
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical approaches to inhibitors of isoprenoid biosynthesis: targeting farnesyl and geranylgeranyl pyrophosphate synthases
Pedro Merino
 *a,
Loredana Maiuolo
*b,
Ignacio Delso
ac,
Vincenzo Algieri
b,
Antonio De Nino
b and
Tomas Tejero
a
*a,
Loredana Maiuolo
*b,
Ignacio Delso
ac,
Vincenzo Algieri
b,
Antonio De Nino
b and
Tomas Tejero
a
aDepartamento de Síntesis y Estructura de Biomoléculas, ISQCH, Universidad de Zaragoza-CSIC, 50009 Zaragoza, Aragón, Spain. E-mail: pmerino@unizar.es
bDipartimento di Chimica, Università della Calabria, 87036 Rende, Italy
cServicio de Resonancia Magnética Nuclear, CEQMA, Universidad de Zaragoza-CSIC, 50009 Zaragoza, Aragón, Spain
First published on 10th February 2017
Abstract
Post-translational lipid modifications farnesylation and geranylgeranylation of proteins (protein prenylation) have been identified to mediate critical events in cancer, cardiovascular disorders, malaria and bone disorders like osteoporosis. To date eight compounds are commercialized for the treatment of bone disorders, and there are considerable efforts to develop selective small molecules that inhibit protein prenylation. This review summarizes the approaches currently employed to synthesize new inhibitors of isoprenoid biosynthesis. Bisphosphonates are mainly prepared through reaction of carboxylic acids with phosphorus reagents, Michael addition to tetraethylvinylidenebisphosphonate and alkylation of tetralkylmethyl bisphosphonate. Approaches to non-bisphosphonate derivatives include a variety of methodologies depending on the structure of the target compound.
 Pedro Merino | Pedro Merino (b. Zaragoza, Spain) received his M.Sc. degree in Organic Chemistry (1986) at the University of Zaragoza. After Ph.D. studies (1989) he moved to Ferrara (Italy) as a post-doctoral associate with Professor Alessandro Dondoni (1989–1992). In 1992 he joined the University of Zaragoza as Assistant Professor. In 1993 he was promoted to Associate Professor and Senior Lecturer in 1994. In 2005 he won national habilitation as full professor in Organic Chemistry. In 2006 he won a Chair in Organic Chemistry at the University of Zaragoza. His research interests include chemical biology, organocatalysis and computational chemistry. |
 Loredana Maiuolo | Loredana Maiuolo (b. Cosenza, Italy) graduated in Chemistry at the University of Calabria (1995) where received her Ph.D (1999). She carried out two post-doctoral positions in Organic Chemistry at the University of Calabria (1999–2004). In 2005 she won a permanent position as Researcher in Organic Chemistry at University of Calabria. In 2014 she spent six months at University of Zaragoza as visiting professor. Her main fields of research include: Asymmetric Synthesis of Biologically Active Molecules by 1,3-Dipolar Cycloaddition; Synthesis of Organic Compounds in non-conventional Solvents; Lewis Acids Catalysts and Organocatalysis; Synthesis of Deuterated Compounds as Standard in Food Chemistry. |
 Ignacio Delso | Ignacio Delso (b. Bilbao, Spain) coursed Chemistry at the University of Zaragoza (2003) and in 2009 he won his Ph.D. In 2004 he spent six months at the University of Florence (Prof. Andrea Goti). In 2008 he carried out a second pre-doctoral stay (three months) at the IGQO, CSIC in Madrid, Spain (Dr Agatha Bastida). In 2008 he won a permanent position as Specialized Research Technician of the CSIC and since then he is the responsible of the NMR Service at CEQMA. His main research interest include: Asymmetric Synthesis. Chemical Biology. NMR and Computational Biochemistry. |
 Vincenzo Algieri | Vincenzo Algieri (b. Acri (CS), Italy) graduated in Chemistry (2014) at the University of Calabria. Currently, he is PhD student in the University of Calabria under the supervision of Prof. Antonio De Nino. He spent a year of his doctorate (2016) at University of Zaragoza in research group of prof. Pedro Merino. His main research interests are: Synthesis and Characterization of Heterocyclic Compounds at Potential Biological Activity, Enantioselective Organocatalysis and Solvent Free 1,3-Dipolar Cycloadditions. |
 Antonio De Nino | Antonio De Nino (b. Capistrano (VV), Italy) received the degree in Chemistry at University of Calabria (1989). From 1993 to 2003, he was researcher in Organic Chemistry at the Chemistry Department of University of Calabria. Since 2003 he is Associate Professor of Organic Chemistry at the same University. His research activities are mainly devoted to study: Asymmetric Synthesis of Biologically Active Molecules by 1,3-Dipolar Cycloaddition; Synthesis of Organic Compounds in non-conventional Solvents; Lewis Acids Catalysts and Organocatalysis; Determination and Characterization of Microcomponents in Food and Natural Products; Reactivity of Enaminones Dianions with Electrophiles. |
 Tomas Tejero | Tomas Tejero (b. Zaragoza, Spain) coursed Chemistry at the University of Zaragoza (1980) where he received his Ph.D. (1985). In 1984 he became Assistant Professor and in 1985 he spent a year in the University Pierre et Marie Curie (Paris) under the supervision of Prof. J. Normant. In 1986 he returned to Zaragoza and in 1987 he was promoted to Senior Lecturer. In 2012 he was appointed Full Professor in the Department of Organic Chemistry at the University of Zaragoza. He is particularly interested in enantioselective processes and in new spectroscopic techniques to the field of the Asymmetric Synthesis. |
Introduction
Isoprenoids (also known as terpenoids) are considered the most ancient and diverse class of natural products.1 They have been found in sediments from 2.5 billion years ago2 and more than 40.000 representatives have been found in all kingdoms of life.3 They participate in a great variety of basic biological functions in plants4,5 (e.g. growth regulation, pigments) and mammals6–8 (e.g. steroids metabolism, cellular signaling, antioxidants), and have been used in the food, pharmaceutical, chemical and biofuel industries.9,10Isoprenoids are biosynthesized ubiquitously in eubacteria, archaebacteria and eukaryotes by the consecutive condensation of the five-carbon monomer isopentenyl diphosphate (IPP) to its isomer dimethylallyl pyrophosphate (DMAPP) (Fig. 1).11,12 Whereas in mammals and yeast IPP is synthesized in the cytosol and endoplasmic reticulum from acetyl-CoA through mevalonic acid (mevalonate pathway), in higher plants and other microorganisms IPP is synthesized in the plastids by the condensation of pyruvate with glyceraldehyde-3-phosphate through 1-deoxyxylulose-5-phosphate (DXP) – also called methylerythritol (MEP pathway). Two consecutive condensations of IPP and DMAPP catalyzed by farnesyl pyrophosphate synthase (FPPS) provide geranyl diphosphate (GPP) and farnesyl diphosphate (FPP). The former is the precursor of monoterpenes and the latter of sesquiterpenes, triterpenes and sterols (via squalene biosynthesis) as well as other important secondary metabolites like ubiquinones and dolichols. An additional condensation of FPP with IPP, catalyzed by the enzyme geranylgeranyl pyrophosphate synthase (GGPPS), furnishes geranylgeranyl pyrophosphate (GGPP) precursor of di- and tetraterpenes and carotenoids (Fig. 1).
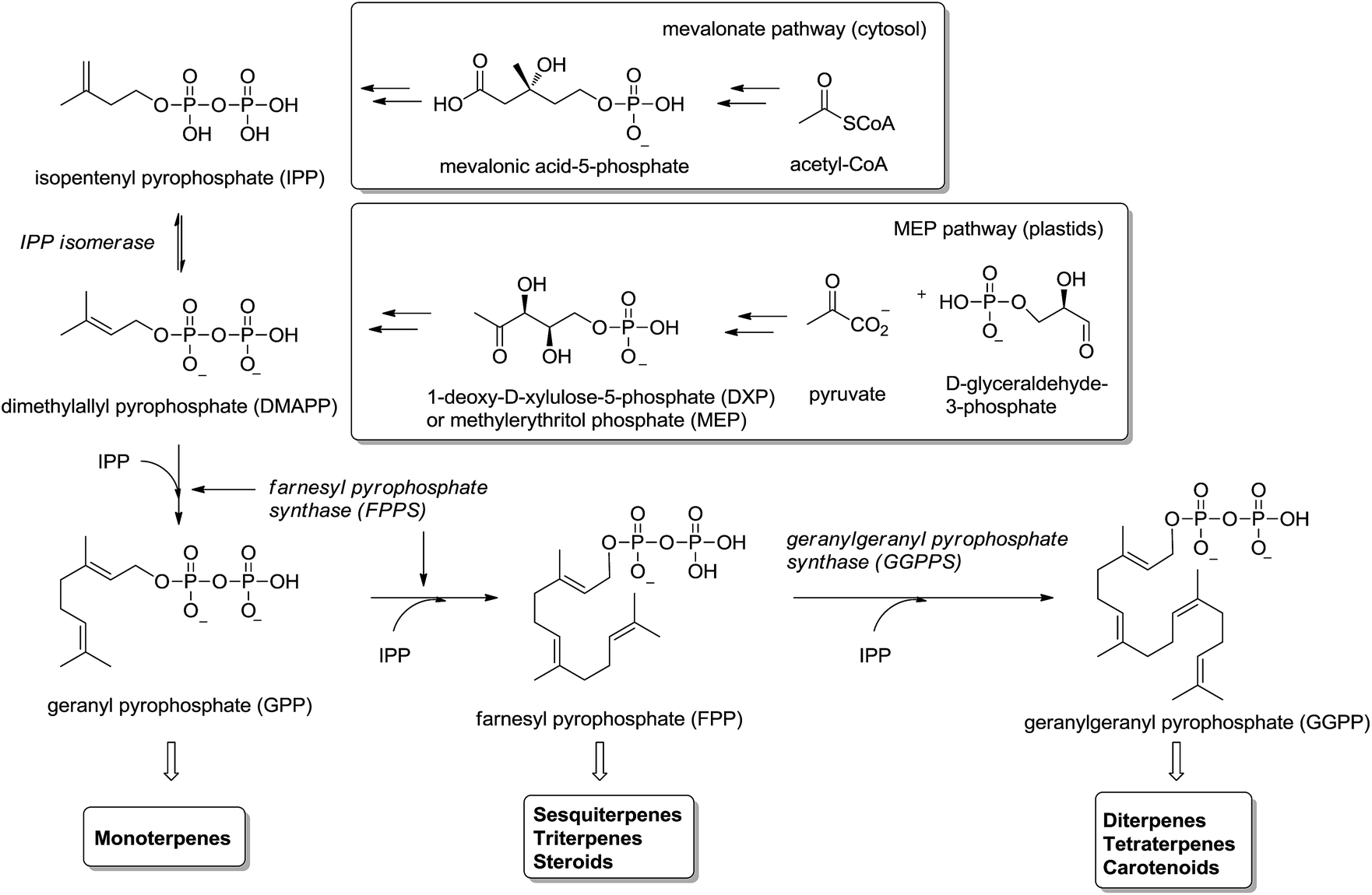 | ||
| Fig. 1 Biosynthesis of isoprenoids. FPPS and GGPPS are essential enzymes. | ||
Given the importance of the metabolites accessible from isoprenoid biosynthesis, the enzymes involved in the process are excellent drug targets.13,14 The non-mevalonate pathway (MEP pathway) is not present in mammalian systems; consequently the enzymes involved in MEP pathway are attractive drug targets15 for the development of herbicides, antimicrobial drugs and fighting against pathogenic microorganisms like P. falciparum (malaria),16,17 T. cruzi (Chagas disease)18,19 and M. tuberculosis.20 Enzymes in the MEP pathway, IspG and IspH, are anti-infective drug targets21 and HMG-CoA reductase involved in the synthesis of IPP is the primary target of hypocholesterolemic drug therapy.22
Protein prenylation, in particular farnesylation and geranylgeranylation, is one of the essential post-translational protein modification in the eukaryote.23 Therefore inhibition and/or modulation of the enzymes FPPS and GGPPS will affect not only to essential secondary metabolites derived from isoprenoid biosynthesis24 but also to the functionality of prenylated proteins.25 FPPS has been identified as a target for a series of drugs acting as anticancer, antimicrobial and antiparasitic agents.26 In particular, human FPPS is a drug target for cancer,27,28 osteoporosis and related diseases,29–31 Paget's disease,32,33 metastatic diseases,34 and cardiovascular disorders.35 Inhibition of human GGPPS has been reported as a new route to bone antiresorption36 and the same enzyme from Plasmodium has been identified as a target against malaria.37 The multiple sequence alignment of FPPS and GGPPS found in different organisms38,39 makes that the same potential inhibitor should be tested against both enzymes from diverse biological sources. The main group of inhibitors is constituted by bisphosphonates (BPs),40 structural analogues of DMAPP considered chemically stable analogues of inorganic pyrophosphate. Bisphosphonates, known from more than 40 years41 are being used clinically in the treatment of osteoporosis and malignant bone diseases (Fig. 2).42
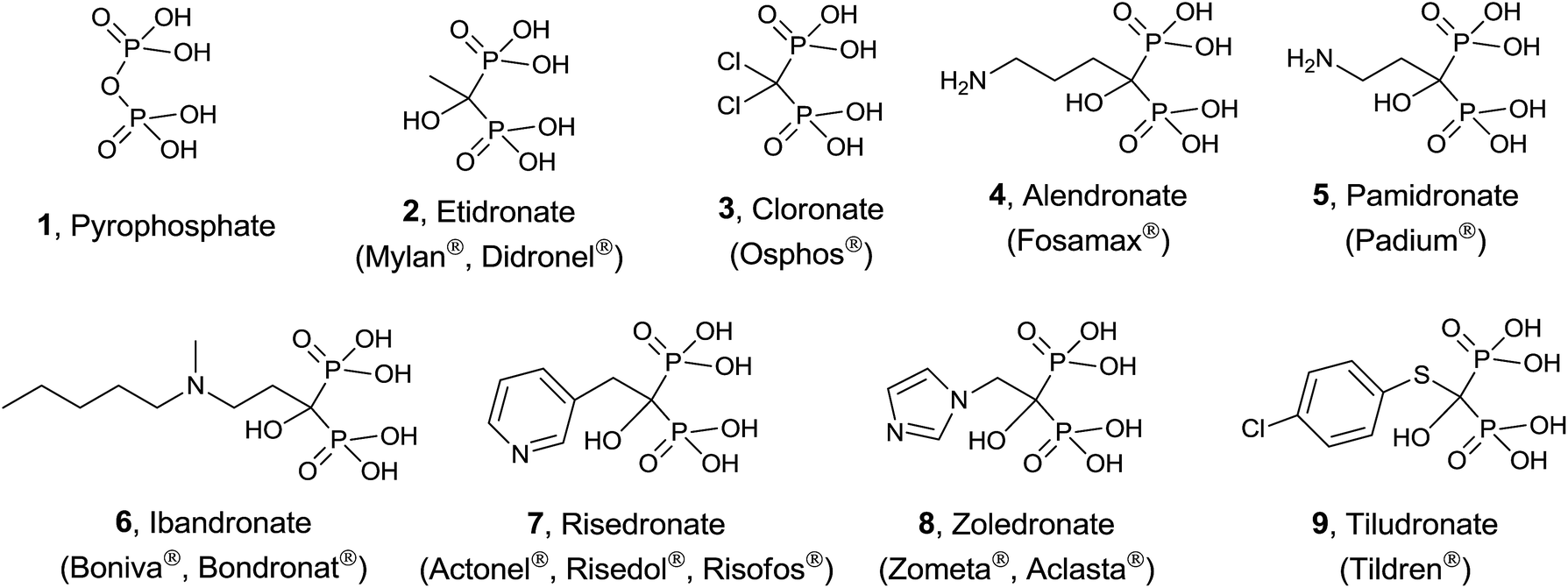 | ||
| Fig. 2 FDA-approved bisphosphonates clinically used for the treatment of bone disorders. Compounds are presented in their protonated forms. | ||
Simple bisphosphonates correspond to analogues of pyrophosphate 1 with therapeutic properties in which the bridging oxygen atom has been replaced by a methylene group that can incorporate non-nitrogenated substituents. Typical examples are etidronate 2 and cloronate 3 and their mechanism of action consists of being incorporated to non-hydrolyzable analogues of ATP, inducing osteoclast apoptosis.43
On the other hand, nitrogen-containing bisphosphonates (e.g. alendronate 4, pamidronate 5, ibandronate 6, risedronate 7, zoledronate 8) showed to be more than 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times more active than non-nitrogenated derivatives. These analogues have a diverse mechanism of action causing (i) disruption of normal function of essential signaling proteins44 and (ii) accumulation of IPP which is incorporated into a toxic nucleotide metabolite.45 More recently, a variety of non-nitrogenated bisphosphonates like tiludronate 9 and others bearing arylsulfonium and phosphonium groups showed cytotoxicity against cancer, inhibition of TpFPPS and stimulation of T-cells in the human immune system revealing that the presence of a nitrogen atom is not strictly necessary. In addition to bisphosphonates, other inhibitors including quinoline and salicylic acid derivatives, have been reported. These non-bisphosphonate inhibitors bind to an allosteric site on FPPS identified by X-ray crystallography.46
000 times more active than non-nitrogenated derivatives. These analogues have a diverse mechanism of action causing (i) disruption of normal function of essential signaling proteins44 and (ii) accumulation of IPP which is incorporated into a toxic nucleotide metabolite.45 More recently, a variety of non-nitrogenated bisphosphonates like tiludronate 9 and others bearing arylsulfonium and phosphonium groups showed cytotoxicity against cancer, inhibition of TpFPPS and stimulation of T-cells in the human immune system revealing that the presence of a nitrogen atom is not strictly necessary. In addition to bisphosphonates, other inhibitors including quinoline and salicylic acid derivatives, have been reported. These non-bisphosphonate inhibitors bind to an allosteric site on FPPS identified by X-ray crystallography.46
The goal of this review is to highlight synthetic methodologies directed to the preparation of FPPS and GPPS inhibitors. Several reviews have been reported elsewhere on the design of inhibitors of isoprenoid synthase enzymes13,14,47 and their mechanism of action.45,48–51 The reader is directed to consult those reviews for details on therapeutic effects. Since the main focus of this survey is discussion on chemical approaches, references to biological activities of the inhibitors is only shortly reviewed here. No patents are considered in this review since they have been recently surveyed.52,53 The review is organized on the basis of the structure of the inhibitor, i.e. bisphosphonates and non-bisphosphonates and then by the synthetic methodologies employed for their synthesis.
Bisphosphonates
Reaction of carboxylic acids with phosphorous reagents
The most general and attractive approach for preparing 1,1-bisphosphonates is the reaction of a carboxylic acid, easily accessible by conventional methods, with an inexpensive phosphorous reagent like phosphorous trichloride. Due to high therapeutic interest of 1,1-bisphosphonates, the first reports regarding their synthesis were published as patents and no clear experimental details were given, the reaction being difficult to scale up and lacking of reproducibility. Indeed, no mechanism of the reaction had been determined and a great variability was observed in reactants ratio, temperature, reaction time and work-up. In 1995, Kieczykowski and Jobson, from Merck and Co., Inc., reported a general procedure for reacting a carboxylic acid with phosphorous acid and phosphorous trichloride in the presence of methanesulfonic acid (Scheme 1).54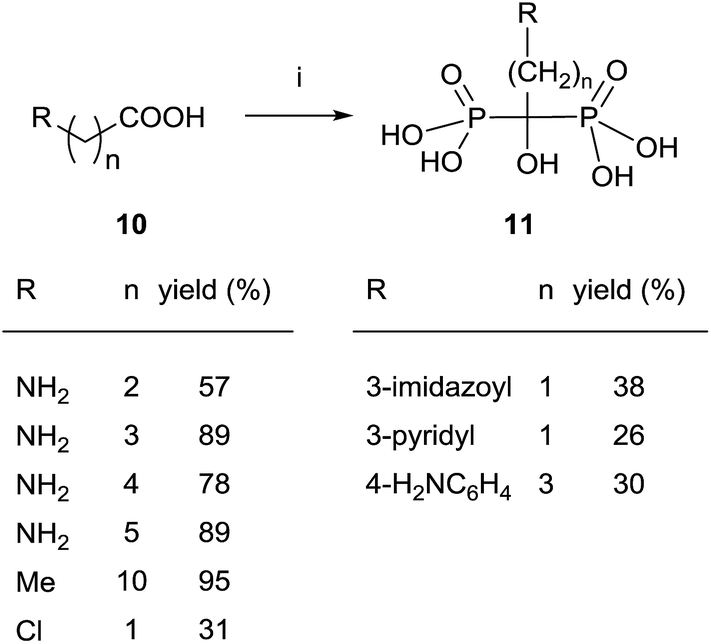 | ||
| Scheme 1 Reagents and conditions: (i) H3PO3 (1 equiv.), MeSO3H (1 equiv.), PCl3 (2 equiv.), 65 °C, 16–20 h; then H2O, reflux; then pH = 4, 50% NaOH. | ||
Since then, this method has been the most widely used for the synthesis of 1-hydroxy-1,1-bisphosphonates although in some particular case it has been reported contamination of the product with the methanesulfonate salt, that can be avoided by performing the reaction without solvent.55 The procedure is amenable of being used with several substrates including those bearing an amino functionality.36,56 For instance, alendronate 4 and pamidronate 5, have been prepared by this route57 and served as precursors of substituted analogues like compounds 13 (Scheme 2).58,59 Similarly, conjugates 14 have been prepared by condensation of 5 with the corresponding carboxylic acid.60 Other conjugates include incorporation to nucleosides61 and oligonucleotides.62 Pamidronate 5 prepared by this route has been further employed for the synthesis of 211At-labeled amidobisphosphonates.63
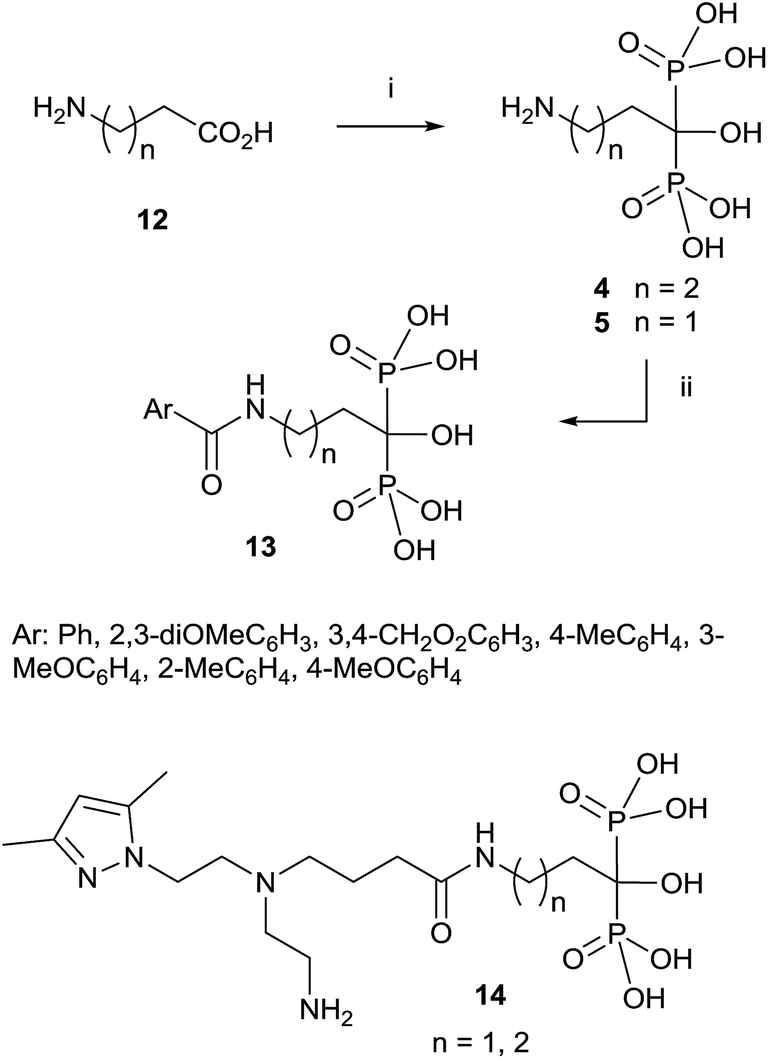 | ||
| Scheme 2 Reagents and conditions: (i) H3PO3, PCl3. (ii) ArCOCl, NaOH. | ||
Alendronate 4 can also be obtained in a straightforward manner from pyrrolidone (Scheme 3).64 Hydrolysis of 15 in aqueous methanesulfonic acid followed by reaction with phosphorous trichloride and hydrolysis with water furnished compound 4.
