
A facile approach for the trifluoromethylthiolation of methylenecyclopropanes†
Min-Tao
Chen
,
Xiang-Ying
Tang
and
Min
Shi
*
State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, University of Chinese Academy of Sciences, 354 Fenglin Road, Shanghai 200032, China. E-mail: mshi@mail.sioc.ac.cn; Fax: +86-21-64166128
First published on 20th October 2016
Abstract
A facile approach for the trifluoromethylthiolation of methylenecyclopropanes (MCPs) has been developed by using AgSCF3/Na2S2O8 as a trifluoromethylthiolation source (SCF3) to give trifluoromethylthiolated 1,2-dihydro-naphthalene derivatives in moderate to good yields, and the reaction has been proven to go through a radical-type pathway. The products can easily be aromatized upon oxidation, offering a new method for the construction of trifluoromethylthiolated naphthalenes.
Introduction
In recent years, fluorinated organic compounds have gained more and more importance, especially in the fields of pharmaceuticals, agrochemicals and materials1 due to their unique biological activity and metabolic stability.2 Compounds with the trifluoromethylthio (SCF3) group have drawn great attention from chemists for their high electronegativity and special lipophilicity, and many synthetic methods3 have been developed to introduce the trifluoromethylthio (SCF3) group into different organic scaffolds. In recent years, a lot of work has been done using AgSCF3 as a source of the trifluoromethylthio (SCF3) moiety owing to its stability and easy availability.4 The process of trifluoromethylthiolation can go through different pathways in which AgSCF3 can act as a nucleophile (−SCF3),4e,5 electrophile (+SCF3)4f,g,l,6 or radical (˙SCF3)4d,i–k,7,8 precursor, and especially in the radical pathway, trifluoromethylthio (SCF3) radical can be conveniently accessed by mixing AgSCF3 and M2S2O8 (Na, K, NH4, etc.) together in polar solvents such as MeCN, DMSO and DMF. For example, in 2013, Wang's group achieved a radical aryltrifluoromethylthiolaton reaction of activated alkenes with AgSCF3/K2S2O8 (Scheme 1a).4d In 2014, Tang's group succeeded in the radical trifluoromethylthiolation of unactivated aliphatic C–H bonds using AgSCF3/Na2S2O8 (Scheme 1b).8 Moreover, following their work, in 2015, Liang's group successfully achieved a radical trifluoromethylthiolation cascade cyclization of 1,6-enynes with AgSCF3/K2S2O8 (Scheme 1c).4i | ||
| Scheme 1 A general review of recent work on trifluoromethylthiolation using AgSCF3 as a trifluoromethylthio radical source. | ||
On the other hand, methylenecyclopropanes (MCPs) are conveniently available organic building blocks9 with special reactivity, which can easily undergo ring-opening or participate in tandem cyclizations when exposed to cationic or radical species. Thus it would be intriguing to bring methylenecyclopropanes (MCPs) and trifluoromethylthio radical (˙SCF3) together to form some fascinating compounds. Inspired by this idea, a first attempt was made and fortunately it works (Scheme 1, this work). Herein, we wish to report a facile approach for the trifluoromethylthiolation of methylenecyclopropanes along with a mechanistic investigation.
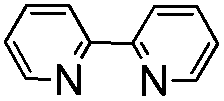
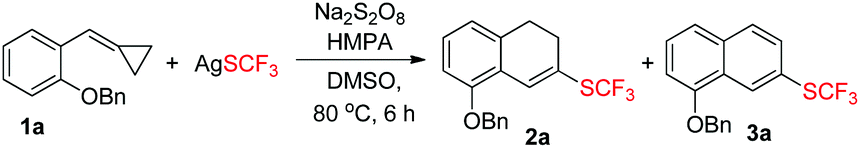
Our exploration was initiated with MCP 1a (0.2 mmol), AgSCF3 (0.3 mmol), K2S2O8 (0.6 mmol), and HMPA (0.1 mmol) in MeCN at 80 °C under an argon atmosphere for 6 h. The isolated yield of 2a was 25%, along with an aromatized product 3a derived from the further oxidation of 2a (see ESI†). The yield was not satisfactory but it strengthened our faith that a further optimization of the reaction conditions was worthwhile.
Results and discussion
In order to find out the optimal conditions, various relevant factors have been investigated and the results are summarized in Table 1. Since AgSCF3 and K2S2O8 are salts, polar solvents such as MeCN, DMSO, and DMF were mainly taken into consideration and DMSO seemed to be the best one, giving 2a in trace amounts and the aromatized product 3a in 46% yield (Table 1, entries 1–4). Using DMSO as the solvent, different oxidants were added into the reaction system to examine their efficiency in producing the trifluoromethylthio (SCF3) radical. We found that Na2S2O8 was more effective than K2S2O8 and (NH4)2S2O8 (Table 1, entries 5 and 6). Different additives also showed great influence on the reaction outcome. Inorganic bases like K2CO3 could partially prevent the oxidation of 2a to 3a and further examination revealed that HMPA was the best additive with which the oxidation could be suppressed to the lowest degree and the overall yield was acceptable (52% total yield) (Table 1, entries 7–11). Adding pyridine or bipyridine as a ligand or organic base did not facilitate the formation of 2a (Table 1, entries 12 and 13). Lowering the reaction temperature and prolonging the reaction time could not improve the yield of 2a or 3a (Table 1, entries 14 and 15).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent (3 mL) | Oxidant (3 eq.) | Additive (0.5 eq.) | Temp./°C |
2a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3ab :3ab |
Yieldc (%) |
| a The reaction conditions: 1a (0.2 mmol), AgSCF3 (0.3 mmol), oxidant (0.6 mmol), and additive (0.1 mmol) were dissolved in 3 mL DMSO and the reaction mixture was stirred at 80 °C for 6 h. b Determined by 19F NMR with p-bromobenzotrifluoride as an internal standard and 1a was taken as a standard for yield evaluation. c The total yield of 2a and 3a. d The reaction time was prolonged to 12 h. | ||||||
| 1 | DMSO | Na2S2O8 | — | 80 | —:46 |
46 |
| 2 | CH3CN | Na2S2O8 | — | 80 | —:13 |
13 |
| 3 | DMF | Na2S2O8 | — | 80 | 37:2 |
39 |
| 4 | Dioxane | Na2S2O8 | — | 80 | 4:— |
4 |
| 5 | DMSO | K2S2O8 | — | 80 | 21:22 |
43 |
| 6 | DMSO | (NH4)2S2O8 | — | 80 | —:44 |
44 |
| 7 | DMSO | Na2S2O8 | K2CO3 | 80 | 10:35 |
45 |
| 8 | DMSO | Na2S2O8 | K3PO4 | 80 | 15:25 |
40 |
| 9 | DMSO | Na2S2O8 | Cs2CO3 | 80 | 4:36 |
40 |
| 10 | DMSO | Na2S2O8 | AgOAc | 80 | —:17 |
17 |
| 11 | DMSO | Na2S2O8 | HMPA | 80 | 47:5 |
52 |
| 12 | DMSO | Na2S2O8 |
|
80 | —:42 |
42 |
| 13 | DMSO | Na2S2O8 |
|
80 | —:48 |
48 |
| 14d | DMSO | Na2S2O8 | HMPA | 60 | 44:7 |
51 |
| 15d | DMSO | Na2S2O8 | HMPA | 40 | 31:4 |
35 |
Now, we were still faced with two problems: the yield was still not satisfactory and the oxidation of 2a to 3a was not under control. Further control experiments demonstrated that substrate 1a was not stable and could be easily oxidized by Na2S2O8 in the reaction system, which might limit the yield of 2a or 3a (see ESI†). To overcome these problems, we decided to change the ratio of 1a and oxidant with regard to AgSCF3 to improve the yield of 2a and the results are shown in Table 2. To our delight, we found that when the employed amounts of substrate 1a and Na2S2O8 (oxidant) were raised to 3.0 equiv. (m/n = 3/3) and AgSCF3 was taken as 1.0 equiv., 2a was obtained in a yield of 56% as a single product (Table 2, entry 3).
|
|
|||
|---|---|---|---|
| Entry | m/nb | 2a:3ac | Yieldd (%) |
| a The reaction conditions: 1a (m equiv.), AgSCF3 (0.2 mmol), oxidant (n equiv.), and additive (0.1 mmol, 0.5 equiv.) were dissolved in 3.0 mL DMSO and the reaction mixture was stirred at 80 °C for 6 h. b The ratio of 1a/Na2S2O8. c Determined by 19F NMR with p-bromobenzotrifluoride as an internal standard. d The yield of 2a and AgSCF3 was taken as a standard for yield evaluation. | |||
| 1 | 1:1 |
36:— |
36 |
| 2 | 2:2 |
53:— |
53 |
| 3 |
3
:3 |
56
:— |
56 |
| 4 | 4:4 |
52:— |
52 |
With the optimized conditions in hand, we then examined the substrate scope of this trifluoromethylthiolation of methylenecyclopropanes as summarized in Scheme 2. Substrates 1 with a variety of substituents at the aromatic ring were successfully converted into the desired products in yields ranging from 25% to 65%.
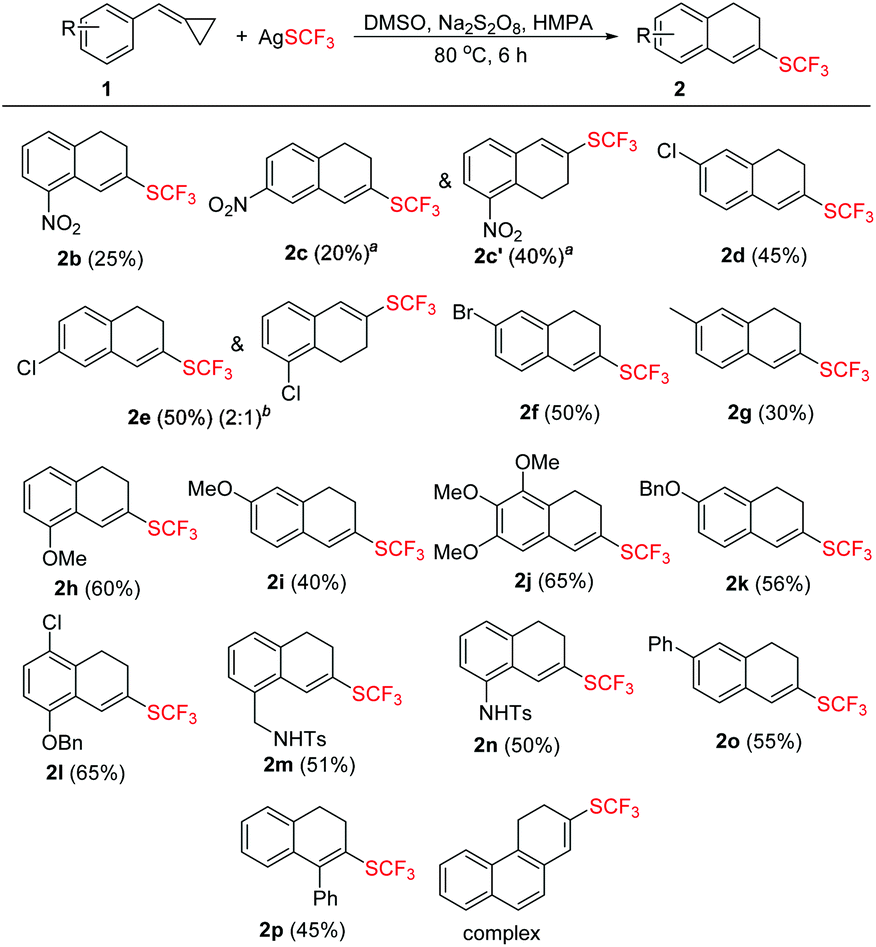 | ||
| Scheme 2 Substrate scope of 1. The reaction conditions: 1 (0.6 mmol), AgSCF3 (0.2 mmol), Na2S2O8 (0.6 mmol), and HMPA (0.1 mmol) were dissolved in 3 mL DMSO and the reaction mixture was stirred at 80 °C for 6 h. All the yields are isolated yields and AgSCF3 was taken as a standard for yield evaluation. aThe two isomers can be separated. bThe two isomers cannot be isolated. | ||
In general, substrates 1b–1f bearing electron-withdrawing substituents gave the desired products in relatively low yields. For example, substrate 1b having a strongly electron-withdrawing nitro group afforded the corresponding product 2b in 25% yield. As for substrates bearing electron-donating groups, the transformations of 1 to 2 are generally favored. For instance, the conversion efficiency of 1j to 2j with three electron-donating methoxy groups was much higher than others. However, in the case of 1i, the desired product 2i was formed in 40% yield, presumably due to its instability in the presence of a large amount of oxidant. It should be also noted that in the cases of 1c and 1e, two trifluoromethylthiolated regioisomers were formed at the same time. The p-toluenesulfonylamino group-containing substrates 1m and 1n can be well tolerated under the reaction conditions, affording the desired products 2m and 2n in good yields. The X-ray crystal structure of 2n has been obtained (Fig. 1) and the CIF data are given in the ESI.† Diphenylmethylenecyclopropane 1p was also compatible in this transformation, but naphthylmethylenecyclopropane only resulted in a complex product mixture under the standard conditions.
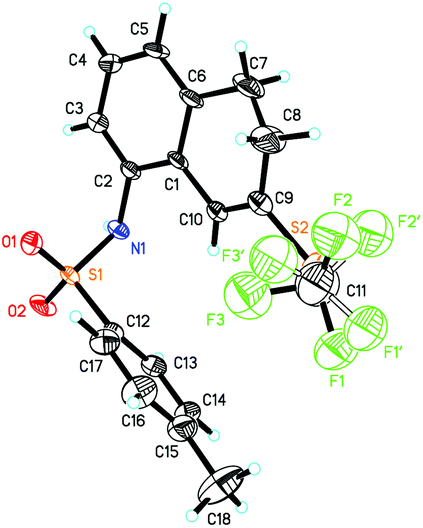 | ||
| Fig. 1 X-ray crystal structure of product 2n. | ||
Product 2 could undergo dehydrogenation to a great tendency in the presence of oxidants and the formation of the aromatized product 3 provided a new synthetic protocol to prepare trifluoromethylthiolated naphthalene derivatives. Inspired by the result that the aromatized compound was obtained while screening the reaction conditions (Table 1, entry 1), we attempted to use the same oxidant Na2S2O8 to achieve the dehydrogenation. After a brief investigation, using 4.0 equiv. of Na2S2O8 alone and stirring the reaction mixture for 4–6 h at 80 °C could produce the aromatized products 3 in moderate to good yields ranging from 40% to 80% and the results are summarized in Scheme 3.
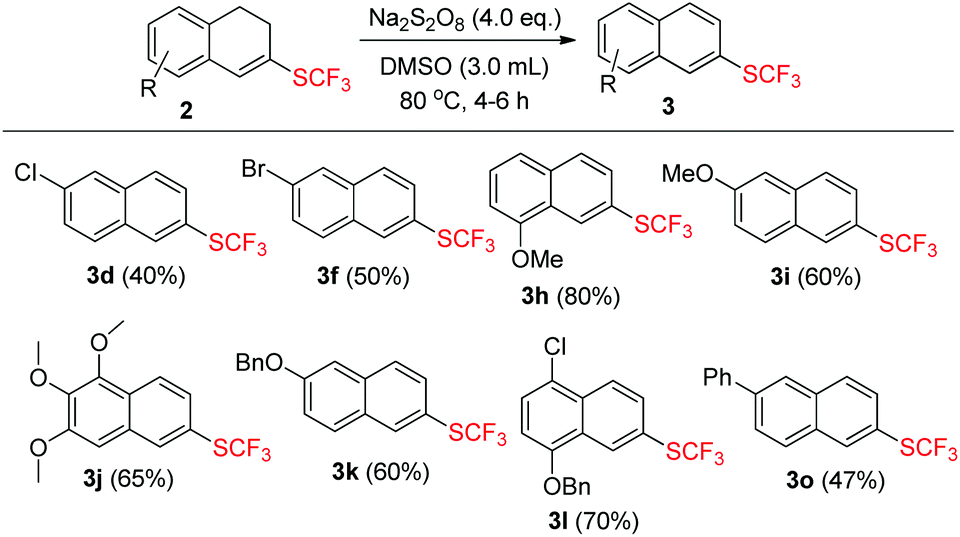 | ||
| Scheme 3 Substrate scope of 2. The reaction conditions: 2 (0.2 mmol), Na2S2O8 (0.8 mmol) in 3.0 mL DMSO. All the yields are isolated yields. | ||
In order to directly obtain 3a, we just needed to mix 1a (1.0 eq.), AgSCF3 (1.5 eq.), and Na2S2O8 (3 eq.) together in DMSO upon heating at 80 °C for about 6 h, directly giving 3a in 46% yield in a one pot manner (Table 1, entry 1 or Scheme 4).
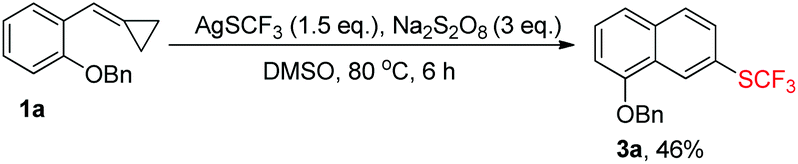 | ||
| Scheme 4 Direct conversion of 1a to 3a. | ||
It was believed that the reaction proceeded via a radical-type pathway on the basis of previously reported literature.4d,i,8 Therefore, control experiments with radical inhibitors TEMPO and BHT were performed as shown in Scheme 5. The formation of the corresponding trifluoromethylthiolated product was significantly suppressed (for more details, see ESI†), rendering a radical process reasonable. However, it is true that AgSCF3 decomposed partially in the presence of a large amount of BHT and even completely decomposed in the presence of a large amount of TEMPO, thus such control experiments can only give mechanistic support to some degree. Moreover, F3CSSCF310 can be detected in the reaction system by 19F NMR spectroscopy resulting from coupling of two SCF3 radicals. Overall, we believe that this reaction goes through a radical process.
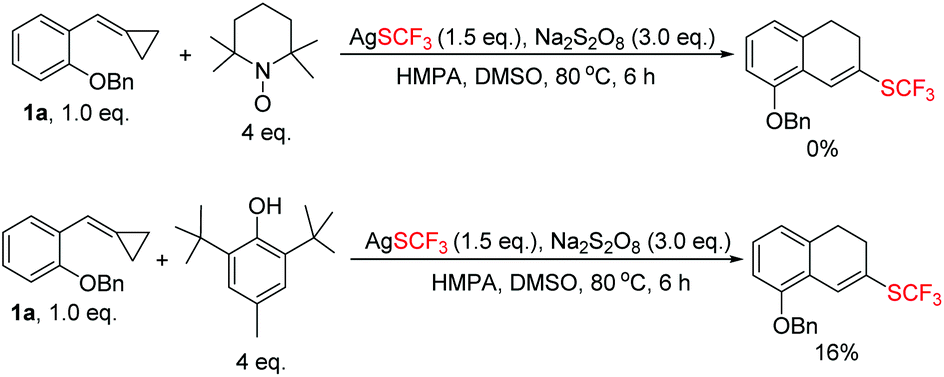 | ||
| Scheme 5 Radical inhibition experiments. | ||
Taking all the above information into consideration, a plausible mechanism of this trifluoromethylthiolation has been proposed in Scheme 6. AgSCF3 can release SCF3 radical upon oxidation with Na2S2O8 and the SCF3 radical can be transformed into its dimer F3CSSCF3, which can also be converted into SCF3 radical with the help of Ag+.4d,i Then, the SCF3 radical adds to the double bond of substrate 1 to form a ring-opening radical intermediate A, followed by a cyclization to give intermediate B. The intermediate B is oxidized by SO4˙− to afford product 2, which can be easily dehydrogenated and aromatized by Na2S2O8 to afford product 3.
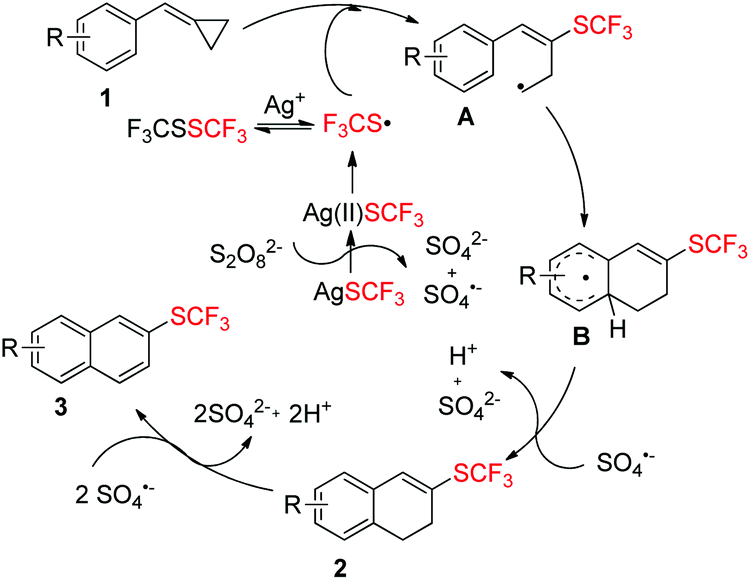 | ||
| Scheme 6 Proposed reaction mechanism. | ||
In summary, we have developed a practical method for the facile trifluoromethylthiolation of methylenecyclopropanes (MCPs) in the presence of AgSCF3/Na2S2O8, and a variety of substrates can tolerate the oxidative conditions to give trifluoromethylthiolated 1,2-dihydronaphthalene derivatives in moderate to good yields. The products can conveniently be further aromatized upon oxidation with Na2S2O8, offering a new synthetic method for the preparation of trifluoromethylthiolated naphthalenes. Efforts are in progress for the application of this new methodology to synthesizing interesting biologically active trifluoromethylthiolated compounds in our laboratory.
Experimental section
General remarks
1H NMR spectra were recorded on a Varian Mercury-300 and 400 spectrometer for solutions in CDCl3 with tetramethylsilane (TMS) as an internal standard; coupling constants J are given in Hz. 13C NMR spectra were recorded on Varian Mercury-300 and 400 spectrophotometers (75 or 100 MHz) with complete proton decoupling (CDCl3: 77.0 ppm). Mass and HRMS spectra were recorded by EI method. Organic solvents used were dried by standard methods where necessary. Infrared spectra were recorded on a Perkin-Elmer PE-983 spectrometer with absorption in cm−1. Melting points were determined on a digital melting point apparatus and temperatures were uncorrected. Commercially obtained reagents were used without further purification. All these reactions were monitored by TLC with silica gel coated plates or 19F NMR. Flash column chromatography was carried out using silica gel at increased pressure.General procedure for the trifluoromethylthiolation of MCPs
Compound 1 (0.6 mmol), AgSCF3 (42 mg, 0.2 mmol), and Na2S2O8 (142 mg, 0.6 mmol) were placed in a Schlenk tube. The tube was evacuated and backfilled with Ar three times and then DMSO (3 mL) and HMPA (0.1 mmol) were injected. Afterwards, the reaction mixture was stirred at 80 °C in an oil bath for 6 h. When the reaction was complete, the product was extracted with EtOAc and washed with water. The organic layer was dried over Na2SO4 and concentrated on a rotary evaporator. The residue was roughly purified by silica gel flash chromatography and further purified by gel permeation chromatography (GPC) to give a pure product.General procedure for the dehydrogenation of 2
The product 2 (0.2 mmol) and Na2S2O8 (191 mg, 0.8 mmol) were placed in a flask and DMSO (3 mL) was added. Then, the reaction mixture was stirred at 80 °C in an oil bath for about 4–6 h. When the reaction was complete, the aromatized product was extracted with EtOAc and washed with water. The organic layer was dried over Na2SO4 and concentrated on a rotary evaporator. The residue was purified by silica gel flash chromatography and if necessary, it was further purified by gel permeation chromatography (GPC) to give a pure product.Acknowledgements
This work was supported by the Joint NSFC-ISF Research Program, jointly funded by the National Natural Science Foundation of China and the Israel Science Foundation. We are also grateful for the financial support from the National Basic Research Program of China (973)-2015CB856603, and the National Natural Science Foundation of China (20472096, 21372241, 21361140350, 20672127, 21421091, 21372250, 21121062, 21302203, 20732008, 21572052 and 21172141).Notes and references
- (a) F. M. D. Ismail, J. Fluorine Chem., 2002, 118, 27 CrossRef CAS; (b) M. Shimizu and T. Hiyama, Angew. Chem., Int. Ed., 2005, 44, 214 CrossRef CAS PubMed; (c) J. Swinson, PharmaChem, 2005, 4, 26 CAS; (d) J.-P. Begue and D. Bonnet-Delpon, J. Fluorine Chem., 2006, 127, 992 CrossRef CAS; (e) C. Isanbor and D. O'Hagan, J. Fluorine Chem., 2006, 127, 303 CrossRef CAS; (f) K. L. Kirk, J. Fluorine Chem., 2006, 127, 1013 CrossRef CAS; (g) G. Theodoridis, Adv. Fluorine Sci., 2006, 2, 121 CAS; (h) K. Mueller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef CAS PubMed; (i) X. Ren, W. Wan, H. Jiang and J. Hao, Mini-Rev. Org. Chem., 2007, 4, 330 CrossRef CAS; (j) J. Swinson, PharmaChem, 2007, 6, 38 CAS; (k) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed; (l) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308 RSC; (m) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (n) R. Filler and R. Saha, Future Med. Chem., 2009, 1, 777 CrossRef CAS PubMed; (o) D. O'Hagan, J. Fluorine Chem., 2010, 131, 1071 CrossRef; (p) R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496 RSC; (q) T. Fujiwara and D. O'Hagan, J. Fluorine Chem., 2014, 167, 16 CrossRef CAS; (r) J. Wang, M. Sanchez-Rosello, J. L. Acena, P. C. del, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed; (s) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315 CrossRef CAS PubMed.
- (a) R. Filler, J. Fluorine Chem., 1986, 33, 361 CrossRef CAS; (b) J. Mann, Chem. Soc. Rev., 1987, 16, 381 RSC; (c) P. Jeschke, ChemBioChem, 2004, 5, 570 CrossRef CAS PubMed; (d) C. Pesenti and F. Viani, ChemBioChem, 2004, 5, 590 CrossRef CAS PubMed; (e) E. N. G. Marsh, Acc. Chem. Res., 2014, 47, 2878 CrossRef CAS PubMed.
- (a) A. Tlili and T. Billard, Angew. Chem., Int. Ed., 2013, 52, 6818 CrossRef CAS PubMed; (b) L. Chu and F.-L. Qing, Acc. Chem. Res., 2014, 47, 1513 CrossRef CAS PubMed; (c) F. Hu, X. Shao, D. Zhu, L. Lu and Q. Shen, Angew. Chem., Int. Ed., 2014, 53, 6105 CrossRef CAS PubMed; (d) K. Kang, C. Xu and Q. Shen, Org. Chem. Front., 2014, 1, 294 RSC; (e) X. Shao, T. Liu, L. Lu and Q. Shen, Org. Lett., 2014, 16, 4738 CrossRef CAS PubMed; (f) F. Toulgoat, S. Alazet and T. Billard, Eur. J. Org. Chem., 2014, 2415 CrossRef CAS; (g) C. Xu and Q. Shen, Org. Lett., 2014, 16, 2046 CrossRef CAS PubMed; (h) X. Shao, C. Xu, L. Lu and Q. Shen, Acc. Chem. Res., 2015, 48, 1227 CrossRef CAS PubMed; (i) X.-H. Xu, K. Matsuzaki and N. Shibata, Chem. Rev., 2015, 115, 731 CrossRef CAS PubMed; (j) X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826 CrossRef CAS PubMed; (k) D. E. Yerien, S. Bonesi and A. Postigo, Org. Biomol. Chem., 2016, 14, 8398 RSC; (l) S. Barata-Vallejo, A. Postigo and S. Bonesi, Org. Biomol. Chem., 2016, 14, 7150 RSC; (m) H. Chachignon and D. Cahard, Chin. J. Chem., 2016, 34, 445 CrossRef CAS; (n) P. Zhang, M. Li, X.-S. Xue, C. Xu, Q. Zhao, Y. Liu, H. Wang, Y. Guo, L. Lu and Q. Shen, J. Org. Chem., 2016, 81, 7486 CrossRef CAS PubMed.
- (a) G. Teverovskiy, D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 7312 CrossRef CAS PubMed; (b) C. Chen, X.-H. Xu, B. Yang and F.-L. Qing, Org. Lett., 2014, 16, 3372 CrossRef CAS PubMed; (c) Q. Xiao, J. Sheng, Q. Ding and J. Wu, Eur. J. Org. Chem., 2014, 217 CrossRef CAS; (d) F. Yin and X.-S. Wang, Org. Lett., 2014, 16, 1128 CrossRef CAS PubMed; (e) K. Zhang, J.-B. Liu and F.-L. Qing, Chem. Commun., 2014, 50, 14157 RSC; (f) S.-Q. Zhu, X.-H. Xu and F.-L. Qing, Eur. J. Org. Chem., 2014, 4453 CrossRef CAS; (g) X.-L. Zhu, J.-H. Xu, D.-J. Cheng, L.-J. Zhao, X.-Y. Liu and B. Tan, Org. Lett., 2014, 16, 2192 CrossRef CAS PubMed; (h) J.-B. Liu, X.-H. Xu, Z.-H. Chen and F.-L. Qing, Angew. Chem., Int. Ed., 2015, 54, 897 CrossRef CAS PubMed; (i) Y.-F. Qiu, X.-Y. Zhu, Y.-X. Li, Y.-T. He, F. Yang, J. Wang, H.-L. Hua, L. Zheng, L.-C. Wang, X.-Y. Liu and Y.-M. Liang, Org. Lett., 2015, 17, 3694 CrossRef CAS PubMed; (j) H. Wu, Z. Xiao, J. Wu, Y. Guo, J.-C. Xiao, C. Liu and Q.-Y. Chen, Angew. Chem., Int. Ed., 2015, 54, 4070 CrossRef CAS PubMed; (k) D.-P. Jin, P. Gao, D.-Q. Chen, S. Chen, J. Wang, X.-Y. Liu and Y.-M. Liang, Org. Lett., 2016, 18, 3486 CrossRef CAS PubMed; (l) L. Ma, X.-F. Cheng, Y. Li and X.-S. Wang, Tetrahedron Lett., 2016, 57, 2972 CrossRef CAS; (m) Y.-F. Qiu, X.-R. Song, M. Li, X.-Y. Zhu, A.-Q. Wang, F. Yang, Y.-P. Han, H.-R. Zhang, D.-P. Jin, Y.-X. Li and Y.-M. Liang, Org. Lett., 2016, 18, 1514 CrossRef CAS PubMed; (n) W. Wu, W. Dai, X. Ji and S. Cao, Org. Lett., 2016, 18, 2918 CrossRef CAS PubMed; (o) Y. Zeng and J. Hu, Org. Lett., 2016, 18, 856 CrossRef CAS PubMed.
- (a) D. J. Adams and J. H. Clark, J. Org. Chem., 2000, 65, 1456 CrossRef CAS PubMed; (b) M. Jiang, F. Zhu, H. Xiang, X. Xu, L. Deng and C. Yang, Org. Biomol. Chem., 2015, 13, 6935 RSC; (c) C. Xu, Q. Chen and Q. Shen, Chin. J. Chem., 2016, 34, 495 CrossRef CAS.
- H. Xiang and C. Yang, Org. Lett., 2014, 16, 5686 CrossRef CAS PubMed.
- Y.-F. Zeng, D.-H. Tan, Y. Chen, W.-X. Lv, X.-G. Liu, Q. Li and H. Wang, Org. Chem. Front., 2015, 2, 1511 RSC.
- (a) S. Guo, X. Zhang and P. Tang, Angew. Chem., Int. Ed., 2015, 54, 4065 CrossRef CAS PubMed; (b) H. Wu, Z. Xiao, J. Wu, Y. Guo, J.-C. Xiao, C. Liu and Q.-Y. Chen, Angew. Chem., Int. Ed., 2015, 54, 4070 CrossRef CAS PubMed.
- (a) A. Brandi and A. Goti, Chem. Rev., 1998, 98, 589 CrossRef CAS PubMed; (b) M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117 CrossRef CAS PubMed; (c) A. Masarwa, A. Fuerstner and I. Marek, Chem. Commun., 2009, 5760 RSC; (d) A. Masarwa and I. Marek, Chem. – Eur. J., 2010, 16, 9712 CrossRef CAS PubMed; (e) H. Pellissier, Tetrahedron, 2010, 66, 8341 CrossRef CAS; (f) M. Shi, J.-M. Lu, Y. Wei and L.-X. Shao, Acc. Chem. Res., 2012, 45, 641 CrossRef CAS PubMed; (g) A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Chem. Rev., 2014, 114, 7317 CrossRef CAS PubMed; (h) H. Pellissier, Tetrahedron, 2014, 70, 4991 CrossRef CAS; (i) D.-H. Zhang, X.-Y. Tang and M. Shi, Acc. Chem. Res., 2014, 47, 913 CrossRef CAS PubMed.
- (a) R. E. A. Dear and E. E. Gilbert, J. Fluorine Chem., 1974, 4, 107 CrossRef CAS; (b) K. Schlosser, Z. Naturforsch., B: Anorg. Chem. Org. Chem., 1982, 37B, 172 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data of new compounds. CCDC 1493132. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00536e |
| This journal is © the Partner Organisations 2017 |