
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New N-acyl amino acid-functionalized biodegradable polyesters for pharmaceutical and biomedical applications†
Vincenzo
Taresco
a,
Jiraphong
Suksiriworapong
b,
Ioanna D.
Styliari
a,
Richard H.
Argent
a,
Sadie M. E.
Swainson
a,
Jonathan
Booth
c,
Eleanor
Turpin
a,
Charles A.
Laughton
a,
Jonathan C.
Burley
a,
Cameron
Alexander
 a and
Martin C.
Garnett
*a
a and
Martin C.
Garnett
*a
aUniversity of Nottingham, School of Pharmacy, University Park, Nottingham, NG7 2RD, UK. E-mail: Martin.Garnett@nottingham.ac.uk
bMahidol University, Department of Pharmacy, Faculty of Pharmacy, Bangkok, 10400, Thailand
cAstraZeneca, Macclesfield, SK10 RNA, UK
First published on 9th November 2016
Abstract
A novel class of biodegradable polyesters has been generated by coupling poly(glycerol adipate) with N-acyl aromatic amino acids. This new set of polymers from this highly versatile polymeric platform may offer unprecedented new opportunities to produce biodegradable and biocompatible polymers with tailorable physical–chemical properties.
Over the past decades, great effort has been made to introduce naturally occurring amino acids (AA) into synthetic polymers.1 Including or grafting an AA along a polymeric backbone may (i) tailor the chirality; (ii) offer new functional groups capable of interacting with drugs, proteins and nucleic acids; (iii) facilitate a further anchoring with other bioactive molecules; (iv) modulate the hydrophilicity/hydrophobicity of the system; or (v) improve the non-covalent interactions leading to the formation of hierarchical ordered superstructures.2 Ring opening polymerization (ROP) of both canonical and non-canonical amino acid N-carboxyanhydrides (NCA)3–5 results in a common strategy to develop synthetic poly peptide/poly(amino acids).6 In order to usually prepare NCAs, side chain protected AAs are routinely phosgenated. The need to functionalize and protect AAs beforehand renders the pre-polymerization steps tedious from the point of view of purification and monomer moisture sensitivity. Classic NCA polymerizations due to side reactions, such as chain transfer and chain termination, suffer crucial limitations in both designing well defined co-polymers,7 and reaching well defined molecular weights. Some alternatives have been developed, such the introduction of organometallic catalyst,8 vacuum conditions,9 and the use of low temperature.7 Despite all these aforementioned pioneering polymerization technologies, and the series of recognised biocompatible properties, poly(amino acids) have found few applications in the biomedical field due to the high crystallinity, low degradation rate, low solubility in common organic solvents, limited thermal stability antigenicity and poor mechanical properties.10 Nevertheless AAs represent a natural font of inspiration in designing multi-responsive, biocompatible complexed macrostructures with enhanced cell interactions and enzymatic biodegradability.11 In order to overcome these limitations different, α-AAS based hybrid polymers have been explored;1,12,13 amongst them two fascinating classes are the poly(α-hydroxy ester)s14 and the poly(ester amide)s (PEA).15 PEAs are generated by replacing the amino group of the AAs with a hydroxyl residue,16 with the aim of broadening the library of polyester carrying stereogenic-centers. These polymers can be synthesised by exploiting different synthetic paths, leading to polyester-based materials with an enhanced hydrophilicity and degradation rate when compared to common polyesters.16 On the other hand this approach requires many non-quantitative pre-polymerization steps and in some cases, a toxic metal-based catalyst, yielding only low molecular weight polymers.14 PEAs normally bear both ester and amide linkages placed in the main backbone thus resulting in intermediate properties between polyesters and polyamides.12 PEAs combine the characteristics such as solubility in organic solvent, degradability at physiological conditions and good flexibility from the ester group with good thermal and mechanical properties, due to a superior web of hydrogen bond interactions, amongst the amide groups.1,17 In spite of these positive points, practically all the polymerization technologies usually employed to produce PEAs, namely ROP and polycondensation, present synthetic limitations in terms of purity of the starting materials, control of temperature, use of metallic catalysts, humidity sensitivity and low yields.1
In the present work, a set of N-acyl-amino acids (NAAs) were coupled to the pendant free hydroxyl group of poly(glycerol adipate) (PGA)18 (Scheme 1). PGA employed in this work presented a degree of tri-substituted glyceryl moiety around 5% mol mol−1 (ref. 19) (Scheme 1, T and T′ as polymer chain and Fig. 1 peak (c′′)), hence the main polymer backbone chain can be considered linear.
