exist in natural antimicrobial products, such as antimicrobial peptides (AMPs),6 and play indispensable roles in asymmetric synthesis,7 but they also have been widely used in fuel cells,5,8,9 antimicrobial,10–12 biology,13,14 medicine,15,16 and catalysis study.17,18 Guanidine groups have unique features that enable their antimicrobial capabilities.9 Guanidine groups are strong bases with a pKa value of 13.8 for pentamethyl guanidine; The positive charges of the guanidine group are delocalized over three nitrogen atoms when guanidine is ionized, which contributes to a high degree of ionization in a wide pH range, a high thermal and basic stability, and a strong interaction with phosphate groups by forming hydrogen bonded ion pairs.19 The bidendate binding between guanidine groups and phosphate groups20 is much stronger than that between amine groups and phosphate groups,21 resulting in a high bactericidal rate at fairly low antimicrobial concentrations of guanidine-based molecules or polymers. Importantly, the toxicity of charge-delocalized cationic compounds, such as pyridinium and guanidine,22 is much lower than that of charge-localized linear amine head group. Guanidine polymers possess both antimicrobial and low hemolytic characteristics.11
Although the applications of the polymers containing guanidine group located in the backbone chain such as polyhexamethylene guanidine oligomer (PHMG), polyhexamethylene biguanide (PHMB), poly(p-xylyleneguanidine) hydrochloride (PPXG) and guanidine and urea copolymer (PGUC) were reported,23–25 the characteristic of high solubility in water restricted their wide application. Grafting guanidine to different polymer chain is a useful method to construct the non-leaching antimicrobial polymers and expands the application of guanidine-rich polymers. Moreover, the contact-killing mode is possible for the guanidine-rich polymer because the interactions of guanidine groups and negatively charged bacterial cell-membrane occur on the surface of cell. One method to prepare contact-killing polymer is physical mixing leaching microbicide and polymer,26,27 which cannot solve the sterilizing effect dropped over time and residual toxicity for environment.28 Accordingly, the linking between polymer and guanidine groups is a promising method to construct non-leaching contact-killing antimicrobial guanidyl-functionalized polymers based on the properties of the guanidine-rich polymer.
At present, the most popular method for fabricating guanidyl-functionalized polymers is the quaternization of penta-substituted guanidines with bromobenzyl (or chlorobenzyl) containing polymers.5 However, this method has a limited reaction rate and efficiency, and lacks of orthogonality and an precise control over the degree and location of functionalization.29 Furthermore, the stability of benzyl guanidines is poor as a result of nucleophilic attack of the benzylic cation by hydroxide8 or methoxyl17 ions and then through ylide formation and rearrangement reactions,30 which will release the guanidine groups and deactivate the guanidine-rich polymers quickly. Additionally, the current synthesis process of penta-substituted guanidines is complicated and not eco-friendly.31,32 It has been a challenge to develop a simple and highly efficient strategy for the preparation of penta-substituted guanidines and stable guanidyl-functionalized polymers.
The growing applications of polyurethanes stem from the ability to tailor their properties via step additional-polymerization between isocyanate and hydroxyl functionalities, which is the foundation of its compatibility with different components. Polyurethane allows for the introduction of functional molecules and the material exhibits excellent thermal and mechanical properties, all of which fuel the use of polyurethanes in adhesives,33 sealants,34 synthetic fibers,35 foams,36 coatings,37 biomedical implants38 and antimicrobial materials.39,40
Click chemistry, especially the favored copper-catalyzed azide-alkyne cycloaddition (CuAAC) process, is a robust, efficient and orthogonal method for the construction of functional molecules.41,42 Recently, we have reported that click chemistry is a powerful tool for the polymer crosslinking and functionalization.43,44 Here, a new facile approach was developed for the synthesis of a click-suitable penta-substituted guanidine (2-propinyl-1,1,3,3-tetramethylguanidine, TMG-Al) via commercially available raw material at room temperature. A TMG-based antimicrobial polyurethane (PU-TMG) was prepared by either clicking TMG-Al onto azide diol (2,2-bis(azidomethyl)propane-1,3-diol, DAzD) to synthesize PU-TMG or by clicking TMG-Al onto the formed azide-containing polyurethane (PU-N3). Meanwhile, the physiochemical properties and the antimicrobial performance of the PU-TMG were investigated.
2 Materials and methods
2.1 Materials
Trimethylolpropane (TMP), 1,4-butanediol (BDO), poly(propylene glycol) (M.W. = 1000, PPG 1000), and isophorone diisocyanate (IPDI) were purchased from J&K chemical Ltd. All other solvents and reagents for the monomer synthesis were purchased from Aldrich or Aladdin chemical Ltd. All other materials were purchased from J&K Acros Organics (Beijing, China). All chemicals were used as received and Milli-Q filtered water was used to prepare bacterial culture media. Polymerizations were performed in dry DMF purchased from Sigma. The two bacteria strains used were Gram-negative Escherichia coli (E. coli, ATCC 25922) and Gram-positive Staphylococcus aureus (S. aureus, ATCC 29213). Bacterial culture media were autoclaved before using to ensure sterility.2.2 Methods
1,1,3,3-Tetramethyl-2-(prop-2-yn-1-yl)guanidine (TMG-Al) was synthesized according to the following procedure. To a 100 mL flask charged with TMG (20 g, 0.17 mol) and 25 mL ethyl acetate, after being cooled by ice bath, propargyl bromide (7.14 g, 0.06 mol) was slowly added with vigorous stirring. Then, stirring was continued for another 16–24 hours at room temperature, and a large amount of precipitate occurred. After completion of the reaction, the solid was filtered. After aqueous workup, extraction with dichloromethane (DCM), wash with brine, and concentration of the reaction mixture, a brown liquid (5.51 g) was obtained in 60% yield.
Diguanidine diol (TMG-OH) was synthesized via CuAAC as follows. A 100 mL flask charged with TMG-Al (1.83 g, 11.84 mmol), DAzD (1.00 g, 5.38 mmol) and 30 mL ethyl acetate was degassed by six freeze–pump–thaw cycles. Subsequently, the prepared catalyst solution, 5 mL dimethylformamide (DMF) solution containing 0.2 g CuBr and 0.36 mL N,N,N′,N′′,N′′-pentamethyl diethylenetriamine (PMDETA), was then transferred the reaction mixture under nitrogen atmosphere. The reaction is completed in 1 hour at room temperature and the viscous solid precipitated. The liquid was removed and, the final solid product was obtained after vacuum dry for at least 48 hours. Yield: 2.44 g (92%).
The click-suitable azide functional polyurethane (PU-N3) was prepared according to the following procedure. Firstly, IPDI (30 g, 135 mmol) and PPG 1000 (27.5 g, 27.5 mmol) were charged into a 250 mL dried four-necked flask equipped with a mechanical stirrer, a thermometer, a condenser, and a nitrogen in/outlet. After stirring for 10 minutes to give a uniform mixture, the temperature was increased to 80 °C and maintained reaction for 2 hours. Then, 140 mL DMF, BDO (4.68 g, 52 mmol), TMP (3 g, 22 mmol) and DAzD (2.75 g, 15 mmol) were added into the flask and the reaction was kept for another 6–7 hours. PU-N3 in DMF (solid content: 34%) was prepared.
Route 1. Polyurethane with side-chain tetramethylgermanium (PU-TMG) was prepared according to the following procedure. The click reaction was performed between PU-N3 and TMG-Al as follows: 15 mL water/DMF mixture (VDMF
:VH2O = 9:1), and a certain amount of TMG-Al (0.030 g for PU-TMG 1, 0.052 g for PU-TMG 2, 0.109 g for PU-TMG 3, and 0.297 g for PU-TMG 4) were added to 15 g solution of PU-N3 in DMF at room temperature under stirring, followed by the addition of 0.01 g CuSO4·5H2O in 1 mL water–DMF mixed solvent (VDMF:VH2O = 9:1). The mixture was degassed for 20 minutes with nitrogen and 0.03 g sodium ascorbate in 1 mL water–DMF mixed solvent was subsequently added in, and then the click reaction was initiated under nitrogen atmosphere. The mixture was stirred for additional 1 hour, then 10 g of the obtained mixture was poured into a 4 cm diameter-teflon disk and dried at 75 °C for 24 hours. The PU-TMGs were precipitated from water and dried. The yields of the final polymer PU-TMGs remained above 98%.
