
Manganese-catalysed hydroperoxidation of carbon–carbon double bonds using molecular oxygen present in air and hydroxylamine under ambient conditions†‡
Daisuke
Yamamoto
,
Masayuki
Soga
,
Hiromasa
Ansai
and
Kazuishi
Makino
*
Department of Pharmaceutical Sciences, Kitasato University, Shirokane, Minato-ku, Tokyo 108-8641, Japan. E-mail: makinok@pharm.kitasato-u.ac.jp; yamamotod@pharm.kitasato-u.ac.jp
First published on 23rd August 2016
Abstract
A highly efficient manganese-catalysed hydroperoxidation of carbon–carbon double bonds of enynes as well as styrene derivatives using N-hydroxyphthalimide, N-hydroxybenzotriazole or N-hydroxysuccinimide was developed. This reaction proceeded at room temperature through the direct incorporation of molecular oxygen present in air. The required catalytic loading of manganese(III) acetylacetonate is extremely low (generally 0.02–0.5 mol%, and a minimum of 0.001 mol%).
Introduction
Developing a new methodology for transition metal-catalysed oxidation reactions has been extensively studied in the recent decade, and molecular oxygen is essentially recognised as an ideal oxidant.1 Despite developing several elegant oxidation processes involving molecular oxygen as a sole oxidant,2 methodologies for directly incorporating molecular oxygen into organic substrates remain a major challenge in synthetic chemistry. The incorporation of an oxygen atom into substrates, such as C–H oxidation and epoxidation, has been observed in several biological processes,3 and aerobic epoxidation is used as an elegant strategy in organic chemistry.4 Recently, several aerobic difunctionalisations of carbon–carbon double bonds, such as oxyazidation,5 oxytrifluoromethylation,6 oxysulfonylation,7 oxysulfoxidation,8 oxysulfurization9 and oxyphosphorylation,10 have been demonstrated. In addition, metal-free or transition metal-catalysed aerobic dioxygenation11–13 of carbon–carbon double bonds using hydroxylamine derivatives, such as N-hydroxyphthalimide (NHPI), N-hydroxybenzotriazole (HOBt), N-hydroxysuccinimide (NHS) and hydroxamic acids, has witnessed significant progress (Scheme 1). The transition metal-catalysed oxidative N-hydroxypthalimidation using molecular oxygen was originally reported by Ozaki et al.13a in 1989 as a side reaction occurring during the manganese(III) tetraphenylporphyrin chloride-catalysed epoxidation of carbon–carbon double bonds present in styrene, 2-norbornene or indene, which resulted in a mixture of β-keto-N-alkoxyphthalimides and β-hydroxy-N-alkoxyphthalimides in moderate yield. Recently, Punniyamurthy et al.13b demonstrated that 10 mol% Cu(OAc)2·H2O-catalysed direct dioxygenation of carbon–carbon double bonds in styrene derivatives using NHPI and molecular oxygen in air at room temperature yields β-keto-N-alkoxyphthalimides in good yield (Scheme 1). Woerpel et al.13c suggested that in addition to NHPI, HOBt should be added to a carbon–carbon double bond in styrene derivatives or conjugated enynes in the presence of 5–20 mol% [Cu(MeCN)4]ClO4 under a pure O2 atmosphere. Furthermore, Lei et al.13d,e reported that using 10 mol% of CuCl or Co(OAc)2·4H2O promoted the oxidative addition of hydroxamic acids to styrene derivatives under a pure O2 atmosphere to selectively yield β-keto or β-hydroxy