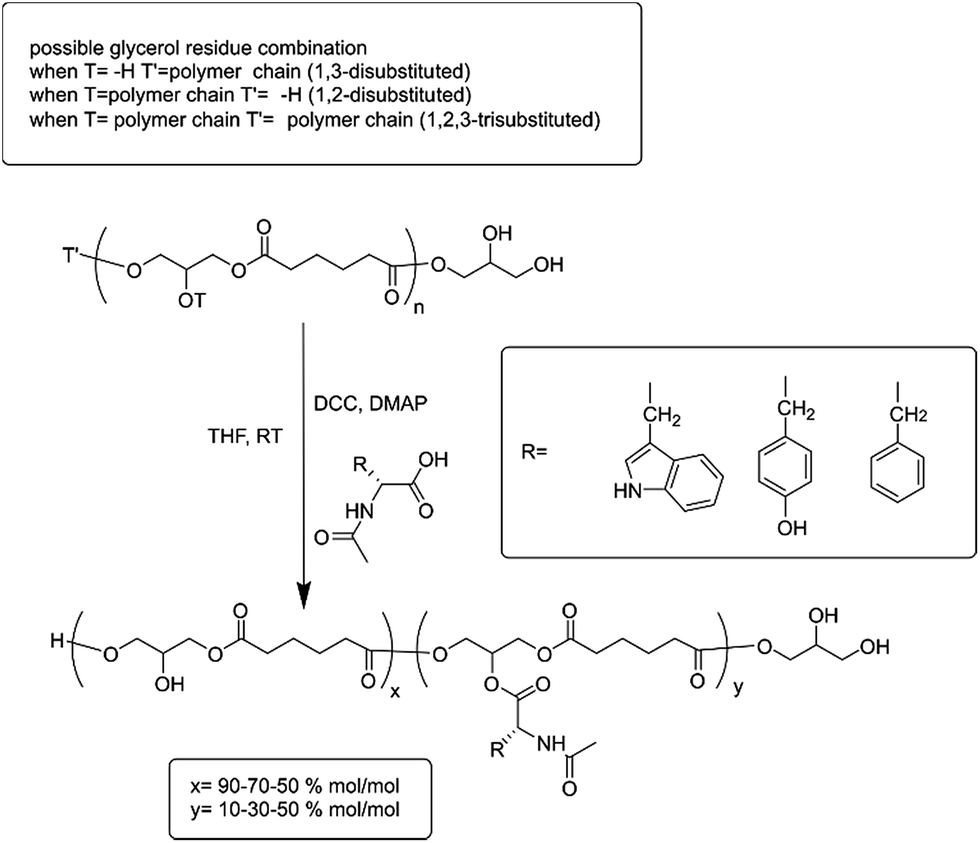 | ||
| Scheme 1 Coupling reaction scheme. PGA backbone is detailed in order to show the possible glyceryl-substitution combinations before Steglich reaction. The three aromatic NAA moieties are reported and also the amount of functionalization in the repetitive units. | ||
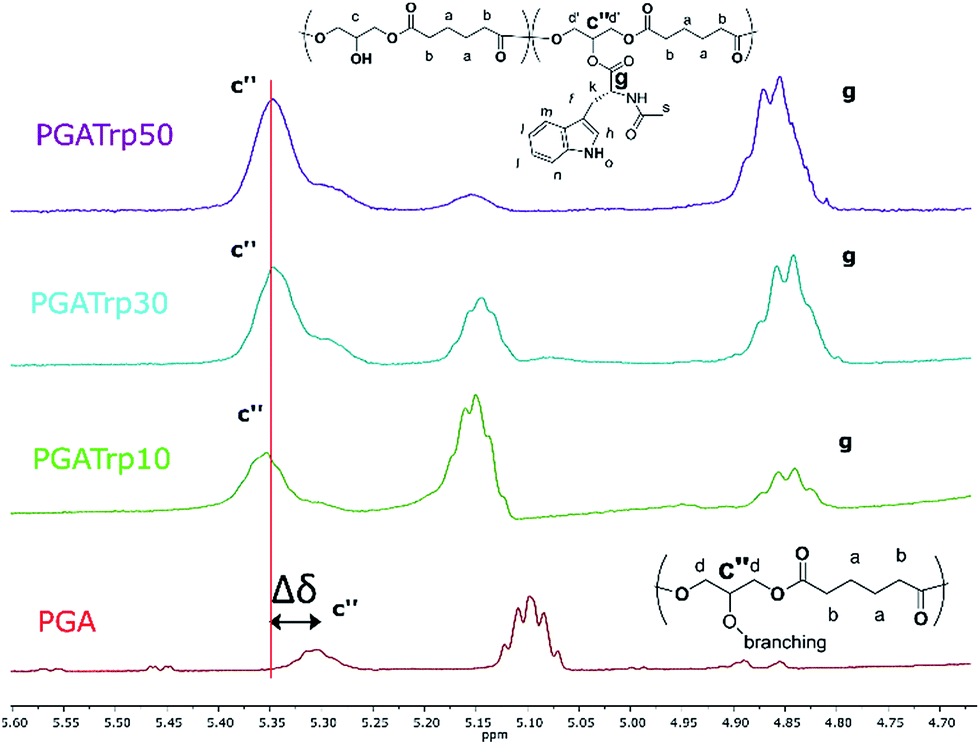 | ||
| Fig. 1 1H NMR inset between 5.60 and 4.70 ppm of PGA and PGATrp10–30 and 50. Polymer backbone peak shifting (c′′) after coupling is highlighted. It is also possible to see that the trisubstituted PGA unit increases with Trp amount (g) supporting that functionalization has occurred. | ||
To the best of our knowledge this is the first time that NAAs have been grafted to a polyester platform, although there are several reported examples of AAs grafted to either acrylic/methacrylic based-backbones synthesised by living radical polymerizations20–22 or by natural biomacromolecules such as polysaccharides.23,24 We previously reported the enzymatic synthesis of PGA,19 which is typically produced from divinyl adipate and unprotected glycerol, yielding polymers with an average Mw of 33 kDa (PDI 2.5). Enzymatic polymerization has emerged as a game-changing development in new synthetic polymers bearing active moieties that can be further modified.25 The use of enzymes in organic solvents to generate polymers with highly specific physical–chemical properties is a very fertile area of research.26 A further advantage of these enzymatically synthesised polymers is their potential for enzymatic degradation in biological systems, which can be exploited in the same way as other smart polymer modifications. These polymers can be synthesized routinely without any post-polymerization deprotection reactions in a one-step synthesis under mild conditions. It is possible to change the average molecular weight and the branching degree of PGA in situ simply by changing the polymerization temperature and thus the lipase (Novozyme 435) selectivity.19
The NAA functionalization was undertaken by a simple Steglich coupling esterification27 of bare PGA in THF at room temperature (Scheme 1), with N-acyl-tryptophan (Trp), N-acyl-tyrosine (Tyr) or N-acyl-phenylalanine (Phe) at three different feed molar ratios: 10, 30 and 50% mol mol−1. The resulting polymers were all analysed by GPC. All the determinations were in DMF containing 0.1% LiBr at 50 °C at a concentration of 2.5 mg ml−1 (chromatograms of PGA and all the modification at 50% are reported as examples of GPC traces in Fig. ESI2†). Polydispersity (Đ) of the PGA amino acyl derivatives ranged from 3.1 for PGATyr10% to 2.1 for PGAPhe50% in comparison to a value for unmodified PGA around 2.5. Variations in PDI could come through a number of possible routes. The additional synthetic step is likely to cause an increase in PDI due to the random nature of the coupling. Similarly changes in polymer intramolecular interactions and flexibility may lead to an extended range of conformations which would influence the elution times of different functionalised polymer molecules. Finally during the precipitation step to collect the functionalised polymers some of the smallest molecules could have been lost leading to an improvement of PDI. Solubility for the polymers was determined for the common polar aprotic solvents acetone, DMF and DMSO. The PGATyr set showed solubility up to 30 mg ml−1 in acetone and DMSO while around 5 to 10 mg ml−1 in DMF. The PGAPhe set showed solubility up to 30 mg ml−1 in DMSO and DMF but only around 5 mg ml−1 or lower in acetone, while the PGATrp set showed solubility up to 30 mg ml−1 in DMSO, DMF as well as in acetone.
1H NMR analysis was used to assess the introduction of the three different NAAs both qualitatively and quantitatively (Fig. ESI1†). In particular, 1H NMR spectra revealed the characteristic peaks of both PGA and the three aromatic NAAs, which indicates a successful coupling esterification.
Interestingly both the peak at 5.30 ppm (Fig. 1c′′) related to the trisubstituted glycerol unit and the peak at 4.35 ppm (Fig. 1, g) of the proton of the chiral carbon of the AAs increase with the amount of coupled NAAs.