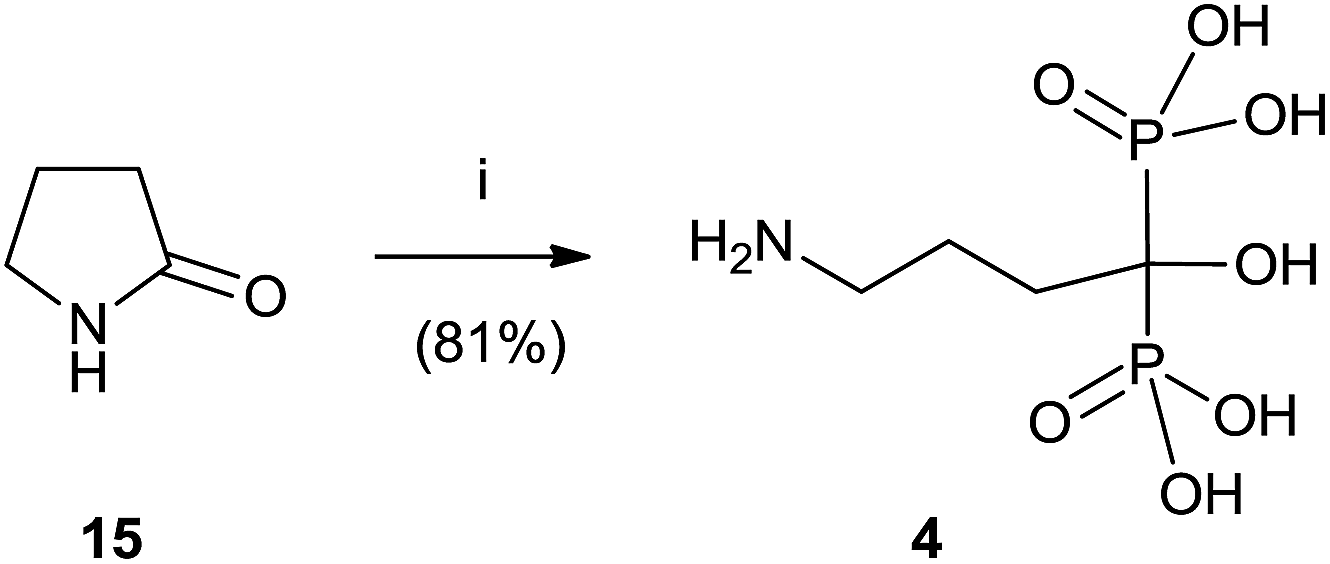 | ||
| Scheme 3 Reagents and conditions: (i) H3PO3, PCl3. (ii) ArCOCl, NaOH. | ||
Good yields in the synthesis of heterocyclic (risedronate 7, zoledronate 8) and aminoalkyl bisphosphonates were obtained in just 20 min using microwaves and sulpholene as a solvent in the first stage of the reaction (Scheme 4).65 A chiral analogue of risedronate 7 was prepared starting from 3-benzyloxybenzaldehyde 18 which was transformed into carboxylic acid 22. Reaction of 22 with phosphorous acid and phosphorous chloride afforded analogue 23 (Scheme 5).66 The complex formed from HsFPPS and racemic 23 revealed that only R-isomer was present in the active site revealing a high enantiospecificity of the enzyme.
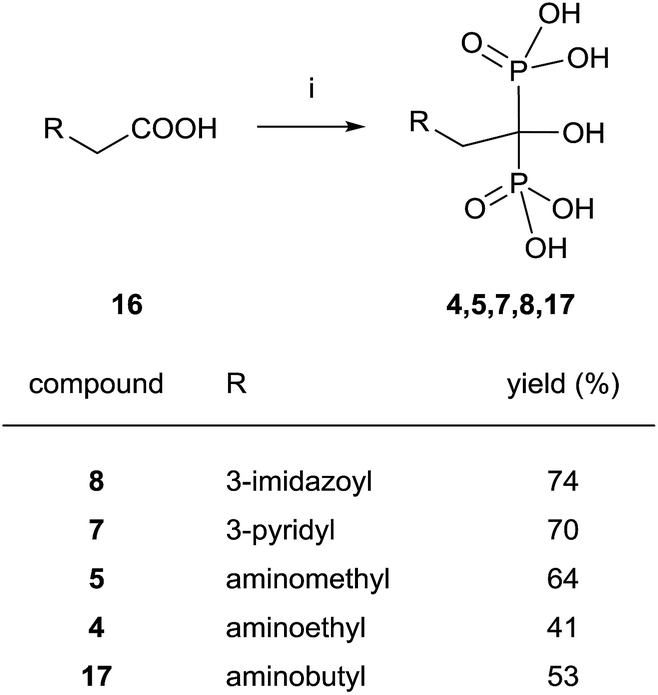 | ||
| Scheme 4 Reagents and conditions: (i) H3PO3 (3 equiv.), PCl3 (4 equiv.), sulpholene, 65 °C, 3–7 min, MWI 200–400 W max.; then H2O, MWI, 450–500 W max., 10 min, 150 °C. | ||
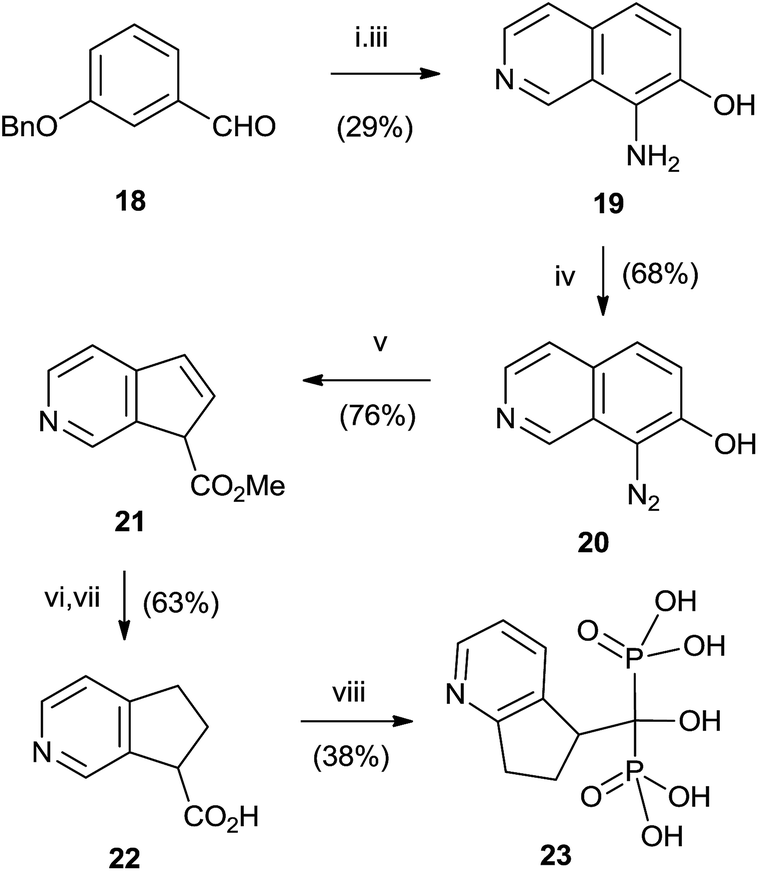 | ||
| Scheme 5 Reagents and conditions: (i) aminoacetaldehyde dimethylacetal, toluene, 6 h, 110 °C; then TFA, BF3·Et2O, <10 °C, 4 days; then Et2O, NH4OH to pH 9. (ii) Sulfolane, nitronium tetrafluoroborate, rt, 6 h. (iii) 10% Pd/C, H2, EtOH. (iv) MeOH·HCl, t-BuONO 0 °C to rt, 4 h. (v) NaHCO3, MeOH, hv, 0 °C, 3 h. (vi) 10% Pd/C, MeOH, rt, 5 h. (vii) NaOH, 58 °C, 4 h. (viii) H3PO3, PCl3, toluene, 110 °C, 4 h; then HCl, 100 °C, overnight. | ||
Oldfield and co-workers have demonstrated that the methodology originally reported by Kieczykowski and Jobson54 allows preparing a huge number of different compounds, including aromatic, aliphatic, sulfur-containing and nitrogen-containing heterocyclic derivatives.67–71 The exact protocol involved a hydrolysis, after the reaction of the carboxylic acid with phosphorous acid and phosphorous trichloride, and pH adjustment to 4.3 with a 50% NaOH solution, followed by recrystallization from water.67
A modified procedure was reported by the same group for the synthesis of pyridinium-1-yl bisphosphonates 27. The corresponding carboxylic acids 26 prepared through a cross-coupling reaction followed by N-alkylation with bromoacetic acid, were made to react with 5 equiv. of phosphorous acid and 5 equiv. of phosphorous trichloride in toluene without methanesulphonic acid. After treating the mixture with 6 N HCl and reflux for 1 h, addition of 2-propanol precipitated the 1,1-bisphosphonates which were further recrystallized from ethanol/water (Scheme 6).72
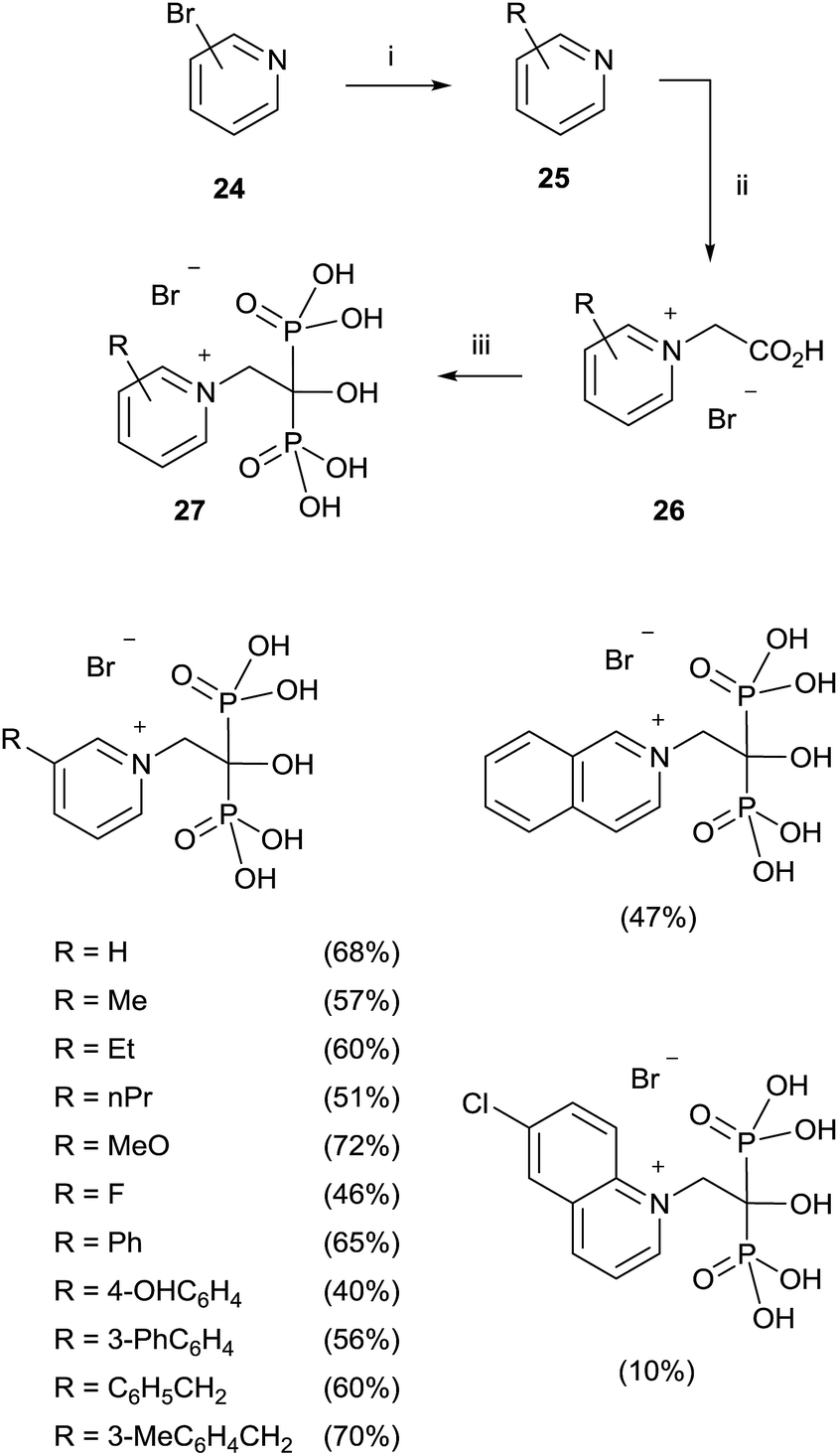 | ||
| Scheme 6 Reagents and conditions: (i) R–B(OH)2, Pd(PPh3)4, K2CO3, toluene, H2O, 10 h, reflux. (ii) BrCH2CO2H, pyridine, EtOAc, 2 days, rt. (iii) H3PO3 (5 equiv.), PCl3 (5 equiv.), toluene, 80 °C, 5 h; then 6 N HCl, 1 h, reflux. | ||
This protocol was also applied to the synthesis of aliphatic and aromatic derivatives with or without heteroatoms, demonstrating a great tolerance with respect to the chemical nature of the substrates.73 Labelled pamidronic acid-13C3,15N, alendronic acid-15N, zoledronic acid-15N2 and risedronic acid-15N were also synthesized by using those conditions.74 Pyridinium fluorescently-labeled conjugates of risedronate 7 were synthesized using an epoxide linker which was bonded to the pyridyl nitrogen. The conjugates were used for fluorescence imaging of Bacillus subtilis.75
Despite this synthetic activity that demonstrate the utility of the carboxylic acid-approach, various inconsistent procedures appeared in patents concerning the synthesis of the most clinically used zoledronic and risedronic acids which has been the matter of some controversy regarding the reagents and the ratio between them to be used. Dedicated reviews have been reported with the aim of clarifying the situation including the mechanism76 of the reaction.77,78 In 2011, Keglevich and co-workers finally established that the best conditions correspond to the use phosphorous trichloride in 1:3.2 molar ratio in methanesulfonic acid.79 Alternatively, it has also been reported the use of benzenesulfonic acid as a solvent.80 When the reaction is carried out in the presence of paraformaldehyde, bisphosphonic acids can be obtained without the addition of water,81 the use of microwaves enhancing the yield of the reaction.82,83 By using their conditions, Keglevich and co-workers reported the synthesis of etidronate 2, alendronate 4, pamidronate 5, ibandronate 6, risedronate and 7 zoledronate acid 8 (Scheme 7).84 The exact treatment of the reaction comprised the use of only 3.2 eq. of phosphorous trichloride as the P-reagent in methanesulfonic acid as a solvent (without addition of phosphorous acid). In fact, it had been reported the use of that solvent for large-scale preparations.85 Time and temperature were adjusted in each case for the first stage of the reaction at which time water was added and the resulting mixture was heated at 105 °C for 5 h. This was followed by hydrolysis with 10 N NaOH and pH adjustment to 1.8–2.0. Notably, the different dronic acids prepared required different purification conditions. Whereas 2, 4 and 6 were purified by direct precipitation in water, 5 was purified by digestion in MeOH followed by precipitation in water, 7 was purified by washing with water and 8 was purified by recrystallization from HCl. Further work in different solvents86 allowed establishing the best conditions for doing the reaction.83
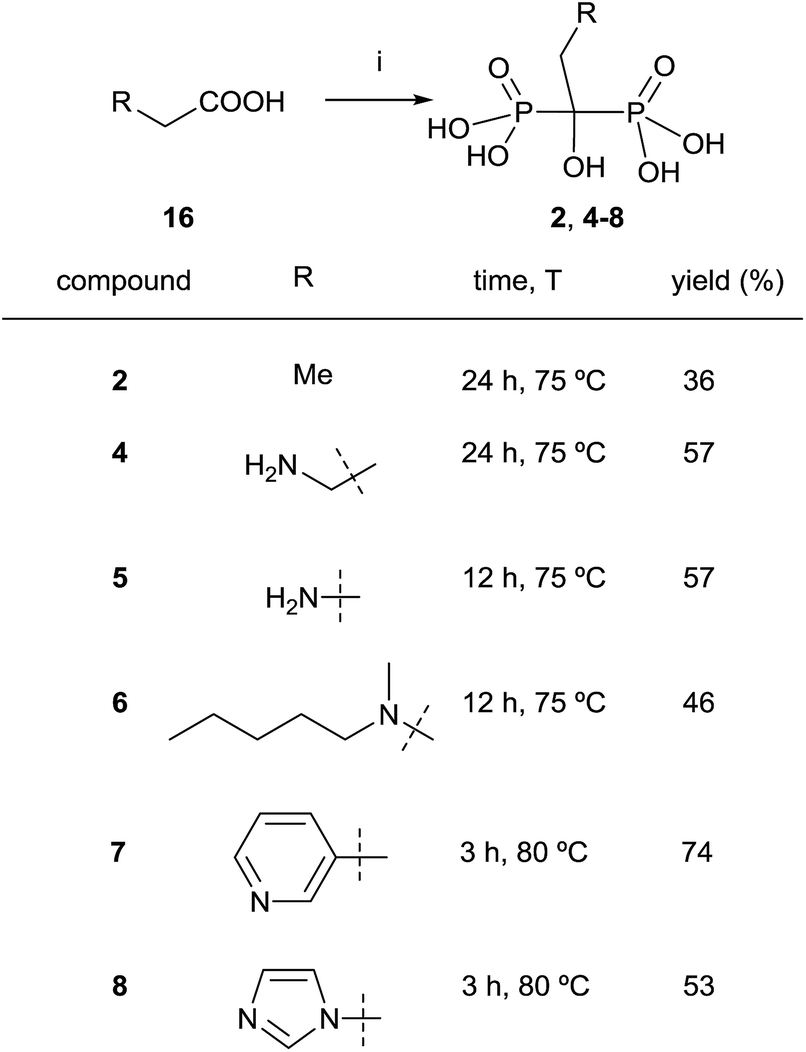 | ||
| Scheme 7 Reagents and conditions: (i) PCl3 (3.2 equiv.), MeSO3H, see scheme for time and temperature; then H2O, 4–5 h, 105 °C; then 50% aq. NaOH to pH = 1.8–2.0. | ||
A variation using phosphorous oxychloride has allowed a one-pot multigram synthesis of zoledronic acid 8 in high yield (Scheme 8).87 The procedure has been carried out with 200 g of compound 28, easy accessible from imidazole.88 The use of phosphorous oxychloride has also been successfully used for the synthesis of aledronate 4 and risedronic acid 7.89 On the other hand, phosphorous trichloride was the preferred reagent in the synthesis of benzidronate.90
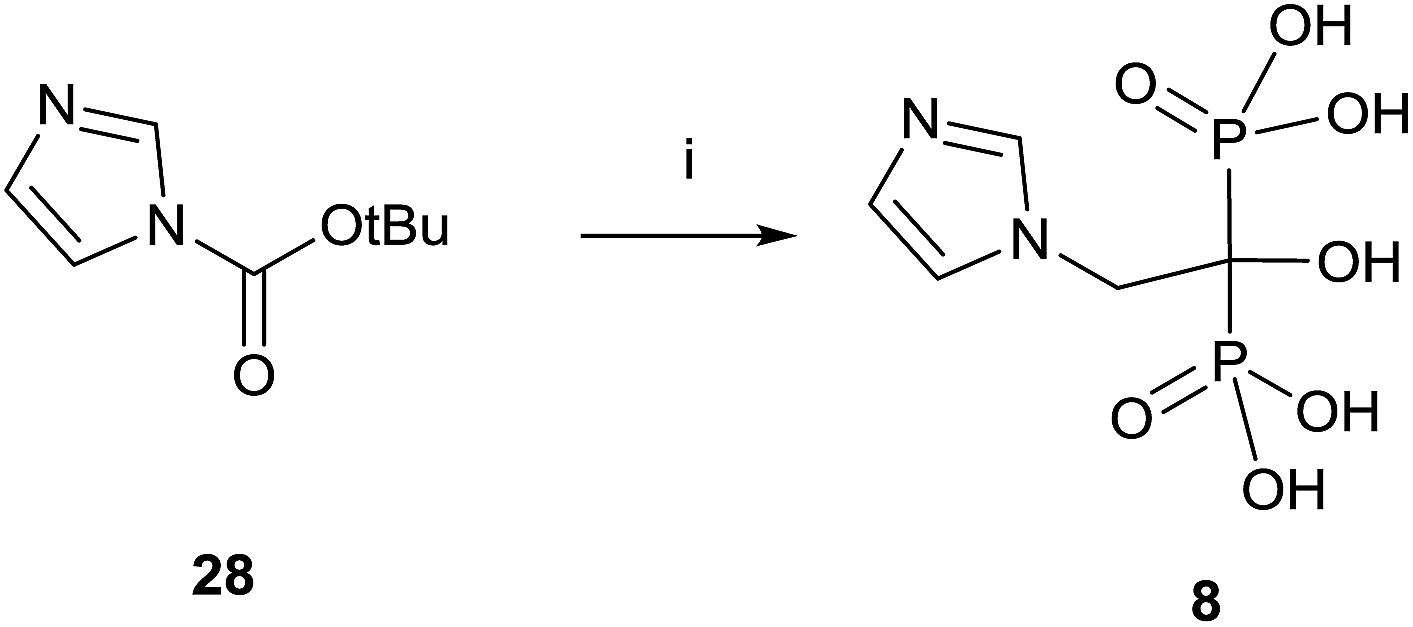 | ||
| Scheme 8 Reagents and conditions: (i) H3PO3, 4-chlorobenzene, 25 °C, 15 min; then CH3SO3H, 30 min, 70 °C; then POCl3, 95 °C, 24 h; then H2O, 6 h, 90 °C; then 30% NaOH, pH = 4.1. | ||
Nitriles 29 can also be used as an alternative to carboxylic acids and the reaction can be performed under similar conditions but without the presence of phosphorous acid (Scheme 9).91 Risedronate 7 prepared by this route was further labeled with 99Tc by forming the corresponding complex.92
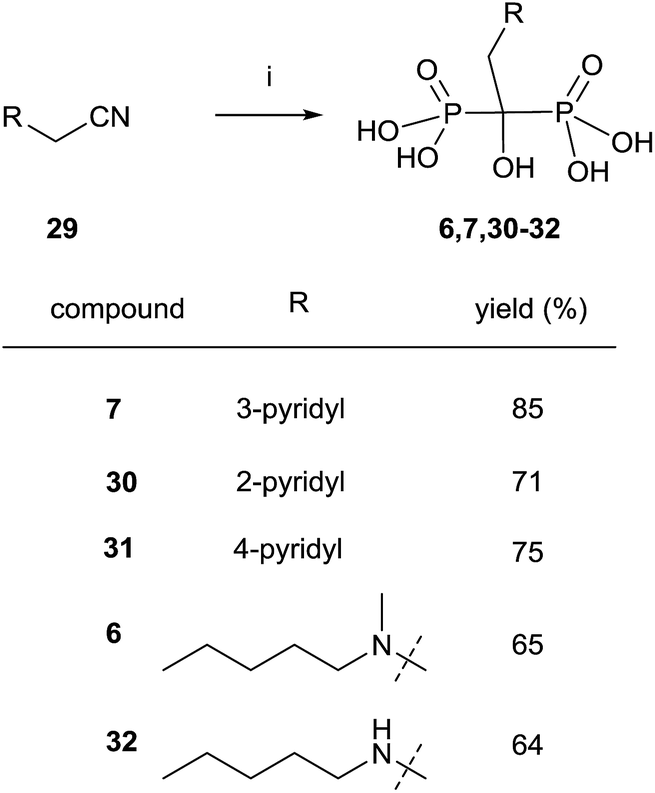 | ||
| Scheme 9 Reagents and conditions: (i) CH3SO3H, 8 h, 100 °C; then PCl3, 70 °C, 5 h; then H2O, 15 h, 98 °C. | ||
Reaction of acylphosphonates with dialkyl phosphites
In some instances, the direct reaction between a carboxylic acid and phosphorus trichloride takes place with low yields or fails completely as in the case of sterically hindered α-aminoacids. A discussion comparing this route with that developed at the Merck company has been reported.93 An alternative is the use of the acyl chloride as starting material. The Arbuzov-type reaction94 with a di-, trialkyl phosphite provides an α-ketophosphonate (acylphosphonates) capable of reacting with another molecule of di-, trialkyl phosphite to give the corresponding 1-hydroxy-1,1-bisphosphonate. The first report on this approach described the use of tris(trimethylsilyl)phosphite as the only phosphorylating reagent (Scheme 10).95 The reaction proceeds under mild conditions rendering the process compatible with labile substrates96 including bile acids.97 Acrylic ester bisphosphonates 35,36 with numerous potential applications in biomedicine are also accessible through this approach.98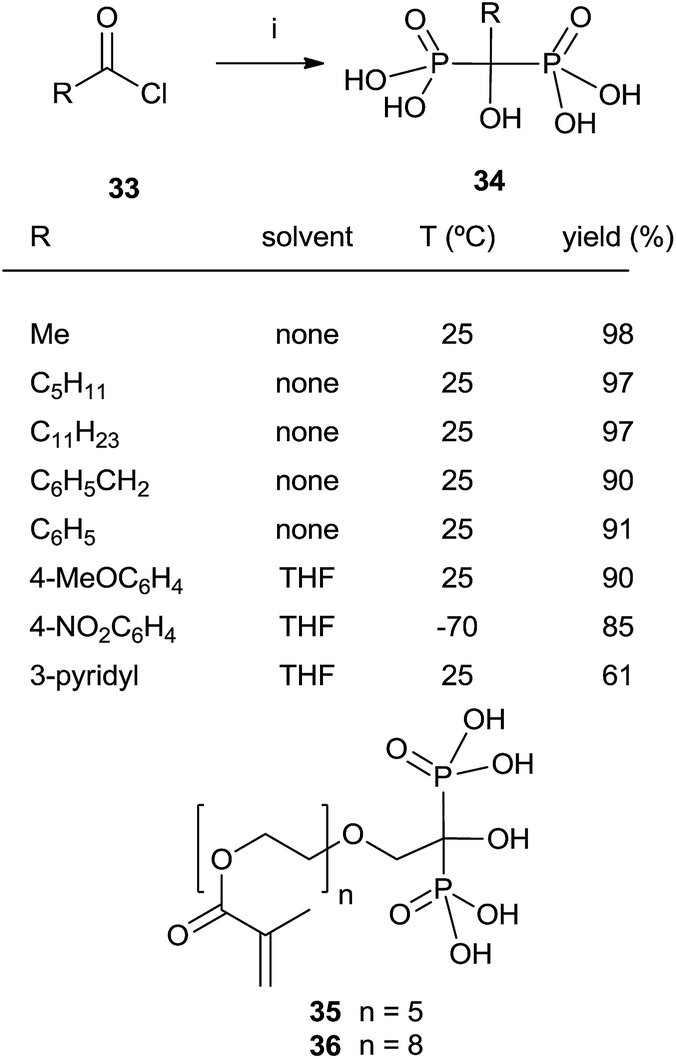 | ||
| Scheme 10 Reagents and conditions: (i) P(OSiMe3)3, 0 °C then rt; then MeOH, 1 h, 25 °C. | ||
The direct use of trialkyl phosphites is also possible. A series of α-amino acid derived bisphosphonates 42 has been prepared in good yield by using as starting materials N-phthalimido-protected amino acids and using sequentially tri- and diethyl phosphite as P-reagents (Scheme 11).99 Although the addition of phosphites is consecutive, the reaction is carried out in a one-pot procedure without isolating the intermediate ketophosphonate 40.