compounds. Xia's group13f reported that the combination of 10 mol% CuBr2 and 4.0 equivalents of di-tert-butyl peroxide worked as a catalyst for the direct dioxygenation of styrene derivatives in air to afford β-keto-N-alkoxyphthalimides in moderate to good yields. On the other hand, Tang et al.13g have demonstrated that 10 mol% of FeCl3 is an effective catalyst for the oxidative N-hydroxypthalimidation of styrene derivatives to afford β-keto-N-alkoxyphthalimides under a pure O2 atmosphere. In the methodologies described above involving the direct incorporation of an oxygen atom into a carbon–carbon double bond using N-hydroxylamine derivatives, the existence of metal hydroperoxide 2 is suggested as an intermediate. However, the corresponding hydroperoxide could not be isolated in acceptable yields due to the instability of the intermediate metal hydroperoxide 2 in metal-catalysed dioxygenation reactions. More recently, Woerpel et al.13h reported a 20 mol% of [Cu(MeCN)4]ClO4-catalysed hydroperoxidation of conjugated enynes using NHPI or HOBt under a pure O2 atmosphere to obtain propargyl hydroperoxides in good yields by lowering the reaction temperature to 0 °C. Furthermore, Punniyamurthy et al.13i also described the hydroperoxidation of styrene derivatives catalysed by 20 mol% Fe(NO3)3·9H2O. While these pioneering research efforts provided new methods for incorporating oxygen atom(s) derived from gaseous dioxygen into organic substrates, several challenges still remain. In particular, developing highly active and robust catalysts capable of directly incorporating molecular oxygen present in air into organic substrates without cooling or heating conditions is in high demand.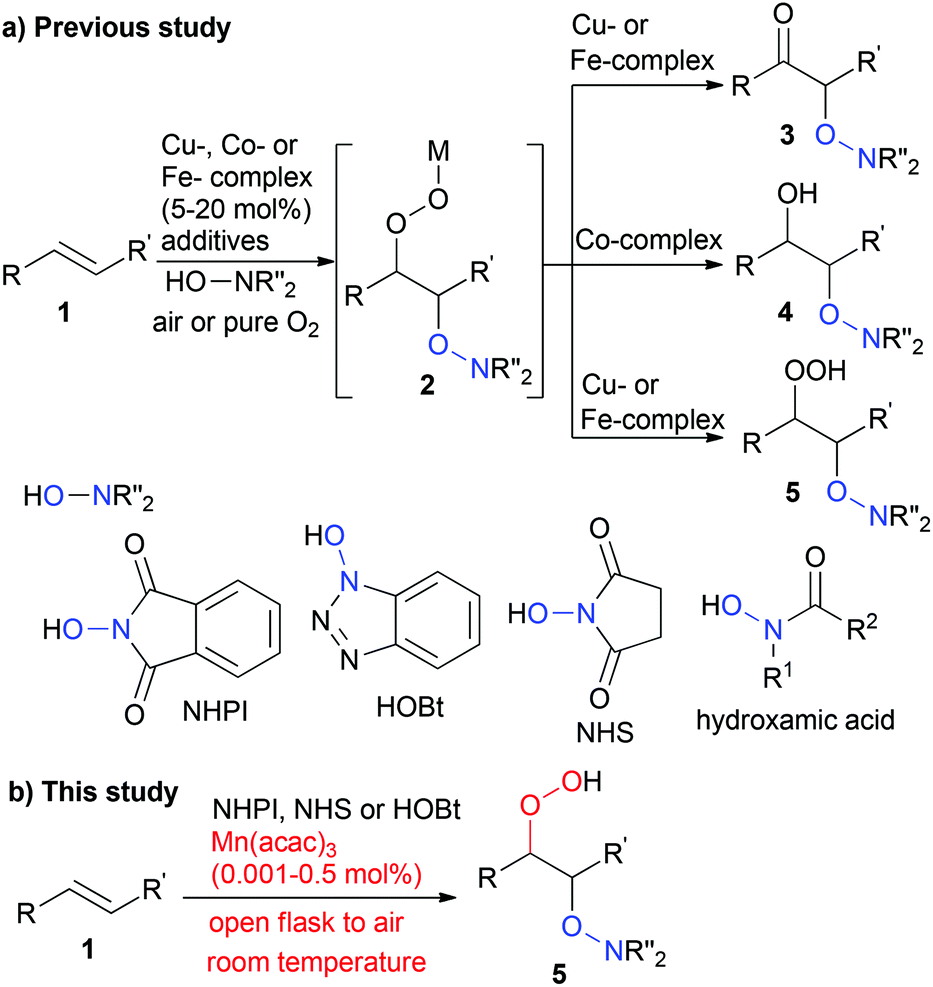 | ||
| Scheme 1 Aerobic hydroperoxidation of a carbon–carbon double bond with N-hydroxyphthalimide. | ||
Herein, we report a highly efficient manganese-catalysed hydroperoxidation of a carbon–carbon double bond using hydroxylamine and molecular oxygen in air (open flask) under ambient conditions.
Results and discussion
The reaction between 4-tert-butylstyrene 1a (1.0 equiv.) and NHPI (1.0 equiv.) in the presence of a metal complex in air at room temperature was initially investigated (Table 1). Using 1.0 mol% of Mn(acac)3 in MeCN as a catalyst for 5 h gave a mixture of hydroperoxide 5a and β-keto-N-alkoxyphthalimide 3a in good yield (61%, 5a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3a = 4:1, entry 1). Decreasing the amount of Mn(acac)3 to 0.02 mol% improved the yield and the product ratio of hydroperoxide 5a and ketone 3a (92%, 5a:3a = >20:1, entry 3). We interpreted this result as being indicative of the fact that further decomposition of hydroperoxide 5a to ketone 3a and other side products by a manganese complex was effectively suppressed due to reduced catalyst loading. To test the scalability of the hydroperoxidation reaction, a gram-scale reaction was performed at 0.02 mol% catalyst loading in air (entry 4). The reaction proceeded in >99% yield at room temperature. Although a further reduction of catalyst loading to 0.001 mol% required a longer reaction time in air at room temperature, the desired hydroperoxide 5a was obtained in high yield with an excellent product ratio (84% yield, 5a:3a = >20:1, entry 6). A series of solvents were screened, and it was observed that MeCN was essential for this reaction, while EtCN, CH2Cl2, benzene, THF, DMF, DMSO and MeOH afforded poor yields (entries 7–13).14
:3a = 4:1, entry 1). Decreasing the amount of Mn(acac)3 to 0.02 mol% improved the yield and the product ratio of hydroperoxide 5a and ketone 3a (92%, 5a:3a = >20:1, entry 3). We interpreted this result as being indicative of the fact that further decomposition of hydroperoxide 5a to ketone 3a and other side products by a manganese complex was effectively suppressed due to reduced catalyst loading. To test the scalability of the hydroperoxidation reaction, a gram-scale reaction was performed at 0.02 mol% catalyst loading in air (entry 4). The reaction proceeded in >99% yield at room temperature. Although a further reduction of catalyst loading to 0.001 mol% required a longer reaction time in air at room temperature, the desired hydroperoxide 5a was obtained in high yield with an excellent product ratio (84% yield, 5a:3a = >20:1, entry 6). A series of solvents were screened, and it was observed that MeCN was essential for this reaction, while EtCN, CH2Cl2, benzene, THF, DMF, DMSO and MeOH afforded poor yields (entries 7–13).14
|
|
|||||
|---|---|---|---|---|---|
| Entry | Mn(acac)3 (mol%) | Solvent | Time (h) | Yieldb (%) | (5a:3a)c |
| a Reaction conditions: 1a (0.50 mmol), metal salt (0.001–1.0 mol%), O2 in air at room temperature. b Isolated yield. c The ratio was determined by 1H NMR analysis of the crude sample. d The reaction was conducted at the 1.04 g (6.50 mmol) scale of 1a. | |||||
| 1 | 1.0 | MeCN | 5 | 61 | (4:1) |
| 2 | 0.1 | MeCN | 16 | 90 | (4:1) |
| 3 | 0.02 | MeCN | 24 | 92 | (>20:1) |
| 4d | 0.02 | MeCN | 72 | >99 | (>20:1) |
| 5 | 0.01 | MeCN | 31 | 83 | (>20:1) |
| 6 | 0.001 | MeCN | 46 | 84 | (>20:1) |
| 7 | 0.01 | EtCN | 24 | 5 | (17:1) |
| 8 | 0.01 | CH2Cl2 | 24 | 7 | (>20:1) |
| 9 | 0.01 | Benzene | 24 | Trace | — |
| 10 | 0.01 | THF | 24 | 11 | (>20:1) |
| 11 | 0.01 | DMF | 24 | Trace | — |
| 12 | 0.01 | DMSO | 24 | Trace | — |
| 13 | 0.01 | MeOH | 24 | Trace | — |
| 14 | none | MeCN | 24 | No reaction | |
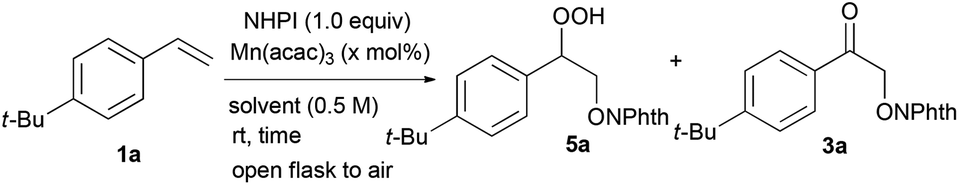
Using these optimal reaction conditions, potential substrates for this manganese-catalysed aerobic hydroperoxidation reaction were evaluated (Table 2). The reaction with styrene 1b gave hydroperoxide 5b in 64% yield. Styrenes substituted with a methyl group at the 4- or 3-position 1c and 1d were transformed into the corresponding hydroperoxides 5c and 5d, respectively, in excellent yield without the oxidation of the methyl group at the benzylic position. The reaction of the styrene derivatives bearing a 4-fluoro, 4-chloro or 4-bromo moiety on the aromatic ring produced the corresponding hydroperoxide 5e, 5f or 5g, respectively, in high yield (88%–94%). The electron-rich styrene derivative 1h with a methoxy group at the 4-position was effective under these oxidation conditions to provide hydroperoxide 5h in good yield (60%). Sterically hindered α-methylstyrene 1i could be efficiently oxidised in >99% yield. In addition, β-substituted styrenes 1j and 1k were transformed into the corresponding hydroperoxides 5j and 5k, respectively, in good yields with moderate diastereoselectivity.
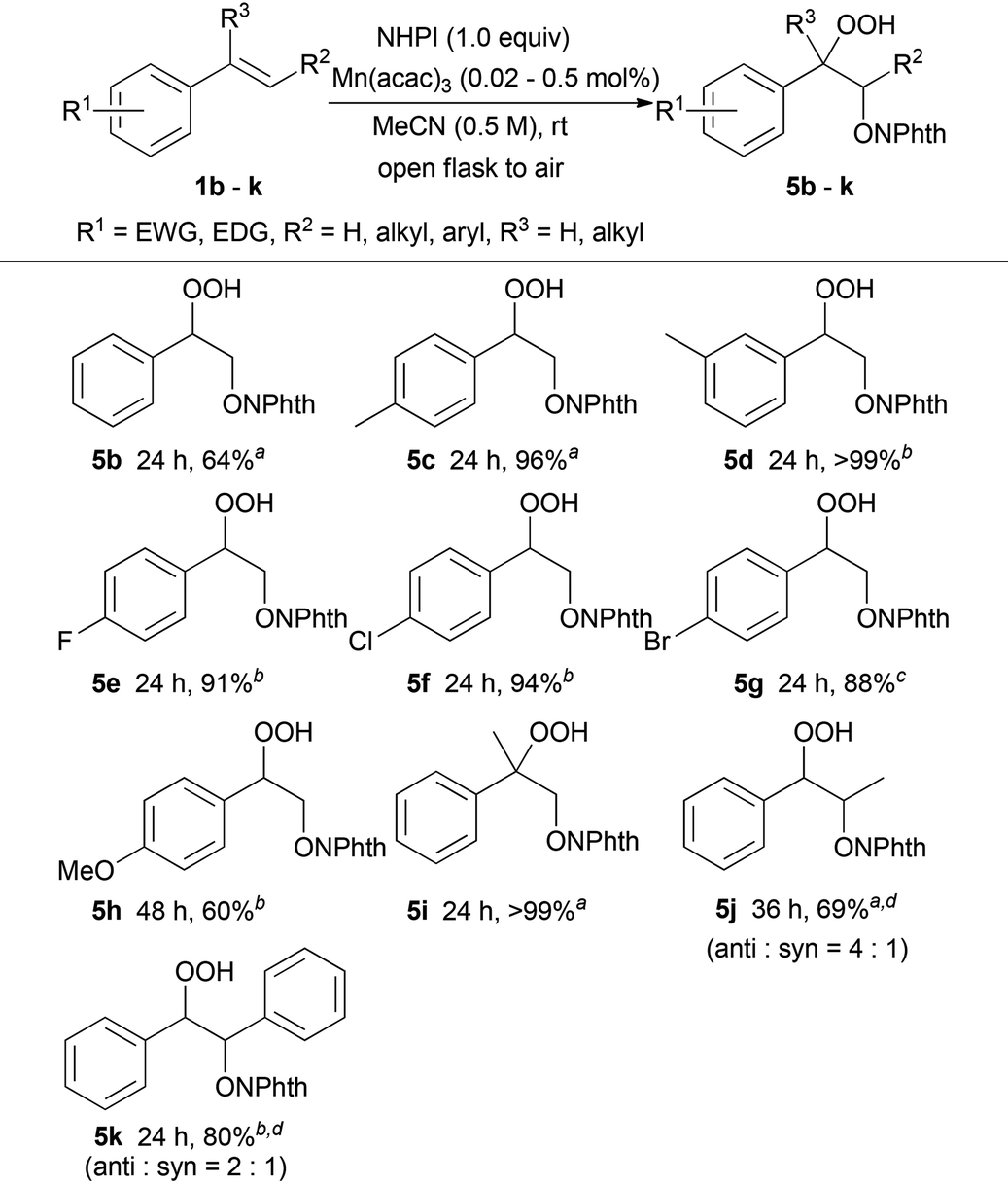
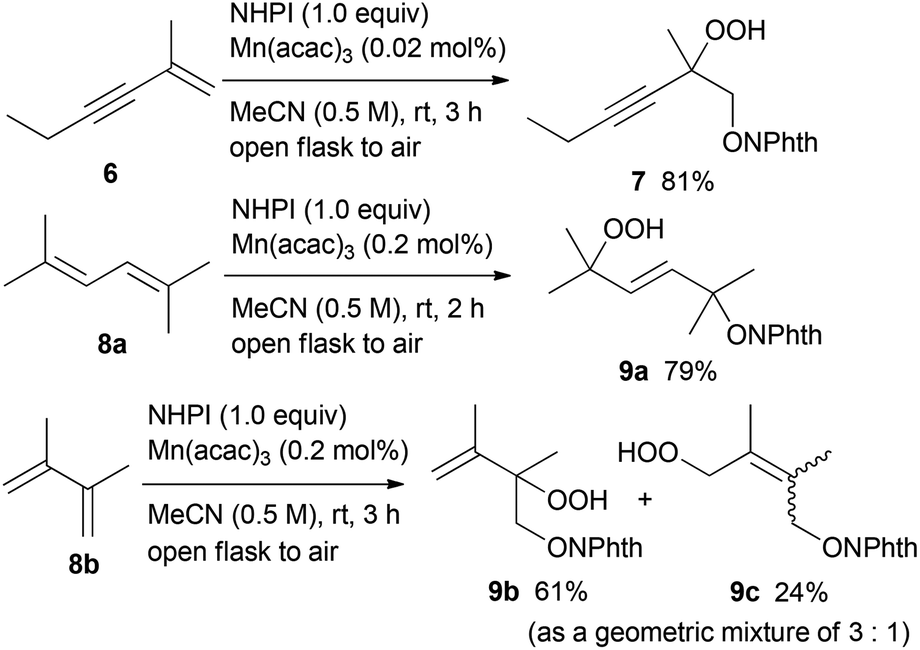
Furthermore, we investigated the hydroperoxidation of the conjugated enyne and dienes (Scheme 2).15 Treatment of enyne 6 with 0.02 mol% of Mn(acac)3 in air at room temperature for 3 h offered hydroperoxide 7 in 81% yield, while the carbon–carbon triple bond remained intact under these oxidation conditions. The conjugated dienes 8a and 8b were also viable substrates. The dioxygenation of 2,5-dimethyl-2,4-hexadiene 8a in the presence of 0.2 mol% of Mn(acac)3 provided the 1,4-dioxygenated product 9a in 79% yield as a single E-isomer. On the other hand, when 2,3-dimethyl-1,3-butadiene 8b was used as a substrate, the 1,2-dioxygenated product 9b and the 1,4-dioxygenated product 9c were obtained in 61% and 24% yield (as a geometric mixture of 3:1), respectively.
 | ||
| Scheme 2 Aerobic hydroperoxidation of the conjugated enyne and dienes. | ||
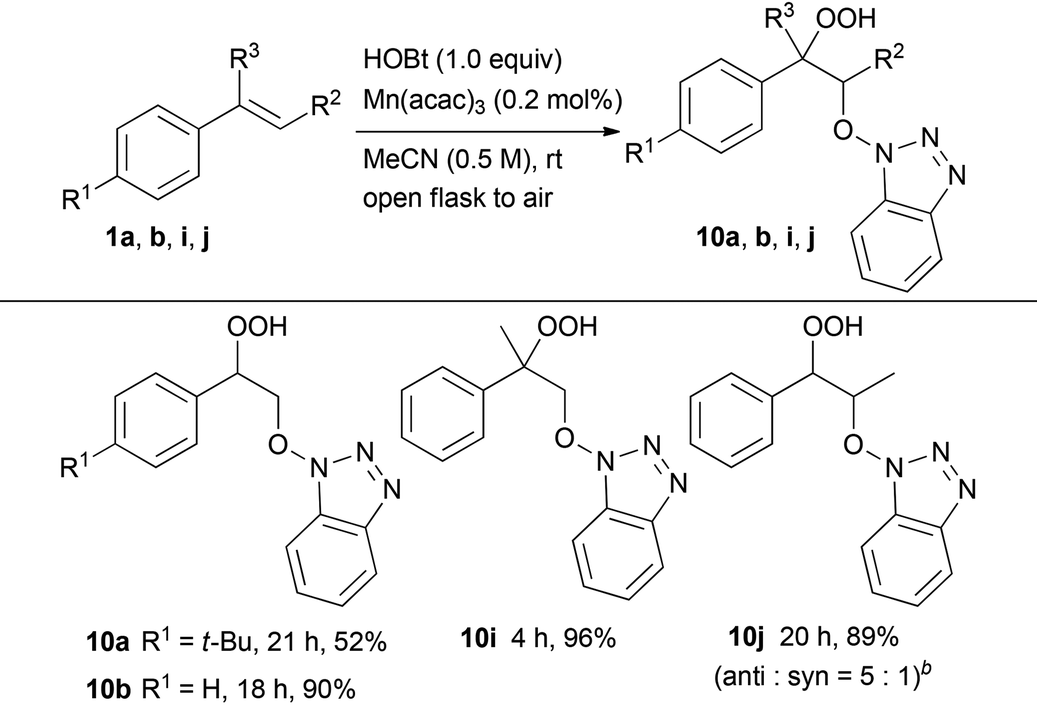
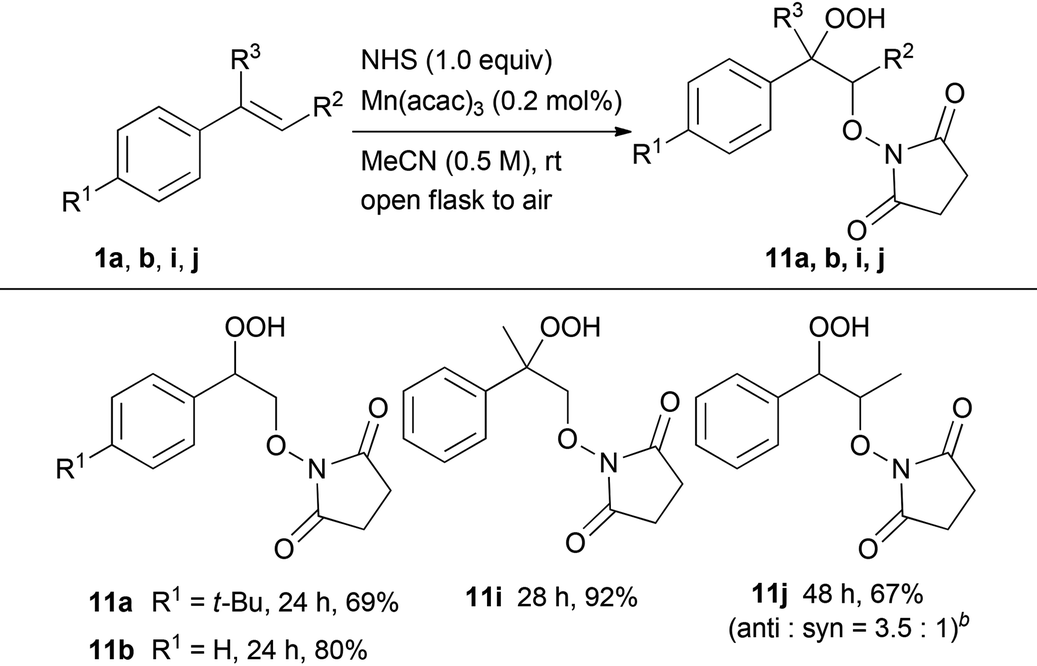
Subsequently, we evaluated the incorporation of molecular oxygen into a carbon–carbon double bond using HOBt or NHS instead of NHPI (Tables 3 and 4). The oxidative addition of HOBt to styrene 1b in the presence of 0.2 mol% of Mn(acac)3 occurred at room temperature in air to give hydroperoxide 10b in excellent yield (90%). On the other hand, the reaction of 4-substituted styrene derivatives, such as 4-tert-butylstyrene 1a, resulted in moderate yield (52%). Both α- and β-alkyl-substituted styrene derivatives 1i and 1j performed well as substrates providing the corresponding hydroperoxides 10i and 10j in 96% and 89% yield (anti:syn = 5:1), respectively. In addition, the oxidative addition of NHS to styrene derivatives, such as 1a, 1b, 1i and 1j, led to similar results yielding the corresponding hydroperoxides 11a, 11b, 11i and 11j, respectively.
Moreover, the manganese-catalysed oxidative addition of HOBt or NHS to the conjugated enyne 6 and diene 8a was also examined (Scheme 3). The dioxygenation of both enyne 6 and diene 8a using HOBt in air proceeded at low catalyst loading to afford hydroperoxides 12 and 14, respectively, in high yields compared to the reaction using NHS.
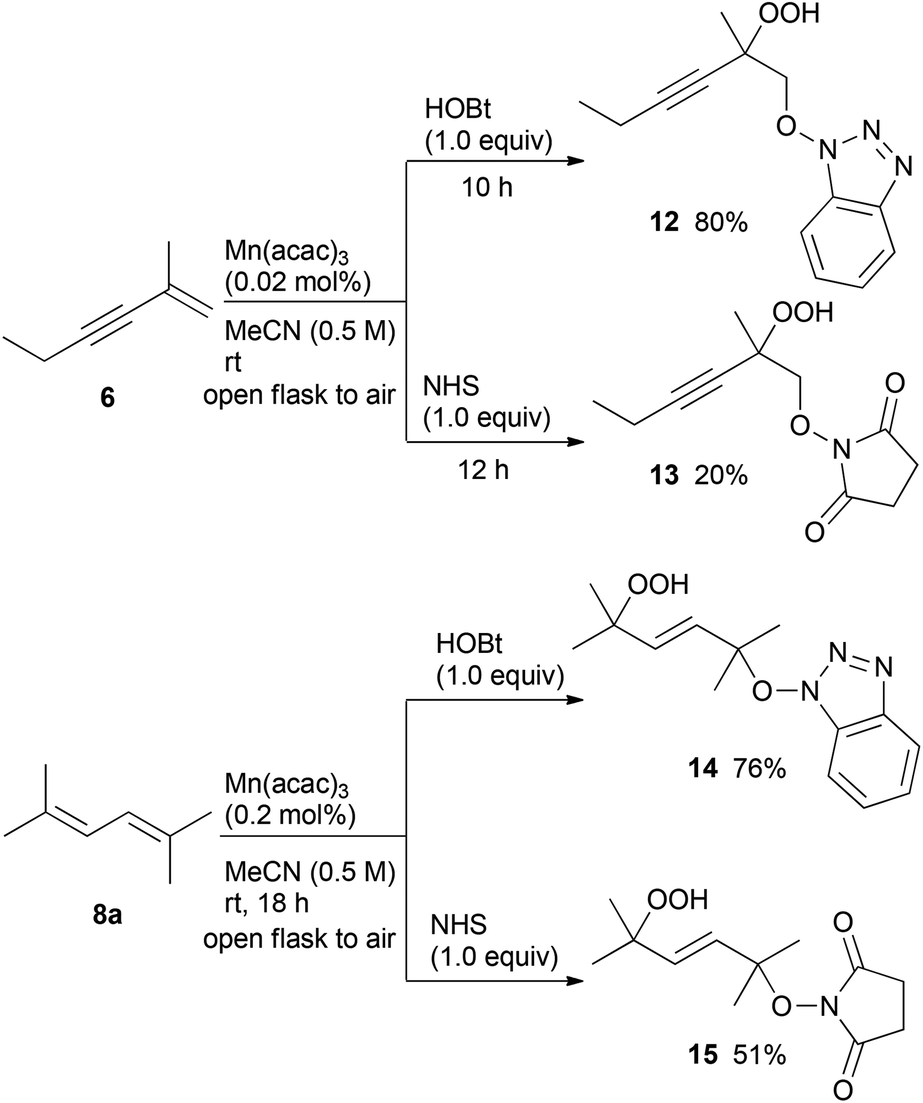 | ||
| Scheme 3 Aerobic hydroperoxidation of conjugated enyne 6 and diene 8a. | ||
Conclusions
To summarise our study, we have developed a manganese-catalysed aerobic hydroperoxidation method for conjugated alkenes. Notably, the unprecedentedly low catalyst loading of Mn(acac)3 promoted the oxidative addition of NHPI, HOBt or NHS to conjugated alkenes through the direct incorporation of molecular oxygen from air (pure oxygen is not required) without any other additives. Considering the desirable features, such as the simplicity of operation, inexpensive catalysts, a wide range of substrates and mild reaction conditions, this novel reaction provides an attractive synthetic approach to produce hydroperoxides. The investigation of the reaction mechanism involved in the manganese-catalysed aerobic hydroperoxidation reactions is currently ongoing in our laboratory.Experimental
Representative procedure for manganese-catalyzed hydroperoxidation with NHPI
To a stirred solution of 4-tert-butylstyrene (80.1 mg, 0.500 mmol) and N-hydroxyphthalimide (81.6 mg, 0.500 mmol) in MeCN (0.9 mL) at room temperature was added a solution of Mn(acac)3 in MeCN (100 μL, 0.10 μmol, 1.0 mM in MeCN) in air (open flask). The solution was stirred for 24 h at room temperature and quenched with saturated aqueous NaCl solution (0.5 mL). The resulting mixture was extracted with ethyl acetate (3 × 1.0 mL). The combined organic phases were washed with brine (1.0 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. The obtained crude material was purified by column chromatography (silica gel, hexane:EtOAc = 3:1) to afford 5a (163 mg, 0.459 mmol, 92%).
Gram-scale synthesis
To a stirred solution of 4-tert-butylstyrene (1.04 g, 6.50 mmol) and N-hydroxyphthalimide (1.06 g, 6.50 mmol) in MeCN (13.0 mL) at room temperature was added a solution of Mn(acac)3 in MeCN (1.0 mL, 1.3 μmol, 1.3 mM in MeCN) in air (open flask). The solution was stirred for 72 h at room temperature and quenched with saturated aqueous NaCl solution (10 mL). The resulting mixture was extracted with ethyl acetate (3 × 14 mL). The combined organic phases were washed with brine (14 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. The obtained crude material was purified by column chromatography (silica gel, hexane:EtOAc = 3:1) to afford 5a (2.31 g, 6.50 mmol, >99% yield).
2-(2-(4-(tert-Butyl)phenyl)-2-hydroperoxyethoxy)isoindoline-1,3-dione (5a)
Colorless oil; 1H NMR (400 MHz, CDCl3) δ: 9.37 (OOH), 7.89–7.84 (m, 2H, Ar–H), 7.80–7.75 (m, 2H, Ar–H), 7.39 (d, J = 8.8 Hz, 2H, Ar–H), 7.33 (d, J = 8.8 Hz, 2H, Ar–H), 5.40 (dd, J = 7.7, 3.8 Hz, 1H, CHOOH), 4.55 (dd, J = 11.6, 3.8 Hz, 1H, CH(OOH)CH2O), 4.50 (dd, J = 11.6, 7.7 Hz, 1H, CH(OOH)CH2O), 1.30 (s, 9H, Ar–C4H9); 13C NMR (100 MHz, CDCl3) δ: 163.8, 152.0, 134.7, 132.6, 128.7, 126.9, 125.7, 123.8, 85.3, 78.9, 34.6, 31.2; IR (neat) 3402, 3098, 3062, 3032, 2962, 1789, 1730, 1466, 1375, 1186, 1132, 1081, 1018, 997, 877, 701 cm−1; HRMS (FAB, NBA) m/z calcd for C20H21NNaO5 [M + Na]+ 378.1317, found 378.1319.Acknowledgements
This work was partially supported by a Grant-in-Aid for Young Scientists B (15K18837) from JSPS and a Kitasato University Research Grant for Young Researchers. We thank Dr K. Nagai and Ms M. Sato of Kitasato University for instrumental analyses.Notes and references
- For selected reviews, see: (a) T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329 CrossRef CAS PubMed; (b) J. Piera and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2008, 47, 3506 CrossRef CAS PubMed; (c) Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381 RSC.
- For a selected review on aerobic oxidation of alcohols, see: B. L. Ryland and S. S. Stahl, Angew. Chem., Int. Ed., 2014, 53, 8824 CrossRef CAS PubMed.
- (a) M. Sono, M. P. Roach, E. D. Coulter and J. H. Dawson, Chem. Rev., 1996, 96, 2841 CrossRef CAS PubMed; (b) F. van Rantwijk and R. A. Sheldon, Curr. Opin. Biotechnol., 2000, 11, 554 CrossRef CAS PubMed; (c) J. T. Groves, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3569 CrossRef CAS PubMed; (d) F. P. Guengerich, J. Biochem. Mol. Toxicol., 2007, 21, 163 CrossRef CAS PubMed.
- (a) T. Mukaiyama, T. Yamada, T. Nagata and K. Imagawa, Chem. Lett., 1993, 22, 327 CrossRef; (b) T. Mukaiyama and T. Yamada, Bull. Chem. Soc. Jpn., 1995, 68, 17 CrossRef CAS; (c) S. Koya, Y. Nishioka, H. Mizoguchi, T. Uchida and T. Katsuki, Angew. Chem., Int. Ed., 2012, 51, 8243 CrossRef CAS PubMed; (d) T. Uchida and T. Katsuki, J. Synth. Org. Chem., Jpn., 2013, 71, 1126 CrossRef CAS; (e) H. Kawai, S. Okusu, Z. Yuan, E. Tokunaga, A. Yamano, M. Shiro and N. Shibata, Angew. Chem., Int. Ed., 2013, 52, 2221 CrossRef CAS PubMed; (f) R. Irie, T. Uchida and K. Matsumoto, Chem. Lett., 2015, 44, 1268 CrossRef.
- X. Sun, X. Li, S. Song, Y. Zhu, Y.-F. Liang and N. Jiao, J. Am. Chem. Soc., 2015, 137, 6059 CrossRef CAS PubMed.
- (a) C.-P. Zhang, Z.-L. Wang, Q.-Y. Chen, C.-T. Zhang, Y.-C. Gu and J.-C. Xiao, Chem. Commun., 2011, 47, 6632 RSC; (b) A. Deb, S. Manna, A. Modak, T. Patra, S. Maity and D. Maiti, Angew. Chem., Int. Ed., 2013, 52, 9747 CrossRef CAS PubMed; (c) Y. Yang, Y. Liu, Y. Jiang, Y. Zhang and D. Vicic, J. Org. Chem., 2015, 80, 6639 CrossRef CAS PubMed; (d) C. Liu, Q. Lu, Z. Huang, J. Zhang, F. Liao, P. Peng and A. Lei, Org. Lett., 2015, 17, 6034 CrossRef CAS PubMed.
- (a) Q. Lu, J. Zhang, F. Wei, Y. Qi, H. Wang, Z. Liu and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 7156 CrossRef CAS PubMed; (b) W. Wei, C. Liu, D. Yang, J. Wen, J. You, Y. Suo and H. Wang, Chem. Commun., 2013, 49, 10239 RSC.
- T. Keshari, V. K. Yadav, V. P. Srivastava and L. D. S. Yadav, Green Chem., 2014, 16, 3986 RSC.
- S.-F. Zhou, X. Pan, Z.-H. Zhou, A. Shoberu and J.-P. Zou, J. Org. Chem., 2015, 80, 3682 CrossRef CAS PubMed.
- (a) W. Wei and J.-X. Ji, Angew. Chem., Int. Ed., 2011, 50, 9097 CrossRef CAS PubMed; (b) T. Taniguchi, A. Idota, S. Yokoyama and H. Ishibashi, Tetrahedron Lett., 2011, 52, 4768 CrossRef CAS.
- For metal-free aerobic dioxygenation of a carbon–carbon double bond using hydroxamic acids, see: (a) V. A. Schmidt and E. J. Alexanian, Angew. Chem., Int. Ed., 2010, 49, 4491 CrossRef CAS PubMed; (b) B. C. Giglio, V. A. Schmidt and E. J. Alexanian, J. Am. Chem. Soc., 2011, 133, 13320 CrossRef CAS PubMed; (c) V. A. Schmidt and E. J. Alexanian, Chem. Sci., 2012, 3, 1672 RSC.
- For metal-free aerobic dioxygenation of 2-norbornene using NHPI, see: Y. Ishii, J. Mol. Catal. A: Chem., 1997, 117, 1231 CrossRef.
- For transition metal-catalysed aerobic dioxygenation of a carbon–carbon double bond using NHPI, HOBt, NHS, or hydroxamic acids, see: (a) S. Ozaki, T. Hamaguchi, K. Tsuchida, Y. Kimata and M. Masui, J. Chem. Soc., Perkin Trans. 2, 1989, 951 RSC; (b) R. Bag, D. Sar and T. Punniyamurthy, Org. Lett., 2015, 17, 2010 CrossRef CAS PubMed; (c) A. A. Andia, M. R. Miner and K. A. Woerpel, Org. Lett., 2015, 17, 2704 CrossRef CAS PubMed; (d) Q. Lu, Z. Liu, Y. Luo, G. Zhang, Z. Huang, H. Wang, C. Liu, J. T. Miller and A. Lei, Org. Lett., 2015, 17, 3402 CrossRef CAS PubMed; (e) Q. Lu, P. Peng, Y. Luo, Y. Zhao, M. Zhou and A. Lei, Chem. – Eur. J., 2015, 21, 18580 CrossRef CAS PubMed; (f) X.-F. Xia, S.-L. Zhu, Z. Gu, H. Wang, W. Li, X. Liu and Y.-M. Liang, J. Org. Chem., 2015, 80, 5572 CrossRef CAS PubMed; (g) J.-Z. Zhang and Y. Tang, Adv. Synth. Catal., 2016, 358, 752 CrossRef CAS; (h) M. R. Miner and K. A. Woerpel, Eur. J. Org. Chem., 2016, 1860 CrossRef CAS; (i) R. Bag, D. Sar and T. Punniyamurthy, Org. Biomol. Chem., 2016, 14, 3246 RSC.
- The solvent effects suggest that the ligation of less hindered acetonitrile to a manganese complex plays an important role in the progress of this hydroperoxidation reaction of the carbon–carbon double bond.
- The oxidation of the conjugated enyne and dienes catalysed by 20 mol% of [Cu(MeCN)4]ClO4 under a pure O2 atmosphere was reported by Woerpel et al., see ref 13c and h.
Footnotes |
| † Celebrating the 75th Birthday of Professor Barry Trost. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00318d |
| This journal is © the Partner Organisations 2016 |