Route 2. Tetramethylgermanium containing polyurethane (PU-TMG 1′–4′) was prepared according to the following procedure. IPDI (30 g, 135 mmol) and PPG 1000 (27.5 g, 27.5 mmol) were charged into a 250 mL dried four-necked flask equipped with a mechanical stirrer, a thermometer, a condenser, and a nitrogen in/outlet. After stirring for 10 minutes to give a uniform mixture, the temperature was increased to 80 °C, and the reaction was maintained for 2 hours. Then, 140 mL DMF, TMP (3 g, 22 mmol), a certain amount of TMG-Al and BDO were added into the flask and the reaction was kept for another 6–7 hours. Finally, the PU-TMG 1′–4′ have the same TMG content as the PU-TMG 1–4 were obtained. The PU-TMGs were precipitated from water and dried. The yields of the final polymer PU-TMGs remained above 98%.
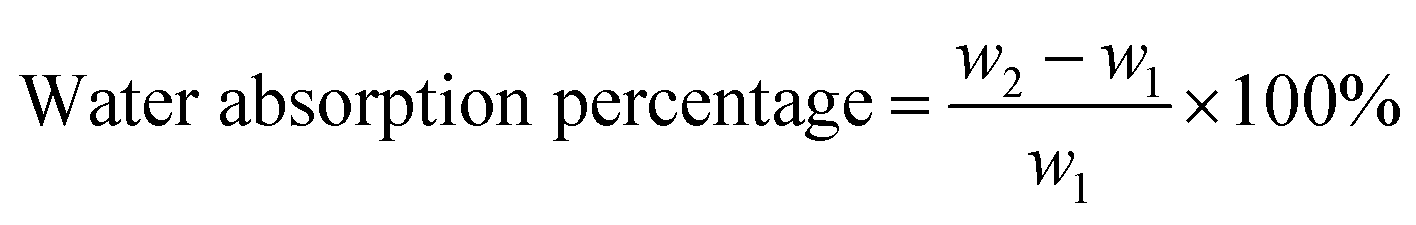 | (1) |
The static contact angles of films were conducted on a PocketGoniometer PGX+ contact angle analyser. The average contact angle was obtained from five tests of each sample.
The disk diffusion of different samples was conducted according to the following procedure. The 24 hours cultured active bacterial suspensions were spreaded on the surface of the solidified agar media. Polymer films with 10 × 10 × 0.5 mm were adhered closely to the medium and the architecture was incubated at 37 °C for 8 hours. The polymer samples were washed with ethanol and sterile double-distilled water and dried at 75 °C before the experiment.
3 Results and discussion
3.1 Monomers synthesis
The basic strategies employed for the synthesis of DAzD, TMG-Al and TMG-OH are based on the nucleophilic substitution. DAzD was synthesized in one step from the 2,2-bis(bromomethyl) propane-1,3-diol in Fig. 1A and the structure was confirmed by 1H/13C NMR and FT-IR spectra shown in Fig. 2A, C and E. The appearance of the characteristic infrared absorption peak of azide group was observed at 2104 cm−1 of FTIR spectrum (Fig. 2E), and the left shift of the chemical shift of the protons on CH2 link to Br groups of the starting material at around 3.20 ppm to 3.70 ppm of the protons on CH2 link to azide groups of DAzD (1H NMR, Fig. 2A).43,44 Generally, penta-substituted guanidine was synthesized in two steps. Briefly, the intermediate product, Vilsmeier salt, was obtained by tetra-alkyl urea and phosgene, and then the active intermediate reacted with primary amine.47 Subsequently, Zhang48 used the phosphorus oxychloride or thionyl chloride instead of hypertoxic phosgene, but the operation was also somewhat complicated due to the sensitivity of phosphorus oxychloride or thionyl chloride to water.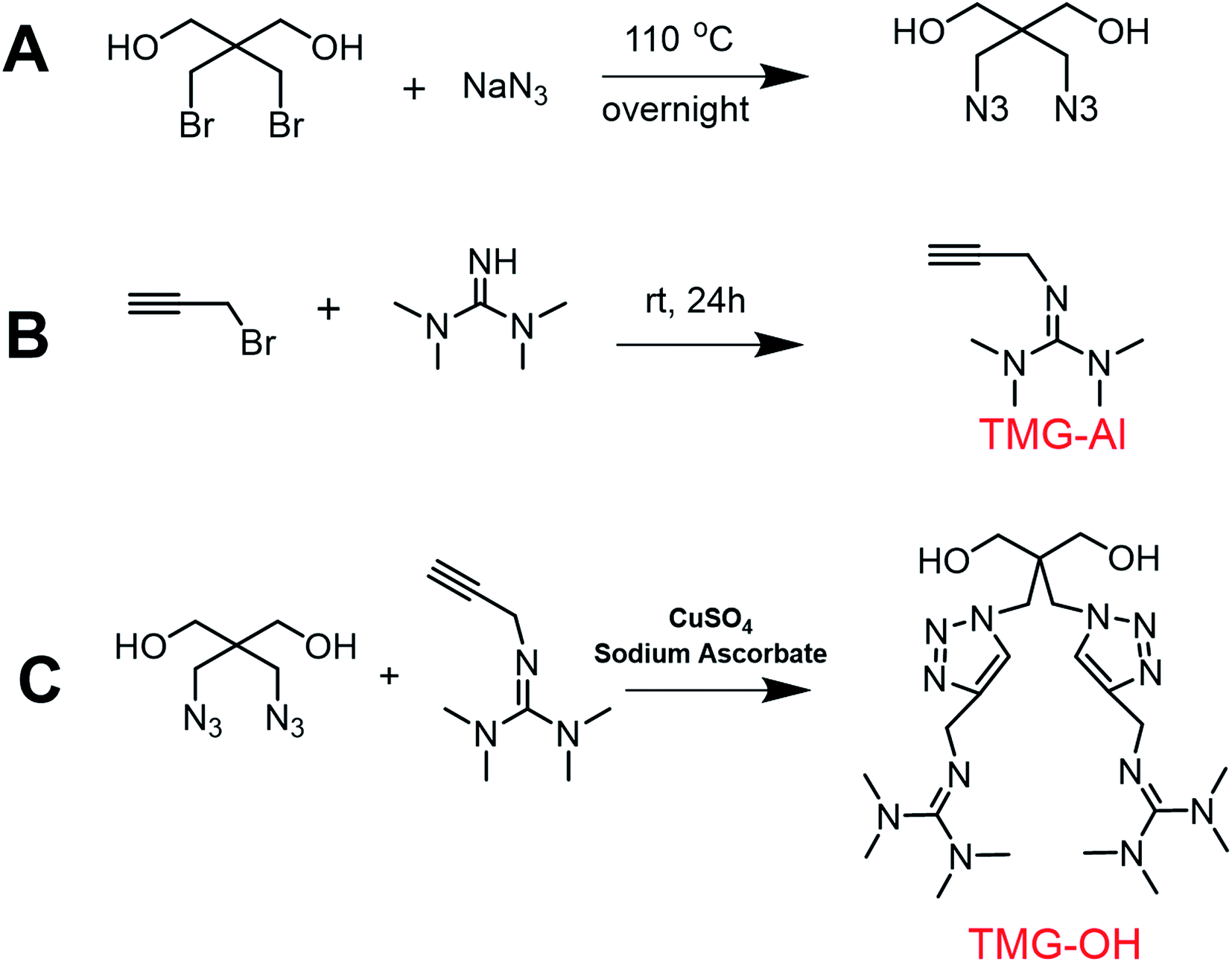 | ||
| Fig. 1 Synthetic routes for the monomers. | ||
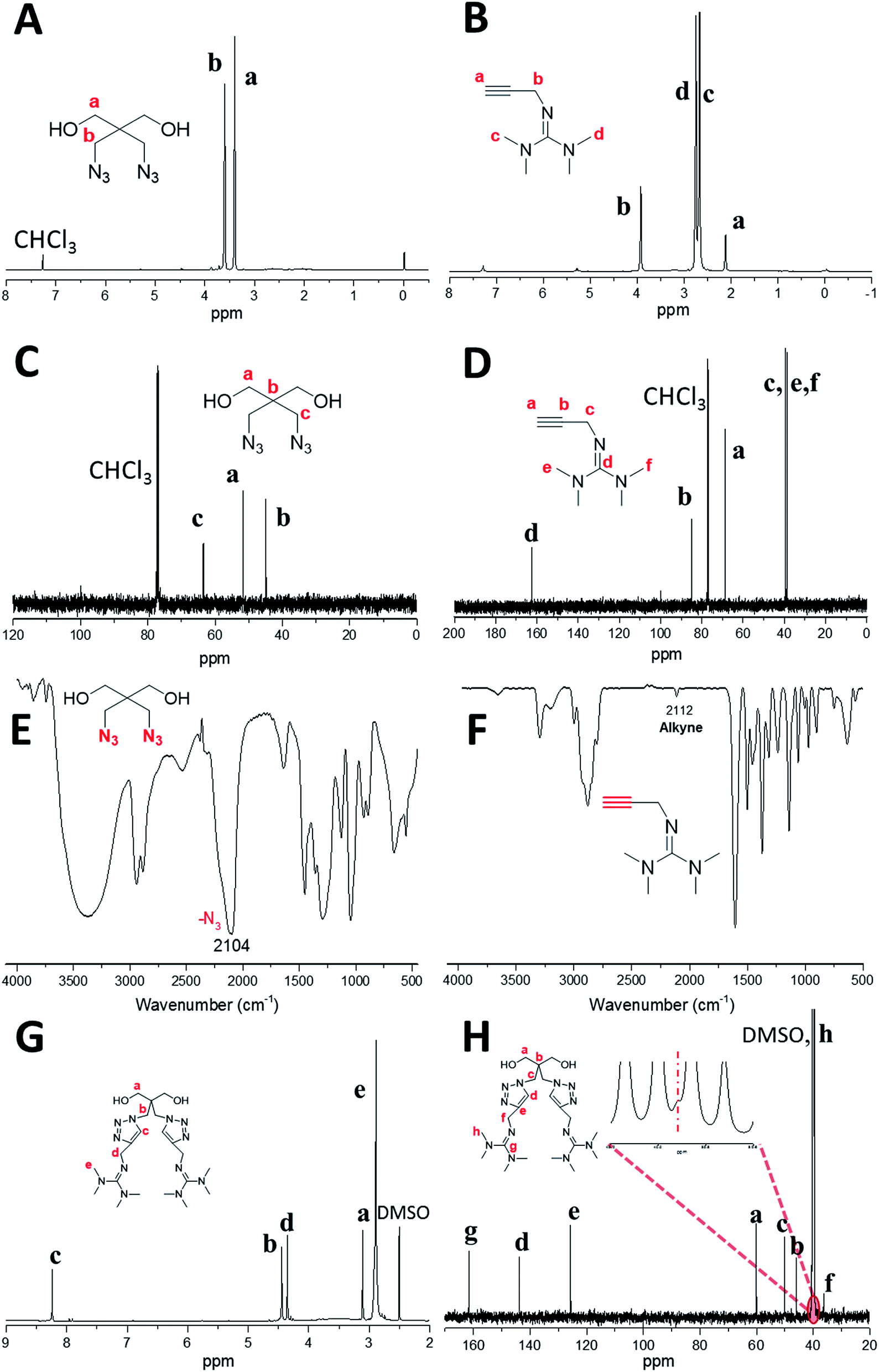 | ||
| Fig. 2 1H NMR, 13C NMR and FT-IR spectra of DAzD (A, C and E), TMG-Al (B, D and F) and 1H NMR, 13C NMR spectra of TMG-OH (G and H) in CDCl3 or DMSO-d6. | ||