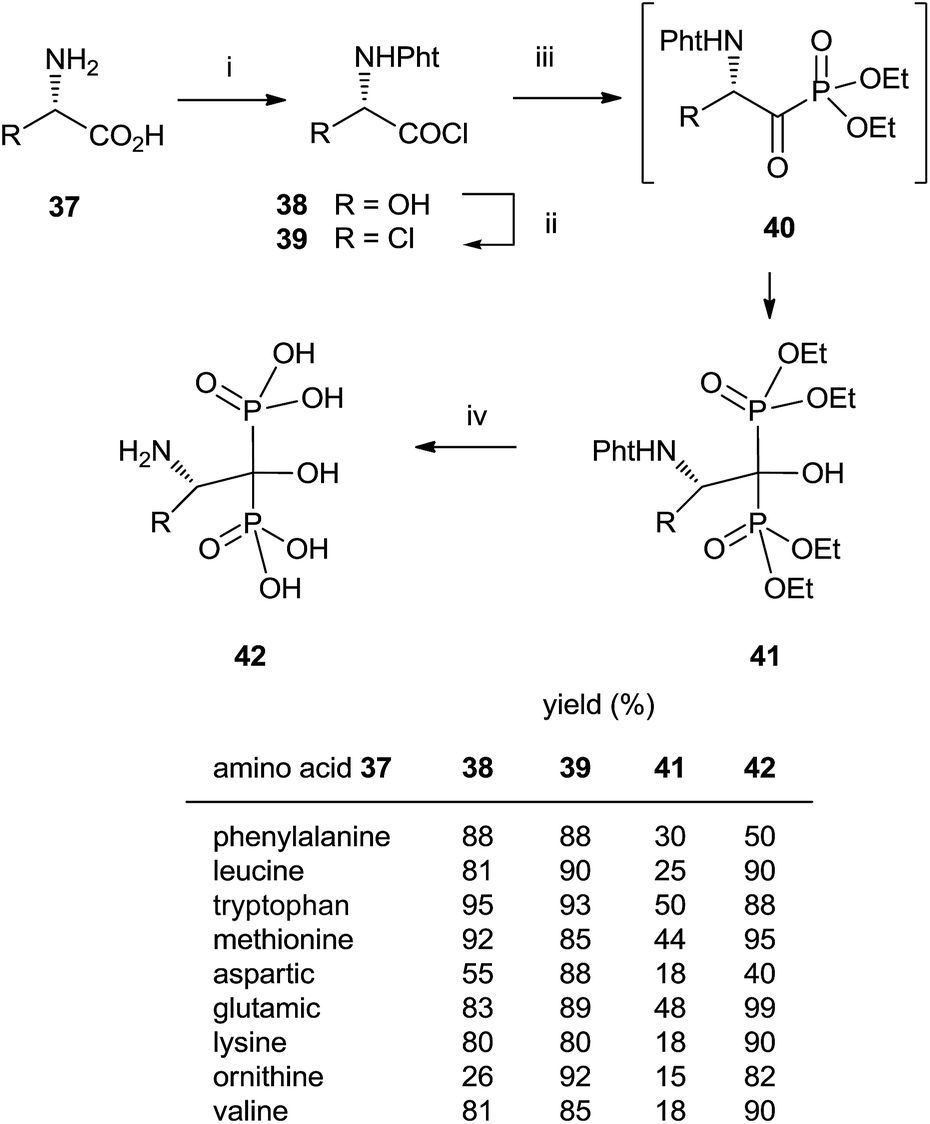 | ||
| Scheme 11 Reagents and conditions: (i) N-(ethoxycarbonyl)phthalimide, NaHCO3, 0 °C, 5 min. (ii) SOCl2, CH2Cl2, 5 h, reflux. (iii) Triethyl phosphite, toluene, 1 h, 0 °C; then diethylphosphite, Et3N, 1–3 h, 0–5 °C. (iv) 6 N HCl, reflux, overnight; then 5 N NaOH, pH = 4.4. | ||
Final deprotection of the amino group was made with 6 N HCl. The use of hydrazine is also possible and better yields are obtained. By using this variation, alendronate 4, pamidronate 5, and neridronate have been prepared as mono- and diesters, which were soluble in water at physiological pH.100 The use of tri- and dimethylphosphite led to shorter reaction times. Conjugation of a fluorophore to methyester-protected pamidronate 45, prepared from β-aminoacid 12, was performed in DMSO in the presence of N-methylmorpholine (Scheme 12). Compound 48 was used as a contrast agent in image-guided surgery of large animals.101
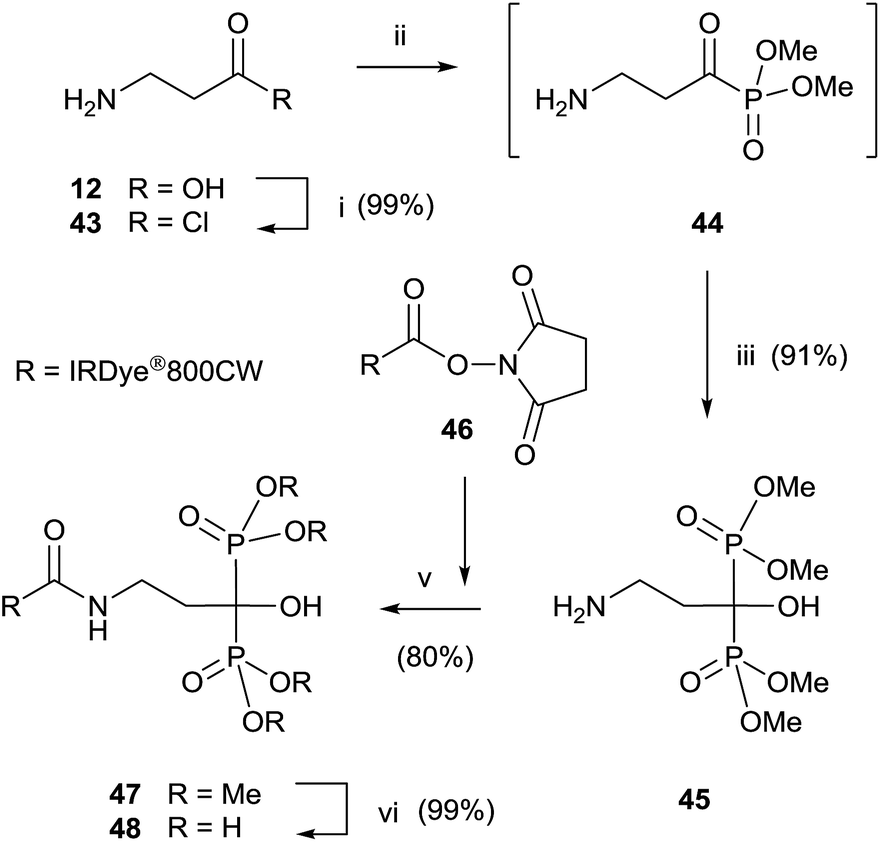 | ||
| Scheme 12 Reagents and conditions: (i) SOCl2, CH2Cl2, 1 h, reflux. (ii) Trimethyl phosphite, toluene, 30 min, 0 °C. (iii) Dimethylphosphite, Et3N, 30 min, 0–5 °C. (iv) DMSO, N-methylmorpholine, 4 h, rt. (v) Me3SiBr, 18 h, rt; then 4:1 MeOH–H2O, 30 min, rt. | ||
The use of dimethylphosphite also afforded better results in the synthesis of alendronate 4 reported by Seki in which both P-reagents were compared. Differences were found in the final hydrolysis of the phosphate esters. Ethyl esters showed a lower reactivity towards hydrolysis (Scheme 13).102
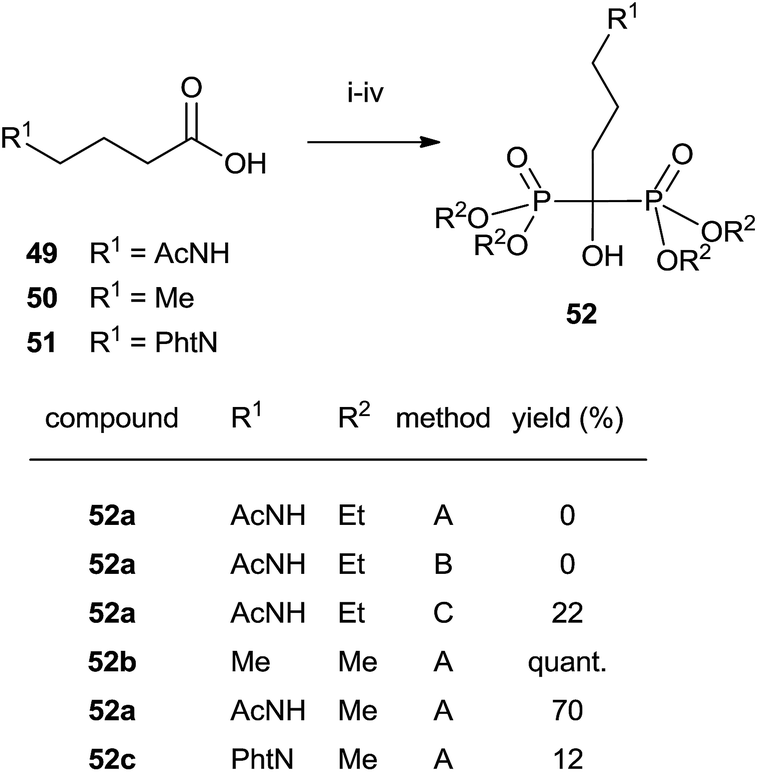 | ||
| Scheme 13 Reagents and conditions: (i) (COCl)2, CH2Cl2, DMF (cat.), 3 h, rt. (ii) (R2O)3P, 1 h, rt. (iii) (R2O)3P, TMSBr, 1 h, rt. (iv) Method A: aq. 6 M HCl, 17 h, reflux; method B: MeSO3H, 17 h, reflux; method C: 48% HBr, 17 h, reflux. | ||
Analogues of alendronic acid 4 have been prepared from the common precursor 53 through a practical one-pot, three step methodology. The protection of the 1-hydroxy group was necessary for avoiding mixtures in the acylation step (Scheme 14).103
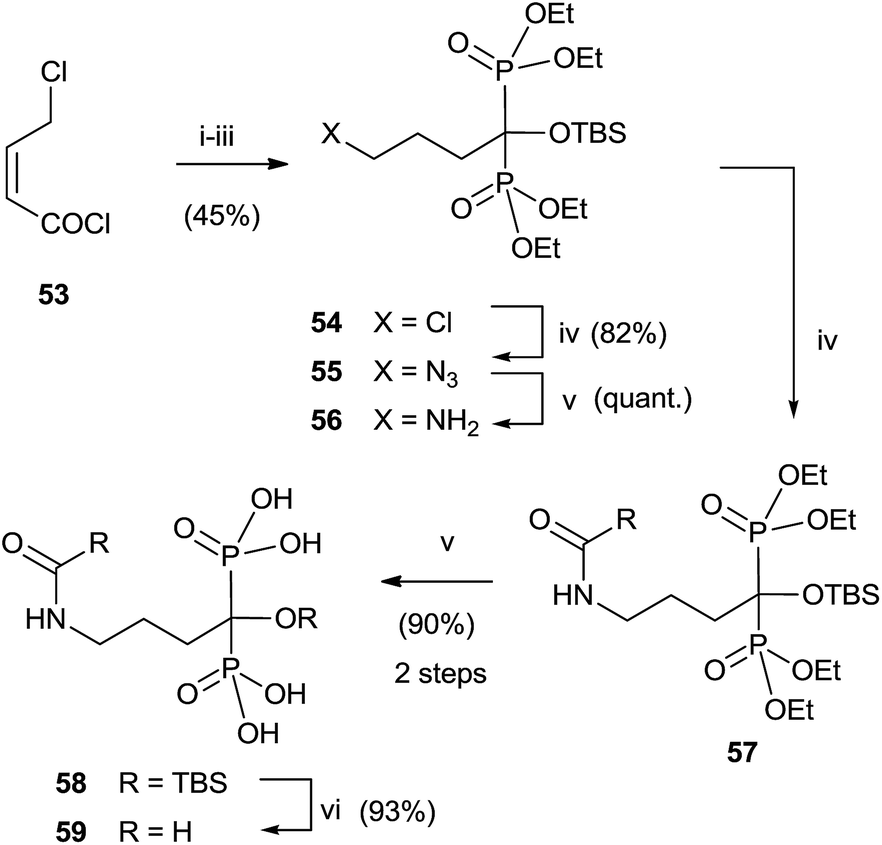 | ||
| Scheme 14 Reagents and conditions: (i) P(OEt)3, 0 °C. (ii) HOP(OEt)2, DMAP, CH2Cl2, room temp, 1 h. (iii) TBSCl, 15 h. (iv) NaN3, DMF, 75 °C, 2 h. (v) Pd–C, 50 psi of H2, AcOEt, 15 h. (vi) RCOCl, Et3N, CH2Cl2 or RCO2H, EDC, DIPEA, MeCN. (v) TMSI, MeCN and then MeOH. (vi) TBAF, THF. | ||
A series of bisphosphonates bearing either the nitrogen-containing NO-donor furoxan (1,2,5-oxadiazole 2-oxide) or the related furazan (1,2,5-oxadiazole) systems in the lateral chain has been prepared by using trimethylphosphite as P-reagent (Scheme 15).104
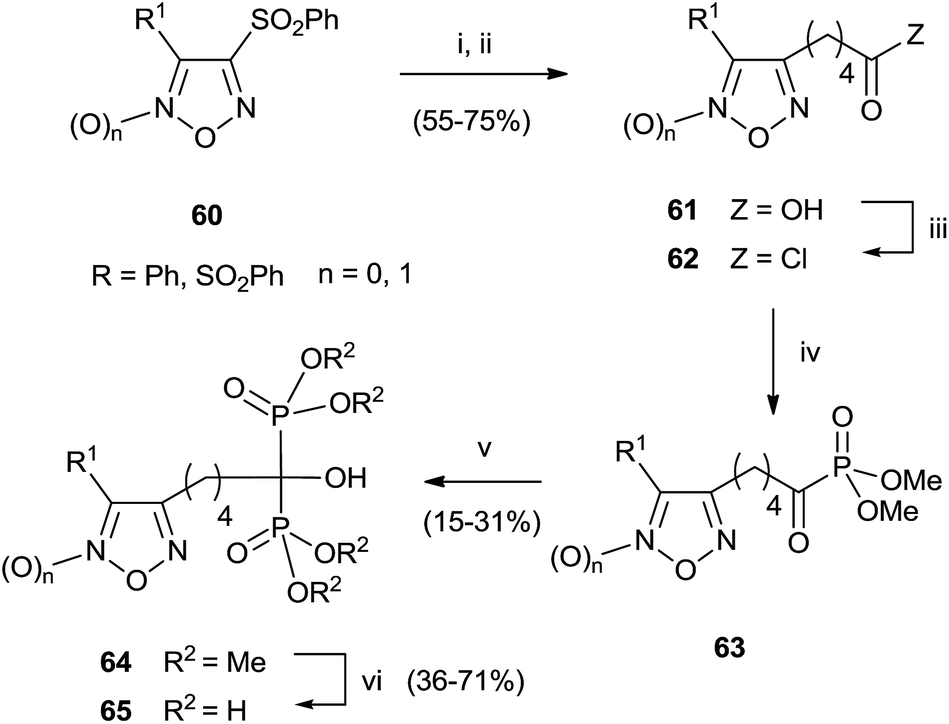 | ||
| Scheme 15 Reagents and conditions: (i) 1,5-pentanediol, NaOH 50% w/w, THF. (ii) Jones reagent, acetone, 0 °C to rt. (iii) SOCl2. (iv) P(OMe)3, dry THF, 0 °C to rt. (v) HPO(OMe)2, Et2NH, dry THF, 0 °C to rt; (vi) TMSBr, CH2Cl2; then MeOH, 0 °C to rt. | ||
Activation of the carboxylic acid as a dioxaborolane can be an alternative to the acid chloride. This approach requires the use of tris(trimethylsilyl)phosphite since dialkylphosphites showed no reaction (Scheme 16).105 Also in this case a poor reactivity was observed for N-hydroxysuccinimide esters towards methyl or phenyl bis(trimethylsilyl)phosphites, in good agreement with previously reported results.100
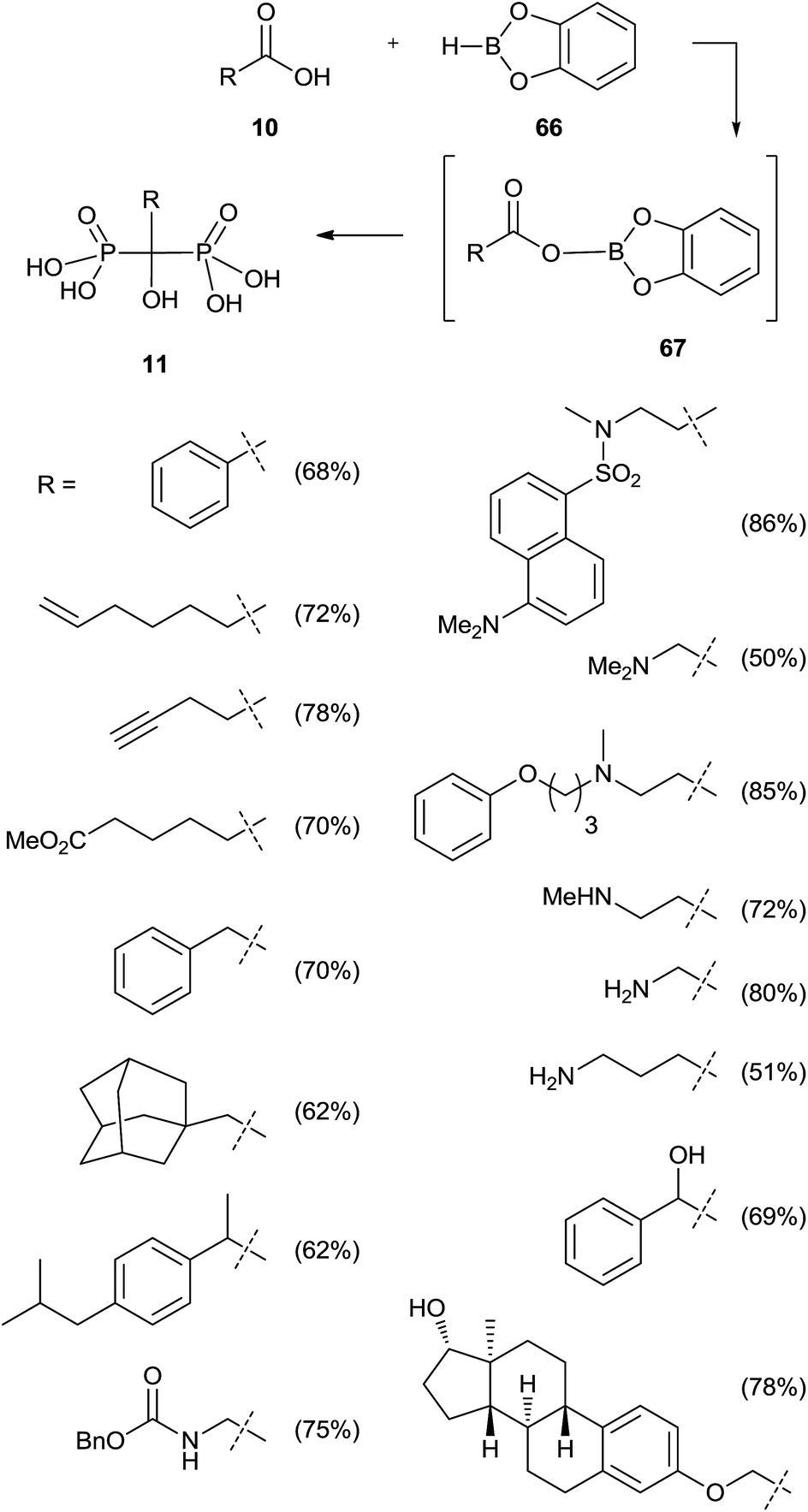 | ||
| Scheme 16 Reagents and conditions: (i) THF, rt. (ii) P(OSiMe3)3, 16 h, 25 °C; then MeOH, 2 h. | ||
Michael addition to tetraethylvinylidenebisphosphonate
Tetraethyl vinylidenebisphosphonate 68 is an easily available Michael acceptor and electron-poor dipolarophile/dienophile that results an excellent synthetic intermediate for the synthesis of 1,1-bisphosphonates (Scheme 17). The synthesis of 68 and its application in the preparation of bisphosphonates has been recently reviewed in 2014 by J. B. Rodriguez.106 The reader is referred to this excellent compilation for the syntheses of bisphosphonates starting from 68 reported until 2013. Here we survey only those reported after publication of the Rodriguez's review.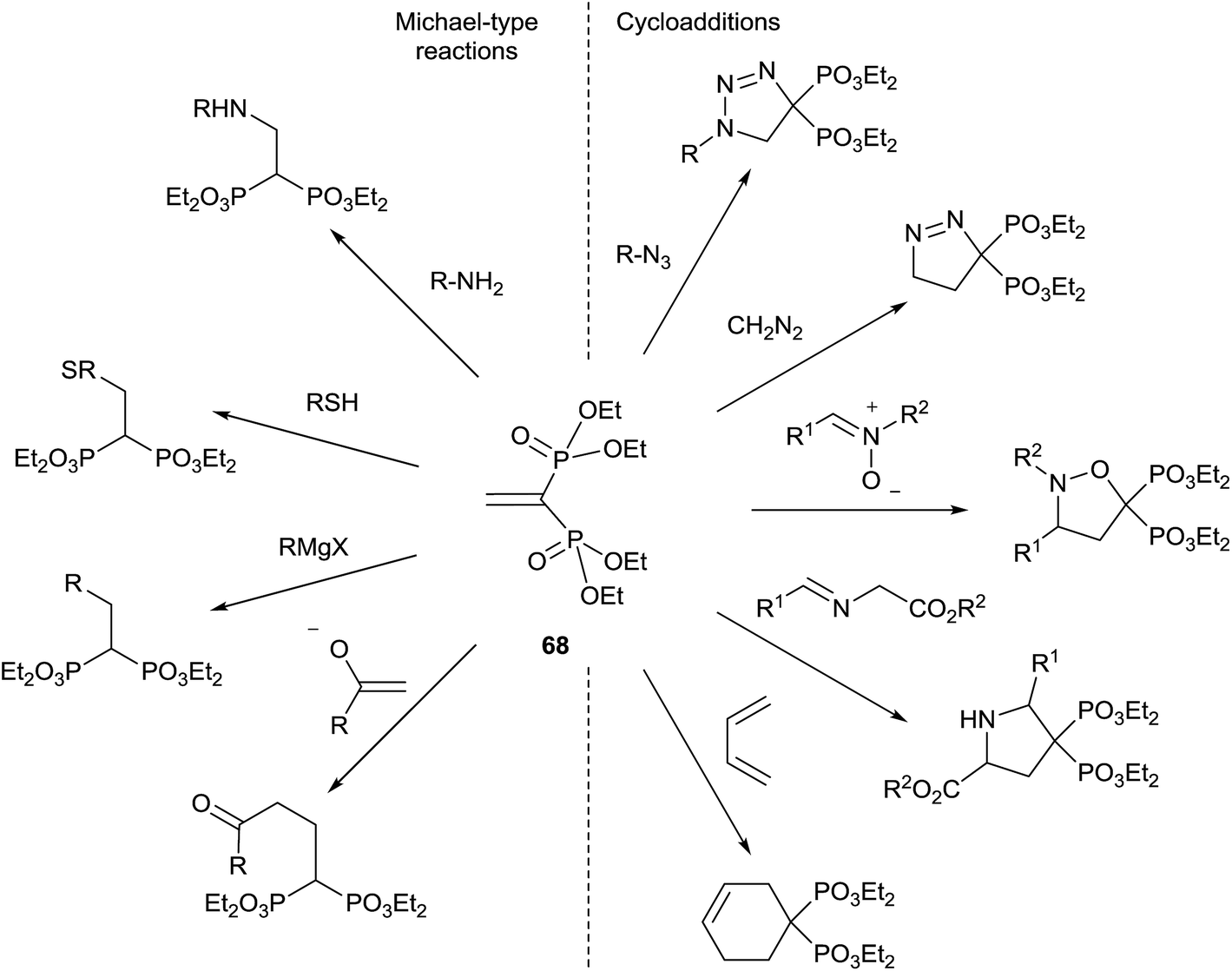 | ||
| Scheme 17 Synthetic utility of tetraethyl vinylidenebisphosphonate 68.106 | ||
The Cu-catalyzed 1,4-conjugate addition of boronic acids and indoles to 68 afforded 1,1-bisphosphonates lacking the gem-OH group (Scheme 18).107 Whereas the reactions with boronic acids proceeded smoothly in toluene, the addition of indoles can be carried out in polar solvents like 1,2-dichloroethane or water with sodium dodecyl sulfate under micellar conditions.