Furthermore, as shown in Fig. 1 there is a shift downfield of the peak at 5.30 of the functionalized PGA to 5.35 (Δδ in Fig. 1), which can be evaluated as a validation of an eventual polymeric backbone modification.
The integral ratio between the polymer peak at 2.39 ppm (b) and the NAAs peak at 4.35 ppm enabled evaluation of the esterification efficacy (Fig. 1). The optimization of reaction conditions led to a functionalization greater than 95%, regardless the nature of the amino acid side chain, highlighting the efficacy of this coupling procedure.
DSC analysis of the functionalised PGA-NAAs was performed to ascertain the effect of the nature and amount of attached amino acid on PGA backbone thermal properties (Table ESI1†). Bare PGA showed a single glass temperature transition (Tg) at −33 °C resulting in a highly viscous liquid at room temperature. Despite the crystalline nature of all the NAAs explored in this work, the resulting substituted PGA-NAAs showed only a Tg step and no detectable melting peak. As shown in Fig. 2 the substitution of the free hydroxyl group along the main chain with the NAAs led to a general Tg value increment. PGATyr and PGAPhe showed similar glass transition values at all degrees of functionalization (Table ESI1†). On the other hand, although PGATrp displayed the same rising Tg trend of the other two polymer sets, absolute higher Tg values were recorded at all the three coupling percentage explored (Fig. 2). Substituting a free OH with a functionalized-aromatic group might drastically modify the nature of intermolecular interactions such as hydrogen bonds and introduce new interactions such as π–π aromatic-stacking. By simply varying the degree of functionalization it has been possible to tune the thermal polymer properties exploring an extended range of temperature from −33 °C for PGA to 49 °C of PGATrp50. This discovery might be extremely useful from the perspective of tailoring the aggregation nature at body or room temperature, or for modulating liquid-like or solid-like behaviour, crucial in drug delivery applications.
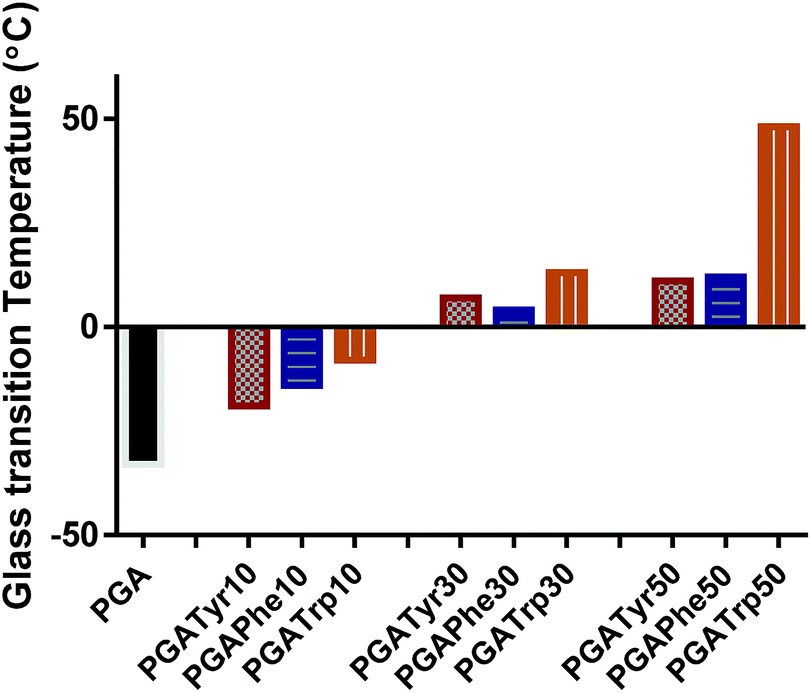 | ||
| Fig. 2 Glass transition temperature (Tg) trend amongst the functionalized polymers and the bare PGA. It is possible to explore a wide and deep shift in Tg from −33 up to 49 °C. | ||
log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P is the most widely used parameter to measure lipophilicity, indicating the partition coefficient of a molecule between aqueous and lipophilic phases. The online ALOGPS 2.1 online software has been used for the estimation of the hydrophobicity of the polymers.16 logP(calc) values were calculated from (PGA)x − (functionalised-PGA)y repetitive units, for x + y = 10 in accordance with the functionalisation degree16 (Table ESI2†). Fig. 3 clearly shows that passing from the bare PGA through the varying NAA degrees of functionalization (from 10 up to 50% mol mol−1) the logP values gradually increase. In particular, PGATrp at all the coupling% explored showed slightly higher logP(calc) values (from 2.3 up to 3.6) compared to PGATyr (ranging from 2.1 up to 2.8) and PGAPhe (2.1–3.3) at the same NAA content, demonstrating that the increase of the hydrophobic nature of the polymers was due to both the hydrophobic nature of the pendant NAA and the loss of a hydroxyl function through coupling (Table ESI2†).
P is the most widely used parameter to measure lipophilicity, indicating the partition coefficient of a molecule between aqueous and lipophilic phases. The online ALOGPS 2.1 online software has been used for the estimation of the hydrophobicity of the polymers.16 logP(calc) values were calculated from (PGA)x − (functionalised-PGA)y repetitive units, for x + y = 10 in accordance with the functionalisation degree16 (Table ESI2†). Fig. 3 clearly shows that passing from the bare PGA through the varying NAA degrees of functionalization (from 10 up to 50% mol mol−1) the logP values gradually increase. In particular, PGATrp at all the coupling% explored showed slightly higher logP(calc) values (from 2.3 up to 3.6) compared to PGATyr (ranging from 2.1 up to 2.8) and PGAPhe (2.1–3.3) at the same NAA content, demonstrating that the increase of the hydrophobic nature of the polymers was due to both the hydrophobic nature of the pendant NAA and the loss of a hydroxyl function through coupling (Table ESI2†).
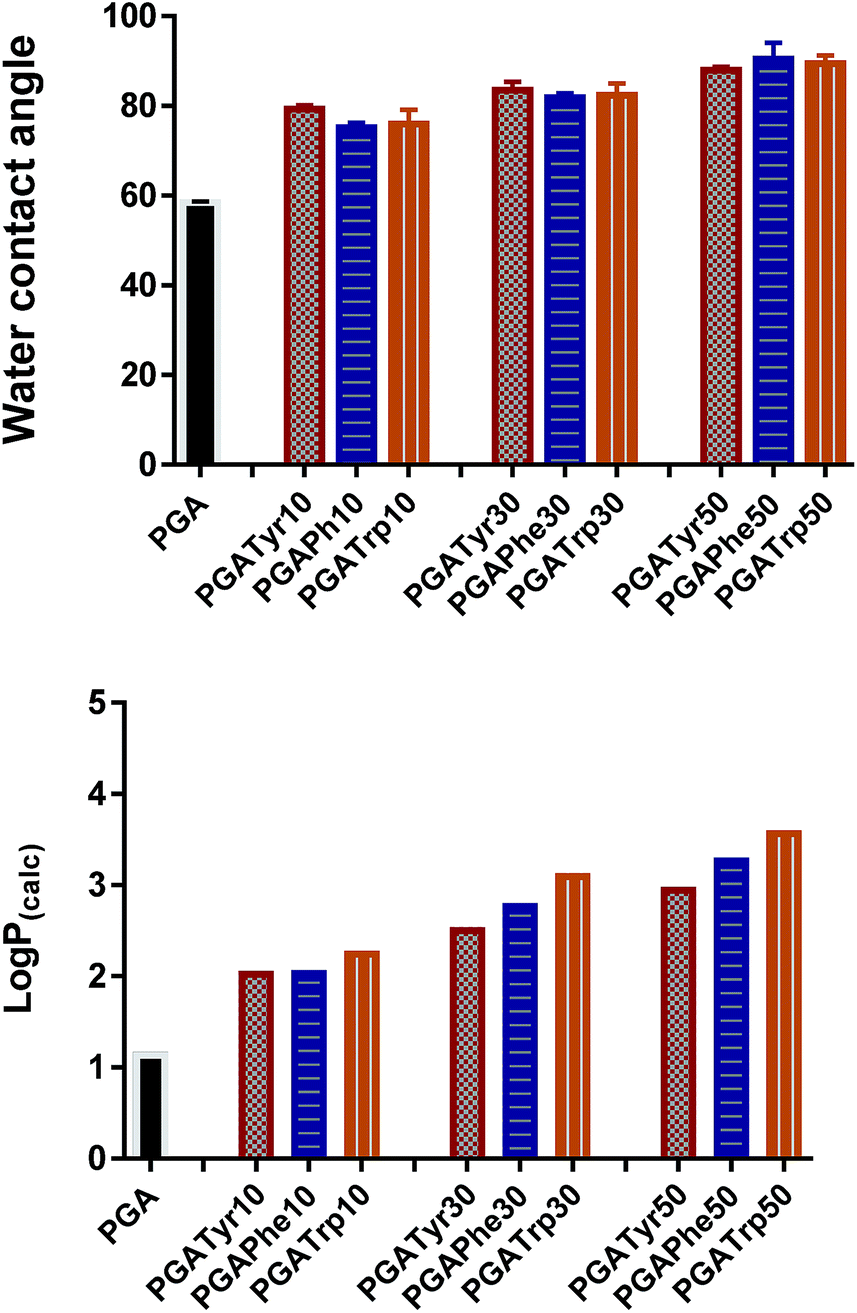 | ||
| Fig. 3 Water contact angle (top graph) and logP(calc) (bottom graph) values show the same trend. A slight increment of both the two parameters is recorded moving from the lowest functionalization up to the highest regardless the nature of the functional NAA group. | ||
Interestingly the same trend is observed in the water contact angle (WCA) values of the polymers, ranging between 75 and 90 degrees. WCA can be consider a simple measurement to assess the hydrophobic/hydrophilic character of the surface. As depicted in Fig. 3 for similar degrees of substitution, mean contact angle values tend to be fairly constant regardless the nature of the NAA substituents. The absence of a pronounced WCA value amongst all the different modifications is likely due to both the nature of the chemistry underlying the coupling reaction and the low similarity of the intermolecular interactions partaken by the three NAAs.
In order to evaluate the formulation properties of these amphiphilic polymers, polymeric NPs were prepared via the nanoprecipitation method, using acetone as the organic phase. In order to maintain a constant and reproducible protocol, the acetone polymer solutions were dispensed in water by means a syringe pump apparatus. The distribution of the size of the NPs as measured via DLS indicates particles with size in diameter varying between 50–100 nm (Fig. 4).
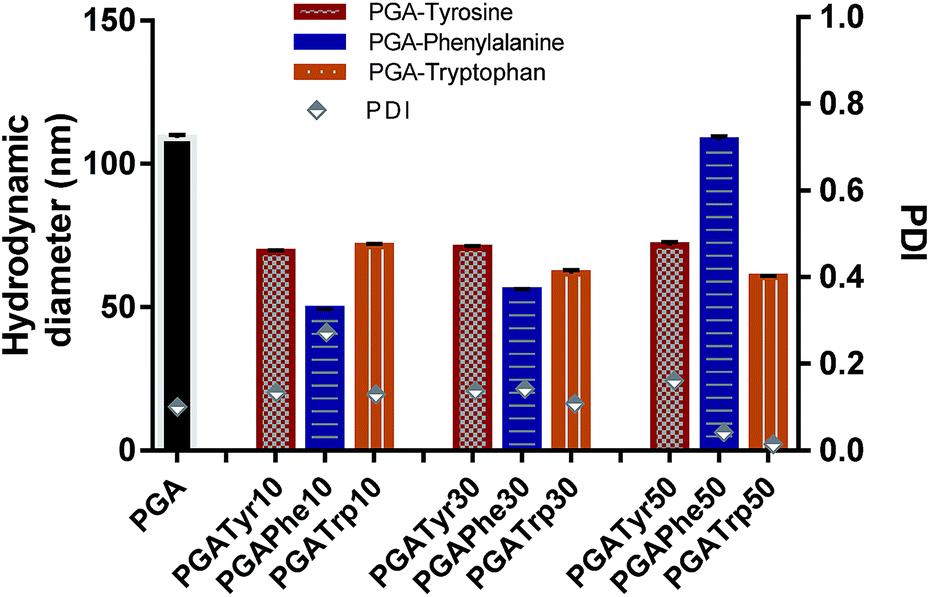 | ||
| Fig. 4 Hydrodynamic diameter and PDI are reported for all the nanoaggregates. Nanoparticles from modified polymers showed a smaller size compared to PGA. | ||
As depicted in Fig. 4 all the NAA modified materials show a smaller nanoparticle size than the bare PGA with really narrow peak width (p-value < 0.05) underlying the enhanced packing effect due to the presence of aromatic functionalization. PGATyr at all the explored degree of modification showed a similar consistent size centred around 70 nm. PGAPhe10 resulted in the smallest colloid (49.3 ± 0.2) while PGAPhe50 (108.1 ± 1.3) gave the highest hydrodynamic diameter mostly due to the low solubility of the material in acetone at the working concentration. On the other hand, PGATrp showed a contraction in sizes passing from around 71.3 ± 0.4 nm (PGATrp10) to 60.5 ± 0.5 nm (PGATrp50) indicating a stronger H-bond network and more settled stacking interactions with increased levels of functionalization.
Interestingly, the more hydrophobic functionalised NAA NPs (Phe and Trp) became more monodisperse with increasing amounts of NAA present in the polymer, a behaviour that could be again attributed to π–π interactions between the aromatic rings replacing the H-bonds between the free OH in the bare PGA.
It might be possible to exploit this feature for producing amorphous polymer–drug blends or viscous-NPs to encapsulate both water soluble drugs and lipophilic drugs.30
In the first attempt to evaluate the biocompatibility of these new materials, a hemolytic assay was performed. All the NPs tested, namely bare PGA and all the NAA alterations at 50% of functionalization, showed a negligible lytic activity (Fig. ESI3†). This activity was lower than 1%, in the concentration range of 0.05 and 0.5 mg ml−1. Some blood cell disruption was observed at 2.5 mg ml−1 of NPs. However, a similar haemolysis was seen in the control sample containing an equivalent amount of water (reported in Fig. ESI3†) so this apparent activity can be simply ascribed to sample preparation.
The biodegradation of the polymers was assessed using a nanoparticle size as, swelling of NPs is recognised as an indicator of enzymatic degradation.28 In order to assess materials degradation, the increase in NP diameter over time following the addition of enzymes can be measured.29 The size of PGA nanoparticles was found to increase after the addition of simulated intestinal fluid, containing pancreatin, in a time dependant manner until a weak signal similar to the enzyme preparation was reached, suggesting that this polymer readily undergoes degradation. However PGAPhe30 reached a plateau at about double the starting NP size suggesting a lower amount of degradation. It is interesting to notice the different swelling trend between the two materials. These differences may either be due to enzymic factors such as specificity and steric accessibility or may be due to nanoparticle packing factors attributable for example to the pi–pi stacking interactions amongst the phenyl groups inside the core of the PGAPhe30 NPs.
Coupling of NAAs offers the intriguing opportunity to introduce a wide range of functionalities into a polyester backbone via trivial coupling reactions with the possibility of using different coupling agents by exploiting the solubility of PGA in different solvents. Moreover as previously demonstrated a plethora of different moieties26,31 can be incorporated into PGA. The remarkably easy enzymatic pathway adopted to produce PGA combined with the possibility to couple both common and uncommon amino acids which are readily available in the market might lead to a new field of materials with highly desirable properties. Both markedly tunable biodegradable functionalized polymers and well-defined, tailor-made nanostructured polymer nanoparticles can be obtained by simply varying grafting density, and the nature of the pendant amino acids.
Data access statement
All raw data created during this research are openly available from the corresponding author (E-mail: martin.garnett@nottingham.ac.uk; ) and at the University of Nottingham Research Data Management Repository (https://rdmc.nottingham.ac.uk/) and all analysed data supporting this study are provided as ESI† accompanying this paper.Acknowledgements
This work was funded by EPSRC grant number EP/L013835/1. Cameron Alexander thanks EPSRC for a Leadership Fellowship grant number EP/H005625/1X. We also thank Christine Grainger-Boultby, Tom Booth and Paul Cooling for technical support.References
- A. C. Fonseca, M. H. Gil and P. N. Simões, Prog. Polym. Sci., 2014, 39, 1291–1311 CrossRef CAS.
- K. Bauri, S. G. Roy and P. De, Macromol. Chem. Phys., 2016, 217, 365–379 CrossRef CAS.
- R. Katakai, M. Oya, K. Uno and Y. Iwakura, J. Org. Chem., 1972, 37, 327–329 CrossRef CAS PubMed.
- R. Hirschmann, H. Schwam, R. G. Strachan, E. F. Schoenewaldt, H. Barkemeyer, S. M. Miller, J. B. Conn, V. Garsky, D. F. Veber and R. G. Denkewalter, J. Am. Chem. Soc., 1971, 93, 2746–2754 CrossRef CAS PubMed.
- R. G. Denkewalter, H. Schwam, R. G. Strachan, T. E. Beesley, D. F. Veber, E. F. Schoenewaldt, H. Barkemeyer, W. J. Paleveda, T. A. Jacob and R. Hirschmann, J. Am. Chem. Soc., 1966, 88, 3163–3164 CrossRef CAS.
- H. Lu, J. Wang, Z. Song, L. Yin, Y. Zhang, H. Tang, C. Tu, Y. Lin and J. Cheng, Chem. Commun., 2014, 50, 139–155 RSC.
- W. Vayaboury, O. Giani, H. Cottet, A. Deratani and F. Schué, Macromol. Rapid Commun., 2004, 25, 1221–1224 CrossRef CAS.
- T. J. Deming, Nature, 1997, 390, 386–389 CrossRef CAS PubMed.
- T. Aliferis, H. Iatrou and N. Hadjichristidis, Biomacromolecules, 2004, 5, 1653–1656 CrossRef CAS PubMed.
- L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762–798 CrossRef CAS.
- H. Sun, F. Meng, A. A. Dias, M. Hendriks, J. Feijen and Z. Zhong, Biomacromolecules, 2011, 12, 1937–1955 CrossRef CAS PubMed.
- U. Edlund and A.-C. Albertsson, Adv. Drug Delivery Rev., 2003, 55, 585–609 CrossRef CAS PubMed.
- K. Kempe, P. a. J. M. de Jongh, A. Anastasaki, P. Wilson and D. M. Haddleton, Chem. Commun., 2015, 51, 16213–16216 RSC.
- N. Cohen-Arazi, A. J. Domb and J. Katzhendler, Polymers, 2010, 2, 418–439 CrossRef CAS.
- X. Pang and C. C. Chu, Biomaterials, 2010, 31, 3745–3754 CrossRef CAS PubMed.
- M. Kolitz, N. Cohen-Arazi, H. Hagag, J. Katzhendler and A. J. Domb, Macromolecules, 2009, 42, 4520–4530 CrossRef CAS.
- A. Rodriguez-Galan, L. Franco and J. Puiggali, Polymers, 2011, 3, 65–99 CrossRef CAS.
- P. Kallinteri, S. Higgins, G. a. Hutcheon, C. B. S. Pourcain and M. C. Garnett, Biomacromolecules, 2005, 6, 1885–1894 CrossRef CAS PubMed.
- V. Taresco, R. G. Creasey, J. Kennon, G. Mantovani, C. Alexander, J. C. Burley and M. C. Garnett, Polymers, 2016, 89, 41–49 CrossRef CAS.
- F. Fernández-Trillo, A. Duréault, J. P. M. Bayley, J. C. M. van Hest, J. C. Thies, T. Michon, R. Weberskirch and N. R. Cameron, Macromolecules, 2007, 40, 6094–6099 CrossRef.
- K. Tappertzhofen, S. Beck, E. Montermann, D. Huesmann, M. Barz, K. Koynov, M. Bros and R. Zentel, Macromol. Biosci., 2016, 16, 106–120 CrossRef CAS PubMed.
- I.-D. Chung, P. Britt, D. Xie, E. Harth and J. Mays, Chem. Commun., 2005, 1046–1048 RSC.
- H. Yu, X. Chen, T. Lu, J. Sun, H. Tian, J. Hu, Y. Wang, P. Zhang and X. Jing, Biomacromolecules, 2007, 8, 1425–1435 CrossRef CAS PubMed.
- T. Heinze, M. Siebert, P. Berlin and A. Koschella, Macromol. Biosci., 2016, 16, 10–42 CrossRef CAS PubMed.
- Y. Yu, D. Wu, C. Liu, Z. Zhao, Y. Yang and Q. Li, Process Biochem., 2012, 47, 1027–1036 CrossRef CAS.
- A. M. Klibanov, Nature, 2001, 409, 241–246 CrossRef CAS PubMed.
- F. Mastrotto, S. Salmaso, Y. L. Lee, C. Alexander, P. Caliceti and G. Mantovani, Polym. Chem., 2013, 4, 4375 RSC.
- C. Caballero-George, E. Marin and M. Briceño, Int. J. Nanomed., 2013, 8, 3071 CrossRef PubMed.
- C. Colombo, L. Dragoni, S. Gatti, R. M. Pesce, T. R. Rooney, E. Mavroudakis, R. Ferrari and D. Moscatelli, Ind. Eng. Chem. Res., 2014, 53, 9128–9135 CrossRef CAS.
- V. M. Weiss, T. Naolou, G. Hause, J. Kuntsche, J. Kressler and K. Mäder, J. Controlled Release, 2012, 158, 156–164 CrossRef CAS PubMed.
- M. Jbeily, T. Naolou, M. Bilal, E. Amado and J. Kressler, Polym. Int., 2014, 63, 894–901 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra21464a |
| This journal is © The Royal Society of Chemistry 2016 |