Herein, we firstly introduced a facile method to fabricate click-suitable penta-substituted guanidine by substitution reaction at room temperature in one step, and tetramethyl guanidine (TMG) is both reactive raw material and acid removal agent of side reaction products (Fig. 1B). The successful synthesis of TMG-Al was confirmed by the appearance of the characteristic infrared absorption peak of alkyne group at 2112 cm−1, the 13C-NMR spectrum characteristic peak of central carbon atom in the guanidyl group (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–) at around 162 ppm and the 1H NMR peaks of protons on –CH2–C
C–) at around 162 ppm and the 1H NMR peaks of protons on –CH2–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH (δ 2.12 ppm, s), –N(CH3)2 (δ 2.67 ppm, s), –N(CH3)2 (δ 2.75 ppm, s) and –CH2–CC (δ 3.92 ppm, s) in Fig. 2B, D and F. For comparison, and to meet various application requirements, hydroxyl-functionalized tetramethyl guanidine (TMG-OH) was prepared in accordance with Fig. 1C and its chemical structural characterizations are shown in Fig. 2G and H.
CH (δ 2.12 ppm, s), –N(CH3)2 (δ 2.67 ppm, s), –N(CH3)2 (δ 2.75 ppm, s) and –CH2–CC (δ 3.92 ppm, s) in Fig. 2B, D and F. For comparison, and to meet various application requirements, hydroxyl-functionalized tetramethyl guanidine (TMG-OH) was prepared in accordance with Fig. 1C and its chemical structural characterizations are shown in Fig. 2G and H.
3.2 Polymer design and synthesis
Herein, we showed two synthetic strategies to fabricate the TMG functionalized polyurethanes. As illustrated in Fig. 3, firstly, IPDI–PPG based polyurethane prepolymer was obtained by step growth polymerization. Two synthetic strategies were then used to create guanidine-functionalized polymers. The first strategy was post-polymerization modification of azide functional polyurethanes, which was accomplished by adding an azide-functional diol (DAzD) directly.49 In contrast, the second strategy is direct polymerization of TMG functional diol that obtained by click reaction of azide diol (DAzD) and alkyne functional TMG, which is a wise choice for the water dispersible polyurethane or polyester dispersions.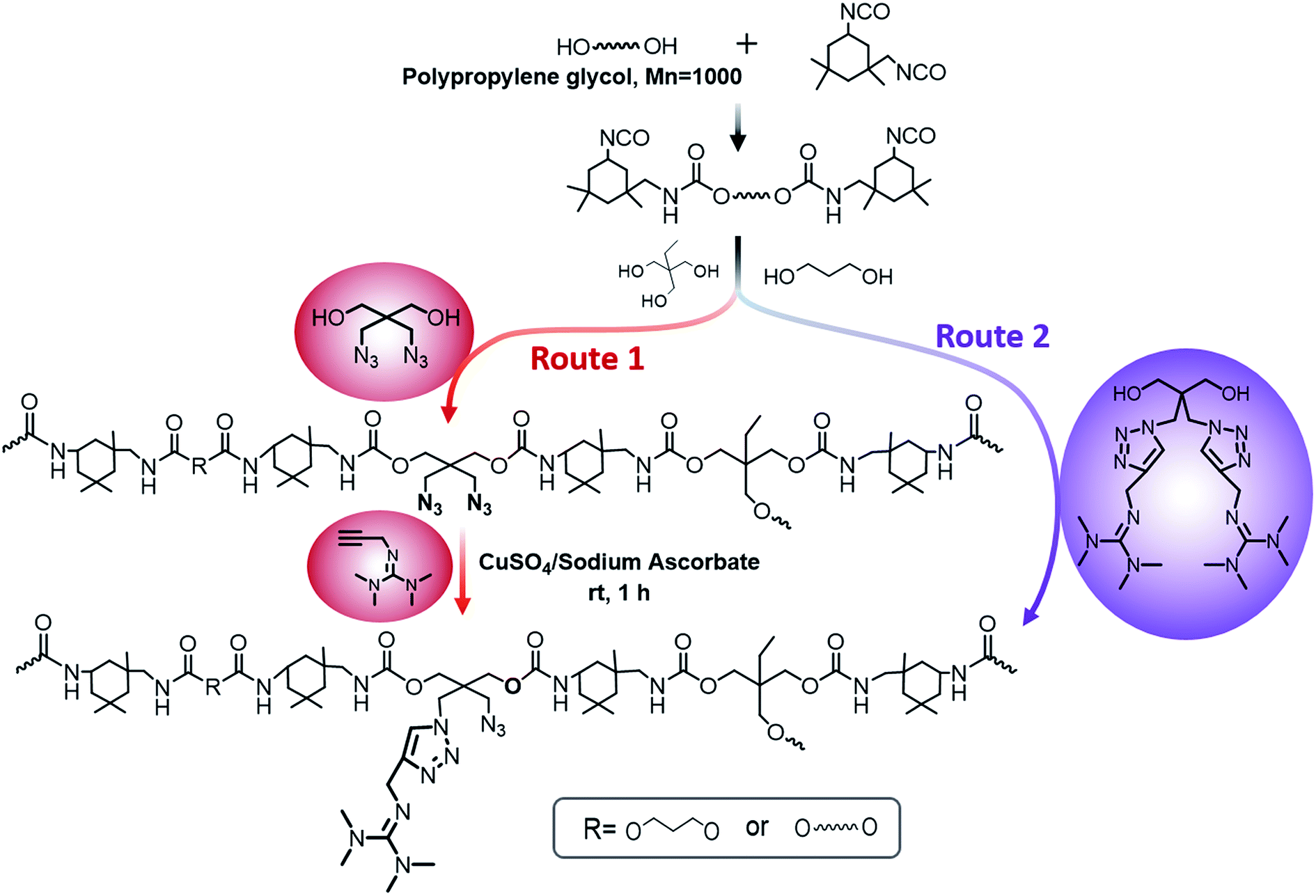 | ||
| Fig. 3 Two synthetic strategies for the polymers. | ||
The structure of the click-suitable polymer (PU-N3) and polyurethane with side-chain tetramethylgermanium (PU-TMG) was confirmed by NMR and IR spectra shown in Fig. 4 and 5. Generally, the characteristic peak of imine group –CN– is at 1675 cm−1,50 but its absorption band overlapped with that of urethane groups –NHCOO–, so no obvious peak emerged in Fig. 4 after the introduction of TMG to PU-N3. As shown in Fig. 4, the characteristic peak of azide groups on PU-N3 at 2106 cm−1 significantly decreased after “click” reaction (CuAAC) between PU-N3 and TMG-Al. Compared with pure PU, in the NMR spectra of PU-TMG polymers shown in Fig. 5, a new peaks appeared at around δ 2.88–2.81 ppm, which could be assigned to the chemical shifts of the protons on the methyl groups, –N(CH3)2, in guanidines,9 and its strength increased with the increase of content of TMG in the polymers. The PU-TMGs were subjected to GPC analysis. The number average molecular weights (Mn) and the polydispersity indexes (PDI) of the PU-TMG 2 (PU-TMG 2′) were 2.203 × 104 g mol−1 (2.115 × 104 g mol−1) and 2.50 (2.74), respectively. It showed that there had no significant difference on the number average molecular weights (Mn) and the polydispersity indexes (PDI) of PU-TMG prepared by route 1 or 2. The results of FTIR, NMR and GPC indicate that the guanidine-functionalized polyurethanes have been synthesized successfully.
