
Synthesis of 2,3-dihydrobenzofurans via the palladium catalyzed carboalkoxylation of 2-allylphenols†
Johnathon T.
Hutt
and
John P.
Wolfe
*
University of Michigan, Department of Chemistry, 930 N. University Ave., Ann Arbor, MI 48109-1055, USA. E-mail: jpwolfe@umich.edu
First published on 22nd August 2016
Abstract
A new Pd-catalyzed alkene carboalkoxylation strategy for the preparation of 2,3-dihydrobenzofurans is described. This method effects the coupling of readily available 2-allylphenol derivatives with aryl triflates to generate a wide range of functionalized 2,3-dihydrobenzofurans in good yields and diastereoselectivities (up to >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Use of newly developed reaction conditions that promote anti-heteropalladation of the alkene is essential in order to generate products in high yield.
:1). Use of newly developed reaction conditions that promote anti-heteropalladation of the alkene is essential in order to generate products in high yield.
2,3-Dihydrobenzofurans are a valuable structural motif present in numerous naturally occurring and biologically active molecules1 with notable examples such as the flavaglines,2 morphine alkaloids,3 and lignans and neolignanans.4 The biological importance of this motif has served as inspiration for numerous synthetic approaches to functionalized derivatives.5 Some examples of these efforts include palladium-catalyzed Wacker-type oxidative cyclizations,6 intramolecular palladium-catalyzed allylic alkylations,7 copper or nickel catalyzed alkene diarylation or alkylarylation of allylphenyl ethers,8 and palladium catalyzed C–H activation/C–O cyclizations.9 Despite the attention these compounds have received in the synthetic community, a general cross-coupling approach employing aryl electrophiles and simple 2-allylphenol derivatives has yet to be reported.10
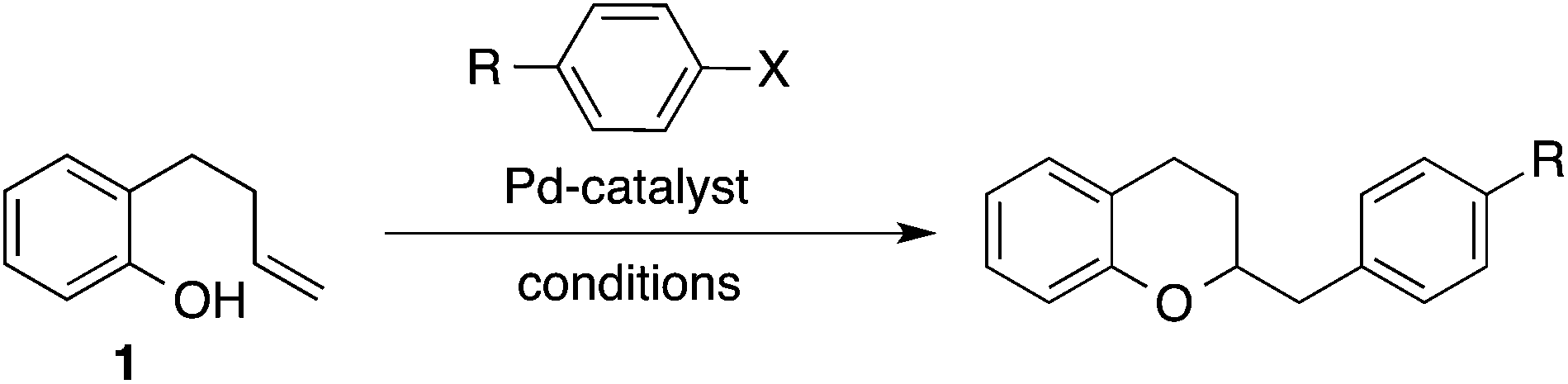
Over the past decade our group has developed a series of Pd-catalyzed alkene carboalkoxylation and carboamination reactions as a means of accessing functionalized, stereodefined heterocycles.11 Despite the broad substrate scope of these transformations, previous attempts to access the 2,3-dihydrobenzofuran core via Pd-catalyzed alkene carboalkoxylation with aryl or alkenyl halide electrophiles were met with limited success. For example, we have previously reported conditions for the construction of chroman derivatives (e.g., 3a) via Pd-catalyzed carboalkoxylation of 2-(but-2-enyl)phenols (e.g., 1), but the analogous generation of 4a from 2-allylphenol 2a proceeded in low yield (37%) (Scheme 1).12,13 The main side product observed in these reactions resulted from base-mediated isomerization of the substrate alkene, which appeared to be relatively fast compared to the rate of catalysis.
 | ||
| Scheme 1 Pd-catalyzed synthesis of chromans vs. dihydrobenzofurans. | ||
Our prior studies on the mechanism of related Pd-catalyzed alkene carboamination reactions illustrated the rate of the key carbon–heteroatom bond-forming step, syn-aminopalladation of the alkene, was highly dependent on the electronic properties of the nucleophile, with electron-rich amines undergoing relatively fast reactions.14 As such, we reasoned that the challenges associated with Pd-catalyzed alkene carboalkoxylations of 2-allylphenols were due at least in part to the relatively poor nucleophilicity of the aromatic alcohol substrate. We have recently demonstrated that Pd-catalyzed alkene carboamination reactions of electron-poor nucleophiles that fail under typical conditions can be conducted in high yield when reaction conditions that favor an anti-aminopalladation mechanistic pathway are employed.15–17 In this communication we describe the application of these conditions to the Pd-catalyzed synthesis of dihydrobenzofurans from 2-allylphenols, which proceeds through a key anti-oxypalladation of the substrate's pendant alkene.18
We began our studies by exploring the coupling of 2-allylphenol 2a and phenyl triflate using conditions analogous to those previously employed in Pd-catalyzed alkene carboamination reactions that proceed via anti-aminopalladation of the alkene.15–17 We were pleased to observe that a number of biaryl phosphine ligands provided the desired product 4a, with CPhos proving optimal to afford 85% yield of 4a (Table 1).
|
|
|||
|---|---|---|---|
| Entry | Solvent | Ligand | NMR yieldb (isolated yield) |
| a Reaction conditions: 1.0 equiv. 2a, 1.2 equiv. PhOTf, 1.4 equiv. LiOtBu, 2 mol% Pd(OAc)2, 5 mol% ligand, solvent (0.125 M), 98 °C, 16 h. b NMR yields were determined using phenanthrene as an internal standard. c The reaction was conducted at 82 °C. d This control reaction was conducted in the absence of Pd(OAc)2 and ligand. | |||
| 1 | PhCF3 | SPhos | 50 |
| 2 | PhCF3 | XPhos | 41 |
| 3 | PhCF3 | RuPhos | 73 |
| 4 | PhCF3 | BrettPhos | 73 |
| 5 | PhCF3 | CPhos | 84 (85) |
| 6 | t BuOH | CPhos | 46c |
| 7 | PhCF3 | Noned | 0 |
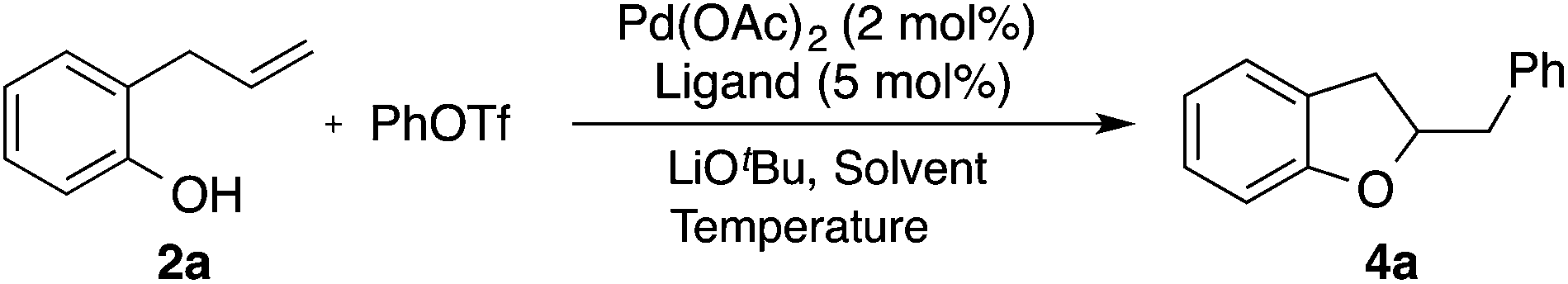
With optimized reaction conditions in hand we explored the scope of this transformation (Table 2). We found that the reaction was fairly general with respect to the aryl triflate component, as electron-rich, electron-poor and ortho-substituted electrophiles all provided the desired dihydrobenzofuran products 4a–4d in good yield. However, alkenyl triflates were found to have limited efficacy in this transformation; product 4e was generated in only 25% yield. Substitution on the aromatic ring of the substrate was generally well-tolerated as substrates bearing halogens, methoxy groups, or fused aromatic rings were all converted to the desired products 4f–4l in good yield. The reactions were also effective with substrates bearing allylic substituents to afford trans-2,3-disubstituted products 4m–4p. The diastereoselectivity of these transformations was dependent on the size of the allylic group. Substrates bearing a methyl group were transformed with moderate stereocontrol (5–8:1 dr), whereas substrates that contain an allylic phenyl group were converted with high selectivity (15–29:1 dr). Finally, substitution at the internal alkene carbon atom was also tolerated to generate products 4q–4r. However, larger loadings of CPhos (7.5 mol%) were necessary to obtain reproducible yields. The synthesis of tricyclic rings from exo-methylenecycloalkane derivatives proceeded with excellent stereocontrol to afford 4s–4v. The consecutive double carboalkoxylation of 2,3-diallylbenzene-1,4-diol proceeded to afford 4w in modest yield, but with low diastereoselectivity.
| a Reaction conditions: 1.0 equiv. 2, 1.2 equiv. R-OTf, 1.4 equiv. LiOtBu, 2 mol% Pd(OAc)2, 5 mol% CPhos, PhCF3 (0.125 M), 98 °C, 16 h. b Yields are isolated yields (average of two or more experiments) of material with >95% purity unless otherwise noted. Diastereomeric ratios were determined by 1H NMR analysis, and diastereomeric ratios of isolated materials were identical to those of the crude products unless otherwise noted in the ESI. c The isolated product contained ca. 10% of unidentified impurities. d The reaction was conducted using BrettPhos as ligand. e The reaction was conducted using a 7.5 mol% loading of CPhos. f The reaction was conducted using 2.4 equiv. PhOTf and 2.4 equiv. LiOtBu in PhCF3 (0.008 M). Diastereomeric ratio determined by GC. |
|---|
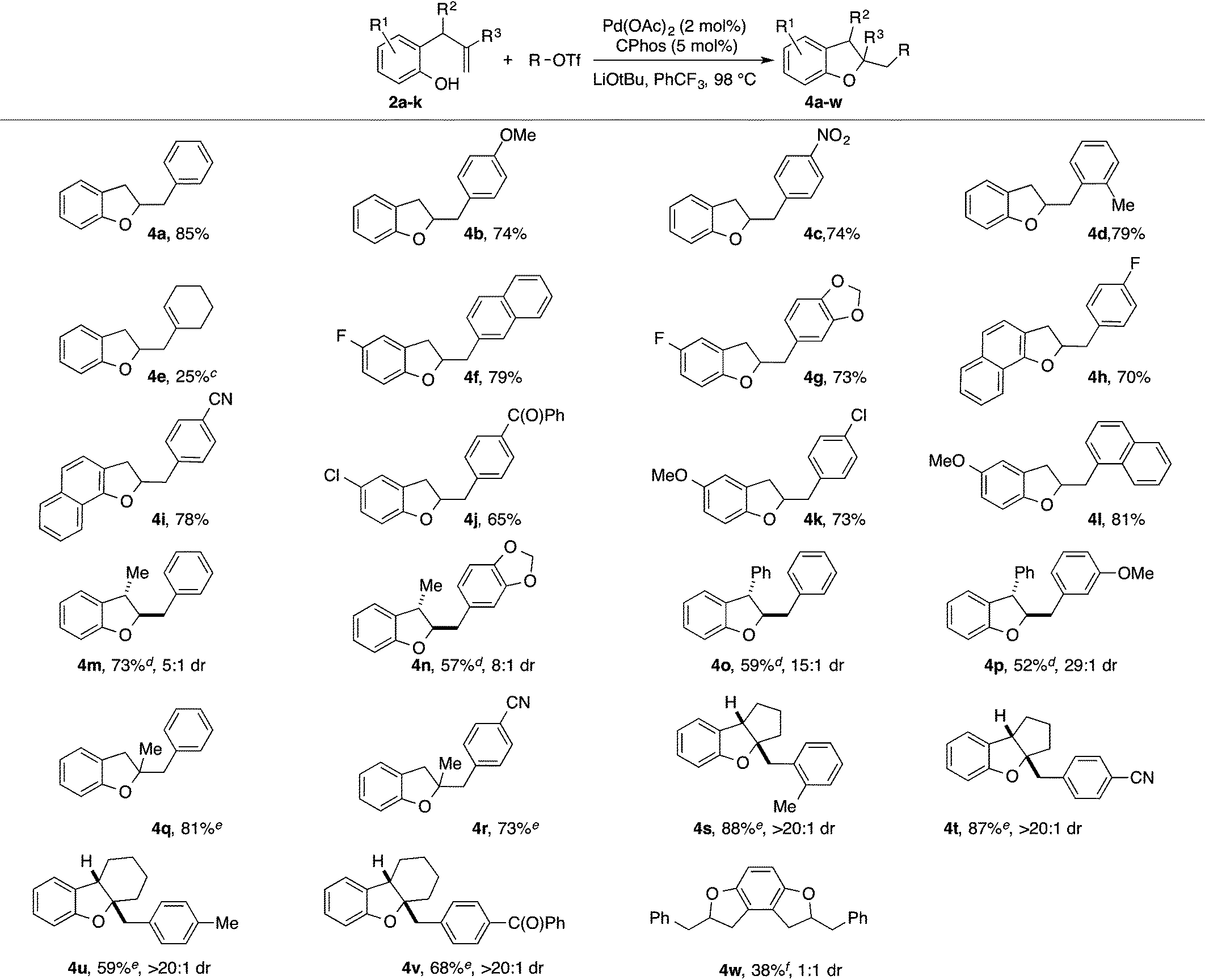
|
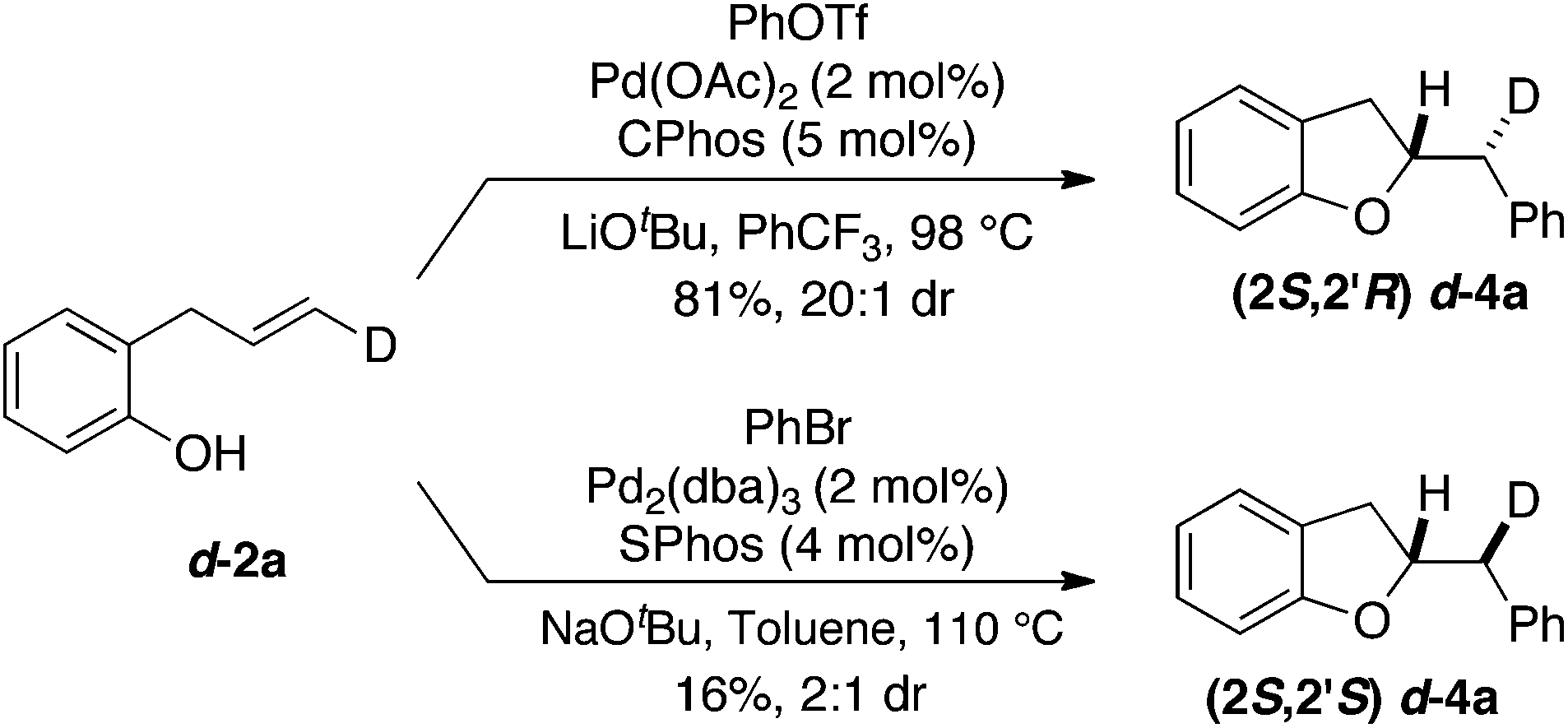
To probe the mechanism of this transformation by examining the stereochemistry of the alkene addition we carried out the Pd-catalyzed carboalkoxylation reaction of deuterated substrate d-2a with phenyl triflate (Scheme 2). This reaction afforded d-4a, which results from anti-addition of the oxygen atom and the aryl group to the alkene, in 81% yield and 20:1 dr. Interestingly, when d-2a was coupled with bromobenzene using the conditions previously employed for the synthesis of chromans the stereoisomeric product d-4a, generated via syn-addition to the alkene, was formed (albeit in low yield with modest 2:1 dr).19 This further illustrates the impact of reaction conditions on the stereochemistry of the heteropalladation step in Pd-catalyzed alkene difunctionalization reactions.15–17,20
 | ||
| Scheme 2 Deuterium labeling experiments. | ||
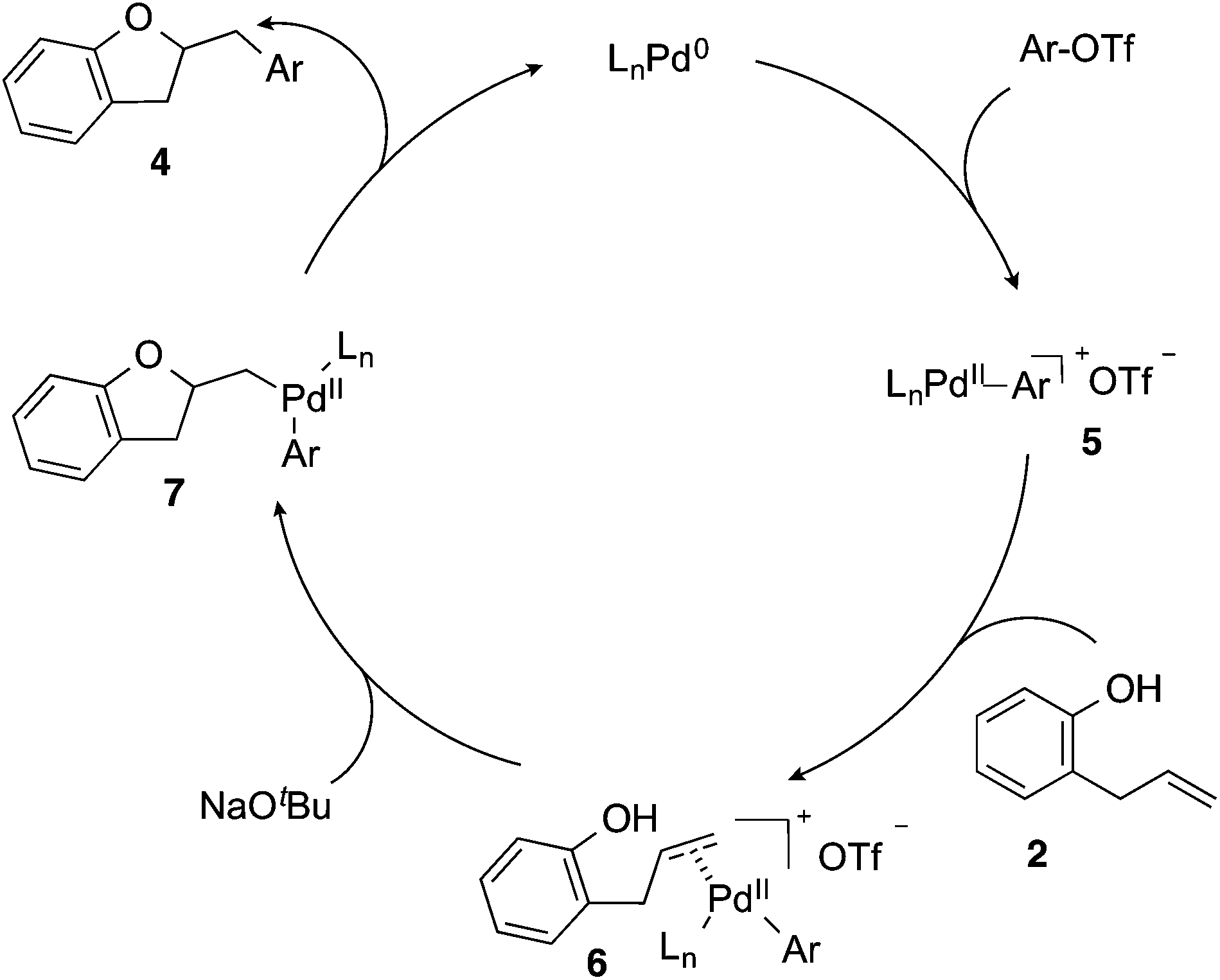
Based on the results of the deuterium labelling experiments, the Pd-catalyzed alkene carboalkoxylation reactions of 2-allylphenols likely proceed through the catalytic cycle illustrated in Scheme 3. Oxidative addition of the aryl triflate to the Pd(0)/CPhos complex (generated in situ) affords intermediate palladium(aryl)triflate complex 5. The cationic Pd-complex then binds to the alkene of substrate 2 to yield 6. Deprotonation of the phenol followed by anti-oxypalladation of the alkene provides intermediate 7, which undergoes reductive elimination to give the product 4 with concomitant regeneration of the Pd(0) catalyst.
 | ||
| Scheme 3 Catalytic cycle. | ||
Given the utility of these new conditions for Pd-catalyzed alkene–carboalkoxylation reactions of 2-allylphenols, we elected to briefly survey the utility of these conditions in chroman-forming reactions. As shown in Table 3, our newly developed conditions successfully promoted the coupling of 1 with three different aryl triflates to afford chromans 3a–c. The yields obtained using the anti-oxypalladation conditions with phenyl triflate or p-methoxyphenyl triflate as the electrophile were lower than the analogous reactions we have previously reported with aryl bromide electrophiles and syn-oxypalladation conditions (entries 1 and 2). However, the anti-oxypalladation conditions provided results superior to those obtained using the syn-oxypalladation conditions with an electron-poor aryl electrophile (entry 3).
|
|
|||
|---|---|---|---|
| Entry | R | X | Yieldb |
| a Reaction conditions for X = OTf: 1.0 equiv. 1, 1.2 equiv. R–OTf, 1.4 equiv. LiOtBu, 2 mol% Pd(OAc)2, 5 mol% ligand, solvent (0.125 M), 98 °C, 16 h. Reaction conditions for X = Br: 1.0 equiv. 1, 2.0 equiv. R–Br, 2.0 equiv. NaOtBu, 2 mol% Pd2(dba)3, 4 mol% S-Phos, toluene (0.25 M), 110 °C. b Yields are isolated yields (average of two or more experiments) of material with >95% purity. c Yields as reported in ref. 12. | |||
| 1 | H | OTf | 68 |
| 2 | H | Br | 76c |
| 3 | OMe | OTf | 41 |
| 4 | OMe | Br | 57c |
| 5 | C(O)Ph | OTf | 78 |
| 6 | C(O)Ph | Br | 51c |
Conclusions
In conclusion, we have developed a new method for the synthesis of 2,3-dihydrobenzofurans via the Pd-catalyzed alkene carboalkoxylation of 2-allylphenols. The reactions proceed in good yields with diastereoselectivities of 5:1 to >20:1 dr, and are effective with a broad range of aryl triflate electrophiles. This provides a new means of rapidly generating many different substituted 2,3-dihydrofurans from the corresponding phenol in a short synthetic sequence. Moreover, this further illustrates the utility of conditions that favor anti-heteropalladation pathways in Pd-catalyzed alkene difunctionalization reactions of relatively weak nucleophiles.
Acknowledgements
The authors acknowledge the NIH-NIGMS (GM 098314) for financial support of this work.Notes and references
- (a) M. Tamada, K. Endo, H. Hikino and C. Kabuto, Tetrahedron Lett., 1979, 10, 873 CrossRef; (b) M. Namikoshi, H. Kobayashi, T. Yoshimoto and S. Meguro, Chem. Lett., 2000, 29, 308 CrossRef; (c) S. Y. Li, H. Fuchino, N. Kawahara, S. Sekita and M. Satake, J. Nat. Prod., 2002, 262 CrossRef CAS; (d) Y. Li, C. Wu, D. Liu, P. Proksch, P. Guo and W. Lin, J. Nat. Prod., 2014, 77, 138 CrossRef CAS PubMed; (e) J.-H. Lee, Y.-G. Kim, S. Y. Ryu, M. H. Choo and J. Lee, J. Nat. Prod., 2014, 77, 168 CrossRef CAS PubMed.
- (a) F. Thuaud, Y. Bernard, G. Turkeri, R. Dirr, G. Aubert, T. Cresteil, A. Baguet, C. Tomasetto, Y. Svitkin, N. Sonenberg, C. G. Nebigil and L. Désaubry, J. Med. Chem., 2009, 52, 5176 CrossRef CAS PubMed; (b) N. Ribeiro, F. Thuaud, C. Nebigil and L. Désaubry, Bioorg. Med. Chem., 2012, 20, 1857 CrossRef CAS PubMed; (c) N. Ribeiro, F. Thuaud, Y. Bernard, C. Gaiddon, T. Cresteil, A. Hild, E. Hirsch, M. P. Michel, C. G. Nebigil and L. Désaubry, J. Med. Chem., 2012, 55, 10064 CrossRef CAS PubMed.
- For recent reviews, see: (a) K. W. Bentley, Nat. Prod. Rep., 2000, 17, 247 RSC; (b) P. R. Blakemore and J. D. White, Chem. Commun., 2002, 11, 1159 RSC; (c) J. Zezula and T. Hudlicky, Synlett, 2005, 388 CAS.
- For a recent review, see: J.-Y. Pan, S.-L. Chen, M.-H. Ynag, J. Wu, J. Sinkkonen and K. Zou, Nat. Prod. Rep., 2009, 26, 1251 RSC.
- For recent reviews, see: (a) T. D. Sheppard, J. Chem. Res., 2011, 35, 10476 Search PubMed; (b) F. Bertolini and M. Pineschi, Org. Prep. Proced. Int., 2009, 11, 385 CrossRef.
- (a) Y. Uozumi, K. Kato and T. Hayashi, J. Am. Chem. Soc., 1997, 119, 5063 CrossRef CAS; (b) R. M. Trend, Y. K. Ramtohul, E. M. Ferreira and B. M. Stoltz, Angew. Chem., Int. Ed., 2003, 42, 2892 CrossRef CAS PubMed.
- S. C. Pelly, S. Govender, M. A. Fernandes, H.-G. Schmalz and C. B. de Koning, J. Org. Chem., 2007, 72, 2857 CrossRef CAS PubMed.
- (a) W. You and M. K. Brown, J. Am. Chem. Soc., 2014, 136, 14730 CrossRef CAS PubMed; (b) H. Cong and G. C. Fu, J. Am. Chem. Soc., 2014, 136, 3788 CrossRef CAS PubMed.
- (a) X. Wang, Y. Lu, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 12203 CrossRef CAS PubMed; (b) H. Wang, G. Li, K. M. Engle, J.-Q. Yu and H. M. L. Davies, J. Am. Chem. Soc., 2013, 135, 6774 CrossRef CAS PubMed.
- Palladium-catalyzed alkene carboalkoxylation reactions between 2-(2-methylallyl)phenols and allyl chloride have been previously reported. These transformations are postulated to proceed via Pd(II)-mediated oxypalladation of the alkene followed by Heck-type carbopalladation of allyl chloride and subsequent β-chloride elimination. See: J. F. Hewitt, H. L. Williams, P. Aggarwal, C. D. Smith and D. J. France, Chem. Sci., 2013, 4, 3538 RSC.
- For recent reviews, see: (a) J. P. Wolfe, Synlett, 2008, 2913 CrossRef CAS; (b) D. M. Schultz and J. P. Wolfe, Synthesis, 2012, 351 CAS; (c) J. P. Wolfe, Top. Heterocycl. Chem., 2013, 32, 1 CAS.
- A. F. Ward, Y. Xu and J. P. Wolfe, Chem. Commun., 2012, 48, 609 RSC.
- In contrast, analogous transformations of 2-allylanilines are quite facile and efficient. See: R. Lira and J. P. Wolfe, J. Am. Chem. Soc., 2004, 126, 13906–13907 CrossRef CAS PubMed.
- For studies on the mechanism of syn-migratory insertion of alkenes into Pd–N bonds, see: (a) J. D. Neukom, N. S. Perch and J. P. Wolfe, J. Am. Chem. Soc., 2010, 132, 6276 CrossRef CAS PubMed; (b) P. S. Hanley, D. Marković and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 6302 CrossRef CAS PubMed; (c) J. D. Neukom, N. S. Perch and J. P. Wolfe, Organometallics, 2011, 30, 1269 CrossRef CAS; (d) P. S. Hanley and J. F. Hartwig, J. Am. Chem. Soc., 2011, 133, 15661 CrossRef CAS PubMed; (e) P. B. White and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 18594 CrossRef CAS PubMed.
- R. M. Fornwald, J. A. Fritz and J. P. Wolfe, Chem. – Eur. J., 2014, 20, 8782 CrossRef CAS PubMed.
- L. J. Peterson and J. P. Wolfe, Adv. Synth. Catal., 2015, 357, 2339 CrossRef CAS PubMed.
- N. R. Babij, G. M. McKenna, R. F. Fornwald and J. P. Wolfe, Org. Lett., 2014, 16, 3412 CrossRef CAS PubMed.
- For reviews on stereochemistry of alkene nucleopalladation reactions, see: (a) P. Kocovsky and J.-E. Bäckvall, Chem. – Eur. J., 2015, 21, 36 CrossRef CAS PubMed; (b) R. I. McDonald and S. S. Stahl, Chem. Rev., 2011, 111, 2981 CrossRef CAS PubMed; (c) K. H. Jensen and M. S. Sigman, Org. Biomol. Chem., 2008, 6, 4083–4088 RSC.
- The deuterated 2-allylphenol starting material was completely consumed in the coupling of d-2a proceeded to completion. The low yield of (2S,2′S)-d-4a is the result of the formation of a complex mixture of unidentifiable side products. Efforts to improve yields in reactions of aryl bromides through use of metal triflate additives has thus far been unsuccessful.
- (a) G. Liu and S. S. Stahl, J. Am. Chem. Soc., 2007, 129, 6328 CrossRef CAS PubMed; (b) A. B. Weinstein and S. S. Stahl, Angew. Chem., Int. Ed., 2012, 51, 11505 CrossRef CAS PubMed; (c) X. Ye, P. B. White and S. S. Stahl, J. Org. Chem., 2013, 78, 2083 CrossRef CAS PubMed; (d) C. Martinez, Y. Wu, A. B. Weinstein, S. S. Stahl, G. Liu and K. Muniz, J. Org. Chem., 2013, 78, 6309 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00215c |
| This journal is © the Partner Organisations 2016 |