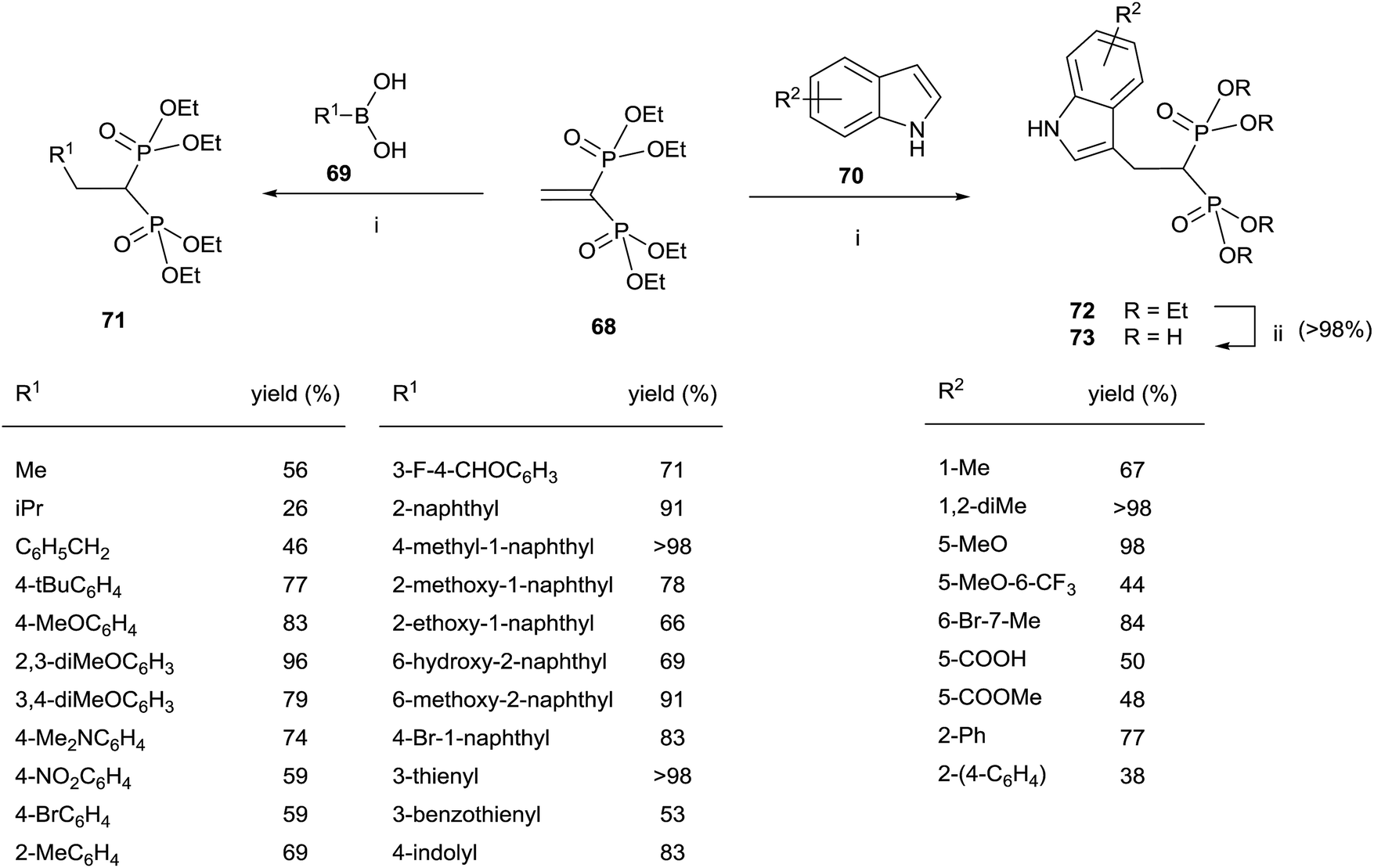 | ||
| Scheme 18 Reagents and conditions: (i) Cu(OTf)2 (10 mol%), toluene, 18 h, 70 °C. (ii) TMSBr, 18 h, rt; then water, 4 h, rt. | ||
A series of spiro[indole-pyrrolizine], spiro[indole-indolizine], and spiro[indole-pyrrolidine] gem-bisphosphonates were synthesized through multicomponent reactions between isatins 74, amino acids 75 and 68 in the presence of montmorillonite (Scheme 19).108 Acyclic aminoacids can also be used.
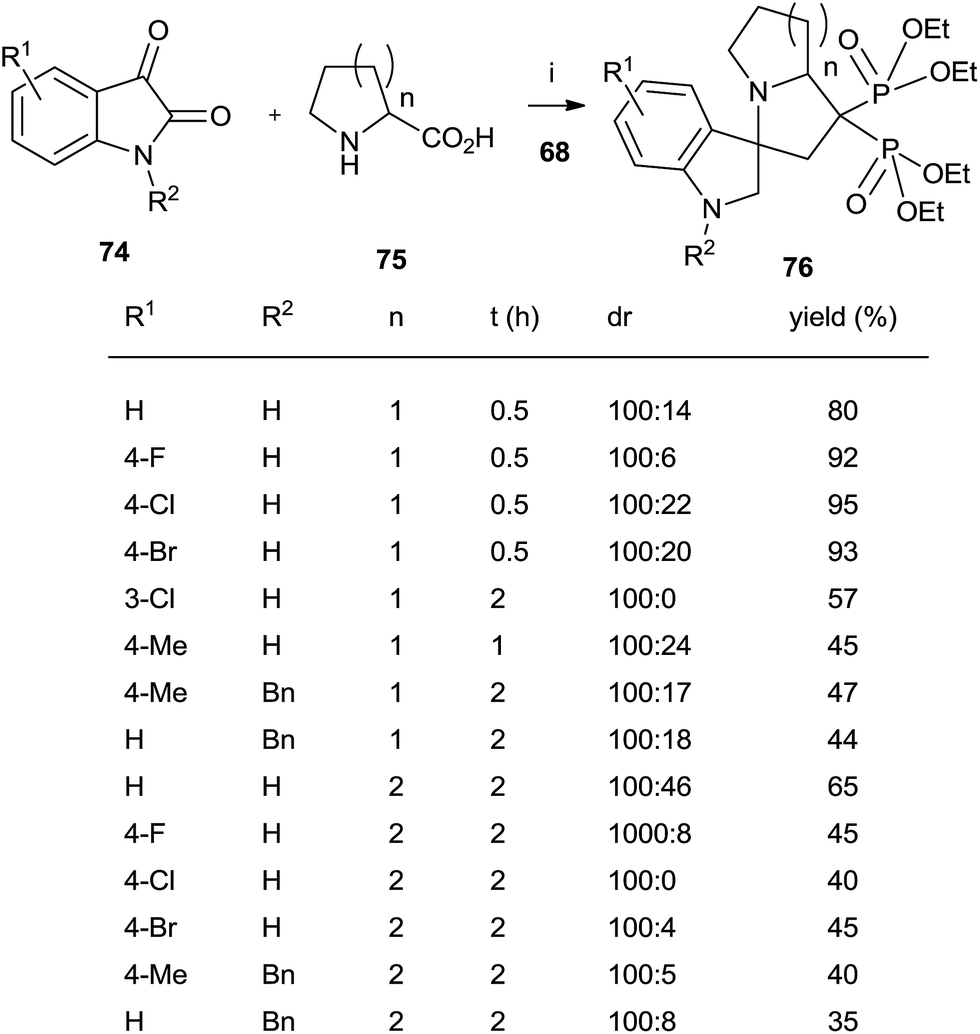 | ||
| Scheme 19 Reagents and conditions: (i) montmorillonite, MeCN, 0.5–2 h, 80 °C. (ii) TMSBr, 18 h, rt; then water, 4 h, rt. | ||
The cycloaddition of aromatic nitrones 77 with 68 furnished spiro(isoxazolino)bisphosphonates 78 (Scheme 20).
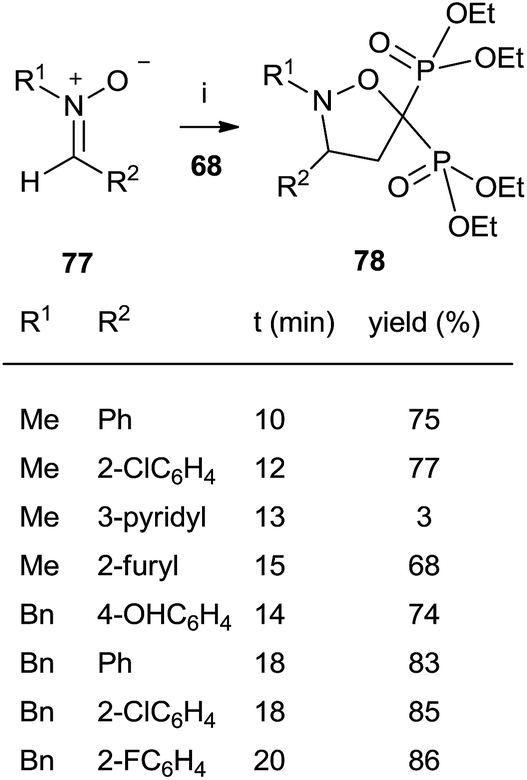 | ||
| Scheme 20 Reagents and conditions: (i) neat, microwave irradiation 200 W. | ||
The reaction, carried out in the absence of solvent and under activation with microwaves takes place in several minutes with good yields.109 In all these cases the bisphosphonates prepared by this route lack the 1-hydroxyl group but consist of interesting structurally constrained analogues.
Alkylation of tetralkylmethyl bisphosphonate
Alkylation of tetraethyl bisphosphonate 79 is an expeditious way of preparing bisphosphonates lacking the 1-hydroxy groups. However, precise reaction conditions must be used in order to avoid elimination reactions.110 Alkylation of 79 with farnesyl and geranyl bromides 80 and 81 using sodium hydride as a base provided bisphosphonates 82a,b. Further hydrolysis furnished free bisphosphonates 83a,b (Scheme 21).111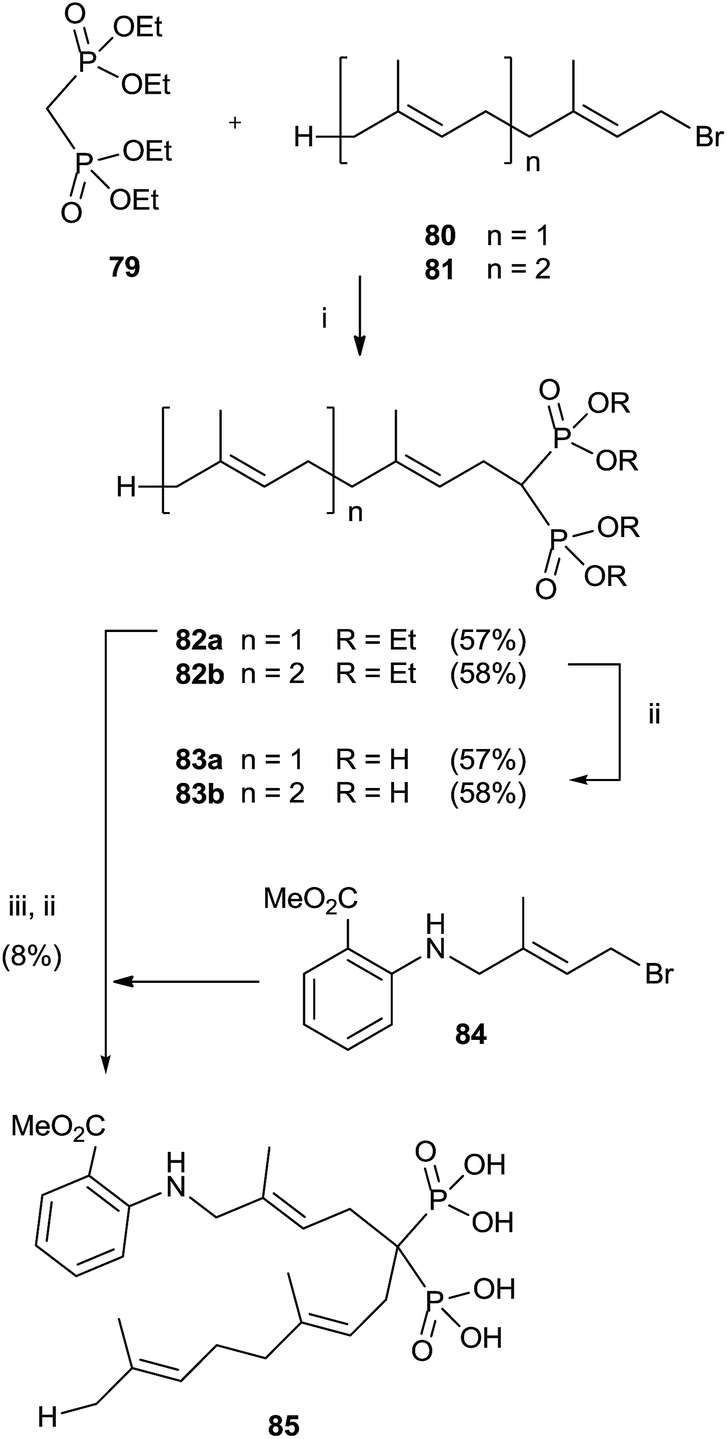 | ||
| Scheme 21 Reagents and conditions: (i) NaH, THF, 0 °C, 1 h; then, 1 day, rt. (ii) TMSBr, collidine, 1 day, rt; then 0.5 M NaOH 16 days, rt. (iii) 84, NaH, THF, 15-crown-5, overnight, rt. | ||
On the other hand, using potassium hydride an undesired elimination reaction was observed.110 By oxidizing the allylic position of the terminal trisubstituted double bond in 80, it was possible to introduce additional substituents at the end of the isoprenoid unit.112 Further alkylation of compound 80 furnished, after hydrolysis, fluorescent anthranilate analogue 85 that showed some inhibition in geranylgeranylation.113
Aminobisphosphonate 86 was alkylated, after protection of the amino group, with methyl 2-bromo acetate. Potassium carbonate in the presence of triethylammonium bromide was used for deprotonating 86. Further deprotection yielded the target bisphosphonate (Scheme 22).114
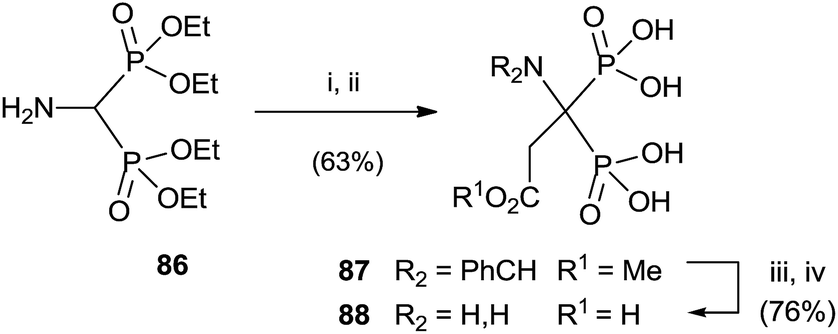 | ||
| Scheme 22 Reagents and conditions: (i) m-ClC6H4CHO, MgSO4, CH2Cl2. (ii) BrCH2CO2Me, Bu4NBr, K2CO3, MeCN. (iii) 1 N HCl, MeCN; then NaOH. (iv) 6 N HCl. | ||
Other methods
Treatment of N-farnesyl lactams with an excess of base and diethyl phosphorochloridite furnished bisphosphonates 90 in good chemical yields. The methodology can be extended to other carbonyl compounds including amides and lactones although in the case of sterically hindered substrates the expected bisphosphonate is not obtained and other byproducts are formed. Whereas lactams can be phosphorylated with either LDA or LHMDS as a base, the former is preferred for amides and lactones (Scheme 23).115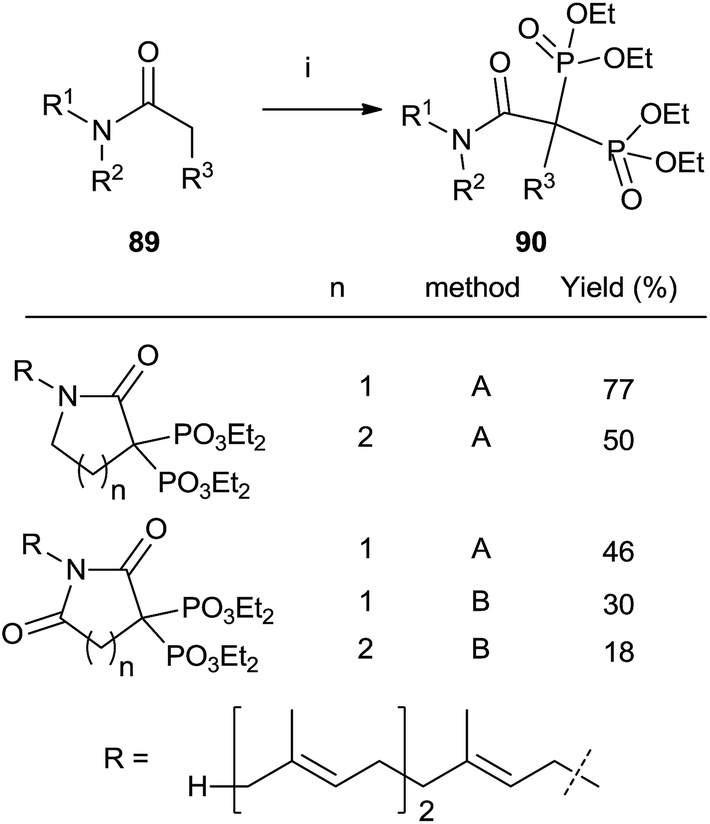 | ||
| Scheme 23 Reagents and conditions: (i) method A: LDA (2.2 equiv.); then ClP(OEt)2 (2.3 eq.); then H2O2 (10 eq.). Method B: LHMDS; then ClP(OEt)2; then LHMDS; then ClP(OEt)2; then H2O2 (20 eq.). | ||
Carboxyamide and sulfonamide bisphosphonates are accessible by treating the appropriate amide with trialkyl orthoformate and dialkylphosphites. Subsequent deprotection under typical conditions furnished bisphosphonates 90 (Scheme 24).116
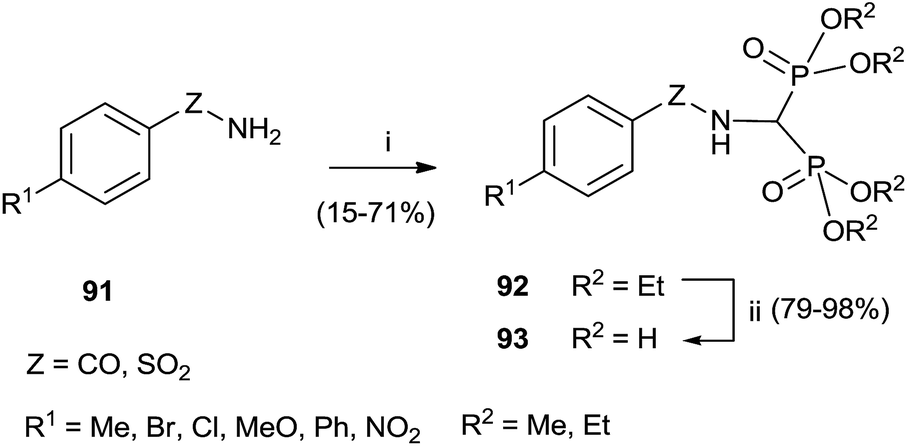 | ||
| Scheme 24 Reagents and conditions: (i) HC(OR)3, HP(OR)2, 150 °C. (ii) BBr3, toluene, MeOH, reflux. | ||
Non-bisphosphonate derivatives
Non-bisphosphonate antiresorptive agents are represented by molecules with variable dimensions and functional groups.The presence of a hydroxamate moiety induces a major attitude in terms of metal chelation.117 Thus, the hydroxamic group is expected to improve the ionic and/or metal chelation interactions with the active site of FPT (Farnesyl Protein Transferase).