 | ||
| Fig. 4 FT-IR spectra of polyurethane (PU), click-suitable polyurethane (PU–N3) and polyurethane with side-chain tetramethylgermanium (PU-TMG 4). | ||
 | ||
| Fig. 5 1H NMR spectra of polyurethane (PU) and polyurethanes with side-chain tetramethylgermanium (PU-TMG 3 and PU-TMG 4) in DMSO-d6. | ||
3.3 Thermal properties
Thermal analysis, including DSC and TGA, was used to investigate the thermal properties of PU, PU-N3 and PU-TMG. The glass transition temperatures (Tgs) for PU, PU-N3, PU-TMG 2, PU-TMG 3 and PU-TMG 4 were 28.0, 25.1, 25.8, 33.6, and 35.1 °C respectively. As shown in Fig. 6, Tg of PU-TMG gradually increased from 25.8 °C to 35.1 °C with the increasing of TMG content, which may be caused by the formation of rigid triazole rings by click reaction (CuAAC)43,44 and the introduction of rigid TMG groups. The decomposition temperature of PU-TMG polymers are around 200 °C.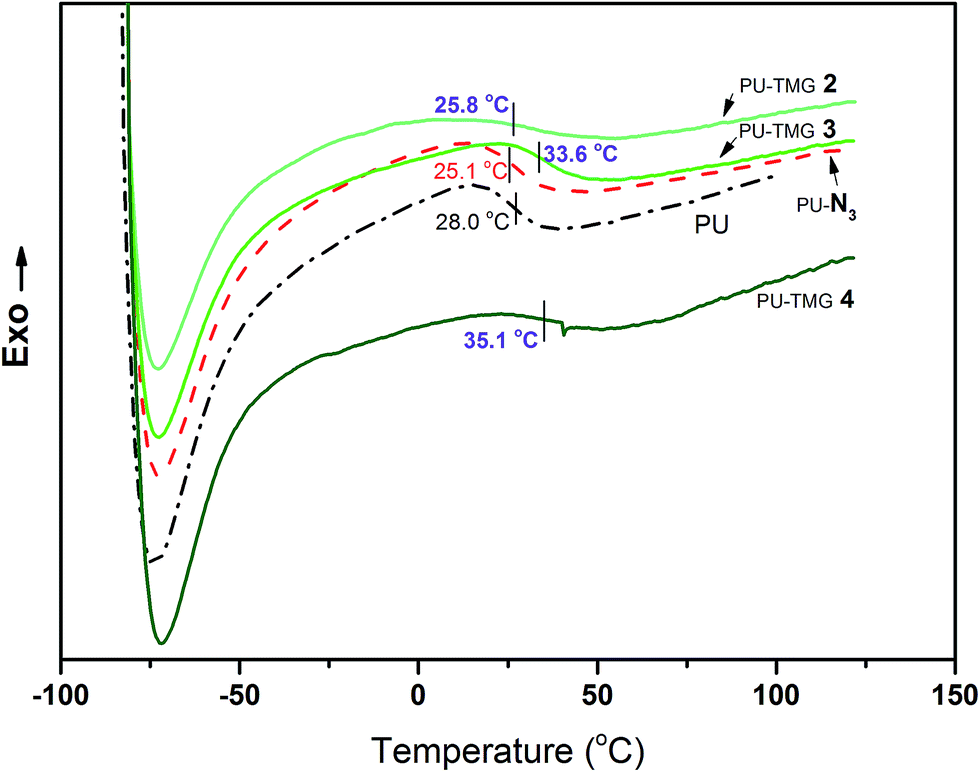 | ||
| Fig. 6 DSC curves of PU, PU-N3, PU-TMG 2 (wt% = 1.0%), PU-TMG 3 (wt% = 2.1%), and PU-TMG 4 (wt% = 5.5%). | ||
Derivative thermogravimetric analysis (DTG) were also performed, which can give important information of polymer system, such as each step of weight loss corresponding to particular degradation process. As described in references,51,52 PU decomposition occurs in two53 or three steps.54,55 In this particular work, all samples underwent a two-step thermal degradation. The first stage is the disintegration of PU polymer chains because the C–NH is the weakest bond in PU polymer and the activation energy is only about 98 kJ mol−1.51 Generally, the urethane and urea bond start to decompose and dissociate into polyol, amine, isocyanate, olefin and carbon dioxide at 200 °C. Subsequently, as shown in Fig. 7B, the degradation is split into two peaks. The first peak is located near about 325 °C for pure PU, at which the amount of weight loss is ∼56% and well correlated with the mass ration of hard segment in polyurethane (see Fig. 7A). At this point, all the urethane and urea bonds were mainly broken. The second stage was found to correlate with the degradation of soft segments. In this stage, the ether bond (C–O), whose bond energy is 326 kJ mol−1, began to break down. In addition, the rate of thermal degradation of PU-N3 and PU-TMG was markedly slower than that of pure PU at first peak, which was attributed to the substitution of BDO with DAzD and the conjugation of TMG groups. As shown in in Fig. 7A, with the converting azide group into the thermal stable triazole ring formed by CuAAC, the thermal stability of PU-TMG became better, whose decomposition temperature reach up to 450 °C.56 The result is in line with the increase of Tg of PU-TMG in comparison to PU-N3. Hence, CuAAC can not only serves as the perfect tool for the introduction of functional molecules onto polymers, but also improves the thermal stability and increases the Tg by the thermal stable and rigid triazole rings.
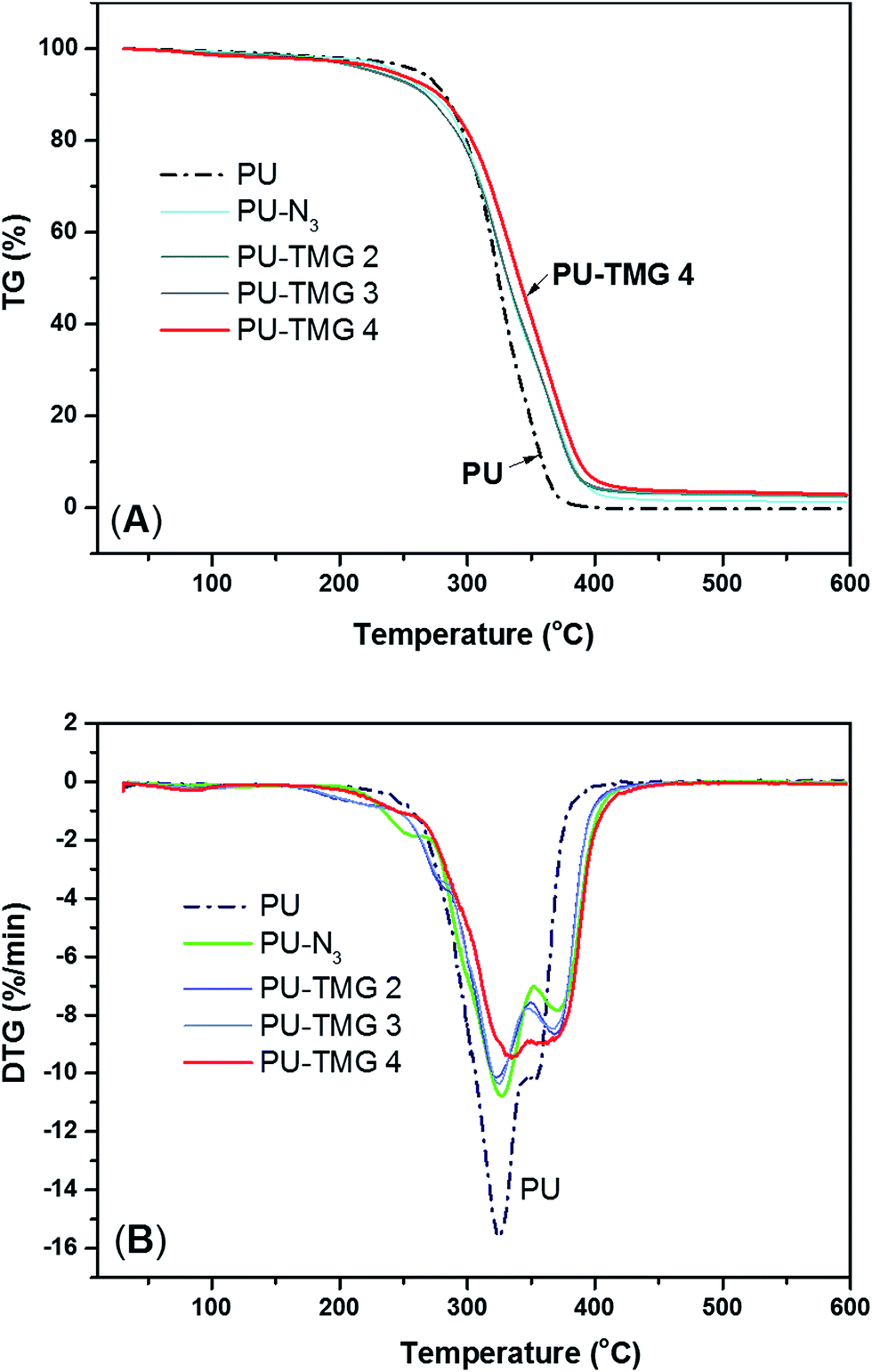 | ||
| Fig. 7 TGA (A) and DTG (B) curves of PU, PU-N3, PU-TMG 2 (wt% = 1.0%), PU-TMG 3 (wt% = 2.1%), and PU-TMG 4 (wt% = 5.5%) under nitrogen. | ||
3.4 Water-resistance
Water absorption is known to have a profound effect on films' antimicrobial durability of and mechanical properties.57 Appropriate water absorption can help hydrophilic groups, such as guanidine group, to rearrange and aggregate to the surface of the films, which is beneficial to the antimicrobial activity of the antimicrobial polymer with a contact-killing mechanism. However, high levels of water absorption can result in the detachment of film from the substrate, reduction of mechanical properties and leaching of a certain amount of antimicrobial polymer. Normally, the water absorption of quaternary ammonium or guanidine-rich polymers is strongly dependent on the density of quaternary ammonium or guanidine groups in polymer chains. The water absorption was determined by the weight percentage of water absorbed by the films when immersed into water at room temperature for 72 hours, in comparison to the dry samples weight. As shown in Fig. 8, the water absorption of PU-N3 is larger than that of pure PU because the azide group is a more hydrophilic group compared with alkyl group. As expected, the water absorption of PU-TMG increased with the rise of the content of TMG in polymer. The water absorptions of PU, PU-N3, PU-TMG 1, PU-TMG 2 were 8.3%, 12.2%, 30.5% and 45.0%, respectively. It is consistent with static contact angle of PU (72.1°), PU-N3 (66.1°), PU-TMG 1 (64.3°), and PU-TMG 2 (61.6°). Furthermore, no obvious film white phenomenon for the PU-TMG films was observed. These results showed that the water-resistance properties of PU-TMGs are acceptable.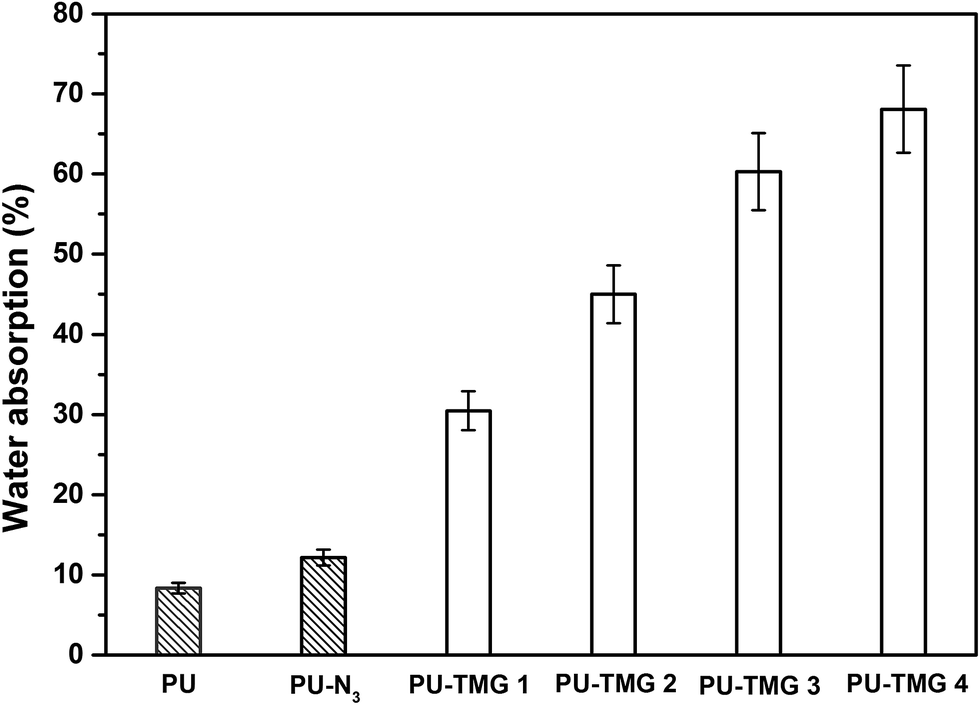 | ||
| Fig. 8 Water absorption properties of PU, PU-N3, PU-TMG 1 (wt% = 0.6%), PU-TMG 2 (wt% = 1.0%), PU-TMG 3 (wt% = 2.1%), and PU-TMG 4 (wt% = 5.5%) at room temperature for 72 hours. | ||
3.5 Antimicrobial activity
To evaluate antimicrobial properties of PU-TMG films, both Gram-positive S. aureus and Gram-negative E. coli were tested using the antibacterial “drop test” method.45,46 PU was used as control. The percentage of bacteria kill was calculated by eqn (2) as follow:
 | (2) |
Here A = CFUs after control (PU) surface contact, B = CFUs after PU-TMG surface contact.
As shown in Table 1 and Fig. 11, the PU-TMG films showed a bacteria kill rate >93% against S. aureus and >60% against E. coli. When the TMG mass content was as low as 0.6% (PU-TMG 1), there was still a percentage of bacteria kill rate >90% against S. aureus. The results demonstrated that TMG containing polymer exhibited excellent antimicrobial characteristic. As shown in Fig. 9 and 10, it can be seen that increasing TMG mass content from 0.6% to 2.1% significantly improved the bacteria contact-killing activities of PU surfaces for either strain of bacteria and the 1.0% TMG content in polymer is sufficient enough to inhibit S. aureus or E. coli. It is in accordance with the mechanism that the key determining factor in the antibacterial efficacy is the density of positive charges on the surface.28,45,58,59 From Table 1, Fig. 8–11, we also observed that PU-TMG films have better antimicrobial activity against S. aureus than against E. coli, which is in line with the results of Saif et al. on quaternary ammonium antibacterial hybrid coating.60 Until now, different mechanisms have been put forward for interpreting the action of killing bacteria28,61–63 and the differentiated antimicrobial activities toward E. coli or S. aureus may stem from the different membrane structures of Gram-positive and Gram-negative bacteria.45
| Surface | % TMG content in polymer | % Bacteria killed | |||
|---|---|---|---|---|---|
| S. aureus (Gram-positive) | E. coli (Gram-negative) | ||||
| Route 1 | Route 2 | Route 1 | Route 2 | ||
| PU | 0 | 0 | 0 | 0 | 0 |
| PU-TMG 1 (1′) | 0.6 | 96.9 ± 1 | 93.2 ± 4 | 62.7 ± 3 | 60.2 ± 4 |
| PU-TMG 2 (2′) | 1.0 | 99.9 | 97.4 ± 2 | 98.0 ± 1 | 97.2 ± 0.6 |
| PU-TMG 3 (3′) | 2.1 | 99.9 | 99.9 | 99.9 | 99.9 |
| PU-TMG 4 (4′) | 5.5 | 99.9 | 99.9 | 99.9 | 99.9 |
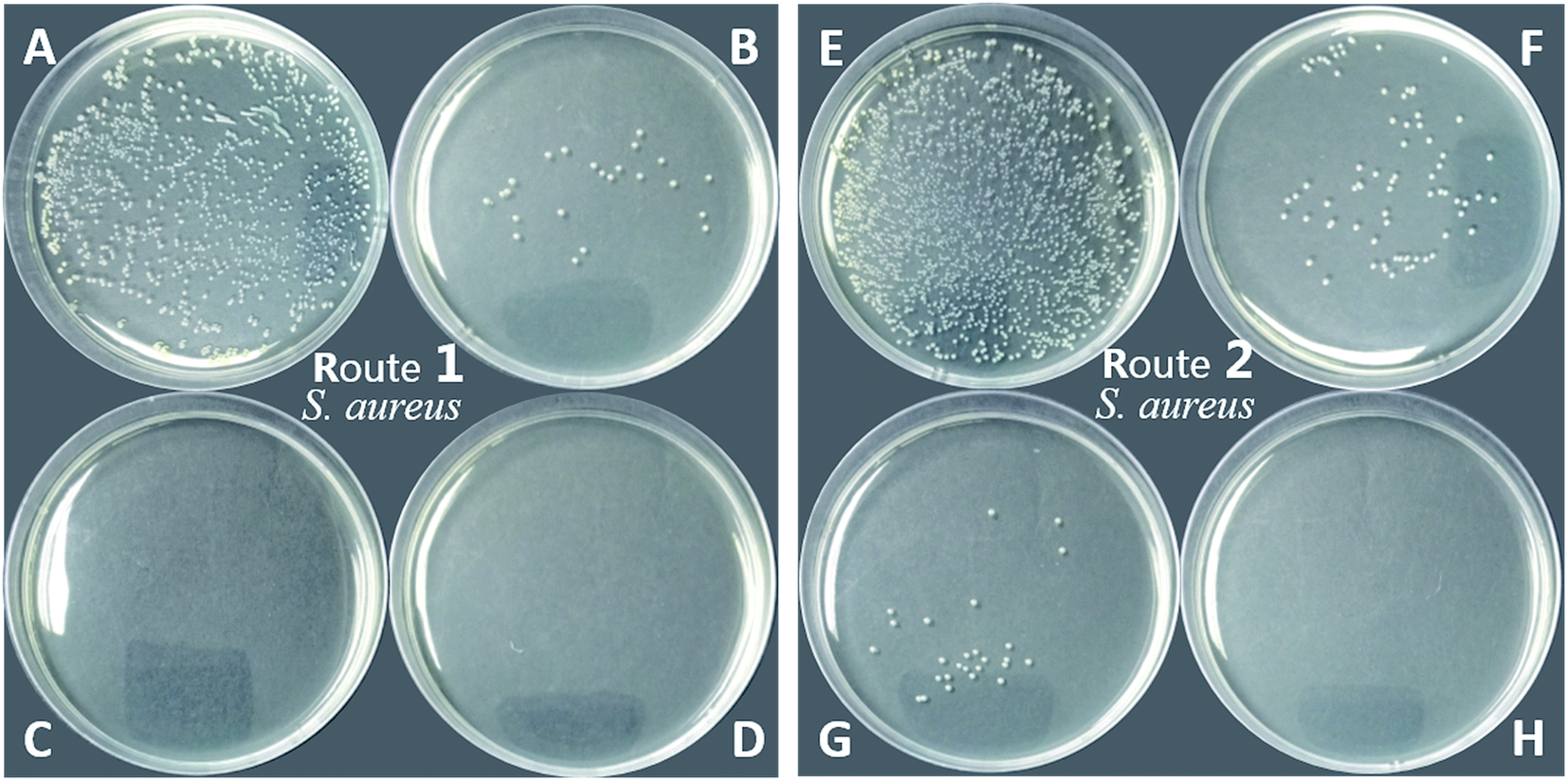 | ||
| Fig. 9 Anti-microbial activity of PU (blank, A and E), PU-TMG 1 (B), PU-TMG 2 (C), PU-TMG 3 (D) (prepared by route 1), and PU-TMG 1′ (F), PU-TMG 2′ (G), PU-TMG 3′ (H) (prepared by route 2) for Staphylococcus aureus. | ||
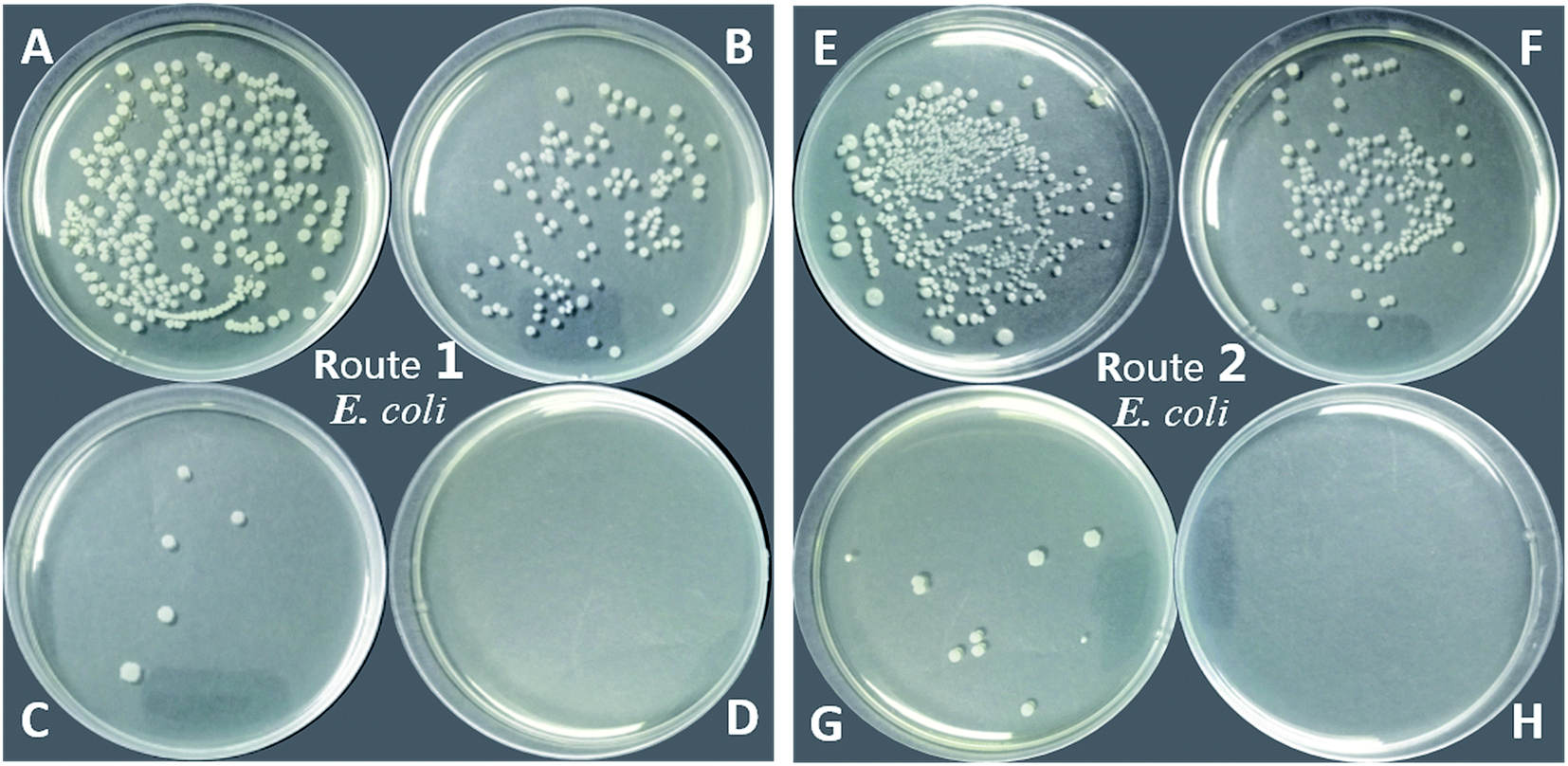 | ||
| Fig. 10 Anti-microbial activity of PU (blank, A and E), PU-TMG 1 (B), PU-TMG 2 (C), PU-TMG 3 (D) (prepared by route 1), and PU-TMG 1′ (F), PU-TMG 2′ (G), PU-TMG 3′ (H) (prepared by route 2) for Escherichia Coli. | ||
 | ||
| Fig. 11 The surviving bacteria (S. aureus or E. coli) counts on the agar after contacting with the PU, PU-TMG 1 (1′), PU-TMG 2 (2′), or PU-TMG 3 (3′). | ||
On the other hand, PU-TMG polymers synthesized through different ways, route 1 and route 2, exhibited almost the same antimicrobial activities (Fig. 9 and 10).
The disk diffusion of PU-TMG and the physical mixture of pure PU and TMG-OH films were carried out on both Gram positive and Gram negative bacterial strains. PU-TMG and physical mixed films (PU and TMG-OH) with 10 × 10 × 0.5 mm were adhered on the Luria Agar medium and the challenged bacteria was incubated at 37 °C for 8 hours. The physical mixed films showed a noticeable inhibition zone as shown in Fig. 12B and D, which means the small molecule TMG-OH can be leached from the mixed film due to non-covalently attaching between the polymer and functional small molecules. On the contrary, the surface of PU-TMG 2 sample was clean and there was no zones of inhibition around PU-TMG film in Fig. 12A and C, which demonstrates that no molecules were leached from PU-TMG film into the environment to kill the bacteria. Instead, the PU-TMG surfaces is capable of killing microorganisms on contact. This again proves that click chemistry is an excellent and practical tool to conjugate functional molecules, both before and after polymerization.
 | ||
| Fig. 12 Disk diffusion of PU-TMG 2 (A and C) and the mixed film (PU + TMG-OH, B and D) for Staphylococcus aureus and Escherichia coli. | ||
4 Conclusions
In summary, as an feasible, efficient chemical reaction that can be efficiently performed under mild conditions in different reaction media, “click” reaction (copper(I)-catalyzed alkyne-azide cycloaddition, CuAAC) was employed to introduce antimicrobial tetramethyl guanidine (TMG) onto polymers before or after polymerization. It is clear that the click-suitable penta-substituted guanidine (TMG-Al) represents an interesting and pretty important new class of molecules. Click reactions among various polymer backbones that contain azide groups and TMG-Al is an extremely efficient method to fabricate antimicrobial materials. The obtained guanidine functionalized polymers exhibited excellent antimicrobial property. Most importantly, unlike polymers mixed with antimicrobial agents/drugs, polymers with covalently bound guanidine groups are contact-killing but non-releasing antimicrobial material. The efficient, robust and orthogonal approach of click chemistry shown here will enable precise control and fine-tune of parameters such as charge-density and functional properties, fabricating a wide variety of well-defined functional macromolecules ultimately leading to a functionally versatile class of all-in-one materials.Acknowledgements
This work was supported by the Aleo BME, Inc. and the National Natural Science Foundation of China (50903031) and the International Science & Technology Cooperation Program of Guangzhou (2012J5100043).References
- O. Cars, A. Hedin and A. Heddini, Drug Resist. Updates, 2011, 14, 68–69 CrossRef PubMed.
- E. Y. Furuya and F. D. Lowy, Nat. Rev. Microbiol., 2006, 4, 36–45 CrossRef CAS PubMed.
- H. F. Chambers and F. R. DeLeo, Nat. Rev. Microbiol., 2009, 7, 629–641 CrossRef CAS PubMed.
- K. Kuroda and G. A. Caputo, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2013, 5, 49–66 CrossRef CAS PubMed.
- L. Liu, Q. Li, J. Dai, H. Wang, B. Jin and R. Bai, J. Membr. Sci., 2014, 453, 52–60 CrossRef CAS.
- A. T. Yeung, S. L. Gellatly and R. E. Hancock, Cell. Mol. Life Sci., 2011, 68, 2161–2176 CrossRef CAS PubMed.
- M. P. Coles, Chem. Commun., 2009, 3659–3676, 10.1039/b901940e.
- D. S. Kim, A. Labouriau, M. D. Guiver and Y. S. Kim, Chem. Mater., 2011, 23, 3795–3797 CrossRef CAS.
- J. Wang, S. Li and S. Zhang, Macromolecules, 2010, 43, 3890–3896 CrossRef CAS.
- K. E. S. Locock, T. D. Michl, N. Stevens, J. D. Hayball, K. Vasilev, A. Postma, H. J. Griesser, L. Meagher and M. Haeussler, ACS Macro Lett., 2014, 3, 319–323 CrossRef CAS.
- G. J. Gabriel, A. E. Madkour, J. M. Dabkowski, C. F. Nelson, K. Nusslein and G. N. Tew, Biomacromolecules, 2008, 9, 2980–2983 CrossRef CAS PubMed.
- J. Budhathoki-Uprety, L. Peng, C. Melander and B. M. Novak, ACS Macro Lett., 2012, 1, 370–374 CrossRef CAS.
- E. M. Kolonko and L. L. Kiessling, J. Am. Chem. Soc., 2008, 130, 5626–5627 CrossRef CAS PubMed.
- C. B. Cooley, B. M. Trantow, F. Nederberg, M. K. Kiesewetter, J. L. Hedrick, R. M. Waymouth and P. A. Wender, J. Am. Chem. Soc., 2009, 131, 16401–16403 CrossRef CAS PubMed.
- R. J. Ono, A. L. Z. Lee, W. Chin, W. S. Goh, A. Y. L. Lee, Y. Y. Yang and J. L. Hedrick, ACS Macro Lett., 2015, 4, 886–891 CrossRef CAS.
- E. A. Dubikovskaya, S. H. Thorne, T. H. Pillow, C. H. Contag and P. A. Wender, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12128–12133 CrossRef CAS PubMed.
- U. Schuchardt, R. M. Vargas and G. Gelbard, J. Mol. Catal. A: Chem., 1996, 109, 37–44 CrossRef CAS.
- C. Savelli and R. Salvio, Chemistry, 2015, 21, 5856–5863 CrossRef CAS PubMed.
- M. Onda, K. Yoshihara, H. Koyano, K. Ariga and T. Kunitake, J. Am. Chem. Soc., 1996, 118, 8524–8530 CrossRef CAS.
- J. B. Rothbard, E. Kreider, C. L. VanDeusen, L. Wright, B. L. Wylie and P. A. Wender, J. Med. Chem., 2002, 45, 3612–3618 CrossRef CAS PubMed.
- K. E. Locock, T. D. Michl, J. D. Valentin, K. Vasilev, J. D. Hayball, Y. Qu, A. Traven, H. J. Griesser, L. Meagher and M. Haeussler, Biomacromolecules, 2013, 14, 4021–4031 CrossRef CAS PubMed.
- H. Lv, S. Zhang, B. Wang, S. Cui and J. Yan, J. Controlled Release, 2006, 114, 100–109 CrossRef CAS PubMed.
- D. Wei, Q. Ma, Y. Guan, F. Hu, A. Zheng, X. Zhang, Z. Teng and H. Jiang, Mater. Sci. Eng., C, 2009, 29, 1776–1780 CrossRef CAS.
- M. Lebrini, F. Bentiss, N.-E. Chihib, C. Jama, J. P. Hornez and M. Lagrenée, Corros. Sci., 2008, 50, 2914–2918 CrossRef CAS.
- W. Li, H. Wang, Y. Ding, E. C. Scheithauer, O.-M. Goudouri, A. Grünewald, R. Detsch, S. Agarwal and A. R. Boccaccini, J. Mater. Chem. B, 2015, 3, 3367–3378 RSC.
- S. D. Worley and G. Sun, Trends Polym. Sci., 1996, 4, 364–370 CAS.
- W. K. Son, J. H. Youk and W. H. Park, Carbohydr. Polym., 2006, 65, 430–434 CrossRef CAS.
- J. Y. Huang, R. R. Koepsel, H. Murata, W. Wu, S. B. Lee, T. Kowalewski, A. J. Russell and K. Matyjaszewski, Langmuir, 2008, 24, 6785–6795 CrossRef CAS PubMed.
- Q. Zhang, S. Li and S. Zhang, Chem. Commun., 2010, 46, 7495–7497 RSC.
- S. Chempath, J. M. Boncella, L. R. Pratt, N. Henson and B. S. Pivovar, J. Phys. Chem. C, 2010, 114, 11977–11983 CAS.
- X. Lin, L. Wu, Y. Liu, A. L. Ong, S. D. Poynton, J. R. Varcoe and T. Xu, J. Power Sources, 2012, 217, 373–380 CrossRef CAS.
- C. H. Zhao, Y. Gong, Q. L. Liu, Q. G. Zhang and A. M. Zhu, Int. J. Hydrogen Energy, 2012, 37, 11383–11393 CrossRef CAS.
- A. I. Bochynska, T. G. Van Tienen, G. Hannink, P. Buma and D. W. Grijpma, Acta Biomater., 2016, 32, 1–9 CrossRef CAS PubMed.
- F. Scognamiglio, A. Travan, I. Rustighi, P. Tarchi, S. Palmisano, E. Marsich, M. Borgogna, I. Donati, N. de Manzini and S. Paoletti, J. Biomed. Mater. Res., Part B, 2016, 104, 626–639 CrossRef CAS PubMed.
- S. Budun, E. İşgören, R. Erdem and M. Yüksek, Appl. Surf. Sci., 2016, 380, 294–300 CrossRef CAS.
- J. O. Vinhal, C. F. Lima and R. J. Cassella, Ecotoxicol. Environ. Saf., 2016, 131, 72–78 CrossRef CAS PubMed.
- N. G. Ireni, R. Narayan, P. Basak and K. V. S. N. Raju, Polymer, 2016, 97, 370–379 CrossRef CAS.
- R. Duarah, Y. P. Singh, B. B. Mandal and N. Karak, New J. Chem., 2016, 40, 5152–5163 RSC.
- D. Filip, D. Macocinschi, E. Paslaru, C. G. Tuchilus and S. Vlad, React. Funct. Polym., 2016, 102, 70–81 CrossRef CAS.
- M. Sahraro, H. Yeganeh and M. Sorayya, Mater. Sci. Eng., C, 2016, 59, 1025–1037 CrossRef CAS PubMed.
- J. M. Spruell, M. Wolffs, F. A. Leibfarth, B. C. Stahl, J. Heo, L. A. Connal, J. Hu and C. J. Hawker, J. Am. Chem. Soc., 2011, 133, 16698–16706 CrossRef CAS PubMed.
- A. Saha, S. De, M. C. Stuparu and A. Khan, J. Am. Chem. Soc., 2012, 134, 17291–17297 CrossRef CAS PubMed.
- J. Guo, Z. Xie, R. T. Tran, D. Xie, D. Jin, X. Bai and J. Yang, Adv. Mater., 2014, 26, 1906–1911 CrossRef CAS PubMed.
- J. Hu, K. Peng, J. Guo, D. Shan, G. B. Kim, Q. Li, E. Gerhard, L. Zhu, W. Tu, W. Lv, M. A. Hickner and J. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 17499–17510 CAS.
- S. Karamdoust, B. Yu, C. V. Bonduelle, Y. Liu, G. Davidson, G. Stojcevic, J. Yang, W. M. Lau and E. R. Gillies, J. Mater. Chem., 2012, 22, 4881 RSC.
- J. J. Lin, W. C. Lin, S. D. Li, C. Y. Lin and S. H. Hsu, ACS Appl. Mater. Interfaces, 2013, 5, 433–443 CAS.
- P. Wang, S. M. Zakeeruddin, M. Grätzel, W. Kantlehner, J. Mezger, E. V. Stoyanov and O. Scherr, Appl. Phys. A, 2004, 79, 73–77 CrossRef CAS.
- H. Xie, S. Zhang and H. Duan, Tetrahedron Lett., 2004, 45, 2013–2015 CrossRef CAS.
- M. A. Gauthier, M. I. Gibson and H. A. Klok, Angew. Chem., Int. Ed. Engl., 2009, 48, 48–58 CrossRef CAS PubMed.
- J. Cheng, G. Yang, K. Zhang, G. He, J. Jia, H. Yu, F. Gai, L. Li, C. Hao and F. Zhang, J. Membr. Sci., 2016, 501, 100–108 CrossRef CAS.
- D. K. Chattopadhyay and D. C. Webster, Prog. Polym. Sci., 2009, 34, 1068–1133 CrossRef CAS.
- H. Sui, X. Ju, X. Liu, K. Cheng, Y. Luo and F. Zhong, Polym. Degrad. Stab., 2014, 101, 109–113 CrossRef CAS.
- K. Pielichowska, J. Bieda and P. Szatkowski, Renewable Energy, 2016, 91, 456–465 CrossRef CAS.
- L. Shufen, J. Zhi, Y. Kaijun, Y. Shuqin and W. K. Chow, Polym.-Plast. Technol. Eng., 2006, 45, 95–108 CrossRef.
- Z. S. Petrovic, Z. Zavarco, J. H. Flynn and W. J. Macknight, J. Appl. Polym. Sci., 1994, 51, 1087–1095 CrossRef CAS.
- K. G. Priyanka, A. K. Mishra, S. Kantheti, R. Narayan and K. V. S. N. Raju, J. Appl. Polym. Sci., 2012, 126, 2024–2034 CrossRef CAS.
- G. H. Zhao, Y. Liu, C. L. Fang, M. Zhang, C. Q. Zhou and Z. D. Chen, Polym. Degrad. Stab., 2006, 91, 703–711 CrossRef CAS.
- J. Y. Huang, H. Murata, R. R. Koepsel, A. J. Russell and K. Matyjaszewski, Biomacromolecules, 2007, 8, 1396–1399 CrossRef CAS PubMed.
- H. Murata, R. R. Koepsel, K. Matyjaszewski and A. J. Russell, Biomaterials, 2007, 28, 4870–4879 CrossRef CAS PubMed.
- M. J. Saif, J. Anwar and M. A. Munawar, Langmuir, 2009, 25, 377–379 CrossRef CAS PubMed.
- R. Kugler, O. Bouloussa and F. Rondelez, Microbiology, 2005, 151, 1341–1348 CrossRef CAS PubMed.
- A. M. Bieser and J. C. Tiller, Macromol. Biosci., 2011, 11, 526–534 CrossRef CAS PubMed.
- K. Lewis and A. M. Klibanov, Trends Biotechnol., 2005, 23, 343–348 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |