
Tandem oxidative radical brominative addition of activated alkynes and spirocyclization: switchable synthesis of 3-bromocoumarins and 3-bromo spiro-[4,5] trienone†
Guanyinsheng
Qiu
*a,
Tong
Liu
b and
Qiuping
Ding
*b
aCollege of Biological, Chemical Science and Engineering, Jiaxing University, 118 Jiahang Road, Jiaxing 314001, China. E-mail: 11110220028@fudan.edu.cn
bCollege of Chemical and Engineering, Jiangxi Normal University, Nanchang 330013, China. E-mail: dqpjxnu@gmail.com
First published on 18th February 2016
Abstract
A K2S2O8-mediated tandem radical brominative addition of alkynoates, oxidative spiro-cyclization, and 1,2-migration of esters is reported for the synthesis of 3-bromocoumarins with high efficiency. The prepared 3-bromocoumarins are successfully converted into 3-alkynyl and 3-phosphite coumarins under mild conditions. The synthesis of a specific bromo-incorporated spiro-[4,5]trienyl-2,8-dione is achieved under the same conditions when N-methyl-N,3-diphenylpropiolate is used as the substrate.
As a privileged scaffold, the coumarin core is ubiquitous in many bioactive natural products and pharmaceuticals.1 Understandably, particular emphasis has been put on the development of methodologies for its synthesis, with an aim to develop facile and efficient protocols to provide coumarin and coumarin-based compound libraries.2 The Pechmann condensation, recognized as one of the most attractive approaches, represents an elegant method to construct the coumarin core.3 Additionally, transitional metal catalysis (especially palladium catalysis) provides an alternative route to various coumarins.4 Despite these achievements, it is still highly desirable to develop novel synthetic methodologies for the preparation of coumarin derivatives, due to the demands of chemical genetic research and the process of coumarin-based drug discovery.
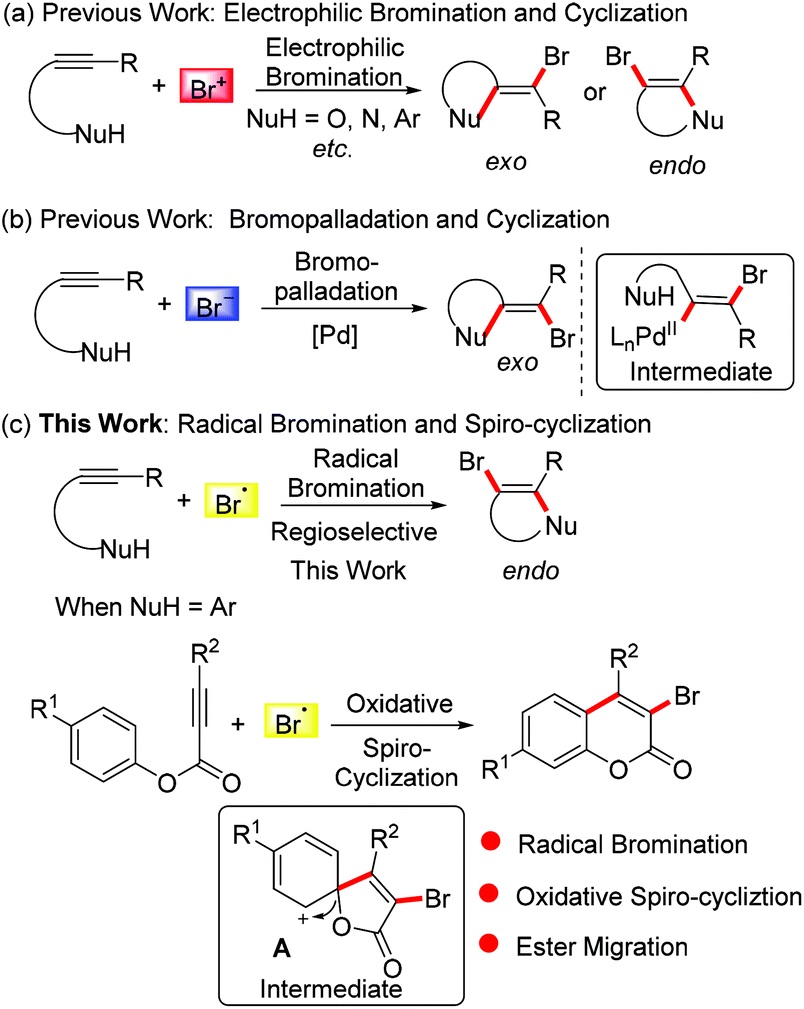
The bromo group is a very important synthon in the field of organic synthesis since it can be elaborated into other versatile building blocks through known nucleophilic substitution and transitional metal-catalyzed coupling reactions. In this respect, tremendous efforts have been made to introduce bromo groups into structures of interest.5 The bromination of alkynes attracts ever-growing interest from chemists. Particularly, numerous heterocyclic compounds have been synthesised via electrophilic bromination of alkynes combined with sequential cyclization. However, the selective formation of exo-dig-cyclization products and endo-dig-cyclization products is still challenging (Scheme 1a).6 With the development of palladium chemistry, bromopalladation has also been employed to incorporate bromo groups into alkynes to provide bromo-elaborated architectures. In these transformations, the bromo group is generally introduced into the product via a selective exo-dig-cyclization (Scheme 1b) and a trans-bromovinylpalladium species is involved as a key intermediate.7
 | ||
| Scheme 1 Reaction design for the synthesis of 3-bromocoumarins via a radical process. | ||
Guided by the above results and considering the versatility of the bromo group in organic synthesis, in this paper we exploit the addition of bromo radicals to alkynes. Combined with cyclization, we envisioned that a radical brominative addition of alkynes may selectively give rise to endo-dig-cyclization products (Scheme 1c). Considering the importance of coumarin derivatives, we turned our attention to alkynoate-based transformations for the synthesis of 3-bromocoumarins, an important synthon for the construction of antimicrobial inhibitors, fluorescent dyes, and mercury detection sensors.8 Traditional methods towards 3-bromocoumarins have been focused on the electrophilic bromination of the prepared coumarins.9 These known protocols often use liquid bromine as a bromo source (also including in situ generated liquid bromine from oxidation of HBr/Et4NBr by oxone/Dess–Martin reagent).9a–c In the method reported herein, we employed tetra-n-butyl ammonium bromide (TBAB) as the bromo source. We assumed that TBAB would be oxidized by K2S2O8 into a bromo radical, and importantly the generated bromo radical did not undergo homocoupling to liquid bromine. According to our previous findings,10 we hypothesized that the bromo radical would add to the triple bond of the alkynoate, followed by oxidative spiro-cyclization and 1,2-migration of the ester group to provide 3-bromocoumarin. In this planned transformation, the bromo-incorporated spiro-bicycle cation A was proposed as a critical intermediate (Scheme 1c).
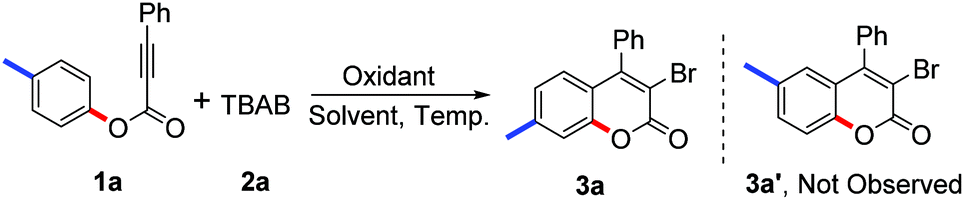
In our preliminary trials, we were pleased to find that the model reaction of tolyl alkynoate 1a with TBAB 2a gave the desired product in 42% yield in the presence of 2.0 equiv. K2S2O8 in 1,2-dichloroethane/H2O (v/v 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 60 °C (Table 1, entry 1). As expected, structure identification by NMR, HRMS, and X-ray diffraction suggested that the desired 3-bromo-7-methylcoumarin 3a had been synthesised.14 The 3-bromo-6-methylcoumarin 3a′, which should be observed according to the results reported by Wu and Wang,11 was not detected in this reaction. From this result, it seemed that our proposed pathway involving a radical bromination, oxidative spiro-cyclization and 1,2-ester migration was reliable. Comparing the product 3a with 3a′, the methyl group in the coumarin core migrated from the C6 position to the C7 position due to the 1,2-migration of the ester group (Scheme 2). This protocol represents a safe and efficient alternative to traditional methods for the synthesis of 3-bromocoumarins.
:1) at 60 °C (Table 1, entry 1). As expected, structure identification by NMR, HRMS, and X-ray diffraction suggested that the desired 3-bromo-7-methylcoumarin 3a had been synthesised.14 The 3-bromo-6-methylcoumarin 3a′, which should be observed according to the results reported by Wu and Wang,11 was not detected in this reaction. From this result, it seemed that our proposed pathway involving a radical bromination, oxidative spiro-cyclization and 1,2-ester migration was reliable. Comparing the product 3a with 3a′, the methyl group in the coumarin core migrated from the C6 position to the C7 position due to the 1,2-migration of the ester group (Scheme 2). This protocol represents a safe and efficient alternative to traditional methods for the synthesis of 3-bromocoumarins.
 | ||
| Scheme 2 Synthetic applications of 3-bromocourmarins. Isolated yield based on alkynoates. | ||
a
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Oxidant | Temp. (°C) | Yieldb (%) |
| a Reaction conditions: alkynoates 1a (0.2 mmol), tetra-n-butyl amonium bromide (TBAB) 2b (1.0 equiv.), K2S2O8 (2.0 equiv.), overnight. b Isolated yield based on alkynoates 1a. c In the presence of 2.0 equiv. TBAB. d 2.5 equiv. TBAB was added. e 2.0 equiv. TEMPO was added as an additive. f The starting material disappeared and many byproducts were observed. | ||||
| 1 | DCE/H2O (v/v = 1:1) |
K2S2O8(2.0 equiv.) | 60 | 42 |
| 2 | MeCN/H2O (v/v = 1:1) |
K2S2O8(2.0 equiv.) | 60 | 27 |
| 3 | DCE | K2S2O8(2.0 equiv.) | 60 | NR |
| 4 | H2O | K2S2O8(2.0 equiv.) | 60 | 11 |
| 5 | MeOH/H2O (v/v = 1:1) |
K2S2O8(2.0 equiv.) | 60 | Messyf |
| 6 | DCE/H2O (v/v = 1:2) |
K2S2O8(2.0 equiv.) | 60 | 36 |
| 7 | DCE/H2O (v/v = 2:1) |
K2S2O8(2.0 equiv.) | 60 | 28 |
| 8 | DCE/H2O (v/v = 1:1) |
K2S2O8(3.0equiv.) | 60 | 41 |
| 9 | DCE/H2O (v/v = 1:1) |
K2S2O8(1.5 equiv.) | 60 | 42 |
| 10 | DCE/H2O (v/v = 1:1) |
(NH4)2S2O8(1.5 equiv.) | 60 | 37 |
| 11 | DCE/H2O (v/v = 1:1) |
K2S2O8(1.5 equiv.) | 80 | 52 |
| 12 | DCE/H2O (v/v = 1:1) |
K2S2O8(1.5 equiv.) | 90 | 58 |
| 13 | DCE/H2O (v/v = 1:1) |
K2S2O8(1.5 equiv.) | 100 | 60 |
| 14 |
DCE/H
2
O (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1) :1)
|
K 2 S 2 O 8 (1.5 equiv.) | 90 | 72 |
| 15d | DCE/H2O (v/v = 1:1) |
K2S2O8(1.5 equiv.) | 90 | 75 |
| 16e | DCE/H2O (v/v = 1:1) |
K2S2O8(1.5 equiv.) | 90 | NR |
Encouraged by these results, we optimized the reaction conditions. Relevant factors including oxidant, solvent, temperature etc. were evaluated. The results are illustrated in Table 1. From the solvent screening, it seemed that a co-solvent was critical in the reaction, and a mixture of DCE and water (v/v = 1:1) was the best choice (entry 1, Table 1). This is probably because water can improve the solubility of the oxidant K2S2O8. The use of a co-solvent probably stabilizes the bromo radical, thus lowering the tendency towards the formation of liquid bromine via homocoupling. The use of DCE or H2O only did not improve the reaction (entries 3 and 4, Table 1). The reactions using other mixtures such as MeOH/H2O or MeCN/H2O did not give better yields (entries 2 and 5, Table 1). Changing the DCE/H2O ratio did not improve the efficiency of the reaction (entries 6 and 7, Table 1). Increasing the oxidant loading did not have significant impact on the yield (entry 8, Table 1). An identical result was observed when the loading of oxidant K2S2O8 was decreased to 1.5 equiv. (entry 9, Table 1). Other oxidants such as (NH4)2S2O8 and TBHP were also explored, and no improvements were detected (entry 10, Table 1). From the results of the temperature screening, the temperature affected the yields significantly. When the reaction was run at 80 °C, the yield improved to 52% (entry 11, Table 1). Further increase of the temperature to 90 °C gave a better yield (58%, entry 12, Table 1). However, when the reaction was carried out at 100 °C, the yield of the reaction did not change significantly (entry 12, Table 1). The loading of TBAB was critical for the reaction efficiency. By altering the amount of TBAB from 1.0 equivalent to 2.0 equivalents, the reaction efficiency improved greatly, leading to the desired product 3a in 72% yield (entry 14, Table 1). A comparable yield was achieved when the loading of TBAB was increased to 2.5 equiv. (entry 15, Table 1). In order to understand the mechanism of the reaction, 2.0 equivalents of TEMPO were added (entry 16, Table 1). The negative result indicated that the transformation probably followed a radical pathway.
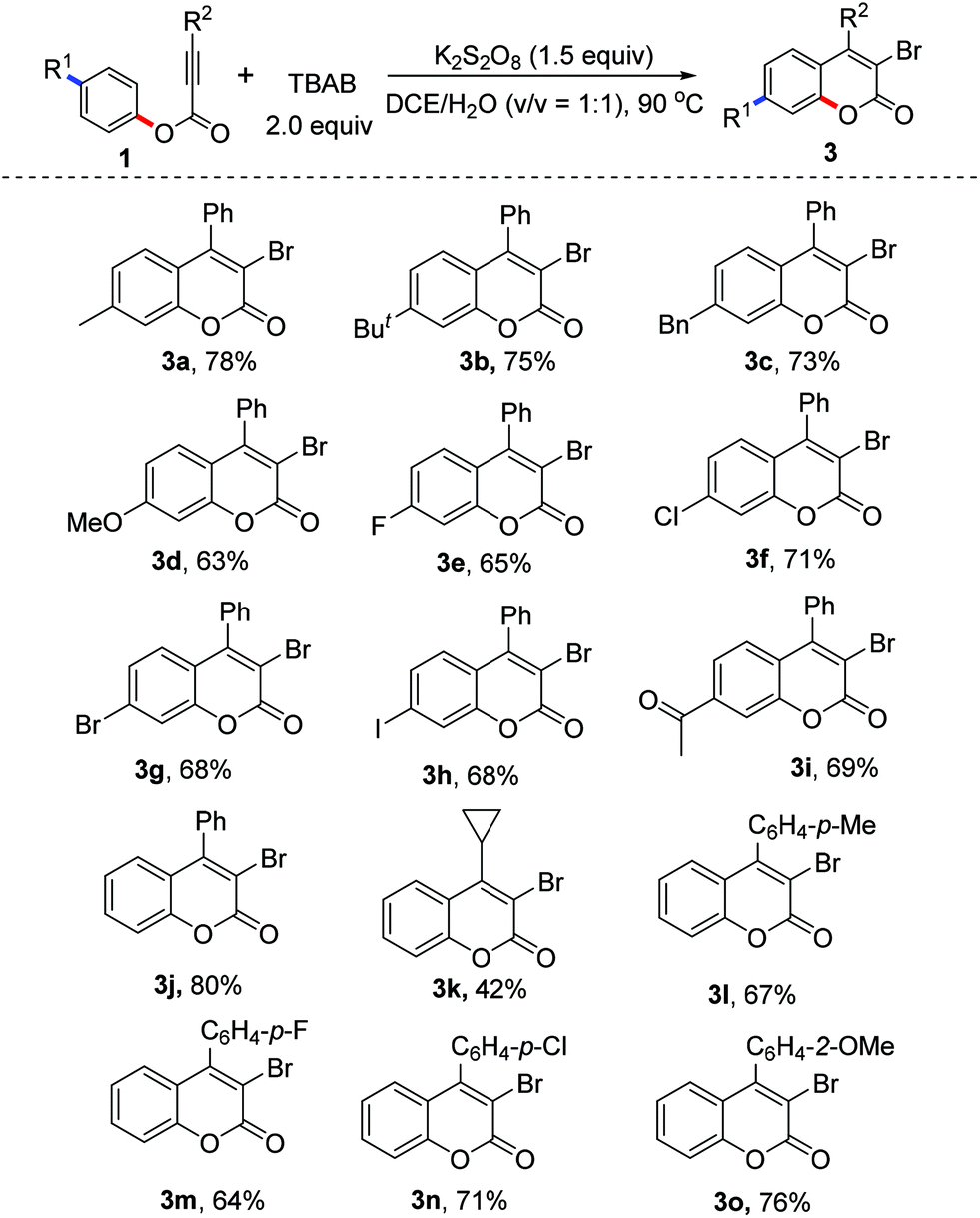
With the optimized conditions in hand (2.0 equiv. TBAB, 2.0 equiv. K2S2O8, in DCE/H2O (v/v 1:1) at 90 °C), we examined the scope of the reaction. The results are presented in Table 2. From the results shown in Table 2, various R1 substituents were suitable for the reaction. The corresponding products 3a–3j were delivered in 63–78% yields. For example, the reactions using bromo, iodo, and acetyl-bearing alkynoates 1g–1i gave the corresponding products 3g–3i in good yields. The scope of the substituents R2 was also explored. From the results, it seems that aryl groups were more favorable for the formation of 3-bromocoumarins. For example, the reaction of phenyl 3-phenylpropiolate provided 3-bromo-4-phenylcoumarin 3j in 80% yield, while the reaction of phenyl 3-cyclopropylpropiolate produced 3-bromo-4-cyclopropylcoumarin 3k in 42% yield. The electronic effect of aryl R2 groups did not affect the yields significantly. The reactions using substrates with methyl, fluoro, chloro, and methoxyl substitutents offered similar yields, providing the corresponding products 3l–3o in good yields. To our surprise, under the standard conditions the reactions using tetra-n-butylammonium fluoride (TBAF), tetra-n-butylammonium chloride (TBAC) and tetra-n-butylammonium iodide (TBAI) as halo sources did not give the desired halogenated coumarins.
| a Isolated yield based on alkynoates 1. |
|---|
|
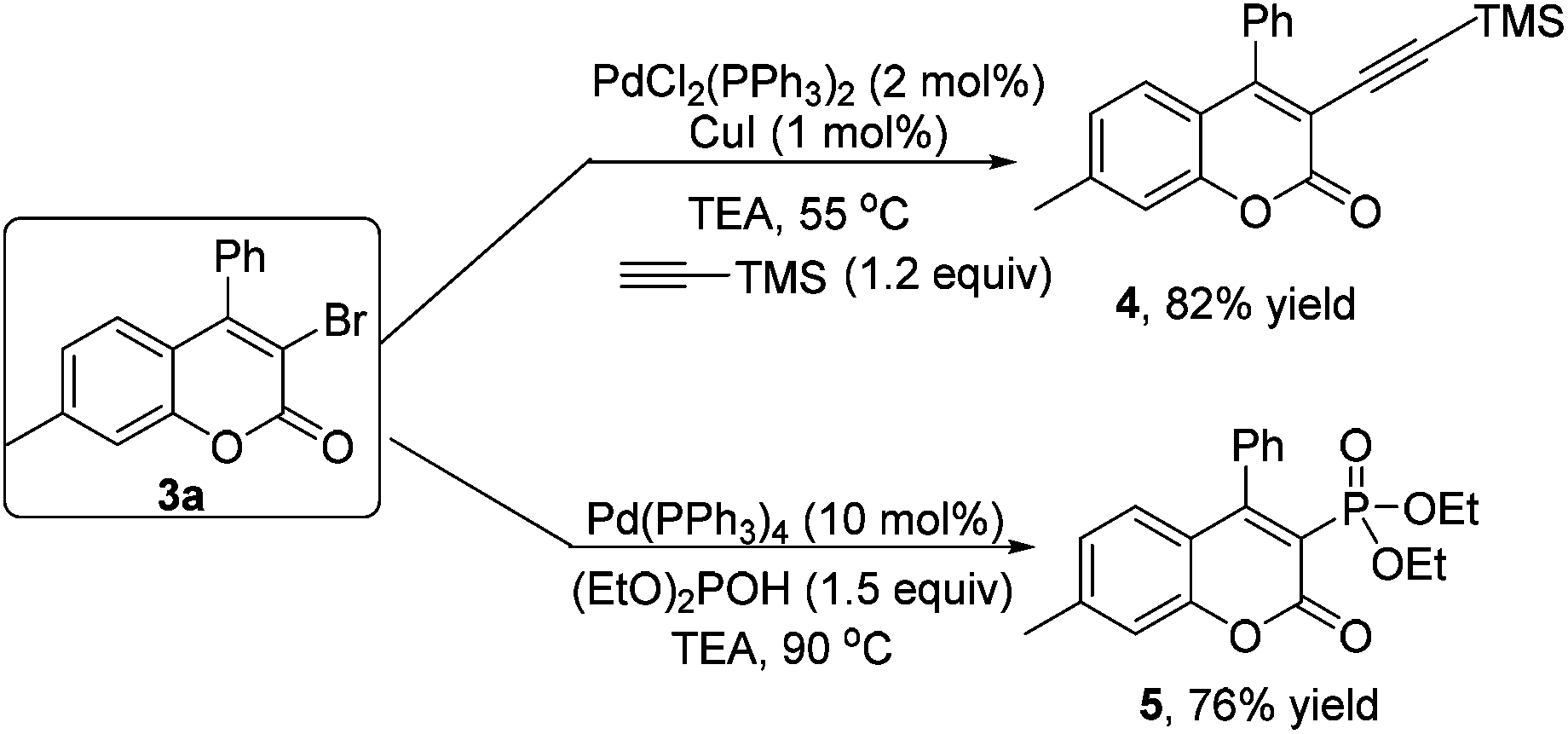
As mentioned above, the bromo group is a versatile building block. Structural elaboration was thus performed. From the results presented in Scheme 2, alkynyl groups and phosphite groups were successfully installed onto the coumarin core through transition-metal-catalyzed coupling reactions in good yields.
In order to expand the application of this radical process, the reaction of N-methyl-N-3-diphenylpropiolamide 1p and TBAB was also carried out. To our surprise, a distinctive spiro [4,5]trienyl-2,8-dione 6 was observed in reasonable yields (Scheme 3). Water from the solvent was incorporated into spiro-[4,5]trienyl-2,8-dione 6.
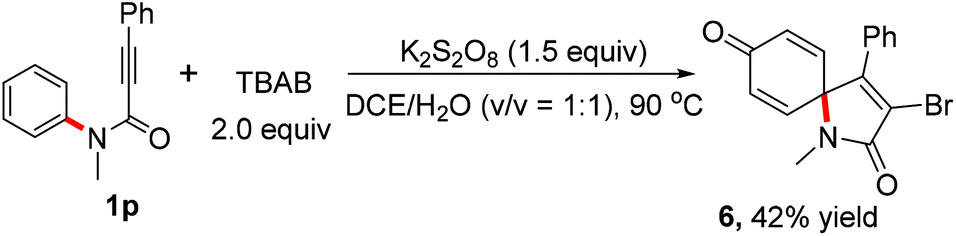 | ||
| Scheme 3 Metal-free based spiro-cyclization of N-methyl-N,3-diphenylpropiolamide 1p and TBAB. Isolated yield based on alkynoates 1. | ||
In light of the above results (especially, the result from entry 16, Table 1), a radical process involving the brominative addition of alkynes, oxidative spiro-cyclization12 and 1,2-ester migration was proposed to account for the formation of 3-bromocoumarins. As illustrated in Scheme 4, a bromo radical was obtained from the treatment of TBAB with K2S2O8.13 The generated bromo radical selectively adds to the α-carbon of propiolate to produce intermediate A. A sequential spiro-cyclization offered intermediate B, which was oxidized to a key intermediate C. 1,2-Ester migration of the intermediate C gave a coumarin cation D. Re-aromatization of the intermediate D provided the desired coumarins 3. The formation of [4,5]trienyl-2,8-dione 6 did not undergo amide migration but cation isomerization of intermediate C, where key intermediate E was formed. Subsequently, intermediate E was trapped by the water from the solvent to give F, which was oxidized to the desired [4,5]trienyl-2,8-dione 6. However, the selective formation of intermediates D and E from the same intermediate C remains to be explained. Related studies are underway in our laboratory.
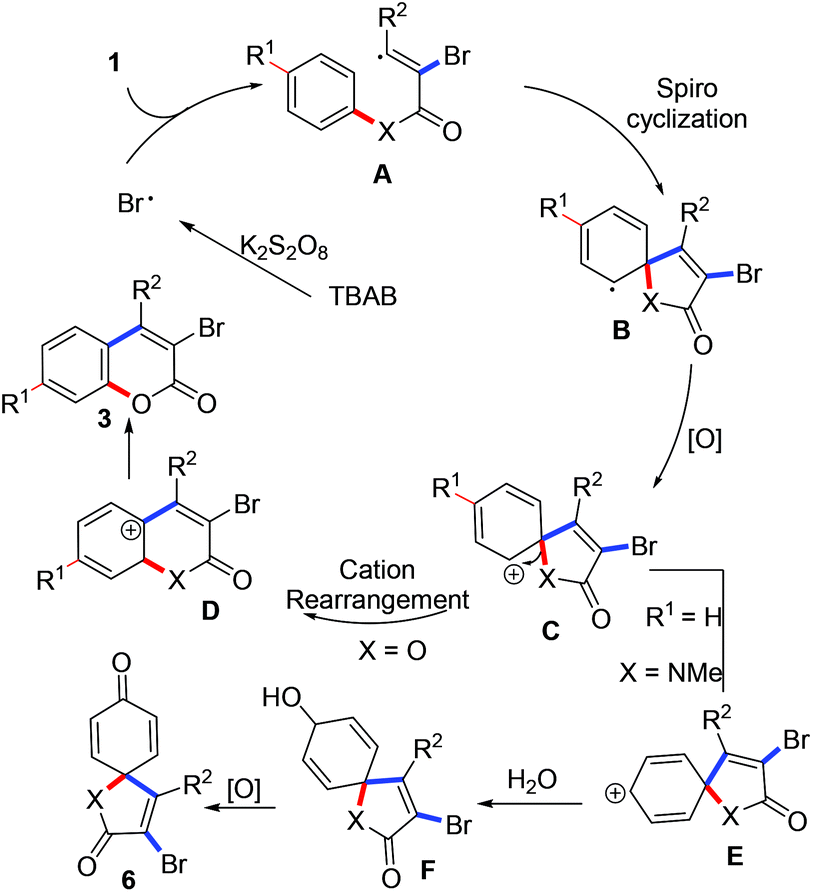 | ||
| Scheme 4 A possible mechanism involving tandem oxidative radical spiro cyclization and carbon cation rearrangement. | ||
In conclusion, we have developed a metal-free and liquid bromine-free protocol for the synthesis of 3-bromocoumarins. In these reactions, tetra-n-butyl ammonium bromide was employed as bromo radical source in the presence of K2S2O8. A preliminary exploration of the mechanism showed that a radical process consisting of a radical brominative addition of alkynoates, oxidative spiro-cyclization and 1,2-migration of esters was involved. Under the same conditions, the reaction of N-methyl-N-3-diphenylpropiolamide 1p and TBAB gave 3-bromo-1-methyl-4-phenyl-1-azaspiro[4.5]deca-3,6,9-triene-2,8-dione 6. More importantly, the prepared 3-bromocoumarins were useful synthons, which were converted into alkynyl, phosphite, aryl and arylamino-substituted coumarins. Applications of this radical process to the synthesis of other heterocycles are ongoing in our laboratory, and the results will be reported in due course.
Experimental section
Alkynoates 1 (0.2 mmol), TBAB 2 (2.0 equiv.), and K2S2O8 (1.5 equiv.) were added into the test tube, and then the solvent mixture DCE/H2O (1:1 v/v, 2 mL) was added. The mixture was stirred at 90 °C overnight. After completion of the reaction as indicated by TLC, the mixture was filtrated, and the filtrate was extracted with EtOAc, and dried by anhydrous Na2SO4. Evaporation of the solvent followed by purification on silica gel provided the products 3 and 6.
Coumarin 3a (0.2 mmol), ethynyltrimethylsilane (1.5 equiv.), CuI (2.5 mol%) and Pd(PPh)3Cl2 (5 mol%) were added into the test tube, and then Et3N was added. The mixture was stirred at 55 °C for 24 h. After completion of reaction as indicated by TLC, evaporation of the solvent followed by purification on silica gel provided the product 4.
Coumarin 3a (0.2 mmol), diethyl phosphonate (2.0 equiv.) and Pd(PPh)4 (10 mol%) were added into a thick-walled pressure bottle, and then Et3N was added. The mixture was stirred at 90 °C for 24 h. After completion of the reaction as indicated by TLC, evaporation of the solvent followed by purification on silica gel provided the product 5.
3-Bromo-7-methyl-4-phenyl-2H-chromen-2-one 3a (78%, 49.0 mg):
1H NMR (400 MHz, CDCl3) δ 7.61–7.50 (m, 3H), 7.30 (d, J = 7.6, 2H), 7.21 (s, 1H), 7.04–6.94 (m, 2H), 2.44 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 157.53, 154.61, 152.47, 143.44, 135.36, 129.22, 128.74, 128.03, 127.26, 125.86, 117.92, 116.86, 111.19, 21.61; HRMS (ESI): m/z [M + Na]+ calcd for C16H11BrNaO2+: 336.9840; found: 336.9835.3-Bromo-7-tert-butyl-4-phenyl-2H-chromen-2-one 3b (75%, 53.4 mg)
1H NMR (400 MHz, CDCl3) δ 7.57–7.51 (m, 3H), 7.42–7.41 (m, 1H), 7.32–7.20 (m, 3H), 7.01 (d, J = 8.5 Hz, 1H), 1.34 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 157.65, 156.71, 154.49, 152.50, 135.40, 129.23, 128.74, 128.06, 127.11, 122.21, 117.86, 113.54, 111.43, 35.24, 30.92; HRMS (ESI): m/z [M + Na]+ calcd for C19H17BrNaO2+: 379.0310; found: 379.0340.7-Benzyl-3-bromo-4-phenyl-2H-chromen-2-one 3c (73%, 56.9 mg)
1H NMR (400 MHz, CDCl3) δ 7.59–7.54 (m, 3H), 7.34–7.24 (m, 6H), 7.20 (d, J = 7.0 Hz, 2H), 7.05–7.02 (m, 2H), 4.06 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 157.41, 154.50, 152.56, 146.52, 139.21, 135.28, 129.25, 128.86, 128.74, 128.70, 128.00, 127.55, 126.61, 125.46, 118.43, 116.77, 111.59, 41.70; HRMS (ESI): m/z [M + Na]+ calcd for C22H15BrNaO2+: 413.0153; found: 413.0148.3-Bromo-7-methoxy-4-phenyl-2H-chromen-2-one 3d (63%, 41.6 mg)
1H NMR (400 MHz, CDCl3) δ 7.59–7.49 (m, 3H), 7.33–7.25 (m, 2H), 6.97 (d, J = 8.9 Hz, 1H), 6.92–6.88 (m, 1H), 6.79–6.71 (m, 1H), 3.88 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 162.89, 157.73, 154.74, 154.18, 135.48, 129.22, 128.74, 128.61, 128.05, 113.94, 112.82, 108.72, 100.59, 55.84; HRMS (ESI): m/z [M + Na]+ calcd for C16H11BrNaO3+: 352.9789; found: 352.9796.3-Bromo-7-fluoro-4-phenyl-2H-chromen-2-one 3e (65%, 41.2 mg)
1H NMR (400 MHz, CDCl3) δ 7.61–7.55 (m, 3H), 7.31–7.29 (m, 2H), 7.15–7.07 (m, 2H), 6.96–6.91 (m, 1H);13C NMR (100 MHz, CDCl3) δ 164.38 (d, 1JCF = 255.3 Hz), 154.14, 153.46 (d, 3JCF = 12.9 Hz), 135.07, 129.51, 129.43, 129.33, 128.94, 127.97, 117.10, 112.81 (d, 2JCF = 22.6 Hz), 111.39, 104.33 (d, 2JCF = 25.8 Hz); HRMS (ESI): m/z [M + Na]+ calcd for C15H8BrFNaO2+: 340.9589; found: 340.9584.3-Bromo-7-chloro-4-phenyl-2H-chromen-2-one 3f (71%, 47.4 mg)
1H NMR (400 MHz, CDCl3) δ 7.63–7.52 (m, 3H), 7.43–7.40 (m, 1H), 7.33–7.26 (m, 2H), 7.20–7.14 (m, 1H), 7.02 (d, J = 8.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 156.71, 153.97, 152.58, 137.95, 134.85, 129.55, 128.97, 128.46, 127.97, 125.28, 118.95, 117.03, 112.54; HRMS (ESI): m/z [M + Na]+ calcd for C15H8BrClNaO2+: 356.9294; found: 356.9288.3,7-Dibromo-4-phenyl-2H-chromen-2-one 3g (68%, 51.4 mg)
1H NMR (400 MHz, CDCl3) δ 7.60–7.53 (m, 4H), 7.32–7.27 (m, 3H), 6.94 (d, J = 8.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 154.06, 152.56, 134.85, 129.59, 129.01, 128.57, 128.16, 128.01, 125.97, 120.04, 119.36, 112.81; HRMS (ESI): m/z [M + Na]+ calcd for C15H8Br2NaO2+: 400.8789; found: 400.8781.3-Bromo-7-iodo-4-phenyl-2H-chromen-2-one 3h (68%, 57.9 mg)
1H NMR (400 MHz, CDCl3) δ 7.79–7.75 (m, 1H), 7.60–7.55 (m, 3H), 7.51 (d, J = 8.4, 1H), 7.30–7.28 (m, 2H), 6.78 (d, J = 8.4 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 156.46, 154.08, 152.11, 134.70, 133.93, 129.52, 128.94, 128.45, 127.95, 125.79, 119.79, 113.00, 97.51; HRMS (ESI): m/z [M + Na]+ calcd for C15H8BrINaO2+: 448.8650; found: 448.8645.7-Acetyl-3-bromo-4-phenyl-2H-chromen-2-one (3i) (69%, 47.2 mg)
1H NMR (400 MHz, CDCl3) δ 7.94–7.92 (m, 1H), 7.76–7.73 (m, 1H), 7.63–7.56 (m, 3H), 7.32–7.29 (m, 2H), 7.19 (d, J = 8.3 Hz, 1H), 2.65 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 196.22, 156.79, 153.71, 152.22, 139.31, 134.74, 129.62, 129.01, 127.97, 123.87, 123.48, 116.65, 115.2, 109.92, 26.77; HRMS (ESI): m/z [M + Na]+ calcd for C17H11BrNaO3+: 364.9789; found: 364.9782.3-Bromo-4-phenyl-2H-chromen-2-one (3j) (80%, 48.0 mg)
1H NMR (400 MHz, CDCl3) δ 7.60–7.55 (m, 4H), 7.42 (d, J = 8.3 Hz, 1H), 7.34–7.30 (m, 2H), 7.23–7.18 (m, 1H), 7.09 (d, J = 8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 157.29, 154.59, 152.41, 135.22, 132.00, 129.31, 128.81, 128.03, 127.57, 124.67, 120.28, 116.76, 112.57; HRMS (ESI): m/z [M + Na]+ calcd for C15H9BrNaO2+: 322.9684; found: 322.9691.3-Bromo-4-cyclopropyl-2H-chromen-2-one (3k) (42%, 22.2 mg)
1H NMR (400 MHz, CDCl3) δ 8.14 (d, J = 8.3 Hz, 1H), 7.60–7.51 (m, 1H), 7.39–7.29 (m, 2H), 1.99–1.85 (m, 1H), 1.43–1.32 (m, 2H), 0.95–0.87 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 157.42, 153.51, 151.91, 131.59, 125.54, 124.42, 120.64, 116.89, 115.53, 14.43, 9.39; HRMS (ESI): m/z [M + Na]+ calcd for C12H9BrNaO2+: 286.9684; found: 286.9678.3-Bromo-4-p-tolyl-2H-chromen-2-one (3l) (67%, 42.1 mg)
1H NMR (400 MHz, CDCl3) δ 7.59–7.55 (m, 1H), 7.42–7.37 (m, 3H), 7.22–7.18 (m, 3H), 7.13 (d, J = 8.0 Hz, 1H), 2.48 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 157.35, 154.76, 152.38, 139.39, 132.24, 131.92, 129.45, 127.99, 127.66, 124.60, 120.38, 116.71, 112.51, 21.40; HRMS (ESI): m/z [M + Na]+ calcd for C16H11BrNaO2+: 336.9840; found: 336.9835.3-Bromo-4-(4-fluorophenyl)-2H-chromen-2-one (3m) (64%, 40.7 mg)
1H NMR (400 MHz, CDCl3) δ 7.61–7.51 (m, 1H), 7.43 (d, J = 8.3 Hz, 1H), 7.35–7.20 (m, 5H), 7.09 (d, J = 8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 163.10 (d, 1JCF = 250.0 Hz), 153.65, 152.44, 132.16, 131.11, 130.28, 130.20, 127.35, 124.78, 120.23, 116.92, 116.15 (d,2JCF = 21.9 Hz), 113.10; m/z [M + Na]+ calcd for C15H8BrFNaO2+: 340.9589; found: 340.9582.3-Bromo-4-(4-chlorophenyl)-2H-chromen-2-one (3n) (71%, 53.7 mg)
1H NMR (400 MHz, CDCl3) δ 7.60–7.55 (m, 3H), 7.42 (d, J = 8.3 Hz, 1H), 7.28–7.25 (m, 2H), 7.24–7.19 (m, 1H), 7.07 (d, J = 8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 157.07, 153.43, 152.44, 135.59, 133.54, 132.22, 129.62, 129.27, 127.27, 124.82, 120.00, 116.94, 112.91; HRMS (ESI): m/z [M + Na]+ calcd for C15H8BrClNaO2+: 356.9294; found: 356.9288.3-Bromo-4-(2-methoxyphenyl)-2H-chromen-2-one (3o) (76%, 50.2 mg)
1H NMR (400 MHz, CDCl3) δ 7.57–7.51 (m, 2H), 7.40 (d, J = 8.3 Hz, 1H), 7.21–7.14 (m, 3H), 7.10 (d, J = 8.4 Hz, 1H), 7.05 (d, J = 8.0 Hz, 1H), 3.78 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 157.46, 155.79, 152.74, 152.33, 131.69, 131.00, 129.20, 127.20, 124.56, 124.11, 120.86, 120.22, 116.64, 113.57, 111.48, 55.65; HRMS (ESI): m/z [M + Na]+ calcd for C16H11BrNaO3+: 352.9789; found: 352.9786.7-Methyl-4-phenyl-3-((trimethylsilyl)ethynyl)-2H-chromen-2-one (4) (82%, 54.4 mg)
1H NMR (400 MHz, CDCl3) δ 7.52–7.50 (m, 3H), 7.42–7.39 (m, 2H), 7.18 (s, 1H), 7.12 (d, J = 8.2 Hz, 1H), 7.01 (d, J = 7.5 Hz, 1H), 2.45 (s, 3H), 0.04 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 159.68, 157.87, 152.92, 143.70, 134.30, 129.12, 128.77, 128.24, 128.04, 127.34, 125.66, 117.06, 109.66, 104.68, 98.30, 21.63, −0.56; HRMS (ESI): m/z [M + Na]+ calcd for C21H20NaO2Si+: 355.1130; found: 355.1125.Diethyl 7-methyl-2-oxo-4-phenyl-2H-chromen-3-ylphosphonate (5) (76%, 64.1 mg)
1H NMR (400 MHz, CDCl3) δ 7.44–7.43 (m, 3H), 7.26–7.25 (m, 2H), 7.10 (s, 1H), 6.92–6.84 (m, 2H), 4.08–3.96 (m, 2H), 3.81–3.89 (m, 2H), 2.39 (s, 3H), 1.07 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 162.32 (d, J = 6.6 Hz), 159.02 (d, J = 16.3 Hz), 153.76, 145.21, 135.01 (d, J = 4.8 Hz), 128.62, 128.58, 127.91, 127.77, 125.40, 117.49 (d, J = 14.2 Hz), 116.57, 115.02 (d, J = 201.0 Hz), 62.40 (d, J = 6.0 Hz), 21.50, 15.89 (d, J = 6.3 Hz); 31P NMR (162 MHz, CDCl3) δ 11.06; HRMS (ESI): m/z [M + Na]+ calcd for C20H21NaO5P+: 395.1024; found: 395.1019.1-Acetyl-3-bromo-4-phenyl-1-azaspiro[4.5]deca-3,6,9-triene-2,8-dione (6) (42%, 30.4 mg)
1H NMR (400 MHz, CDCl3) δ 7.48–7.35 (m, 3H), 7.23–7.17 (m, 2H), 6.56 (d, J = 10.1 Hz, 2H), 6.40 (d, J = 10.6 Hz, 2H), 2.64 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 183.69, 168.50, 164.41, 156.22, 142.81, 132.49, 130.53, 128.57, 128.11, 119.15, 68.49, 25.59; HRMS (ESI): m/z [M + Na]+ calcd for C17H13BrNO3+: 358.0079; found: 358.0072.Acknowledgements
Financial support from the National Natural Science Foundation of China (no. 21502069) and the Natural Science Foundation of Zhejiang Province (No. LQ15B020006) is gratefully acknowledged.Notes and references
- For selected examples, see: (a) B. Naser-Hijazi, B. Stolze and K. Zanker, Second Proceedings of the International Society of Coumarin Investigators, Springer, Berlin, 1994 Search PubMed; (b) L. Santana, E. Uriarte, F. Roleira, N. Milhazes and F. Borges, Curr. Med. Chem., 2004, 11, 3239 CrossRef CAS PubMed; (c) F. Borges, F. Roleira, N. Milhazes, L. Santana and E. Uriarte, Curr. Med. Chem., 2005, 12, 887 CrossRef CAS PubMed; (d) B. M. Trost and F. Tost, J. Am. Chem. Soc., 1996, 118, 6305 CrossRef CAS; (e) X. Peng, G. Damu and C. Zhou, Curr. Pharm. Des., 2013, 19, 3884 CrossRef CAS PubMed.
- For selected reviews of the synthesis of coumarins, see: (a) R. Vekariya and H. Patel, Synth. Commun., 2014, 44, 2756 CrossRef CAS; (b) S. Trenor, A. Shulty, B. Love and T. Long, Chem. Rev., 2004, 104, 3059 CrossRef CAS PubMed; (c) S. Sethna and N. Shah, Chem. Rev., 1945, 36, 1 CrossRef CAS.
- S. Sethna and R. Phadke, Organomet. React., 1953, 7, 1 Search PubMed.
- For selected examples see: (a) B.-M. Trost and F.-D. Toste, J. Am. Chem. Soc., 1996, 118, 6305 CrossRef CAS; (b) J. Ferguson, F. Zeng and H. Alper, Org. Lett., 2012, 14, 5602 CrossRef CAS PubMed; (c) M. Amézquita-Valencia and H. Alper, Org. Lett., 2014, 16, 5827 CrossRef PubMed.
- For selected reviews, see: (a) P. S. Skell and J. G. Traynham, Acc. Chem. Res., 1984, 15, 160 CrossRef; (b) M. F. Ruass, Acc. Chem. Res., 1990, 23, 87 CrossRef; (c) C. J. Easton and C. A. Hutton, Synlett, 1998, 457 CrossRef CAS.
- For selected reviews, see: (a) R. S. Brown, Acc. Chem. Res., 1997, 30, 131 CrossRef CAS; (b) N. Suryakiran, Synlett, 2008, 1267 CrossRef CAS; (c) S.-H. Yang, Synlett, 2009, 1351 CAS; (d) G. Qiu, Y. Kuang and J. Wu, Adv. Synth. Catal., 2014, 356, 3483 CrossRef CAS.
- For selected examples, see: (a) G. Zhu and Z. Zhang, J. Org. Chem., 2005, 70, 3339 CrossRef CAS PubMed; (b) X.-C. Huang, F. Wang, Y. Liang and J.-H. Li, Org. Lett., 2009, 11, 1139 CrossRef CAS PubMed; (c) X. Lu, Handbook of Organopalladium Chemistry for Organic Synthesis, John Wiley & Sons, Inc, 2002, vol. 2, p. 2267 Search PubMed; (d) X. Lu, Z. Wang and A. Lei, Current Trends In Organic Synthesis, Proceedings of the international Conference on Organic Synthesis, 12th, Venezia, June 28–July 2, 1998, 275.
- For selected examples, see: (a) S. S. Soman and T. H. Thaker, Med. Chem. Res., 2013, 22, 4223 CrossRef CAS; (b) T. H. V. Huynh, B. Abrahamsen, K. K. Madsen, A. Franquesa-Gonzalez, A. A. Jensen and L. Bunch, Bioorg. Med. Chem., 2012, 20, 6831 CrossRef CAS PubMed; (c) L. Meimetis, J. Carlson, R. Giedt, R. Kohler and R. Weissleder, Angew. Chem., Int. Ed., 2014, 53, 7531 CrossRef CAS PubMed; (d) M.-S. Schiedel, C. A. Briehn and P. Bauerle, Angew. Chem., Int. Ed., 2001, 40, 4677 CrossRef CAS; (e) L. Zhang, T. Meng, R. Fan and J. Wu, J. Org. Chem., 2007, 72, 7279 CrossRef CAS PubMed.
- For selected examples, see: (a) K.-M. Kim and I.-H. Park, Synthesis, 2004, 2641 CrossRef CAS; (b) G. V. Ramanarayanan, V. G. Shukla and K. G. Akamanchi, Synlett, 2002, 2059 CAS; (c) M. A. Kumar, C. N. Rohitha, S. J. Kulkarni and N. Narender, Synthesis, 2010, 1629 CAS; (d) C.-G. Cho, J.-S. Park, I.-H. Jung and H. Lee, Tetrahedron Lett., 2001, 42, 1065 CrossRef CAS; (e) R. M. Kelkar, U. K. Joshi and M. V. Paradkar, Synthesis, 1986, 214 CrossRef CAS.
- (a) T. Liu, Q. Ding, Q. Zong and G. Qiu, Org. Chem. Front., 2015, 2, 670 RSC; (b) T. Liu, Q. Ding, G. Qiu and J. Wu, Tetrahedron, 2016, 72, 279 CrossRef CAS.
- (a) W. Yang, S. Yang, P. Li and L. Wang, Chem. Commun., 2015, 51, 7520 RSC; (b) W. Fu, M. Zhu, G. Zou, C. Xu, Z. Wang and B. Ji, J. Org. Chem., 2015, 80, 4766 CrossRef CAS PubMed; (c) K. Yan, D. Yang, W. Wei, F. Wang, Y. Shuai, Q. Li and H. Wang, J. Org. Chem., 2015, 80, 1550 CrossRef CAS PubMed; (d) W. Wei, J. Wen, D. Yang, M. Guo, Y. Wang, J. You and H. Wang, Chem. Commun., 2015, 51, 768 RSC; (e) Y. Li, Y. Lu, G. Qiu and Q. Ding, Org. Lett., 2014, 16, 4240 CrossRef CAS PubMed; (f) X. Mi, C. Wang, M. Huang, J. Zhang, Y. Wu and Y. Wu, Org. Lett., 2014, 16, 3356 CrossRef CAS PubMed.
- For selected examples of electrophilic spiro-cyclizations of alkynoates, see: (a) T. Okitsu, D. Nakazawa, A. Kobayashi, M. Mizohata, Y. In, T. Ishida and A. Wada, Synlett, 2010, 203 CAS; (b) X. Zhang and R. C. Larock, J. Am. Chem. Soc., 2005, 127, 12230 CrossRef CAS PubMed; (c) T. Dohi, D. Kato, R. Hyodo, D. Yamashita, M. Shirao and Y. Kita, Angew. Chem., Int. Ed., 2011, 50, 3784 CrossRef CAS PubMed; (d) B.-X. Tang, D.-J. Tang, S. Tang, Q.-F. Yu, Y.-H. Zhang, Y. Liang, P. Zhong and J.-H. Li, Org. Lett., 2008, 10, 1063 CrossRef CAS PubMed; (e) B.-C. Tang, Y.-H. Zhang, R.-J. Song, D.-J. Tang, G.-B. Deng, Z.-Q. Wang, Y.-X. Xie, Y.-Z. Xia and J.-H. Li, J. Org. Chem., 2012, 77, 2837 CrossRef CAS PubMed; (f) B.-X. Tang, Q. Yin, R.-Y. Tang and J.-H. Li, J. Org. Chem., 2008, 73, 9008 CrossRef CAS PubMed; (g) J. Wen, W. Wei, S. Xue, D. Yang, Y. Lou, C. Gao and H. Wang, J. Org. Chem., 2015, 80, 4966 CrossRef CAS PubMed; (h) Y. Chen, X. Liu, M. Lee, C. Huang, I. Inoyatov, Z. Chen, A. C. Perl and W. H. Hersh, Chem. – Eur. J., 2013, 19, 9795 CrossRef CAS PubMed; (i) L.-J. Wang, A.-Q. Wang, Y. Xia, X.-X. Wu, X.-Y. Liu and Y.-M. Liang, Chem. Commun., 2014, 50, 13998 RSC; (j) W.-T. Wei, R.-J. Song, X.-H. Ouyang, Y. Li, H.-B. Li and J.-H. Li, Org. Chem. Front., 2014, 1, 484 RSC; (k) H. Cui, W. Wei, D. Yang, J. Zhang, Z. Xu, J. Wen and H. Wang, RSC Adv., 2015, 5, 84657 RSC.
- (a) K. Moriyama, Y. Nakamura and H. Togo, Org. Lett., 2014, 16, 3812 CrossRef CAS PubMed; (b) U. Dutta, A. Deb, D. Lupton and D. Maiti, Chem. Commun., 2015, 51, 17744 RSC.
- CCDC 1439381 contains the supplementary crystallographic data for this paper.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, characterization data, 1H and 13C NMR spectra of compounds 3. CCDC 1439381 (compound 3a). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00041j |
| This journal is © the Partner Organisations 2016 |