The synthesis of N-methyl substituted hydroxamic acid 103 was carried out starting from N-methyl-O-benzyl-hydroxylamine hydrochloride 94 and aspartic acid derivative 95 (Scheme 25).118 Deprotection of 96 and successive coupling of resulting 97 with tripeptide H-Val-Leu-Ser-OMe 98 furnished tetrapeptide 99.
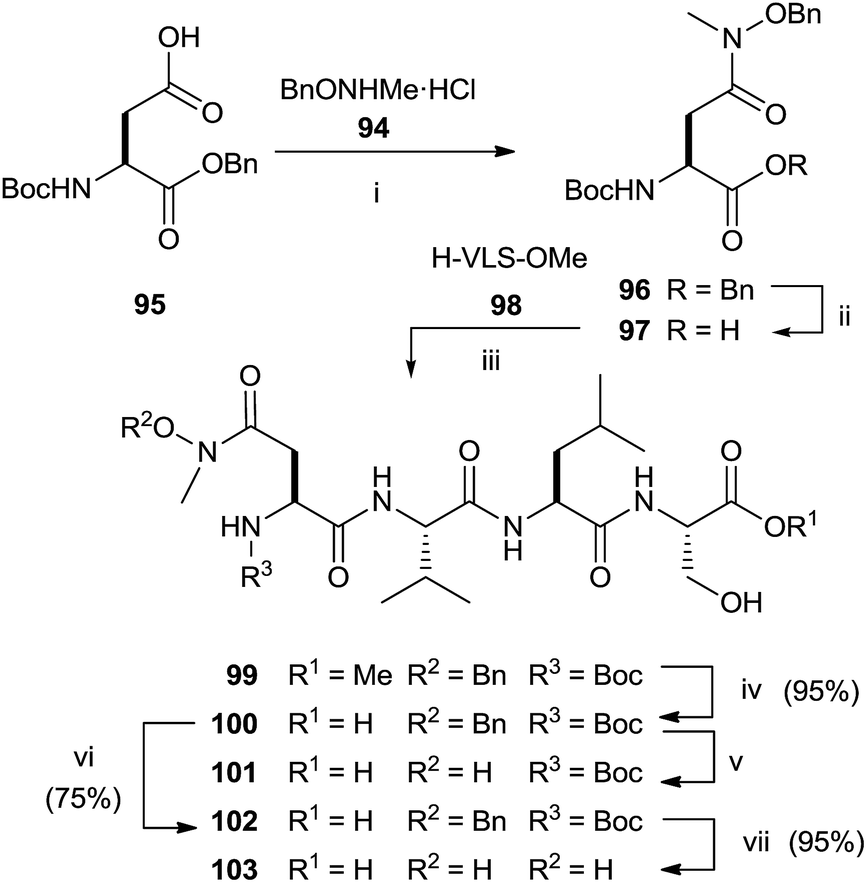 | ||
| Scheme 25 Reagents and conditions: (i) CD1, iPr2NEt. (ii) 1 N NaOH, MeOH, (iii) EDC, HOBT, iPr2NEt, HVLS-OMe, (iv) Na2CO3 (1.0), 2:1 MeOH, H2O. (v) H2, 10% Pd/C, MeOH. (vi) Anhydrous HCl/dioxane, EtOAc. (vii) H2, 10% Pd/C, MeOH. | ||
Basic hydrolysis with NaOH of the ester group in 99 provided 100 with 20% of the free aspartic acid derivative resulting from cleavage of hydroxamic function. The use of sodium carbonate in 2:1 MeOH–water avoided the formation of byproducts maintaining unchanged the chirality. Further deprotection steps gave provided 103 in good yields. In the same work, the authors prepared hydroxamic bisubstrate analogs 114, 116, 118 and 120 by introducing a full farnesyl group on the hydroxamic portion to improve the inhibition of FPT (Scheme 26).
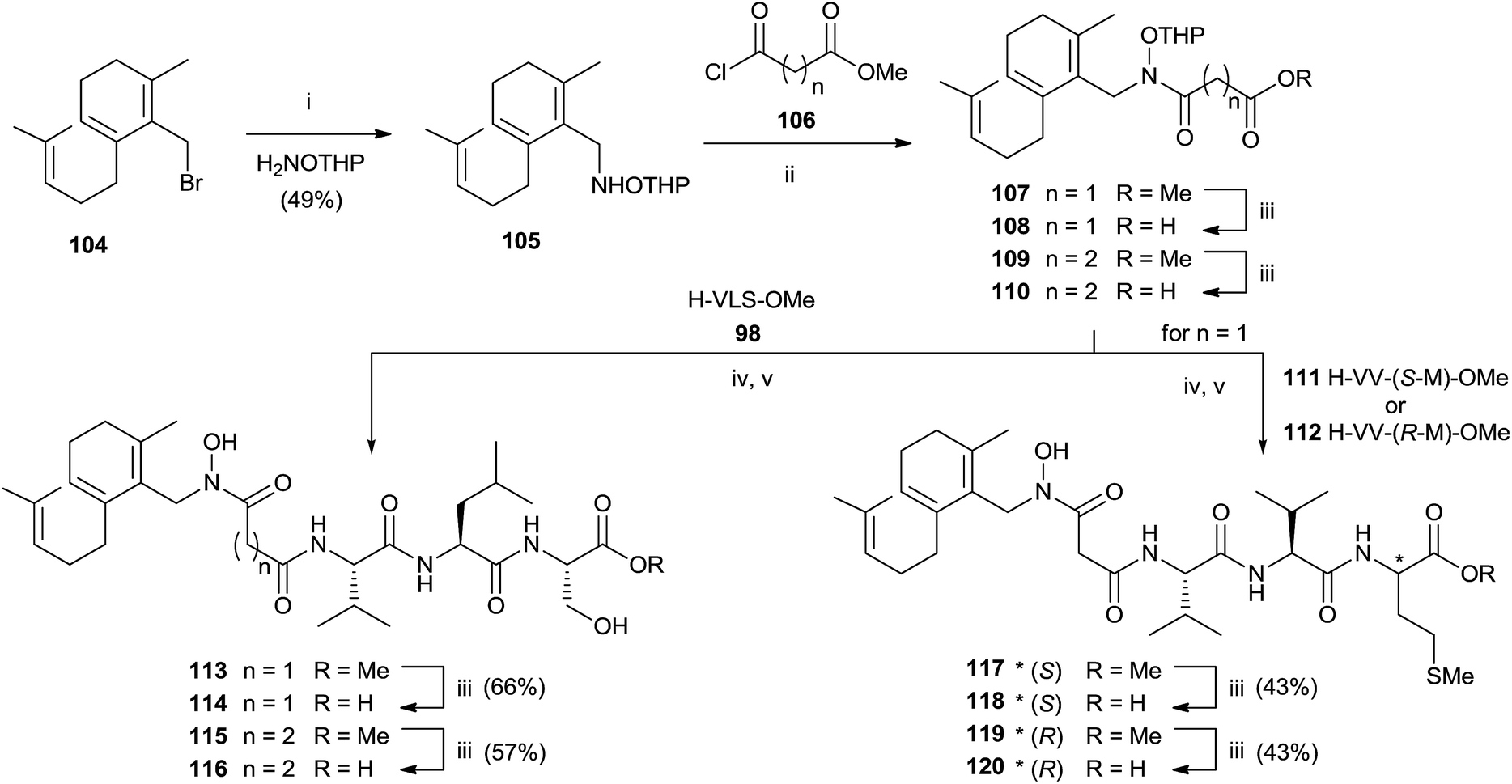 | ||
| Scheme 26 Reagents and conditions: (i) NH2OTHP, THF. (ii) iPr2NEt, THF. (iii) 1 N NaOH, MeOH. (iv) EDC, HOBT, iPr2NEt. (v) pTsOH, MeOH. | ||
The synthesis was carried out by a multistep sequence utilizing farnesyl bromide 104 as starting material which was transformed into 105 in moderate yield. Acylation of 105 required 2 eq. of the 3-carbomethoxy-propionyl chloride and 3 eq. of DIPEA. Saponification of the resulting 107 furnished intermediate 108 which was coupled with various tripeptides to give bisubstrates 114, 116, 118 and 120 after opportune removal of protective groups. A critical step in the synthesis was the deprotection of THP group in the presence of farnesyl chain. The problem was solved by using p-TsOH although the yields were modest.
In 1993, the chaetomellic acids A and B 121 and 122, classified as alkyl cis-dicarboxylates, were discovered to be potent inhibitors of FPTase because of analogy with the active site of FPP (Fig. 3).119,120
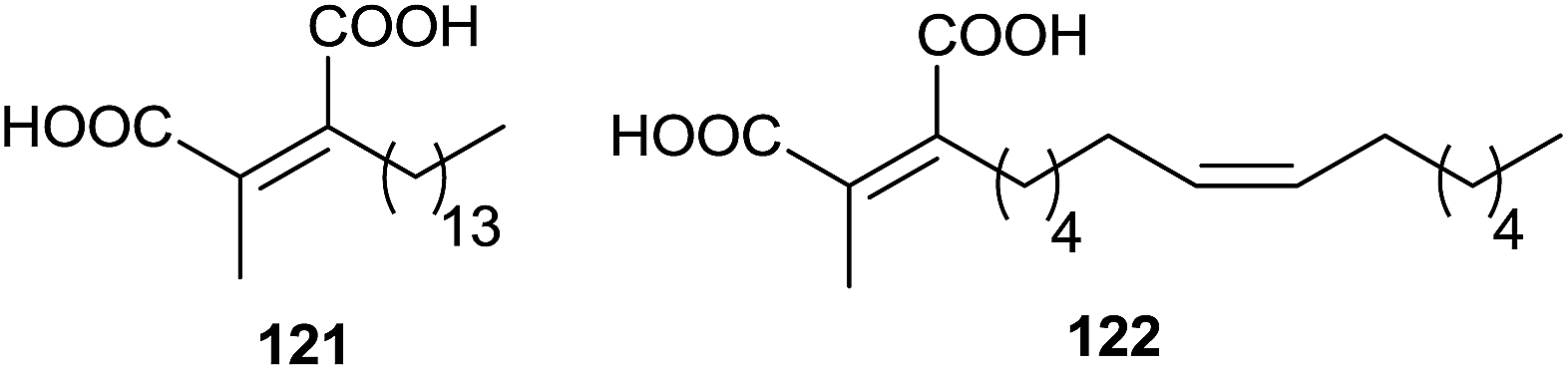 | ||
| Fig. 3 Chaetomellic acids. | ||
Singh and collaborators carried out the synthesis of various chaetomellic acids derivatives through a sequence of three steps.121,122 Starting from fatty acid esters 123–125, the reaction with methylpyruvate in the presence of LDA at −78 °C furnished a 1:1 diastereomeric mixture of aldol products 126 and 127 (Scheme 27). A β-elimination reaction of aldol substrates – opportunely protected with a tosyl group produced the tetra-substituted olefins 130–132 via anti or syn periplanar elimination. In the final step, the hydrolysis of ester derivatives by refluxing with a NaOH solution gave chaetomellic acid analogs 133–135 in moderate yield.
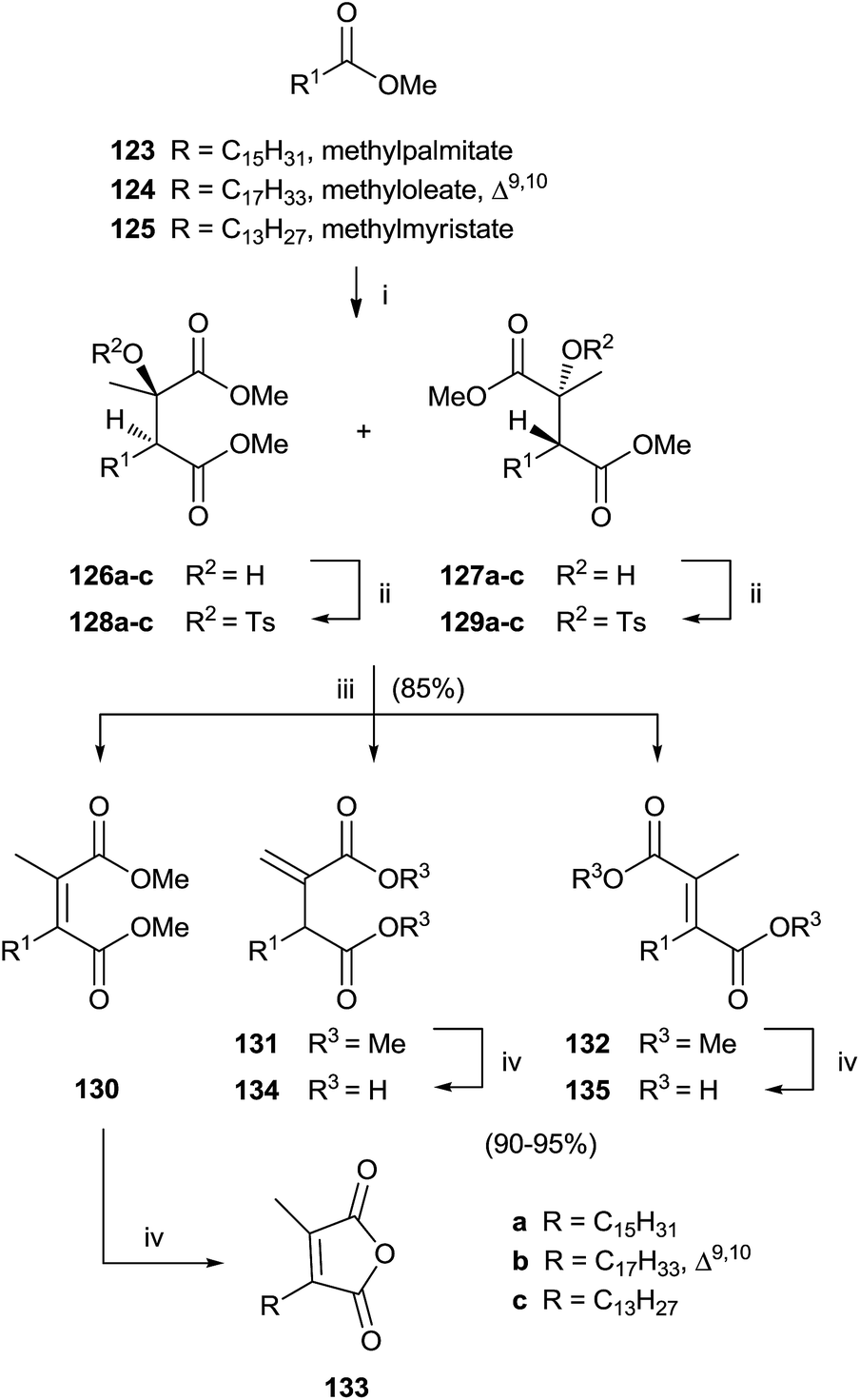 | ||
| Scheme 27 Reagents and conditions: (i) LDA, methylpyruvate, THF, −78 °C. (ii) p-Toluenesulfonic anhydride, CH2Cl2, C5H5N, 2,6-di-tert-butyl-4-methylpyridine, 40 °C. (iii) DBU, toluene, reflux. (iv) a: 1 N NaOH, MeOH, THF, H2O, 80 °C. b: 4 N HCl. | ||
Tucker et al. showed that a large arylthio or aryloxy group adjacent to the cyano function provided compounds with high activity against GGPP and FPT enzymes.123,124
An approach to the synthesis of inhibitors 139–155 was developed by initial reductive amination of aldehydes 136 with N-Boc-piperazine, titanium iso-propoxide and NaBH3CN in THF–EtOH (Scheme 28).
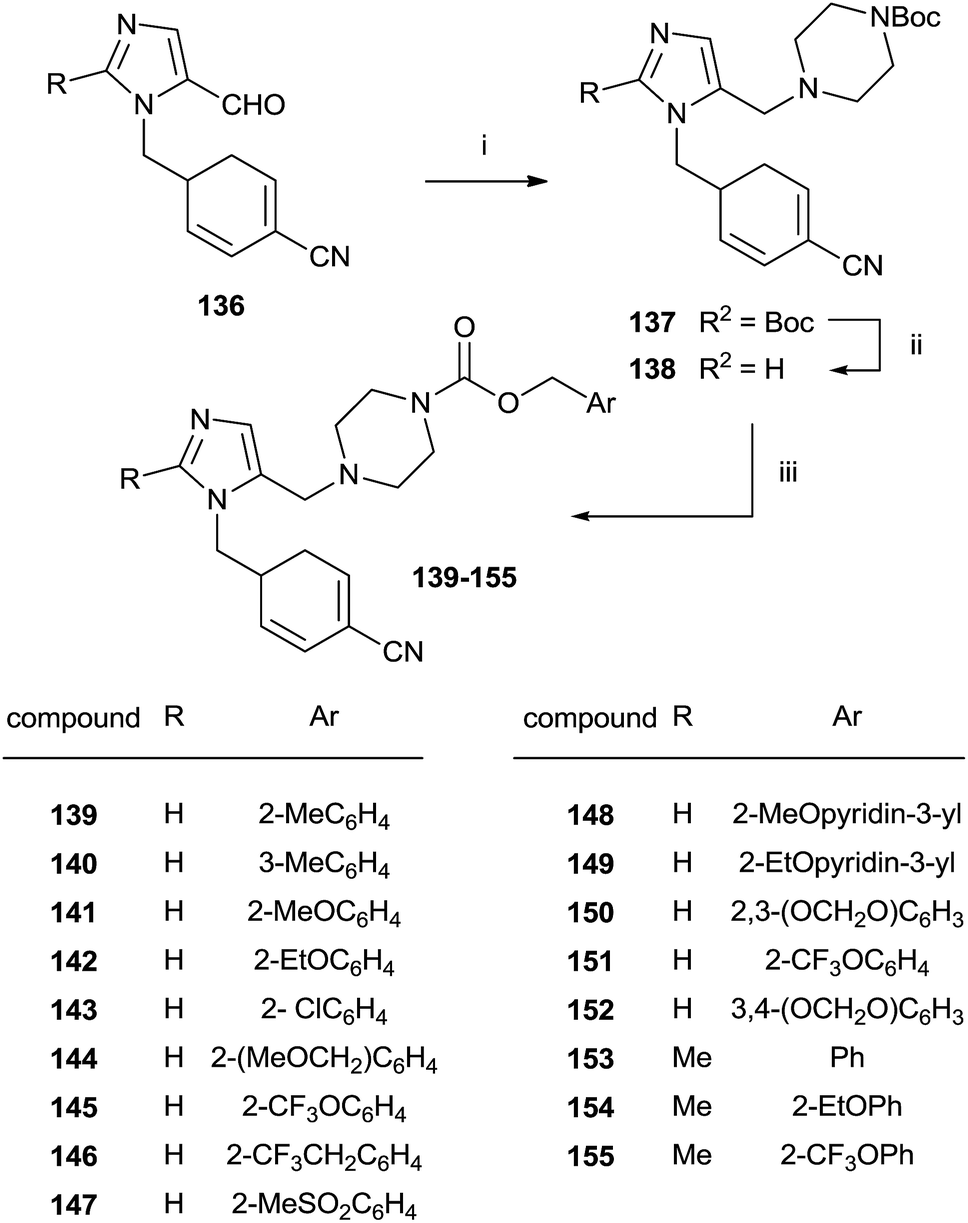 | ||
| Scheme 28 Reagents and conditions: (i) Ti(OPr)4, NaBH3CN, N-Boc-piperazine/THF–EtOH. (ii) TFA/CH2Cl2. (iii) Benzyl-(p-nitrophenyl)carbonate, DIEA/DMF, 80 °C. | ||
The resulting compounds 137 were deprotected by TFA furnishing the free amines 138 in good yields. The reaction of 138 with appropriate p-nitrophenyl carbonate yielded target compounds 139–155 in good yields.
Phosphonocarboxylate (PC), analogues of N-BP, characterized by a carboxylic and a phosphonic group on the same carbon, exhibit a chiral structure in contrast to the respective bisphosphonates, increasing the possibility of stereospecificity in their biological activity.125–128
Minodronic acid 156 (Fig. 4) was the first bisphosphonate developed and approved for osteoporosis treatment in Japan and today is available in a number of countries worldwide.
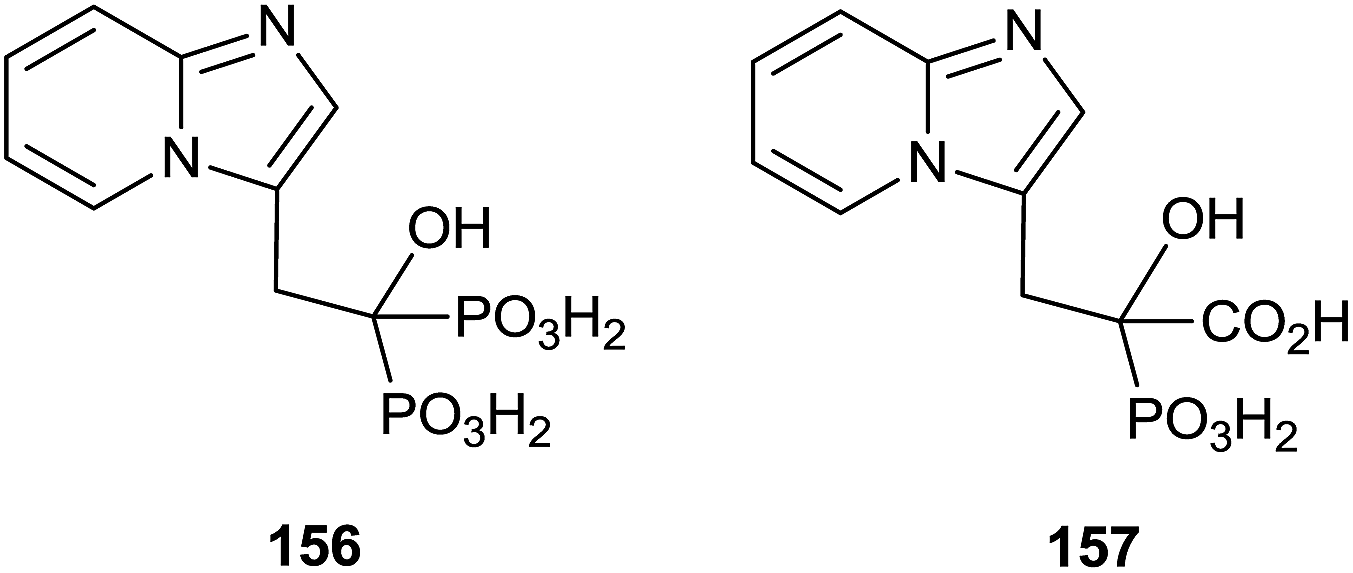 | ||
| Fig. 4 Minodronic acid and analog. | ||
McKenna and co-workers reported, in 2010, the synthesis of the analogue 157 starting from imidazo[1,2-a]pyridine 158 (Scheme 29).129 A Vilsmeier–Haack formylation of 158 furnished aldehyde 159 which was transformed into 160 and then the dehydroaminoester 162. Hydrolysis of 162 and further addition of diethyl phosphite to the resulting 163 furnished protected bisphosphonate 164 that was conveniently deprotected into 156. Resolution of 156 enantiomers by chiral HPLC furnished the (+)-156 isomer, which revealed a potent inhibitory activity of RGGT.
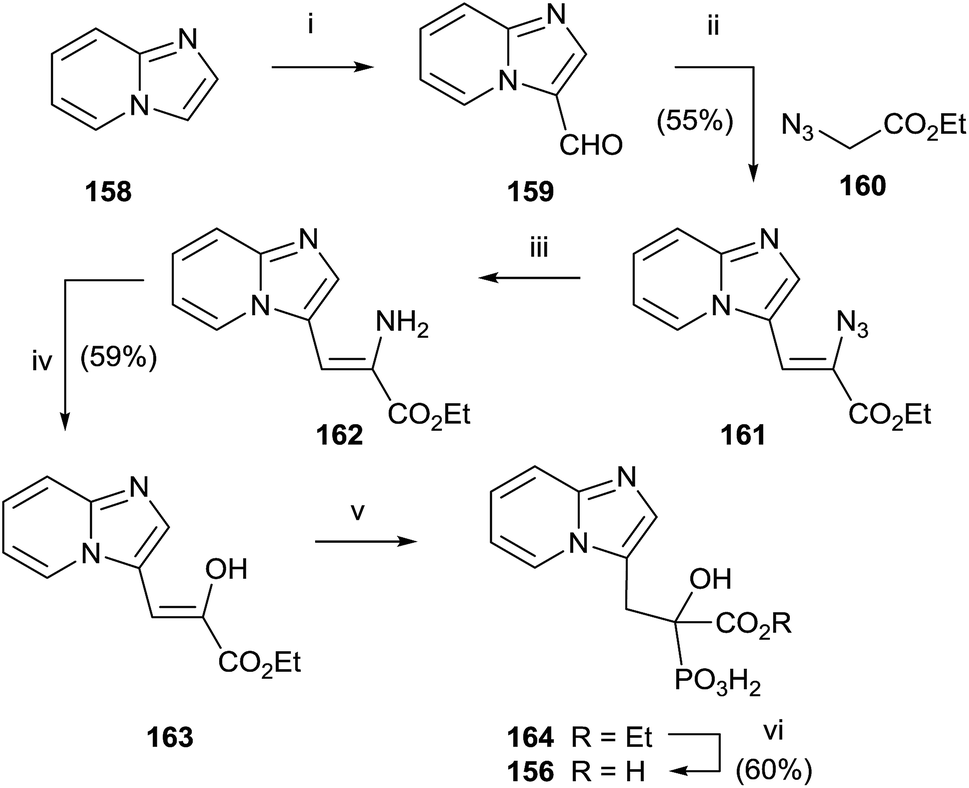 | ||
| Scheme 29 Reagents and conditions: (i) Vilsmeier reagent, 140 °C. (ii) EtONa/EtOH, from −30 °C to rt, 4 h. (iii) H2/10% Pd/C, MeOH, 2.5 h, rt. (iv) AcOH/H2O (7/1 v/v), 1.5 h, 0 °C. (v) (EtO)2P(O)H, 70 °C, 21 h. (vi) 6 N HCl, 6 h, reflux, (v) and (vi) combined. | ||
Replacement of the geminal hydroxyl moiety of PC with a halogen atom allowed to obtaining three α-halo derivatives 172–174 with potential biological activity against RGGT.130 The choice of this substitution had a significant impact to studying the role of the heterocyclic base respect to inhibition of enzyme activity. Starting from 159, a multi-step approach was carried out to synthesize common precursor 167. Halogenation with Selectfluor, N-chlorosuccinimide or N-bromosuccinimide provided haloderivatives 169–171. Further hydrolysis of the ester groups afforded the free acids 172–174 (Scheme 30).
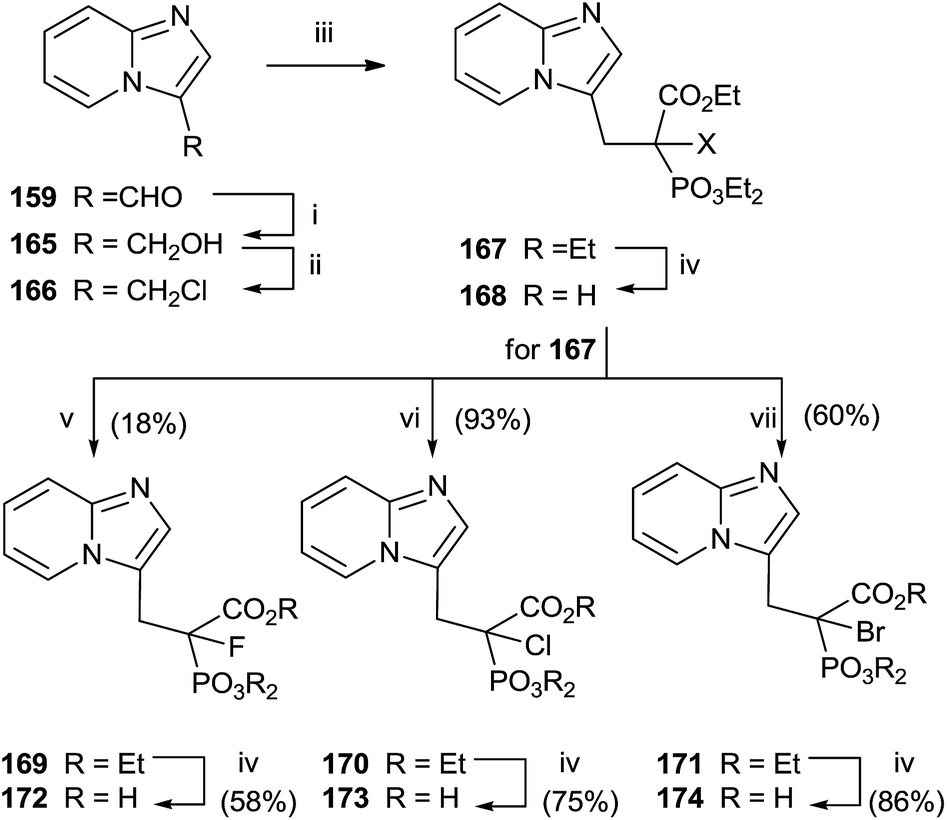 | ||
| Scheme 30 Reagents and conditions: (i) NaBH4, MeOH, reflux. (ii) SOCl2, reflux, (iii) triethyl phosphonoacetate, NaH, DMF, THF, 0 °C to rt. (iv) 12 M HCl, reflux. (v) Selectfluor, NaH, THF. (vi) N-Chlorosuccinimide, NaH, THF. (vii) N-Bromosuccinimide, NaH, THF. | ||
Phosphonocarboxylates can also be approached by routes commonly used for preparing bisphosphonates such as Arbuzov–Michaelis reaction of trialkyl phosphite with α-bromoesters,131 reaction of diethyl phosphite with α-ketoesters,129 and reaction of enolates and chlorodialkyl phosphites.132 In addition, the insertion of carboxylic function can be performed by using lithium alkylphosphonate and diethyl carbonate133 or CO2.134 Another methods include alkylation of trialkyl phosphonoacetate135 and functionalization of trialkyl 2-phosphonoacrylate via Michael-type addition.136
Recently, Coxon and colleagues used various approaches among those described to synthesize phosphonocarboxylates 175–178 showing some structural diversity (Fig. 5).137
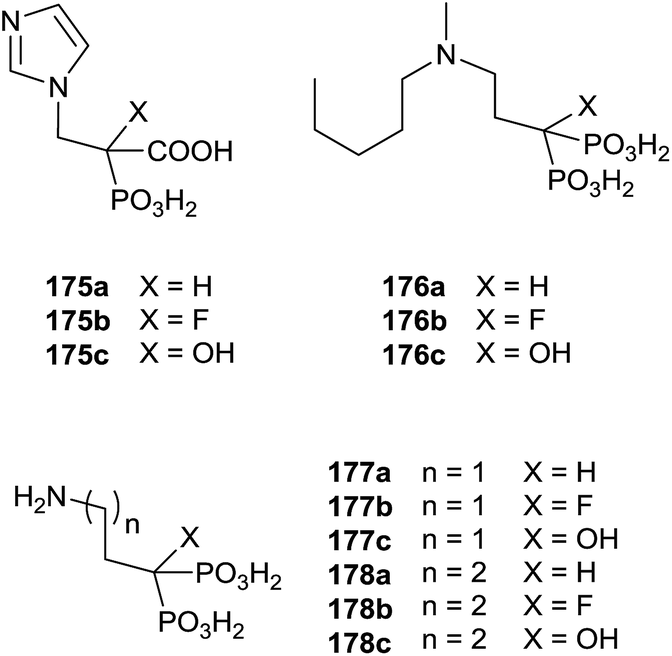 | ||
| Fig. 5 Phophonocarboxylates. | ||
Interestingly, the exchange of hydroxyl group with an alkyl chain of different length increased in the hydrophobicity enhancing the activity against GGPPS and FPPS. The synthesis of derivatives 181 was carried starting from α-alkyl phosphonoacetate 179 through an Arbuzov reaction and subsequent alkylation with picolyl chloride (Scheme 31). The corresponding free acids 181 were obtained upon hydrolysis with a 12 M solution of HCl at reflux.
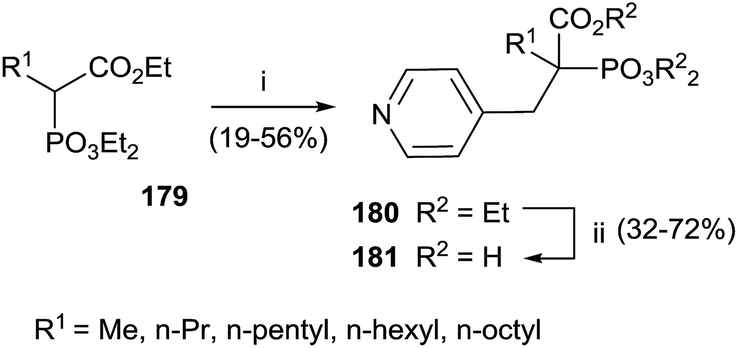 | ||
| Scheme 31 Reagents and conditions: (i) picolyl chloride, NaH, DMF/THF. (ii) 12 M HCl, reflux. | ||
A category of FPP analogues is constituted from inhibitors with a chain that mimic farnesyl diphosphate or farnesyl group in position C-3. Gibbs et al. have synthesized 3-cyclopropyl-3-desmethyl FPP (3-cpFPP) 187 and 3-tert-butyl-3-desmethyl FPP (3-tbFPP), 193 as potential irreversible inactivators of FTPase.138 The synthesis of 187 proceeded from 182. Coupling with cyclopropyl cyanocuprate 183 gave 184 in 71% yield. Reduction of the ester group with DIBALH produced the corresponding alcohol 185 that was chlorinated and immediately treated with tris(tetrabutyl ammonium) hydrogen diphosphate to give 187 (Scheme 32).
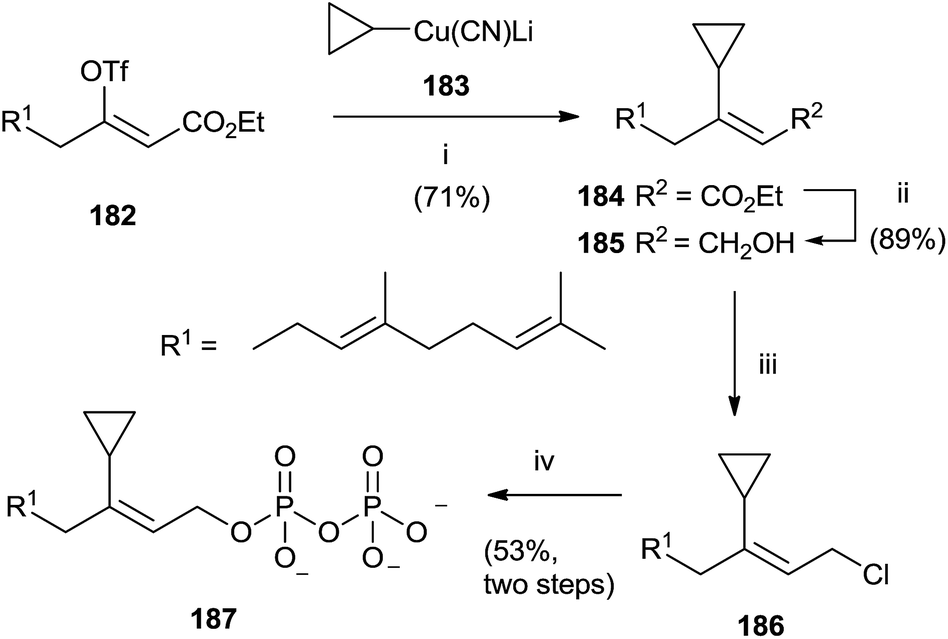 | ||
| Scheme 32 Reagents and conditions: (i) cyclopropyl cianocuprate, Et2O, −78 °C. (ii) DIBALH, PhMe, −78 °C. (iii) NCS, Me2S, CH2Cl2. (iv) (Bu4N)3HP2O7, CH3CN. | ||
Similarly, the synthesis of 193 was carried out through an initial reaction between 182 and the t-butyl cyanocuprate 188. Further steps of reduction, chlorination and pyrophosphorylation furnished 191 (Scheme 33).
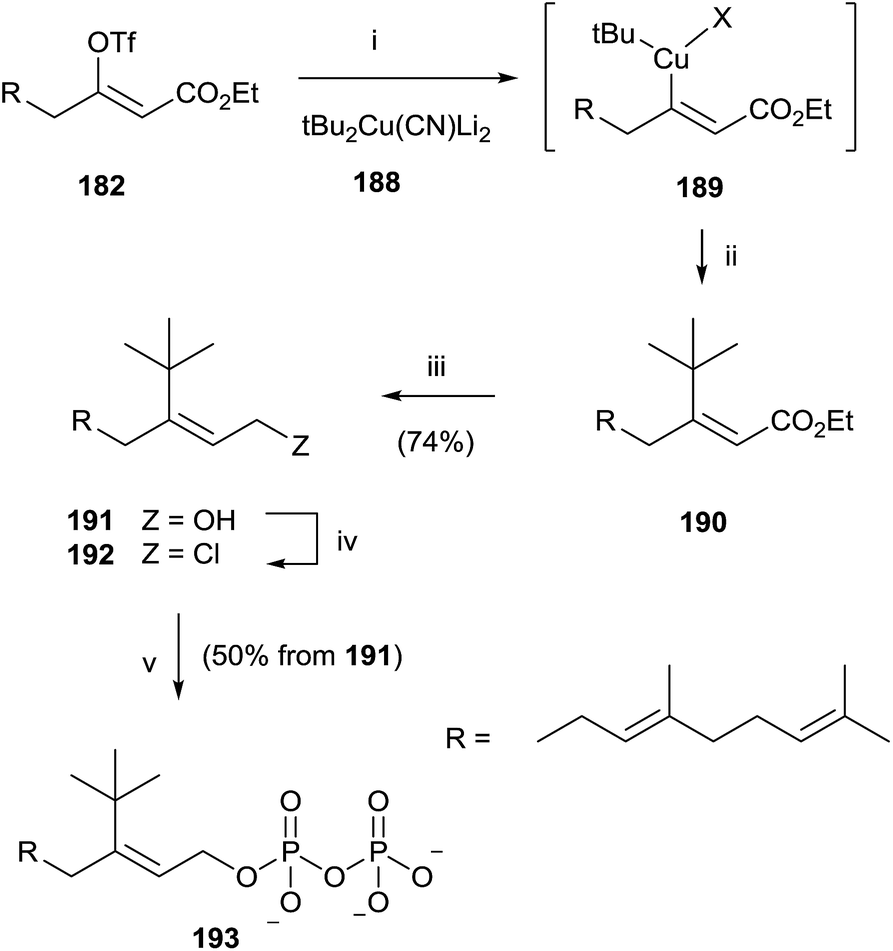 | ||
| Scheme 33 Reagents and conditions: (i) tBu2Cu(CN)Li2, −78 °C. (ii) Reductive elimination. (iii) DIBALH, PhMe, −78 °C. (iv) NCS, Me2S, CH2Cl2. (v) (Bu4N)3HP2O7, CH3CN. | ||
Gibbs and co-workers developed a stereoselective synthesis of cis-isoprenoid analogues such as 199 using the vinyl triflate method (Scheme 34).139,140 The fundamental step was the stereoselective preparation of triflate derivative 196 from the enolate of β-ketoester 194. The choice of solvent was found to have a significant impact on the trend of the reaction, not only in terms of yield, but also in the stereoselectivity. In fact, the use of DME instead of THF resulted in a loss of stereocontrol, while DMF as reaction solvent promoted only the stereoisomer 196 in excellent yield (93%). Coupling of 196 with tetramethyltin furnished the ester 197 that was reduced with DIBALH to the alcohol 198. Bromination and pyrophosphorylation gave 199.
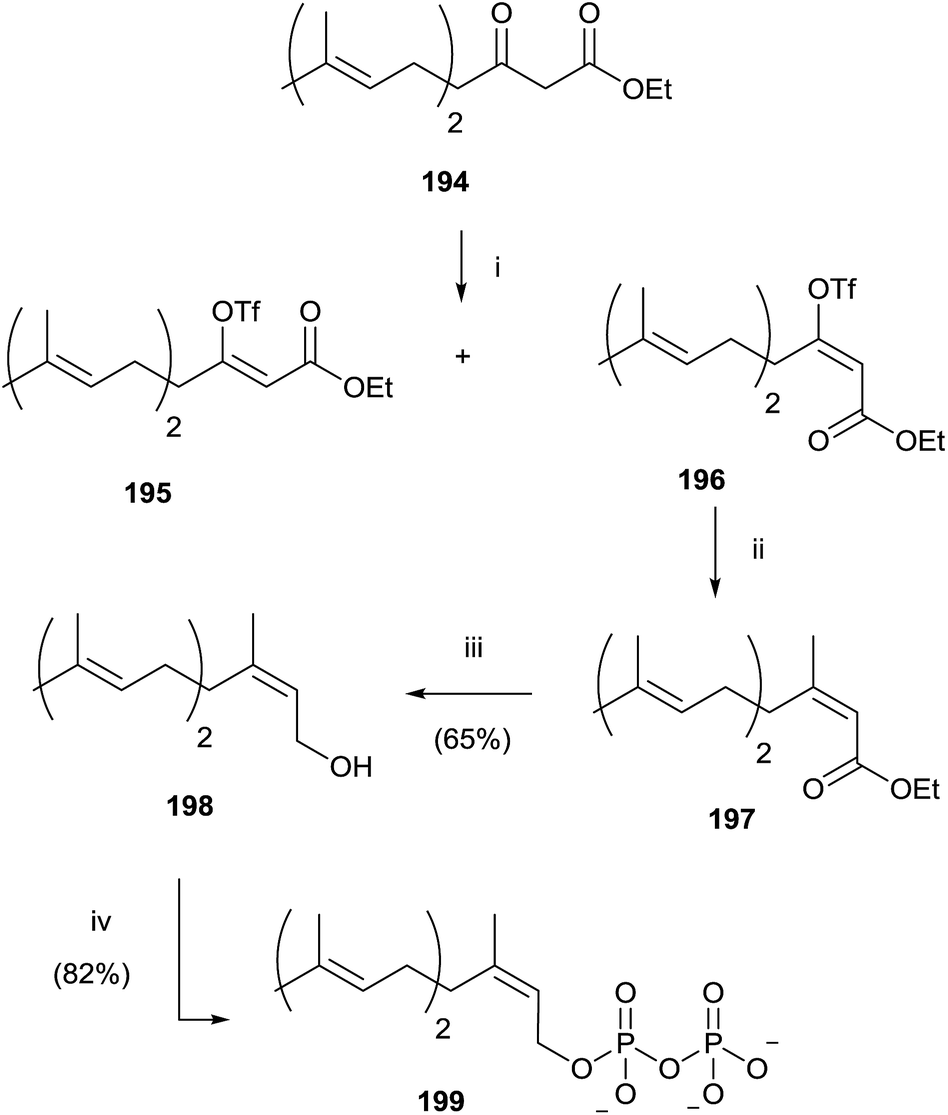 | ||
| Scheme 34 Reagents and conditions: (i) (Me3Si)2NK, solvent, −78 °C, PhN(SO2CF3)2. (ii) Me4Sn, CuI, Pd(AsPh3)2, NMP, 100 °C. (iii) DIBALH, toluene, −78 °C. (iv) a: NBS, CH2Cl2, Me2S; b: (Bu4N)3HP2O7, CH3CN. | ||
Compound 194 was employed to synthesize FPP analogues 202 with an alkyl or haloaryl chain in C-3 (Scheme 35).141
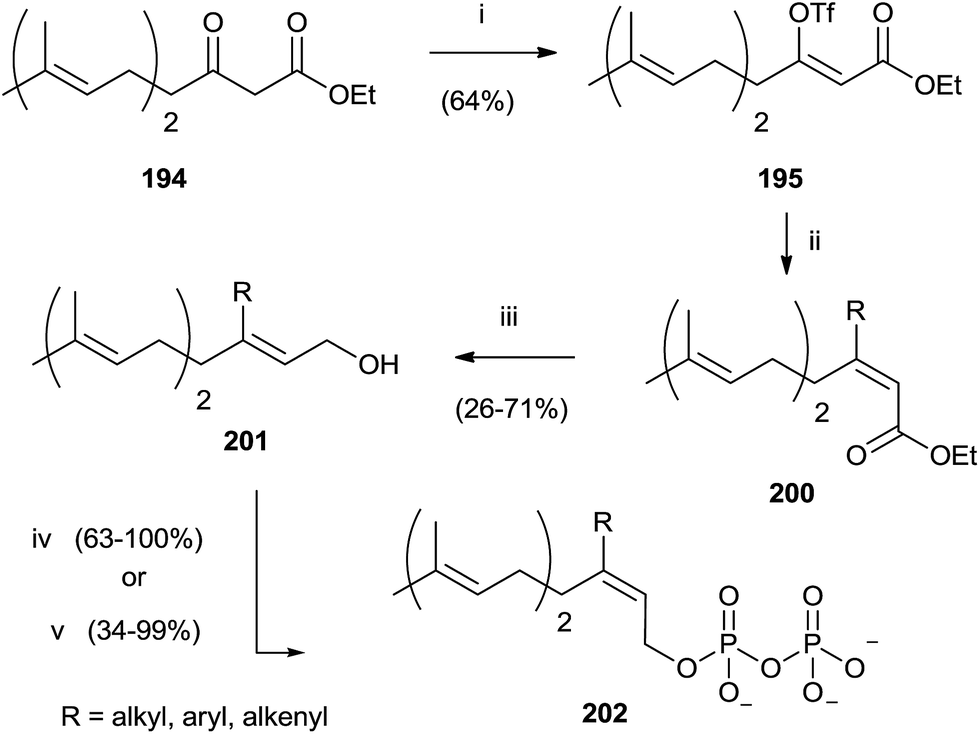 | ||
| Scheme 35 Reagents and conditions: (i) (Me3Si)2NK, THF, PhN(SO2CF3)2. (ii) RMgX, CuCN, ether. (iii) DIBALH, toluene. (iv) a: NBS, CH2Cl2, Me2S; b: (Bu4N)3HP2O7, CH3CN. | ||
Grignard reagents were made to react with triflate 195 in the presence of copper cyanide with yields from poor to high (39–91%). Further elaboration of intermediates 200 furnished pyrophosphates 202.
The presence of a sulfur atom as thiodiphosphate seems to promote the (S)-alkyl thiodiphosphates regioselectivity. Therefore, (S)-alkyl isopentenyl and allylic thiodiphosphates 204 and 206–209 were obtained by the procedure illustrated in Scheme 36.142 The reaction proceeded through slow addition of the isoprenoid derivatives 203 and 205a–d to an acetonitrile solution containing tris(tetra-n-butylammonium)thio-pyrophosphate (SPPi). The residue was passed through an ion-exchange column, replacing the tetra-n-butylammonium cation with ammonium in order to purify the final product by cellulose chromatography (57–89% yield).
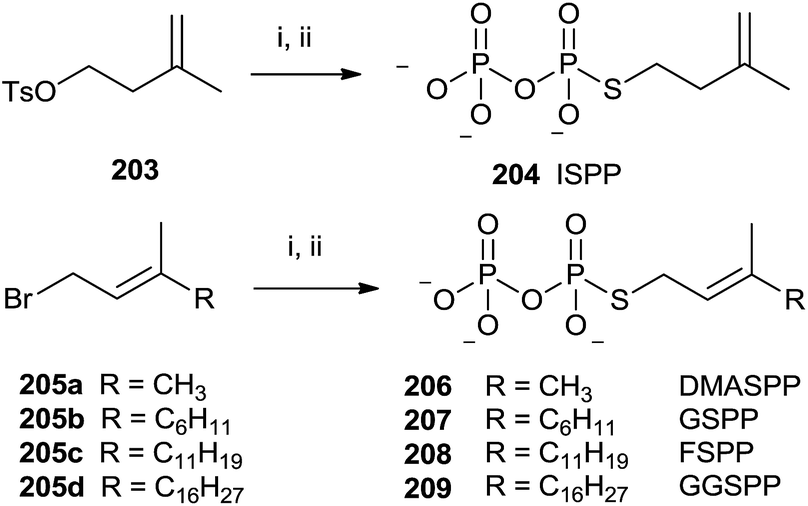 | ||
| Scheme 36 Reagents and conditions: (i) Tris-(tetra-n-butylammonium)thiopyro-phosphate, CH3CN. (ii) Dowex AG 50W-X8 (NH4+ form). | ||
Quinolines and salicylic derivatives have also been shown to be inhibitors of FFPS. In particular, the combination of quinolines with zoledronate seems to amplify the inhibition effect respect to the individual inhibitor. Therefore, a series of quinoline derivatives was synthesized starting from aniline 210 and ethyl 4-chloroacetoacetate 211 in PPA at 130 °C (Scheme 37).143
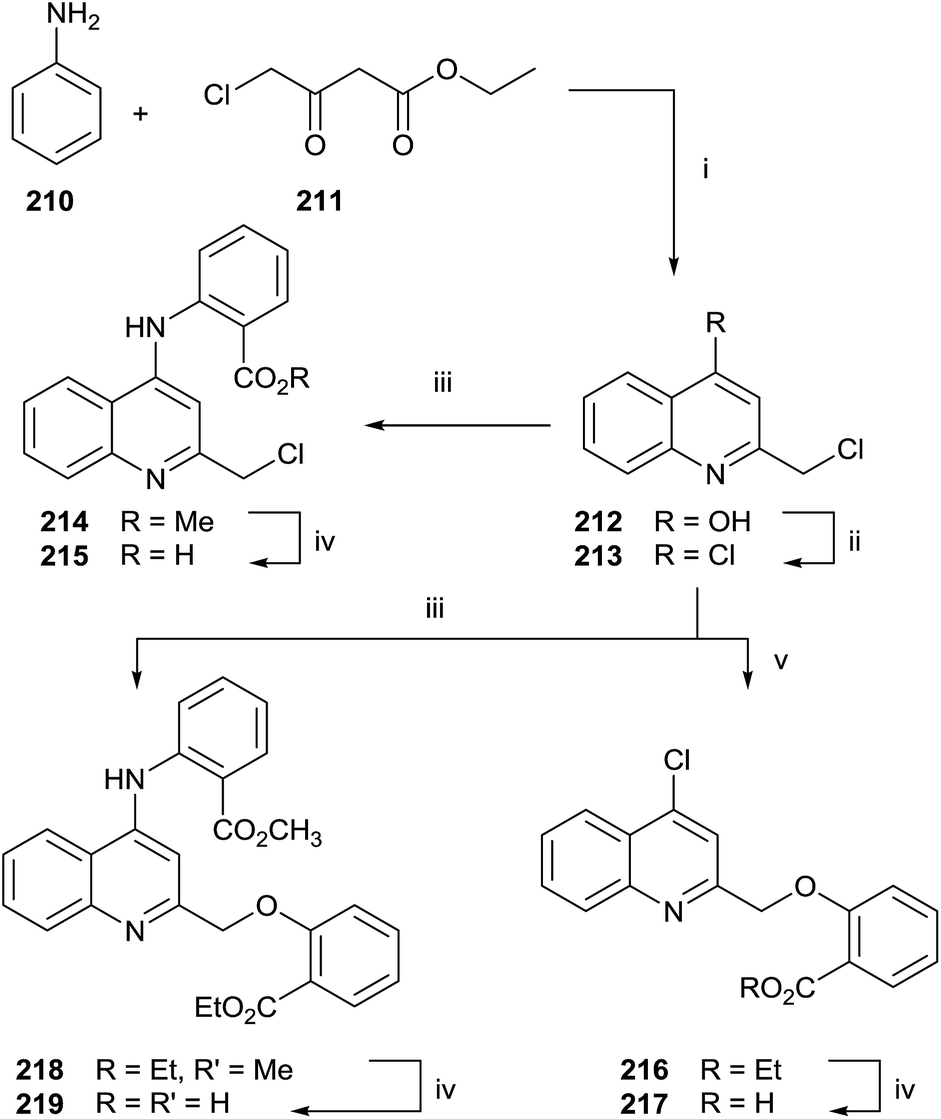 | ||
| Scheme 37 Reagents and conditions: (i) PPA, 130 °C, 1 h. (ii) POCl3, 100 °C, 3 h. (iii) Methyl 2-aminobenzoate, C2H5OH, conc. HCl, 5 h. (iv) THF/MeOH, LiOH, 3 h. (v) Ethyl-2-hydroxybenzoate, K2CO3, CH3CN, 90 °C, 5 h. | ||
The crude mixture was directly chlorinated by POCl3 at 100 °C. The resulting intermediate 213 was used without further purification in the following reaction with methyl 2-aminobenzoate 214 or ethyl 2-hydroxybenzoate 216. Different reaction routes allowed obtaining three different quinoline analogues 215, 217 and 219.
In 2015, Marzinzik et al. synthesized a library of salicylic acid derivatives exploiting the presence of a bromine atom in the para position of the carboxylic acid in the phenyl ring.144 Compound 220 represented the starting point to synthesize a variety of salicylic acid analogues through an initial reaction with boronic acid by microwave irradiation. The final step with LiOH in MeOH/THF was promoted by the use of microwaves yielding substrates 221 (Scheme 38).
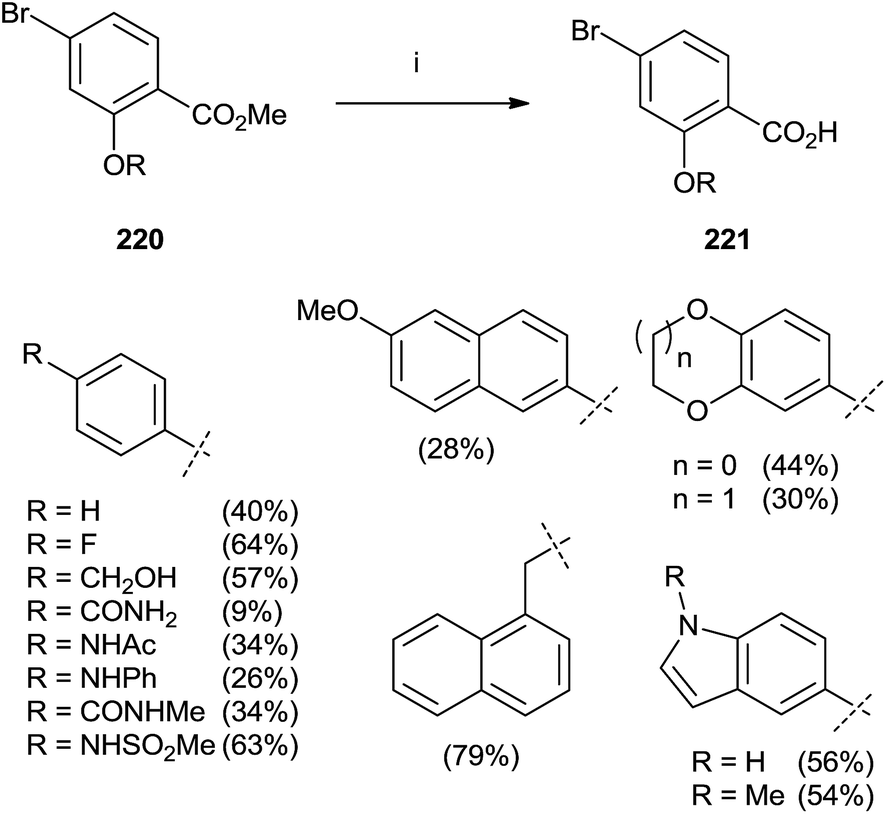 | ||
| Scheme 38 Reagents and conditions: (i) 1: boronic acid, Pd(PPh3)Cl2, Na2CO3, DME/EtOH, H2O, MW (110 °C), 10 min. (ii) LiOH, MeOH/THF, MW (110 °C), 12 min. | ||
The same authors prepared a variety quinoline substrates 222–228 via different synthetic strategies (Fig. 6).
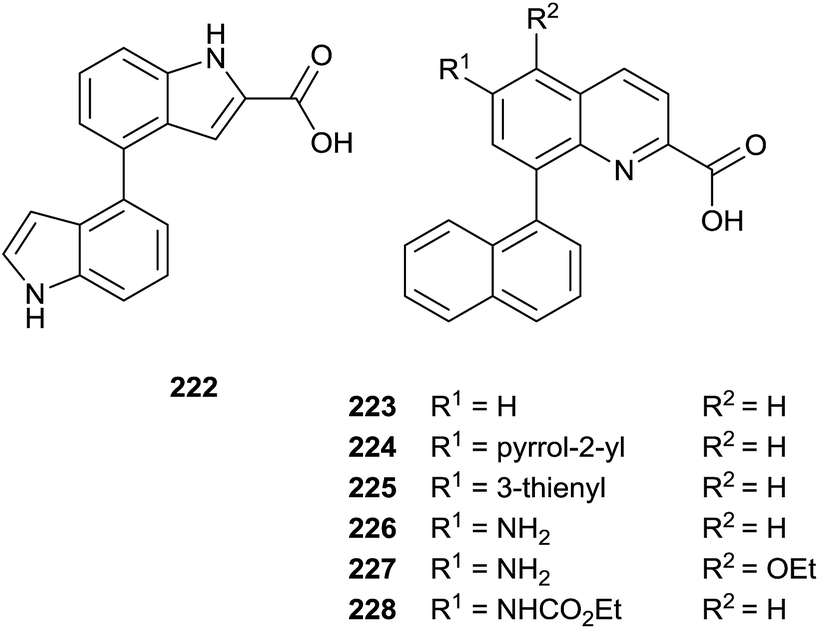 | ||
| Fig. 6 Quinoline substrates. | ||
In particular, a unique method allowed the simultaneous synthesis of three active quinoline compounds 226–228 starting from nitration of 8-bromoquinoline-2(1H)-one 229 (Scheme 39).
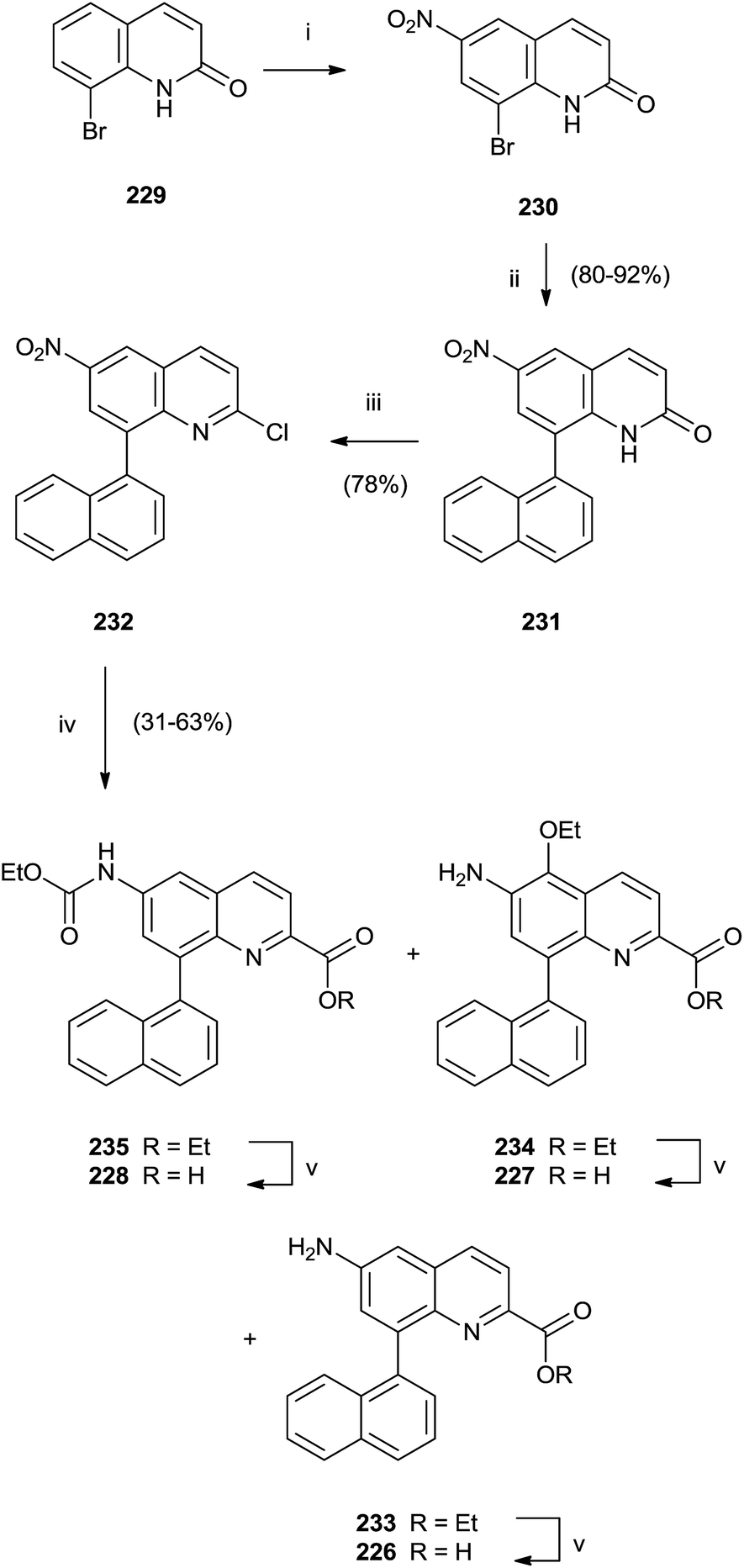 | ||
| Scheme 39 Reagents and conditions: (i) fuming HNO3, TFA, from 0 °C to rt. (ii) K2CO3, boronic acid, [(C6H5)3P]2PdCl2, DMF/H2O, 80–92 °C. (iii) POCl3, Et4NCl, N,N-dimethylaniline, CH3CN. (iv) CO, [(C6H5)3P]2PdCl2, Et3N, EtOH, 110 °C. (v) LiOH, H2O/dioxane. | ||
Suzuki coupling of the crude of the nitro-derivatives 230 with naphthyl boronate gave 231 that was converted in 232 with POCl3 in the presence of N,N-dimethylaniline and Et4NCl. Palladium-catalyzed carbonylation and reduction of nitro group of 232 produced the intermediate esters 233–235 in moderate yield that were hydrolyzed with LiOH to perform quinolines 226–228.
Concluding remarks
Historically, bisphosphonates are benchmark drugs for the treatment of a variety of bone disorders including osteoporosis and bone metastasis. Their inhibitory activity of the isoprenoid biosynthesis resulted in other important applications as modulators of the metabolism of several protozoa parasites thus also being potential therapeutic agents for the treatment of trypanosomiasis (Chagas disease), leishmaniasis, toxoplasmosis and malaria. Bisphosphonates could also be useful in the treatment of other diseases such as breast cancer, myeloma multiple and progeria. Both clinical success in bone disorders and expectative for other diseases prompted the enormous synthetic activity directed to the preparation of bisphosphonates and more recently, nitrogen-containing bisphosphonates which are demonstrated better biological properties. In general, the reaction of carboxylic acids with phosphorous reagents like POCl3 is the preferred approach to bisphophonates. The reaction, however, is very sensitive to steric hindrance and in such cases an Arbuzov-type reaction results more advisable. More recently, the use of tetraethylvinylidenebisphosphonate as the source of phosphorylated part of the molecule has facilitated enormously the access to a variety of bisphophonates. Moreover, the use of such a reagent presented a high tolerance to a variety of functional groups. For the particular case of bisphosphonates lacking the hydroxyl group the alkylation of tetralkylmethyl bisphosphonate is preferred; however, elimination reactions are common undesired lateral processes that, on the other hand, can be eliminated by using precise reaction conditions.The presence of two phosphate units, in addition to difficult manipulation and purification, limits the oral bioavailability and contribute to undesired side-effects. In this respect, novel bisphosphonate analogues that selectively target FPPS and GGPPS enzymes might provide notable advantages over the currently used drugs and this is now the subject of intense investigation in medicinal chemistry and chemical biology. Achieving this target implies to have at disposition a series of synthetic strategies that allow not only the preparation of the target compound but also structural variations of the parent compound that provide lead compounds for further studies for treatment of the several diseases related to isoprenoid biosynthesis.
Acknowledgements
This work was supported by the MINECO and FEDER Program (Madrid, Spain, project CTQ2016-76155-R) and the Gobierno de Aragon (Zaragoza, Spain. Bioorganic Chemistry Group. E-10). We thank the Italian Ministry of University and Scientific Research (MIUR) for a doctoral grant and the University of Calabria for financial support.Notes and references
- B. M. Lange, T. Rujan, W. Martin and R. Croteau, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13172–13177 CrossRef CAS PubMed.
- R. E. Summons, L. L. Jahnke, J. M. Hope and G. A. Logan, Nature, 1999, 400, 554–557 CrossRef CAS PubMed.
- J. C. Sacchettini and C. D. Poulter, Science, 1997, 277, 1788–1789 CrossRef CAS PubMed.
- M. Rodriguez-Concepcion, Methods Mol. Biol., 2014, 1153, 1–5 CAS.
- D. Tholl, Adv. Biochem. Eng./Biotechnol., 2015, 148, 63–106 CrossRef CAS PubMed.
- P. Sakthivel, N. Sharma, P. Klahn, M. Gereke and D. Bruder, Curr. Med. Chem., 2016, 23, 1549–1570 CrossRef CAS PubMed.
- Natural products and cancer signaling: isoprenoids, polyphenols and flavonoids, ed. S. Z. Bathaie and F. Tamanoi, Academic Press, London, 2014 Search PubMed.
- T. Kuzuyama and H. Seto, Proc. Jpn. Acad., Ser. B, 2012, 88, 41–52 CrossRef CAS PubMed.
- M. Galata and S. Mahmoud, Stud. Nat. Prod. Chem., 2012, 37, 135–171 CAS.
- Biotechnology of Isoprenoids, ed. J. Schrader and J. Bohlmann, Springer, Heidelberg, 2015 Search PubMed.
- A. Boronat and M. Rodriguez-Concepcion, Adv. Biochem. Eng./Biotechnol., 2015, 148, 3–18 CrossRef CAS PubMed.
- D.-K. Ro, in Plant Metabolism and Biotechnology, ed. H. Ashihara, A. Crozier and A. Komamine, John Wiley & Sons, Ltd, Chichester, UK, 2011, pp. 217–240 Search PubMed.
- E. Oldfield, Acc. Chem. Res., 2010, 43, 1216–1226 CrossRef CAS PubMed.
- J. Park, A. N. Matralis, A. M. Berghuis and Y. S. Tsantrizos, Front. Chem., 2014, 2, 1–21 Search PubMed.
- I. Hale, P. M. O'Neill, N. G. Berry, A. Odom and R. Sharma, MedChemComm, 2012, 3, 418–433 RSC.
- A. M. Guggisberg, R. E. Amthor and A. R. Odom, Eukaryotic Cell, 2014, 13, 1348–1359 CrossRef PubMed.
- T. Qidwai, F. Jamal, M. Y. Khan and B. Sharma, Biochem. Res. Int., 2014, 657189 Search PubMed.
- V. G. Duschak, Recent Pat. Anti-Infect. Drug Discovery, 2011, 6, 216–259 CrossRef CAS.
- M. Sanchez-Sanchez, G. Rivera, E. A. Garcia and V. Bocanegra-Garcia, Mini-Rev. Org. Chem., 2016, 13, 227–243 CrossRef CAS.
- M. O. Kim, X. Feng, F. Feixas, W. Zhu, S. Lindert, S. Bogue, W. Sinko, C. de Oliveira, G. Rao, E. Oldfield and J. A. McCammon, Chem. Biol. Drug Des., 2015, 85, 756–769 CAS.
- W. Wang and E. Oldfield, Angew. Chem., Int. Ed., 2014, 53, 4294–4310 CrossRef CAS PubMed.
- J. S. Burg and P. J. Espenshade, Prog. Lipid Res., 2011, 50, 403–410 CrossRef CAS PubMed.
- U. T. T. Nguyen, A. Goodall, K. Alexandrov and D. Abankwa, Protein Rev., 2011, 13, 1–37 CAS.
- J. Desai, Y. Wang, K. Wang, S. R. Malwal and E. Oldfield, ChemMedChem, 2016, 11, 2205–2215 CrossRef CAS PubMed.
- S. A. Holstein and R. J. Hohl, Enzymes, 2011, 30, 301–319 CAS.
- A. Srivastava, P. Mukherjee, P. V. Desai, M. A. Avery and B. L. Tekwani, Infect. Disord.: Drug Targets, 2008, 8, 16–30 CrossRef CAS.
- K. M. Swanson and R. J. Hohl, Curr. Cancer Drug Targets, 2006, 6, 15–37 CrossRef CAS PubMed.
- T. Todenhoefer, J. Hennenlotter, U. Kuehs, V. Gerber, G. Gakis, U. Vogel, S. Aufderklamm, A. Merseburger, J. Knapp, A. Stenzl and C. Schwentner, World J. Urol., 2013, 31, 345–350 CrossRef CAS PubMed.
- M. K. Tsoumpra, J. R. Muniz, B. L. Barnett, A. A. Kwaasi, E. S. Pilka, K. L. Kavanagh, A. Evdokimov, R. L. Walter, F. Von Delft, F. H. Ebetino, U. Oppermann, R. G. G. Russell and J. E. Dunford, Bone, 2015, 81, 478–486 CrossRef CAS PubMed.
- K. L. Kavanagh, K. Guo, J. E. Dunford, X. Wu, S. Knapp, F. H. Ebetino, M. J. Rogers, R. G. G. Russell and U. Oppermann, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 7829–7834 CrossRef CAS PubMed.
- R. Eastell, J. S. Walsh, N. B. Watts and E. Siris, Bone, 2011, 49, 82–88 CrossRef CAS PubMed.
- I. R. Reid and D. J. Hosking, Bone, 2011, 49, 89–94 CrossRef CAS PubMed.
- M. Colina, G. Ciancio and F. Trotta, Clin. Med.: Ther., 2009, 1, 1451–1456 CAS.
- M. Wilke, A. Gobel, M. Rauner, P. Benad-Mehner, D. Rachner Tilman, N. Schutze, S. Fussel, P. Hadji and C. Hofbauer Lorenz, Journal of Bone Oncology, 2014, 3, 10–17 CrossRef PubMed.
- C.-Q. Du, X.-W. Liu, G.-Z. Zeng, H.-F. Jin and L.-J. Tang, Int. J. Mol. Med., 2015, 35, 1767–1772 CAS.
- C. M. Szabo, Y. Matsumura, S. Fukura, M. B. Martin, J. M. Sanders, S. Sengupta, J. A. Cieslak, T. C. Loftus, C. R. Lea, H.-J. Lee, A. Koohang, R. M. Coates, H. Sagami and E. Oldfield, J. Med. Chem., 2002, 45, 2185–2196 CrossRef CAS PubMed.
- J. D. Artz, A. K. Wernimont, J. E. Dunford, M. Schapira, A. Dong, Y. Zhao, J. Lew, R. G. G. Russell, F. H. Ebetino, U. Oppermann and R. Hui, J. Biol. Chem., 2011, 286, 3315–3322 CrossRef CAS PubMed.
- K. L. Kavanagh, J. E. Dunford, G. Bunkoczi, R. G. G. Russell and U. Oppermann, J. Biol. Chem., 2006, 281, 22004–22012 CrossRef CAS PubMed.
- M. F. Mabanglo, M. A. Hast, N. B. Lubock, H. W. Hellinga and L. S. Beese, Protein Sci., 2014, 23, 289–301 CrossRef CAS PubMed.
- R. G. G. Russell, Ann. N. Y. Acad. Sci., 2006, 1068, 367–401 CrossRef CAS PubMed.
- R. G. G. Russell, Bone, 2011, 49, 2–19 CrossRef CAS PubMed.
- L. Widler, W. Jahnke and J. R. Green, Anti-Cancer Agents Med. Chem., 2012, 12, 95–101 CrossRef CAS PubMed.
- S. Nishida, Y. Fujii, S. Yoshioka, S. Kikuichi, M. Tsubaki and K. Irimajiri, Life Sci., 2003, 73, 2655–2664 CrossRef CAS PubMed.
- S. E. Sen, L. Wood, R. Jacob, A. Xhambazi, B. Pease, A. Jones, T. Horsfield, A. Lin and M. Cusson, Insect Biochem. Mol. Biol., 2015, 63, 113–123 CrossRef CAS PubMed.
- M. J. Rogers, J. C. Crockett, F. P. Coxon and J. Monkkonen, Bone, 2011, 49, 34–41 CrossRef CAS PubMed.
- W. Jahnke, J.-M. Rondeau, S. Cotesta, A. Marzinzik, X. Pelle, M. Geiser, A. Strauss, M. Goette, F. Bitsch, R. Hemmig, C. Henry, S. Lehmann, J. F. Glickman, T. P. Roddy, S. J. Stout and J. R. Green, Nat. Chem. Biol., 2010, 6, 660–666 CrossRef CAS PubMed.
- S. L. Graham, Expert Opin. Ther. Pat., 1995, 5, 1269–1285 CrossRef CAS.
- A. J. Roelofs, K. Thompson, F. H. Ebetino, M. J. Rogers and F. P. Coxon, Curr. Pharm. Des., 2010, 16, 2950–2960 CrossRef CAS PubMed.
- K. Thompson and M. J. Rogers, Clin. Rev. Bone Miner. Metab., 2007, 5, 130–144 CrossRef CAS.
- M. J. Rogers, Curr. Pharm. Des., 2003, 9, 2643–2658 CrossRef CAS PubMed.
- M. T. Drake, B. L. Clarke and S. Khosla, Mayo Clin. Proc., 2008, 83, 1032–1045 CrossRef CAS PubMed.
- J. B. Rodriguez, B. N. Falcone and S. H. Szajnman, Expert Opin. Drug Discovery, 2016, 11, 307–320 CrossRef CAS PubMed.
- S. Sun and C. E. McKenna, Expert Opin. Ther. Pat., 2011, 21, 1433–1451 CrossRef CAS PubMed.
- G. R. Kieczykowski, R. B. Jobson, D. G. Melillo, D. F. Reinhold, V. J. Grenda and I. Shinkai, J. Org. Chem., 1995, 60, 8310–8312 CrossRef CAS.
- E. Maltezou, M. Stylianou, S. Roy, C. Drouza and A. D. Keramidas, Bioinorg. Chem. Appl., 2010, 563875 Search PubMed.
- M. B. Martin, J. S. Grimley, J. C. Lewis, H. T. Heath III, B. N. Bailey, H. Kendrick, V. Yardley, A. Caldera, R. Lira, J. A. Urbina, S. N. J. Moreno, R. Docampo, S. L. Croft and E. Oldfield, J. Med. Chem., 2001, 44, 909–916 CrossRef CAS PubMed.
- E. Migianu-Griffoni, I. Chebbi, S. Kachbi, M. Monteil, O. Sainte-Catherine, F. Chaubet, O. Oudar and M. Lecouvey, Bioconjugate Chem., 2014, 25, 224–230 CrossRef CAS PubMed.
- Y. Xie, H. Ding, L. Qian, X. Yan, C. Yang and Y. Xie, Bioorg. Med. Chem. Lett., 2005, 15, 3267–3270 CrossRef CAS PubMed.
- P. A. Turhanen and J. J. Vepsalainen, Beilstein J. Org. Chem., 2006, 2(2) DOI:10.1186/1860-5397-2-2.
- E. Palma, J. D. G. Correia, B. L. Oliveira, L. Gano, I. C. Santos and I. Santos, Dalton Trans., 2011, 40, 2787–2796 RSC.
- H. Schott, D. Goltz, T. C. Schott, C. Jauch and R. A. Schwendener, Bioorg. Med. Chem., 2011, 19, 3520–3526 CrossRef CAS PubMed.
- M. Lecouvey, C. Dufau, D. El Manouni and Y. Leroux, Nucleosides Nucleotides, 1999, 18, 2109–2120 CAS.
- Y. Yang, N. Liu, J. Liao, M. Pu, Y. Liu, M. Wei and J. Jin, J. Radioanal. Nucl. Chem., 2010, 283, 329–335 CrossRef CAS.
- G. Xu, Y. Xie and X. Wu, Org. Prep. Proced. Int., 2004, 36, 185–187 CrossRef CAS.
- D. A. Mustafa, B. A. Kashemirov and C. E. McKenna, Tetrahedron Lett., 2011, 52, 2285–2287 CrossRef CAS.
- S. Deprele, B. A. Kashemirov, J. M. Hogan, F. H. Ebetino, B. L. Barnett, A. Evdokimov and C. E. McKenna, Bioorg. Med. Chem. Lett., 2008, 18, 2878–2882 CrossRef CAS PubMed.
- M. B. Martin, J. M. Sanders, H. Kendrick, K. de Luca-Fradley, J. C. Lewis, J. S. Grimley, E. M. Van Brussel, J. R. Olsen, G. A. Meints, A. Burzynska, P. Kafarski, S. L. Croft and E. Oldfield, J. Med. Chem., 2002, 45, 2904–2914 CrossRef CAS PubMed.
- J. M. Sanders, A. O. Gómez, J. Mao, G. A. Meints, E. M. Van Brussel, A. Burzynska, P. Kafarski, D. González-Pacanowska and E. Oldfield, J. Med. Chem., 2003, 46, 5171–5183 CrossRef CAS PubMed.
- S. Ghosh, J. M. W. Chan, C. R. Lea, G. A. Meints, J. C. Lewis, Z. S. Tovian, R. M. Flessner, T. C. Loftus, I. Bruchhaus, H. Kendrick, S. L. Croft, R. G. Kemp, S. Kobayashi, T. Nozaki and E. Oldfield, J. Med. Chem., 2004, 47, 175–187 CrossRef CAS PubMed.
- J. M. Sanders, S. Ghosh, J. M. W. Chan, G. Meints, H. Wang, A. M. Raker, Y. Song, A. Colantino, A. Burzynska, P. Kafarski, C. T. Morita and E. Oldfield, J. Med. Chem., 2004, 47, 375–384 CrossRef CAS PubMed.
- Y. Ling, G. Sahota, S. Odeh, J. M. W. Chan, F. G. Araujo, S. N. J. Moreno and E. Oldfield, J. Med. Chem., 2005, 48, 3130–3140 CrossRef CAS PubMed.
- J. M. Sanders, Y. Song, J. M. W. Chan, Y. Zhang, S. Jennings, T. Kosztowski, S. Odeh, R. Flessner, C. Schwerdtfeger, E. Kotsikorou, G. A. Meints, A. O. Gomez, D. Gonzalez-Pacanowska, A. M. Raker, H. Wang, E. R. van Beek, S. E. Papapoulos, C. T. Morita and E. Oldfield, J. Med. Chem., 2005, 48, 2957–2963 CrossRef CAS PubMed.
- C. K. M. Chen, M. P. Hudock, Y. Zhang, R.-T. Guo, R. Cao, J. H. No, P.-H. Liang, T.-P. Ko, T.-H. Chang, S.-c. Chang, Y. Song, J. Axelson, A. Kumar, A. H. J. Wang and E. Oldfield, J. Med. Chem., 2008, 51, 5594–5607 CrossRef CAS PubMed.
- J. Mao, S. Mukherjee, Y. Zhang, R. Cao, J. M. Sanders, Y. Song, Y. Zhang, G. A. Meints, Y. G. Gao, D. Mukkamala, M. P. Hudock and E. Oldfield, J. Am. Chem. Soc., 2006, 128, 14485–14497 CrossRef CAS PubMed.
- L.-S. Zhou, K.-W. Yang, L. Feng, J.-M. Xiao, C.-C. Liu, Y.-L. Zhang and M. W. Crowder, Bioorg. Med. Chem. Lett., 2013, 23, 949–954 CrossRef CAS PubMed.
- G. Keglevich, A. Grun, R. Kovacs, K. Koos, B. Szolnoki, S. Garadnay, J. Neu, L. Drahos and I. Greiner, Lett. Drug Des. Discovery, 2012, 9, 345–351 CrossRef CAS.
- H. R. Hudson, N. J. Wardle, S. W. A. Bligh, I. Greiner, A. Grun and G. Keglevich, Mini-Rev. Med. Chem., 2012, 12, 313–325 CrossRef CAS PubMed.
- R. Kovacs, A. Gruen, S. Garadnay, I. Greiner and G. Keglevich, Green Process. Synth., 2014, 3, 111–116 CAS.
- G. Keglevich, A. Gruen, K. Aradi, S. Garadnay and I. Greiner, Tetrahedron Lett., 2011, 52, 2744–2746 CrossRef CAS.
- M. Recher, A. P. Barboza, Z.-H. Li, M. Galizzi, M. Ferrer-Casal, S. H. Szajnman, R. Docampo, S. N. J. Moreno and J. B. Rodriguez, Eur. J. Med. Chem., 2013, 60, 431–440 CrossRef CAS PubMed.
- K. Troev, P. Todorov, E. Naydenova, V. Mitova and N. Vassilev, Phosphorus, Sulfur Silicon Relat. Elem., 2013, 188, 1147–1155 CrossRef CAS.
- G. Keglevich, A. Gruen, I. G. Molnar and I. Greiner, Heteroat. Chem., 2011, 22, 640–648 CrossRef CAS.
- R. Lenin, R. M. Raju, D. V. N. S. Rao and U. K. Ray, Med. Chem. Res., 2013, 22, 1624–1629 CrossRef CAS.
- G. Keglevich, A. Grun, S. Garadnay and I. Greiner, Phosphorus, Sulfur Silicon Relat. Elem., 2015, 190, 2116–2124 CrossRef CAS.
- L. Widler, K. A. Jaeggi, M. Glatt, K. Mueller, R. Bachmann, M. Bisping, A.-R. Born, R. Cortesi, G. Guiglia, H. Jeker, R. Klein, U. Ramseier, J. Schmid, G. Schreiber, Y. Seltenmeyer and J. R. Green, J. Med. Chem., 2002, 45, 3721–3738 CrossRef CAS PubMed.
- R. Kovacs, A. Gruen, O. Nemeth, S. Garadnay, I. Greiner and G. Keglevich, Heteroat. Chem., 2014, 25, 186–193 CrossRef CAS.
- S. S. Ratrout, A. e. M. Al Sarabi and K. A. Sweidan, Pharm. Chem. J., 2015, 48, 835–839 CrossRef CAS.
- S. K. Singh, N. Manne, P. C. Ray and M. Pal, Beilstein J. Org. Chem., 2008, 4(42) DOI:10.3762/bjoc.4.42.
- A. Grun, R. Kovacs, S. Garadnay, I. Greiner and G. Keglevich, Lett. Drug Des. Discovery, 2015, 12, 253–258 CrossRef CAS.
- A. Grun, R. Kovacs, D. I. Nagy, S. Garadnay, I. Greiner and G. Keglevich, Lett. Drug Des. Discovery, 2015, 12, 78–84 CrossRef CAS.
- D. V. N. S. Rao, R. Dandala, R. Lenin, M. Sivakumaran, S. Shivashankar and A. Naidu, ARKIVOC, 2007, 34–38 CAS.
- M. A. Motaleb, A. S. A. Adli, M. El-Tawoosy, M. H. Sanad and M. Abd Allah, J. Labelled Compd. Radiopharm., 2016, 59, 157–163 CrossRef CAS PubMed.
- M. Lecouvey and Y. Leroux, Heteroat. Chem., 2000, 11, 556–561 CrossRef CAS.
- Actually, the Arbuzov reaction (or Michaelis–Arbuzov reaction) is the reaction of a trialkyl phosphite with an alkyl halide to form a phosphonate. In this case, the reaction takes place with an acyl chloride to give a ketophosphonate.
- M. Lecouvey, I. Mallard, T. Bailly, R. Burgada and Y. Leroux, Tetrahedron Lett., 2001, 42, 8475–8478 CrossRef CAS.
- E. Guenin, D. Ledoux, O. Oudar, M. Lecouvey and M. Kraemer, Anticancer Res., 2005, 25, 1139–1145 CAS.
- O. Bortolini, G. Fantin, M. Fogagnolo, S. Rossetti, L. Maiuolo, G. Di Pompo, S. Avnet and D. Granchi, Eur. J. Med. Chem., 2012, 52, 221–229 CrossRef CAS PubMed.
- S. Kachbi Khelfallah, M. Monteil, J. Deschamp, O. Gager, E. Migianu-Griffoni and M. Lecouvey, Org. Biomol. Chem., 2015, 13, 11382–11392 CAS.
- D. M. Mizrahi, T. Waner and Y. Segall, Phosphorus, Sulfur Silicon Relat. Elem., 2001, 173, 1–25 CrossRef CAS.
- E. Guenin, M. Monteil, N. Bouchemal, T. Prange and M. Lecouvey, Eur. J. Org. Chem., 2007, 3380–3391 CrossRef CAS.
- K. R. Bhushan, E. Tanaka and J. V. Frangioni, Angew. Chem., Int. Ed., 2007, 46, 7969–7971 CrossRef CAS PubMed.
- M. Seki, Synthesis, 2012, 44, 1556–1558 CrossRef CAS.
- P. Vachal, J. J. Hale, Z. Lu, E. C. Streckfuss, S. G. Mills, M. MacCoss, D. H. Yin, K. Algayer, K. Manser, F. Kesisoglou, S. Ghosh and L. L. Alani, J. Med. Chem., 2006, 49, 3060–3063 CrossRef CAS PubMed.
- M. L. Lolli, B. Rolando, P. Tosco, S. Chaurasia, A. Di Stilo, L. Lazzarato, E. Gorassini, R. Ferracini, S. Oliaro-Bosso, R. Fruttero and A. Gasco, Bioorg. Med. Chem., 2010, 18, 2428–2438 CrossRef CAS PubMed.
- M. Egorov, S. Aoun, M. Padrines, F. Redini, D. Heymann, J. Lebreton and M. Mathe-Allainmat, Eur. J. Org. Chem., 2011, 7148–7154 CrossRef CAS.
- J. B. Rodriguez, Synthesis, 2014, 46, 1129–1142 CrossRef.
- A. Chiminazzo, L. Sperni, M. Damuzzo, G. Strukul and A. Scarso, ChemCatChem, 2014, 6, 2712–2718 CrossRef CAS.
- G. Li, M. Wu, F. Liu and J. Jiang, Synthesis, 2015, 47, 3783–3796 CrossRef CAS.
- O. Bortolini, I. Mulani, A. De Nino, L. Maiuolo, M. Nardi, B. Russo and S. Avnet, Tetrahedron, 2011, 67, 5635–5641 CrossRef CAS.
- S. A. Holstein, D. M. Cermak, D. F. Wiemer, K. Lewis and R. J. Hohl, Bioorg. Med. Chem., 1998, 6, 687–694 CrossRef CAS PubMed.
- A. R. P. M. Valentijn, O. van den Berg, G. A. van der Marel, L. H. Cohen and J. H. van Boom, Tetrahedron, 1995, 51, 2099–2108 CrossRef CAS.
- L. W. Shull and D. F. Wiemer, J. Organomet. Chem., 2005, 690, 2521–2530 CrossRef CAS.
- M. A. Maalouf, A. J. Wiemer, C. H. Kuder, R. J. Hohl and D. F. Wiemer, Bioorg. Med. Chem., 2007, 15, 1959–1966 CrossRef CAS PubMed.
- J. Beck, S. Gharbi, A. Herteg-Fernea, L. Vercheval, C. Bebrone, P. Lassaux, A. Zervosen and J. Marchand-Brynaert, Eur. J. Org. Chem., 2009, 85–97 CAS.
- Y. Du, K.-Y. Jung and D. F. Wiemer, Tetrahedron Lett., 2002, 43, 8665–8668 CrossRef CAS.
- M. T. Rubino, M. Agamennone, C. Campestre, P. Campiglia, V. Cremasco, R. Faccio, A. Laghezza, F. Loiodice, D. Maggi, E. Panza, A. Rossello and P. Tortorella, ChemMedChem, 2011, 6, 1258–1268 CrossRef CAS PubMed.
- E. W. J. Petrillo and M. A. Ondetti, Med. Res. Rev., 1982, 2, 1–41 CrossRef CAS PubMed.
- D. V. Patel, M. G. Young, S. P. Robinson, L. Hunihan, B. J. Dean and E. M. Gordon, J. Med. Chem., 1996, 39, 4197–4210 CrossRef CAS PubMed.
- S. B. Singh, D. L. Zink, J. M. Liesch, M. A. Goetz, R. G. Jenkins, M. Nallin-Omstead, K. C. Silverman, G. F. Bills and R. T. Misley, Tetrahedron, 1993, 49, 5917–5926 CrossRef CAS.
- R. B. Lingham, K. C. Silverman, G. F. Bills, C. Cascales, M. Sanchez, R. G. Jenkins, S. E. Gartner, I. Martin, M. T. Diez, F. Peláez, S. Mochales, Y.-L. Kong, R. W. Burg, M. S. Meinz, L. Huang, M. Nallin-Omstead, S. D. Mosser, M. D. Schaber, C. A. Omer, D. L. Pompliano, J. B. Gibbs and S. B. Singh, Appl. Microbiol. Biotechnol., 1993, 40, 370–374 CrossRef CAS PubMed.
- S. B. Singh, Tetrahedron Lett., 1993, 34, 6521–6524 CrossRef CAS.
- S. B. Singh, H. Jayasuriya, K. C. Silverman, C. A. Bonfiglio, J. M. Williamson and R. B. Lingham, Bioorg. Med. Chem., 2000, 8, 571–580 CrossRef CAS PubMed.
- T. J. Tucker, M. T. Abrams, C. A. Buser, J. P. Davide, M. Ellis-Hutchings, C. Fernandes, J. B. Gibbs, S. L. Graham, G. D. Hartman, H. E. Huber, D. Liu, R. B. Lobell, W. C. Lumma, R. G. Robinson, J. T. Sisko and A. M. Smith, Bioorg. Med. Chem. Lett., 2002, 12, 2027–2030 CrossRef CAS PubMed.
- J. M. Bergman, M. T. Abrams, J. P. Davide, I. B. Greenberg, R. G. Robinson, C. A. Buser, H. E. Huber, K. S. Koblan, N. E. Kohl, R. B. Lobell, S. L. Graham, G. D. Hartman, T. M. Williams and C. J. Dinsmore, Bioorg. Med. Chem. Lett., 2001, 11, 1411–1415 CrossRef CAS PubMed.
- F. P. Coxon, M. H. Helfrich, B. Larijani, M. Muzylak, J. E. Dunford, D. Marshall, A. D. McKinnon, S. A. Nesbitt, M. A. Horton, M. C. Seabra, F. H. Ebetino and M. J. Rogers, J. Biol. Chem., 2001, 276, 48213–48222 CAS.
- Y.-L. Liu, R. Cao, Y. Wang and E. Oldfield, ACS Med. Chem. Lett., 2015, 6, 349–354 CrossRef CAS PubMed.
- K. W. Cheng, J. P. Lahad, J. W. Gray and G. B. Mills, Cancer Res., 2005, 65, 2516–2519 CrossRef CAS PubMed.
- A. J. Roelofs, P. A. Hulley, A. Meijer, F. H. Ebetino, R. G. G. Russell and C. M. Shipman, Int. J. Cancer, 2006, 119, 1254–1261 CrossRef CAS PubMed.
- C. E. McKenna, B. A. Kashemirov, K. M. Blazewska, I. Mallard-Favier, C. A. Stewart, J. Rojas, M. W. Lundy, F. H. Ebetino, R. A. Baron, J. E. Dunford, M. L. Kirsten, M. C. Seabra, J. L. Bala, M. S. Marma, M. J. Rogers and F. P. Coxon, J. Med. Chem., 2010, 53, 3454–3464 CrossRef CAS PubMed.
- K. M. Blazewska, F. Ni, R. Haiges, B. A. Kashemirov, F. P. Coxon, C. A. Stewart, R. Baron, M. J. Rogers, M. C. Seabra, F. H. Ebetino and C. E. McKenna, Eur. J. Med. Chem., 2011, 46, 4820–4826 CrossRef CAS PubMed.
- B. Fiszer and J. Michalski, Rocz. Chem., 1954, 28, 185–195 CAS.
- K. Lee and D. F. Wierner, Phosphorus, Sulfur Silicon Relat. Elem., 1993, 75, 87–90 CrossRef CAS.
- M. Ferella, Z.-H. Li, B. Andersson and R. Docampo, Exp. Parasitol., 2008, 119, 308–312 CrossRef CAS PubMed.
- P. Coutrot and G. A., Synthesis, 1986, 661–664 CrossRef CAS.
- K. Hackeloer, G. Schnakenburg and S. R. Waldvogel, Eur. J. Org. Chem., 2011, 6314–6319 CrossRef.
- L. Albrecht, B. Richter, H. Krawczyk and K. A. Jorgensen, J. Org. Chem., 2008, 73, 8337–8343 CrossRef CAS PubMed.
- F. P. Coxon, L. Joachimiak, A. K. Najumudeen, G. Breen, J. Gmach, C. Oetken-Lindholm, R. Way, J. E. Dunford, D. Abankwa and K. M. Blazewska, Eur. J. Med. Chem., 2014, 84, 77–89 CrossRef CAS PubMed.
- Y. Mu, R. A. Gibbs, L. M. Eubanks and C. D. Poulter, J. Org. Chem., 1996, 61, 8010–8015 CrossRef CAS PubMed.
- R. A. Gibbs, U. Krishnan, J. M. Dolence and C. D. Poulter, J. Org. Chem., 1995, 60, 7821–7829 CrossRef CAS.
- Y. Shao, J. T. Eummer and R. A. Gibbs, Org. Lett., 1999, 1, 627–630 CrossRef CAS PubMed.
- T. J. Zahn, C. Weinbaum and R. A. Gibbs, Bioorg. Med. Chem. Lett., 2000, 10, 1763–1766 CrossRef CAS PubMed.
- R. M. Phan and C. D. Poulter, J. Org. Chem., 2001, 66, 6705–6710 CrossRef CAS PubMed.
- J. Liu, W. Liu, H. Ge, J. Gao, Q. He, L. Su, J. Xu, L.-q. Gu, Z.-s. Huang and D. Li, Biochim. Biophys. Acta, Gen. Subj., 2014, 1840, 1051–1062 CrossRef CAS PubMed.
- A. L. Marzinzik, R. Amstutz, G. Bold, E. Bourgier, S. Cotesta, J. F. Glickman, M. Goette, C. Henry, S. Lehmann, J. C. D. Hartwieg, S. Ofner, X. Pelle, T. P. Roddy, J.-M. Rondeau, F. Stauffer, S. J. Stout, A. Widmer, J. Zimmermann, T. Zoller and W. Jahnke, ChemMedChem, 2015, 10, 1884–1891 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |